


Abstract
Estrogens are critical mediators of multiple and diverse physiologic effects throughout the body in both sexes, including the reproductive, cardiovascular, endocrine, nervous, and immune systems. As such, alterations in estrogen function play important roles in many diseases and pathophysiological conditions (including cancer), exemplified by the lower prevalence of many diseases in premenopausal women. Estrogens mediate their effects through multiple cellular receptors, including the nuclear receptor family (ERα and ERβ) and the G protein–coupled receptor (GPCR) family (GPR30/G protein–coupled estrogen receptor [GPER]). Although both receptor families can initiate rapid cell signaling and transcriptional regulation, the nuclear receptors are traditionally associated with regulating gene expression, whereas GPCRs are recognized as mediating rapid cellular signaling. Estrogen-activated pathways are not only the target of multiple therapeutic agents (e.g., tamoxifen, fulvestrant, raloxifene, and aromatase inhibitors) but are also affected by a plethora of phyto- and xeno-estrogens (e.g., genistein, coumestrol, bisphenol A, dichlorodiphenyltrichloroethane). Because of the existence of multiple estrogen receptors with overlapping ligand specificities, expression patterns, and signaling pathways, the roles of the individual receptors with respect to the diverse array of endogenous and exogenous ligands have been challenging to ascertain. The identification of GPER-selective ligands however has led to a much greater understanding of the roles of this receptor in normal physiology and disease as well as its interactions with the classic estrogen receptors ERα and ERβ and their signaling pathways. In this review, we describe the history and characterization of GPER over the past 15 years focusing on the pharmacology of steroidal and nonsteroidal compounds that have been employed to unravel the biology of this most recently recognized estrogen receptor.
I. Introduction
The pharmacology and physiology of estrogen and its receptors are particularly complex (Dahlman-Wright et al., 2006; Prossnitz and Barton, 2011), with the origins of the estrogen signaling system dating back more than 500 million years in evolutionary history (Thornton, 2001, 2003; Callard et al., 2011) and those of G protein–coupled estrogen receptor (GPER) more than 200 million years (Thomas et al., 2010). Although estrogens are recognized predominantly for their function in female mammalian reproduction and the development of secondary sex characteristics, namely uterine and mammary effects, they also play important roles in almost every physiologic system of the body (Edwards, 2005) in both women and men (Lombardi et al., 2001; Finkelstein et al., 2013). As pharmaceutical targets, estrogens and their varied antagonists have been particularly important in contraception (Benagiano et al., 2006) and breast cancer therapy (Jensen and Jordan, 2003), with an increasing appreciation of their therapeutic value in the nervous (McEwen et al., 2012), immune (Cunningham and Gilkeson, 2011), vascular (Knowlton and Lee, 2012), skeletal (Imai et al., 2013), and endocrine systems (Mauvais-Jarvis et al., 2013). For decades, the actions of estrogen(s) were thought to be mediated by a single estrogen receptor first identified in the 1960s (Jensen and Jacobson, 1962; Jensen and DeSombre, 1973), that is, until the discovery of a second highly homologous estrogen receptor in 1996 (Kuiper et al., 1996), whereupon the first estrogen receptor was renamed estrogen receptor α (ERα) and the new receptor ERβ. Although some of the first described cellular/tissue activities of estrogen were cAMP production (Szego and Davis, 1967) and calcium uptake (Pietras and Szego, 1975), many of the physiologic functions of estrogen receptors were subsequently best understood as ligand-activated transcription factors (Carroll and Brown, 2006; Schultz-Norton et al., 2011), belonging to the family of nuclear hormone receptors, which includes receptors for other steroids such as progesterone, androgen, glucocorticoid, and mineralocorticoid (Burris et al., 2013). However, as described above, estrogens, as well as other steroids, had also been demonstrated to mediate rapid cellular and physiologic responses, inconsistent with the time frame of transcriptional mechanisms (Falkenstein et al., 2000). The discovery and characterization of a third estrogen receptor in the 2000s (Filardo et al., 2000; Revankar et al., 2005; Thomas et al., 2005), namely GPR30/GPER, belonging to the family of 7-transmembrane G protein–coupled receptors (GPCRs), which classically mediate rapid responses such as kinase activation and ion mobilization, have expanded our understanding of the varied and complex activities of estrogenic compounds throughout the body (Prossnitz, 2008, 2012; Prossnitz and Barton, 2009, 2011, 2014; Prossnitz and Maggiolini, 2009b; Filardo and Thomas, 2012; Han et al., 2013; Srivastava and Evans, 2013; Lappano et al., 2014; Barton and Prossnitz, 2015).
The rapid cellular effects of estrogen include the production of cAMP, the mobilization of intracellular calcium, and the activation of multiple kinases, such as extracellular signal-regulated kinase (ERK) and phosphoinositide 3-kinase (PI3K), as well as ion channels and endothelial nitric oxide synthase (eNOS) among other pathways. Although classic ERs (predominantly ERα) have been reported to activate such pathways (Edwards, 2005), GPER has also been demonstrated to stimulate each of these pathways in various cell types (Prossnitz et al., 2008). Although it has been suggested that GPER is in fact not an estrogen receptor (Otto et al., 2008; Levin, 2009), but is nevertheless critical in membrane-initiated signaling of estrogen via presumably classic ERs, extensive data over many years (e.g., estrogen-mediated actions and binding in ERα/β-negative cells that are GPER dependent) now largely refute such assertions and are presented in this review. However, in cells that express both ERα and GPER there certainly exists the possibility of inter/codependent signaling, with some evidence supporting this (Albanito et al., 2007). Existing data do not preclude a complex scenario in which, for example, binding of 17β-estradiol (E2; and referred to throughout as estrogen, with the term estrogens referring to any one or a combination of physiologic forms of estrogen and/or its derivatives) to an extranuclear ER that associates with GPER is blocked by antagonist binding to GPER in cells that coexpress both receptors. However, in the absence of ERα expression, this mechanism would seem to be precluded.
With the existence of at least three estrogen receptors (ERα, ERβ, and GPER), the roles of individual receptors have been particularly difficult to establish, all the more so as a result of the overlapping and often confounding effects of both natural and synthetic ligands. The list of estrogenic (including antiestrogenic) compounds is truly immense (Lorand et al., 2010; Paterni et al., 2013), with many classified as endocrine disrupting chemicals (EDCs) due to their harmful (often reproductive and developmental) effects on humans and other animals (Casals-Casas and Desvergne, 2011; Schug et al., 2011). In addition to the endogenous physiologic ligands (estrone, estradiol, estriol), plants and fungi produce a large array of compounds (phytoestrogens and mycoestrogens, respectively, both subsets of natural xenoestrogens) that can mimic the actions of estrogen (Ososki and Kennelly, 2003; Lorand et al., 2010). Plastic precursors and pesticides represent two important categories of anthropogenic estrogenic nonsteroidal compounds (synthetic xenoestrogens) produced in large quantities by industry (Singleton and Khan, 2003; Fucic et al., 2012). Environmental estrogens, even at low doses, are now recognized to mediate developmental reprogramming through nongenomic signaling mechanisms such as PI3K/Akt, leading to epigenetic alterations and increased lifetime risks of multiple diseases including cancer (Wong and Walker, 2013).
Finally, because of their role in multiple aspects of health and disease, pharmaceutical companies have synthesized vast collections of compounds, some of which have found great success as contraceptives or therapies for cancer and postmenopausal conditions including osteoporosis, depression, and hot flashes (Paterni et al., 2013). Some of these drugs fall into the categories of tissue-dependent mixed agonists/antagonists (selective estrogen receptor modulators [SERMs]) or full antagonists (selective estrogen receptor downregulators [SERDs]) based on their activities in the breast, uterus, and bone. However, with the discovery and characterization of GPER, the activities of such compounds on GPER must be taken into account to achieve a more complete understanding of their physiologic functions and side effects, which can include endocrine effects, uterine cancer, and deep vein thrombosis. In this review, we will first provide a comprehensive overview of our understanding of the functions of GPER in relation to the classic estrogen receptors, highlighting physiologic, pathologic, and therapeutic implications of GPER function, followed by a discussion of the pharmacology of the vast array of estrogenic substances that have been reported to bind and modulate the activity of GPER.
II. Estrogen and Its Receptors
A. Estrogens
The most active natural physiologic form of estrogen is E2 (see Fig. 1), formed from the aromatization of testosterone in multiple tissues but predominantly in the ovaries (of premenopausal women). Additional forms of estrogen include estrone (E1), the least abundant estrogen, derived from aromatization of androstenedione, and estriol (E3), produced primarily during pregnancy from 16-hydroxydehydroepiandrosterone sulfate in the fetal liver and adrenal glands. Throughout this review, the term estrogen will refer to E2, unless otherwise stated. Total serum estrogen (E1 + E2) levels in premenopausal women generally range from ∼0.1 to 1.3 nM, whereas postmenopausal women (∼0.02–0.2 nM) and men (∼0.07–0.25 nM) display significantly lower levels (Kushnir et al., 2008; Blair, 2010); however, local tissue concentrations of E2 may be considerably higher, up to eightfold (Huhtinen et al., 2012), particularly in the tissue microenvironment (Simpson et al., 1999). In situations of autocrine or juxtacrine signaling with limited diffusion from the site of synthesis, effective E2 concentrations could be far higher (Owen et al., 1999; Singh and Harris, 2005). E2 exhibits affinities (measured in various systems, including whole or permeabilized cells, cell or tissue homogenates, in vitro synthesized or purified full-length protein, or most often purified ligand-binding domain) for ERα and ERβ of ∼0.1–0.4 nM (Anstead et al., 1997; Kuiper et al., 1997; Bologa et al., 2006), whereas the affinity of E2 (measured in permeabilized cells or membrane preparations) for GPER is ∼10-fold lower, in the range of 3–6 nM (Revankar et al., 2005; Thomas et al., 2005) (Table 1). E1 and E3 display affinities for ERα of 0.3 and 1.4 nM, respectively and for ERβ of 0.4 and 0.7 nM, respectively, employing in vitro–synthesized protein (Kuiper et al., 1997). On the contrary, GPER exhibits binding affinities of >10 μM and >1 μM for E1 and E3, respectively (Thomas et al., 2005), with E3 reported to act as a low-affinity antagonist for GPER in the ∼1–10 μM range (Lappano et al., 2010), demonstrating the pharmacologic differences between GPER and the classic estrogen receptors (see Table 1).
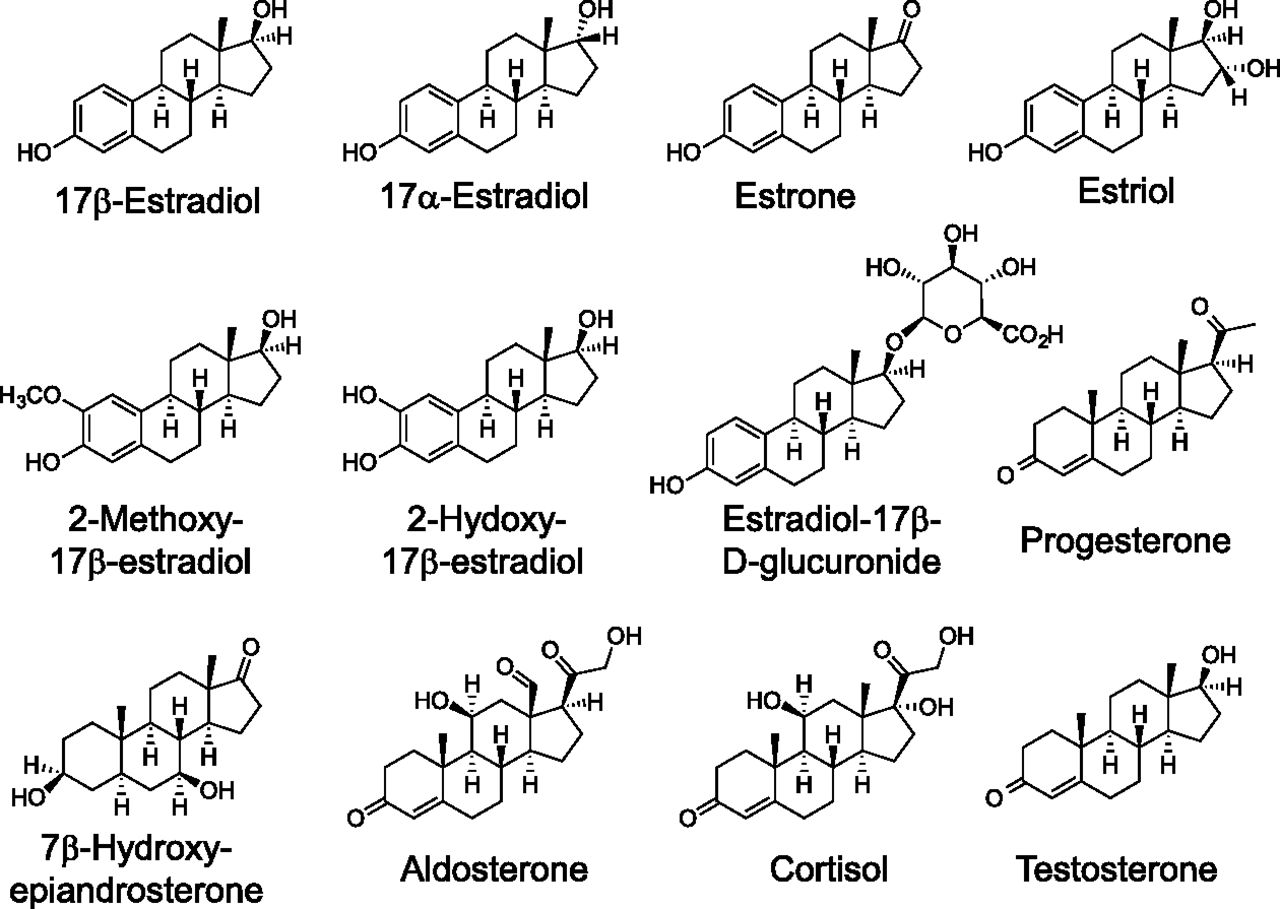
Structures of steroids.
Binding affinities of estrogenic ligands to estrogen receptors
B. Classic Estrogen Receptors
1. Structure.
The classic estrogen receptors (ERα and ERβ) are members of the steroid hormone receptor family, which itself belongs to the nuclear receptor superfamily (Dahlman-Wright et al., 2006; Huang et al., 2010; Burris et al., 2013). The latter function predominantly as ligand (hormone)-dependent transcription factors, consisting of two major domains, a carboxy-terminal ligand-binding domain and a central DNA-binding domain (Heldring et al., 2007). Additional regions are involved in transcriptional activation, namely the constitutively active amino-terminal AF-1 domain and the ligand-dependent carboxy-terminal AF-2 domain, contained within the ligand-binding domain, that mediates the wide range of functional responses to diverse ligands (agonists, SERMs, SERDs, etc.). Whereas the ligand-binding and DNA-binding domains are conserved within the family, the activation domains are highly variable. Finally, a hinge domain between the ligand- and DNA-binding domains gives the proteins flexibility, presumably important in their function but that has limited crystal structure determinations of the holoproteins (Helsen and Claessens, 2014). In addition to the full-length 66-kDa protein (of ERα, designated ER66), splice variants have been described (Taylor et al., 2010), resulting in proteins of 46 kDa (resulting from an amino-terminal truncation due to an alternate intron-localized start site) (Kim and Bender, 2009) and 36 kDa (generated from the same start site as ER46 but with an additional truncation at the carboxy terminus) (Lin et al., 2013; Chaudhri et al., 2014). Missing the amino terminal transcription activation domain, these proteins have been shown to act as inhibitors of ERα-mediated transcription and to mediate rapid signaling pathways (Wang et al., 2006). Multiple splice variants of ERβ have also been identified but are less well characterized (Heldring et al., 2007).
2. Localization.
Nuclear hormone receptors that act as transcription factors must by definition be localized to the nucleus to mediate their gene expression effects, although all such proteins dynamically traffic into and out of the nucleus via nuclear localization and export sequences (Kumar et al., 2006). However, whereas unliganded androgen receptors are localized predominantly in the cytoplasm and translocate to the nucleus upon agonist binding (Saporita et al., 2003), unactivated estrogen receptors are localized predominantly (∼95%) in the nucleus with the remainder in the cytoplasm (Hager et al., 2000). Ligand activation typically results in dimerization after monomer dissociation from chaperones (Hsp90) and translocation of cytosolic receptors to the nucleus. Of the cytosolic fraction of ERα, and in particular its splice variants, a fraction is further localized to the plasma membrane, and in particular caveolae, where upon agonist binding, the receptor mediates rapid signaling through such pathways as PI3K/Akt and eNOS (Chambliss et al., 2010; Banerjee et al., 2014). Localization to the membrane is thought to occur through palmitoylation (La Rosa et al., 2012), or possibly phosphorylation (Mintz et al., 2008), as well as through the recent characterization of a transmembrane domain in the ER46 splice variant (Kim et al., 2011). Both the existence of splice variants and post-translational modifications have been shown to affect ligand affinity and specificity (Lin et al., 2013).
3. Function.
As with other steroid hormone receptors, ERs function in transcription as dimers (both homodimers and heterodimers), binding to palindromic DNA sequences (estrogen response elements [EREs]) and acting through the recruitment of coregulators, both coactivators to stimulate and/or corepressors to inhibit gene expression (Smith and O'Malley, 2004; McDonnell and Wardell, 2010; Burris et al., 2013). Activated ERs also bind to DNA indirectly through associations with other transcription factors, such as AP-1 and Sp-1, and in the absence of ligands can regulate transcription through post-translational modifications (Dahlman-Wright et al., 2006). The diverse array of ligands capable of binding to ERs produces multiple conformational states of the receptor ligand-binding domain (in particular helix 12) that in turn generate multiple protein binding sites for coregulators and other proteins, which, with differential expression patterns in specific tissues, results in the complex physiology of ERs and their ligands (McDonnell and Wardell, 2010; Burris et al., 2013). Thus, although E2 is a full agonist in all tissues, 4-hydroxytamoxifen (the active metabolite of tamoxifen, a SERM; see Fig. 2) is an antagonist in the breast presumably because of low coactivator expression levels, but an agonist in the endometrium where coactivator expression is higher (Burris et al., 2013). Raloxifene, another SERM, however, is a weaker agonist in the endometrium because of the differential stabilization of alternative ER conformations (Burris et al., 2013). Importantly, both tamoxifen and raloxifene also act as partial agonists/antagonists on a number of distinct E2-regulated genes, further complicating the categorization of their activity (Frasor et al., 2004; Margueron et al., 2004). ICI 182,780 (7α,17β-[9-[(4,4,5,5,5-pentafluoropentyl)sulfinyl]nonyl]estra-1,3,5(10)-triene-3,17-diol), an SERD and full antagonist, on the other hand promotes Hsp90 dissociation but prevents ER dimerization and coactivator recruitment, resulting in the ligand-bound receptor being targeted for degradation (Heldring et al., 2007). Thus, depending on the ligand and the tissue, ERs regulate both positively and negatively the expression of thousands of genes (Katzenellenbogen et al., 2000; Charn et al., 2010).

Structures of synthetic steroid derivatives, analogs, and therapeutics.
In addition to transcriptional/genomic regulation, E2 mediates a multitude of rapid/nongenomic cellular signaling events including cAMP production, calcium mobilization, ion channel activation, and protein kinase activation with the resulting activation of secondary signaling cascades and effectors, which can also regulate transcription through or independent of ERs. These rapid effects are believed to originate at least in part from membrane-bound populations of ERs and have been explored recently using various forms of transgenic mice expressing mutant forms of ERα, altering membrane localization (Pedram et al., 2013, 2014; Adlanmerini et al., 2014) and membrane-impermeable estrogens (Banerjee et al., 2014). ERα-mediated mechanisms of rapid signaling have been reported to involve direct binding of ERα to heterotrimeric G proteins, c-Src, and the regulatory subunit of PI3K, leading to the latter’s activation (Banerjee et al., 2014). Some of these associations are promoted by striatin, which results in the localization of ERα to caveolae, particularly important in the activation of eNOS in endothelial cells (Wu et al., 2011a).
C. G Protein–Coupled Estrogen Receptor
1. Structure.
GPER was originally identified by a number of laboratories in the late 1990s as an orphan receptor (a cloned receptor with no known ligand) and soon named GPR30 (based on the sequential numbering scheme for orphan receptors), belonging to the family of 7-transmembrane–spanning GPCRs. The cDNA was identified from multiple sources including B lymphocytes (Owman et al., 1996; Kvingedal and Smeland, 1997), ER-expressing breast cancer cells (Carmeci et al., 1997), and endothelial cells exposed to sheer stress (Takada et al., 1997), as well as database mining (O'Dowd et al., 1998) and degenerate oligonucleotide screening of genomic DNA (Feng and Gregor, 1997). Sequence homology suggested GPR30 was most similar to the chemoattractant/chemokine subfamily of GPCRs, but an extensive screen of chemokines yielded no activating ligands (Owman et al., 1996). Since these original attempts to identify a ligand for this orphan receptor, more than 700 articles have been published, the vast majority since 2005. The existing data overwhelmingly demonstrate that GPR30 specifically binds estrogens and thereby activates intracellular signaling cascades commonly associated with G protein–coupled receptors, leading to its designation as G protein–coupled estrogen receptor (GPER) by the International Union of Basic and Clinical Pharmacology in 2007 (Alexander et al., 2013). In the following sections, we will discuss the different structural classes of naturally occurring and synthetic compounds that bind and either activate or inhibit GPER. Subtle features of structure and activity are evident from comparing the binding and activity profiles of such compounds, and special emphasis is placed on the identification of compounds that exhibit selectivity for GPER over ER.
The organization of the seven transmembrane domains of GPCRs is such that the amino terminus is localized to the cell exterior, where it is often glycosylated, and the carboxy terminus is localized to the cytoplasm, where it plays an important role in receptor desensitization and internalization through phosphorylation by G protein–coupled receptor kinases (Gurevich et al., 2012) and arrestins, which also initiate secondary signaling cascades (Liggett, 2011; Luttrell and Miller, 2013). Cytoplasmic loops are involved in the selective binding and activation of heterotrimeric G proteins, with many GPCRs able to activate multiple G proteins (Wong, 2003; Moreira, 2014). With recent high-resolution structural determinations of a number of GPCRs (Rosenbaum et al., 2009; Kruse et al., 2014), the structure-function relationship within this receptor family has been greatly advanced.
2. Localization.
The classic view of membrane-localized receptor function involves the transmission of signals from the cell exterior across the plasma membrane to the cell interior, and GPCRs and other receptors (for cytokines, growth factors, etc.) are almost exclusively depicted as functioning at the plasma membrane. This model is consistent with the majority of GPCR ligands that are charged and do not passively permeate membranes. Although early graphical depictions of GPER function suggested its placement in the plasma membrane at the cell surface (Filardo, 2002; Filardo et al., 2002), subsequent experimentation demonstrated that in many cell types, the majority of receptors (as determined through confocal microscopy) under steady-state conditions was localized to intracellular membranes, including the endoplasmic reticulum and Golgi apparatus (Revankar et al., 2005) with contradictory reports subsequently published (Thomas et al., 2005; Funakoshi et al., 2006; Filardo et al., 2007). Although the localization of GPER remained controversial, a majority of studies using both cellular and tissue samples yielded results consistent with a predominantly intracellular cytoplasmic membrane localization (Sakamoto et al., 2007; Albanito et al., 2008; Matsuda et al., 2008; Otto et al., 2008; Liverman et al., 2009; Terasawa et al., 2009), although nuclear localization has also been observed (Smith et al., 2009; Madeo and Maggiolini, 2010; Pupo et al., 2013). E2 is freely membrane permeable and cell surface expression is not required of an estrogen receptor, as exemplified by the predominantly nuclear localization of ERα (Hager et al., 2000). Furthermore, it is now widely recognized that many GPCRs (Luttrell and Miller, 2013), in particular those for lipid mediators (Zhu et al., 2006b), as well as other transmembrane receptors can signal from intracellular locations (Platta and Stenmark, 2011). That GPER could function from intracellular membranes was supported by studies that demonstrated only cell-permeable E2 derivatives could rapidly activate GPER (Revankar et al., 2007) and that intracellular injection of a GPER-selective ligand resulted in a more rapid and potent calcium response compared to extracellularly applied ligand (Deliu et al., 2012). Despite these results, other studies suggest that GPER can indeed be detected at the surface of certain cell types (Cheng et al., 2014). A resolution to this controversy may derive from recent observations that GPER undergoes constitutive clathrin-mediated internalization from the cell surface to intracellular membranes, ultimately the trans Golgi network (Cheng et al., 2011a,b), and that GPER trafficking to the cell surface is regulated by coexpression of receptor activity-modifying protein 3 (Lenhart et al., 2013). Thus, ineffective trafficking to the cell surface and constitutive internalization of GPER likely account for the low to undetectable equilibrium levels of GPER found at the cell surface of many cell types. However, regulation of these trafficking mechanisms in other cell types could lead to significant levels of GPER at the cell surface (Filardo and Thomas, 2012), which could result in cellular signaling distinct from that of intracellular receptors, in part due to differences in the associated signaling partners.
3. Function.
GPER, as a member of the GPCR superfamily, couples to heterotrimeric G proteins, which subsequently regulate a multitude of downstream effectors within the cell. Evidence exists for GPER coupling to both Gi/o and Gs proteins. The first activity of GPER to be demonstrated involved the E2-mediated rapid activation of ERK1/2 in a pertussis toxin-sensitive manner, indicating the involvement of Gi/o proteins (Filardo et al., 2000). The downstream signaling pathway involved the Src-mediated activation of metalloproteinases, which liberate heparin-binding EGF, with the ensuing transactivation of the EGFR and thereafter ERK1/2. The ability of GPER to activate adenylyl cyclase was demonstrated next, first as a mechanism involved in the attenuation of ERK1/2 activation (Filardo et al., 2002) and most recently in vascular dilation (Lindsey et al., 2014). GPER also activates the PI3K/Akt axis in response to E2 and although ERα also activates this pathway, the two receptors differ in the mechanisms employed; only GPER-mediated PI3K activation involves EGFR transactivation, with ERα employing an EGFR-independent mechanism (Revankar et al., 2005). In addition, GPER activates eNOS to produce nitric oxide within the vasculature (Meyer et al., 2012a; Lindsey et al., 2014) and sphingosine kinase to yield sphingosine 1-phosphate in cancer cells (Sukocheva et al., 2006). Additional studies have revealed GPER-mediated calcium mobilization (Revankar et al., 2005; Haas et al., 2009) and the regulation of potassium channels (Yu et al., 2011; Dong et al., 2013). In addition to, and as a consequence of, these rapid signaling events, GPER also regulates gene expression, although not to the same extent as ERα (Prossnitz and Maggiolini, 2009a). Among the genes whose expression is regulated by GPER are c-fos, cyclin A and D1 (Vivacqua et al., 2006a; Albanito et al., 2015), connective tissue growth factor (Pandey et al., 2009; Madeo and Maggiolini, 2010), fatty acid synthase (Santolla et al., 2012), and vascular endothelial growth function (De Francesco et al., 2013, 2014). Phosphatidylinositol 3,4,5 trisphosphate production as a result of PI3K activation by GPER (stimulated by E2, genistein, 4-hydroxytamoxifen, and 2-(4-hydroxyphenyl)-3-phenylpent-2-enoic acid [4-(2-dimethylaminoethoxy)phenyl]amide [E isomer; STX]; see section IV) also leads to activation of the transcription factor SF-1, resulting in aromatase Cyp19a1 expression and increased E2 production and proliferation (Lin et al., 2009). Together, these varied GPER-activated pathways regulate diverse cellular functions from proliferation to metabolism to migration to secretion, with profound implications for the role of GPER in normal physiology and disease. When combined with the actions of classic ERs at the cellular level, ERs and GPER may act either in concert (synergistically or with a requirement for the other receptor) or to antagonize aspects of the others activity, with the ultimate cellular output being dependent on the integration of all the stimulated and inhibited pathways.
III. Physiologic and Pathophysiologic Functions of G Protein–Coupled Estrogen Receptor
Because of the multiple estrogen receptors (ERα, ERβ, their many splice variants, and GPER) expressed throughout the body, determining the functions of individual receptors in normal physiology and disease has been particularly challenging. Despite this complexity, through the use of GPER knockout mice and highly selective GPER agonists and antagonists, roles for GPER have been described in virtually every physiologic system of the human body. In addition, regulation of GPER activity has been shown to ameliorate pathophysiology in a growing number of diseases, with implications for neurologic diseases including stroke and traumatic brain and spinal cord injury, cardiovascular and renal diseases (including hypertension, atherosclerosis and myocardial infarct), metabolic diseases such as diabetes and obesity, autoimmune diseases such as multiple sclerosis, cancer, and many more. The widespread involvement of GPER in such a wide array of pathophysiologies suggests that GPER-targeted therapies could represent an important new approach to the treatment of these diseases.
A. Reproduction
The physiologic functions of E2 and ERα are perhaps best understood in the development and function of the reproductive system, specifically the uterus, ovary, and the breast, with ERα knockout mice displaying several reproductive defects resulting in infertility. On the contrary, GPER knockout mice (Wang et al., 2008a; Otto et al., 2009) and ERβ knockout mice (Couse and Korach, 2001) have been reported to be fertile, although the most recent ERβ knockout mouse, devoid of alternately spliced variants, was reported to be sterile (Antal et al., 2008). These differences are further evidenced by the lack of effects of the selective GPER agonist G-1 (1-[4-(6-bromobenzo[1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl]-ethanone) (see section IV.H and Fig. 7) on classic assays of uterine function (Hewitt et al., 2003). Whereas E2 induces robust uterine water imbibition in ovariectomized mice, G-1 displays no such activity (Dennis et al., 2009). In addition, the acute proliferative response of uterine epithelial cells observed upon E2 treatment is greatly reduced with G-1 treatment (Dennis et al., 2009), whereas the E2-mediated response is inhibited by simultaneous high doses of G-1, suggesting GPER may even oppose ERα function in the uterus (Gao et al., 2011). Furthermore, the fundamental structure and function of the mammary gland in GPER knockout mice appears largely normal (Otto et al., 2009). Taken together, these results suggest that the majority of E2’s reproductive functions are mediated by ERα and that GPER and G-1 lack the classic feminizing effects of ERα and E2. Nevertheless, G-1 has been demonstrated to increase the frequency and amplitude of rat myometrial contractions (Tica et al., 2011), as well as the oxytocin-induced contractile response of human myometrium explants (Maiti et al., 2011), suggesting limited specific functions of GPER in the uterus. A role for GPER in mammalian primordial follicle formation has been reported (Wang et al., 2008b), and in nonmammalian vertebrates, G-1 has also been shown to reduce both spontaneous and progestin-induced oocyte maturation, suggesting a role for GPER in maintaining oocyte meiotic arrest (Pang et al., 2008; Peyton and Thomas, 2011). In terms of female pathophysiology, increased GPER expression has been associated with endometriosis (Heublein et al., 2012; Plante et al., 2012; Samartzis et al., 2012; Yuguchi et al., 2013) and is more frequent in malignant versus benign ovarian endometriotic cysts (Long et al., 2012). GPER expression and its activity have also been demonstrated to be of importance in endometrial and ovarian cancers (see section III.F).
Although the functions of GPER in the male reproductive system are far from clear, GPER is expressed in both normal germ cells and somatic cells and is involved in mediating certain actions of E2 in spermatogenesis, regulating both proliferative and apoptotic events (Chimento et al., 2014b). GPER expression and function have been reported in spermatogonia and spermatids (Sirianni et al., 2008; Chimento et al., 2011; Sheng and Zhu, 2011), Sertoli cells (Lucas et al., 2010), Leydig cells (Chimento et al., 2014a; Vaucher et al., 2014), and gubernaculum testis cells (Zhang et al., 2014c). Furthermore, seminoma-associated GPER genetic variants (Chevalier et al., 2014), GPER overexpression in human seminoma (Franco et al., 2011; Chevalier et al., 2012b), and other testicular germ cell tumors (Franco et al., 2011), as well as GPER-mediated tumor cell proliferation in response to estrogenic compounds, such as bisphenol A (Chevalier et al., 2012a), have been reported.
B. Endocrine/Neuroendocrine System
Many of the physiologic effects of E2, which impinge upon virtually all aspects of the endocrine system, are reproduced to varying extents by selective GPER activation. In addition to direct effects on the reproductive system described above, these include actions within the nervous system on the hypothalamus-pituitary-gonadal axis, where GPER is expressed in the anterior and posterior pituitary as well as in the paraventricular, ventromedial, and supraoptic nuclei of the hypothalamus (Brailoiu et al., 2007). GPER exhibits distinct expression patterns compared with classic ERs (Hazell et al., 2009), exemplified by expression in magnocellular oxytocin but not vasopressin neurons (Sakamoto et al., 2007). Selective GPER activation with G-1 attenuates oxytocin and ACTH responses (Xu et al., 2009) and GPER is required for E2-mediated desensitization of serotonin signaling in the paraventricular nucleus (McAllister et al., 2012). GPER is also implicated in the rapid E2-mediated release of luteinizing hormone-releasing hormone via enhanced Ca2+ oscillations in primate luteinizing hormone-releasing hormone neurons (Noel et al., 2009) as well as negative feedback by E2 of gonadotropin-releasing hormone–induced luteinizing hormone secretion (Rudolf and Kadokawa, 2013). Roles for GPER in the hypothalamus-pituitary-gonadal axis of males were recently reviewed (Chimento et al., 2014b).
The increased prevalence of obesity, insulin resistance, and diabetes after menopause reveals a strong influence of E2 on energy balance and glucose homeostasis (Meyer et al., 2011a; Mauvais-Jarvis et al., 2013; Rettberg et al., 2014). Although roles for the classic ERs are well documented in these conditions (Faulds et al., 2012), GPER knockout mice also exhibit insulin resistance, glucose intolerance, dyslipidemia, obesity, and an elevation of proinflammatory cytokines with reduced adiponectin levels (Martensson et al., 2009; Sharma et al., 2013; Davis et al., 2014), despite displaying no differences in food intake or locomotor activity (Sharma et al., 2013; Davis et al., 2014). G-1, like E2, stimulates insulin secretion from the islets of mice (Sharma and Prossnitz, 2011) and humans (Kumar et al., 2011), with both G-1– and E2-mediated insulin secretion absent in islets of GPER knockout mice (Sharma and Prossnitz, 2011). Patterns of G-1–mediated inhibition of glucagon and somatostatin release are identical to those of E2 (Balhuizen et al., 2010). In addition to mediating pancreatic hormone secretion, GPER is also important in the protection and survival of β-cells under conditions of stress (Liu et al., 2009a), with G-1 improving survival of transplanted islets in a murine model of type I diabetes (Liu et al., 2013). Interestingly, both male and female GPER knockout mice display decreased energy expenditure and increased brown fat lipid accumulation, yet only female GPER knockout mice show a deficit in leptin- and cholecystokinin-mediated anorexis (Davis et al., 2014). In ovariectomized female mice, the E2-mediated improvements in weight and fat reduction, glucose homeostasis, and adipocyte size were absent or mitigated in GPER knockout mice (Davis et al., 2014). The recent demonstration of the regulation of GPER expression by insulin suggests an additional potential linkage between GPER and metabolism (De Marco et al., 2014).
Uptake of GPER-targeted radiolabeled ligands in vivo by the adrenal gland (Ramesh et al., 2010) and immunohistochemical localization of GPER to the adrenal medulla (Baquedano et al., 2007; Hazell et al., 2009) and zona glomerulosa (Baquedano et al., 2007) suggest possible functions in the secretion of hormones from this gland. Expression of GPER in multiple other tissues/organs with endocrine functions including liver (Hsieh et al., 2007), kidney (Lindsey et al., 2011b; Cheng et al., 2014), adipose (Gavin et al., 2013), and muscle (Baltgalvis et al., 2010) suggest multiple additional potential roles for GPER in endocrine function throughout the body.
C. Immune System
Estrogen (Bonds and Midoro-Horiuti, 2013; Sakiani et al., 2013), as well as therapeutic “antiestrogens,” such as tamoxifen and raloxifene (Ray and Ficek, 2012), exerts diverse effects upon multiple aspects of immune system development and function. A role for GPER in E2-mediated thymic atrophy (Pernis, 2007) was first suggested through the use of ERα, ERβ, and GPER knockout mice (Wang et al., 2008a), where ERα expression was required for the early developmental blockage of thymocyte development and GPER expression was necessary for apoptosis of T-cell receptor double-positive thymocytes. In addition, G-1 treatment induced thymic atrophy and thymocyte apoptosis but had no effect on the developmental blockage of thymocytes. Lower frequencies of T cells (particularly those expressing CD62L) in both sexes of GPER knockout mice have also been reported, consistent with impaired production of T cells in the thymus (Isensee et al., 2009).
Estrogens (Palaszynski et al., 2004; Niino et al., 2009) and estrogenic compounds, such as genistein (De Paula et al., 2008), are also receiving greater attention as potential anti-inflammatory agents for autoimmune diseases, such as multiple sclerosis. Employing the murine experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis, E2-mediated protection was significantly decreased in GPER knockout mice (Wang et al., 2009), whereas G-1 treatment mediated an equivalent protection against the clinical and histologic manifestations of EAE to that of E2 (Blasko et al., 2009; Wang et al., 2009). The protective effects of G-1 were absent in GPER knockout mice, confirming the selectivity of G-1 for GPER in vivo (Wang et al., 2009). Furthermore, the therapeutic efficacy of ethynyl estradiol in established disease required expression of GPER but not ERα and were associated with anti-inflammatory cytokine interleukin (IL)-10 production (Yates et al., 2010). Mechanistic studies revealed that G-1 not only enhanced the suppressive activity of CD4+Foxp3+ T regulatory cells through upregulation of programmed death 1 (Wang et al., 2009), but also inhibited inflammatory cytokine production by macrophages (Blasko et al., 2009), suggesting multiple coordinated or perhaps independent effects on the immune system.
In terms of direct effects on T-cell differentiation and function, G-1 was shown not only to elicit de novo IL-10 production and secretion in Th17-polarized cells ex vivo as well as following G-1 administration in vivo (Brunsing and Prossnitz, 2011), but to induce Foxp3 expression (a marker of natural and induced regulatory T cells) in purified CD4+ T cells under Th17-polarizing conditions, which are prevalent in autoimmune diseases (Brunsing et al., 2013). G-1 was also recently shown to modulate the respiratory burst in vertebrate granulocytes (neutrophils) as well as pro- and anti-inflammatory gene expression profiles (Cabas et al., 2013). A direct immunomodulatory function of G-1 in the endothelium was also demonstrated through attenuation of the tumor necrosis factor α–induced upregulation of proinflammatory adhesion molecules intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 (Chakrabarti and Davidge, 2012). Interestingly and perhaps unexpectedly, G-1 reversed the immunosuppression of the peripheral immune system that follows experimental stroke in ovariectomized female mice (Zhang et al., 2010). Together, these results suggest that GPER induces predominantly immunoprotective effects on multiple classes of immune and other cells.
D. Nervous System
Estrogens, both gonadally and brain derived (Zhang et al., 2014b), mediate extensive effects in the central and peripheral nervous system, including regulation of the hypothalamus-pituitary-gonadal axis (discussed above), sexual behavior, synaptic plasticity, mood, memory, cognition, and pain sensation (Hammond and Gibbs, 2011). Although many of these effects likely involve genomic and rapid signaling by ERα and potentially ERβ (Bean et al., 2014), as well as a multitude of additional pharmacologically defined but otherwise unidentified estrogen receptors (e.g., ER-X, Gq-mER) (Toran-Allerand, 2004; Kelly and Ronnekleiv, 2013), increasing evidence indicates that predominantly rapid signaling via GPER has multiple roles in E2-mediated neurologic functions (Raz et al., 2008; Srivastava and Evans, 2013). GPER (mRNA and protein) is expressed throughout the central and peripheral nervous system (although not universally) of both female and male rodents, including the cortex, hippocampus, hypothalamus, specific nuclei of the midbrain, the trigeminal nuclei and cerebellum Purkinje layer of the hindbrain, the anterior, intermediate and neural lobes of the pituitary, as well as the spinal cord and dorsal root ganglia (Brailoiu et al., 2007; Dun et al., 2009; Hazell et al., 2009). The activation of ERK1/2 in trigeminal ganglion neurons and the increased allodynia induced by PPT [4,4′,4′′-(4-propyl-[1H]-pyrazole-1,3,5-triyl) trisphenol] and G-1 has led to the conclusion of roles for both ERα and GPER in peripheral sensitization (Liverman et al., 2009); however, with the recent demonstration that PPT can also function as a GPER agonist (Petrie et al., 2013), it is possible that both responses, in fact, were mediated by GPER, because independent methods to assess receptor involvement were not employed. G-1 also depolarizes spinal cord neurons (Dun et al., 2009), stimulates mechanical hyperalgesia via protein kinase Cε activation (Kuhn et al., 2008), and mediates visceral hypersensitivity in the absence of inflammation (Lu et al., 2009).
The protective effects of E2 are well documented in the brain and spinal cord and include reducing neuronal loss after stroke and traumatic injury (Stein and Hoffman, 2003; Prossnitz, 2012) as well as increasing neuronal connectivity and improving cognitive performance (Hammond and Gibbs, 2011). GPER has been implicated in E2-mediated effects on cholinergic neurons in the basal forebrain, which suggests that GPER might be an important regulator of cognitive function, particularly important after menopause (Hammond et al., 2011). By using immortalized hippocampal cell lines, GPER was implicated in the protective effects of E2 in glutamate-induced injury (Gingerich et al., 2010). In vivo studies have also demonstrated that G-1 replicates the effects of E2 in promoting neuronal survival after global or local ischemia in the brain (Lebesgue et al., 2009a, 2010), improves cerebral microvascular function after hypoxia and reperfusion (Murata et al., 2013), and improves immunosuppression after stroke (Zhang et al., 2010), suggesting that GPER agonists could represent a new therapeutic approach for stroke as well as chronic neurodegenerative diseases (Etgen et al., 2011). Interestingly, stroke induces GPER expression in the brain (Broughton et al., 2013), with G-1 shown to reduce neurologic deficit, apoptosis, and infarct volume (Broughton et al., 2014), in a sex-specific manner. Finally, the protective effects of raloxifene on dopamine neurons in a murine model of Parkinson’s disease have been suggested to occur through GPER, based on the inhibitory effects of a selective GPER antagonist (Bourque et al., 2014).
G-1 (like E2) also attenuates serotonin receptor signaling in the paraventricular nucleus of the hypothalamus and reduces responses to oxytocin and adrenocorticotropic hormone, suggesting that GPER could have a role in mood disorders (Xu et al., 2009). Consistent with this, G-1, like E2, exhibited antidepressant properties in a mouse model of depression, effects that were inhibited by the GPER-selective antagonist G15 [4-(6-bromo-benzo[1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolone] (Dennis et al., 2009). A reduced sensitivity to leptin- and cholecystokinin-induced anorexia in female but not male GPER knockout mice was correlated with a lack of E2-induced ERK activation in the basal medial hypothalamus of ovariectomized female mice, suggesting a role for GPER in the E2-mediated aspects of satiety (Davis et al., 2014). In primates, GPER contributed to the E2-mediated regulation of luteinizing-hormone-releasing hormone neurons, which maintain gonadal function and fertility (Noel et al., 2009). However, whereas GPER activation promoted short latency prolactin secretion, G-1 did not alter the E2-mediated negative feedback inhibition of luteinizing hormone secretion and knockdown of GPER in the mediobasal hypothalamus did not alter lordosis behavior in rats (Lebesgue et al., 2009b), although a recent report demonstrated that G-1 promotes lordosis in mice (Anchan et al., 2014), suggesting that although GPER is sufficient to promote lordosis, it may not be necessary. The extent and complexity of neurologic effects of GPER activation suggest that selective ligands could play an important role in multiple neurologic conditions and diseases.
E. Cardiovascular System
Estrogen is an important regulator of cardiovascular function and is associated with the decreased incidence of hypertension and coronary artery disease in premenopausal women compared with age-matched men and postmenopausal women (Barton and Meyer, 2009; Chakrabarti et al., 2014; Maric-Bilkan et al., 2014). G-1 action through GPER mimics many of the actions of E2 in regulating vascular tone and providing protection from myocardial ischemia/reperfusion injury (Lindsey and Chappell, 2011; Meyer et al., 2011b; Han et al., 2013; Holm and Nilsson, 2013; Chakrabarti et al., 2014; Prossnitz and Barton, 2014). G-1–mediated GPER activation results in endothelial NO-mediated vasodilation in multiple vessels (Meyer et al., 2011b; Li et al., 2012; Lindsey et al., 2014), although a role for cAMP production in vascular smooth muscle cells has recently been identified (Lindsey et al., 2014). Importantly, G-1 lacks activity in arteries isolated from GPER knockout mice, further establishing in vivo selectivity of this compound (Haas et al., 2009). Inhibition of GPER with the selective GPER antagonist G15 results in vasoconstriction (Lindsey et al., 2011a; Yu et al., 2011; Meyer et al., 2012a), suggesting that GPER exhibits basal vasodilatory activity, representing either constitutive activity in the absence of ligand or in response to endogenous physiologic ligands. Furthermore, GPER knockout mice display increased vasoconstrictor responses in part as a result of enhanced cyclooxygenase-derived endothelium-dependent contracting factor activity as well as enhanced calcium sensitivity (Meyer et al., 2010, 2012a,b). Consistent with studies on isolated vessels, acute G-1 administration to healthy rats (Haas et al., 2009), as well as well as chronic G-1 administration in an E2-deficient model of hypertension (Lindsey et al., 2009), has been shown to lower blood pressure.
Activation of GPER by G-1 or E2 is also protective in multiple models of heart disease and damage. Chronic G-1 administration attenuates diastolic dysfunction and left ventricular remodeling in hypertensive ovariectomized rats independent of changes in blood pressure (Wang et al., 2012) and improves cardiac function through the modulation of adrenergic receptor expression in isoproterenol-induced heart failure (Kang et al., 2012). GPER knockout mice have also been reported to exhibit impaired left ventricular cardiac function (Delbeck et al., 2011). G-1 has been shown by many groups to reduce ischemia/reperfusion-induced cardiac injury as measured by infarct size as well as contractile function (Deschamps and Murphy, 2008, 2009; Bopassa et al., 2010; Patel et al., 2010). Recent reports have also demonstrated that specifically GPER expression, but not that of ERα or ERβ, is required for the protective effects of E2 on myocardial reperfusion injury (Bopassa et al., 2011, 2012a,b). Finally, GPER knockout mice also show increased atherosclerosis and vascular inflammation when fed a high-fat atherogenic diet, with G-1 treatment of wild-type mice reducing both plaque formation and macrophage infiltration (Meyer et al., 2014).
In the kidney, GPER is expressed predominantly in distal convoluted tubules and the loop of Henle, with lower expression in proximal convoluted tubules and no detectable expression in collecting ducts, with extensive regulation of expression levels and patterns throughout the estrous cycle (Cheng et al., 2014). For example, GPER is upregulated on cortical epithelia during the secretory phase, localized to the basolateral surface during proestrus with intracellular redistribution occurring during estrus, and downregulation on the surface of renal epithelia during the luteal phase. Together, these complex spatial and temporal expression patterns of GPER during the estrous (menstrual) cycle suggest that GPER could play a physiologic role in kidney function (e.g., water reabsorption), particularly during reproduction (Cheng et al., 2014).
Like E2, G-1 also exhibits renoprotective effects (Perez-Torres et al., 2011). Estrogen and G-1 as well as the SERD ICI 182,780 (fulvestrant), an ER antagonist, were shown to stimulate rapid calcium signaling and H+-ATPase activity in renal tubules and isolated intercalating cells (Hofmeister et al., 2012). All three ligands were without effect in tubules and cells isolated from GPER knockout mice, once again demonstrating the critical role of GPER in these activities. In an E2-replete (i.e., ovary intact) model of salt-induced hypertension with renal damage, G-1 reduced renal hypertrophy, improved creatinine clearance, and reduced proteinuria, although, presumably due to the presence of E2, G-1 had no effect on blood pressure (Lindsey et al., 2011b). The actions of G-1 led to a reduction of tubular oxidative stress and induction of megalin expression leading to improved protein reabsorption (Lindsey et al., 2011b). The combined effects of G-1 on the cardiovascular and renal systems, as well as on glucose metabolism, suggest that GPER could represent an important target in multiple aspects of metabolic syndrome and cardiovascular diseases.
F. Cancer
Estrogen plays an important role in the development, diagnosis, prognosis, and treatment of breast cancer (Williams and Lin, 2013) as well as an increasingly recognized role in other cancers (Fucic et al., 2012; Gallo et al., 2012). Although the effects are often clearly linked to the expression of ERα, particularly in breast cancer, studies over the last decade have begun to reveal important functions for GPER in multiple cancers (Lappano et al., 2014). In a study of 361 breast tumor samples, GPER was expressed in approximately half of breast cancers, regardless of their ER status, and correlated with clinical and pathologic biomarkers of poor outcome, such as increased tumor size and metastasis (Filardo et al., 2006). However, in a study of 164 primary breast cancers and matched normal tissues, decreased GPER expression was reported to represent an unfavorable factor in overall survival (Ignatov et al., 2013b). Yet in breast cancer patients treated only with tamoxifen, GPER protein expression increased with treatment and survival was significantly lower in patients with initially GPER-positive tumors, consistent with the activity of tamoxifen as a GPER agonist (Ignatov et al., 2011a). Furthermore, recent studies demonstrate that GPER expression is increased in metastases, relative to matched primary tumors, and that tamoxifen-induced resistance can be reversed with a GPER antagonist (Mo et al., 2013). GPER activation also leads to rapid signaling and proliferation in MCF10A cells, considered normal (though immortalized but not tumorigenic) human breast epithelial cells (Scaling et al., 2014). Recently, the first evidence for a role for GPER in breast tumorigenesis and metastasis in vivo has been provided employing GPER knockout mice crossed to the widely used transgenic mouse model of mammary tumorigenesis MMTV-PyMT (Lin et al., 2003). Tumors from mice deficient in GPER expression were smaller, of lower histologic grade, and exhibited decreased proliferation and importantly metastasis (Marjon et al., 2014). Similarly, the first studies to examine the effects of GPER activation and inhibition on primary human breast tissue (from reduction mammoplasty) and primary breast cancer tissue reveal that G-1, like E2, stimulates proliferation and that the GPER antagonist G36 [4-(6-bromo-benzo[1,3]dioxol-5-yl)-8-isopropyl-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolone] not only completely inhibits G-1–mediated proliferation, but also inhibits ∼80% of E2-mediated proliferation, suggesting a critical and potentially complex costimulatory role for GPER in breast cancer growth (Scaling et al., 2014).
In addition to breast cancer cell lines and primary tumors of the breast (Carmeci et al., 1997; Filardo et al., 2000; Revankar et al., 2005; Albanito et al., 2008), GPER is also expressed in cancers and cell lines of the endometrium (Vivacqua et al., 2006a; Leblanc et al., 2007; Smith et al., 2007; He et al., 2009; Petrie et al., 2013; Dai et al., 2014), ovaries (Albanito et al., 2007, 2015; Henic et al., 2009; Smith et al., 2009; Liu et al., 2014), thyroid (Vivacqua et al., 2006a), lung (Siegfried et al., 2009), prostate (Chan et al., 2010), and testes (Franco et al., 2011). In cell lines of thyroid, ovarian, endometrial, and breast cancers, stimulation of GPER with E2 (Vivacqua et al., 2006a,b; Albanito et al., 2007) or other estrogenic compounds, such as genistein (Vivacqua et al., 2006a), bisphenol A (Dong et al., 2011; Chevalier et al., 2012a), or tamoxifen (Vivacqua et al., 2006b) activates signaling mechanisms that typically promote proliferation. Furthermore, in endometrial (Smith et al., 2007) and ovarian cancer (Smith et al., 2009), high GPER expression correlated with poor survival, although a recent study reported the opposite in ovarian cancer (Ignatov et al., 2013a). GPER is also highly expressed in postpubertal testicular germ cell tumors (intratubular germ cell tumors, seminomas, and embryonal carcinomas) with little expression in teratomas (Franco et al., 2011).
As normal breast tissue is highly sensitive to E2, inducing proliferation during puberty and pregnancy, a majority of breast cancers involve E2-dependent signaling pathways in cancer initiation, progression, and metastasis (Cordera and Jordan, 2006). This has led to development of drugs that target E2 binding to ERα and E2 synthesis, including the SERMs tamoxifen and raloxifene (Jordan, 2007; Sengupta and Jordan, 2008), SERDs (such as fulvestrant), and aromatase inhibitors (Orlando et al., 2010). Many of these agents, particularly SERMs such as tamoxifen (Revankar et al., 2005) and raloxifene (Petrie et al., 2013) and fulvestrant (Filardo et al., 2000), which also function as GPER agonists, produce complex physiologic and therapeutic actions. Multiple studies now reveal that long-term E2 deprivation of the E2-dependent human breast cancer cell line MCF-7, mimicking treatment of women with antiestrogens or aromatase inhibitors, increased expression of GPER (Craig Jordan et al., 2007), with tamoxifen treatment of such resistant cells stimulating proliferation via GPER (Ignatov et al., 2010). Prolonged tamoxifen treatment also leads to increased aromatase activity and expression via GPER signaling, suggesting a possible mechanism involved in the development of tamoxifen resistance (Catalano et al., 2014). Despite the generally stimulatory effects of GPER stimulation on cancer cell line growth, G-1, particularly at high doses (generally ≥1μM), has also been shown to inhibit the proliferation of certain cancer cell lines in vitro (Ariazi et al., 2010; Chimento et al., 2014a; Weißenborn et al., 2014a,b), which may be a result of reported effects on microtubules at high concentrations (Holm et al., 2012; Wang et al., 2013), or possibly protein kinase Cε–mediated destabilization of microtubules (Goswami et al., 2011). Furthermore, in both androgen-dependent and -independent prostate cancer cells, G-1 inhibited growth via a sustained activation of ERK leading to G2 cell cycle arrest, resulting in a substantial reduction in tumor xenograft size (Chan et al., 2010). Importantly, G-1 induced no growth or histologic changes in the prostate and did not inhibit growth of an immortalized benign prostatic epithelial cell line. More recently, in a model of castration-resistant prostate cancer, G-1 induced substantial growth inhibition and neutrophil-induced necrosis of castration-resistant but not androgen-sensitive prostate cancer and was shown to be expressed at high levels in 80% of castration-resistant prostate cancer metastases, suggesting GPER may be a novel therapeutic target in this disease (Lam et al., 2014). Thus, the functions of GPER, like those of E2 (Lewis-Wambi and Jordan, 2009), in the dysregulated signaling of cancer cell lines are clearly complex, with both growth-promoting and -inhibiting actions reported (even in the same cell lines), likely depending on the specifics of the altered signaling pathways in a given cancer cell line, as well as the extent (magnitude/doses and length) of receptor activation. Clearly, to gain a better understanding of GPER function in carcinogenesis, it will be important to continue examining the roles of GPER in more relevant systems, including transgenic mouse models (Marjon et al., 2014) and primary human cancers (Scaling et al., 2014), opening the door to the development of GPER-targeted drugs for cancer therapy.
IV. G Protein–Coupled Estrogen Receptor Ligands and Pharmacology
Estrogenic activities are modulated by a multitude of organic compounds with myriad natural and synthetic origins. Binding affinity and potency can vary over a wide range, and structure-activity relationships within a series typically exhibit large changes in potency associated with relatively minor structural or stereochemical modifications. The plasticity of the nuclear estrogen receptors in binding structurally diverse ligands has been documented through extensive crystallographic and computational modeling studies (Huang et al., 2010; Nilsson and Gustafsson, 2011; Nilsson et al., 2011). Because of the rigid (conformationally inflexible) nature of E2 and the requirements of a ligand-binding pocket that recognizes E2, it is perhaps not surprising that GPER exhibits a similar but not identical capacity for “promiscuous” binding to many of the same compounds that bind ERα/β (Lathe and Kotelevtsev, 2014). With the developing recognition of the clinical relevance for ERα/β-selective pharmacologic agents (Minutolo et al., 2011) and the involvement of multiple receptors and pathways in physiologic responses, there is increasing awareness of the importance of evaluating the interactions of these compounds with respect to GPER binding and activity (Prossnitz and Barton, 2014). The issues of receptor selectivity, cross-reactivity, and multiple signaling pathways are intimately connected with the effective concentrations of a compound used to elicit pharmacologic responses, which can result in nonclassic or biphasic dose responses (Calabrese, 2001; Lebedeva et al., 2012). The following section describes important considerations for the experimental determination of binding affinities and reviews representative examples of compounds from the different individual classes that exhibit reported activities involving GPER (Tables 1 and 2). It should be noted that in some instances, significant discrepancies exist between reported binding affinities and EC50 values, which may suggest that low receptor occupancy is needed to elicit a given cellular effect. Alternatively, apparently disparate results for a given substance may be due to the fact that different systems are used for binding and functional experiments. Finally, many compounds exhibit very high (i.e., poor) binding affinities and/or EC50 values, raising the question of their biological significance. We nevertheless provide these values with the goal of exploring the structure-activity relationship of GPER. In all these areas, further studies are required to address these issues.
Binding and function of estrogenic compounds toward GPER
A. Receptor Binding Characteristics and Assays for G Protein–Coupled Estrogen Receptor
The quantitative determination of ligand affinities for their receptors is essential for characterizing the biological properties and defining the structure-activity relationships of pharmacologic agents. Binding equilibria are typically represented by the dissociation constant Kd, the reciprocal of the binding (association) constant Ka, with several methods available for their experimental determination. Relative binding affinities (RBA) are frequently employed in the steroid hormone (but typically not the GPCR) field to describe ligand affinities relative to the native hormone for a given receptor, 17β-estradiol in the case of ERα/β (RBA = 100% for E2 for both ERα and ERβ, despite the fact that the Kd values for E2 are different for the two receptors). The use of RBA values also facilitates comparisons between reports employing different binding assays, where the affinity for binding of E2 itself may vary many fold. The RBA values are typically obtained from IC50 values determined using competitive ligand-binding assays with the relationship:  Ligand affinities can also be expressed as Ki using the Cheng–Prusoff equation (Cheng and Prusoff, 1973; Munson and Rodbard, 1988):
Ligand affinities can also be expressed as Ki using the Cheng–Prusoff equation (Cheng and Prusoff, 1973; Munson and Rodbard, 1988):  with more complex analyses required for certain ligands (Giraldo et al., 2007). Significant differences exist between the nuclear hormone receptors and GPCRs that result in varying methodologies being used for measuring Kd values for ERα/β and GPER. Nuclear steroid receptors are generally soluble and have much greater inherent structural stability, which allows the use of homogenates of receptor-rich tissues or cells as sources of receptor for binding studies, although depending on the purity of the preparation, such assays likely do not allow a distinction to be made between ERα and ERβ (Blair et al., 2000). In addition, many assays for ERα/β binding, particularly commercially available assays, currently employ only the ligand-binding domain of the receptor, usually expressed in heterologous systems and in a purified state (Witkowska et al., 1997).
with more complex analyses required for certain ligands (Giraldo et al., 2007). Significant differences exist between the nuclear hormone receptors and GPCRs that result in varying methodologies being used for measuring Kd values for ERα/β and GPER. Nuclear steroid receptors are generally soluble and have much greater inherent structural stability, which allows the use of homogenates of receptor-rich tissues or cells as sources of receptor for binding studies, although depending on the purity of the preparation, such assays likely do not allow a distinction to be made between ERα and ERβ (Blair et al., 2000). In addition, many assays for ERα/β binding, particularly commercially available assays, currently employ only the ligand-binding domain of the receptor, usually expressed in heterologous systems and in a purified state (Witkowska et al., 1997).
In contrast to soluble receptors, the measurement of ligand binding to membrane-associated receptors (mERα/β and GPER) is considerably more challenging due to relatively low levels of receptor expression, and the high nonspecific background binding of hydrophobic ligands to lipid-rich membranes (Filardo and Thomas, 2012), the latter being required to maintain receptor activity in the absence of receptor solubilization (Sklar et al., 2000; Bennett et al., 2001; Key et al., 2001; Schuler et al., 2013). Importantly, unless solubilized and subsequently purified, GPCR binding assays are carried out with crude or variously enriched membrane preparations, which in either case contain a vast array of additional proteins. The lability and cellular localization of GPCRs can present additional experimental challenges for ligand binding assays (Baneres and Mouillac, 2012). Furthermore, the association of GPCRs with multiple signaling proteins can affect ligand affinity (Key et al., 2001, 2003; Prossnitz and Sklar, 2006). It is widely recognized that GPCRs often express low levels of receptor on the cell surface, as a result of regulated export via chaperones and escort proteins (Shirvani et al., 2012) and internalization by endocytosis, an important step in receptor desensitization, degradation, and recycling (Maestes et al., 1999; Prossnitz, 2004; Claing and Laporte, 2005). GPER has been shown to have a short half-life on the plasma membrane (<1 hour), with internalization occurring independently of stable arrestin association yet via clathrin-coated pits, followed by trafficking to the trans-Golgi network and receptor degradation subsequently occurring in the 26S-proteasome (Cheng et al., 2011a,b). In general, three different systems have been used to measure GPER ligand binding employing competitive (or direct for E2) radioligand (or fluorescent ligand) binding: (1) plasma membrane fractions, (2) whole cells, or (3) permeabilized cells. Computational docking studies based on the presumed ligand-binding site of GPER have also been employed to explore ligand binding properties (Rosano et al., 2012). A brief summary of each of these physical methods listed above and the important experimental considerations and constraints/limitations are discussed in the following sections.
1. Competitive Radioligand Binding to G Protein–Coupled Estrogen Receptor in Membrane Preparations.
Tritiated 17β-estradiol ([3H]E2) is commercially available and is the most commonly used tracer for competitive binding assays with estrogen receptors. Tritium emits a weak beta particle and has a half-life of 12.3 years. Labeling with isotopic hydrogen is ideal because this exchange does not affect the steric or electronic features of the ligand, and highly sensitive detection of [3H]E2 is possible with scintillation counting. The Kd values for 17β-estradiol binding to nuclear estrogen receptors (ERα/β) in cellular isolates are typically subnanomolar, ranging from 0.1–1.0 nM (Blair et al., 2000). The major difficulty in the isolation and purification of functionally active GPCRs results from the extensive interactions with phospholipid membranes that maintain functional structural conformations. Centrifugation methods have been employed for the preparation of subcellular fractions containing GPER that exhibit specific binding with filtration assays typically used to separate free from bound ligand in membrane fractions. This approach was first demonstrated in ER-negative cell lines using the human wild-type and recombinant receptor in SKBR3 and HEK293 cells, respectively (Filardo and Thomas, 2005), and later in human urothelial cell membranes (Teng et al., 2008) and with the zebrafish ortholog (Liu et al., 2009b). The lability of GPER requires short incubation times, low temperature (4°C), and the inclusion of protease inhibitors for reproducible ligand binding assays (Filardo and Thomas, 2012). Measured Kd values for 17β-estradiol binding to GPER in membrane preparations from divergent species including zebrafish (Liu et al., 2009b), croaker (Pang et al., 2008), and recombinant human (Filardo and Thomas, 2005) are very similar (2.3–3.3 nM) (Filardo and Thomas, 2012). The description of [3H]E2 binding to GPER in membrane preparations as “limited capacity” (Thomas et al., 2005; Pang et al., 2008) likely reflects the low abundance or instability of the receptor in such preparations but has no functional implications. In particular, as the majority of GPER is typically found expressed in intracellular membranes, depending on the cell type, the more enriched the plasma membrane preparation used, the less GPER-mediated binding one would expect to be present. The use of nonstandard assays in whole cells and conditions that provide incomplete saturation of GPER binding sites or that promote receptor degradation (low concentrations of [3H]E2, high temperature and long incubation times) has been identified as potential issues in reports where 17β-estradiol binding to GPER was not observed (Pedram et al., 2006; Otto et al., 2008) as recently discussed (Thomas et al., 2010; Filardo and Thomas, 2012).
2. G Protein–Coupled Estrogen Receptor Binding Using Radioligands in Whole Cells.
A number of studies have employed competitive ligand binding assays using [3H]E2 in ER-negative GPER-expressing whole cells (Teng et al., 2008; Lappano et al., 2010, 2012b; Chimento et al., 2014a). In one example, GPER-expressing SKBR3 cells were incubated with [3H]E2 in the presence or absence of competing ligands for 2 hours at 37°C. The cells were washed with ice-cold phosphate-buffered saline, extracted with 100% ethanol and the radioactivity of the extracts measured by liquid scintillation counting, with competitor binding expressed as a percentage of maximal specific binding (Lappano et al., 2012b). Alternatively, trichloroacetic acid-based protein precipitation followed by solubilization in 0.1 N NaOH was employed (Chimento et al., 2014a). Such intact cell assays typically report Ki values ≈100-fold higher (≈1 μM) than membrane-based assays, possibly due to incomplete removal of unbound or nonspecifically bound intracellular [3H]E2 (Filardo and Thomas, 2012).
An alternative approach for competitive ligand binding assays in GPER-expressing cell lines employing radiolabeled synthetic ligands that are selective for GPER has been described (Ramesh et al., 2010). Two iodinated tetrahydro-3H-cyclopenta[c]quinoline derivatives (see section IV.H.3 and Fig. 7) exhibit selective binding for GPER (≈ 2–8 nM) with the radioiodinated [125I] analogs prepared using the iodogen method from tributylstannyl precursors. [125I] is a low energy gamma emitter, with a half-life of 59.4 days. Competition binding studies were performed on adherent monolayers of ER-negative GPER-expressing human endometrial Hec50 cancer cells. This binding assay was verified by evaluating the receptor binding affinity for the GPER agonist G-1 (IC50 ≈ 7 nM), which compared favorably with the value obtained using a competitive binding assay with a fluorescent E2 ligand (see below) and recombinant GPER (IC50 ≈ 11 nM) and reported values for E2 (IC50 ≈ 3–6 nM). The GPER antagonist G15 exhibited slightly weaker binding (IC50 ≈ 20 nM). This approach was further used to evaluate the binding of GPER-ligand-tetraazacyclododecanetetraacetic acid In(III) complex (G-DOTA-In) (nonradioactive) in Hec50 cells (Nayak et al., 2010). The binding affinity of the neutral 113In-G-DOTA complex was found to be ≈ 34 nM. These results illustrate the potential for using radiolabeled GPER ligands for routine binding studies in whole cells. No commercial sources of these agents are available at this time, necessitating access to a synthetic source or commercial radiolabeling service. In principle, this GPER-selective binding approach could also be used with a tritiated G-1 derivative, but has not been reported to date.
3. Fluorescent Ligands Employing Permeabilized Cells.
Competitive binding assays employing fluorescent ligands offer practical advantages by avoiding radioactive waste, with versatility for applications using varied instrumentation platforms. The estradiol-Alexa Fluor dye conjugates (E2-Alexa) synthesized from 17α-ethynylbenzylamine-estradiol (17α-[4-aminomethyl–phenyl-ethnyl]-estra-1,3,5(10)-triene 3,17β-diol) (Arterburn et al., 2000) were used for ligand binding assays in COS7 cells (that express neither ERα, ERβ, nor GPER endogenously) transiently transfected to express GFP-tagged GPER, employing both microscopy to assess subcellular distribution and colocalization as well as flow cytometry to achieve quantitative binding analyses. Cells expressing the nuclear estrogen receptors ERα/β-GFP were used to compare ligand binding characteristics and evaluate selectivity for the individual receptor types (Revankar et al., 2005). Colocalization of E2-Alexa Fluor 546 with a human GPER-GFP fusion protein in the endoplasmic reticulum and Golgi apparatus was consistent with the observed localization demonstrated by immunofluorescent staining of endogenous GPER. Because of the charged state of the Alexa dyes, permeabilization with saponin was required to enable entry of the charged E2-Alexa 633 or E2-Alexa 546 dye conjugates to the intracellular compartments, with concomitant loss of soluble cytoplasmic proteins from the cell. The binding affinity of 17β-estradiol calculated from this heterologous competition assay (≈ 6 nM) (Revankar et al., 2005) was comparable to that determined from equilibrium binding analysis of [3H]E2 binding to cell membranes (≈3 nM) (Thomas et al., 2005), demonstrating that the measured binding affinities of 17β-estradiol for GPER using either direct binding of [3H]E2 to membrane preparations or competition binding of fluorescent E2 derivatives in permeabilized cells are remarkably similar.
B. Steroids
The observation of specific, high-affinity, competitive binding of 17β-estradiol (E2) with GPER established the experimental connection of this important hormone with the functional biology and physiology of this transmembrane receptor. Considering the general structural similarities exhibited by the naturally occurring steroid hormones (Fig. 1) and therapeutically important synthetic analogs (Fig. 2), evaluation of GPER interactions with this family of compounds was conducted during the early stages of GPER characterization (Thomas et al., 2005). The position of the 17β-hydroxy group in E2 is critical for binding affinity with key amino acid contacts with His524 in ERα and His475 in ERβ. The unnatural diastereomer 17α-estradiol (17α-E2) exhibits very low affinity for GPER (Kd, >>10 μM; Tables 1 and 2) and because this ligand does not initiate rapid signaling pathways, 17α-E2 can be used as a negative control for GPER binding and function. The hormone estrone (E1) possesses an oxo group at the 17-position, has low binding affinity for GPER (Kd, >10 μM), and has not been reported to display any functional activity toward GPER. The other major endogenous estrogen hormone estriol (E3), produced primarily during pregnancy by the placenta, possesses the 17β-hydroxy and an additional 16α-hydroxy group. E3 exhibits higher binding affinity for ERβ/ERα [RBA = 80/29, respectively (Kuiper et al., 1997)] and exhibits low binding affinity for GPER (IC50 > 1 μM), but functions as an antagonist in GPER-expressing, ER-negative SKBR3 cells at concentrations greater than 1 μM (Lappano et al., 2010).
Estrogen is metabolized by several different processes that include oxidation by cytochrome P450s at the C-2, C-4, and C-16 positions of E2 and E1 and transformation to ionic glucuronate or sulfate derivatives with increased water solubility facilitating excretion and elimination (Yager and Davidson, 2006). The 2- and 4-hydroxy catechol estrogens can undergo further oxidation to quinones with mutagenic properties, a process that is prevented by methylation at these positions. Limited characterization of the GPER binding properties of these numerous E2 metabolites has been reported; however, the existing evidence suggests that differential binding may provide mechanisms for the selective activation or inhibition of different estrogen receptors (Lappano et al., 2010). Intriguing relationships associating oxidized E2 metabolites with increased risks for breast cancer have been described (Yager and Davidson, 2006; Fuhrman et al., 2012). The oxidized E2 metabolite 2-methoxy-estradiol (2-MeO-E2) is effective in tumor growth inhibition in a variety of cell lines, with evidence of apoptotic and antiangiogenic activity as well as other molecular mechanisms, including microtubule stabilization, having been reported (Lakhani et al., 2003). The E2 metabolite 2-hydroxyestradiol exhibits high-affinity binding for recombinant human ERα/β with relative binding affinities reduced fivefold and threefold relative to E2, respectively (Zhu et al., 2006a). Methylation of the 2-hydroxy group to yield 2-MeO-E2 attenuates the binding affinity for ERα/β by 50- and 100-fold, respectively, compared with E2 (Zhu et al., 2006a). 2-MeO-E2 was recently identified as a high affinity (Kd = 10 nM) agonist of GPER (Koganti et al., 2013). This A-ring modified compound 2-MeO-E2 exhibited specific saturable binding [3H-2-MeO-E2] in cell membranes that was sensitive to pertussis toxin, with the specific binding of 2-MeO-E2 inhibited by GPER-selective agonist (G-1) and antagonist (G15) ligands, suggesting a mode of action via GPER. 2-MeO-E2 has been investigated clinically under the trade name Panzem (EntreMed, Inc., Rockville, MD) for ovarian cancer and other indications (Verenich and Gerk, 2010). In addition, 2-hydroxyestradiol has recently been shown to act as a GPER antagonist, acting to promote the resumption of meiosis in zebrafish oocytes, with a binding affinity in the range of 0.1–1 μM (Chourasia et al., 2015). The GPER binding of the other oxidized E2 metabolites 4-hydroxy-E2 and 4-methoxy-E2 has not been reported.
Glucuronidation and sulfonation of E2 can occur at both the 3-phenolic and 17-hydroxy positions, enhancing solubilization for excretion. The glucuronic acid metabolite 17β-estradiol-17-d-glucuronide was evaluated for agonism of GPER-mediated effects with high concentrations (50 μM) increasing cAMP levels and protein kinase A activity, both of which were blocked by the GPER-selective antagonist G15. Knockdown with siRNA targeting GPER also strongly prevented 17β-estradiol-17-d-glucuronide–induced impairment of canalicular transporter function and localization (Zucchetti et al., 2014).
GPER binds E2 with high selectivity (>1000×) over the androgen testosterone, the progestogen progesterone, and the glucocorticoid cortisol (Thomas et al., 2005). These non–GPER-binding steroid structures all possess an unsaturated 4-ene-3-one functional group in the A-ring and also have a methyl group at C-10, whereas the A-ring of E2 is a planar aromatic phenol. Testosterone possesses a 17β-hydroxy group, similar to E2, whereas progesterone and cortisol have acetyl and hydroxyacetyl groups in the 17β-position, respectively. Cortisol possesses additional hydroxyl groups at the 17α- and 11β-positions. Dehydroepiandrosterone (DHEA) is a highly abundant circulating human steroid hormone that serves as an important metabolic intermediate in steroid biosynthesis and has multiple biological effects. The A-ring of DHEA features 3β-hydroxyl and C10-methyl groups, a C5-6 alkene, and a 17-oxo group similar to estrone. The alkene group of DHEA undergoes metabolic reduction to the saturated endogenous hormone epiandrosterone, which exhibits weak androgenic activity; however, no reports of GPER binding or activity with DHEA or epiandosterone have been reported. In contrast, the endogenous metabolite 7β-hydroxy-epiandrosterone (7β-OH-EpiA) was recently reported to antagonize GPER-mediated estrogenic responses with high potency (Sandra et al., 2012). Subnanomolar concentrations of 7β-OH-EpiA countered the agonist effects of G-1 on the growth of SKBr3 and MCF7 cancer cell lines, consistent with 7β-OH-EpiA antagonism of GPER. The A-ring in 7β-OH-EpiA is fully saturated, with two hydroxyl groups located at the 3β- and 7β-positions and the ketone functionality at the 17-position analogous to E1. No GPER binding data for 7β-OH-EpiA have been reported and therefore, without binding data, other mechanisms of growth inhibition cannot be excluded.
Aldosterone (Aldo) is a mineralocorticoid steroid hormone that plays an important role in the regulation of blood pressure. The structure of aldosterone is similar to cortisol, although lacking the 17α-hydroxyl group and possessing an aldehyde group in the C18 compared with the methyl group of cortisol in that position. Certain rapid actions of Aldo have been correlated with GPER expression (Ding et al., 2009; Gros et al., 2011, 2013), but recently published binding studies of Aldo to membrane and cytosolic kidney preparations revealed no direct binding of [3H]Aldo to membrane preparations containing functional GPER or competition of [3H]E2 binding to GPER by unlabeled Aldo, whereas Aldo binding to soluble MR was easily detected (Cheng et al., 2014). These results suggest that, whereas GPER expression appears to be involved in aspects of rapid Aldo function, binding to GPER appears not to be involved (Prossnitz and Barton, 2014). The mechanisms of rapid signaling by Aldo remain to be fully elucidated.
C. Synthetic Steroid Derivatives, Analogs, and Therapeutics
A variety of synthetic estrogen derivatives and nonsteroidal compounds with agonist or antagonist activities have been developed for different therapeutic applications (Fig. 2). Many of these compounds were investigated before the 1996 discovery of ERβ and have been the subject of subsequent studies to characterize the affinity and selectivity of ligand binding to the different receptor subtypes (Kuiper et al., 1997, 1998). The classic estrogen receptors ERα and ERβ have highly similar ligand-binding domains but very different tissue distribution and physiologic roles (Nilsson and Gustafsson, 2011). This scenario presented the dual challenge of identifying the roles of the individual receptors in ligand-induced responses and the need for the development of subtype-selective agonists and antagonists, which continues to be an active area of research (Minutolo et al., 2011; Nilsson et al., 2011). The discovery of GPER has further complicated this scenario of differences in ligand-binding selectivity, affinity, and type of responses mediated through the estrogen receptor subtypes and classes and raises additional issues such as receptor cross-talk that affect the interpretation of experimental results and experimental designs that involve measurement of ligand-mediated functional responses (Prossnitz et al., 2008). The successful development of truly selective agonists/antagonists, able to distinguish between both ERα/β receptor subtypes and membrane-localized receptors such as GPER (as well as membrane-localized ERα and potentially ERβ and their splice variants), would provide more useful research tools and may ultimately lead to improved therapeutics.
The structurally simple phenolic stilbene compound diethylstilbestrol (DES; Fig. 2), approved by the Food and Drug Administration in 1941 for a number of indications (Newbold, 2004), exhibits higher binding affinities than E2 for ERα/β by approximately fourfold (Kuiper et al., 1997) but only weak binding for GPER (∼1 μM) (Thomas et al., 2005). The selectivity of DES for ERα/β has been used in studies to distinguish biological responses mediated by ERα/β from GPER. In ER-positive MCF7 breast cancer cells, G-1 and DES selectively activate GPER and ER, respectively, and both E2 and DES transiently downregulated both ER and GPER, whereas G-1 did not affect this expression, indicating that this process was ER mediated. Furthermore, DES did not induce calcium mobilization in MCF7 or SKBr3 cells, whereas both 4-hydroxytamoxifen (4-OHT) and G-1 stimulated this response (Ariazi et al., 2010). Recent studies, however, suggest that DES-induced activation of CREB downstream of protein kinase A, Src, and ERK1/2 in mouse gubernaculum testis cells was mediated to some extent by GPER (Zhang et al., 2014c). Recognized metabolites of DES such as indenestrol have not yet been assessed for GPER binding or activity, but further studies are warranted considering their documented estrogenic activity (Korach et al., 1978, 1989).
Tamoxifen is a triphenylethylene compound that was originally investigated for potential contraceptive applications but is now widely used as antiestrogen adjuvant therapy for the treatment of both early and advanced ERα-positive breast tumors in pre- and postmenopausal women (Jordan, 2000; Ariazi et al., 2006). The active metabolite 4-OHT antagonizes ERα in breast tissue but behaves as an agonist in other tissues such as the endometrium, and therefore 4-OHT is referred to as an SERM. Although 4-OHT exhibits binding affinities for ERα/β similar to E2 (Blair et al., 2000), it has also been demonstrated to exhibit significant binding toward GPER (in the range of 30 nM for SKBr3 cell membranes to 1 μM for GPER-transfected HEK293 cell membranes) (Thomas et al., 2005). However, in contrast to its actions as an antagonist in breast cancer cells, 4-OHT acts as a GPER agonist in breast and other cancer cells (Revankar et al., 2005; Petrie et al., 2013). Consistent with this agonist activity toward GPER, increasing numbers of studies are finding roles for GPER in tamoxifen resistance in breast cancer and the increased incidence of endometrial cancer after tamoxifen treatment (Craig Jordan et al., 2007; Ignatov et al., 2010, 2011a,b; Mo et al., 2013; Petrie et al., 2013; Catalano et al., 2014).
Raloxifene (RAL or Evista; Eli Lilly and Company, Indianapolis, IN) is a phenolic benzothiophene benzoketone substituted with a basic piperidin-1-yl ethoxy side chain and acts as a SERM with respect to ERα/β. The ability of RAL to elicit estrogenic actions on bone has been used therapeutically for the prevention of osteoporosis in postmenopausal women, and the antiestrogenic actions of RAL in the uterus and breast have demonstrated efficacy in reducing the incidence of breast cancer with a lower risk of uterine cancers and blood clots than women taking tamoxifen (Pinkerton and Goldstein, 2010). RAL exhibits strong GPER efficacy (EC50 = 10–100 nM), acting as a GPER agonist (Petrie et al., 2013). A series of structurally related benzothiophene derivatives including desmethylarzoxifene, the active bisphenolic metabolite of arzoxifene, was evaluated for the ability to provide neuroprotection of primary cortical neurons in response to oxygen-glucose deprivation (Abdelhamid et al., 2011). The ER antagonist ICI 182,780 at 1 μM did not block the neuroprotection afforded by RAL or desmethylarzoxifene, although it did for E2, whereas G15 at 100 nM prevented the neuroprotection elicited by both RAL and E2. Thus, the neuroprotective effects of the benzothiophenes were found to be mediated by GPER and to be independent of ER. Finally, the ability of RAL and E2 to stimulate proliferation in human fetal osteoblast cells that express GPER but lack ERα was prevented by GPER siRNA, again demonstrating a mechanism involving GPER (Noda-Seino et al., 2013).
ICI 182,780 (ICI), also known as fulvestrant or Faslodex (AstraZeneca, London, UK) is a steroidal E2 antagonist developed from a series of derivatives with substituents at the 7α-position, optimized for potency with an extended nonyl-pentafluoropentylsulfinyl chain (Wakeling and Bowler, 1992). ICI was designed to lack the undesired tissue-dependent estrogenic agonist activity that occurs with the SERM tamoxifen and is therefore often referred to as a pure antiestrogen (Howell et al., 2000). Binding of ICI to the ERα/β monomer causes dissociation of the stabilizing Hsp90 protein but prevents ER dimerization, leading to instability and subsequent receptor degradation (downregulation) and its designation as an SERD (McDonnell and Wardell, 2010). Despite its antagonistic activity toward ERα/β, ICI functions as a GPER agonist (Filardo et al., 2000) with a binding affinity of ∼30–50 nM (Thomas et al., 2005). Thus, although ICI is defined as a SERD based on its ER activity, it has been shown to mediate many of the same stimulatory functions as E2 in certain systems (Mercier et al., 2003; Wong et al., 2003; Zhao et al., 2006; Kamanga-Sollo et al., 2008; Meyer et al., 2010; Baranda-Avila et al., 2013). In many cases, these occur through GPER activation as exemplified by the observation that both E2 and ICI enhance MCF7 adhesion to matrigel, effects that were suppressed by G15 or knockdown of GPER (Chen et al., 2014).
17α-Ethynyl-17β-estradiol (EE, also called ethinyl estradiol) is a synthetic estrogen used in oral contraceptives because of its prolonged bioactivity in the body, a result of decreased metabolism. It exhibits an affinity for ERα/β approximately twofold better than E2 (Blair et al., 2000), but its affinity for GPER has not been reported. In the animal model of multiple sclerosis (EAE), E2 can prevent onset of disease but it cannot treat established disease, whereas EE can reduce the severity of existing disease (Yates et al., 2010). The protective effects of E2 have been reported to be reduced in both ERα knockout (Morales et al., 2006) as well as GPER knockout mice (Wang et al., 2009). In subsequent studies the effects of EE were shown to be entirely mediated through GPER, as they were completely absent in GPER knockout mice but maintained in ERα knockout mice (Yates et al., 2010). The lack of disease improvement in EE-treated GPER knockout mice indicates a crucial role for GPER in altering disease severity, likely through the production of IL-10 and associated anti-inflammatory effects (Bodhankar and Offner, 2011). Beyond its direct use, EE also represents an ideal scaffold for the elaboration of pendent linkages, permitting retained binding to all classes of ERs (see below).
D. Membrane-Impermeable Estrogen Probes
Because E2 is a highly cell-permeable ligand, passively diffusing through membranes (Muller et al., 1979), the need to distinguish responses mediated by intracellular/nuclear receptor pools from those involving plasma membrane-localized receptors has stimulated the development of E2-derived membrane-impermeable probes (Fig. 3). This approach uses molecular structure and/or charge to impede intracellular uptake (i.e., membrane permeability) of the ligand. In general, three design strategies have been employed: 1) conjugation to large proteins, such as bovine serum albumin (E2-BSA); (2) attachment to oligomeric/polymeric dendrimers (E2-PAMAM) or cyclodextrins; and (3) ionic E2 derivatives (E2-NMe3+). The use and cautionary experimental considerations for the use of conjugates to investigate rapid E2 signaling was recently reviewed (Shearer et al., 2012).

Selectively impermeable estrogen derivatives.
E2-BSA and its fluorescent derivative, E2-BSA-fluorescein isothiocyanate (FITC), represent the first forms of membrane-impermeable E2 designed to activate and visualize nonnuclear estrogen receptors (Binder, 1984; De Goeij et al., 1986), consisting of conjugates of E-6-CMO (17β-estradiol-6-carboxymethyloxime), or 17β-estradiol-17-hemisuccinate, and FITC (for E2-BSA-FITC), with each linked to the ε-amino groups of lysine residues of BSA. Figure 3 shows the conjugation of E-6-CMO and the approximate scale of E2 conjugated to BSA, as well as a free lysine side chain. Differential effects with BSA conjugates attached to different positions on E2 have been observed (Temple and Wray, 2005). Commercial preparations of E2-BSA (E-6-CMO) usually contain ∼30 E2 molecules per BSA (representing derivatization of ∼50% of the lysine residues in the protein), whereas E2-BSA-FITC contains 5–10 conjugated E2 and 3–5 FITC per BSA molecule. The molecular mass of such derivatives exceeds ∼75 kDa. Although widely used, E2-BSA suffers from a number of drawbacks, requiring caution in its use. First, E2-BSA preparations also contain a significant amount of free and noncovalently bound E2 derivative, due to BSA’s primary transport function of hydrophobic small molecules (De Goeij et al., 1986; Stevis et al., 1999). This results in high levels of free E2 derivatives and a continuous dissociation of membrane-permeable E2 during incubation with cells and upon freeze/thaw cycles. Size filtration immediately before use has been found to mitigate these effects (Taguchi et al., 2004); however, E2-BSA devoid of free E2 was nevertheless unable to compete for radiolabeled E2 binding to ERα or ERβ (Stevis et al., 1999). Second, the common routes of conjugation at C-6 of E2 can result in a significant loss in binding affinity toward ERα/β (De Goeij et al., 1986; Anstead et al., 1997). Third, the high density of conjugated E2s and polymerization of BSA through chemical conjugation can result in receptor clustering, resulting in the often-observed large membrane-localized puncta in microscopy images employing E2-BSA-FITC (Wu et al., 2011b). Finally, E2-BSA has been demonstrated to mediate cellular signaling events that are not mediated by E2 itself, raising concerns regarding their ability to mimic the effects of E2 on membrane receptors (Stevis et al., 1999). The use of fluorescently labeled E2-BSA is prone to similar caveats (Binder, 1984), often requiring high concentrations (e.g., 15 μM) of E2 for displacement (Wu et al., 2011b), although prolonged preincubation (4 hours) with E2-BSA can prevent subsequent E2 binding, suggesting binding kinetics may play an important role in the observed binding properties (Taguchi et al., 2004) or that additional E2 dissociates or hydrolyzes from the BSA in that time frame, the latter being possible given the potential for cleavage of the covalent linkage by acidic hydrolysis, and the low pH found in lysosomes and endosomes. The activation of plasma membrane-localized ERs in an immortalized hypothalamic cell line with a fluorescent E2-BSA conjugate was recently shown to result in ligand internalization through endocytosis (Kisler et al., 2013). Furthermore, an E2-activated process that results in localization of ERα to lysosomes and endosomes as a pathway for degradation was recently reported, identifying that lysosome function was required for E2-dependent cell proliferation but not for ERα-mediated ERE-containing gene transcription (Totta et al., 2014). As an alternative to BSA, peroxidase conjugation of E2 has also been employed to investigate membrane-initiated signaling (Bulayeva et al., 2004), with similar caveats to E2-BSA.
The next generation of membrane-impermeable ligands sought to overcome the limitations of E2-BSA through the use of large, abiotic, nondegradable poly(amido)amine dendrimer macromolecules conjugated to multiple (typically 18–20) E2 molecules (as well as fluorophores) with hydrolytically stable linkages at the 17α position (Harrington et al., 2006; Kim and Katzenellenbogen, 2006). As a result of their charge and size (Generation 6 PAMAM contains 256 primary amines and has a molecular mass of ∼58 kDa), such E2-dendrimer conjugates (E2DCs) remain outside the nucleus, and in fact the cell (i.e., cytoplasm), with reported cytoplasmic localization in punctate structures likely the result of receptor-mediated endocytosis or pinocytosis. E2DCs have been shown to mediate multiple nonnuclear/rapid signaling events such as ERK1/2, Src, and eNOS activation through both ERs (Alyea et al., 2008; Chambliss et al., 2010) and GPER (Noel et al., 2009) but not nuclear events such as transcription of endogenous E2 target genes or MCF7 cell proliferation because E2DCs are 10,000-fold less potent than E2 in genomic actions (Harrington et al., 2006; Chambliss et al., 2010). Although the function of E2DCs through ERα is well established, it is unclear how the E2 moiety of E2DCs (or E2-BSA) gains access to nonnuclear ERα located either in the cytosol or at the inner face of the plasma membrane. Although a single transmembrane spanning domain has been identified in the ERα splice variant ER46 (Kim et al., 2011), with the carboxy-terminal domain being located on the cytoplasmic face, it is believed that membrane-localized ERα is entirely cytosolic, appearing to prevent access to charged extracellular E2DCs. For ERα to be activated by an E2DC or E2-BSA, the receptor (or at least the ligand binding domain) must be extracellular, or the E2 moiety with its associated linker would have to pass through the membrane to the cytoplasmic face of the membrane. With a total diameter of ∼8.8 nm and an internal core of ∼5.6 nm (Kim and Katzenellenbogen, 2006), the conjugated E2 on an E2DC, extending ∼1.6 nm beyond the core, is unlikely to be able to pass through the plasma membrane [∼4 nm for the phospholipid bilayer (Mitra et al., 2004) and 7–10 nm with associated (glyco)proteins (Hine, 1999)]. Access to the extracellular surface of plasma membrane-localized GPER would however be unimpeded assuming that the site of E2 conjugation did not prevent GPER binding. However, as the majority of GPER appears to be intracellular in most cell types examined to date, it is unlikely that E2DCs would be capable of activating rapid signaling via GPER. Finally, an alternative scaffold for conjugation of multiple (up to 7) E2 moieties employed the hydrophilic, cyclic heptasaccharide β-cyclodextrin (yielding a heptavalent conjugate with final molecular weight ∼3400) and was used to examine estrogenic signaling (Kim et al., 2010).
The third approach for impeding membrane permeability is based on the slow rates with which ions diffuse through the hydrophobic interior of the lipid membrane bilayer. Early examples include the positively charged quaternary ammonium salts of the basic amine side chain of tamoxifen that were found to exhibit similar binding affinity in cytosolic receptor extracts but greatly diminished binding and reduced cytostatic activity in the E2-responsive MCF-7 breast cancer cell line (Jarman et al., 1986). Subsequent studies have demonstrated utility and revealed potential pharmacologic applications for permanently charged tamoxifen derivatives (Biegon et al., 1996; Somjen et al., 1996; Allen et al., 2000; Dick et al., 2002; Dietze et al., 2004). The time course of experiments employing this electrostatic approach is an important consideration and should be reserved for rapid signaling events occurring with short duration (seconds–minutes), because slow passive membrane permeability is possible given longer exposure times (Rickert et al., 2010; Rivera-Guevara et al., 2010).
Along these lines, we have synthesized a series of neutral and charged E2 monomers as permeability-restricted E2 probes consisting of substituted 17α-ethynylbenzene derivatives. The design of these probes was based on the EE scaffold and derivative fluorescent dye conjugate E2-Alexa to contain either neutral or charged appendages as a simple physicochemical parameter for controlling membrane permeability through passive diffusion (Revankar et al., 2007). As discussed previously, the large anionic charged dye of E2-Alexa inhibited diffusion and required permeation of cellular membranes with saponin to enable intracellular uptake. Competitive ligand binding experiments in permeabilized cells of the synthetic E2s containing a series of para-substituents including the neutral tert-butyl benzylcarbamate E2-BnNHBoc, cationic N,N,N-trimethylanilinium E2-PhNMe3+, and negatively charged benzoate E2-PhCO2−, demonstrated they were able to bind ERα (Ki values: 2.7, 1.4, and 2.0 nM, respectively) and GPER (Ki values: 16, 17, and 30 nM, respectively) with similar affinities. The capacity of these probes to activate rapid signaling in intact, nonpermeabilized cells expressing ERα or GPER was assessed using rapid intracellular calcium mobilization (on the time scale of seconds). The neutral probe E2-PhNHBoc initiated calcium mobilization at 10 nM that was indistinguishable from that of E2, whereas the charged compounds E2-PhNMe3+ and E2-PhCO2− failed to stimulate measureable calcium mobilization. These results are attributed to the reduced ability of the charged compounds to enter the cell by passive diffusion and are consistent with a model of GPER signaling from an intracellular location. It is important to note that over a 24-hour period, all compounds were able to activate ER-mediated transcription, suggesting that with extended time these derivatives are able to access nuclear ERs. Therefore, these permeability-restricted E2 probes should be reserved for experiments that involve relatively short time frames (< ∼15 minutes).
In conclusion, based on the caveats associated with each type of “membrane-impermeable” probe, great care must be taken in the design of experimental protocols including appropriate controls (Chambliss et al., 2010) as well as in the interpretation of results based on their use.
E. Phytoestrogens
Phytoestrogens are (xeno) estrogenic compounds of plant origin that occur naturally in a variety of foods (Moutsatsou, 2007) (Fig. 4). There has been considerable interest in the purported beneficial health effects of phytoestrogens as dietary supplements, including antioxidant and anti-inflammatory properties, as well as concern for potential adverse responses from their modulation/disruption of endocrine function (Ososki and Kennelly, 2003; Yoon et al., 2014). Fungi constitute another natural source of estrogenic mycotoxins and other endocrine-disrupting compounds, with representative examples discussed within this section. The multiple biological effects associated with these compounds are mediated in part through estrogen receptors, and considerable effort has been focused on assessing the interactions of phytoestrogens with ERα/β. In response to the discovery of GPER, new studies have evaluated the interactions of phytoestrogens with this receptor. Several structurally distinct classes of phytoestrogens have been characterized, including flavones, isoflavones, lignans, coumestans, and stilbenes. Many phytoestrogens occur in foods as glycoside conjugates; however, the aglycones frequently exhibit more potent bioactivity and these are often the constituents included in nutritional supplements. Endogenous and exogenous metabolism of phytoestrogens by gut bacteria provides additional routes for the generation of bioactive compounds. The characterization of the GPER pharmacology of this class of compounds is incomplete and relatively few phytoestrogen-GPER binding affinities have been reported at this time; however, several recent publications have implicated phytoestrogens in GPER-mediated pathways and these are included in this discussion to provide preliminary indications and stimulate further studies.
Structures of phytoestrogens.
The chromen-4-one core is common to both the flavones and isoflavones, which differ in the position of phenyl substituents at the 2- and 3-position, respectively. The isoflavones genistein and daidzein, occurring in soy-based foods, are recognized as potent phytoestrogens (Ososki and Kennelly, 2003). Genistein is a 5,7-dihydroxy-isoflavone and a prototypical example of an ERβ-selective compound, exhibiting >20-fold higher binding affinity for ERβ (7–16 nM) than for ERα (330–910 nM) (Kuiper et al., 1997, 1998). Genistein also exhibits high binding affinity for GPER (IC50 = 133 nM; RBA=13% versus E2), exhibiting a selective binding preference for GPER that is in between the classic ERs (Thomas et al., 2005) and functioning as a GPER agonist (Maggiolini et al., 2004). The structurally related isoflavone daidzein, a 7-hydroxy-isoflavone, exhibits both estrogenic and antiestrogenic effects. No GPER binding affinities for daidzen have been reported, but this compound has been reported to mediate glutamate-induced effects via GPER (Kajta et al., 2013). Daidzen can be metabolized to equol in the presence of certain intestinal bacteria, a net reduction of the chromene-4-one core to a dihydrobenzopyran, with two enantiomeric forms of equol possible, the 3S-enantiomer exhibiting selectivity for ERβ. No GPER binding affinities for equol have been reported, but this compound has been suggested to mediate mitochondrial reactive oxygen species generation via GPER-mediated EGFR transactivation (Rowlands et al., 2011). Tectoridin is the 7-glucoside of the isoflavone tectorigenin that has been implicated in the stimulation of estrogenic effects through GPER (Kang et al., 2009). No other studies of the interaction of the aglycone tectorigenin with GPER have been reported.
Quercetin is a 3,5,7-trihydroxy-flavone that possesses a catechol (3,4-dihydroxyphenyl) in the 2-position of the chromen-4-one core. Quercetin naturally occurs as a variety of glycosides depending on the plant source. Like genistein, quercetin was found to exhibit activity as a GPER agonist (at 1 μM) stimulating c-fos expression in ER-negative GPER-expressing SKBr3 cells (Maggiolini et al., 2004). The 4′,5,7-trihydroxyflavone apigenin exhibits anticancer properties and has been associated with protective effects on endothelial function. The inhibitory effects of apigenin on the tumor necrosis factor α–induced expression of eNOS have, however, been ascribed to ERα (Palmieri et al., 2012).
A variety of other phenolic phytoestrogens have been implicated in GPER-mediated pathways. Resveratrol is a phenolic stilbene derivative that inhibited neuronal potassium channels, and this response was reduced by the GPER antagonist G15 or by short hairpin RNA–mediated knockdown of GPER (Dong et al., 2013). The simple phenylethanoid hydroxytyrosol and the glycosylated elenolic acid conjugate oleuropein are isolated from olives and have been reported to act as GPER agonists (Chimento et al., 2014a). Zearalenone (also called RAL and F-2 mycotoxin) is a mycotoxin produced by some Fusarium and Giberella species that infect cereal crops and has been associated with infertility, abortion, and other breeding problems in domestic farm animals. Zearalenone, which consists of a phenolic 14-membered lactone with a ketone in the 7-position and 11-E alkene, was found to exhibit a moderate binding affinity for GPER (IC50 = 763 nM), with selectivity (8–20 fold) for ERα/β over GPER (Thomas et al., 2005; Thomas and Dong, 2006). The reduced 7R-hydroxy alcohol zearalanol (also referred to as zeranol) is approved for use as a growth promoter in livestock in the United States and has been reported to have more potent estrogenic properties than zearalenone (Gajecki, 2002; Stephany, 2010). However, no GPER binding or activity data for zearalanol or the semisynthetic derivatives, such as the unsaturated alcohols α/β-zearalenol, or the saturated ketone zearalanone have been reported. Among the wide variety of additional potential phytoestrogen ligands for GPER, the lignans are polyphenolic phenylpropanoid compounds, many of which possess 3,4-dioxygenated substituted phenyl groups. Because they represent significant components of plant cell walls found in seeds, grains, nuts, and fruits, further investigation of these compounds and their metabolites may reveal new examples of GPER-binding compounds.
F. Xenoestrogens
Synthetic xenoestrogens include a wide variety of nonsteroidal industrial chemicals and products (Fig. 5) associated with endocrine disrupting activities, affecting reproductive function, immune and nervous systems, behavior, metabolism, and cancer through their interactions with estrogen and potentially other receptors (Fucic et al., 2012; Wong and Walker, 2013). Human exposures may occur through direct or indirect routes, including food and water supplies and contact with materials intentionally or inadvertently introduced in the environment. Consumer products including detergents, surfactants, resins, lubricants, plasticizers, fire retardants, and pesticides have been identified as sources of xenoestrogens, and scrutiny has focused on chemical classes such as alkyl phenols and esters of parahydroxybenzoic acid, phthalates, polybrominated diphenyl ethers, organophosphates, chlorinated hydrocarbons, and biphenyls (Shanle and Xu, 2011). Although many xenoestrogens exhibit low binding affinities to the nuclear ERs and often require relatively high concentrations (≥1 μM) to affect genomic pathways, recent studies have focused on xenoestrogen effects on rapid nongenomic signaling pathways where significantly more potent, low-dose effects have been observed, including those that involve GPER-mediated processes (Thomas and Dong, 2006; Shanle and Xu, 2011; Marino et al., 2012; Watson et al., 2012, 2014).

Structures of synthetic xenoestrogens.
Bisphenol A (BPA) is produced in vast quantities globally, primarily for use in polycarbonate plastics and epoxy resins, with the safety of human exposures to BPA the subject of intensive scientific assessment and regulatory consideration (Hengstler et al., 2011; Shelnutt et al., 2013). BPA binds relatively weakly to ERα (IC50 = 1–10 μM) (Blair et al., 2000) with a similar affinity for ERβ (Kuiper et al., 1998) but with a much greater affinity for estrogen-related receptor γ (IC50 = 13 nM) (Takayanagi et al., 2006). The binding affinity of BPA for GPER (IC50 = 0.6 μM) is higher than that for the nuclear receptors (Thomas and Dong, 2006) (Table 2), with numerous recent reports demonstrating bioactivities of BPA to be mediated at least in part by GPER. Multiple studies have examined the expression of GPER and the mechanisms of BPA signaling in the testes. Somatic (Sertoli cells) and germ cells (spermatogonia and spermatocytes) in normal adult human testes express GPER, which was overexpressed in testicular germ cell cancer seminomas (Chevalier et al., 2012a,b). Nanomolar concentrations of BPA (1–10 nM) stimulated the proliferation of immature murine Sertoli cells, with reduced proliferation observed at higher concentrations of BPA (>1 μM) and with both G15 and ICI 182,780 inhibiting the BPA-induced proliferation, suggesting the involvement of GPER and ERα/β (Ge et al., 2014). The proliferation of human seminoma JKT-1 cells in response to E2-BSA was inhibited by both G15 and GPER siRNA (Chevalier et al., 2012b), and the proliferation of spermatogonial GC-1 cells was stimulated by low concentrations (nM) of BPA through the GPER/EGFR/ERK/c-fos pathway (Sheng and Zhu, 2011; Sheng et al., 2013). BPA also affects proliferation and migration in SKBR3 human breast cancer cells and cancer-associated fibroblasts that lack ERα and ERβ (Pupo et al., 2012) as well as the migration and invasion of lung cancer cells (Zhang et al., 2014a).
A variety of alkylated phenols are employed as additives, lubricants, components, and chemical feedstocks (Sharma et al., 2009). These products contain alkyl groups of varying length and branching substituents typically as mixtures of isomers. The nonylphenols are a representative family of compounds that exhibit estrogenic responses, although the binding affinities of constituents for ERα and ERβ are relatively weak (RBA = 0.05 and 0.09, respectively). Nonylphenols exhibit higher binding affinity for GPER (IC50 = 0.8 μM; RBA = 2.2) and are reported to initiate nongenomic responses (Thomas and Dong, 2006). Increased resistance to oxidative insult was observed in Swiss 3T3 fibroblasts exposed to either nonylphenol or BPA (0.1–100 nM) for extended times (30–45 days), suggesting a possible role for GPER in oxidative stress resistance (Nishimura et al., 2014).
Chlorinated hydrocarbons and their metabolites and degradation products represent another source of xenoestrogens that persists in the environment (Rosner and Markowitz, 2013). Commercial dichlorodiphenyltrichloroethane (DDT) products typically consist of a mixture of compounds with the p,p′-isomer as the major component (∼77%), followed by the o,p′-isomer (∼15%). The o,p′-DDT isomer exhibits ∼100-fold higher binding affinity for ERα than the p,p′-DDT and has been considered to be the more estrogenic isomer (Kojima et al., 2004); however, the respective GPER binding affinities are reversed (IC50 > 10 versus 2.8 μM) (Thomas and Dong, 2006). In an embryonic mouse neuronal cell model of neural development, p,p′-DDT and o,p′-DDT were found to induce apoptosis through interaction with nuclear ER but also GPER-dependent transcriptional activation (Kajta et al., 2014). The degradation of DDT produces isomers of dichlorodiphenyldichloroethylene (DDE) p,p′-DDE and o,p′-DDE, which exhibit similar weak binding to ER (Zhuang et al., 2012). The o,p′-DDE isomer exhibits stronger binding to GPER (7.1 μM) and was found to exhibit agonist activity, whereas the other isomer had reduced affinity p,p′-DDE (>10 μM) (Thomas and Dong, 2006). Methoxychlor shares the bis(phenyl)-trichloroethane structure of DDT, with methoxy groups replacing the chlorine substituents in the latter, and has low affinity for both ERα and ERβ (RBA < 0.01) (Kuiper et al., 1998) as well as GPER (>10 μM) (Thomas and Dong, 2006). Hydroxylated derivatives of methoxychlor have increased affinity for ER as expected for phenolic compounds, and the bis-phenolic species 2,2-bis-(p-hydroxyphenyl)-1,1,1-trichloroethane functions as an agonist for ERα and an antagonist for ERβ (Gaido et al., 2000); however, no information about this compound interacting with GPER has been reported.
Polychlorinated biphenyls (PCBs) are another class of environmental xenoestrogen with significant concerns. The phenolic derivative 2,2′,5′-PCB-4-OH exhibits low binding affinity for GPER (IC50 = 3.8 μM; RBA 0.47) (Thomas and Dong, 2006); however, the activity toward GPER is unknown. The GPER binding of other chlorinated endocrine disrupting chemicals such as the insecticide kepone (also called chlordecone) (IC50 = 1.4 μM) and the chloro-s-triazine herbicide atrazine (IC50 > 10 μM) has been reported (Thomas and Dong, 2006). Although atrazine did not induce transcriptional activation through ERα, it did induce ERK1/2 activation and the expression of E2 target genes, as well as stimulating the proliferation of ovarian cancer cells through GPER with a corequirement for ERα expression (Albanito et al., 2015). Although the Kd and EC50 values of many xenoestrogens for GPER are high, reflecting poor binding, the potential effects of chronic/long-term low-level exposure are not currently known. Thus, considering the diverse structural variety represented by xenoestrogens, as well as the metabolites and environmental degradation products, many of which are remarkably persistent and bioavailable, and the complexity of the cellular context and receptor signaling networks involved, it is likely that further study will elucidate other examples in which GPER mediates important physiologic responses to xenoestrogens.
G. Estrogen Receptor–Selective Ligands
With the identification of ERβ in 1996, the need became apparent for pharmacologic agents for the selective activation or inhibition of ERα and ERβ. However, because of the extensive homology in the ligand-binding sites of the two receptors (with only two conservative amino acid differences), identifying compounds with selectivity between ERα and ERβ has been challenging (Hubbard et al., 2000; Dahlman-Wright et al., 2006). Tetrasubstituted pyrazoles were identified as high-affinity ligands for the estrogen receptor in 2000, with the compound PPT (Fig. 6) found to be a potent ERα agonist, activating gene transcription through ERα and considered to be the first ERα-selective agonist (EC50 ∼200 pM). PPT binds to ERα with high affinity (within twofold that of estradiol), and displays 410-fold selectivity for ERα over ERβ (Stauffer et al., 2000). PPT prevents ovariectomy-induced weight gain and loss of bone mineral density and induces gene expression in the hypothalamus after systemic administration in vivo (Harris et al., 2002). In Hec50 endometrial cancer cells (ER-negative/GPER-positive) several compounds initiated GPER-mediated PI3K and ERK1/2 activation, including E2, G-1, 4OHT, RAL, ICI, as well as PPT (with submaximal effects for PPT observed at 10 nM). This report provided the first evidence that PPT also functions as a GPER agonist at higher concentrations, demonstrating that although PPT remains selective for ERα over both ERβ and GPER, care must be taken in the interpretation of results employing receptor-selective ligands, particularly with respect to the concentrations and doses of these compounds used in experimental systems (Washburn et al., 2013). Notably, the ERα-selective antagonist (methyl-piperidino-pyrazole) (Sun et al., 2002; Harrington et al., 2003), which has not been as widely used as PPT, has not been evaluated for selectivity toward GPER.

Structures of ER-selective compounds.
The diarylpropionitrile derivative 2,3-bis(4-hydroxyphenyl)-propionitrile (DPN; Fig. 6) is one of the most potent and selective ERβ agonists known, exhibiting 70-fold selectivity over ERα in binding assays (racemic mixture: RBA of 0.25% for ERα and 18% for ERβ) and 170-fold selectivity in transcriptional assays (Meyers et al., 2001). The R-enantiomer of DPN exhibits 3-fold higher ERβ binding affinity and about 2-fold higher ERβ/ERα selectivity (∼300-fold) compared to the S-enantiomer (Carroll et al., 2012; Paterni et al. 2014). The GPER-mediated activation of ERK1/2 and PI3K in Hec50 cells was not stimulated by DPN at concentrations up to 10 μM, where DPN activated PI3K activation in cells cotransfected with ERβ at nM concentrations (Petrie et al., 2013). Thus, although these early studies indicate that DPN and to a lesser extent PPT remain selective for their intended receptors, the selectivity of PPT with respect to GPER is limited, indicating that future development of receptor-selective ligands is warranted and must include GPER in the determination of overall estrogen receptor selectivity.
H. G Protein–Coupled Estrogen Receptor–Selective Ligands
1. Agonists.
With the lack of receptor specificity of E2 and the majority of compounds discussed above toward the classic estrogen receptors (ERα and ERβ) and GPER, there was a great need for the identification of truly specific pharmacologic agents for assessing the specific roles of individual receptors, particularly in complex systems expressing multiple estrogen receptors. Through a combination of virtual and biomolecular screening in an academic setting (Huryn et al., 2013), we identified a small molecule, G-1 (Fig. 7), that inhibited binding of a fluorescent E2 probe to GPER (Bologa et al., 2006). Competition binding studies revealed a Ki for GPER of approximately 10 nM for G-1, compared to ∼6 nM for E2. Functional analysis of this compound revealed that it acted as an agonist toward GPER for calcium mobilization and PI3K activation and that it exhibited no binding toward either ERα or ERβ at concentrations up to 10 μM. Further functional studies also demonstrated that G-1 exhibited no stimulatory transcriptional activity toward an ER/ERE-based reporter assay in MCF7 cells (Dennis et al., 2011). Although minor inhibition of E2-mediated ERE activation was noted at 1 and 10 μM G-1, selectivity over E2 was over 104-fold. G-1 has been found to reproduce the effects of E2 in a wide array of cellular and physiologic systems (see section III), as well as in a diverse array of species beyond humans including zebrafish (Pang and Thomas, 2010), croaker (Pang et al., 2008), mouse (Wang et al., 2009), rat (Deschamps and Murphy, 2009), cow (Kamanga-Sollo et al., 2008), and Rhesus monkey (Macaca mulatta) (Noel et al., 2009). It is important to note that G-1 and related analogs (e.g., G15 and G36) possess three chiral centers, resulting in the presence of both endo (syn) and exo (anti) diastereomers, each of which also consists of a pair of enantiomers. Efficient diasterioselective syntheses of the tetrahydro-3H-cyclopenta[c]quinoline ring system of G-1 and related analogs have been described, with the endo (syn) structure of the predominant G-1 diastereomer having been verified by single crystal X-ray diffraction (Burai et al. 2010). To date, there have been no reports of enantioselective syntheses or characterization of the activity of individual enantiomers.
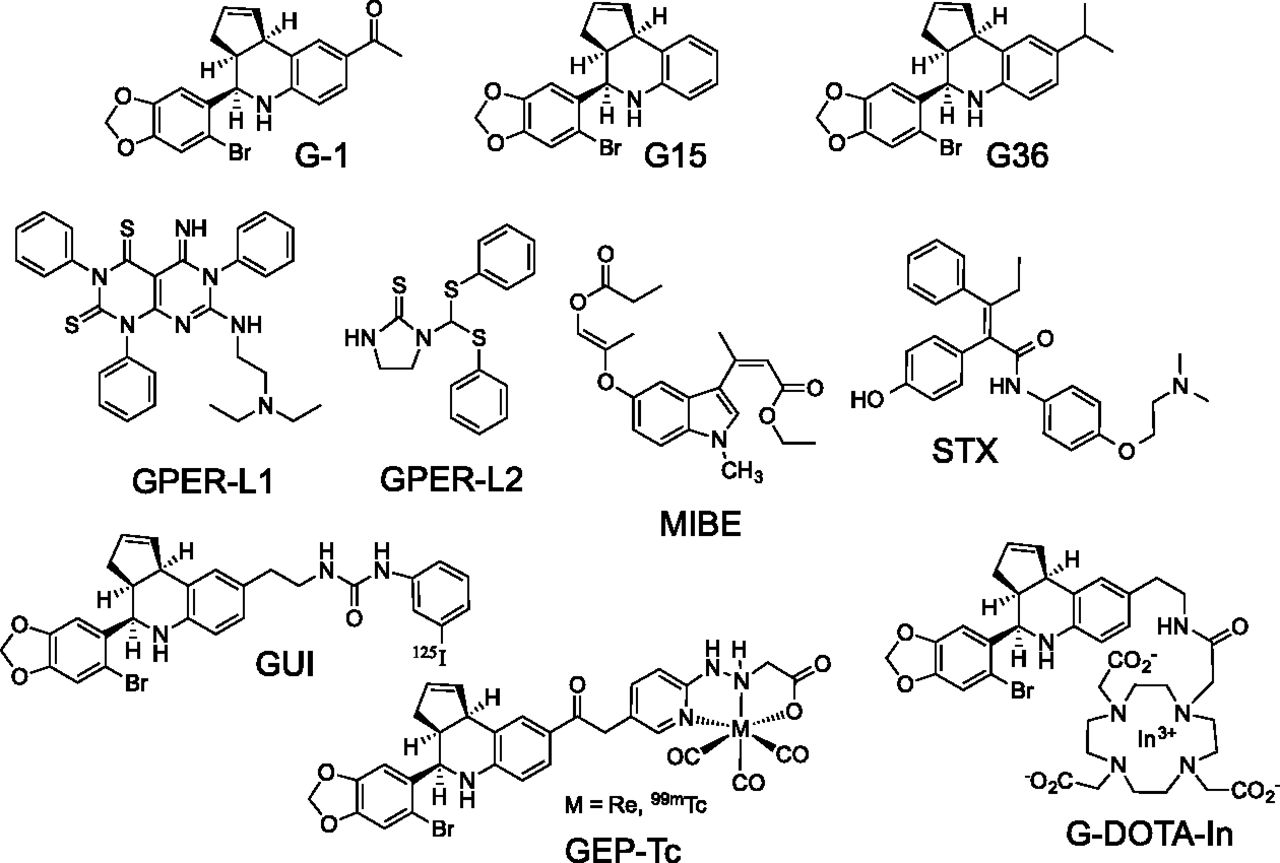
Structures of GPER-selective compounds and imaging agents.
Two additional compounds, GPER-L1 and GPER-L2, have been reported to function as GPER-selective agonists, with binding affinities of ∼100 nM for GPER and no detectable binding to ERα (Lappano et al., 2012a). They were identified based on in silico screening of a GPER homology model based on the structure of bovine rhodopsin and a hypothetical binding site from a library of 300 compounds. GPER-L1 is an iminodihydropyrimido-pyrimidine-dithione that is structurally related to the thiobarbituric acid topoisomerase II inhibitor merbarone. GPERL2 is an imidazolidine-2-thione derivative containing an N-substituted bis(phenylthio)methyl group. Interestingly, neither compound exhibits any obvious structural similarity with E2, reminiscent of the previously discussed scenario involving xenoestrogens. Structural homology models provide an initial entry point for the computational analysis of binding affinity and functional responses of GPER-selective ligands, with continuing advances in the crystallography of GPCRs providing additional structural insights that may be effectively coupled with computational studies (Rosano et al., 2012).
The diphenylacrylamide STX was designed by combining structural elements of 4-hydroxytamoxifen and raloxifene, using the transformation of the central ketone group in RAL to a carboxamide. Although STX does not bind to, activate, or antagonize ERα/β, it has been reported to elicit nongenomic E2 responses (ERK and Akt activation) in endometrial cancer cell lines that lack ERα/β but express GPER (Lin et al., 2009). STX, as well as tamoxifen and genistein, also increased SF-1 transcription, promoted Ishikawa cell proliferation, and induced the SF-1 target gene aromatase in a GPER-dependent manner. STX has also been demonstrated to mediate rapid signaling events in the hypothalamus (similar to E2) via a membrane-associated Gq-coupled estrogen receptor that is neither ERα nor ERβ (Qiu et al., 2006; Micevych and Kelly, 2012). Although GPER appears to be a likely candidate for this activity, STX continues to exhibit activity in GPER knockout mice, as well as ERα and ERβ knockout mice (Roepke et al., 2009), suggesting the presence of an entirely distinct STX target in the brain. Thus, STX may nevertheless function as a GPER-selective agonist.
2. Antagonists.
With the identification and wide use of G-1 to explore GPER function and physiology, the development of GPER-selective antagonists became of paramount importance. Based on the structural similarity of G-1 and E2, with hydrogen bond acceptors (cyclodioxo and keto moieties) in G-1 in a similar position to E2, we hypothesized that removal of the keto group could prevent receptor activation with a limited effect on GPER binding (Dennis et al., 2009). Synthesis of the proposed compound G15 yielded a G-1 derivative with only slightly reduced binding (∼2-fold) compared with G-1, while maintaining high selectivity against ERα and ERβ (>104-fold). Importantly, G15 inhibited the actions of both G-1 and E2 via GPER, as determined by calcium mobilization and PI3K activation. In vivo studies revealed that G15 completely inhibited the minimal effects of G-1 on uterine epithelial cell proliferation while inhibiting ∼50% of the robust E2 response. G15 also reversed the effects of both G-1 and E2 in a murine model of depression (Dennis et al., 2009).
Subsequent studies of transcriptional activation employing an ERE-reporter system revealed that upon a 24-hour stimulation at 10 μM, G15 exhibited a weak stimulatory effect (∼25% that of the maximal E2 effect, EC50 ∼0.1 nM), nevertheless maintaining selectivity of >106-fold over E2 (Dennis et al., 2011). Interestingly, 10 μM G15 also exhibited a small inhibitory effect on E2-stimulated ERE activation, suggesting its mode of action on ERα is actually that of a partial agonist. As 10 μM G-1 exhibited no such stimulatory effect, we speculated that the reduced size of G15 compared with G-1 (removal of the ethanone moiety), allowed it limited access to the ERα ligand binding pocket. To test this, we synthesized G36, in which the bulk of the ethanone moiety of G-1 is restored but with a methyl group replacing the keto functionality. The functional properties of G36 were identical to G15 with respect to GPER inhibition in multiple assays, and the minimal effects of G15 on E2 binding to GPER and ERE activity at 10 μM were almost completely eliminated, demonstrating that G36 possessed significantly improved counterselectivity against ERα and ERβ compared to G15 (Dennis et al., 2011).
As an alternative to GPER antagonists with high counterselectivity against ERα/β, Lappano et al. (2012b) identified an indole derivative named MIBE (ethyl3-[5-(2-ethox-ycarbonyl-1-methylvinyloxy)-1-methyl-1H-indol-3-yl]but-2-enoate) that acts as an antagonist of both GPER and ERα. Although MIBE exhibited binding affinities of ∼5 μM for GPER in SKBr3 cells and ∼100 μM for purified recombinant ERα, the transcriptional activity of ERα and ERβ reporters in HEK293 cells was surprisingly inhibited by much lower MIBE concentrations of ∼500 nM and ∼1 μM (IC50 values), respectively, with inhibition of ER-mediated transcription in MCF7 cells occurring at ∼200 nM. Inhibition of functional cellular responses to GPER activation was observed at MIBE concentrations between 1 and 10 μM (Lappano et al., 2012b). Although the pharmacologic profile of MIBE as a dual antagonist of GPER and ERα is intriguing, MIBE contains potentially reactive butenoate and aryloxypropenyl propionate appendages with hydrolyzable ester groups. It is therefore unclear what the functional form of MIBE is or would be, particularly in vivo, where the potential chemical reactivity and metabolism of MIBE could alter efficacy.
3. Imaging Agents.
Radionuclide isotopes that decay with emission of energetic gamma rays or positron particles coupled to targeting agents enable the highly sensitive in vivo detection and imaging of targets and metabolic processes using single photon emission computed tomography (SPECT) or positron emission tomography, respectively. The profound clinical significance of E2 in normal and diseased states including cancer has stimulated the development of diagnostic and therapeutic imaging agents targeting the nuclear ER in pursuit of the ultimate goal of effective noninvasive imaging for improved detection and management of therapy (Katzenellenbogen, 1980, 1995, 1997; Kiesewetter et al., 1984; Skaddan et al., 2000; Yoo et al., 2005; Ramesh et al., 2006; Seo et al., 2007; Fowler et al., 2012). This approach typically involves labeling the native steroid hormone, receptor ligand, or drug with a short-lived isotope (e.g., 18F for positron emission tomography or 99mTc for SPECT). The position of attachment, the structure of the radionuclide element or associated chelate, and connecting linkage are important parameters that can affect the overall physicochemical properties, receptor binding, biodistribution, and targeting characteristics of the agent. Because these studies were initiated before the discovery of GPER, agents that have reached the clinic such as 16α-[18F]-fluoro-17β-estradiol were developed with a focus on nuclear receptors and, as the previous discussion of selectivity has outlined, are incapable of distinguishing receptor subtypes. We described in 2006 the first characterization of binding properties of a series of 17α-conjugated-17β-estradiol derivatives with a panel of estrogen receptors (ERα, ERβ, and GPER), demonstrating the effects of the connecting linkage to the steroid on the binding affinity and selectivity of E2 [ERα/ERβ/GPER RBA (versus E2): ethyne 20/5/42; (Z)-ethene 10/24/64; ethane 1/0.5/ND] (Ramesh et al., 2006). As a next step toward the design and characterization of E2-based Tc-labeled tracers with improved targeting and SPECT imaging characteristics, we evaluated a [99mTc]estradiol-pyridin-2-yl hydrazine tricarbonyltechnetium[I] chelate, which exhibited a Kd for GPER of 11 nM (Nayak et al., 2008). In vivo studies revealed that receptor-mediated uptake occurred in all phases of the estrous cycle in reproductive organs and mammary glands but was highest during the diestrous phase. Although high nonspecific uptake was present in the liver, the receptor-mediated uptake in target tissues and estrogen receptor–expressing tumors was significant (0.7% injected dose (ID)/g for MCF7 tumors and 0.8% ID/g for endometrial tumors). Tumor uptake was reduced by ∼50% upon coinjection of unlabeled E2, as expected for competition in a receptor-mediated process. With the subsequent results from numerous recent studies supporting the significance of GPER expression as a potential diagnostic biomarker and therapeutic target, there is a need for the development of new improved imaging agents that are either highly selective for GPER, to further enable in vivo studies, or have enhanced selectivity for ER (anti-GPER) to provide enhanced imaging of nuclear estrogen receptors.
The potential for developing a GPER-selective radiohalogenated derivative was investigated using a series of synthetic iodo-substituted analogs and 125I-radiolabeled agents based on the tetrahydro-3H-cyclopenta[c]quinolone scaffold of G-1/G15/G36 (Ramesh et al., 2010). The affinity and specificity of binding, in addition to associated rapid signaling responses, were determined and used to select iodo-substituted urea- and hydrazone-linked tetrahydro-3H-cyclopenta[c]quinolones (GUI and GHI, respectively) that exhibited high GPER binding affinity (IC50 < 20 nM) in competitive cell binding assays. Both compounds functioned as GPER antagonists. The radiotracer ligands exhibited GPER-mediated uptake in tumor, adrenal, and reproductive organs in ovariectomized female athymic mice bearing GPER-expressing human endometrial tumors. Both GUI and GHI were rapidly metabolized and eliminated through the hepatobiliary system, with some significant differences such as nonspecific accumulation in the intestine versus stomach, respectively. The high lipophilicity of the compounds [log Po/w = 7.00 (GUI) and 6.35 (GHI)] adversely affected the in vivo biodistribution and clearance, suggesting that more polar compounds may have improved targeting characteristics.
An alternative design employed a series of 111In-radiolabeled G-1 analogs with polyamino-polycarboxylate chelate conjugates to assess the influence of ionic charge on cell binding, cellular permeability, and in vivo tumor imaging (Nayak et al., 2010). In cultured GPER-expressing cells, only the neutral G-DOTA-In complex activated GPER-mediated rapid signaling pathways. The charged anionic complexes did not activate these rapid responses, consistent with the model of activation involving functional intracellular receptor rather than a primarily extracellular membrane localization. The conformationally flexible carboxamide group in the linkage can serve as an H-bond acceptor, analogous to the ketone functionality in G-1 that is associated with agonism of GPER; however this activity profile was not observed for the similar urea linkage in GUI. The GPER binding affinity of G-DOTA-In was measured to be 34 nM, which is slightly decreased relative to G-1 and G15, and lipophilicity as measured by log Po/w = 4.8 was reduced relative to iodides GUI and GHI. In vivo studies using mice bearing GPER-expressing human endometrial Hec50 tumors revealed receptor-mediated uptake of the radiotracer in tumors and target organs. Tumors were visualized but images were affected by nonspecific localization of the G-DOTA-In in the intestines and gall bladder. The relatively rapid clearance from tumor and target tissues suggests that improved agents would benefit from reduced rates of metabolism and excretion.
A series of GPER-targeted organometallic tricarbonyl complexes was evaluated in cell-based assays to identify novel imaging agents with improved targeting capabilities (Burai et al., 2012). The in vitro biological characterization studies were conducted using stable isotope rhenium complexes, and the corresponding ligands were efficiently radiolabeled with 99mTc in aqueous media to provide the radioligands with high radiochemical yields and purity (Nayak et al., 2014). The ethanone conjugate GEP-Tc with a favorable log Po/w = 4.6 and potent agonist properties analogous to G-1 was identified as the most promising agent. All complexes exhibited high selectivity and affinity for GPER (10–30 nM) without binding to the nuclear estrogen receptors ERα and ERβ. In vivo biodistribution and imaging studies using GEP-Tc in mice bearing human endometrial and breast cancer cell xenograft tumors demonstrated significant tumor uptake (0.4–1.1%ID/g). The uptake was specific and receptor-mediated in multiple organs (adrenals, uterus, mammary tissue) and tumor, with similar levels of blocking achieved by competition with either E2 or G-1, demonstrating that GPER functions as an estrogen receptor in vivo and revealing the potential for GPER visualization in whole animals. Taken together, these results provide a molecular basis to evaluate GPER expression and function as an estrogen receptor through in vivo imaging, in addition to the further study of GPER in E2-mediated carcinogenesis and physiology. These first generation GPER-selective imaging agents also provide a foundation for the continued development of highly selective GPER-targeted agents for diagnostics, prognostic, therapeutic, and image-guided drug delivery.
V. Conclusions and Future Directions
It is clear that the actions of E2 in the human body are highly complex and multifaceted, involving multiple receptors that signal via both transcriptional regulation as well as rapid nongenomic pathways. At present, there are at least three major receptors (ERα, ERβ, and GPER), belonging to two distinct receptor families (Huang et al., 2010; Katritch et al., 2013), that are known to mediate these effects through both nuclear-localized genomic effects and membrane-initiated rapid signaling events. It is important to note that these three receptors display distinct expression and activity profiles throughout the body, with both overlapping and unique functions, in which ERα plays a major role in reproductive (proliferative) function, whereas ERβ and GPER may be thought of as playing more homeostatic roles (Matthews and Gustafsson, 2003). The classic pharmacology of estrogenic compounds has been based predominantly on the stimulatory actions (or lack thereof) of these compounds on the breast, uterus, and bone, with less regard for the molecular mechanisms involved (Kangas, 1992). Whereas the actions of full agonists (and antagonists) would seem relatively straightforward based on their agonism (or antagonism) in all tissues, the actions of full agonists are nevertheless complex as exemplified by their distinct effects in multiple tissues [uterine imbibition (Hewitt et al., 2003), breast cancer/cell proliferation (Ariazi et al., 2006), bone calcification (Nelson et al., 2013)]. In contrast, the actions of SERMs were defined based on differential activity within each of these three tissues (Kangas, 1992). These differential activities were subsequently understood at least in part at the molecular level based on the stabilization of distinct ER conformations (involving helix 12 of the ligand binding domain), which in turn result in differential recruiting of transcriptional coactivators and corepressors (Smith and O'Malley, 2004), in a ligand- and tissue-dependent manner (McDonnell and Wardell, 2010; Burris et al., 2013). Far less is understood regarding the mechanisms and pharmacology of rapid signaling via ERα and ERβ (Banerjee et al., 2014), including their many splice variants (Kim et al., 2011; Chaudhri et al., 2014), and in particular, the agonistic rapid cellular effects of SERMs and SERDs, which can now at least be partially understood through their actions on GPER. However, just as our understanding of the actions of agonists, SERMs and SERDs for ERs has expanded over the years, GPCRs are now understood to exist in a number of activated conformations (Katritch et al., 2013), stabilized to varying extents by different ligands, that induce distinct subsets of downstream signaling pathways, processes termed functional selectivity or biased agonism (Kenakin, 2012, 2013; Seifert, 2013). At a minimum, GPCRs can couple to multiple heterotrimeric G proteins (Woehler and Ponimaskin, 2009), G protein–coupled receptor kinases (Evron et al., 2012; Gurevich et al., 2012), and arrestins (Key et al., 2003; Luttrell and Miller, 2013), all of which can be differentially engaged by a given ligand (Andresen, 2011; Reiter et al., 2012; Mary et al., 2013). GPCR activity can be regulated further by (hetero)dimerization (Goupil et al., 2012), desensitization (Walther and Ferguson, 2013), trafficking (export, internalization, and degradation) (Shirvani et al., 2012; Van Craenenbroeck, 2012; Walther and Ferguson, 2013), allosteric ligands (Wootten et al., 2013), and post-translational modifications (Grimsey et al., 2011), as well as the spectrum of interacting proteins expressed in a given cell. In principle, many of these mechanisms (e.g., heterodimerization, allosteric ligands and interacting proteins, post-translational modifications) are equally relevant to the pharmacology and regulation of ERα/β activity. Finally, the differential expression of ERα, ERβ and GPER in a given cell will further determine the integrated result of stimulation by a given compound.
Given the lack of selectivity of current ER-targeted therapeutic agents toward GPER, there exists an important opportunity for the development of such drugs, which would complement the activity of GPER-selective agonists and antagonists (Paterni et al., 2013). Furthermore, as the development of ERβ-selective (versus ERα) agonists is being actively sought (Mohler et al., 2010; Minutolo et al., 2011), consideration must be paid to the activity of such agents with regard to genomic versus rapid signaling through ERs as well as activity toward GPER. Whether the pharmacology of membrane-localized ERα (and potentially ERβ), which can be targeted with cell-impermeable E2 conjugates (Wu et al., 2011a), is distinct from nuclear ERs remains unknown. If one considers the presence of at least three differentially expressed estrogen receptors, each of which can be individually targeted with respect to agonism, partial and/or mixed agonism/antagonism, or antagonism in multiple cellular signaling pathways, the number of potential new drug opportunities is truly astonishing. However, to assess which of these activity profiles is worth pursuing clinically, an improved understanding of the individual role(s) of each receptor in human physiology as well as the interactions between these receptors (e.g., synergism, antagonism, cross-activation, and cross-inhibition) within a cell and throughout the body is needed. Nevertheless, at this point, the unique pharmacologic properties of GPER-selective ligands and the evolving physiologic functions of GPER in health and disease make this receptor an intriguing therapeutic target with great promise.
Footnotes
This work was supported by the National Institutes of Health National Cancer Institute [Grants R01-CA163890 (to E.R.P.) and R01-CA127731 (to E.R.P. and J.B.A.)]; with support from the University of New Mexico Cancer Center (P30-CA118100) and the New Mexico Cowboys for Cancer Research Foundation. NC-IUPHAR is supported in part by Wellcome Trust Grant 099156/Z/12/Z.
Abbreviations
- 4-OHT
- 4-hydroxytamoxifen
- 7β-OH-EpiA
- 7β-hydroxy-epiandrosterone
- Aldo
- aldosterone
- BPA
- bisphenol A
- BSA
- bovine serum albumin
- DDE
- dichlorodiphenyldichloroethylene
- DDT
- dichlorodiphenyltrichloroethane
- DES
- diethylstilbestrol
- DHEA
- dehydroepiandrosterone
- DPN
- 2,3-bis(4-hydroxyphenyl)-propionitrile
- E1
- estrone
- E2
- 17β-estradiol (also referred to throughout as estrogen)
- E2DC
- E2-dendrimer conjugate
- E3
- estriol
- E-6-CMO
- 17β-estradiol-6-carboxymethyloxime
- EAE
- experimental autoimmune encephalomyelitis
- EDC
- endocrine disrupting chemical
- EE
- 17α-ethynyl-17β-estradiol
- eNOS
- endothelial nitric oxide synthase
- ER
- estrogen receptor
- ERE
- estrogen response element
- ERK
- extracellular signal-regulated kinase
- FITC
- fluorescein isothiocyanate
- G-1
- 1-[4-(6-bromobenzo[1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl]-ethanone
- G15
- 4-(6-bromo-benzo[1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolone
- G36
- 4-(6-bromo-benzo[1,3]dioxol-5-yl)-8-isopropyl-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolone
- G-DOTA-In
- GPER-ligand-tetraazacyclododecanetetraacetic acid In(III) complex
- GPCR
- G protein–coupled receptor
- GPER
- G protein–coupled estrogen receptor
- ICI 182,780
- 7α,17β-[9-[(4,4,5,5,5-pentafluoropentyl)sulfinyl]nonyl]estra-1,3,5(10)-triene-3,17-diol
- IL
- interleukin
- MeO-E2
- 2-methoxy-estradiol
- MIBE
- ethyl3-[5-(2-ethox-ycarbonyl-1-methylvinyloxy)-1-methyl-1H-indol-3-yl]but-2-enoate
- PCB
- polychlorinated biphenyls
- PPT
- 4,4′,4′′-(4-propyl-[1H]-pyrazole-1,3,5-triyl) trisphenol
- RAL
- raloxifene
- RBA
- relative binding affinity
- SERD
- selective estrogen receptor downregulator
- SERM
- selective estrogen receptor modulator
- SPECT
- single photon emission computed tomography
- STX
- 2-(4-hydroxyphenyl)-3-phenylpent-2-enoic acid [4-(2-dimethylaminoethoxy)phenyl]amide
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵





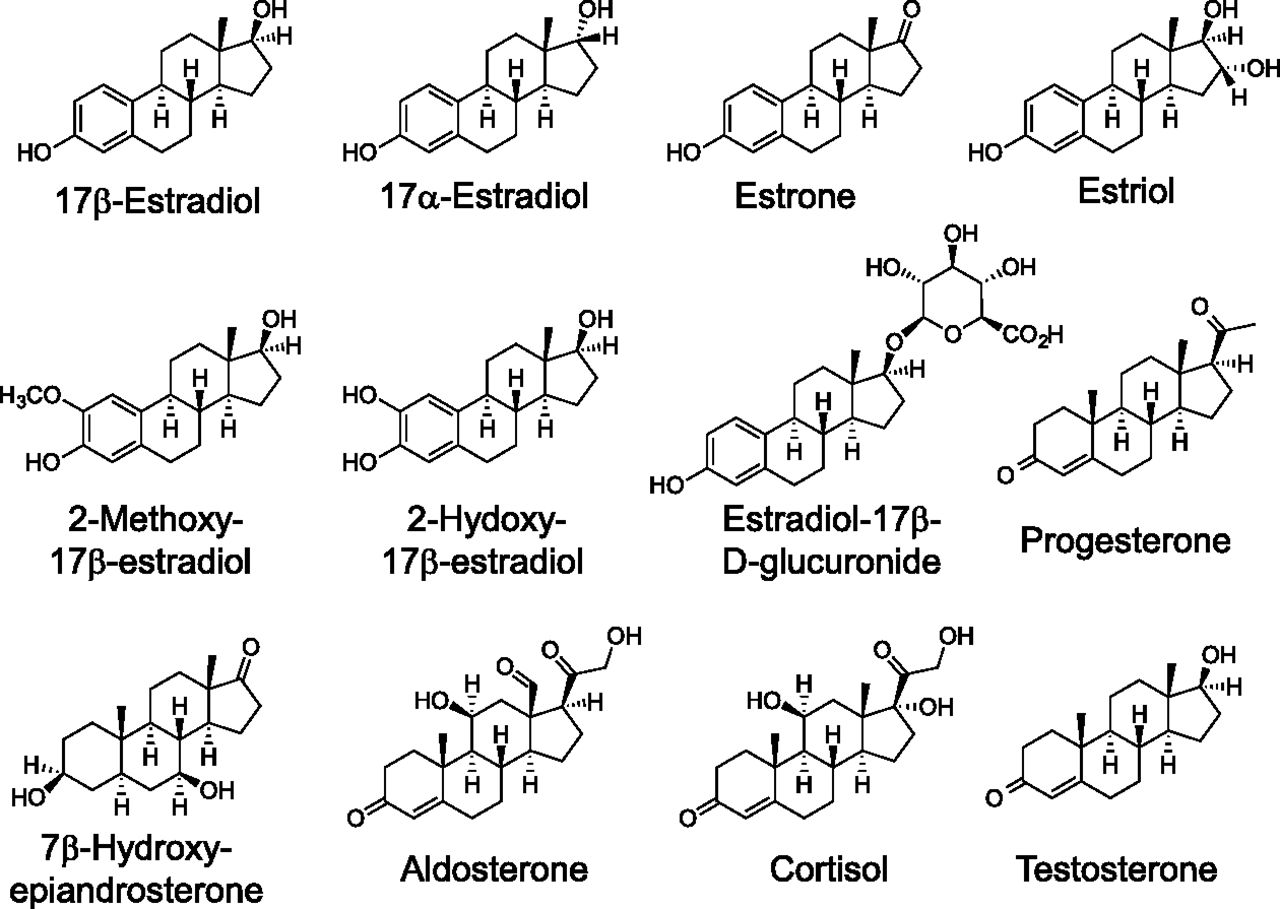{kind=link}
{kind=link}
{kind=link}
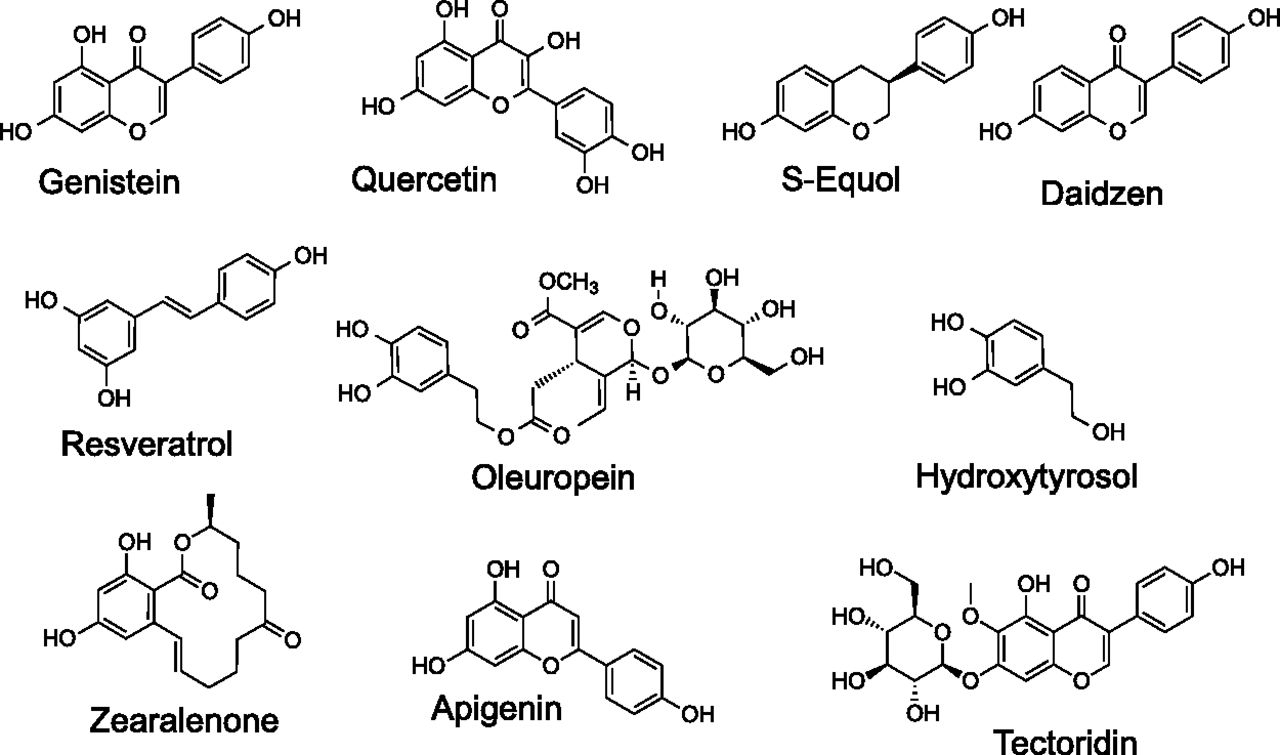{kind=link}
{kind=link}
{kind=link}
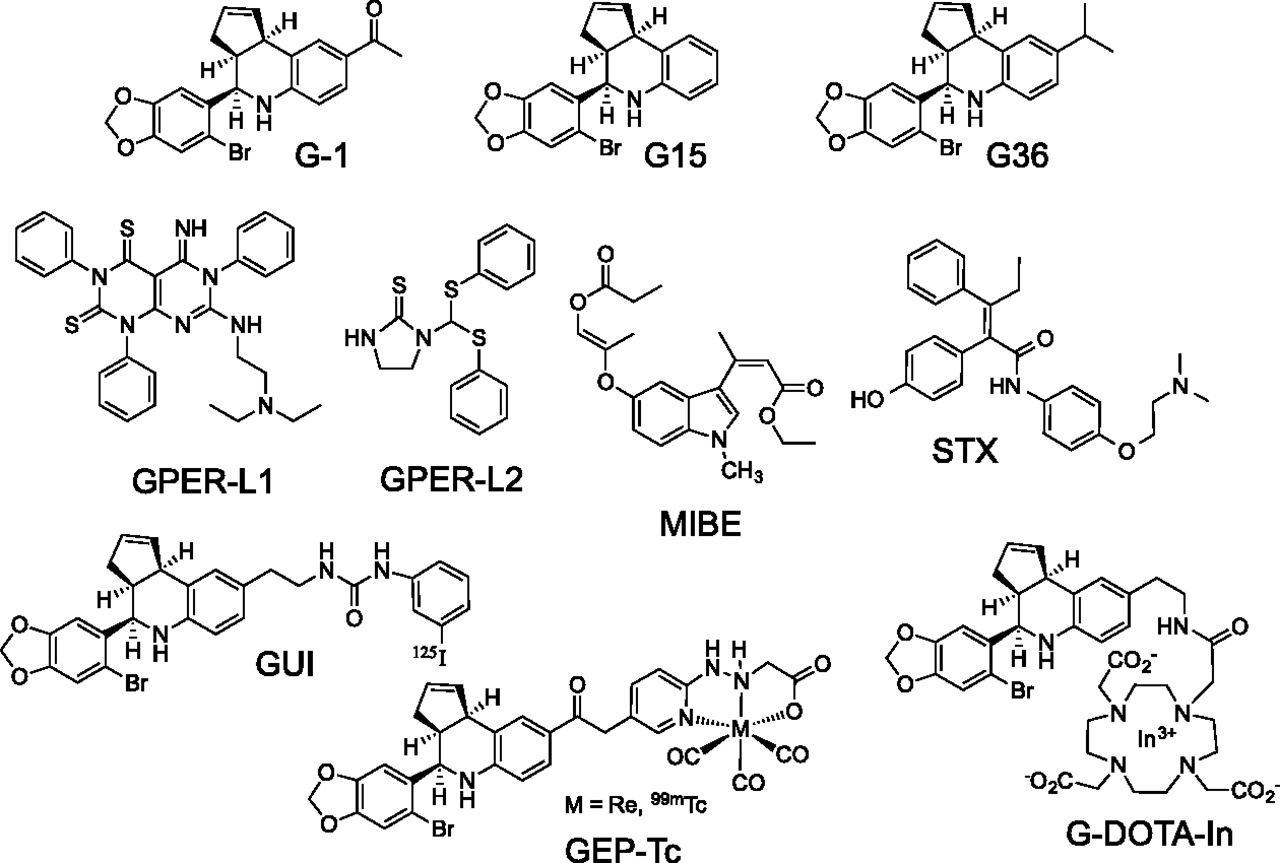{kind=link}