


Abstract
The activation of the complement cascade, a cornerstone of the innate immune response, produces a number of small (74–77 amino acid) fragments, originally termed anaphylatoxins, that are potent chemoattractants and secretagogues that act on a wide variety of cell types. These fragments, C5a, C4a, and C3a, participate at all levels of the immune response and are also involved in other processes such as neural development and organ regeneration. Their primary function, however, is in inflammation, so they are important targets for the development of anti-inflammatory therapies. Only three receptors for complement peptides have been found, but there are no satisfactory antagonists as yet, despite intensive investigation. In humans, there is a single receptor for C3a (C3a receptor), no known receptor for C4a, and two receptors for C5a (C5a1 receptor and C5a2 receptor). The most recently characterized receptor, the C5a2 receptor (previously known as C5L2 or GPR77), has been regarded as a passive binding protein, but signaling activities are now ascribed to it, so we propose that it be formally identified as a receptor and be given a name to reflect this. Here, we describe the complex biology of the complement peptides, introduce a new suggested nomenclature, and review our current knowledge of receptor pharmacology.
I. Introduction
A. Production of Complement Peptides
Complement is a vital part of the host defense system, capable of reacting to foreign material and damaged or altered host tissues (Carroll and Sim, 2011). Complement-like genes are present in organisms that diverged more than 1.3 billion years ago (reviewed in Pinto et al., 2007); animals that are incapable of mounting an adaptive immune response possess complement, including the sea-squirt Ciona intestinalis (Pinto et al., 2003) and the “living fossil” horseshoe crab, Carcinoscorpius rotundica (Zarkadis et al., 2001). The complement system consists of a network of soluble and cell-surface proteins that can recognize potential threats and then undergo an amplification phase to produce a response of sufficient proportions to neutralize the perceived danger. The overall response is a balance between positive and negative regulators that allows very fine control of what is a potentially hazardous system: misdirected or excessive activation of complement can be rapidly lethal to the host.
There are three major pathways for the initiation of a complement response (Fig. 1). The first or classic pathway usually requires the formation of immune complexes of IgM or IgG1 antibodies. The closely related second or lectin pathway relies on the direct recognition of foreign material by a series of soluble pattern-recognition receptors. The third or alternative pathway depends on the continual turnover of one component, C3. When complement activation occurs in the fluid phase or proximal to host tissues, inhibitory factors such as CD59 or decay-accelerating factor ensure that no further complement response happens. However, on a receptive surface lacking the appropriate control factors (microbes or xenografts, for example), complement activation is greatly magnified to become a full-blown response even in the absence of any positive triggers.

Pathways for the production of complement peptides. The three major routes of complement activation and the extrinsic pathways for the direct production of complement peptides by protease activities are shown.
The complement cascade is based on a series of proteolytic events, with inactive complement proteins successively cleaved to form the next active protease in the chain. The terminal event is the formation of the membrane attack complex (MAC), a lytic pore in the membrane that can cause the death of some cell types. The MAC is inefficient at the cytolysis of nucleated cells, primarily because of defense mechanisms that prevent MAC formation or remove MAC from the cell surface (Tegla et al., 2011). Sublytic MAC, however, is a proinflammatory stimulus for some cell types (reviewed in Dobrina et al., 2002) that has also been linked with the control of hemopoiesis (Ratajczak et al., 2010). The large pool of precursor complement proteins allows a rapid and sizeable response to detected threats; conversely, any failure to control the complement system can lead to inflammatory disease. As a result of proteolysis, a series of small, biologically active protein fragments are produced: C3a and C5a from all three pathways and C4a primarily from the classic pathway. These protein fragments are rapidly metabolized by carboxypeptidases (Section I.D), forming des-arginated fragments (Burger and Zilow, 1993). Other fragments—for example, derived from the cleavage of C2 or factor B—are also produced, but these are structurally unrelated to C3a, C4a, and C5a (Krishnan et al., 2009) and are outside the scope of this review.
B. Concentrations of Complement Peptides in Health and Disease
Determining the “physiologic” levels of complement peptides is problematic, with wide variations in the reported values (Table 1). This is due in part to the different assay techniques and in part to the nature of the biologic sample tested. Serum, obtained after clotting of plasma, generally contains higher levels of the fragments due to the actions of the proteases in the clotting cascade on C3, C4, and C5 (Amara et al., 2008) (see Section I.C). Thus, plasma is a more reliable indicator of circulating complement peptide levels, particularly where EDTA has been used, effectively blocking all three major routes of complement activation. Plasma concentrations in healthy human subjects have been reported to be 119, 219, and 5.2 ng/ml for C3a, C4a, and C5a, respectively (Table 1). It is likely that all these studies are reporting the des-arginated forms of the fragments because the assay methods cannot discriminate between the two forms. In addition, neoepitope-specific antibodies, which can discriminate between C3 and its cleavage product, detect usually the des-arginated form of C3a with higher sensitivity. Complement peptide levels are clearly elevated in inflammatory diseases and even in pregnancy (Table 1). Elevation of C3a but not C4a suggests activation of the alternative pathway, which does not involve the proteolysis of C4, whereas very high levels of both C4a and C3a suggest that both the classic/lectin and alternative pathways are constantly in operation. Both C5a and C5a des-Arg can be rapidly cleared by receptor-mediated endocytosis (Oppermann and Gotze, 1994), unlike C4a des-Arg and C3a des-Arg, which therefore accumulate to much higher levels.
Complement peptide concentrations in normal and diseased states
Reported concentrations of complement peptides are shown as nanogram per milliliter.
C. C3a and C5a Generation outside the Complement Cascade
Although the production of complement peptides is usually linked to the activation of the complement cascade, biologically relevant amounts are also generated by the so-called extrinsic pathways. This was first reported in 1968, when proteolysis of C5 by trypsin was observed to produce anaphylatoxic activity (Cochrane and Muller-Eberhard, 1968). This type of activation is now known to be widespread. Gingipain-1, a cysteine proteinase from Porphyromonas gingivalis, cleaves C3 and C5 to produce leukocyte chemoattractant activities that strongly resemble C3a and C5a (Wingrove et al., 1992), albeit with higher molecular weights than predicted for these fragments. Extrinsic pathway activation may be a common feature of many pathogenic bacteria, being exploited by unrelated organisms such as Tanarella forsythia (Jusko et al., 2012) and Aeromonas sobria (Nitta et al., 2008). For P. gingivalis, the production of C5a has been proposed to inhibit production mediated by Toll-like receptor 2 (TLR2) interleukin-12 (IL-12), allowing escape from immune clearance (Liang et al., 2011). The feces of the house dust mite Dermatophagoides farinae contain a protease, DerP1, which can generate active complement peptides from C3 and C5 (Maruo et al., 1997). Asbestos and silica can also cause the cleavage of C5 (Governa et al., 2000, 2002, 2005). More recently, cross-talk between the complement and the coagulation cascades has been demonstrated to result in the robust generation of C3a and C5a, through the actions of factors Xa/XIa, plasmin, and thrombin (Amara et al., 2010). Factor VII–activating protease can also activate C3 and C5 (Kanse et al., 2012), producing C3a identical to that formed by complement cascade activation but with the N-terminal 4 residues missing from C5a. Enzymes released by activated or damaged host cells can also cause C3a and C5a generation: the pro-apoptotic aspartic acid protease cathepsin D, elevated in trauma (Huber-Lang et al., 2012); β-tryptase, secreted by mast cells (Fukuoka et al., 2008); and granzyme B, produced by leukocytes (Perl et al., 2012). The relative importance of these fragment-generation systems that lie outside the complement cascade is not yet clear. The increasing availability of inhibitors of complement peptide generation (Qu et al., 2009; Woodruff et al., 2011) that inhibit either common pathways (e.g., compstatin, which acts on C3) or more specific pathways (e.g., eculizumab, which acts on C5 proteolysis) will help to answer this question.
D. Deactivation of Complement Peptides
Human serum contains a potent deactivator of complement peptides, shown to be a carboxypeptidase B–like activity that removes the C-terminal Arg residues of C3a and C5a (Bokisch et al., 1969; Bokisch and Muller-Eberhard, 1970). Inhibition by a carboxypeptidase inhibitor, dl-2-mercaptomethyl-3-guanidinoethylthiopropanoic acid, made administration of ordinarily survivable doses of cobra venom factor (CVF) or C3a lethal (Huey et al., 1983). Two major carboxypeptidases control the activity of the fragments. The zinc metalloprotease carboxypeptidase N (CPN) is released in an active form (Levin et al., 1982), and carboxypeptidase R (CPR) is an acute-phase protein, up-regulated in inflammation and secreted in an inactive form, proCPR (reviewed in Campbell et al., 2001). ProCPR is bound to plasminogen and activated by plasmin or thrombin (Wang et al., 1994; Sato et al., 2000; Nishimura et al., 2007; Leung et al., 2008). It preferentially degrades C5a over C3a (Campbell et al., 2002) and can remove Lys residues from fibrin clots, preventing plasminogen binding (Bajzar et al., 1995). CPR is also known as plasma carboxypeptidase B, activated thrombin-activatable fibrinolysis inhibitor (TAFI), and carboxypeptidase U (Campbell et al., 2001). CPB is a more active carboxypeptidase than CPN and also acts on bradykinin and osteopontin as substrates. TAFI carboxypeptidase is protective in allergic asthma (Fujiwara et al., 2012) and down-regulates inflammation in rheumatoid arthritis (RA) (Song et al., 2011). Both CPR and CPN can also inactivate C3a and C5a octapeptides (Section III.C.4) (Campbell et al., 2002).
Defense against host responses by microorganisms can also involve complement peptide degradation. Brugia malayi and Trichinella spiralis, parasitic nematodes, release a metallocarboxypeptidase that inactivates C5a (Rees-Roberts et al., 2010), presumably to prevent eosinophil chemotaxis and activation. Streptococcus pyogenes produces a C5a peptidase (Wexler et al., 1985) that cleaves between His67 and Lys68 (Cleary et al., 1992). This peptidase, ScpA, is a cell wall–anchored serine protease that is important for virulence (Husmann et al., 1997). The virulence of the enterobacterium Serratia marcescens, depends in part on the activity of a 56-kDa protease that can inactivate C5a, thus inhibiting neutrophil influx (Oda et al., 1990).
II. The Role of Complement Peptides in Pathophysiology
There is a very fine balance that must be maintained whenever the complement cascade is activated because of the destructive nature of the effector systems: too little may result in incomplete clearance of immune complexes, leading to autoimmune diseases such as systemic lupus erythematosus (SLE) and failure to control infections, but too much causes damage to healthy tissues (Carroll and Sim, 2011). The uncontrolled or inappropriate production of complement peptides has been implicated in many inflammatory disorders, and, although it is beyond the scope of this review to discuss the roles of C3a and C5a in any detail, a list of these disorders is shown in Tables 2 and 3. It should also be noted that C3a and C5a do not always act as partners in crime, both promoting inflammation. In the development of asthma, the generation of C5a during sensitization appears to inhibit the further development of lung disease whereas C3a promotes disease progression, through skewing of the subsequent T-cell response. However, both fragments have deleterious effects at later stages (reviewed in Wills-Karp, 2007). C4a has no known role in human disease.
Pathologic conditions involving complement peptide C3a
Pathologic conditions involving complement peptide C5a
A. Complement Peptides Are Important Biomarkers of Disease
The stability of C3a des-Arg and C4a des-Arg has made these complement peptides in particular potentially useful as biomarkers in a number of disorders, even those not traditionally seen as inflammatory in nature such as cancer. Examples are the increased serum levels of C3a that indicate the presence of colorectal tumors (Habermann et al., 2006), hepatitis C virus–related hepatocellular carcinoma (Lee et al., 2006a; Kanmura et al., 2010), benign prostatic hyperplasia (Xie et al., 2011), and ductal carcinoma in situ of the breast (Solassol et al., 2010). C4a has recently been shown to be elevated in patients with chronic hepatitis C infection and active disease but is even higher in infected but asymptomatic individuals (Imakiire et al., 2012). Both C4a and C3a levels are predictive of the responses of esophageal cancer patients to chemoradiation (Maher et al., 2011). In more obviously inflammatory disorders, C3a (and to a lesser extent C4a) have been shown to be potentially useful markers in dermatomyositis (Campo et al., 2007), aneurysmal subarachnoid hemorrhage (Mack et al., 2007), acute Lyme disease in tick-bite patients (Shoemaker et al., 2008; Stricker et al., 2009), the exudative form of age-related macular degeneration (Machalinska et al., 2009), endometriosis (Fassbender et al., 2009), adverse pregnancy outcomes (Lynch et al., 2011), chronic obstructive pulmonary disease (Marc et al., 2004; Zhang et al., 2011), cryptogenic and large-vessel disease subtypes of stroke (Stokowska et al., 2011), heart failure (Gombos et al., 2012), cerebral arteriovenous malformations (Haque et al., 2011), asthma (Joks et al., 2008), gestational diabetes mellitus (Lappas, 2011), SLE (Wild et al., 1990), acute relapses in multiple sclerosis (Ingram et al., 2010), IgA nephropathy/Henoch–Schonlein nephritis (Abou-Ragheb et al., 1992) and impaired renal function (Abou-Ragheb et al., 1991), atopic dermatitis (Sergeev Iu et al., 1989), psoriasis (Takematsu et al., 1986) and psoriatic arthritis (Muto et al., 1991), idiopathic pulmonary arterial hypertension (Abdul-Salam et al., 2006), postexercise malaise in myalgic encephalomyelitis/chronic fatigue syndrome (Nijs et al., 2010), AIDS-associated retinitis (Mondino et al., 1990), and grafted corneas (Mondino and Sumner, 1990). In addition, C4a and C5a levels decrease after liver resection whereas C3a levels increase (Strey et al., 2009); and C3a and C4a are elevated in liver transplant recipients (Pfeifer et al., 2000). Finally, elevated C3a and C4a in sepsis are associated with a fatal outcome, whereas C5a levels are not correlated (Hack et al., 1989).
B. Functions of the Complement Peptides beyond Innate Immunity
The complement system has evolved a number of nonimmunologic functions. Homeostasis is maintained by the action of complement on cellular debris, which is important in the prevention of autoimmune responses. In development, complement has roles in bone metabolism, hemopoiesis, angiogenesis, and tissue repair. Complement is also involved in liver and lens regeneration (reviewed in Rutkowski et al., 2010a). This range of activities makes the complement system and its fragments important players in neoplasia (Rutkowski et al., 2010b) and a wide variety of other functions, some of which we will detail.
1. Cell Migration and Homing
In the central nervous system, C3a (and to a lesser extent C5a) is active during the development of the rat cerebellum (Benard et al., 2008) and the in vitro differentiation and migration of neural progenitor cells (Shinjyo et al., 2009). C3a is known to be the coattractant that organizes neural crest cells when they migrate during early development (Carmona-Fontaine et al., 2011). The C3a receptor (C3a receptor, also known as C3aR; see Section IV.B) is a key mediator of insulin resistance and functions by modulating macrophage infiltration and activation in adipose tissue (Mamane et al., 2009). The homing of hemopoietic stem and progenitor cells to bone marrow also depends on the C3a-C3a receptor axis (Reca et al., 2003).
2. Adaptive Immunity
In addition to well-known roles in innate immunity, the complement peptides also act to modulate adaptive immunity (Chenoweth et al., 1982; Ottonello et al., 1999). The presence of complement peptide receptors on certain subsets of B and T cells has been reported (Martin et al., 1997; Werfel et al., 2000). Expression levels are much higher on antigen-presenting cells (Sacks, 2010). C3a and C5a produced by dendritic cells (DC) are required for optimal CD4 T-cell help for CD8 T cells in murine allograft rejection (Vieyra et al., 2011). Similarly, locally produced C5a and C3a provide costimulatory and survival signals for naive CD4+ T cells (Strainic et al., 2008), and γδT-cell function in sepsis can be modulated by C5a (Han et al., 2011) due to increased production of IL-17 after blockade of DC C5a receptor (C5a1 receptor, also known as C5R1, CD88; Section IV.C) (Xu et al., 2010a). Conversely, preventing C5a stimulation of DC by ablation of the C5a1 receptor or with a receptor antagonist induces Treg and TH17 cells by increasing the production of transforming growth factor-β (TGF-β) (Weaver et al., 2010). In experimental autoimmune encephalomyelitis, a model of multiple sclerosis, interferon-γ (IFN-γ) and IL-17 production in autoreactive T cells depends on local production of C3a and C5a (Liu et al., 2008). C5a-mediated TH17 differentiation has been proposed to underlie some autoimmune and inflammatory disorders such as autoimmune arthritis (Hashimoto et al., 2010), and blockade of this pathway may be beneficial for the control of these diseases. However, C5a can negatively regulate TH17 cell differentiation in asthma (Lajoie et al., 2010), so this therapeutic strategy may be of limited use. In contrast to the many reports of DC activation by complement peptides, T-cell-expressed C5a1 receptor is required for enhanced T-cell expansion, as a result of inhibition of apoptosis (Lalli et al., 2008).
3. Hemopoiesis
In contrast to the effects of C3a on stem-cell homing to bone marrow as mentioned previously, C5a and C5a des-Arg disrupt the CXCL12 (SDF-1α)/CXCR4 axis and increase the mobilization of hemopoietic stem and progenitor cells (Jalili et al., 2010). Mobilization of these cells is also impaired in C5-deficient mice (Lee et al., 2009). The roles of complement peptides in stem-cell mobilization have been reviewed elsewhere (Ratajczak et al., 2012).
4. Regeneration and Other Functions
Human mesenchymal stem cells (MSC), which are involved in the repair of various tissues, express complement peptide receptors and are chemoattracted to C3a and C5a (Schraufstatter et al., 2009). Interestingly, MSC have been found to modulate innate immunity by activating the complement cascade (Moll et al., 2011). Complement C3a and C5a can modulate bone biology in inflammation (Ignatius et al., 2011b). C5a1 receptor has been shown to control osteoblast migration during fracture healing (Ignatius et al., 2011a), and efficient osteoclast differentiation requires local complement activation (Ignatius et al., 2011b). Normal heart function appears to depend on the C5a/C5a1 receptor axis, with C5 deficiency and receptor knockout or blockade causing a “state of distress” (Mullick et al., 2011). C3a also has positive effects on food intake regulation, with the administration of C3a to the central nervous system leading to suppression of appetite (Ohinata et al., 2002, 2007, 2009; Ohinata and Yoshikawa, 2008).
III. Structure of Complement Peptides
Complement components C3, C4, and C5 are thought to have arisen by gene duplication events predating the emergence of cartilaginous fish (Terado et al., 2003). It is therefore not surprising that the complement peptides derived from these proteins all have similar primary structures, with 74 to 79 amino acids (Fig. 2, A–C) and largely conserved Cys residues that suggest similar patterns of disulfide bridges in the tertiary structures. Nuclear magnetic resonance (NMR) and X-ray crystallographic studies have confirmed that the three-dimensional structures of C3a (Nettesheim et al., 1988) and C5a (Zuiderweg et al., 1988) are very similar overall.

Complement peptide sequence alignments. Complement peptides were aligned using ClustalW (Larkin et al., 2007) in JalView (Waterhouse et al., 2009). Conserved residues are colored according to physicochemical properties (http://www.ebi.ac.uk/Tools/msa/clustalw2/help/faq.html#24).
A. C3a
1. Sequence
All C3a sequences found to date have six Cys residues (numbered 23, 24, 37, 50, 57, and 58 in guinea pig C3a) (Gerard et al., 1988), apart from the deuterostome, C. intestinalis, where the outermost pair of Cys residues are missing, implying that it has only two disulfide bridges (Fig. 2A). It is noteworthy that the disulfide bridges in human C3a appear to be unusually labile (Chang et al., 2008), a phenomenon that may provide additional regulation of the activity of C3a in vivo. Guinea pig C3a has 70% identity with rat (Jacobs et al., 1978), human, porcine (Corbin and Hugli, 1976), and mouse (Hugli et al., 1975b), and C3a from these species have identical activity in smooth muscle contraction, histamine release, and vascular permeability assays. Among all C3a sequences, the overall arrangement of basic residues is also highly conserved, with a ubiquitous C-terminal Arg. There is an almost invariant C-terminal Leu-Gly-Leu-Ala-Arg sequence across species, again not wholly conserved in C. intestinalis. Despite these differences, Ciona C3a does bind to a specific receptor, which has significant homology to the mammalian C3a receptor (Melillo et al., 2006). The Ciona C3a-C3a receptor axis is responsible for the chemotaxis of hemocytes, suggesting a role for C3a in inflammation in this organism.
Interestingly, the des-Arg form of Ciona C3a also stimulates chemotaxis of hemocytes (Pinto et al., 2003) whereas C3a des-Arg is inactive in the majority of assays across other species. However, C3a and C3a des-Arg are equipotent in the killing of Gram-positive (e.g., Enterococcus faecalis) and Gram-negative (e.g., Pseudomonas aeruginosa) organisms and are actually more potent than the classic antimicrobial peptide LL37 (Nordahl et al., 2004). It has been suggested that C3a (and C4a) evolved initially as antimicrobial peptides (Pasupuleti et al., 2007), with chemoattractant activity emerging later. Antifungal activity of C3a and C3a des-Arg has also been demonstrated for Candida albicans (Sonesson et al., 2007). The mechanism appears to be the disruption of the plasma membrane and is dependent on Arg residues although the C-terminal Arg is dispensable, demonstrating that the chemotactic and antimicrobial activities of C3a are separate. Heparin can also bind to the same regions of C3a (Andersson et al., 2004) and inhibits antimicrobial activity (Nordahl et al., 2004). Structure-activity studies show that the formation of an amphipathic helix with a high net positive charge is critical for antimicrobial activity (Pasupuleti et al., 2008).
2. Structure
A crystal structure was obtained for human C3a before one was available for the other complement peptides (Huber et al., 1980; Paques et al., 1980), and it has also been investigated by NMR (Nettesheim et al., 1988). The N terminus of C3a is not well ordered, and the first 12 residues are not visible in the crystal structure although the NMR data also indicate some order at the C3a N terminus (Nettesheim et al., 1988), suggesting that the overall structure of C3a may be very similar to that of C5a (Fig. 3). In contrast, the C terminus of C3a appears to be well ordered in the crystal structure and forms a loop that turns toward the fourth helix. It has been proposed that the N-terminal portion (residues 1–21) and the disulfide-linked core region (residues 22–57) in intact C3a serve primarily to stabilize ordered conformation in the C-terminal region (residues 58–77) (Lu et al., 1984). The des-argination of C3a prevents interaction with C3a receptor but does not apparently alter the structure (Hugli et al., 1975a). As shown for C5a, the C terminus of C3a is involved in receptor activation although the more complete effects of des-argination on C3a suggest that it is also critical for receptor binding.

C5a structure. The structure of human C5a shows the major receptor interacting residues, based on 1KJS (Zhang et al., 1997b). Structures were visualized using the UCSF Chimera package, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California–San Francisco, funded the National Institutes of Health National Center for Research Resources and the National Institute of General Medical Sciences (Pettersen et al., 2004).
3. Peptide Analogs of C3a
The pentapeptide Leu-Gly-Leu-Ala-Arg is the minimal sequence required for receptor activation, albeit with only 0.2% molar activity of intact C3a (Caporale et al., 1980), whereas the 13-mer sequence C3a 65-77 has 6% and the 21-mer C3a 57-77 100% activity (Huey et al., 1984). Modifications at the N termini of these peptides with hydrophobic moieties such as 9-fluorenylmethyloxycarbonyl or acryloyl-amino hexanoyl improve agonist potency (Gerardy-Schahn et al., 1988; Kohl et al., 1990), and a superagonist (Trp-Trp-Gly-Lys-Lys-Tyr-Arg-Ala-Ser-Lys-Leu-Gly-Leu-Ala-Arg) with 200%–1500% of the potency of C3a has been reported (Ember et al., 1991), but it may not be selective for the human C3a receptor (Scully et al., 2010).
A series of hexapeptides has selective agonist and antagonist activity for the C3a receptor (Scully et al., 2010). One of these peptides, Phe-Leu-Thr-Leu-Ala-Arg (Fig. 4A), was shown by NMR to have a β-turn motif in dimethylsulfoxide (DMSO), which could be an important structural determinant of receptor activation. Several peptides originally developed as C5a1 receptor agonists (e.g., Tyr-Ser-Phe-Lys-Pro-Met-Pro-Leu-dAla-Arg) are also potent agonists at the C3a receptor (Scully et al., 2010), suggesting that the same turn motifs may be required for the activation of both types of receptor.

C3a receptor antagonists. (A) C3a hexapeptide (His-Leu-Gly-Leu-Ala-Arg). (B) SB290157. (C) Arg-substituted derivative of SB290157. (D) Aminopiperidine derivative of SB290157.
Gly-Tyr-Pro-Met-Tyr-Pro-Leu-Pro-Arg (oryzatensin) has antianalgesic and antiamnesic properties (Jinsmaa et al., 2000; Jinsmaa et al., 2001). An analog, [Trp5]-oryzatensin (Trp-Pro-Leu-Pro-Arg), is an orally available appetite suppressant (Ohinata et al., 2002, 2007) although both of these peptides have only low affinities for human C3a receptor (Scully et al., 2010). Bovine κ-casein peptide, casoxin C (Tyr-Ile-Pro-Ile-Gln-Tyr-Val-Leu-Ser-Arg), is also reported to have agonist properties at the C3a receptor (Takahashi et al., 1997).
4. Peptidomimetic Analogs of C3a
High-throughput screening and optimization resulted in the identification of N(2)-[(2,2-diphenylethoxy)acetyl]-l-arginine (SB290157) (Fig. 4B), a competitive antagonist for the C3a receptor (Ames et al., 2001) with a pIC50 of 6.7 at RBL-2H3 cells transfected with human C3a receptor. This compound is selective for the C3a receptor and has no activity at the C5a1 receptor or a number of other chemotactic G protein-coupled receptors (GPCR). SB290157 blocks Ca2+ mobilization in human neutrophils with pIC50 of 7.6 and is also active at the guinea pig and mouse C3a receptor (Ames et al., 2001), making it potentially very useful in animal models of disease, including arthritis, allergic asthma, and ischemia-reperfusion injury (Qu et al., 2009). However, this antagonist has partial agonist properties in some systems (Mathieu et al., 2005), possibly dependent on the receptor expression levels.
An Arg-containing derivative of SB290157 has also been reported (Denonne et al., 2007a) that has a higher affinity for the C3a receptor (Fig. 4C). Like SB290157, this compound has a short half-life because of the lability of the Arg moiety, so it has had very limited use in animal models (Denonne et al., 2007a). An amino-piperidine antagonist developed by the same group (Fig. 4D), which lacks the Arg but has poorer antagonist activity (pIC50 = 5.8) and derivatization, has so far resulted only in higher affinity compounds with agonist activity (Denonne et al., 2007b).
B. C4a
1. Sequence
Unlike C3a and C5a, C4a appears to have little, if any, activity in humans although C4a has been reported to be a major mediator in inner ear damage (Harada et al., 1992). The amino acid sequences of C4a obtained to date—such as rat (Cui et al., 1988), human (Moon et al., 1981), and bovine (Smith et al., 1982)—show conservation of the 6 Cys residues likely to form the disulfide knot and the basic residues that are present in C3a and C5a (Fig. 2B). Rodent but not human or bovine C4a is glycosylated (Cui et al., 1988). From this sequence similarity, the three-dimensional structure of C4a is inferred to be very similar to that of C3a and C5a (Fig. 3). Structure-function studies are lacking for C4a.
C4a is a potent agonist of guinea pig but not human C3a receptor (Ames et al., 1997; Lienenklaus et al., 1998). Guinea pig macrophages undergo a Ca2+ response to C4a and are desensitized to subsequent stimulation by C3a (Murakami et al., 1993) although the purity of serum-derived C4a may be an issue because human C4a is 100- to 1000-fold less active as a spasmogen than human C3a in guinea pig ileum (Hugli, 1981). For these activities, the C-terminal Arg residue has been shown to be critical (Gorski et al., 1979), and, as for C3a, the agonist activity of C4a is located in the C-terminal pentapeptide (Hugli et al., 1983). C4a also appears to share a similar range of antimicrobial activities with C3a (Pasupuleti et al., 2007).
C. C5a
1. Sequence
C5a, the most intensively studied of the fragments, is a 74–79 amino acid polypeptide (Fig. 2C), although the classic anaphylatoxin is actually the des-arginated form, C5a des-Arg (Gerard and Hugli, 1981). Human C5a has an N-linked glycosylation site (Fernandez and Hugli, 1978) that is not found in C5a from other species. This glycosylation may be inhibitory for C5a des-Arg, as porcine C5a has no glycosylation sites (Fig. 2C) and porcine C5a des-Arg is more active than the human form. In addition, the enzymatic de-glycosylation of human C5a des-Arg increases its activity (Gerard et al., 1981). More recently, discrepancies have been observed between the activities of recombinant (aglycosylated) and purified (glycosylated) C5a des-Arg, suggesting that control of the activity of the des-Arg form is complex (Reis et al., 2012).
2. Structure
The NMR structure of human and porcine C5a has revealed a four-helix core (Zuiderweg et al., 1989; Williamson and Madison, 1990; Zhang et al., 1997a,b), stabilized by three disulfide bridges (Cys21–Cys47, Cys22–Cys54, Cys34–Cys55) (Fig. 3). The structure of bovine C5a, determined by NMR, is similar to human and porcine (Zarbock et al., 1988). The disruption of these disulfide bridges by reduction or mutation causes a loss of function (Mollison et al., 1989). The crystal structure of C5 reveals a similar structure for the C5a-moiety of the uncleaved molecule (Fredslund et al., 2008). The sequence of human C3a is 35% identical to human C5a and has disulfide linkages in comparable locations, so C5a modeled on the C3a crystal structure appears to be nearly identical (Greer, 1985).
In contrast with these studies, a recently published crystal structure of C5a des-Arg has a three-helix core with the N-terminal domain able to adopt different conformations (Cook et al., 2010). C5a des-Arg also appeared to form dimers in these crystals, with the interaction interface potentially blocking some of the proposed ligand interacting residues we will discuss later. However, there is no empirical evidence to support dimer formation.
The C terminus of C5a/C5a des-Arg (residues 64–73/74) appears to be a disordered structure and is not observed in the crystallographic study or most NMR studies, perhaps due to a pH-dependent lability in this region (Zhang et al., 1997b). In the one study where C-terminal structure was observed, a short helix (residues 69–74) connected by a loop to the fourth helix of the core of C5a was present (Zhang et al., 1997b).
3. Receptor Interacting Residues of C5a
Unlike C3a, extensive mutagenesis studies on the entire C5a molecule have been performed that highlight basic residues, concentrated in the loops that connect the helices, and these are important in forming the interaction sites with C5a1 receptor (Fig. 3). These can be divided into three separate clusters (Mollison et al., 1989; Huber-Lang et al., 2003). Site 1 is formed by residues 12–20, including His15, Lys19 and 20, and Arg46. Site 2 includes Asp24, Arg37, Arg40, and possibly Lys49. Site 3 comprises residues 67–74, including His67, Lys68, Arg69, and the C-terminal Arg74 (Mollison et al., 1989; Bubeck et al., 1994; Toth et al., 1994; Vlattas et al., 1994). Other important residues are hydrophobic: Val18, Leu43, Met70, and Leu72; the latter residue when mutated to either Lys or Asp causes big loss in binding affinity (Mollison et al., 1989).
The unpaired Cys27 is unique to human C5, and mutation to Arg has been found to be necessary to display C5a on phage (Cain et al., 2003; Heller et al., 1999), presumably by preventing aberrant cross-linking. However, this residue has been exploited to help map the ligand-binding site on the C5a1 receptor (Section IV.C.4), and the data suggest that this part of C5a lies close to the receptor N terminus.
Human C5a (1–69), lacking the C-terminal loop-helix structure, can still bind to cell-surface receptors, albeit with considerably lower affinity than intact C5a, but cannot activate the receptor (Chenoweth et al., 1982). The substitution of the C-terminal pentapeptide sequence of human C3a (Leu-Gly-Leu-Ala-Arg) to C5a (1–69) (Bautsch et al., 1992) gives a ligand that binds to both the C5a1 receptor and C3a receptor, whereas adding a modified C5a C-terminal sequence through a new disulfide linkage makes a C5a1 receptor antagonist (Zhang et al., 1997a). These observations suggest that C5a (1–69) provides a recognition site for the receptor whereas the C terminus provides the activation signal.
4. Peptide Analogs of C5a
a. Agonists
Although the pentapeptide Met-Gln-Leu-Gly-Arg that is analogous to the C terminus of C5a is inactive at the C5a1 receptor (Chenoweth and Hugli, 1980), the discovery that the C-terminal octapeptide of human C5a, His-Lys-Asp-Met-Gln-Leu-Gly-Arg, has weak agonist activity (Kawai et al., 1991, 1992) has provoked intense research into the mechanism of receptor activation and the development of antagonists.
A series of decapeptide analogs first reported by Ember et al. (1992), substituting Phe for His67, had increased potency over the native sequence. These were later developed into a large series of constrained peptides. In some of these, the insertion of Pro into the sequence has resulted in greater agonist activity (Sanderson et al., 1994) but also in a loss of selectivity (Scully et al., 2010). EP-54 (Tyr-Ser-Phe-Lys-Pro-Met-Pro-Leu-dAla-Arg) is an agonist at both the C5a1 receptor and C3a receptor but has little or no activity at the second C5a receptor C5a2 receptor (also known as C5R2, C5L2, GPR77) (Scola et al., 2007). In contrast, EP-67 (Tyr-Ser-Phe-Lys-Asp-Met-Pro-(Me)Leu-dAla-Arg) is a weak agonist at both the C5a1 receptor and the C3a receptor but may have greater activity at the C5a2 receptor (Kawatsu et al., 1996; Short et al., 1999; Taylor et al., 2001; Vogen et al., 1999a, 1999b).
Peptides such as EP-54 and EP-67 have been described as “response selective,” producing different responses at the C5a1 receptor depending on the cell type under study (reviewed in Taylor et al., 2001). However, a more likely explanation is the nonselectivity of these peptides, with varying affinities for two or more of the known complement peptide receptors. Peptide ligands have been found to have conserved turn structures (Tyndall et al., 2005), and C5a and its analogs have a β/γ turn motif that has also been observed in bradykinin, enkephalin, and vasopressin. The conservation of the turn structure may be linked to the mechanism of receptor activation because a two-step binding mechanism is a common feature of secretin family GPCR (Tyndall et al., 2005).
b. Antagonists
Further development of C-terminal peptides resulted in a family of hexapeptide analogs of the form N(Me)-Phe-Lys-Pro-dCha-Xxx-dArg, which were potent agonists and antagonists depending on the nature of the fifth residue (Konteatis et al., 1994). Increasing aromaticity at this position was found to enhance antagonist activity, with Trp providing complete antagonism. From these studies, a cyclic peptide antagonist (PMX53; 3D53, (Ac)Phe-[Orn-Pro-dCha-Trp-Arg]) has been developed that shows improved affinity (Wong et al., 1999) (Fig. 5A) and has a β-turn structure similar to the C5a C terminus, as determined by NMR (Zhang et al., 2008).

C5a1 receptor peptide antagonists. (A) PMX53 (AcPhe-[Orn-Pro-dCha-Trp-Arg]). (B) PMX205 hydrocinnamic acid substituted derivative of PMX53. (C) JPE-1375. (D) L156,602.
PMX53 (pIC50 = 7.05 in human neutrophil membranes) is an insurmountable antagonist for C5a1 receptor (Strachan et al., 2000, 2001) with no discernible activity at the C5a2 receptor (Scola et al., 2007). Like many peptides, PMX53 has low oral bioavailability, but its long-lasting effect means that even once-daily oral dosing is sufficient to maintain circulating levels in the rat (Morgan et al., 2008). The development of PMX53 has been discontinued (http://www.evaluatepharma.com/Universal/View.aspx?type=Story&id=178099), although PMX205, a more stable derivative (Delisle Milton et al., 2011) with a hydrocinnamic acid moiety at the N terminus (Fig. 5B), is now being used in animal models of disease, including rat and mouse models of neurologic diseases such as Huntington’s disease, Alzheimer’s disease, and amyotrophic lateral sclerosis (ALS) (Fonseca et al., 2009; Woodruff et al., 2006, 2008). However, PMX205 has been reported as unlikely to be of benefit in ALS patients (ALS-TDI http://www.als.net/ALS-Research/PMX205/ALS-Topics/).
PMX53 also has limited selectivity, in common with the related compound JPE-1375 (Hoo-Phe-Orn-Pro-(d-HLeu)-Phe4F-Phe) (Fig. 5C), and it also binds to other receptors such as NK2 and Mas-related gene 2 receptor (Schnatbaum et al., 2006; Subramanian et al., 2011). Despite this, PMX53 (and, to a lesser extent, JPE-1375) has been of immense value in defining the role of C5a in a wide range of animal models of disease. Described in more detail in reviews (Monk et al., 2007; Klos et al., 2009; Qu et al., 2009; Woodruff et al., 2011), these include inflammatory bowel disease, ischemia-reperfusion injuries, sepsis, and arthritis. Unfortunately, PMX53 and JPE-1375 have limited activity at the rodent C5a1 receptor in transfected cell lines, particularly when used with recombinant rodent C5a (Waters et al., 2005; P. N. Monk, unpublished observations), making the reported in vivo activities more difficult to analyze.
JPE-1375 was developed as a linear analog of PMX53 by replacing the Arg with Phe and adding hydroorotic acid (Hoo) in place of the N-acetyl group (Proctor et al., 2006; Woodruff et al., 2006). The potency of JPE-1375 is comparable with PMX53, but the former has increased stability in liver microsome preparations. JPE-1375 has been discontinued (Qu et al., 2009) despite showing some promise in AMD (Ricklin and Lambris, 2007), renal allograft survival (Gueler et al., 2008), experimental tubulointerstitial fibrosis (Boor et al., 2007), and atherosclerotic plaque stabilization (Shagdarsuren et al., 2010). The hexadepsipeptide L156,602 (Fig. 5D) is a nonselective antibiotic antagonist for the C5a1 receptor from Streptomyces with a pIC50 = 5.7 at the C5a1 receptor, although extensive modification failed to improve selectivity (Hensens et al., 1991; Tsuji et al., 1992a,b, 1995). The C terminus of C5a has also been exploited by the use of phage display to allow selection of libraries of C5a mutants to produce a potent antagonist, A8Δ71–73 (Heller et al., 1999). This polypeptide binds both the C5a1 receptor and the C5a2 receptor (Otto et al., 2004) and has some activity in vivo (e.g., Zhang et al., 2009), although its size and protein derivation make it unsuitable for drug development.
5. Naturally Occurring Non-Complement-Derived Analogs of C5a
There are three naturally occurring ligands for C5a1 receptor that do not derive from C5. Skp, also known as OmpH (outer membrane protein H), is a major structural porin of enteric bacteria such as Escherichia, Klebsiella, Salmonella, and Yersinia (Koski et al., 1989). Skp acts as a cavity chaperone to prevent aggregation (Walton et al., 2009) after forming trimers (Walton and Sousa, 2004) and is also a chemoattractant for both monocytes and polymorphonuclear neutrophils (PMN) (Shrestha et al., 2004). However Skp is not a secretagogue for these cells, unlike C5a, suggesting that it is only a partial agonist. The region of Skp proposed to interact with C5a1 receptor is structurally unrelated to C5a (Fig. 6A). Mutagenesis experiments have identified residues required for C5a1 receptor interaction, namely, Gln103–Arg105 (Jia et al., 2010). Gln103 of Skp may be equivalent in function to Leu72, a critical residue in C5a. Skp has been proposed for a novel method of vaccination against pathogenic bacteria, targeting C5a1 receptor on intestinal M cells (Luo et al., 1999).

Noncomplement derived C5a1 receptor ligands. (A) Skp (OmpH). (B) RP-S19. (C) CHIPS in complex with a peptide mimic of the N terminus of the C5a1 receptor (Ippel et al., 2009). Dark blue sections on A and B represent the sequences analogous to the C terminus of C5a. The pale blue section of A represents the sequence thought to antagonist activity on neutrophils. In C, CHIPS is shown in dark blue, with the N-terminal peptide of C5a1 receptor in gray. Structures were visualized using the UCSF Chimera package.
A considerable body of work, mainly from the Yamamoto laboratory, has demonstrated the complex activity of S19 at C5a1 receptor. S19 is a 145-amino-acid component of the ribosome, released by apoptotic cells in the form of homodimers, cross-linked by plasma transglutaminase (Semba et al., 2010). Overall, it has only 4% sequence identity with C5a (Yamamoto, 2000) and is structurally unrelated (Fig. 6B). In the extracellular milieu, it can act as an agonist at monocytes, promoting chemotaxis, and as an antagonist for neutrophils. This activity can be blocked by antibodies against the C5a1 receptor (Nishiura et al., 1996, 1998). The monocytic infiltrate acts to phagocytose apoptotic cells (Horino et al., 1998; Nishimura et al., 2001) although S19 itself can also promote apoptosis in fibroblasts (Nishiura et al., 2005). Cross-linked S19 is proposed to activate C5a1 receptor in the same way as C5a, through a two-step mechanism (Shibuya et al., 2001).
The first binding site is a cluster of basic residues (Lys41-His42-Lys43), and the second, which directly causes receptor activation, is Leu131-Asp132-Arg133, 12 residues from the C terminus of S19. These binding sites are divided between the two components of the homodimer (Nishiura et al., 2010a). The dual agonist/antagonist activity of S19 is located in a “switch” region located between Leu134 and Lys144, at the C terminus beyond the second activation site (Revollo et al., 2005; Shrestha et al., 2003). Recombinant C5a (or peptide analogs) bearing the C-terminal protein of S19 also assume this dual activity (Jia et al., 2010; Oda et al., 2008). S19 appears to bind to the second C5a receptor, the C5a2 receptor, but this does not explain the dual activity (Nishiura et al., 2010b), proposed to be due to a diminution of Gαi coupling to the C5a1 receptor in neutrophils by an as yet unidentified cofactor (Nishiura et al., 2011).
The third noncomplement molecule that binds C5a1 receptor is the chemotaxis inhibitory protein of Staphylococcus aureus (CHIPS) (de Haas et al., 2004). CHIPS (Fig. 6C) has been intensively studied and has been found to bind only to C5a1 receptor and the formyl peptide receptor FPR1. The C5a1 receptor is bound by CHIPS at the N terminus, at a site that overlaps with the C5a binding, meaning that CHIPS is a potent antagonist for the C5a1 receptor (Kd = 1.1 nM) (de Haas et al., 2004; Haas et al., 2004; Postma et al., 2004, 2005; Ippel et al., 2009). CHIPS is too immunogenic for therapeutic use (Wright et al., 2007), but ADC-1004, a derivative of CHIPS produced by directed evolution, has minimal immunogenicity and may have therapeutic potential (Gustafsson et al., 2009a,b, 2010; van der Pals et al., 2010). More recently, a smaller peptide derivative of CHIPS has been reported (Bunschoten et al., 2011). A cofactor of C5a activity, vitamin D-binding protein, is apparently required for maximal chemotactic activity but does not interact with either C5a or the C5a1 receptor (DiMartino et al., 2001).
6. Nonpeptidic Analogs of C5a
Substituted 4,6-diaminoquinolines (structure not shown) were selected from a screen of positively charged compounds as analogs of the core regions of C5a but were found to be weak antagonists (pIC50 = 5.5) and were not amenable to modification (Lanza et al., 1992). Most of the other analogs listed are mimics of the C terminus of C5a. W54011 [N-[(4-dimethylaminophenyl)methyl]-N-(4-isopropylphenyl)-7-methoxy-1,2,3,4-tetrahydronaphthalen-1-carboxamide hydrochloride] (Fig. 7A) is an orally active and potent C5a1 receptor antagonist (pIC50 = 8.7) developed by the Mitsubishi Pharmaceuticals Company by library screening and optimization. W54011 inhibits human neutrophil chemotaxis and superoxide production with low nanomolar activity (Sumichika et al., 2002) but has no detectable activity at the C5a2 receptor (Scola et al., 2007). Although originally described as inactive in the mouse and rat, it has been shown to have some effect on mouse C5a1 receptor expressed on DC in vitro (Peng et al., 2009a).

C5a1 receptor nonpeptide antagonists. (A) W54011. (B) NDT 9520492. (C) NTD 9513727. (D and E) Aniline-substituted tetrahydroquinolines. (F) CPP 447,697. (G) RPR 121154. (H) Example of bis-amide series. (I) JSM 7717. (J) Example of (R) arylalkylamino series. (K) Example of bis-sulfonamide series.
NDT 9520492 (Fig. 7B) is a substituted tetrahydroisoquinoline with a pIC50 of 7.5 at the human C5a1 receptor that also inhibits gerbil and primate C5a receptors but not rat or mouse. This pattern is also seen with W54011 and PMX53, suggesting that they bind to a similar site on the C5a1 receptor (Waters et al., 2005) (Section IV.C.4). NGD 2000-1 (structure unknown) is a derivative of NDT 9520492 which has been tested in phase II trials on asthma and RA. For the primary end points—forced expiratory volume in 1 second (FEV1) and C-reactive protein (CRP) levels, respectively—the drug did not show any benefits although there were some positive effects on the Subject Global Assessment of the disease in RA (Powers et al., 2011). Because of inhibition of cytochrome P450 3A4, no further development of this drug occurred (Lee et al., 2008).
NDT 9513727 [N,N-bis(1,3-benzodioxol-5-ylmethyl)-1-butyl-2,4-diphenyl-1H-imidazole-5-methanamine] (Fig. 7C) is an inverse agonist at the human, primate, and gerbil C5a1 receptor with little activity at the rodent C5a1 receptor (Brodbeck et al., 2008). NDT 9513727 has a pIC50 = 6.9 at that C5a1 receptor but is not active at the C5a2 receptor and is also orally bioavailable. 5,6,7,8-tetrahydroquinoline (Barbay et al., 2008) and aniline-substituted tetrahydroquinolines (Gong et al., 2008) have been reported as C5a1 receptor antagonists (Fig. 7, D and E), with one compound having a pIC50 = 7.7 and an IC50 = 20 nM for C5a-stimulated Ca2+ flux in human neutrophils. Closely related compounds were shown to be orally bioavailable with reasonable pharmacokinetics in the rat.
CP-447,697 (Fig. 7F) was isolated by library screening and optimization and has a pIC50 = 7.5 (Blagg et al., 2008b). Attempts to reduce toxicity and improve availability by increasing polarity resulted in compounds with reduced binding affinity, suggesting that the binding site on the C5a1 receptor did not tolerate polar or basic groups (Blagg et al., 2008a). A modified phenylguanidine, RPR121154 (Fig. 7G), was reported to have a pIC50 = 6.1 and could inhibit the respiratory burst in C5a-stimulated human neutrophils (Astles et al., 1997). A bis-amide compound (Fig. 7H), isolated by high-throughput screening for inhibition of C5a binding (Sanganee et al., 2009), has a pIC50 = 7.6 but has no activity at the rodent or dog C5a1 receptor or the human C5a2 receptor. An inability to improve stability and availability without loss of potency has led to the discontinuation of this compound.
CCX168 (Xiao et al., 2010) (undisclosed structure) is reported to antagonize the human (but not mouse) C5a1 receptor, pIC50 = 9.2, and to inhibit Ca2+ mobilization in monocytes at subnanomolar concentrations (Powers et al., 2011). JSM-7717, with a structure similar to that shown in (Fig. 7I), was developed by Jerini AG as a small-molecule C5a1 receptor antagonist, with a pIC50 = 8.5 and activity in vivo in a gerbil model of neutropenia (Powers et al., 2011).
A series of (R)-arylalkylamino compounds (e.g., Fig. 7J) has been produced by Dompe SpA, derived from a dual C5a and IL-8 antagonist, which were found to be selective for the C5a1 receptor, pIC60 = 8 (Allegretti et al., 2008; Powers et al., 2011). Bis-sulfonamides, exemplified by Fig. 7K, identified by screening and optimization, have also been reported as effective C5a1 receptor antagonists (pIC50 = 7.2 in human neutrophils) (Chen et al., 2010).
IV. Receptors
A. Introduction
The three known complement peptide receptors (http://www.iuphar-db.org/DATABASE/FamilyMenuForward?familyId=5) are all members of the GPCR superfamily, in family A, subgroup A8, with the formyl peptide receptor family (Joost and Methner, 2002). They have the classic GPCR structure, comprising an extracellular N terminus, seven helical transmembrane domains connected by intracellular and extracellular loops, and an intracellular C terminus (Findlay and Pappin, 1986) (Fig. 8). Ligand binding and signaling by two of the receptors, the C5a1 receptor and the C3a receptor, are well characterized, but the third receptor, the C5a2 receptor, remains enigmatic.

Complement peptide receptors. Sequences and domain structures are shown of the three human complement peptide receptors. Putative additional intracellular loops formed by S-acylation of cysteine residues are shown for the C5a2 receptor and the C3a receptor. Glycosylation sites are indicated by black ellipses. Phosphorylation sites in intracellular domains and tyrosine sulfation sites in extracellular domains are indicated by gray circles; residues at the extracellular face of the transmembrane domain shown to influence ligand binding in mutagenesis experiments are indicated by hexagons.
B. C3a Receptor
1. Sequence and Genetics
In contrast to the other receptors, the C3a receptor (Ames et al., 1996; Crass et al., 1996) has a much smaller N-terminal domain and a greatly enlarged second extracellular domain (Fig. 8) that is conserved across all species so far sequenced (Melillo et al., 2006). In humans, C3AR is present in a single copy located on chromosome 12p13.2-3, with the entire coding sequence present in a single exon (Paral et al., 1998). Transcriptional control of C3ar expression in murine myeloid cells is mediated by activator protein 1 (AP-1), nuclear factor kappa B (NF-κB), Ets, and GATA (Martin and Martin, 2005); in murine glial cells, transcription is controlled by AP-1 but not Ets binding (Martin et al., 2007b). Human C3AR does not have a functional NF-κB site, but AP-1 and Ets control C3AR expression in monocytic cells (Schaefer et al., 2005). A 1526G/A single-nucleotide polymorphism (SNP) in human C3AR is associated with severity of childhood bronchial asthma and 1595 A/G with atopic dermatitis (Hasegawa et al., 2004). Other polymorphisms (http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?locusId=719) (Sherry et al., 2001) have not so far been associated with disease.
2. Post-translational Modifications
a. Glycosylation
The human C3a receptor is reported to be highly glycosylated (Mizuno et al., 2007) and has two N-linked glycosylation sites (Asn9, Asn194), in the N terminus and second extracellular domain, respectively. In contrast, the mouse C3a receptor has four potential sites in this domain (Tornetta et al., 1997), but it is not clear what functional role this glycosylation plays.
b. Tyrosine Sulfation
Five or six tyrosine residues in the 172-amino-acid second extracellular loop (174, 184, 188, 317/318) of the human C3a receptor are sulfated in vivo (Gao et al., 2003). Tyr174 sulfation is critical for high-affinity binding of C3a but not for receptor activation by C-terminal peptide analogs of C3a, suggesting that Tyr174 directly interacts with the core of C3a.
c. Phosphorylation
The C3a receptor is known to undergo serine/threonine phosphorylation after ligand binding, mediated by G protein-coupled receptor kinases (Langkabel et al., 1999), probably GRK2 and GRK3. The C terminus of the C3a receptor contains 10 serine/threonine residues that may undergo phosphorylation. Mutation of these to alanine indicates that disruption of phosphorylation of Ser465/470 and Thr463/466 inhibits ligand-induced internalization. Ser449 is not involved in internalization but is involved in signal transduction (Settmacher et al., 2003).
d. S-acylation
A potential S-palmitoylation site is present at the intracellular C terminus (Cys468) of the human C3a receptor and also in the rat and mouse but not guinea pig C3a receptor. It is not known if this Cys is actually modified in vivo. A potential eighth helix at the C terminus (Asn431–Gln451) is suggested by sequence similarities to bradykinin R2 and other receptors (Feierler et al., 2011). Interestingly, the C5a1 and C5a2 receptors do not have this basic sequence and also contain proline, a potential helix blocker.
3. Expression
The human and mouse C3a receptor appears to be broadly expressed, with mRNA detected in most tissues, albeit at widely varying levels (Ames et al., 1996; Tornetta et al., 1997), with high levels of expression in the lung, spleen, ovary, placenta, small intestine, spinal cord, and brain. In contrast to the C5a1 receptor, expression in leukocytes is relatively low (Ames et al., 1996), and no C3a receptor is detectable on naive B and T lymphocytes (Martin et al., 1997; Zwirner et al., 1999; Werfel et al., 2000;). However, treatment of human T cells with IFN-γ up-regulates C3a receptor expression in vitro, and this receptor can be detected on T cells from patients with atopic dermatitis (Werfel et al., 2000).
Myeloid cells that express the C3a receptor are eosinophils, DC, monocytes/macrophages, microglia, and mast cells. Nonmyeloid cells that express the C3a receptor are activated astrocytes, endothelial cells, and epithelial and smooth muscle cells from asthma patients (reviewed in Klos et al., 2009). The C3a receptor has also been detected on neurons (Davoust et al., 1999), and the neuronal C3a receptor may have a role in central nervous system inflammation (reviewed in Yanamadala and Friedlander, 2010) and during development (Benard et al., 2004, 2008).
4. Ligand Binding by C3a Receptor
Relative to the C5a1 receptor, little is known about the mechanism of ligand binding and receptor activation for the C3a receptor. The agonist activity of C-terminal peptide analogs of C3a suggest that, as for the C5a1 receptor, multiple sites are involved. Most of the data concerning the location of these sites come from an investigation using a series of chimeras between the C5a1 receptor and the C3a receptor (Crass et al., 1999a). The substitution of the C3a receptor N terminus by that of the C5a1 receptor has little effect on the binding of C3a but produces a receptor that both binds and is activated by C5a, suggesting that the primary binding site for C3a lies outside of the N-terminal domain and confirming that this site on the C5a1 receptor is indeed at the N terminus. When the second extracellular loop of the C3a receptor was substituted by the much smaller C5a1 receptor loop, this chimera could not bind or be activated by C3a, indicating that this domain rather than the N terminus forms the primary binding site (Chao et al., 1999). The loss of up to 65% of this loop, however, failed to inhibit C3a binding, with the significant residues being 162–183 and 309–322, at the N and C termini of this loop, respectively.
The mutation of a series of aspartate residues in this region to lysine effectively inhibited C3a binding, suggesting that these acidic residues make up a binding site for the basic core of C3a (Chao et al., 1999). The sulfated tyrosine residue (Tyr174) also appears to form part of this binding site (Gao et al., 2003). Several charged residues at the tops of TM2, TM3, TM4, TM5, and TM7 have been mutated and found to affect C3a binding and receptor activation by both C3a and peptide analogs of the C3a C terminus (Sun et al., 1999). These data suggest that these residues form part of a binding pocket at the extracellular face of the transmembrane helical cluster of C3a receptor, similar to that predicted for the C5a1 receptor (Section IV.C.5).
5. C3a Receptor Signaling
a. G protein mediated
By comparison with C5a, C3a is generally a much weaker chemotactic stimulus in leukocytes (Fernandez et al., 1978), despite sharing many of the same signaling mechanisms. Following the binding of C3a to C3a receptor, the primary signaling mechanism activated is through the pertussis toxin (PT)-sensitive G protein Gαi in human and mouse immune cells such as neutrophils (Norgauer et al., 1993), eosinophils (Elsner et al., 1994), and microglia (Moller et al., 1997). C3a has also been reported to decrease intracellular cAMP levels in murine dendritic cells (Li et al., 2008), which might be dependent on Gαi. C5a has been shown to have this effect as well in cell lines (Vanek et al., 1994) and in DC (Peng et al., 2009b). However, in endothelial cells, C3a signaling caused a PT-insensitive cytoskeletal response and ERK1/2 activation, attributed to activation of Gα12 and/or Gα13 despite the availability of Gαi for coupling to C5a1 receptor in the same cells (Schraufstatter et al., 2002). The promiscuous PT-insensitive G protein Gα16 can also couple to C3a receptor in cotransfected cell lines (Crass et al., 1996). Differences have also been observed in the subsequent changes in intracellular free Ca2+ (Ca2+i), with C3a typically stimulating a smaller and transient increase in intracellular Ca2+i in neutrophils solely due to the influx of extracellular Ca2+ whereas C5a also causes the release of Ca2+ from intracellular stores and a more prolonged elevation due to the activation of phospholipase C (PLC) (Norgauer et al., 1993). However, in microglia, the Ca2+i responses are equal (Moller et al., 1997), and in human MSC both C3a and C5a cause a prolonged activation of protein phosphorylation (Schraufstatter et al., 2009). Similarly, in a human astrocytic cell line, both C3a and C5a stimulated the release of Ca2+ from intracellular stores, although the response to C3a was smaller (Sayah et al., 2003).
b. Arrestin mediated
In mast cells, although C3a provokes only a transient increase in Ca2+i, the activation of ERK1/2 and Akt phosphorylation is sustained (Venkatesha et al., 2005). This disparity in the temporal aspects of C3a signaling in mast cells has been explored in great detail, and it is known that degranulation is uncoupled from other responses, such as de novo production of the chemokine CCL2 (Ahamed et al., 2001). Here, the use of a C3a receptor mutant, truncated at the C terminus to remove potential serine/threonine phosphorylation sites, has shown that production is absolutely dependent on receptor phosphorylation whereas degranulation was not affected. The mutant receptor failed to bind β-arrestin 2, suggesting that arrestins can act as mediators of additional signaling mechanisms for the C3a receptor. However, both degranulation and the production of CCL2 were equally sensitive to PT, suggesting that Gαi activation is still critical, most likely through ERK1/2 activation. Arrestins were originally described as adaptors that link ligand-activated GPCR to the cellular internalization machinery (Wilden et al., 1986; Lohse et al., 1990), an important part of the complex desensitization process (Jalink and Moolenaar, 2010). The Gq-coupled protease-activated receptor PAR2 couples to ERK1/2 through two pathways, one G protein dependent and one dependent on β-arrestin/Src kinase that results in ERK1/2 activation by endocytosed receptors in a particular subcellular location (DeFea et al., 2000). However, the early phase of ERK1/2 activation by C3a receptor is not dependent on Src (Ahamed et al., 2001), so the mechanism by which β-arrestin mediates C3a receptor signaling is still unclear. For the C3a receptor, at least, different arrestins appear to play different roles. The silencing of β-arrestin 2 in human mast cells prevents the desensitization and internalization of C3a receptor, leading to a prolonged increase in Ca2+i (Vibhuti et al., 2011) but has no effect on degranulation whereas silencing of β-arrestin 1 could actually inhibit degranulation. Interestingly, both arrestins were observed to suppress ERK1/2 activation by the C3a receptor, in contrast to results observed with other GPCR (Defea, 2008). Phosphorylation of the C3a receptor by GRK2, 3, 5, and 6 has been implicated in the control of the association with arrestins (Guo et al., 2011), with different GRK involved in different aspects of the response to C3a. Thus, signaling by C3a receptor, at least in mast cells, is complex and is critically dependent on arrestins as well as G proteins. This sensitivity to arrestins may explain why C3a receptor signaling is often more transient and weaker in nature relative to that of C5a1 receptor.
C. C5a1 Receptor
1. Sequence and Genetics
C5AR1 was cloned in 1991 (Gerard and Gerard, 1991) and is located on chromosome 19q13.3–13.4 (mouse chr 7), adjacent to the gene for C5AR2 and close to genes for formyl peptide receptors (Gerard et al., 1993). C5AR1 is encoded in two exons, the first with the 5′-untranslated region (UTR) and the initiating Met codon separated by 9 kb from the second exon that has the remainder of the coding sequence. Myeloid-cell specific promoter activity was detected just upstream of the initiating codon (−49 to −82) and suppressing activity at −225 to −346 (Gerard et al., 1993). Distinct transcriptional control mechanisms appear to exist in murine myeloid and nonmyeloid cells (Martin et al., 2007a), although an LPS-response element, a CCAAT box/NF-Y binding site at −96, a GATA site at −298, and a CP2 site at −155 are active in both macrophages and endothelial cells (Hunt et al., 2005). More recently, NF-κB, CCAAT, and NFAT sites have been identified in the first 200 bp of the 5′-UTR of human C5AR1, with two adenylate-uridylate-rich (AU-rich) elements in the 3′-UTR (Palmer et al., 2012). The latter have no effect on basal or stimulated expression levels. Although a number of SNP in the C5AR1 gene have been described (reviewed in Monk et al., 2007; http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?locusId=728; Sherry et al., 2001), there are no associations with disease yet reported. In detailed studies, no association with C5AR1 SNP was found in AMD (Skeie et al., 2010) or bronchial asthma (Hasegawa et al., 2004) although a C5 haplotype was protective. SNP rs17611 in C5 has been associated with plasma C5a levels and is a risk factor for adverse cardiac events (Hoke et al., 2012).
2. Post-translational Modifications
a. Glycosylation
There is one potential site for N-linked glycosylation on human C5a1 receptor, at Asn2. Removal of N-linked glycosylation by endoglycosidase F or by mutation of Asn2 had little effect on either affinity for the ligand or expression of the receptor at the cell surface (Pease and Barker, 1993).
b. Tyrosine sulfation
There are three tyrosine residues at the N terminus of C5a1 receptor, two of which (Tyr11, Tyr14) are proximal to Asp residues, making them potential sulfation sites (Rosenquist and Nicholas, 1993). Both Tyr11 and Tyr14 are sulfated, and this modification is essential for the binding of C5a (Farzan et al., 2001), C5a des-Arg (Scola et al., 2007), and CHIPS (Ippel et al., 2009; Liu et al., 2011c).
c. Phosphorylation
Phosphorylation of the C5a1 receptor after ligand binding or phorbol ester treatment has been linked to desensitization (Ali et al., 1993). The major phosphorylation sites are at the C terminus (Ser314, Ser317, Ser327, Ser332, Ser334, and Ser338) (Giannini et al., 1995) although the third intracellular loop also contains a potential PK-C site (Lys239-Thr-Leu-Lys) (Bock et al., 1997). Mutation of Ser332, Ser334, and Ser338 to Ala reduces phosphorylation by 80% (Giannini et al., 1995) and also inhibits receptor internalization (Naik et al., 1997). This inhibition was thought to be due to a loss of association with molecules important for internalization such as β-arrestin, dynamin, and clathrin. Interestingly, β-arrestin 1 and 2 were still able to bind to phosphorylation-deficient C5a1 receptor but much more weakly than to a wild-type receptor (Braun et al., 2003). However, internalization of the receptor, one mechanism for desensitization, was found to be dependent only on amino acids 335–350 in a series of truncated C5a1 receptor mutants, beyond the majority of the phosphorylation sites (Bock et al., 1997). Similarly, another study found that although Ser334, Ser327, Ser332, and Ser338 could be phosphorylated by PK-Cβ, these sites were functionally redundant for internalization and desensitization and even that, at high concentrations of C5a, β-arrestin binding to the C terminus could actually inhibit phosphorylation (Pollok-Kopp et al., 2007). Thus, the role of phosphorylation in the control of receptor function remains unclear.
3. Expression
Although typically expressed at high levels on cells of myeloid origin, the C5a1 receptor is expressed at low-to-moderate levels on a wide variety of cell types (reviewed in Monk et al., 2007). Expression on lymphocytes remains controversial, with some reports showing expression on inactivated B and T cells with others failing to find this or to show expression restricted to small subsets (reviewed in Klos et al., 2009). Reports of expression in epithelial cells have been shown to be probably artifactual (Klos et al., 2009).
4. Ligand Binding by C5a1 Receptor
a. C5a1 receptor binds both C5a and C5a des-Arg
Determining the locations and mechanisms of interaction between C5a1 receptor and its ligands is vital to the production of effective therapeutics. As for most members of the GPCR family, there is no available crystal structure for the C5a1 receptor, so structural knowledge of the receptor has been based on extensive ligand binding, receptor chimera, and mutagenesis studies. Molecular modeling of the receptor now plays a major role in our understanding, with a recent model of a ligand-bound C5a1 receptor providing novel insights into the mechanisms of ligand and receptor interaction (Nikiforovich et al., 2008). The most extensively studied interaction to date is between the receptor and the full-length natural ligand C5a. In vivo, C5a is rapidly converted into C5a des-Arg by carboxypeptidase enzymes, which remove the terminal arginine residue (Bokisch and Muller-Eberhard, 1970). Experimentally, C5a binds the C5a1 receptor with a Kd of 1 nM, whereas the truncated form has a binding affinity which is 10- to 100-fold lower than that of C5a and is a partial agonist at the C5a1 receptor (Higginbottom et al., 2005). Interactions with this truncated form will also be considered here, where data are available.
5. Two-Site Binding Paradigm
It is now widely accepted that there are at least two sites of interaction between the C5a and C5a1 receptors, a binding paradigm common to members of the GPCR family that bind large macromolecular ligands (Kristiansen, 2004). The first interaction is between acidic residues in the N terminus of the C5a1 receptor and basic residues in the core of C5a; a second binding interaction is thought to occur between the C terminus of C5a and the transmembrane domains and charged residues at the base of the C5a1 receptor extracellular loops. This binding model mainly stems from mutagenesis studies, the creation of chimeric receptors, and interactions with C5a analogs. Initially, experiments showing the C-terminal octapeptide of C5a could alone bind C5a1 receptor led to the proposal of a binding site in the C terminus of C5a (Kawai et al., 1991) (Section III.C.4). It was also shown that C-terminal peptide analogs could act as full agonists of the C5a1 receptor (Ember et al., 1992), leading to the hypothesis that not only does the C terminus of C5a possess a receptor-binding site, but it is also critical for activation of the receptor. However, while removing the terminal pentapeptide of C5a reduced receptor activity, the binding of ligand to the receptor was unaltered, indicating the existence of a second binding site. This site was identified by the demonstration that antibodies directed against the N-terminal domain of the C5a1 receptor reduced binding and activation in response to C5a (Oppermann et al., 1993). In a similar way, deletion of the first 22 residues of the receptor dramatically reduced binding of intact C5a, but the mutated receptor could still be activated by peptide analogs of the C5a C terminus, thus confirming the presence of two distinct binding sites (DeMartino et al., 1994). Critical residues and sequences within these general binding sites have been more challenging to identify, as presented here for each binding site.
a. Binding site one: C5a1 receptor N terminus
Yeast random saturation mutagenesis (RSM) and NMR studies on the N terminus of the C5a1 receptor have indicated the importance of the many acidic residues in C5a binding (Chen et al., 1998; Hagemann et al., 2006). In general, single/double mutations of these have had little effect on binding whereas various multiple mutations within the N terminus of the C5a1 receptor have significantly reduced the affinity of the C5a1 receptor for C5a. For example, the C5a1 receptor(D15,16,18,21N) reduced C5a affinity 40-fold, and the C5a1 receptor(D10,15,16,18,21N) reduced affinity by 133-fold (DeMartino et al., 1994), whereas the C5a1 receptor(D10N), C5a1 receptor(D27N), and C5a1 receptor(D21,27N) had no effect on C5a binding (Mery and Boulay, 1994). The lack of importance of individual residues in forming the binding site was supported by the yeast RSM study, which concluded that no single residues were essential for ligand activation of the C5a1 receptor (Hagemann et al., 2006). More recently, site-specific disulfide-trapping experiments performed in yeast have identified several potential specific points of contact between C5a and the N terminus of the C5a1 receptor (Hagemann et al., 2008).
The N terminus of the C5a1 receptor is highly flexible, resulting in many possible low-energy conformations for the interaction between the C5a and C5a1 receptors (Nikiforovich et al., 2008). Some interactions were consistent across many of the possible conformations and can be rationalized using available mutagenesis data, especially as these data were not used in the building of the model. The aspartate residues Asp15, Asp16, and Asp21 were predicted in most conformations to form a salt bridge with Lys17 within the N terminus of C5a1 receptor, indicating that the reductions in C5a binding affinity induced by multiple aspartate replacements likely result from changes to the overall conformation of the receptor N terminus. Asp27 was predicted to form a less important salt bridge than neighboring Asp28, in accordance with the lack of disruption in binding upon single mutation of this residue to asparagine (Mery and Boulay, 1994). Asp18 was predicted not to be involved in salt bridge formation within the C5a1 receptor, but in a small number of conformations was predicted to interact with Lys19 or Lys20 of C5a, supported by mutagenesis data showing a reduction in binding when these lysine residues within C5a are mutated (Bubeck et al., 1994; Toth et al., 1994). Other important potential contact points include Arg46 (the side chain of which in certain conformations could interact electrostatically with Asp10 or Asp16) and Cys27, which has also been predicted to contact fragment 24–30 of the C5a1 receptor by RSM (Hagemann et al., 2006) and the modeling study (Nikiforovich et al., 2008).
The importance of tyrosine sulfation in forming the ligand-binding site in other GPCRs (Hsu et al., 2005) led to the investigation of Tyr11 and Tyr14 in C5a1 receptor, which have been shown to be sulfated. The C5a1 receptor mutations Y14F and Y11F induced a 50% reduction or complete loss of binding affinity for C5a, respectively, indicating the importance of these sulfations in the formation of the N-terminal binding site (Farzan et al., 2001). The binding of CHIPS, which acts as an antagonist for the C5a1 receptor, has also been shown to be dependent on the sulfation of tyrosine residues 11 and 14 (Ippel et al., 2009; Liu et al., 2011c) (Section IV.C.2).
b. Binding site two: transmembrane/extracellular loop regions of C5a1 receptor
One approach used to ascertain the general regions of C5a1 receptor involved in the second binding site was the construction of chimeric C5a1 receptor/FPR1. Replacing the first extracellular loop of the C5a1 receptor with the corresponding region of the FPR1 had no effect on binding affinity for C5a whereas replacement of EC2 or EC3 abolished binding (Pease et al., 1994). A lack of direct interactions between EC1 and C5a was also supported in the 2008 model (Nikiforovich et al., 2008), but the effects of several point mutations within EC1 that did alter ligand interactions indicate that this region is important for overall structural conformation (Cain et al., 2001b); the presence of the Trp-Phe-Xxx-Gly motif in EC1, which is highly conserved among GPCR, supports this (Klco et al., 2006).
Mutagenesis studies identified Ile116, Arg175, Arg206, Glu199, Asp282, and Val286 as potential interaction sites in ligand binding (Fig. 8). A recent model of C5a bound to the C5a1 receptor has suggested a more extensive list of potential interaction sites with residues 59–74 of C5a being predicted to interact with the side chains of Leu117, Met120, Tyr121 (TM3); Leu167, Phe172 (TM4); Leu187, Cys188, Asp191, His194 (EC2); Glu199, Arg200, Ala203, Arg206, Leu207, Leu209, Pro214 (TM5); Met265 (TM6); and Leu277, Asn279 (EC3) (Nikiforovich et al., 2008). Some of these residues had already been identified as potential ligand-binding sites in mutagenesis studies.
Analysis of mutations of residue Glu199 at the top of TM5 has produced some conflicting biologic results regarding receptor activation. An interaction between Glu199 of the C5a1 receptor and Lys68 of C5a has been suggested in several studies and has been shown to be important for activation of the C5a1 receptor by C5a des-Arg, but not C5a (Crass et al., 1999c; Monk et al., 1995). Specifically, an E199K mutation of the C5a1 receptor produced a lower binding affinity for wild-type C5a and a higher binding affinity for the C5a mutant K68E, indicating the presence of a salt bridge between these two residues of ligand and receptor (Crass et al., 1999c). The recent model of C5a bound to the C5a1 receptor does indeed predict salt bridge formation between Lys68 of C5a and the side chain of residue Glu199 of the C5a1 receptor, with the E199K mutation also disrupting hydrogen bonding between residues Glu199 and His194/Gln71 of the C5a1 receptor (Nikiforovich et al., 2008). In addition to the interaction with Lys68, Glu199 has also been predicted to interact with Arg74 after a lack of response of an E199K mutant to agonists lacking a C-terminal arginine (Crass et al., 1999b; Higginbottom et al., 2005).
Another residue at the extracellular face of TM5 that has been proposed to interact with the C-terminal arginine of C5a (Arg74) is Arg206 (DeMartino et al., 1995; Gerber et al., 2001); however, subsequent studies found only a small effect on receptor activation of the R206A mutation (Cain et al., 2001a). This, along with the fact that the truncated ligand C5a des-Arg binds to the R206A mutant but does not activate it, led to the hypothesis that this mutation simply alters the global structure of the receptor (Monk et al., 2007). R206A mutants have been reported to have varying effects on C5a binding affinities from no change (DeMartino et al., 1995) to significant reductions in binding (Raffetseder et al., 1996). As stated previously, Arg206 has been predicted from modeling to interact with C5a. Within this model, the R206A mutation disrupts the predicted interaction between Arg206 and Arg74 of C5a but induces no major changes in residue-residue interactions and does not support experimental data indicating a complete absence of C5a binding (Nikiforovich et al., 2008).
Buried deep in TM5, Trp214 in the human C5a1 receptor is conserved in the gerbil C5a1 receptor but not in rodent receptors or in human C5a2 and C3a receptors. If substituted by its murine equivalent, Leu, the resulting C5a1 receptor mutant (W213L) is no longer antagonized by W54011 or NDT9520492. Conversely, the murine C5a1 receptor could be sensitized to these antagonists by the reciprocal substitution L214W (Waters et al., 2005). Interestingly, PMX53 showed no activity at either the wild-type murine receptor or the mutant L214W, suggesting that this antagonist binds at a different site.
Other residues that have been predicted to interact with the C terminus of C5a include Arg175 and Asp282. Arg175 is located in EC2, and it too has been predicted to interact with Arg74 of C5a with R175A and R175D mutations, both causing a reduction in C5a binding affinities and activation of C5a1 receptor (Cain et al., 2003; Higginbottom et al., 2005). In the C5a bound model of C5a1 receptor, no major disruptions in binding were predicted with an R175D mutant, indicating no specific interaction between Arg175 and C5a (Nikiforovich et al., 2008). However, the mutagenesis data can be explained by the loss of a salt bridge between Arg175 and Glu179 in the mutant receptors, an interaction that may be crucial in stabilizing the “open” conformation of EC2. Asp282 is at the extracellular face of TM7 and is predicted to form an important interaction with Arg74 of C5a. In support of this, the D282R mutant is relatively unresponsive to C5a but sensitive to C5a des-Arg and analogs (Cain et al., 2001a, 2003). However, again minimal changes were detected in the C5a bound model of the C5a1 receptor for the D282R mutant. Although both Arg175 and Asp282 were not predicted to interact directly with C5a in this model, they have previously been predicted to interact with the cyclic hexapeptide antagonist PMX53, along with Glu199, Arg206, and Ile116 among others (Higginbottom et al., 2005).
Ile116 in TM3 of the C5a1 receptor was predicted to be part of an activation switch after an I116A mutation altered the activity of the C5a peptide C-terminal analog (Phe-Lys-Pro-dCha-Trp-dArg) from antagonist to agonist (Gerber et al., 2001). Further studies mutating this residue found an increased affinity for agonists but a decreased activation of the C5a1 receptor, indicating that the mutation reduced the activation efficiency of the C5a1 receptor (Higginbottom et al., 2005). In more recent modeling studies, the side chains of C5a fragment 59–74 have been predicted to contact the nearby Leu117 residue (Nikiforovich et al., 2008) but not Ile116 itself, whereas modeling based on the binding of hexapeptide antagonist has predicted interactions with Ile116 (Gerber et al., 2001; Higginbottom et al., 2005). Val286, in TM7, has also been proposed to be involved in the activation switch mechanism, with its side chain being proposed to point toward that of Ile116, allowing the two residues to form a binding cleft for ligands. In recent models, Val286 too has been predicted to interact with peptide ligands but not intact C5a (Higginbottom et al., 2005; Nikiforovich et al., 2008).
Much of the information now available indicates that different ligands bind the C5a1 receptor in different ways; even just the cleaving of the terminal arginine residue in C5a des-Arg changes the predicted interactions between receptor and ligand. Through modeling of the ligand-receptor interaction, two important differences in the way in which C5a des-Arg binds compared with C5a were the lack of an interaction between Arg206 and the missing Arg74 of the ligand, and the lack of an interaction between Arg200 and Asp69. In addition to these specific missing interactions, the C terminus of C5a des-Arg was predicted to bind in an entirely different orientation to C5a (Nikiforovich et al., 2008). Available antagonists and analogs of the native ligand are again predicted to interact with different residues of C5a1 receptor; identifying the mechanisms important in eliciting desired effects, such as antagonism, will be crucial in directing the future development of therapeutic agents.
In addition to identifying ligand-binding positions, many mutations in the second extracellular loop of the C5a1 receptor have also been found to result in constitutive receptor activity, identifying a negative regulatory role of EC2 (Klco et al., 2005). As constitutively active GPCR are thought to most likely represent the activated GPCR state, the study of such mutants could provide vital insight into the activation switch of GPCR in general. In modeling studies, the EC2 loops of constitutively active C5a1 receptor mutants were found to contact the EC3 loops, leading to interaction of EC2 domains with TM3 (Nikiforovich and Baranski, 2012). This in turn triggers the movement of TM7 toward TM2 and TM3 with a resultant change in hydrogen bonding between these regions known to be crucial for C5a1 receptor activation (Nikiforovich et al., 2011).
c. Comparison with C5a2 receptor
The second identified receptor for C5a, the C5a2 receptor, shares ∼35% identity with the C5a1 receptor; in contrast with the C5a1 receptor, the C5a2 receptor binds C5a des-Arg and C5a with similar affinities (Scola et al., 2007), implying different binding mechanisms. Interfering with the N terminus of the C5a2 receptor using an antibody did not alter the binding of C5a to the C5a2 receptor, but it did significantly inhibit the binding of C5a des-Arg to the C5a2 receptor (Scola et al., 2007). Replacing the N terminus of the C5a2 receptor with that of the C5a1 receptor did not affect the affinity for C5a des-Arg. Many of the residues at the extracellular face of the transmembrane domain of C5a1 that have been shown to influence ligand binding are also present in the C5a2 receptor (Fig. 8).
6. C5a1 Receptor Signaling
a. G protein mediated
Often the most potent of the complement peptides, C5a has also been the most intensively studied with regard to signal transduction (reviewed in Monk et al., 2007; Klos et al., 2009). The C5a1 receptor couples primarily to the PT-sensitive G protein Gαi2 (Sheth et al., 1991; Skokowa et al., 2005) in cells such as neutrophils and mast cells or, less frequently, to PT-insensitive G proteins such as Gα16/Gα15 (Amatruda et al., 1993; Monk and Partridge, 1993; Davignon et al., 2000) in cells of the monocytic lineage. The C5a1 receptor is unusual among GPCR in being precoupled to G proteins (Siciliano et al., 1990); mutants that are unable to precouple in this way have reduced affinity for C5a (Raffetseder et al., 1996). Precoupling is also seen in a mutant of the α2B adrenoreceptor, where it increases agonist potency (Ge et al., 2003); it is tempting to speculate that this may also be the case for the C5a1 receptor, allowing the response to C5a to predominate over responses to other chemoattractants such as C3a.
As with the C3a receptor, the downstream response to receptor ligation is cell type dependent. For example, the Ca2+ response in C5a-activated neutrophils is predominantly due to release from intracellular compartments, whereas in monocyte-like cells a much greater contribution from extracellular influx is observed (Monk and Partridge, 1993). Similarly, mast cells respond to C5a with a rapid and transient increase in Ca2+i (Hartmann et al., 1997) whereas microglial cells respond with a more prolonged response (Moller et al., 1997). In neutrophils and macrophages, sphingosine-1-phosphate (S-1-P) production by sphingosine kinase 1 is required for the C5a-stimulated release of Ca2+ from intracellular stores (Ibrahim et al., 2004; Melendez and Ibrahim, 2004). S-1-P also up-regulates the expression of the C5a2 receptor (the second C5a receptor) in mouse neutrophils, which is thought to be a protective response against endotoxemia (Bachmaier et al., 2012).
b. Arrestin signaling
Phosphorylation of the C5a1 receptor leads to association with arrestins and subsequent targeting to clathrin-coated pits for internalization (Braun et al., 2003). PK-CβII and GRK2 interact with intracellular loop 3 and the C-terminal domain of the C5a1 receptor and may be responsible for the serine/threonine phosphorylation of the receptor (Suvorova et al., 2008). A dileucine motif in the C terminus of the C5a1 receptor is thought to stabilize the “eighth helix” (Gln305–Arg320) proximal to the membrane (Suvorova et al., 2008). This structure, sometimes stabilized by a palmitoylated cysteine residue, often acts as a protein interaction site in GPCR (Huynh et al., 2009) and may be involved in receptor internalization (Thomas et al., 1995). Interestingly, the other receptors—the C3a receptor and C5a2 receptor—both have cysteine residues in the C-terminal domains whereas the C5a1 receptor does not (Fig. 8). In some GPCR with cysteine residues in the C-terminal domain, such as bradykinin B2, helix 8 is critical for the association with β-arrestins, so the lack of a cysteine residue in this position in the C5a1 receptor may be responsible for some of the differences in signaling observed for the complement peptide receptors. Unlike the C3a receptor, there is no evidence that the C5a1 receptor signals through arrestins (Gripentrog and Miettinen, 2008), although an association occurs (Braun et al., 2003; Kalant et al., 2005; van Lith et al., 2009). Kinase activation by the C5a1 receptor is almost entirely Gαi dependent (Buhl et al., 1994; Gripentrog and Miettinen, 2008), with little evidence to date that arrestins are directly involved. It is also interesting that the C terminus of the C5a1 receptor can be substantially deleted (from residue 311) without a negative effect on signaling (Matsumoto et al., 2007a,b; Monk et al., 1994), although it is required for trafficking to the cell surface and for internalization (Bock et al., 1997).
The ability of the C5a1 receptor to form homodimers and heterodimers has been demonstrated (Geva et al., 2000; Huttenrauch et al., 2005; Klco et al., 2003). In both cases, ligation of a dimerized C5a1 receptor can cause the phosphorylation and/or internalization of the partner receptor. The functional consequences of this ability to form oligomers are unknown but may be linked to the cross-modulation of the response to chemoattractants or the rapid down-regulation of the C5a1 receptor in severe conditions such as sepsis.
D. C5a2 Receptor as a C5a and C5a des-Arg Receptor
1. Sequence and Genetics
In 2000, a seven-transmembrane domain receptor sharing high sequence identity with human C5AR1 was cloned and designated human C5a receptor-like 2 (C5L2, GPR77) (Ohno et al., 2000). The C5a2 receptor was found to be encoded for by the human GPR77 gene, which is located within the respective gene cluster on chromosome 19q13 (Lee et al., 2001). GPR77, now to be described as C5AR2, shows the two-exon structure characteristic of chemoattractant receptor family members, with the 5′-UTR and initiating methionine encoded in the first exon, and the actual coding sequence and 3′-UTR in the second (Gerard et al., 1993). Message size heterogeneity was later described for the human and mouse C5a2 receptors and was attributed to alternative splicing in the UTR (Okinaga et al., 2003). Two SNP possibly linked to fatty acid metabolism have been identified in the human C5AR2 gene. First, SNP 968G/T, causing an amino acid exchange of S323I, was found associated with inheritable combined hyperlipemia in a French Canadian family, but could not be identified in a screening that involved Han, Uygur, and Kazakh subjects with familial hyperlipemia or type 2 diabetes (Cui et al., 2009b; Marcil et al., 2006; Zheng et al., 2011a). Second, a recently discovered 698C/T substitution, leading to a P233L amino acid exchange, appears to be a genetic marker of coronary artery disease and type 2 diabetes mellitus in the Han and Uygur populations in northwestern China (Zheng et al., 2011b, 2012). Furthermore, C5a2R displays two synonymous SNP at 614G/A and 860C/T without any known association with human disease (Birney et al., 2006). Other SNP have not yet been associated with disease (http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?locusId=27202; Sherry et al., 2001). Due to its homology with the chemoattractant receptors C5a1 receptor, C3a receptor, FPR, and ChemR23, the C5a2 receptor was categorized into the GPCR subfamily A8 (Joost and Methner, 2002). Correspondingly, the C5a2 receptor was clustered with A8 subfamily members and other chemoattractant receptors such as the type-2 angiotensin-II receptor, bradykinin receptor, and several orphan receptors in a study that compared the transmembrane domain-binding cavities of various human GPCR (Surgand et al., 2006).
2. Post-translational Modification
a. Glycosylation
A potential N-linked glycosylation site exists at Asp3 of the human C5a2 receptor, and, although the glycosylation status of this residue has not been formally investigated, the apparent molecular weight observed by Western blot analysis suggests that the receptor is glycosylated in vivo (Okinaga et al., 2003).
b. Tyrosine sulfation
Like the C5a1 receptor, there are three tyrosine residues at the N terminus of the C5a2 receptor, all of which (Tyr8, Tyr10, and Tyr13) are proximal to acidic residues, making them potential sulfation sites (Rosenquist and Nicholas, 1993). Mutation of Tyr10 or Tyr13 but not Tyr8 can affect ligand binding to the C5a2 receptor, but only the binding of C5a des-Arg is significantly inhibited (Scola et al., 2007).
c. Phosphorylation
The C5a2 receptor has only seven serine/threonine residues at the C terminus, in comparison with the C5a1 receptor which has 11. A low level of phosphorylation of the C5a2 receptor has been observed after C5a treatment of transfected cells (Okinaga et al., 2003), suggesting that few of these residues are phosphorylated, perhaps because of the lack of the PK-CβII binding site found on the third intracellular loop of the C5a1 receptor (Section IV.C.2).
d. S-acylation
Similar to the C3a receptor, the C5a2 receptor has a cysteine in the C-terminal domain that is a potential S-acylation site. It is not known whether any modification of this residue occurs in vivo.
3. C5a2 Receptor Expression
In most tissues, the C5a2 receptor is coexpressed with the C5a1 receptor, with overall levels tending to be lower than those of the C5a1 receptor (Okinaga et al., 2003; Lee et al., 2008; Bamberg et al., 2010). When the C5a2 receptor was first described, its transcripts were found to be most abundant in human leukocytes and the spleen by Northern blot analysis; using polyclonal antiserum, the receptor protein could be detected on immature but not mature DC (Ohno et al., 2000). In mice, bone marrow and peripheral blood leukocytes were identified as the most abundant sources of the C5a2 receptor message (Okinaga et al., 2003). Subsequently, C5a2 receptor expression on the mRNA or protein level was described for a broad range of immune and nonimmune mouse, rat, and human cells and tissues, including neutrophils, macrophages, thyroid, adipocytes, skin fibroblasts, neurons and astroglia of the CNS, spinal cord, adrenal gland, lung, heart, liver, kidney, ovary, and testis (Lee et al., 2001; Okinaga et al., 2003; Otto et al., 2004; Gao et al., 2005; Gavrilyuk et al., 2005; Kalant et al., 2005; Bamberg et al., 2010).
a. Intracellular or extracellular localization for C5a2 receptor
Although it is generally accepted that mouse, rat, and human PMN express the C5a2 receptor, controversy exists about C5a2 receptor localization in this cell type. Some investigators did not detect significant amounts of the receptor on the cell surface (Otto et al., 2004; Bamberg et al., 2010), but others could verify surface expression in binding assays or by flow cytometry (Okinaga et al., 2003; Gao et al., 2005; Rittirsch et al., 2008). Finally, Scola et al. (2009) suggested a wide natural variation in C5a2 receptor surface expression between human individuals. In some cases, opposing findings were subsequently published by the same group (Okinaga et al., 2003; Bamberg et al., 2010). These discrepancies may in part be due to differential regulation of C5a2 receptor expression depending on the activation status of the cells. Also, not all of the polyclonal antisera or monoclonal antibodies used may have been suitable to detect the C5a2 receptor on primary cells. Some antibodies raised against antigenic peptides or against transfected cells fail to recognize their antigen on primary cells, despite being able to detect it on transfected cells. This should be taken into account whenever newly developed antibodies are used for detection of this receptor on primary cells (Jorg Köhl, personal communication).
b. Regulation of expression
The regulation of C5a2 receptor expression is influenced by proinflammatory and anti-inflammatory signals. Several cell lines have been shown to express the C5a2 receptor either constitutively or after induction with certain substances. On HeLa cells, the C5a2 receptor but not the C5a1 receptor is detected at low levels, and it decreases upon treatment with IFN-γ or tumor-necrosis factor α (TNF-α). Both the C5a1 and C5a2 receptors are absent from the surface of native HL-60 or U937 cells, but their expression can be induced with Bt2-cAMP or IFN-γ, whereas TNF-α has no effect (Johswich et al., 2006). In rat astrocytes, C5a2 receptor mRNA and protein were found to be up-regulated by noradrenaline, associated with suppression of nitric oxide synthase 2 and NF-κB genes (Gavrilyuk et al., 2005). Huber-Lang et al. (2005) could show that prolonged exposure of rat and human PMN to C5a in vitro decreased C5a2 receptor expression. Likewise, C5a2 receptor is reduced on PMN isolated from septic patients or from rats subjected to sepsis induced by cecal ligation and puncture (CLP). TLR activation in human PBMCs was found to inhibit C5a2 receptor expression, thereby increasing cellular responsiveness to C5a1 receptor signaling. Thus, the C5a2 receptor appears to be differentially regulated in different cell types, and its expression level may influence cellular functions, especially in the context of inflammation.
4. Ligand Binding by C5a2 Receptor
a. C5a and C5a des-Arg
The C5a2 receptor was initially speculated to be the long sought-after C4a receptor (Ohno et al., 2000; Lee et al., 2001), but in fact turned out to be a second human receptor for C5a, binding both C5a and C5a des-Arg with high affinity (Cain and Monk, 2002; Okinaga et al., 2003; Johswich et al., 2006). The Kd values of C5a2 receptor binding were in the range of ∼3–10 nM for C5a, and ∼12–36 nM for C5a des-Arg, respectively, with C5a2 receptor having 10- to 100-fold higher affinity for C5a des-Arg in comparison with the C5a1 receptor. Although the affinity of the human C5a2 receptor is about equal for C5a and C5a des-Arg, both mouse and rat C5a2 receptor homologs clearly prefer species-matched C5a des-Arg over C5a, with mouse C5a2 receptor displaying an over 4000-fold higher affinity for C5a des-Arg than for C5a. As in humans, the rodent C5a2 receptor outmatches the rodent C5a1 receptor in its affinity for C5a des-Arg (Scola et al., 2007). Moreover, in one study, the human C5a2 receptor exhibited distinctly (∼100-fold) slower on-rates and slightly (∼3-fold) slower off-rates for C5a at 0°C and in the presence of azide (Okinaga et al., 2003).
b. Other complement peptides
Whether the C5a2 receptor can bind C3a/C3a des-Arg or C4a/C4a des-Arg is a matter of controversy. Moderate to low binding of C3a and C4a to a site distinct from the C5a binding site was first detected in 2002 (Cain and Monk, 2002). Shortly thereafter, Kalant et al. (2003) reported binding of the des-arginated ligands C3a des-Arg and C4a des-Arg to the C5a2 receptor, and observed increased triglyceride synthesis in C5a2 receptor-transfected cells after stimulation with C3a des-Arg, also referred to as acylation-stimulating protein (ASP), or with C3a. In contrast, others were unable to detect binding of any ligands apart from C5a and C5a des-Arg to C5a2 receptor (Okinaga et al., 2003; Johswich et al., 2006), and provided evidence that C3a and C3a des-Arg interact with plastic surfaces in a pattern highly suggestive of specific receptor-ligand binding (Johswich et al., 2006). The complex issue of C5a2 receptor as a functional receptor for C3a des-Arg is reviewed in more detail in Section IV.E.
c. Comparison with C5a1 Receptor ligand binding
C5a and C5a des-Arg binding by the C5a2 receptor differs from that of the C5a1 receptor, despite the two receptors sharing patterns of tyrosine and acidic N-terminal residues as well as charged and hydrophobic residues in the extracellular loops and transmembrane regions (Scola et al., 2007). In the C5a1 receptor, these residues are known to interact with the core and C terminus of C5a, respectively (Oppermann et al., 1993; Pease et al., 1994). However, the well-characterized antagonist PMX53 (Wong et al., 1999; Taylor et al., 2001) and other effective peptidic and nonpeptidic ligands for the transmembrane hydrophobic pocket of the human C5a1 receptor are moderate-to-poor inhibitors of ligand binding to the C5a2 receptor, suggesting that the C5a core segment is the most relevant for C5a binding to the C5a2 receptor (Okinaga et al., 2003; Otto et al., 2004; Scola et al., 2007). An antibody raised against the N terminus of human C5a2 receptor inhibited the binding of C5a des-Arg to the C5a2 receptor but left C5a binding unaffected, while substituting the N terminus of the C5a2 receptor with that of the C5a1 receptor had little effect on the high affinity of the C5a2 receptor for C5a des-Arg (Scola et al., 2007). Finally, mutational analyses revealed three N-terminal residues of the C5a2 receptor that were essential for C5a des-Arg binding but had little involvement in C5a binding (Scola et al., 2007). Taken together, these findings indicate that the N terminus is an important but not the only determinant of the high affinity of the human C5a2 receptor for C5a des-Arg, and that, unlike the C5a1 receptor, critical N-terminal residues of the C5a2 receptor interact with C5a des-Arg only, not with C5a.
d. Modulation of ligand binding
While a number of blocking antibodies specific for the mouse, rat, or human C5a2 receptor have been developed and employed by different investigators in vitro and in vivo (Gao et al., 2005; Cui et al., 2007; Lee et al., 2008; Bamberg et al., 2010), to our knowledge the only antagonists for the human C5a2 receptor are the C5a mutant jun/fos-A8, and its derivative jun/fos-A8Δ71–73, which inhibit ligand binding to both the C5a1 receptor and C5a2 receptor (Heller et al., 1999; Otto et al., 2004).
5. C5a2 Receptor Signaling
a. G protein mediated
Despite sharing the 7TM structure of typical GPCR, the C5a2 receptor is widely accepted to be uncoupled from G proteins (Cain and Monk, 2002; Kalant et al., 2003; Okinaga et al., 2003; Johswich et al., 2006; Scola et al., 2009). This is believed to be due to changes in conserved sequences shared by most functional chemoattractant or chemokine receptors, including the C5a1 receptor. Primarily, both mouse and human C5a2 receptors lack the highly conserved “DRX” motif usually found at the end of TM3 (Oliveira et al., 1994; Ballesteros et al., 1998), actually Asp-Arg-Phe in the human C5a1 receptor. In the C5a2 receptor, it reads Asp-Leu-Cys, thus lacking the arginine residue that is most important for the interaction with G proteins (Savarese and Fraser, 1992; Scheer et al., 1996; Cain and Monk, 2002; Okinaga et al., 2003). Mutating the respective arginine in FPR, CCR5, histamine H2 receptor, and other receptors greatly decreases or altogether abolishes their ability to couple to G proteins (Prossnitz et al., 1999; Alewijnse et al., 2000; Lagane et al., 2005; Rovati et al., 2007). Conversely, C5a binding to a C5a2 receptor in which Asp-Leu-Cys is mutated to Asp-Arg-Cys induces a low level of intracellular Ca2+ mobilization in HEK293 cells cotransfected with Gα16, indicative of a partial gain-of-function in coupling to this rather promiscuous G protein subtype (Okinaga et al., 2003). However, RBL-2H3 cells transfected with the C5a2 receptor mutated to resemble the C5a1 receptor at this motif did not respond to C5a or C5a des-Arg in an intracellular free Ca2+ assay (Scola et al., 2009). Given that rat basophilic leukemia (RBL) cells do not endogenously express Gα16, variations in the amount and selectivity of the two G proteins may explain these differing results.
Further residues in the mouse and human C5a2 receptors that differ from conserved sequences in the C5a1 receptor and other GPCR are in the “NPXXY” motif (Asn-Pro-Leu-Met-Phe in C5a2 receptor) in TM7, and deletions of serine/threonine residues and a basic region (239Lys-Thr-Leu-Lys in C5a1 receptor) that truncate the third intracellular domain (Cain and Monk, 2002; Scola et al., 2009). Substitution of the analogous serine/threonine residues with alanine in the C5a1 receptor does not interfere with ligand binding but abolishes downstream signaling, suggesting that this region is also involved in G protein coupling (Bock et al., 1997). Different mutations of the NPXXY motif have been shown to inhibit the activation of Gq by the cholecystokinin B receptor and to disturb internalization, ligand binding, or coupling to selected downstream pathways in the case of FPR (Gales et al., 2000).
In an effort to further elucidate the regions responsible for uncoupling the C5a2 receptor from G proteins, a recent study compared the C5a1 receptor and the C5a2 receptor with four substitution mutants of the human C5a2 receptor: DRX (Asp-Arg-Phe), NPXXY (Asn-Pro-Met-Leu-Tyr), DRX/NPXXY double mutant, a chimeric receptor carrying the third intracellular loop of C5a2 receptor, and a triple mutant containing all of the mentioned modifications. Unlike the C5a1 receptor, none of the mutants exhibited ligand-induced internalization, Ca2+ mobilization, or translocation of endogenous β-arrestin 1 in RBL-2H3 cells, and neither did the wild-type C5a2 receptor (Scola et al., 2009). Taken together, multiple structural features contribute to effectively restrict G protein-coupled signaling by the C5a2 receptor.
In accordance, several groups have consistently found that binding of C5a, C3a, C4a, or their des-arginated derivatives to the wild-type C5a2 receptor in vitro does not support G protein-mediated cellular responses such as Ca2+ mobilization, degranulation, chemotaxis, or activation of the mitogen-activated protein kinase (MAPK) pathway, in striking contrast to the diverse effects induced by the C5a1 receptor (Cain and Monk, 2002; Kalant et al., 2003; Okinaga et al., 2003; Johswich et al., 2006; Scola et al., 2009). Pretreatment of human C5a2 receptor-expressing RBL-2H3 cells with C5a, C5a des-Arg, C3a, or C4a could facilitate FcεRI-mediated secretory responses (Cain and Monk, 2002), implying that, at the most, the C5a2 receptor exhibits very limited capacities for signal transduction under particular conditions.
b. Internalization of C5a2 receptor
The C5a2 receptor has been suggested to act as a decoy receptor, limiting the availability of C5a or C5a des-Arg to competing receptors, especially the C5a1 receptor (Okinaga et al., 2003; Scola et al., 2009). This concept was first described in 1993 for IL-1RII (Colotta et al., 1993), and has since then been extended to GPCR including D6, DARC, US28, CCX CKR (CCRL-1), CRAM-B (CCRL-2), and CXCR7 (Nibbs et al., 1997; Gosling et al., 2000; Randolph-Habecker et al., 2002; Lee et al., 2003; Burns et al., 2006; Hartmann et al., 2008; Leick et al., 2010). By sequestering ligands and targeting them for degradation, these receptors negatively regulate cellular responses to inflammatory mediators. Decoy receptors are commonly uncoupled from G proteins, most of them due to lack of the DRX motif. Unlike signaling receptors, they do not undergo ligand-induced internalization, but some of them recycle constitutively between the cell surface and intracellular compartments, thus efficiently removing ligands from the extracellular space. This may lead to a relatively high proportion of intracellular receptors, as demonstrated for D6 (Weber et al., 2004).
It has been demonstrated by the use of human PMN or transfected cell lines, including RBL-2H3, that significant amounts of C5a2 receptor are located intracellularly, and that human C5a2 receptor is neither phosphorylated nor internalized upon binding of C5a or C5a des-Arg (Cain and Monk, 2002; Okinaga et al., 2003; Scola et al., 2009; Bamberg et al., 2010). Instead, clathrin-dependent constitutive internalization occurs, resulting in net transport of ligand into the cell and in ligand degradation (Scola et al., 2009). This appeared to be more efficient for C5a des-Arg than for C5a, suggesting that the C5a2 receptor might be responsible for removing the more abundant des-arginated form from the circulation (Scola et al., 2009). Interestingly, a predominantly intracellular location and constitutive internalization were recently reported for FPR3, which, despite being capable of eliciting cellular responses, does not seem to activate G proteins (Rabiet et al., 2011). In the respective study, the N-terminal extracellular region and TM1 were identified to contain the structural determinants of this unusual recycling behavior, which appears to be clathrin- and β-arrestin-independent. Based on these results, the investigators hypothesized that FPR3 may serve a ligand scavenging or intracellular receptor function, in analogy to what has been described for the C5a2 receptor (Scola et al., 2009; Bamberg et al., 2010). Again, contrasting data have come from studies investigating the C5a2 receptor as a receptor for C3a des-Arg /ASP: here, ligand-induced clathrin-dependent receptor endocytosis and recycling were observed in HEK293 cells, and were impaired by an S323I mutation, indicating that this residue is vital for C3a des-Arg /ASP-induced C5a2 receptor activation (Cui et al., 2009b).
c. Arrestin-mediated signaling
Because there is no evidence for the involvement of G proteins in the observed C5a2 receptor-mediated effects, the idea emerged that the C5a2 receptor uses G protein-independent signaling, such as pathways targeted by β-arrestins (reviewed in Defea, 2008). A β-arrestin 1-GFP fusion protein was translocated to the human C5a2 receptor in transfected HEK293 cells after stimulation with C5a, C5a des-Arg, C3a, or C3a des-Arg (Kalant et al., 2005). The presence of large amounts of exogenous protein or varying preferences of the C5a2 receptor for β-arrestin subtypes depending on the cell type may account for the divergence between these and other results (Scola et al., 2009). Later, two independent studies of C5a2 receptor-transfected cells concordantly reported C5a-induced translocation of β-arrestin 2 (Cui et al., 2009b; van Lith et al., 2009). In addition, the role attributed to the C5a2 receptor as a functional receptor for C3a des-Arg (ASP) in lipid metabolism seems to be associated with recruitment of β-arrestin 2-GFP (Cui et al., 2009a). Furthermore, it was recently shown in the human mast cell line HMC-1 that G protein-mediated ERK1/2 activation downstream of the C3a receptor may be negatively regulated by β-arrestins (Vibhuti et al., 2011). Colocalization of β-arrestin 1 with the C5a1 and C5a2 receptors occurs in endosomal vesicles of C5a-stimulated human PMNs and was found to be dependent on C5a1 receptor activation (Bamberg et al., 2010). Assuming a regulatory interplay of the C5a2 receptor with the C5a1 receptor, as they had already hypothesized earlier (Gerard et al., 2005), the investigators showed that two putative signaling events downstream of β-arrestin 1, ERK1/2 activation and chemotaxis, are enhanced after antibody blockade of the C5a2 receptor. However, it remains to be confirmed that β-arrestin 1 mediates C5a1 receptor-induced functions in human primary cells or transfected cells, as they may as well result from G protein activation (Buhl et al., 1994; Nilsson et al., 1996; Haribabu et al., 1999; Schraufstatter et al., 2009; Nishiura et al., 2010b;). In summary, there are credible indications of an active signaling function of the C5a2 receptor involving β-arrestin, but further confirmation should be aimed at elucidating how this modulates the cellular response to C5a.
d. Dimerization
For the C5a1 receptor, homodimerization as well as heterodimerization with CCR5 have been observed, and have been shown to lead to homologous or heterologous co-internalization of phosphorylation-deficient C5a1 receptor or of CCR5, respectively, upon C5a binding to the wild-type C5a1 receptor (Huttenrauch et al., 2005; Rabiet et al., 2008). Interestingly, the chemokine receptor CXCR7, which is known to associate with Gαi but does not elicit any intracellular signaling (Levoye et al., 2009), has been found to heterodimerize with CXCR4. The dimers were constitutively associated with β-arrestin and concomitantly decoupled from Gαi, resulting in potentiated CXCL12-induced signaling via MAPK pathways to promote cell migration (Decaillot et al., 2011). It is tempting to speculate that a similar interaction could take place between the C5a1 receptor and the C5a2 receptor, shifting or switching downstream signaling from G protein-dependent to β-arrestin-mediated events, and thus providing at least some explanation for the unresolved controversies about the functions of the C5a2 receptor. This possibility has been considered, but no evidence was seen in mouse cells (Chen et al., 2007), and it was not reported in human PMN, even when modulation of C5a1 receptor signaling was observed (Bamberg et al., 2010). The investigators considered differential regulation of downstream pathways rather than dimerization as the underlying mechanism (Chen et al., 2007; Bamberg et al., 2010). However, more recent evidence points to the formation of hetero- and homodimers by C5a1 and C5a2 (Croker et al., 2012; Poursharifi et al., 2013), although the functional significance is still unclear.
e. Cross-talk with Toll-like receptors
Apparently, nonsignaling 7TM receptors may also act as chemokine chaperones or transporters (Middleton et al., 1997; Nibbs et al., 2003), but to date, none of these functions have been attributed to the C5a2 receptor. Lately, a cross-talk between TLR and the C5a1 receptor/C5a2 receptor has been described, in which TLR activation reduces C5a2 receptor activity, thereby releasing the C5a1 receptor from negative modulation and enhancing the inflammatory response to C5a (Raby et al., 2011). This study suggests that in the course of every inflammatory reaction, a balance between TLR-induced cellular hypersensitivity to C5a and the C5a2 receptor counteracting this effect has to be established, and that the net outcome of the process varies considerably among individuals.
6. Pro- and Anti-inflammatory Effects of C5a2 Receptor
The human C5a2 receptor shares many features with the well-characterized chemokine scavenging receptor D6, clearly implying they have analogous functions. The C5a2 receptor should thus be expected to have anti-inflammatory effects in vivo and to negatively regulate C5a1 receptor-mediated responses (Locati et al., 2005; Borroni et al., 2006). In fact, when compared with wild-type animals, mice with a targeted deletion of C5ar2 mounted an exaggerated inflammatory reaction, with a 2- to 3-fold increased neutrophil influx and higher levels of IL-6 and TNF-α in a model induced by ovalbumin (OVA) of pulmonary immune complex injury; the chemotaxis of murine C5ar2−/− bone marrow cells was also enhanced in vitro (Gerard et al., 2005). Up-regulation of the C5a2 receptor in rat astrocytes is associated with suppression of proinflammatory genes (Gavrilyuk et al., 2005); upon stimulation with C5a and LPS, rat neutrophils pretreated with a C5a2 receptor-blocking antibody produced higher amounts of IL-6 than untreated control cells (Gao et al., 2005). Similarly, a study of human septic patients found that low C5a2 receptor expression on PMN correlated with multiorgan failure and nonsurvival, and corresponding results were obtained from rats subjected to CLP-induced sepsis (Huber-Lang et al., 2005).
The decoy receptor hypothesis has been contrasted by a recent report that the C5a2 receptor can positively modulate C5a1 receptor- and C3a receptor-induced inflammatory responses (Chen et al., 2007). The C5ar2−/− mouse strains on C57BL/6 and BALB/c background used here were generated independently of the animals used for the aforementioned studies (Gerard et al., 2005), and several disease models were investigated. In comparison with wild-type mice, C5ar2−/− animals showed reduced macrophage infiltration in peritonitis induced by thioglycolate (TGC), and less pronounced cellular infiltration and activation after injection of C5a and TGC in a dorsal air-pouch model of inflammation (Chen et al., 2007). C5ar2−/− mice were also less affected by OVA-induced airway hyperresponsiveness than wild-type mice, as characterized by one lung function parameter and histologic evaluation of the inflammatory infiltrate. After in vitro stimulation with C3a or C5a, up-regulation of cell-surface markers and activation of downstream MAPK pathways were diminished in neutrophils and macrophages from C5ar2−/− animals. Then again, deletion of C5ar2 seems to render mice more susceptible to LPS-induced shock, as indicated by elevated IL-1β serum levels and high mortality after i.p. injection of LPS (Chen et al., 2007).
Likewise, Rittirsch et al. (2008) support a proinflammatory role for the C5a2 receptor: according to their observations in C5ar1−/− and C5ar2−/− mice subjected to CLP-induced sepsis, and treated or not with blocking antibodies, the synergistic action of both receptors is required for the development of full-blown sepsis. Only a combined pretreatment with both anti-C5a1 receptor and anti-C5a2 receptor antibodies could lower the plasma levels of proinflammatory mediators and reduce lethality, with the role of the C5a2 receptor being linked to the release of HMGB1 (Rittirsch et al., 2008). It may be assumed that the animals used in this study were provided by the Gerard group, although this was not explicitly specified.
Recently, a complex role for the C5a2 receptor has emerged in the pathogenesis of experimental allergic asthma in mice. Here, the C5a2 receptor acts at the mDC/T cell interface to suppress TH1 and TH17 polarization, while concomitantly driving TH2 cytokine production in other pulmonary cells (Zhang et al., 2010).
E. C5a2 Receptor as a Possible Receptor for C3a des-Arg /Acylation Stimulating Protein
1. Mediators of Metabolic Activity
The adipose tissue is an active endocrine organ that secretes mediators such as adiponectin, leptin, or resistin, which are prototypical adipokines (or adipocytokines) (Kershaw and Flier, 2004). This term describes a group of factors secreted by the white adipose tissue. Adiponectin is a member of the pattern-recognition family of defense collagens, and it interacts with a number of target molecules, including surface structures of damaged endothelium and apoptotic cells. It can bind to complement factor C1q, thus activating the classic pathway of the complement system (Peake et al., 2008), and it is regulated by factor H, a soluble complement inhibitor (Peake and Shen, 2010). Other adipokines include cytokines and chemokines such as IL-1, IL-6, IL-8, IL-18, TNF-α, MCP-1 (CCL2), CXCL5, and proteins involved in the acute phase response such as PAI-1 (Kershaw and Flier, 2004; Ouchi et al., 2011). Adipsin, also known as factor D of the alternative pathway, was one of the first recognized adipokines (Cook et al., 1987; White et al., 1992). Adipokines influence energy metabolism, appetite, body weight, and insulin resistance. Most of them stimulate inflammation, but some (adiponectin and SFRP5) are anti-inflammatory. They play an important role in obesity-related metabolic dysfunction, the pathogenesis of which implies chronic low-grade inflammation (Ouchi et al., 2011), as well as in classic inflammatory diseases such as sepsis (Hillenbrand et al., 2012). Hence, there is a growing interest in adipokines in biomedical sciences.
2. Acylation-Stimulating Protein
ASP has been proposed mainly by the Sniderman and Cianflone group as a potent mediator of triglyceride synthesis, simultaneously increasing glucose transport, and as an adipokine with both metabolic and immune functions. Purification from human serum and sequence analysis have revealed that ASP is identical to the des-arginated form of C3a: C3a des-Arg (Cianflone et al., 1987, 1989b, 2003). Functional impairment of ASP has been suggested as major cause of familial hyperapobetalipoproteinemia (Sniderman et al., 1985; Cianflone et al., 1988, 1990; Sniderman and Cianflone, 1994). Moreover, it has been claimed that the C5a2 receptor serves as a signaling receptor for ASP on adipocytes and other cell types (Baldo et al., 1993; Kalant et al., 2003; Kalant et al., 2005; Maslowska et al., 2005; MacLaren et al., 2008). Although dozens of elaborate studies have already been published, these issues remain highly controversial.
3. The Challenges in Designing Studies Involving Acylation-Stimulating Protein
Due to the complexity of this topic, the involved research groups face a variety of challenges with study design and data interpretation. The following points must be considered to critically assess the respective publications and our knowledge of ASP.
a. Lack of an acylation-stimulating protein knockout model
C3a des-Arg /ASP as a cleavage product of C3 is generated in all situations where the complement cascade is activated by the main activation routes. Unfortunately, it is impossible to generate a knockout mouse with an isolated deletion of C3a des-Arg /ASP without interfering with other functions of the complement system.
b. Problems with C3 knockout animals
Although mice deficient in the precursor molecule C3 have erroneously been described as “ASP knock-out mice” or “ASP-deficient mice” (Cianflone et al., 1999), beyond lacking C3a des-Arg /ASP (Murray et al., 1999c,d, 2000; Xia et al., 2002, 2004) these animals are deficient in main complement effector functions downstream of C3—that is, opsonization due to C3b, inflammation and immune modulation mediated by C3a and C5a, sublytic cell activation, and cell lysis of bystander cells caused by MAC. Thus, C3 knockout mice cannot be considered an adequate model to specifically investigate the role of C3a des-Arg (ASP).
c. Receptor for acylation-stimulating protein
As described previously, the C5a2 receptor is widely accepted as a receptor for C5a and C5a des-Arg. Its role as a signaling receptor for C3a des-Arg /ASP, however, is controversial (Kalant et al., 2005; Johswich et al., 2006; Johswich and Klos, 2007; Klos et al., 2009). Thus, mice with a C5ar2 gene knockout might be more appropriate for the analysis of ASP-related issues than C3 knockout mice, the caveat being that it remains to be proven that the C5a2 receptor is indeed a functional receptor for ASP.
d. C3a des-Arg regarded as inert
Traditionally, a large number of researchers working in the complement field consider C3a des-Arg as a relatively stable metabolite without any biologic potential, making it a useful marker for complement activation (Burger et al., 1988; Klos et al., 1988; Hartmann et al., 1993; Stove et al., 1995). Although C5a des-Arg retains some affinity for its receptors, most complement researchers assume that C3a des-Arg does not interact with the C3a receptor (Klos et al., 1992; Ames et al., 1996; Crass et al., 1996; Johswich et al., 2006). It is thus neither internalized nor degraded, remaining detectable in the bloodstream. In line with this point of view, correlations of an increase in C3a des-Arg /ASP in body fluids of humans or animals with a certain genotype or a disease suggest a role of complement activation and inflammation in general, and not necessarily a role of this particular complement cleavage product and its assumed function. This is further stressed by the fact that unbalanced obesity and insulin resistance are thought to be linked to chronic inflammation in adipose tissue (Ouchi et al., 2011), and that complement activation is a typical early component of an inflammatory response (Manabe, 2011).
e. Difficulties working with C3a/C3a des-Arg
Partially due to their charge, C3a and C3a des-Arg are difficult to handle, more difficult than C5a and C5a des-Arg (Hugli, 1975). C3a and C3a des-Arg /ASP show a significantly higher nonspecific binding to cells and surfaces (Burg et al., 1996). For this reason, plastic materials have to be coated with silicone, albumin, or other inert proteins when handling C3a or C3a des-Arg /ASP. Synthetic C-terminal peptides of C3a with high purity and more favorable physiochemical properties have been used to study binding and activation of C3a receptor by C3a (Ames et al., 1996; Ember et al., 1991; Hugli and Erickson, 1977; Kretzschmar et al., 1992). However, such peptides have rarely been used to study the interaction between the C5a2 receptor and C3a des-Arg, and they mimic the effects of ASP only partially (Murray et al., 1999a).
f. Simulation of specific binding
In addition, an easily misleading, rare phenomenon can simulate specific binding in the absence of any cellular receptor. This occurs when binding studies with adherent cells are performed in cell culture dishes or 96-well plates, or when membranes instead of a sucrose gradient are used to separate free from cell-bound labeled ligands. This binding of 125I-C3a or 125I-C3a des-Arg (ASP) to the surface of materials occurs despite the presence of albumin or dry milk powder (but because of albumin: Cui et al., 2009a), and can be effectively and dose-dependently competed with nonlabeled C3a or C3a des-Arg, thereby imitating specific binding with a Kd of the “plastic C3a/C3a des-Arg receptor” in the nanomolar range (EC50 of ∼20 nM). This phenomenon is not observed using 125I-C5a or C5a des-Arg. To prevent this pseudo-specific nonreceptor binding of 125I-C3a and 125I-C3a des-Arg /ASP, all plates and materials have to be preincubated with protamine sulfate (or histones) in addition to albumin (Johswich et al., 2006). To address this easily misleading feature of C3a or C3a des-Arg /ASP in binding studies, the use of control cells that do not express the ASP receptor in question are essential. Likewise, when investigating primary cells, the experiment must also be performed in the absence of any cells.
g. Purification of acylation-stimulating protein
The purity of plasma-derived C3a des-Arg (pASP) is another critical issue to be considered, especially in the context of the high concentrations used to show ASP-mediated functions. Human C5a and C5a des-Arg bind to and activate the C5a1 receptor with an EC50 of ∼1 nM (Burg et al., 1995); the EC50 of C3a needed for C3a receptor activation is only slightly higher (Klos et al., 1992; Settmacher et al., 2003). The first biologic effects of these fragments can even be observed at ∼0.1 nM. Of note, in experiments on metabolic responses, pASP has been used at concentrations ranging from 30 nM to 10 μM, with an EC50 of one to several hundred nanomolars. As C3a/C3a des-Arg and C5a/C5a des-Arg are related cationic peptides sharing common features like molecular weight, isoelectric point, or acid stability (Hugli et al., 1975b, 1981a; Paques et al., 1980; Daumy et al., 1988), it is rather difficult to obtain highly pure preparations of C3a des-Arg from plasma without low contaminations of other fragments still being present. Thus, one must exclude even minor contaminations of C5a/C5a des-Arg or C3a to rule out the possibility of a (still intriguing) effect of these polypeptides on lipid and glucose metabolism. Additionally, when observed more than 20 years ago, mast-cell activation by C3a and C3a des-Arg at concentrations exceeding 10 μM was proposed to be based on “a nonspecific mechanism similar to that of other polybasic compounds” (Fukuoka and Hugli, 1990).
h. Recombinant acylation-stimulating protein
The use of recombinant C3a des-Arg (rASP) could solve this problem. However, recombinant purified protein has been consequently applied only in some experiments of more recent publications (Cui et al., 2009b; Paglialunga et al., 2010). Formerly, it seemed to have been difficult to produce in large amounts (K. Cianflone, personal communication) and was therefore reserved for a few key experiments that revealed rASP-induced effects in the micromolar range (Murray et al., 1997). In any case, adequate controls are needed to exclude contaminations such as LPS, which might interfere with biologic responses in cell culture or in animal models, when rASP is prepared from Escherichia coli (Roy et al., 2011).
4. Review of Evidence for C5a2 Receptor as the Receptor for Acylation-Stimulating Protein
a. Introduction
Based on these considerations, and to identify the studies providing the best evidence for the proposed role of C3a des-Arg /ASP and the C5a2 receptor, this review will address the following questions:
How credible is the evidence that ASP is identical with C3a des-Arg, and that it influences energy metabolism?
How indisputably has it been demonstrated that the C5a2 receptor plays a role in energy metabolism?
How convincing is the evidence that C3a des-Arg /ASP binds to and signals via the C5a2 receptor?
Can the C3a/C3a receptor or the C5a/C5a des-Arg /C5a1 receptor be ruled out as confounding mediators and signaling components?
Do the effects observed and the concentrations of ASP used in cell culture represent a physiologic situation in vivo?
Is the effect on lipid metabolism a main physiologic function of the complement system and ASP? Or should it be considered as one component of “inflammatory” diseases including obesity?
b. Changes after application of an oral fat load in diets
Using a competitive enzyme-linked immunosorbent assay (ELISA), it was shown in 1989 that the serum levels of C3a/C3a des-Arg increased significantly within hours after an oral fat load, but not after challenge with glucose (Cianflone et al., 1989a, 1992). However, this was not affirmed by others (Charlesworth et al., 1998; van Oostrom et al., 2004). In moderately hypercholesterolemic women, ASP levels were altered after hydrogenated fat consumption (Matthan et al., 2001). Plasma ASP concentration and adipose tissue C3 mRNA expression were higher in obese than in lean men, and fasting plasma ASP concentration and C3 mRNA expression negatively correlated with insulin sensitivity (Koistinen et al., 2001). Moderate weight loss of obese women or a prolonged fast led to decreased ASP plasma levels (Sniderman et al., 1991; Cianflone et al., 1995). After gastric bypass surgery in morbidly obese subjects, body weight, ASP, and leptin decreased and adiponectin increased, while plasma lipids and insulin resistance improved (Faraj et al., 2003). Thus, diet and obesity can influence ASP concentrations in plasma.
c. Modified acylation-stimulating protein levels in various diseases and during a modified hormonal status
Plasma ASP was significantly higher in patients with coronary artery disease, and there was an association with plasma triglyceride, very low-density lipoprotein (VLDL) cholesterol, VLDL, apolipoprotein B (apoB), and certain apolipoprotein E (apoE) phenotypes (Cianflone et al., 1997). Children with Prader-Willi syndrome frequently suffer from high ASP levels and dyslipidemia (de Lind van Wijngaarden et al., 2010). Increased levels of plasma ASP, CRP, insulin, cholesterol, and nonesterified fatty acids are present in women with polycystic ovary syndrome compared with controls (Wu et al., 2009). In pregnant women, ASP levels were markedly elevated. They correlated with elevated levels of triglycerides, apoB, low-density lipoprotein cholesterol, and body mass index (BMI) (Saleh et al., 2007). In neonates, cord blood ASP showed a positive correlation with fetal birth weight above average, and with maternal plasma TG (Saleh et al., 2008).
For a complementologist, an increase of C3a des-Arg /ASP in body fluids indicates that an activation of the complement system has taken place. One can assume that besides this (surrogate) marker, all effector molecules that are released during activation of the complement cascade, such as C3a, C5a, C5a des-Arg, C3b, or the MAC, were at least transiently present as well. A direct conclusion regarding a biologic function of C3a des-Arg should thus not be drawn. In any case, it is remarkable that a correlation of complement activation and lipid metabolism has been observed in various settings. This supports the concept that unbalanced obesity is linked to chronic low-grade inflammation, and suggests that drastic fat uptake can rapidly trigger a low inflammatory response.
d. Animal models, complement factor C3, acylation-stimulating protein, and energy metabolism
Serum from obese rats was found to contain almost twice as much C3 as serum from lean rats (Boggs et al., 1998). In C3−/− mice, which lack significant parts of the complement system, including ASP, a marked delay in postprandial TG clearance, reduced body fat, and lower leptin levels were observed in comparison with wild-type animals. Also, postprandial nonesterified fatty acids were increased in serum/plasma independently of low or high fat diets, and modest changes in insulin/glucose metabolism implied increased insulin sensitivity (Murray et al., 1999d). In contrast and of note, the Wetsel group found no significant differences in the plasma TG, cholesterol, or free fatty acid concentrations of fasted C3−/− mice compared with their wild-type litter mates. Plasma lipoprotein analyses indicated no significant differences in various lipoproteins and triglycerol in the absence of ASP. The C3−/− animals were not impaired in their ability to clear TG and free fatty acids from their circulation after an oral fat load (Wetsel et al., 1999). These contradicting results might be caused by differences in the C3−/− and wild-type mouse strains bred and used by the two research groups in different facilities. It suggests that the genetic background of these animals should be checked thoroughly (e.g., by SNP analysis) to prove sufficient and effective backcrossing.
A third research group cross-bred C3−/− mice to mice deficient in both apoE and the low-density lipoprotein receptor (Apoe−/− Ldlr−/− mice). Compared with controls, mice additionally lacking C3 had higher serum TG levels and a more proatherogenic lipoprotein profile, and lower body weight and fat content. In contrast, there were no differences between factor B-deficient mice, being additionally analyzed where complement activation of the alternative pathway is hampered, and wild-type animals. Moreover, the size of lesions in the aorta was significantly higher in C3−/− mice than in controls. The investigators concluded that complement activation by the classic or lectin pathway exerts atheroprotective effects, possibly through the regulation of lipid metabolism (Persson et al., 2004).
e. C5ar2−/− mice and blockade of C5a2 receptor by antibodies
In studies conducted by or in cooperation with the Cianflone group, C5ar2−/− mice also showed changes in their metabolism as compared with wild-type controls: these mice are hyperphagic on a low-fat diet. Nevertheless, their body weight and adipose tissue mass did not differ from that of wild-type litter mates. On a high-fat diet, average adipocyte size and adipose tissue triglyceride/DNA content were significantly reduced. Postprandial clearance of TG was delayed in C5ar2−/−animals, and the synthesis of adipose tissue triglycerides, lipolysis, and fatty acid re-esterification were significantly reduced. Indirect calorimetry measurements suggested preferential fatty acid utilization over carbohydrate (Paglialunga et al., 2007).
In vivo, application of anti-ASP and anti-C5a2 receptor antibodies did not alter body weight, adipose tissue mass, food intake, or levels of insulin, leptin, or adiponectin. However, the blockade particularly of the anti-C5a2 receptor induced a significant delay in TG and nonesterified fatty acids clearance and decreased perirenal, hepatic, and muscle TG mass. Adenosine monophosphate-activated protein kinase (AMPK) and lipoprotein lipase (LPL) activity were increased by this treatment. Thus, the ASP/C5a2 receptor neutralizing antibodies that effectively block the ASP-C5a2 receptor interaction altered lipid distribution and energy utilization. Moreover, continuous administration of anti-ASP antibodies as compared with an IgG negative control had effects on energy expenditure and glucose storage, increasing in vivo whole-body energy utilization (Cui et al., 2007; Paglialunga et al., 2010).
f. Systemic application of purified acylation-stimulating protein or recombinant acylation-stimulating protein
Injection of 500 μg i.p. of ASP accelerated TG clearance after a fat load in vivo in C57BL/6 mice, and reduced plasma glucose within a few hours (Murray et al., 1999b). Untreated male and female C3−/− mice had elevated oxygen consumption, increased fatty acid oxidation in liver and muscle, and increased inguinal fat and muscle mRNA expression. Fatty acid incorporation into lipids was decreased but could be normalized by administration of ASP together with oral fat intake (Xia et al., 2004). However, assuming complete resorption of 500 μg of ASP from the peritoneum, its redistribution into an estimated 2 ml of total blood volume should lead to ∼25 μM ASP in the circulation. For pASP, a purity of 99% was ascertained by mass spectrophotometry, potentially permitting up to 250 nM of contaminating proteins with a molecular weight similar to ASP in the bloodstream. Of note, this impedes interpretation of data from the corresponding experiments because ∼10–50 nM C3a, C5a, or C5a des-Arg would already result in maximal cellular responses mediated by the C3a receptor or C5a1 receptor. Thus, when applying 500 μg of pASP, it cannot be excluded that contaminating factors could be responsible for the observed effects.
Yet there are more convincing studies with a continuous administration of recombinant instead of purified ASP over up to 4 weeks that clearly demonstrate decreased fasting levels of nonesterified fatty acids and decreased energy expenditure in C3−/− mice, but only small (if any) changes in body weight or food uptake (Paglialunga et al., 2010). As a negative control, saline is applied instead of an inert recombinant protein purified from E. coli in parallel with the rASP. In differentiated adipocytes, 100 nM rASP resulted in a drastic increase in fatty acid uptake in vitro. This was blocked in vitro by incubation with an anti-ASP antibody, excluding the possibility that contaminations, such as LPS, are responsible for the observed effect on adipocytes. The interpretation of the data obtained in the mouse is more difficult due to the absence of a similar specificity control (i.e., inhibition by antibody directed against ASP) and also due to the fact that, surprisingly, the achieved and effective serum steady-state levels of rASP delivered by an osmotic pump were very small (i.e., only in the range of 0.25 nM). This large difference in concentrations needed between the in vitro assay to see cellular responses and the in vivo experiment makes it more difficult to draw conclusions about a common underlying mechanism.
On the other hand, continuous application of antibodies against endogenous ASP with corresponding IgG controls in the animal model resulted in clear effects on the lipid and glucose metabolism (Paglialunga et al., 2010). Moreover, injection of rASP into the third ventricle of the brain inhibits food intake and locomotor activity in rats (Roy et al., 2011). Again, data interpretation is limited by the absence of a negative control which could exclude that impurities in the rASP might account for effects in this system. Further, it is difficult to judge what concentrations of rASP are reached in the cerebrospinal fluid, and whether they are in the physiologic range or rather correspond to a situation of severe neurologic disease such as meningitis. Nevertheless, these are interesting data as they indicate additional functions for the complement system and its cleavage products.
g. In vitro data using purified acylation-stimulating protein, synthetic C3a-related peptides, or recombinant acylation-stimulating protein
There are a variety of older publications using human pASP. They have been presented and reviewed in detail elsewhere (Johswich and Klos, 2007; Klos et al., 2009). Therefore, this review focuses on a selection of key data from more recent publications, preferentially based on the use of recombinant ASP or synthetic peptides. For in vitro studies on human skin fibroblasts (HSF) and murine 3T3 cells, ASP was chemically and enzymatically modified; additionally, synthetic C3a-analogous C-terminal peptides, known ligands of the human C3a receptor, were analyzed (Murray et al., 1999a). The results show that the N-terminal region of ASP plays a minor role in receptor binding (determined by inhibition of 125I-ASP binding) and triacylglycerol synthesis. An intact disulfide-linked core region is essential for lipid synthesis but not for receptor interaction. The C3a-analogous C-terminal peptide P117 (containing a C-terminal Arg) can inhibit ASP binding and can partially activate lipid synthesis, suggesting “that this region participates but is not sufficient for complete receptor binding” (Murray et al., 1999a), whereas smaller C-terminal peptides were ineffective.
On the human C3a receptor, P117 has an EC50 in the range of 50 nM (while ASP is completely ineffective) (Martin et al., 1997; Johswich and Klos, 2007; Klos et al., 2009). Triacylglycerol synthesis was stimulated by P117 at 1000-fold higher concentrations of 4–40 μM, similar to the molar concentrations of ASP needed. Surprisingly, in competition studies using 1 nM 125I-ASP as a tracer on HSF, unlabeled P117 and ASP were equally potent, with an IC50 in the range of only 100 nM (pIC50 = 7). This behavior is rather surprising as the ligand concentrations needed for binding and dose-response curves are usually quite similar.
As mentioned previously, C3a and C3a des-Arg show high nonspecific binding as well as “pseudo-specific nonreceptor binding” to various surfaces in the absence of any cell, with a Kd close to that observed in binding studies (Johswich et al., 2006). This suggests that the binding of 125I-ASP and the surprisingly low Kd observed in the Cianflone group, at least in earlier studies on adipocytes of fibroblasts, are best explained by this effect. Of note, in experiments of the Klos group, neither 100 nM fluorescence-labeled (FL) pC3a nor the des-arginated peptide (FL-ASP) bound to C5a2 receptor expressed on human embryonic kidney (HEK) cells as compared with HEK control cells, whereas FL-C3a was still able to bind to the C3a receptor (Johswich et al., 2006). In contrast, when similar studies were performed by the Cianflone group, using FL-rASP, HEK cells stably expressing C5a2 receptor bound and internalized this ligand more strongly than nontransfected HEK control cells (Cui et al., 2009a). Both studies using FL-ligands seem to be thoroughly performed, so there is presently no explanation for the differing results.
With an EC50 of ∼1.5 μM, the rASP used here was much more active in stimulating lipid synthesis and fatty acid uptake than the ASP purified from blood and former preparations. In the same study, C5a2 receptor-expressing HEK cells bound 1 nM of 125I-ASP much better than nontransfected cells or the no-cell control with a Kd of 34 nM. HEK and Chinese hamster ovary (CHO) cells expressing C5a2 receptor bound FL-rASP in a dose-dependent manner at similar concentrations (Cui et al., 2009a).
h. Summary and assessment of the role of C3a des-Arg/ASP and C5a2 receptor
The pathogenesis of obesity-related metabolic dysfunction is considered to include chronic low-grade inflammation (Ouchi et al., 2011). Various cytokines and chemokines act as adipokines, simultaneously influencing inflammation and metabolism (Kershaw and Flier, 2004). The complement system appears to be linked with adipokine signaling in multiple ways: cells in the adipose tissue can produce various complement factors (Choy et al., 1992; White et al., 1992), and adiponectin itself can trigger the classic pathway of complement activation (Peake et al., 2008). Moreover, complement factor D has been identified as an adipokine. Considering this connection between obesity and inflammation, it does not seem unlikely that other components of the activated complement system might play a similar dual role as proinflammatory mediators and metabolic modulators. In accordance, activation of the complement cascade as evidenced by the increase of the “activation marker” C3a des-Arg /ASP is associated with changes in lipid and glucose metabolism. In this context the publications of the Cianflone group are very informative.
The majority of investigations using ASP purified from blood (or synthetic C3a-related peptides) and most data from animal models are in accordance with a potential role of ASP in metabolism, but they do not provide final proof that C3a des-Arg is indeed the responsible and active mediator. In particular, they do not exclude that contaminations with C3a or C5a des-Arg, and their interactions with the C3a receptor or C5a1 receptor, respectively, might be causing some of the observed effects of pASP. A few recent key publications using recombinant C3a des-Arg (ASP) provide much stronger evidence for an active role of ASP and for the C5a2 receptor as its receptor. However, there are some contradictory results from both animal studies (Wetsel et al., 1999) and in vitro experiments (Johswich et al., 2006) that should be borne in mind when assessing the role of C3a des-Arg as a metabolic regulator.
V. Complement Peptide Functions Not Mediated by the Three Known Receptors
A. C4a
Although C4a has receptor-mediated effects in the guinea pig (see Section III.B.1), its effects on human cells have not been shown to be receptor mediated, and serum-derived C4a may be contaminated with C3a. Studies showing that C4a can enhance vascular permeability in human skin (Gorski et al., 1979), desensitize smooth muscle contraction by C3a (Hugli et al., 1981b), and stimulate mast cells to secrete (Hugli and Muller-Eberhard, 1978; Moon et al., 1981) should be repeated with recombinant C4a. However, recombinant C4a has been demonstrated to inhibit C3a- and C5a-stimulated degranulation of human mast cells by increasing cAMP levels through an unidentified receptor (Xie et al., 2012). A derivative of C4 in synovial fluid has been shown to indirectly inhibit the chemotaxis of human monocytes but not neutrophils (Matsubara et al., 1991). This was later shown to be C4a, which worked at concentrations as low as 10−16 M, but this could be inactivated by conversion to C4a des-Arg (Tsuruta et al., 1993). An anti-inflammatory role for C4a in glomerulonephritis has also been proposed (Welch et al., 2001). C3a and C4a can both form complexes with IgG (Nezlin and Freywald, 1992), one molecule for each H and L chain. One gram of IgG can bind up to 0.3 mg C3a (1:200 molar ratio). The function of this is unknown but has been proposed to eliminate complement peptides from the circulation.
B. C3a
In many instances, a biologic activity is shared by both C3a and its metabolite C3a des-Arg. It is clear that C3a des-Arg does not interact with the C3a receptor (Wilken et al., 1999; Johswich et al., 2006), so these activities are presumed to be not receptor mediated. The homing of bone-marrow hemopoietic cells in response to CXCL12 is enhanced by both C3a and C3a des-Arg (Honczarenko et al., 2005b) in mice that are C3a receptor or C5a2 receptor deficient (Honczarenko et al., 2005a). The LPS-mediated production of proinflammatory cytokines by PBMC mediated by LPS is also enhanced by both C3a and C3a des-Arg (Takabayashi et al., 1996) whereas B-cell cytokine production is diminished (Fischer and Hugli, 1997). Both polypeptides have been shown to stimulate pituitary cell cultures to secrete a variety of hormones (Francis et al., 2003), with the C3a des-Arg response being insensitive to pertussis intoxication.
Another shared activity is the inhibition of bacterial and fungal growth. These antimicrobial properties are conserved from invertebrates to humans (Pasupuleti et al., 2007), and C3a and C4a (but not C5a) have been suggested to have evolved primarily as antibacterial agents (Malmsten and Schmidtchen, 2007).
C3a-derived peptides have also been shown to bind to the high affinity IgE receptor, FcεRI, and can inhibit signaling and cytokine secretion by mast cells (Peterfy et al., 2008). C3a and C3a des-Arg can also enhance CXCL12-mediated platelet production (Wysoczynski et al., 2007). The receptor for advanced glycation end products (RAGE) can bind C3a, CpG oligos, and DNA/C3a complexes, resulting in the stimulation of IFN-γ production in human PBMC (Ruan et al., 2010).
Oxidized LDL and C3a colocalize in atherosclerotic plaques as a result of C3a binding to modified lysine side chains. C3a also recognizes oxidized lipids on apoptotic cells in a similar way (Veneskoski et al., 2011), possibly aiding the removal of C3a from the circulation. As a cationic amphiphile, C3a can also activate G proteins independent of the receptor by cross-linking sialic acid residues displayed on a variety of membrane proteins (Emadi-Khiav et al., 1995). Finally, C3a has been shown to bind to GPCR Mas-related genes MrgX1 and MrgX2 (Kashem et al., 2011) on human mast cells, allowing C3a to stimulate degranulation in a non-C3a receptor-dependent manner (Kashem et al., 2011; Subramanian et al., 2011).
C. C5a
C5a has also been shown to be bound by immunoglobulins (Basta et al., 2003), and F(ab)′2 fragments of irrelevant specificity can neutralize the ability of both C3a and C5a to activate the human mast cell line HMC-1. To our knowledge, no other nonreceptor-mediated functions of C5a or C5a des-Arg have been reported.
D. Conclusions, Future Directions
1. Nomenclature
Although originally termed anaphylatoxins, the C5a, C4a, and C3a act only indirectly to mediate an anaphylactic response (Taylor et al., 1994), and it is now clear that they have many more functions inside and outside of the immune system. For this reason, we would suggest the use of the term “complement peptides” rather than anaphylatoxins. The second C5a receptor, sometimes described as a binding protein (Okinaga et al., 2003) or as a “receptor-like” protein (Kalant et al., 2003), has clearly been shown to transduce signals, particularly in mouse cells, so we propose that the symbol C5a2 receptor is ascribed to this receptor (human gene name C5AR2).
2. Drugs That Act at Complement Peptide Receptors
Despite many years of intensive investigation, there are still no useful drugs that act directly at the complement peptide receptors. It is not clear as to whether this is due to peculiar molecular mechanisms associated with receptor binding and activation or to the complex biology of the receptors. The former is becoming increasingly well understood as satisfying models, at least for C5a and the C5a1 receptor, are being produced. However, the relatively low affinities of many of the antagonists for rodent fragment receptors has troubled the development of many drug leads and may have provided some misleading information about the roles of the complement peptides in disease. It is possible that inhibition of complement peptide formation by agents that act on the complement cascade may prove to be the most effective drugs. Here, however, the extrinsic pathways of fragment generation may confound this expectation, so further work on the consequences of these pathways is required.
3. Signaling by Complement Peptide Receptors
The events that occur after receptor binding are also proving difficult to untangle. Although signaling through G proteins is well understood, the role of arrestins in signal transduction remains obscure. Work on the C3a receptor suggests that these proteins have complex roles in the modulation of signaling, but for the C5a1 and C5a2 receptors little is yet known. Signaling by the C5a2 receptor is still controversial, and the question of ligand selectivity has not been finally settled. Even if limited to just C5a and C5a des-Arg, the C5a2 receptor may be a signaling receptor equivalent to the C5a1 receptor; it may provide “negative” signals, through arrestins, to counteract the C5a1 receptor; or it may cause ligand sequestration and degradation. The role of homo- and heterodimerization of C5a1 and C5a2 in the control of signaling also awaits elucidation. Interestingly, although the C5a2 receptor binds C5a des-Arg with the same affinity as C5a, the des-arginated form is rarely used in experiments with this receptor, resulting in the potential loss of important information. Further, the reported activity of the C5a2 receptor may depend on the species under investigation, or perhaps it is the expressing cell type (or cellular activation status) that determines the role of this receptor. Selective agonists and antagonists are urgently required for the C5a2 receptor, and it is to be hoped that such compounds, once identified, can be used to solve the enigma that this receptor represents.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Klos, Wende, Wareham, Monk.
Footnotes
This study was funded in part by the British Heart Foundation [Project Grant PG/09/018/25279] (to P.N.M., for K.J.W.); by the Hannover Biomedical Research School (HBRS/ZIB) (to E.W.); the Helmholtz International Research School for Infection Biology (HIRSIB) (to E.W.); and BMBF Verbund Zoonotischer Chlamydien, TP9 [Project Grants 01 Kl 1011F] (to E.W.), SFB587, TP A16 (to A.K.)].
Abbreviations
- ALS
- amyotrophic lateral sclerosis
- AMD
- age-related macular degeneration
- AMPK
- adenosine monophosphate-activated protein kinase
- AP-1
- activator protein 1
- apoB
- apolipoprotein B
- apoE
- apolipoprotein E
- ASP
- acylation-stimulating protein
- AU-rich
- adenylate-uridylate-rich
- BMI
- body mass index
- Cha
- cyclohexylalanine
- CHIPS
- chemotaxis inhibitory protein of Staphylococcus aureus
- CHO
- Chinese hamster ovary
- CLP
- cecal ligation and puncture
- CPB
- carboxypeptidase B
- CPN
- carboxypeptidase N
- CPR
- carboxypeptidase R
- CRP
- C-reactive protein
- DC
- dendritic cell
- DMSO
- dimethyl sulfoxide
- EC
- extracellular domain
- EDTA
- ethylenediaminetetraacetic acid
- ELISA
- enzyme-linked immunosorbent assay
- ERK
- extracellular signal-regulated kinase
- FEV1
- forced expiratory volume in 1 second
- FL
- fluorescence labeled
- FPR
- formyl peptide receptor
- GPCR
- G-protein-coupled receptor
- HEK
- human embryonic kidney
- HMGB1
- high-mobility group box 1
- Hoo
- hydroxyorotic acid
- HSF
- human skin fibroblast
- IC
- inhibitory concentration
- IFN
- interferon
- IL
- interleukin
- JPE-1375
- Hoo-Phe-Orn-Pro-(d-HLeu)-Phe4F-Phe
- LDL
- low-density lipoprotein
- LPL
- lipoprotein lipase
- LPS
- lipopolysaccharide
- MAC
- membrane attack complex
- MAPK
- mitogen-activated protein kinase
- MeLeu
- methyl-leucine
- NDT 9513727
- N,N-bis(1,3-benzodioxol-5-ylmethyl)-1-butyl-2,4-diphenyl-1H-imidazole-5-methanamine
- NF-κB
- nuclear factor κB
- NMR
- nuclear magnetic resonance
- MSC
- mesenchymal stem cells
- NDT 9513727
- N,N-bis(1,3-benzodioxol-5-ylmethyl)-1-butyl-2,4-diphenyl-1H-imidazole-5-methanamine
- OmpH
- outer-membrane protein H
- OVA
- ovalbumin
- PBMC
- peripheral blood mononuclear cell
- PLC
- phospholipase C
- PMN
- polymorphonuclear neutrophilic granulocyte
- PMX53
- (3D533D53) (Ac)Phe-[Orn-Pro-dCha-Trp-Arg]
- PT
- pertussis toxin
- RBL
- rat basophilic leukemia
- RSM
- random saturation mutagenesis
- S-1-P
- sphingosine-1-phosphate
- SB290157
- N(2)-[(2,2-diphenylethoxy)acetyl]-l-arginine
- SLE
- systemic lupus erythematosus
- SNP
- single-nucleotide polymorphism
- TAFI
- thrombin-activatable fibrinolysis inhibitor
- TG
- triglyceride
- TGC
- thioglycolate
- TLR
- Toll-like receptor, TM, transmembrane domain
- TNF-α
- tumor-necrosis factor α
- UTR
- untranslated region
- VLDL
- very low-density lipoprotein
- W54011
- N-[(4-dimethylaminophenyl)methyl]-N-(4-isopropylphenyl)-7-methoxy-1,2,3,4-tetrahydronaphthalen-1-carboxamide hydrochloride
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵





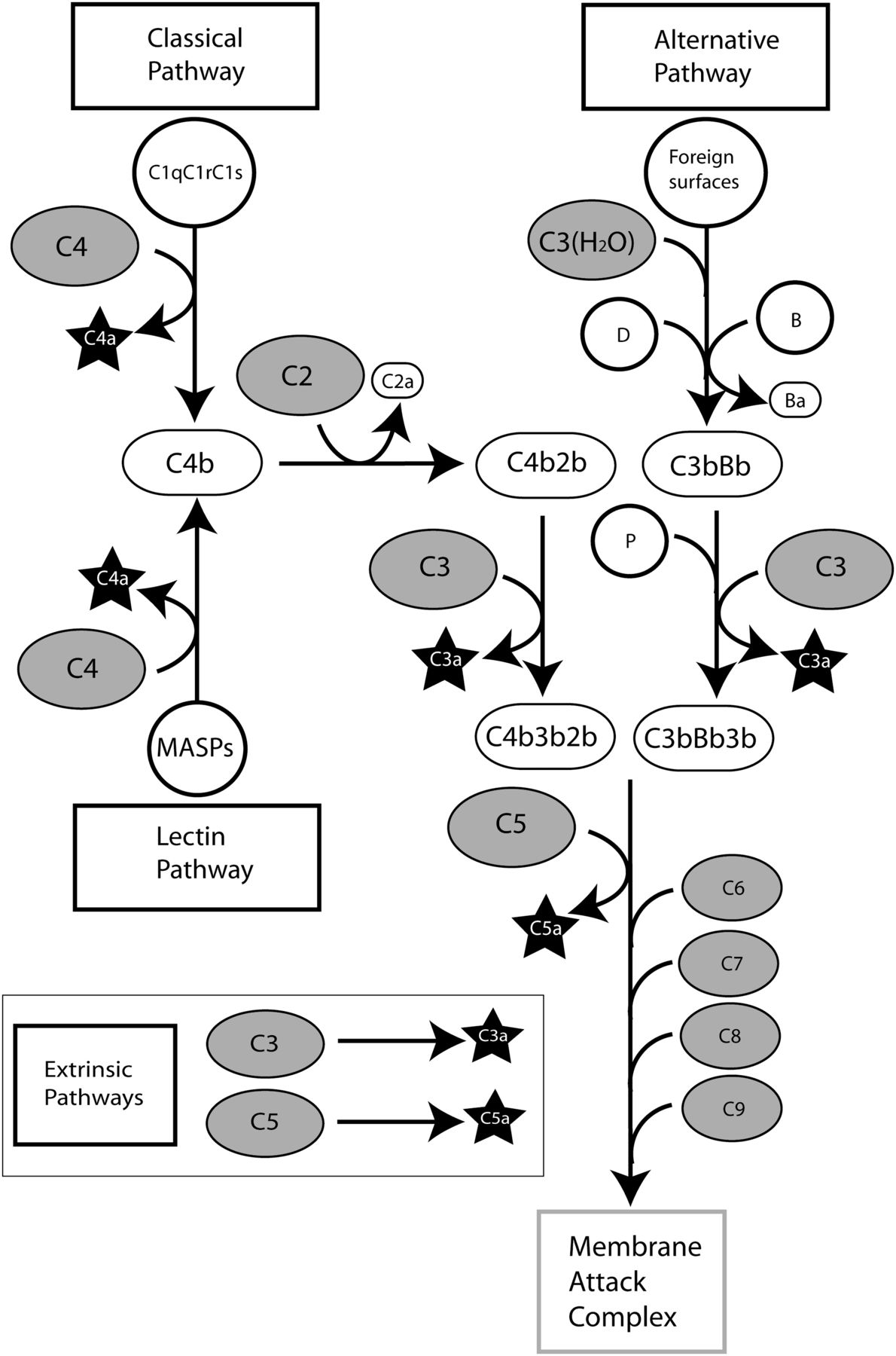{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
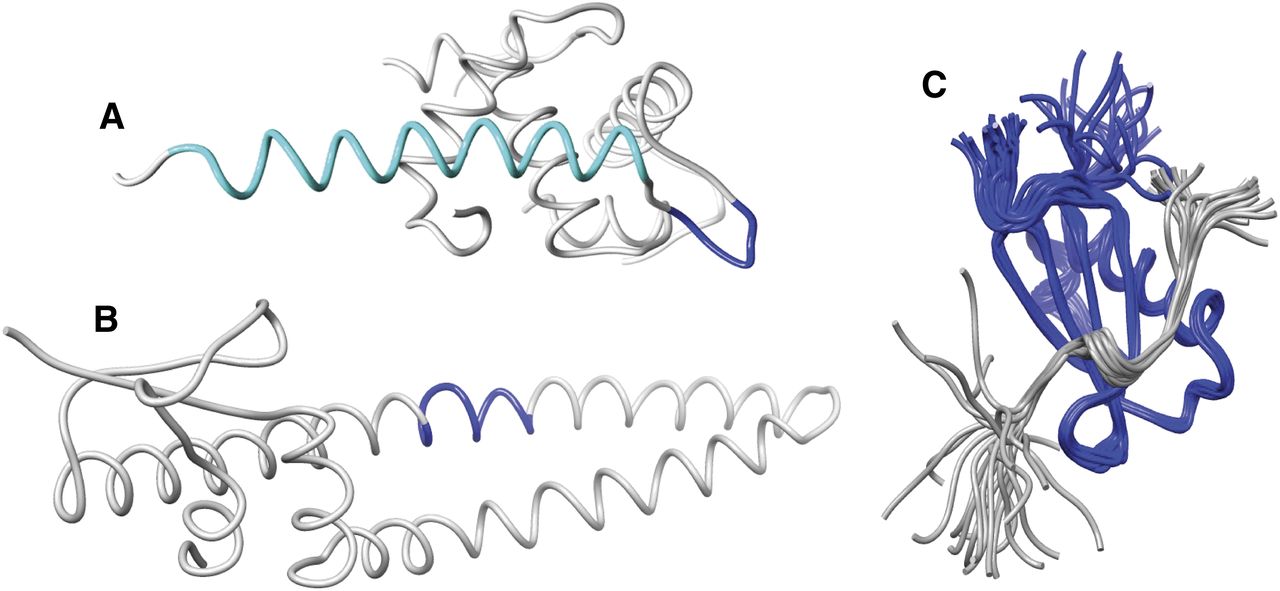{kind=link}
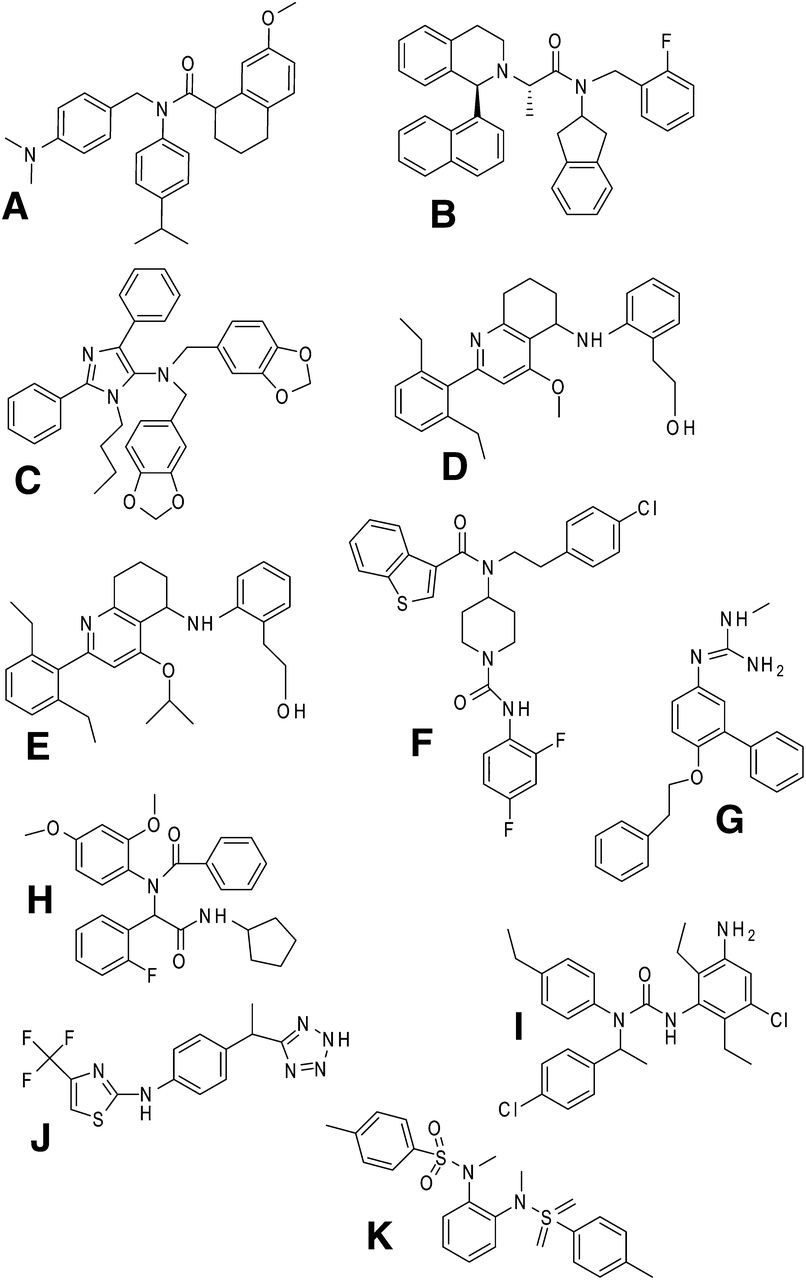{kind=link}
{kind=link}