


Abstract
Major depressive disorder is one of the most prevalent and life-threatening forms of mental illnesses and a major cause of morbidity worldwide. Currently available antidepressants are effective for most patients, although around 30% are considered treatment resistant (TRD), a condition that is associated with a significant impairment of cognitive function and poor quality of life. In this respect, the identification of the molecular mechanisms contributing to TRD represents an essential step for the design of novel and more efficacious drugs able to modify the clinical course of this disorder and increase remission rates in clinical practice. New insights into the neurobiology of TRD have shed light on the role of a number of different mechanisms, including the glutamatergic system, immune/inflammatory systems, neurotrophin function, and epigenetics. Advances in drug discovery processes in TRD have also influenced the classification of antidepressant drugs and novel classifications are available, such as the neuroscience-based nomenclature that can incorporate such advances in drug development for TRD. This review aims to provide an up-to-date description of key mechanisms in TRD and describe current therapeutic strategies for TRD before examining novel approaches that may ultimately address important neurobiological mechanisms not targeted by currently available antidepressants. All in all, we suggest that drug targeting different neurobiological systems should be able to restore normal function but must also promote resilience to reduce the long-term vulnerability to recurrent depressive episodes.
I. Introduction
Major depressive disorder (MDD) is a chronic debilitating illness that represents a major economic and medical burden for our society. It is characterized by different and heterogeneous symptoms that lead to functional disability in affected individuals.
Although a large number of antidepressant drugs have been developed over the last 50–60 years, the therapeutic response is often partial, and around 20%–30% of patients are considered treatment resistant or they do not respond adequately to two successive antidepressant treatments under a proper therapeutic regimen (McIntyre et al., 2014). Treatment-resistant depression (TRD) is associated with a significant impairment of cognitive function, higher risk for comorbidity, and an increased suicidality (Gaynes, 2016). On these bases, there is a great deal of interest in identifying the elements that may contribute to TRD to improve clinical outcomes.
The present review will provide an up-to-date description of key issues in TRD and how the comprehension of specific aspects related to MDD could be instrumental for a proper selection of the therapeutic approaches and may ultimately lead to the development of novel therapeutic strategies. In particular, we will discuss how etiological mechanisms and a better definition of the neurobiological dysfunction in MDD patients can provide key information to identify altered genes and pathways that are not a direct target of the current antidepressants and may therefore represent potential “limiting factor” of the effectiveness of pharmacological intervention. Moreover, following a description of the current therapeutic strategies, we will discuss novel approaches that address important neurobiological mechanisms and may ultimately offer new hopes for a more thoroughly impact on TRD patients.
II. The New Neuroscience-Based Nomenclature and the Classification of Antidepressant Drugs: Implications for Drug Discovery and Clinical Practice
The classification of psychotropic medications represents an essential tool for the clinician and it should always reflect contemporary knowledge, informing the clinician about rational evidence-based prescribing strategies. It has become clear that the WHO’s anatomic-therapeutic-chemical (ATC) classification system shows different limitations when applied to clinical practice, because it does not always reflect the most recent advances in the field of neuropsychopharmacology. The ATC classification system was established in the 1960s, when the development of psychotropic drugs was only in an early phase and the drugs were classified according to the first indication obtained by the regulatory agencies. This explains why this system classifies psychotropic drugs to only one of five classes: antipsychotics, antidepressants, anxiolytics, hypnotics, and mood stabilizers. Unfortunately, the ATC nomenclature for psychotropic drugs fails to describe pharmacological domains or mechanisms of action and also does not indicate all the potential clinical uses of a particular agent developed different years after the first approval. For example, under the ATC classification, “antidepressants” may be prescribed for anxiety disorders, and “second generation antipsychotics” such as aripiprazole or quetiapine are used for treating depressed patients with no signs or symptoms of psychosis, but with a history of treatment resistance.
Starting from this evidence, the European College of Neuropsychopharmacology, the American College of Neuropsychopharmacology, the Asian College of Neuropsychopharmacology, the International College of Neuropsychopharmacology, and the International Union of Basic and Clinical Pharmacology joined forces to design a more precise and descriptive nomenclature for psychotherapeutics with the aim of developing the new neuroscience-based nomenclature (NbN) able to overcome the limitations of the ATC classification system (Zohar et al., 2015) (http://nbnomenclature.org). The aim of this approach is to provide physicians with clearer alternatives than the ATC system when deciding the proper therapeutic strategy. Furthermore, the NbN nomenclature system has been developed to accommodate the discovery of new psychotropic drugs with different pharmacodynamic profiles and different mechanisms of action.
The NbN is focused on the pharmacology and the molecular mechanism of action (Caraci et al., 2017a) and identifies pharmacological domains, modes of action as well as additional dimensions beyond basic pharmacology, including approved indications, efficacy, and side effects, practical notes and neurobiology [see Caraci et al. (2017a) for further details]. Currently, the NbN nomenclature classifies 109 psychotropic drugs representing a broad range of agents and indications. The drug target or receptor nomenclature has been developed according to the International Union of Basic and Clinical Pharmacology/British Pharmacological Society nomenclature available on www.guidetopharmacology.org or the Concise Guide to Pharmacology (Alexander et al., 2015). The NbN classification can therefore provide the scientific basis to differentiate the molecular mechanism of action of the different antidepressant drugs currently used in clinical practice.
According to the ATC classification, tricyclic antidepressants (TCAs) are classified as nonselective monoamine reuptake inhibitors (N06AA) and monoamine oxidase inhibitors are grouped in nonselective (N06AF), such as tranylcypromine, or selective, such as moclobemide (N06AG). When we consider second-generation antidepressant drugs, only selective serotonin reuptake inhibitors (SSRIs) constitute a separate class (N06AB), whereas all the other second-generation antidepressants, serotonin and noradrenaline reuptake inhibitors (SNRIs) (duloxetine, venlafaxine, desvenlafaxine); the noradrenaline and dopamine reuptake inhibitors bupropion, agomelatine, trazodone, hypericum perforatum; and the new multimodal antidepressants vilazodone and vortioxetine are all included in a heterogeneous class (N06AX). This example demonstrates the limits of ATC classification, where different antidepressant drugs are present in N06AX class without considering relevant differences in their pharmacodynamic profile and their clinical use. Unfortunately, the ATC classification of antidepressant drugs has been developed according to the monoaminergic hypothesis of depression and has not been designed to include recent advances in drug discovery processes in depression. As we will discuss in this review, new relevant pharmacological targets were recently identified in major depression, with the aim of developing novel and rapidly acting compounds, especially for patients with treatment-resistant depression (Ionescu and Papakostas, 2017).
According to the NbN classification, it is now possible to differentiate the molecular mechanism of action of the different first-generation antidepressants versus second-generation antidepressants (Zohar et al., 2015). For example, nortriptyline can be described as follows: 1) norepinephrine and serotonin reuptake inhibitor, 2) approved for the treatment of major depressive disorder, 3) a cytochrome P450 2D6 substrate with antidepressant efficacy that displays side effects expected from an agent that interacts with multiple neurotransmitter receptors, and 4) interacts with a host of secondary targets with multiple effects on brain chemistry and signaling. This profile differs from another TCA, such as clomipramine, which is known to be more selective in blocking 5-HT reuptake compared with previously launched TCAs (Millan et al., 2001), and it is particularly effective in the treatment of obsessive compulsive disorder by a mechanism that is still poorly understood (Pizarro et al., 2014; Millan et al., 2015).
A major effort has been done to include in the section “practical notes” essential information on the drug interaction profile of old and newer antidepressants that, considering the significant differences between antidepressants (Spina et al., 2012), may represent an important criteria for drug selection with respect to the long-term treatment of MDD patients in the presence of comorbid psychiatric or somatic disorders.
The NbN also describes the multimodal pharmacodynamic profile of recently approved antidepressants, such as vortioxetine [reuptake inhibitor, receptor partial agonist (5-HT1A), receptor antagonist (5-HT3 and 5-HT7)], compared with other second-generation antidepressants such as duloxetine, a reuptake inhibitor (serotonin transporter and NET). For example, the NbN describes the specific clinical efficacy of vortioxetine in the treatment of cognitive dysfunction in MDD. Recent clinical studies do not suggest a global greater efficacy of multimodal antidepressants such as vortioxetine compared with SSRIs or SNRIs, but an improved efficacy on specific clinical domains (e.g., deficits in memory and executive functioning) where SSRIs or SNRIs are less effective (Thase et al., 2016). The multimodal profile of vortioxetine described on NbN classification is consistent with the results of these clinical studies (Caraci et al., 2017b).
This new nomenclature also shows specific advantages in incorporating the most relevant advances in drug discovery for depression (Zohar et al., 2015; Caraci et al., 2017b). For example, if a new target or an innovative mechanism of action is identified behind the classic monoaminergic hypothesis of depression [e.g., modulation of mammalian target of rapamycin (mTOR) pathway by ketamine in TRD], NbN can be expanded in a meaningful way to incorporate such new advances in drug development. As we will discuss in this review, the discovery of rapid-acting glutamatergic drugs represents a major advance in the field of TRD, and a new class of antidepressants will be developed in the next years starting from ketamine. Presently the old ATC classification would incorporate ketamine-like drugs in a heterogeneous class (N06AX), which also includes SNRIs, agomelatine, and vortioxetine, without considering the neurobiology of TRD and the relevance of glutamatergic system as a new pharmacological target in TRD. Opposite of the ATC system, the NbN classification will describe the basic pharmacology of ketamine and will also summarize in an additional dimension (“neurobiology”) how this drug interferes with recently identified pathways in TRD and the clinical relevance of these effects.
On these bases, we suggest that new antidepressant drugs, as described in this review, which will target different molecular mechanisms for the treatment of TRD, might easily be incorporated in the new NbN classification.
III. Clinical Phenotypes of Treatment-resistant Depression
One of the international research consortiums that most comprehensively studied the topic of clinical phenotypes of patients with TRD represents the “European Group for the Study of Resistant Depression (GSRD)” (Schosser et al., 2012b; Dold et al., 2016). For nearly two decades, this study group has sought to elucidate clinical as well as genetic factors contributing to treatment resistance in major depressive disorder (Table 1).
Clinical factors significantly associated with treatment resistance in unipolar depression according to the studies of the GSRD (European Group for the Study of Resistant Depression)
A. Definition of Treatment Resistance and Staging Models
In 1999, the GSRD implemented a staging method for treatment-resistant depression (Souery et al., 1999). According to their definition, the criteria for treatment resistance are fulfilled if a patient is resistant to at least two consecutive adequate antidepressant trials independently from the class of antidepressant (including augmentation and combination medications) administered. The different stages of treatment resistance correspond to the number of the following failed antidepressant trials (Souery et al., 1999).
Similarly, the European Medicines Agency (http://www.ema.europa.eu) defines treatment resistance as a nonresponse to at least two adequate antidepressant trials. In detail, the European Medicines Agency states: “TRD is considered, when treatment with at least two different antidepressant agents (of the same or a different class) prescribed in adequate dosages for adequate duration and adequate affirmation of treatment adherence showed lack of clinically meaningful improvement in the regulatory setting” (http://www.ema.europa.eu). Another staging model has been suggested by Thase and Rush (1997), considering a hierarchy of efficacy of different therapeutic strategies including also electroconvulsive therapy (ECT).
In the context of identifying treatment-resistant MDD conditions, it should be critically taken into account that some patients are considered to be treatment resistant even if they exhibit so-called “pseudoresistance” (i.e., a merely alleged resistance to the current antidepressant pharmacotherapy). Therefore, the debarment of “pseudoresistance” represents the first measure in case of insufficient response to the initial antidepressant monotherapy trial. Potential reasons for “pseudoresistance” can be, for instance, an inadequate dose and treatment duration of the antidepressant, insufficient plasma levels of the administered drugs, noncompliance of the patient with respect to medication intake, or relevant (nontreated) psychiatric and/or somatic comorbidities (Dold and Kasper, 2017) (Table 2).
“Checklist” with potential reasons for an only alleged resistance to the initial antidepressant medication (“pseudo-resistance”) based on Dold and Kasper (2017)
B. Features Contributing to Treatment Resistance
In a comprehensive multicenter study of the GSRD, Souery et al. (2007) analyzed sociodemographic and clinical characteristics of 702 patients with MDD and found the following variables to be significantly associated with the presence of treatment resistance: comorbid anxiety disorders (panic disorder and social phobia), comorbid personality disorder, suicide risk, high depressive symptom severity, melancholic features, more than one previous hospitalization due to MDD, recurrent depressive episodes, nonresponse to the first administered antidepressant, and an age at onset of ≤18 (Table 3).
Clinical factors associated with treatment resistance in unipolar depression according to the European multicenter study (n = 702) of Souery et al. (2007)
Treatment resistance was defined by a failure of at least two consecutive trials with antidepressant drugs.
Applying machine learning algorithms to the above-mentioned GSRD patient sample, Kautzky et al. (2017) determined the timespan between first and last depressive episode, age at first antidepressant treatment, response to first antidepressant treatment, symptom severity, suicidality, melancholia, number of lifetime depressive episodes, patients’ admittance type, education, occupation, and comorbid diabetes, panic, and thyroid disorder to be the most useful predictors for treatment outcome.
In a recent further study of the GSRD, Balestri et al. (2016) investigated predictors for a very high degree of treatment resistance. Hereby, sociodemographic and clinical variables were examined in 98 patients with inadequate treatment response to at least three different antidepressants, including escitalopram and venlafaxine. In this clinical study, long duration and high severity of the current depressive episode, outpatient status, high suicidal risk, higher rate of the presence in the family history of psychiatric disorders, and the occurrence of adverse effects during the pharmacotherapy served as clinical predictors for severe treatment resistance. In another GSRD survey, Zaninotto et al. (2013) identified the following factors to be associated with treatment resistance: longer hospitalization over lifetime, longer duration of the current depressive episode, comorbid panic disorder, presence of melancholic and psychotic features, and suicide risk. However, it should be considered that this study (N = 699) was the first of all designed to determine differences between patients exhibiting psychotic or melancholic features (Zaninotto et al., 2013). Furthermore, Mandelli et al. (2016) investigated the impact of occupational levels on response patterns in 654 patients with MDD, whereby three occupational levels (high, middle, low) were compared with regard to the achievement of treatment response versus resistance. The analyses revealed a significant association between high occupational level and poorer treatment response in comparison with medium and low occupational levels (Mandelli et al., 2016). With respect to somatic comorbidities in MDD, no significant differences were found between responders and treatment-resistant patients in a sample of 702 patients with MDD (Amital et al., 2013). In terms of family history, Serretti et al. (2014) reported no statistically significant differences between patients with and without a family history of MDD when analyzing overall depressive symptoms. However, nonresponders with a family history of MDD showed higher core depressive symptoms compared with nonresponders without a family history of MDD (Serretti et al., 2014) (Table 3).
In addition to the aforementioned multicenter studies carried out by the GSRD, the finding of significantly poorer treatment outcome for patients with unipolar depression and concurrent anxiety was replicated in a study of the German Algorithm Project comprising 429 inpatients with MDD (Wiethoff et al., 2010). Similar findings were found in the large North American multicenter STAR*D (Sequenced Treatment Alternatives to Relieve Depression) study (Trivedi et al., 2006) investigating altogether 2876 outpatients with depression. Furthermore, largely corresponding findings stem from recent evidence derived from the STAR*D sample suggesting a final predictive model with accuracy about 70% and sensitivity about 88%, relating delayed remission rates to unemployment and severe baseline depression for instance (Falola et al., 2017).
IV. Genetics of Treatment-resistant Depression
Genetic variants explain ∼42% of variance in antidepressant response (Tansey et al., 2013), and genotyping can easily be implemented in clinical settings (saliva or blood sample, quite rapid and cost affordable). Thus, individual genetic makeup may be used for providing personalized antidepressant treatments that would reduce the rates of treatment-resistant depression.
Some issues have delayed the identification of genetic predictors with clinical validity, but promising technological evolutions have recently raised hopes, such as the drop of genotyping costs and improved analysis facilities. The growing of international consortia helps to overcome one of the main issues in the genetic analysis of complex traits, i.e., the lack of statistical power. For example, in a genome-wide association study (GWAS), a sample size ∼2000 subjects provides adequate power to identify individual variants associated with a binary trait with heritability ∼40% (Visscher et al., 2014). Unfortunately, the diagnosis of TRD is not as easy to determine as just the diagnosis of major depression interferes with the collection of large samples. A GWAS including 1311 TRD patients failed to identify common variants associated with TRD in the 23AndMe study (Li et al., 2016a). The top variants did not reach the genome-wide significance threshold (all P > 2e−07, while the standard genome-wide significance threshold is 5e−08) and they were within genes having unclear biologic connection with TRD (FAM98A, MYADML, and dipeptidyl peptidase like 10). Another GWAS investigated rare variants and it identified non-significant enrichment of duplications in TRD and a deletion spanning the PABPC4L gene (O’Dushlaine et al., 2014).
Alternative strategies to very large GWAS can provide meaningful insights in TRD genetics. These include: 1) a priori selection of strong candidate genes and use of complementary approaches (e.g., gene expression analysis, animal models), 2) detailed phenotype characterization (MDD is clinically and biologically heterogeneous), and 3) aggregated approaches that test the effect of many variants in a gene or pathway. The power increase in an aggregated approach can be attributed to the reduction in the number of tests performed and the capture of the cumulative effects of a number of variants (the disruption of a gene or pathway functioning is the result of cumulative effects of variants within it) (Li et al., 2017a).
The GSRD has been working for over 15 years to study methodological issues, operational criteria, and clinical and genetic variables associated with TRD (Schosser et al., 2012b). GSRD applied the strategies listed in points 1–3 to maximize the power of identifying genetic variants associated with TRD. Genes of interest included those involved in glutamatergic and monoaminergic neurotransmission as well as synaptic plasticity, as suggested by the antidepressant efficacy of the N-methyl-d-Aspartate (NMDA) receptor antagonist ketamine (de Sousa et al., 2017) and ECT in TRD (Kellner et al., 2012). GRIK4 gene (glutamate ionotropic receptor kainate type subunit 4) was proposed as candidate gene for TRD (Serretti et al., 2012; Milanesi et al., 2015) and ECT response in TRD (Minelli et al., 2016). Ketamine rapidly activates synaptic plasticity mediated by glutamatergic receptors, leading to increased number and function of new spine synapses (Li et al., 2010). Protein phosphatase 3 catalytic subunit gamma is involved in the induction of glutamatergic-mediated synaptic plasticity through the modulation of calcineurin (Yu et al., 2013) and variants in this gene were associated with TRD (Fabbri et al., 2014). An aggregated analysis approach, including variants in protein phosphatase 3 catalytic subunit gamma, 5HTR2A, and brain-derived neurotrophic factor (BDNF) genes demonstrated good prediction of TRD (Kautzky et al., 2015). A gene set (GO: 0006942) including the CACNA1C gene showed interesting prediction of TRD using machine learning models (mean sensitivity of 0.83, specificity of 0.56, positive predictive value = 0.77, and negative predictive value = 0.65 after 10-fold cross validation repeated 100 times) (Fabbri et al., 2018). CACNA1C encodes for the α-1C subunit of the L-type voltage-dependent calcium channel, and it is involved in the modulation of synaptic plasticity. This gene has been associated with multiple psychiatric phenotypes, including schizophrenia, bipolar disorder, and MDD, suggesting it plays a pleiotropic role in psychiatric disorders (Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013). Other genes involved in monoaminergic neurotransmission and synaptic plasticity were associated with TRD in GSRD samples. Catechol-O-methyltransferase is one of the main enzymes responsible for monoamine metabolism, and variants in this gene were associated with increased risk of suicide in TRD (Schosser et al., 2012a). Growth-associated protein 43 and cell adhesion molecule L1 like are pivotal genes in synaptic plasticity; polymorphisms in these genes and the growth-associated protein 43 pathway were proposed as candidates for TRD risk (Fabbri et al., 2015, 2017). Both genes showed gene expression differences in human lymphoblastoid cells displaying high versus low paroxetine sensitivities (Morag et al., 2011).
Evidence from complementary approaches makes it worth mentioning a couple of other genes. The first one codes for a potassium channel (KCNK2 or TREK1) that is involved in a reciprocal regulation with the serotonin transporter. In mice, the deletion of KCNK2 led to an increased efficacy of serotonin neurotransmission and a resistance to depression in five different models (Heurteaux et al., 2006). KCNK2 variants were associated with the risk of nonresponse to the second and third antidepressant trials in the STAR*D study (Perlis et al., 2008). Complementary research demonstrated that in cultured hippocampal neurons TREK1 channel blockers upregulated genes involved in BDNF signal transduction, they increased the firing rate of serotonergic neurons in relevant mice brain areas, and they showed antidepressant-like effect (Ye et al., 2015). The second gene is ABCB1 that codes P-glycoprotein (P-gp), a drug efflux pump at the blood-brain barrier. Acute and chronic P-gp inhibition yields elevated antidepressant brain concentrations (O’Brien et al., 2015), thus genetic variants increasing P-gp activity may be involved in TRD. Some antipsychotics used as antidepressant augmentation in TRD inhibit P-gp, and they were associated with increased antidepressant concentration in the brain, suggesting an additional efficacy mechanism of this treatment strategy (O’Brien et al., 2012). Variants in the ABCB1 gene were associated with antidepressant response and remission, and the implementation of ABCB1 genotyping as a diagnostic tool led to an improvement of treatment outcome (Breitenstein et al., 2014). A case series suggested that subjects carrying variants associated with P-gp increased activity may develop TRD when treated with normal doses of antidepressants that are targets of P-gp (e.g., venlafaxine, paroxetine) (Rosenhagen and Uhr, 2010). Furthermore, ABCB1 gene expression was associated with TRD (Breitfeld et al., 2017).
Currently several pharmacogenetic tests that claim to predict antidepressant response are on the market, despite no demonstration of validity and cost effectiveness performed independently from the producing companies (Fabbri et al., 2016). If validated, they may contribute to the reduction of TRD rates thanks to targeted antidepressant prescription. In the future, the improvement of genotyping techniques and analysis methods are expected to improve our knowledge of TRD genetics. GSRD recently completed the collection of the largest sample with detailed characterization of TRD (n∼1400) genotyping is ongoing using a combination of exome sequencing and genome-wide arrays. Analyses will be focused on pathways to point out which biologic functions may be disrupted in TRD.
V. From the Neurobiology of Depression to Treatment Resistance
A. Etiological Mechanisms of Major Depression
Depression is a multifactorial disease characterized by a heterogeneous group of symptoms associated with functional disability. A better understanding of the mechanisms and factors that contribute to disease onset is crucial not only for a better definition of different disease dimensions, but also to establish the molecular and functional alterations that may sustain specific symptoms. Such information may allow the identification of specific subgroups of patients with different sensitivity and responsiveness to pharmacological intervention. This possibility is in line with the NIMH Research Domain Criteria that represents a new way to classify mental disorders based on behavioral dimensions and neurobiological measures (Woody and Gibb, 2015). Indeed, the NIMH Research Domain Criteria initiative has the explicit goal of linking classification of psychopathology to the advances in genetics and neuroimaging across traditional diagnostic boundaries (Cuthbert and Insel, 2013). This may be further integrated with environmental and contextual influences to take into account the development or progression of a specific disease (Woody and Gibb, 2015). We postulate that different etiological mechanisms and their combination can lead to selected dysfunction that may be more or less sensitive to pharmacological intervention with classic drugs.
Despite strong evidence of heritability (Hyde et al., 2016), as described above, the efforts to identify the genetic underpinning of MDD have been largely unsuccessful, possibly due to disease heterogeneity and the absence of a biologic gold-standard for diagnosis. However, it is feasible to postulate that the major reason for the unsuccessfulness of genetic studies is represented by the strong contribution of environmental factors that interact and modulate genetic susceptibility (Klengel and Binder, 2013). With this respect, stress, in all its multiple forms, represents a key element for the risk to develop depression. There is, however, another important element that modulates the classic gene X environment interaction, which is time. Indeed, we know that genetic susceptibility factors may reshape developmental trajectories leading to altered ability to cope or respond to the environment within specific time frames (Babenko et al., 2015). Moreover, it is well known that exposure to stressful adverse events may have a different impact on brain function according to the timing of exposure, which can be clearly related to the maturation of different neuronal circuits that participate in stress response and that may serve to develop proper coping strategies.
It may be inferred that the functional outcome of stress exposure will not only depend upon the genetic background, but on the timing of stress exposure resulting in different effects in terms of circuits and brain structures affected, as well as molecular mechanisms that may sustain long-lasting modifications of brain function.
On a speculative basis, it may be possible to delineate specific vulnerability signatures as a result of stress exposure, based on the type and timing of the stressful experience, the genetic settings as well as other factors, including sex differences. These combinations may be associated with specific features (symptoms or dimension) of the disorder and may show a preferential responsiveness to a given therapeutic strategy.
A further complexity may be due to the fact that such “etiological” mechanisms may not necessarily produce a pathologic phenotype, but could represent a predisposing condition to develop MDD if reexposed to challenging precipitating traumatic experiences (Arloth et al., 2015; Pena et al., 2017). With this respect, epigenetic changes (see below) represent one important mechanism, through which a given system or cell population may keep the “memory” of early adversities, setting the stage for the onset of the disease later in life. The characterization of these epigenetic mechanisms is opening new possibilities to associate a given genotype with a specific pathologic phenotype.
A number of studies have been conducted all over the world with the purpose of reproducing selected etiological mechanisms to identify downstream changes relevant for specific behavioral and functional alterations associated with MDD. These studies have clearly defined some core mechanisms that are associated with MDD and that may contribute to different aspects of disease vulnerability and manifestation. The focus of these studies has now shifted from “classic” concepts, closely related to monoamine alterations, to more complex mechanisms, including reduced neuronal plasticity, synaptic dysfunction, enhanced inflammation, and altered hypothalamic-pituitary-adrenals (HPA) axis function and responsiveness. Although some of these aspects will be described in more detail in the next paragraphs, it is important to consider that there may be a close link between these alterations pointing to a cascade of events that may have a different origin based, for example, on etiological mechanisms, and will then propagate to affect global brain functioning. We believe that the ability to interfere with such network of changes represents a critical element for therapeutic response to pharmacological intervention, which should not only be able to restore normal function, but, more importantly, must promote resilience and reduce the long-term susceptibility to recurrent depressive episodes (relapse prevention).
In summary, the characterization of the etiologic mechanisms for MDD represents a key strategy to associate a given phenotype (for example, specific clinical features or specific symptoms) with a specific set of molecular and functional alterations. Although currently available drugs are known to interfere with synaptic mechanisms by blocking monoamine transporters or acting on different monoamine receptors, these agents may differ extensively with respect to their ability in modulating downstream mechanisms, which may be differentially affected in depressed subjects. This possibility can be particularly pertinent for treatment resistance that may be due to the inability of a given compound to effectively modulate one or more of these systems.
B. The Glutamatergic System
Depression has been historically defined a “monoaminergic disorder” according to the idea that the disease is due and sustained by a deficit of different monoamines, primarily noradrenaline and serotonin (Nestler et al., 2002). However, this view, while maintaining its importance, has been challenged by evidence and a number of questions that have brought about a revision of such a “hypothesis” that is now framed within more general concepts related to miswiring, deficits of neuronal plasticity, and cell-cell communication (Berton and Nestler, 2006). In this respect, evidence has pointed to glutamate as a crucial player in the etiology of depression and in treatment response (Mathews et al., 2012). Glutamate is the main excitatory neurotransmitter in the mammalian brain, and it plays a central role in memory processes and synaptic plasticity (Machado-Vieira et al., 2012) as well as in emotion regulation. Glutamate can affect neuronal activity and function in two different ways: 1) rapid actions, exerted via ligand-gated ion channels namely NMDA (N-methyl-d-aspartate), alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainate receptors and 2) slow modulatory actions, exerted via the eight G-protein coupled metabotropic receptors (mGluRs). When released in the synaptic cleft, its concentration is tightly regulated by glutamate transporters localized in both neurons and astrocytes (Danbolt, 2001). As mentioned above, preclinical and clinical evidence have associated glutamate with major depression. As an example, it has been hypothesized that a dysregulated excitatory (glutamatergic) neurotransmission in the ventral anterior (subgenual) cingulate cortex may result into a functional hypoactivity of the ascending monoamine systems (serotoninergic, noradrenergic, and dopaminergic) that contribute to the onset of affective and cognitive symptoms in MDD (Artigas, 2015). In particular, increased concentrations of glutamate and glutamine have been found in occipital cortex from MDD patients, whereas a decrease of the same neurotransmitters was detected in prefrontal regions (Hasler et al., 2007). An abnormal glutamate/glutamine/GABA cycling has been demonstrated in TRD patients (Price et al., 2009), where elevated Glx:GABA ratios were observed in occipital cortex, thus suggesting an impairment of glutamate-glutamine neuronal-glial cycling resulting in excessive buildup of extracellular glutamate and decreased glutamate release, leading to a reduction in cortical GABA (Sanacora et al., 2003; Price et al., 2009). A number of significant changes in the expression of glutamate receptors have been demonstrated in human postmortem studies (Gibbons et al., 2012). Altered glutamatergic function may contribute to reduced neuroplasticity and structural alterations that have been reported in the brains of subjects with depression as well as in animals exposed to chronic stress, which recapitulates a key etiological mechanism for major depression (Kim and Na, 2016).
Different studies in the last 20 years have demonstrated that a chronic treatment with “monoaminergic” antidepressants, such as tricyclics, strongly affects NMDA binding profiles and receptor function (Mjellem et al., 1993; Nowak et al., 1998). Moreover, the antidepressant-like effects of the SSRI escitalopram are prevented by NMDA receptor activation (Zomkowski et al., 2010). Coadministration of TCAs, such as imipramine, with amantadine (a noncompetitive NMDA receptor antagonist) reduced immobility time in the forced swim test (FST) in rats to a much greater extent than either treatment alone (Rogoz et al., 2002). Similar synergistic interactions have been observed between SSRIs and SNRIs and different uncompetitive NMDA receptor antagonists (Ates-Alagoz and Adejare, 2013), thus suggesting that NMDA receptor antagonists can enhance the preclinical efficacy of currently used monoaminergic antidepressants. On these bases, the glutamatergic system currently represents a field of major interest for drug discovery in TRD and for the development of new and more efficacious antidepressant drugs. Indeed, as described below (see Glutamatergic System), recent groundbreaking clinical studies have demonstrated that targeting the glutamatergic transmission is an effective and useful approach for treatment-resistant depression (Berman et al., 2000; Zarate et al., 2006; Sanacora and Schatzberg, 2015).
It has been hypothesized that by blocking NMDA receptors on GABAergic interneurons, ketamine causes a rapid, but transient, increase in extracellular glutamate in the prefrontal cortex (Duman, 2013). This process seems to involve spontaneous glutamate release, rather than typical evoked synaptic glutamate release. The consequent activation of AMPA receptors causes depolarization of postsynaptic neurons, leading to L-type voltage-gated calcium channels activation. As a consequence, BDNF released from vesicles activates the mammalian target of rapamycin, a signaling system that plays a central role in synaptic plasticity (Duman, 2013; Machado-Vieira et al., 2017) and that is known to be impaired in the prefrontal cortex of patients with TRD (Jernigan et al., 2011). Furthermore, blockade of NMDA receptors by ketamine can result in the inhibition of eukaryotic elongation factor 2 (eEF2) kinase, dephosphorylation of eEF2, which may lead to a desuppression of BDNF translation (Autry et al., 2011).
The role of mTOR as a target of ketamine has been observed also in humans, where ketamine is able to rescue mTOR signaling, as assessed in peripheral cells after acute administration of ketamine in MDD patients (Denk et al., 2011).
Recently, Zanos et al. (2016) claimed that the production of a distinct metabolite of ketamine [(2R,6R)-hydroxynorketamine (HNK)] is necessary and sufficient to produce the antidepressant effects of ketamine in mice through an NMDAR-independent pathway via sustained activation of AMPAR. However Suzuki et al. (2017) recently demonstrated that (2R,6R)-HNK inhibits synaptic NMDARs, triggering the same signaling pathways activated by ketamine (i.e., decreased eEF2 phosphorylation) and proposing that the sustained inhibition of NMDARs by (2R,6R)-HNK can explain the long-lasting antidepressant effects of ketamine in TRD, despite its relatively short half-life, which cannot be observed with other NMDAR antagonists. Nevertheless it should be noted that the potency of HNK for the NMDAR is much lower than ketamine and a functional inhibition of the NMDAR is observed only at concentrations that are higher than those reported as pharmacologically relevant for its antidepressant action in mice (Zanos et al., 2017). Moreover, ketamine and HNKs show comparable pharmacokinetic profiles, therefore ruling out the contribution of HNK for the protracted antidepressant activity of ketamine (Zanos et al., 2017). Future studies are needed to disentangle the complexity of the pharmacological and clinical activities of ketamine and its metabolites.
Ketamine has been promoting drug discovery processes in depression and TRD with the aim of developing ketamine-like molecules with the same clinical efficacy, but without psychotomimetic effects and an improved safety profile (Iadarola et al., 2015; Caraci et al., 2017b) (see Glutamatergic System).
C. Synaptic Plasticity and Neurotrophic Mechanisms
Antidepressant drugs have classically been associated with the ability to increase synaptic monoamines, mainly serotonin and noradrenaline, to restore the diminished levels that may contribute to specific symptoms of depression. However, as mentioned above, it is now well accepted that synaptic dysfunction may represent a core element for the pathologic phenotype (Calabrese et al., 2016). Indeed, structural alterations, including neuronal atrophy, reduced number of spines, and dendritic arborization have been consistently reported in the prefrontal cortex and hippocampus of depressed patients (Rajkowska et al., 1999; McEwen and Lasley, 2003; Stockmeier et al., 2004; Duman and Aghajanian, 2012). A link between structural alterations in depressed subjects and exposure to stress or traumatic experiences early in life has also been demonstrated (Duman and Duman, 2015), suggesting that such adverse events may lead to a protracted impairment of synaptic function and in cell-cell communication, thus leading to functional alterations of specific neuronal circuits (Negron-Oyarzo et al., 2016; Nephew et al., 2017). Interestingly, an inverse relationship between the total hippocampal volume and the duration of untreated depression has been demonstrated (Sheline et al., 1999). These alterations may result from toxic mechanisms, related to excessive glutamate, as well as to an overactivity of the glucocorticoid system (HPA dysfunction) (McEwen, 2001). Moreover, the extent of such structural alterations may be associated with and are paralleled by alterations of genes encoding proteins involved in synapse formation and function (Kang et al., 2012).
There are two important implications of these structural changes in relation to antidepressant response. On one end, regional brain volumes might be associated with rate and extent of clinical response to antidepressant medication (MacQueen et al., 2008). On the other end, smaller hippocampal volumes may predict lower response/remission rates in patients with depression treated with antidepressant drugs (Colle et al., 2016). A recent study has shown that patients with current depression had bilaterally reduced gray matter in the hippocampus compared with healthy control or untreated patients in stable remission. An increase in gray matter was observed in the hippocampus following treatment with citalopram in currently depressed patients. Moreover gray matter reduction in the hippocampus appears specific to the depressed state and can be considered a potential biomarker for a depressive episode (Arnone et al., 2013).
Although more information is needed to better define the relationship between such mechanisms, depressive state, and treatment responsiveness, these data suggest that the enhancement of both synaptic plasticity and synaptic strength may represent a critical aspect for the therapeutic effect of antidepressant drugs (Duman and Aghajanian, 2012). Although different mechanisms may contribute to structural and synaptic alterations of depressed patients, one class of proteins that play an important role in the maintenance of synaptic structure and function are neurotrophic factors, in particular the neurotrophin BDNF (Kuipers and Bramham, 2006; Greenberg et al., 2009; Park and Poo, 2013).
The role of BDNF in the pathophysiology of depression and in the mechanism of action of antidepressant drugs is well known (Altar, 1999; Duman, 2002; Duman and Monteggia, 2006; Bjorkholm and Monteggia, 2016; Calabrese et al., 2016; Cattaneo et al., 2016a). Several lines of evidence have shown that the expression of the neurotrophin is reduced in selected brain structures, as well as at the peripheral level, of subjects with depression (Shimizu et al., 2003; Karege et al., 2005; Thompson Ray et al., 2011; Reinhart et al., 2015). Similarly, reduced BDNF levels are found in chronically stressed rats and also in experimental models that show depressive-like behavioral alterations (Urani et al., 2005; Duman and Monteggia, 2006; Calabrese et al., 2009, 2015; Molteni et al., 2010a,b; Chourbaji et al., 2011; Luoni et al., 2014a,b; Berry et al., 2015). Interestingly, chronic treatment with different antidepressants can promote the expression of the neurotrophin and normalize its alterations in animal models (Calabrese et al., 2007, 2010; Molteni et al., 2009; Duric and Duman, 2013; Luoni et al., 2014b; Castren and Kojima, 2017). More importantly antidepressant treatments can normalize the alterations of peripheral BDNF levels observed in MDD patients (Cattaneo et al., 2010), an effect that correlates with symptomatology improvement (Sen et al., 2008; Cattaneo et al., 2013), suggesting a potential relationship between drug response and the ability to modulate neurotrophic mechanisms (Molendijk et al., 2011). Moreover, since it has been reported that pretreatment BDNF levels are directly correlated with antidepressant responses, the neurotrophin expression may also predict the response to antidepressants (Wolkowitz et al., 2011; Cattaneo et al., 2013).
Clinical studies have indeed reported alterations of BDNF system in TRD patients. For example, reduced BDNF gene expression was found in the blood of TRD patients (Hong et al., 2014). However, the effect of therapeutic intervention in TRD is not necessarily associated with the modulation of peripheral BDNF levels. Indeed, although it has been demonstrated that TRD patients who respond to ketamine show an elevation of serum BDNF (Duncan et al., 2013; Haile et al., 2014), other authors found that the effects of ketamine or ECT in TRD were not associated with changes in blood neurotrophin levels (Allen et al., 2015; Rapinesi et al., 2015).
All in all, these studies indicate that structural, synaptic alterations and changes in neuroplastic players, such as BDNF, play a crucial role in the pathophysiology of depression and may be highly relevant for TRD. Given that both synaptic dysfunction and deficits in BDNF system reflect compromised neuronal plasticity and consequently increased vulnerability to environmental risk factors, we might speculate that one potential therapeutic strategy for TRD should be to stimulate BDNF expression as well as synaptic mechanisms to promote resilience and counteract core alterations found in subjects with depression. In this respect, it must be kept in mind that the alterations of BDNF associated with depression are strictly dependent on the brain region considered. Indeed, although BDNF is downregulated at cortical and hippocampal level, opposite changes can be found in the mesolimbic system (Berton et al., 2006; Krishnan and Nestler, 2008; Wook Koo et al., 2016), suggesting that effective therapeutic intervention should be able to modulate the neurotrophin expression and function with anatomic selectivity. Recently, both tropomyosin receptor kinase B (TrkB) agonists and antagonists have shown a relevant antidepressant activity in animal models by rescuing BDNF signaling in the hippocampus and in the prefrontal cortex (TrkB agonists) or reducing its activity in the nucleus accumbens (TrkB antagonists) (Zhang et al., 2016). An alternative approach in this field might be the development of TrkB partial agonists able to rescue BDNF signaling in prefrontal cortex and hippocampus and to reduce the activity of this pathway in the nucleus accumbens, which may indeed lead to a significant antidepressant efficacy.
D. Hypothalamic-Pituitary-Adrenal Axis Dysfunction
Epidemiologic evidence supports a role for stress as a risk factor for depression (Cowen, 2010). Indeed, chronic exposure to stress can promote the development of major depression (Czeh and Lucassen, 2007; Pariante, 2017). Stressful events are known to activate the HPA axis, which finally stimulates the release of glucocorticoids from the adrenal cortex (de Kloet et al., 2005). Glucocorticoids are steroid hormones that readily cross the blood-brain barrier and bind to low-affinity glucocorticoid receptors (GR) and high-affinity mineralocorticoid receptors, exerting a physiologic negative feedback on HPA axis (de Kloet et al., 2005). It has been hypothesized that both high cortisol levels and the activation of immune system in MDD can be explained with the development of the so called “glucocorticoid receptor resistance” found in depressed patients. According to this idea, GR dysfunction may lead to an impaired function of the negative feedback, resulting in HPA axis hyperactivity and elevated cortisol levels (de Kloet et al., 2005; Maes et al., 2016; Pariante, 2017). GR resistance is particularly evident in patients with TRD (Bauer et al., 2003). Indeed, severe TRD is associated with an imbalance in the normal physiology of the HPA axis, with glucocorticoid receptor resistance combined with an increased mineralocorticoid receptor sensitivity (Juruena et al., 2013).
Different hypotheses have been made to explain the molecular links between HPA axis dysfunction, hypercortisolemia, and TRD pathogenesis. Cortisol increases the activity of tryptophan 2,3 dioxygenase (TDO), with an ensuing reduction in available tryptophan and a significant decrease of serotonin levels. Hypercortisolemia can also reduce neurogenesis in the hippocampal dentate gyrus (Krishnan and Nestler, 2008) and may lead to structural abnormalities, such as retraction of hippocampal apical dendrites (McLaughlin et al., 2007). Moreover, rats or mice exposed to social defeat show an activated response of the HPA axis (Keeney et al., 2006; Razzoli et al., 2009), which can be reversed by antidepressant treatments (Becker et al., 2008). Interestingly mice that are susceptible to social defeat stress show hypercortisolemia as well as significantly less GR protein expression and nuclear translocation in the hippocampus compared with resilient mice (Han et al., 2017). Accordingly, animals exposed to chronic mild stress (CMS) show increased expression of the chaperone protein FKBP5 as well as enhanced cytoplasmic levels of GR in hippocampus and prefrontal cortex (Guidotti et al., 2013).
Glucocorticoids can also contribute to MDD pathogenesis by reducing synaptic plasticity and increasing the vulnerability to neuronal death in the hippocampus (Yu et al., 2008). In particular glucocorticoids induce the expression of Dickkopf-1 (Dkk-1), an inhibitor of the canonical Wnt pathway, in hippocampal neurons and this may contribute to stress-induced structural alterations within the hippocampus (Matrisciano et al., 2011), which have also been consistently observed in MDD patients with a history of treatment resistance (Abdallah et al., 2015).
On these bases, preventing hypercortisolemia has been recently considered as a novel pharmacological strategy for MDD and, in particular, for TRD (Henter et al., 2017). Mifepristone, a glucocorticoid receptor antagonist, seems to be efficacious in the treatment of psychotic depression, a subtype of depression characterized by hypercortisolemia (Blasey et al., 2011), with a rapid improvement of depressive and psychotic symptoms (Belanoff et al., 2001, 2002). Moreover, one 6-week pilot study found that 600 mg/day of mifepristone improved depressive symptoms and cognition in patients with treatment-resistant bipolar depression, and this clinically relevant improvement was inversely associated with basal cortisol levels (Young et al., 2004). An alternative pharmacological approach to target hypercortisolemia is the use of metyrapone, an inhibitor of 11βhydroxylase, the enzyme that catalyzes the conversion of 11-deoxycortisol to cortisol (Sigalas et al., 2012). Preliminary clinical studies have shown the clinical efficacy of metyrapone in TRD. In particular a controlled, randomized, double-blind trial found that adjunctive metyrapone therapy to fluvoxamine was superior to placebo and accelerated the onset of antidepressant action (Jahn et al., 2004). A multicenter, placebo-controlled, randomized, phase 3 trial was conducted to evaluate the effects of metyrapone augmentation in patients with TRD, although the results of this study are not yet available (NCT01375920).
All in all, the ability to modulate HPA axis dysfunction as well as glucocorticoid receptor resistance represents an important aspect for the therapeutic action of antidepressant drugs and, together with other mechanisms, may be a key issue for treatment resistance (Pariante, 2017).
E. Immune System Dysregulation and Neuroinflammation
Over the last two decades several studies have demonstrated that inflammation and dysfunction of the immune system play a key role in the pathophysiology of major depression and may therefore contribute to treatment resistance (Caraci et al., 2010; Capuron and Miller, 2011; Maes et al., 2016; Remus and Dantzer, 2016; Bhattacharya and Drevets, 2017; Pariante, 2017). An altered activation of the immune system and the ensuing state of “peripheral and central inflammation” seem to be strictly correlated to HPA axis dysfunction observed in depressed patients (Remus and Dantzer, 2016; Pariante, 2017).
Depressed patients show higher levels of proinflammatory cytokines, such as interleukin-1 (IL-1), interleukin-2 (IL-2), interleukin-6 (IL-6), interleukin-8 (IL-8), interleukin-12 (IL-12), interferon-γ and tumor necrosis factor-α (TNF-α) (Dowlati et al., 2010; Capuron and Miller, 2011), as well as increased acute phase proteins, chemokines, and cellular adhesion molecules (Maes et al., 2016). In particular, recent meta-analyses found the most relevant longitudinal association between two inflammatory markers, namely C-reactive protein and IL-6, and depressive disorders, suggesting that indeed inflammation may contribute to the development of the disease (Valkanova et al., 2013; Smith et al., 2018). Moreover, elevated markers of microglial activation (measured by translocator protein binding in vivo with positron emission tomography) have been found in MDD patients (Setiawan et al., 2015). Interestingly, a positive correlation between the severity of the symptoms of depression and the increase in the inflammatory status has been demonstrated (Maes, 1999; Maes et al., 2016).
A number of preclinical studies have provided support for the role of immune/inflammatory dysfunction in depression (Remus and Dantzer, 2016; Leonard, 2018). As an example, lipopolysaccharide administration in rodents increases peripheral (and central) cytokines, such as IL-1 and TNF-α, leading to sickness behavior (reduced motor activity and a decrease in food and water intake) followed, 24 hours later, by depressive-like behavioral alterations (Ge et al., 2015; Remus and Dantzer, 2016; Sulakhiya et al., 2016). Accordingly, neuroinflammation and altered cytokine expression have been demonstrated in animal models of depression. Indeed, exposure to CMS leads to an increase of proinflammatory cytokines (IL-1β, TNF-α) and a decrease of anti-inflammatory cytokines (IL-10, IL-4, and TGF-β1) in different brain regions, as well as to enhanced markers of microglia activation (You et al., 2011; Hinwood et al., 2013; Rossetti et al., 2016). Interestingly the increase in inflammatory markers in dorsal hippocampus was inversely related to sucrose consumption, thus suggesting that the anhedonic-like phenotype in CMS rats may be linked to neuroinflammation (Rossetti et al., 2016).
Proinflammatory cytokines can interfere with many of the pathophysiological mechanisms relevant of depression (Maes et al., 2016). For example, interferon-γ and TNF-α induce the expression of the tryptophan-metabolizing enzyme indoleamine 2,3-dioxygenase (Campbell et al., 2014), the rate limiting step of the kynurenine pathway, and TDO (Leonard, 2007; Remus and Dantzer, 2016). Although it was proposed that such effects may lead to decreased serotonin levels (Remus and Dantzer, 2016), it was demonstrated that the reduction in peripheral blood TRP has no effects on CSF TRP concentrations (Raison et al., 2010). However, activation of indoleamine 2,3-dioxygenase and TDO leads to an increased production of the neurotoxins 3-hydroxykynurenine and quinolinic acid, which can contribute to the pathophysiology of MDD by activating the NMDA receptor (Myint and Kim, 2003; Leonard, 2007; Bay-Richter et al., 2015; Remus and Dantzer, 2016). Inflammatory cytokines strongly influence glutamate metabolism in astrocytes and microglia, and markers of inflammation correlate with dysfunction of glutamatergic system in the dorsal anterior cingulate cortex and are associated with anhedonia and psychomotor retardation. These studies suggest a strong neurobiological link between inflammation-induced depression and the dysfunction of glutamatergic system in TRD (Haroon and Miller, 2017).
It has been hypothesized that immune system activation and neuroinflammation can also lead to a deficit in dopaminergic mesolimbic pathway combined with a dysfunction in prefrontal glutamatergic system finally leading to the onset of anhedonia, loss of motivation, fatigue, psychomotor retardation, and cognitive deficits (Eisenberger et al., 2010; Leggio et al., 2013; Pan et al., 2017).
Neuroinflammation is also associated with a reduced response to the treatment with SSRIs (Kulmatycki and Jamali, 2006; Maes et al., 2016), and it may also account for the complex interaction of depression and cognitive deficits in older adults (Ownby, 2010).
Antidepressant drugs exert immune-regulatory effects, reducing the production of “peripheral” proinflammatory cytokines and stimulating the synthesis of anti-inflammatory cytokines such as IL-10 and TGF-β1 in patients with depression (Caraci et al., 2010; Maes et al., 2016). Recent studies suggest that antidepressant drugs may also exert direct anti-inflammatory effects on microglia (Tynan et al., 2012), which is known to be overactivated in MDD patients (Setiawan et al., 2015). Different antidepressants, including SSRIs, SNRIs, and the melatonergic drug agomelatine, are able to inhibit the production of different proinflammatory cytokines in vitro and in vivo (Tynan et al., 2012; Molteni et al., 2013; Ohgi et al., 2013). Moreover imipramine, agomelatine, and the novel antipsychotic drug lurasidone possess anti-inflammatory properties in the CMS model (Rossetti et al., 2016). Interestingly different preclinical and clinical studies have shown that ketamine is endowed with anti-inflammatory activity (De Kock et al., 2013) and reverses inflammation-induced depression in the lipopolysaccharide model by decreasing brain levels of inflammatory cytokines (Yang et al., 2013) or by blocking the effects of quinolinic acid, a downstream product of the kynurenine pathway, on NMDA receptors (Walker et al., 2013b). Furthermore, recent in vitro studies have shown that fluoxetine and venlafaxine increase the release of TGF-β1, an anti-inflammatory cytokine that is reduced in nonresponder MDD patients (Vollmar et al., 2008; Caraci et al., 2016).
However, although some markers of immune activation have been validated in TRD patients, some caution should be used when translating preclinical results with antidepressant drugs to the clinical setting. Nevertheless, immune parameters may predict treatment response in MDD patients. Indeed, nonresponder MDD patients show significant elevations in a variety of proinflammatory immunologic markers (Carvalho et al., 2013; Cattaneo et al., 2013; Kiraly et al., 2017), such as IL-1, macrophage inhibiting factor (MIF), and TNF-α (Cattaneo et al., 2013; Strawbridge et al., 2015). One of the most relevant studies in this field was conducted by Cattaneo et al. (2013) based on a large European Union-funded study, the Genome-based Therapeutic Drugs for Depression study (GENDEP). The authors provide evidence that MDD patients who do not respond to first- or second-generation antidepressants had higher baseline mRNA levels of IL-1, MIF, and TNF-α (Cattaneo et al., 2013), the levels of these three pro-inflammatory cytokines being able to predict about 50% of the variance in antidepressant response. The same group replicated these results in a second cohort of patients, showing that IL-1 and MIF mRNA levels can accurately predict antidepressant response in MDD patients with positive predictive values and specificity for nonresponders of 100% (Cattaneo et al., 2016b).
The link between inflammation and treatment resistance appears to be highly relevant for late-life depression (Alexopoulos and Morimoto, 2011). Geriatric depression and in particular “vascular depression” represents a specific clinical subtype of depression characterized by a low rate of response to “monominergic” antidepressants (Alexopoulos and Morimoto, 2011) and it is characterized by high levels of proinflammatory cytokines, such as IL-1β, IL-8, and IL-6 (Diniz et al., 2010; Taylor et al., 2013). Reduced levels of anti-inflammatory cytokines, such as IL-4, IL-10, and TGF-β1, have been found in the plasma of patients with depression (Maes, 1999; Myint et al., 2005; Musil et al., 2011; Rush et al., 2016) and can significantly contribute to treatment resistance in MDD (Musil et al., 2011). Interestingly responder and remitter patients with MDD had higher initial TGF-β1 levels at baseline compared with patients who did not respond to treatment (Musil et al., 2011). Patients with melancholic depression with a recent history of treatment resistance had significantly higher levels of the proinflammatory cytokine IL-6, and lower levels of the regulatory cytokine TGF-β1 than healthy controls (Rush et al., 2016). Deficit of TGF-β1 signaling is a common pathophysiological event both in depression and cognitive decline (Caraci et al., 2012), and the presence of cognitive symptoms in patients with depression might predict a low rate of response to current antidepressant drugs (Silverstein and Patel, 2011). We recently identified a key role for TGF-β1 in recognition memory formation demonstrating that this neurotrophic factor is essential for the transition from early to late long-term potentiation (Caraci et al., 2015). We hypothesize that a deficit of TGF-β1 may contribute to treatment resistance in elderly patients with MDD by increasing Aβ accumulation and the development of the so-called “amyloid-related depression,” a recently identified clinical phenotype in which the response to “monoaminergic” antidepressants is low (Li et al., 2017b).
F. Epigenetic Mechanisms
Epigenetic literally means “above genetics” and refers to changes in DNA structure without alterations of nucleotide sequence. The major epigenetic mechanisms are represented by DNA methylation, posttranscriptional histone marking, and by the control of mRNA processing and translation through noncoding RNAs (miRNAs) (Tsankova et al., 2007; Luoni and Riva, 2016).
Epigenetic processes are important mechanisms for experience-dependent changes in brain function and responsiveness and are considered key players for “long-term” maintenance of the effects produced by exposure to adverse events, particularly during early life stages. Epigenetic alterations may therefore represent a permanent scar of such events, thus contributing to the increased vulnerability for psychiatric disorders, such as major depression. This issue has been the topic of different excellent reviews that addressed several mechanisms linking environmental factors to mental illness through the complex interplay of epigenetic modifications (Tsankova et al., 2007; Graff and Tsai, 2013; Nestler, 2014; Han and Nestler, 2017). Moreover, although negative events occurring after brain maturation may exert limited and transient effects, insults experienced in a key period of development could reprogram the epigenome, and, being incorporated in the germ cells, the consequences of these events can also be transmitted to the progeny (Champagne, 2008; Bohacek et al., 2013; Babenko et al., 2015; Bale, 2015).
Epigenetic mechanisms appear to play an important role in major depression. On one end, the expression of different epigenetic regulators is altered in subjects with depression. As an example, histone deacetylases (HDACs) levels are significantly increased in peripheral blood cells (Iga et al., 2007; Hobara et al., 2010) and in the nucleus accumbens (Covington et al., 2009) of depressed patients, as well as in mice exposed to chronic stress (Renthal et al., 2007; Covington et al., 2009).
Furthermore, the dysregulation of important players in MDD appears to be sustained by epigenetic mechanisms. Indeed, in human brain the promoter of the GR gene is hypermethylated in men abused during childhood (McGowan et al., 2009) and in infants from mothers who self-reported depression during pregnancy (Braithwaite et al., 2015), as well as in a rat model of reduced maternal care (Weaver et al., 2004). Moreover, epigenetic mechanisms underlie the reduction of BDNF expression in an animal model of chronic stress (Tsankova et al., 2006). Similarly, a hypermethylation of BDNF promoter IV was observed in the Wernicke’s area of suicide completers (Keller et al., 2010). Finally, changes in DNA methylation and chromatin modification were reported in promoter regions of genes involved in protein synthesis (McGowan et al., 2008), polyamines (Fiori et al., 2012), and in neurotransmission (Abdolmaleky et al., 2006; Poulter et al., 2008).
While the investigation of selected genes does provide useful information with respect to the specific contribution of epigenetic mechanisms for functional alterations in MDD, genome-wide studies (epigenome-wide association studies) provide novel and important information on the epigenetic signatures that may be associated with a depressive phenotype (Sabunciyan et al., 2012; Labonte et al., 2013; Dempster et al., 2014).
It is therefore feasible to hypothesize that such mechanisms, which are not directly targeted by classic pharmacological intervention, may contribute to drug resistance. Accordingly, clinical evidence reports that HDAC2 and HDAC5 expression is upregulated in leukocytes of patients during the depressive phase but not in remission (Hobara et al., 2010) and that the DNA methylation profile of BDNF (exon I) allows for distinguishing between depressed and control patients (Fuchikami et al., 2011). On these bases, it is feasible to hypothesize that patients characterized by such abnormalities may require pharmacological interventions able to “act” at this level to show significant clinical improvement.
Epigenetic make up may also predict the response to antidepressant therapy. In particular, it has been reported that patients showing hypomethylation of the promoter of BDNF exon IV at plasma level are unlike to benefit from antidepressant therapy (Tadic et al., 2014), whereas responders to treatment showed a decrease of trimethylation of the histone 3 (Lopez et al., 2013). Accordingly, remitters/responders after ECT treatment showed a significantly lower methylation of exon I at the peripheral level compared with nonremitter/nonresponder subjects (Kleimann et al., 2015).
The relevance of these mechanisms in TRD is also suggested by the observation that drugs that may be effective in TRD act at epigenetic level (Tsankova et al., 2006; Vialou et al., 2013; Menke and Binder, 2014). For example, clinical doses of the valproic acid (VPA), a mood stabilizer used for the treatment of bipolar disorder, inhibit class I HDAC (Gottlicher et al., 2001; Kramer et al., 2003). In a small cohort of patients with severe TRD, antidepressant augmentation with VPA provides substantial clinical improvement and maintenance (Ghabrash et al., 2016). In rodents, chronic treatment with VPA alone significantly increases the gene expression of HDAC5, while combination with the antipsychotic lurasidone leads to a significant decrease of HDAC1 and two mRNA levels (Calabrese et al., 2013).
On another note, chronic treatment with suberoylanilide hydroxamic acid (also known as vorinostat), a class I and II HDAC inhibitor, partially rescues the molecular alterations and the depressive-like behavior of CRCT1−/− (CREB-regulated transcription coactivator 1) mice, whereas conventional antidepressants, such as desipramine, do not show any effect (Meylan et al., 2016). Accordingly, infusion in the nucleus accumbens of suberoylanilide hydroxamic acid or another HDAC inhibitors (MS-275) rescues the depressive phenotype and the molecular alteration observed in the social defeat stress paradigm (Covington et al., 2009).
Moreover, HDAC inhibitors possess antidepressant properties in rodents as well as complementary procognitive actions also associated with neurodegenerative disease (Graff and Mansuy, 2008; Covington et al., 2009; Day and Sweatt, 2011; Lin et al., 2012; Yamawaki et al., 2012; Graff and Tsai, 2013; Noh and Seo, 2014). This is particularly important in depression, because cognitive dysfunctions have long been recognized as an intrinsic characteristic of major depressive disorder (Conradi et al., 2011; Millan et al., 2012) and represent the most common residual symptoms in partial responders (Gotlib and Joormann, 2010).
Interestingly, natural products with the ability to interfere with epigenetic mechanisms, such as folic acid and S-adenosylmethionin (fundamental for DNA formation and methylation), are lacking in depression and may be useful as adjunctive antidepressant therapy (Gilbody et al., 2007; Gomez-Pinilla, 2008; Sharma et al., 2017). Accordingly, L-acetylcarnitine, probably influencing the acetylation of H3K27 (Nasca et al., 2013), is able to normalize the phenotype of endogenously depressed Flinders sensitive line rats (Bigio et al., 2016).
However, it is worth mentioning that a major problem for any pharmacological intervention acting on epigenetic mechanisms is the lack of selectivity, and this may require additional studies before becoming an effective therapeutic strategy.
Although most of the results obtained at the peripheral level are in line with the changes observed in post mortem samples as well as in animal models, it remains to be established to what extent peripheral tissue/blood measures may be a proxy of brain changes in living humans.
On these bases, even if additional studies are needed, epigenetic mechanisms may represent an important target to develop new pharmacological treatments with potentially higher and more persistent efficacy in patients with TRD, although the lack of selectivity remains a major problem to be considered with such approach.
VI. Current Therapeutic Strategies for Treatment-resistant Depression
As nonresponse to an initial antidepressant monotherapy trial emerges frequently in the pharmacotherapy of MDD (Trivedi et al., 2006; Souery et al., 2007), the question of the next therapeutic measures within an algorithm to achieve sufficient treatment response arises (Dold and Kasper, 2017). In patients nonresponding to the initial antidepressant treatment, the assessment of eventual pseudoresistance including therapy drug monitoring is strongly suggested prior to further psychopharmacotherapeutic optimization (Hiemke et al., 2011; Dold and Kasper, 2017). Strategies that are widely applied in clinical routine care usually imply 1) increasing the dose of the currently dispensed antidepressant compound (dose escalation, high-dose pharmacotherapy), 2) switching to another new antidepressant, 3) combination of two or more antidepressants, 4) augmentation of the ongoing antidepressant trial with compounds of other substance classes [e.g., second-generation antipsychotics (SGAs), lithium, or thyroid hormones (T3/T4)], and 5) utilization of innovative psychopharmacotherapy, such as ketamine, as well as (nonpharmacological) biologic treatment options [e.g., therapeutic sleep deprivation, light therapy, transcranial magnetic stimulation (or ECT)] (Thase et al., 2016; Bauer et al., 2017; Dold and Kasper, 2017). In terms of conventional psychopharmacotherapy, augmentation with SGAs and lithium as well as combination treatment with two antidepressant compounds with different receptor-binding properties (e.g., SSRIs and mirtazapine) have been shown to be the most effective treatment strategies in TRD (Mojtabai and Olfson, 2010; Seemuller et al., 2010; Dold et al., 2016; Thase et al., 2016) and hence can be regarded as evidence-based treatment of TRD according to the current international treatment guidelines (Bauer et al., 2017).
A. Dose Increase
With respect to dose increase strategies, a recent meta-analysis by Dold et al. (2017) found no evidence that nonresponders to an initial antidepressant trial benefit from a dose escalation of the same antidepressant drug. However, it should be taken into account that most of the included individual trials in this meta-analysis investigated high-dose treatment with SSRIs. This observation corresponds with a recent prospective study reporting that increasing escitalopram above the therapeutic range of serum escitalopram concentration seems to be useless with respect to improvement of antidepressant efficacy (Florio et al., 2017). It is noteworthy that these findings are in line with the current international treatment guidelines, suggesting that dose escalation cannot be currently regarded as a general evidence-based strategy for TRD (Bauer et al., 2017), although evidence based on open-label trials currently exists for a potential dose-response relationship of some tricyclic antidepressants (Hiemke et al., 2011) and the irreversible monoamine oxidase inhibitor tranylcypromine (Adli et al., 2008). Moreover, it should be considered that patients with polymorphisms in the cytochrome P450 enzyme system provoking an accelerated elimination of drugs (the so-called “ultra-rapid metabolizers”) probably require higher doses of the antidepressant to achieve treatment response if their plasma drug concentration is below the effective therapeutic range in a standard dose (Hiemke et al., 2011).
B. Switch of the Antidepressant Drug
According to available evidence and the current international treatment guidelines, a switch from one antidepressant drug to another new antidepressant after insufficient symptom improvement to the initial compound cannot generally be regarded as evidence-based treatment strategy despite potential advantages of continuing monotherapy (Bauer et al., 2017). In a recent meta-analysis of randomized controlled trials, switching was not superior to maintaining the pharmacotherapy with the initial antidepressant (Bschor et al., 2018). Following theoretical pharmacological considerations, it appears advantageous to choose preferably as a second, new antidepressant an agent with a different mechanism of action compared with the first administered compound. The World Federation of Societies of Biologic Psychiatry guidelines for instance advise explicitly that switching from an SSRI to venlafaxine or tranylcypromine appears justified within a treatment algorithm (Bauer et al., 2017). These recommendations are mainly based on the findings of a meta-analysis comparing a switch from an SSRI to either a second course of an SSRI or a switch to another class of antidepressants (Papakostas et al., 2008). As main result, slight but statistically significantly higher pooled remission rates for the latter strategy could be shown (28% for the across-class switch vs. 23.5% for the within-class switch) (Papakostas et al., 2008). In contrast to this meta-analytic finding, switching to a different subclass of an antidepressant (across-class switch) was not significantly superior to a within-class switch in a large European multicenter study (n = 340) when evaluating response and remission rates (Souery et al., 2011). In a recent meta-analysis of randomized controlled trials, switching was not superior to maintaining the pharmacotherapy with the initial antidepressant (Bschor et al., 2018). Accordingly, switching is generally not recommended as an appropriate treatment option for TRD and should only be employed in cases of absolutely no response or intolerable adverse effects (Bauer et al., 2017; Dold and Kasper, 2017). It is noteworthy that a careful selection of the initial antidepressant along with consistent therapy drug monitoring might minimize this risk (Serretti, 2018).
C. Antidepressant Combination Medication
Although antidepressant combination strategies are frequently used in the pharmacological management of TRD, the evidence for this measure is rather sparse, and meta-analytic findings on this topic were inconclusive (Rocha et al., 2012; Lopes Rocha et al., 2013). As the efficacy of this strategy depends primarily on the concurrently prescribed agents, treatment guidelines consistently advise establishing antidepressant combination preferably with reuptake inhibitors such as SSRIs or SNRIs on the one hand and inhibitors of presynaptic autoreceptors (e.g., mirtazapine) or serotonin antagonist and reuptake inhibitors (e.g., trazodone) on the other hand (Bauer et al., 2017). In this case, synergistic antidepressant effects can be anticipated due to the complementary mechanisms of action of these compounds (Moller et al., 2014). Furthermore, these drug combinations appear beneficial from a clinical viewpoint as presynaptic autoreceptor inhibitors are for instance—in contrast to SSRIs/SNRIs—characterized by meaningful sedating properties.
D. Augmentation Strategies
With regard to psychopharmacotherapeutic augmentation strategies in TRD, the most compelling evidence is available for SGAs and the mood stabilizer lithium (Dold and Kasper, 2017). The efficacy of SGA augmentation could be shown in a large number of randomized clinical trials and meta-analyses (Nelson and Papakostas, 2009; Zhou et al., 2015). For instance, in their meta-analysis of 16 placebo-controlled SGA augmentation trials (n = 3480), Nelson and Papakostas (2009) found significant superiority of adjunctive aripiprazole, olanzapine, quetiapine, and risperidone over placebo in response and remission rates. Moreover, some SGAs received the official approval as add-on medication after nonresponse to antidepressant monotherapy by regulatory authorities. For instance, quetiapine extended release (XR) is licensed in the United States and the European Union, aripiprazole in the United States, and olanzapine has the regulatory approval in the United States in combination with fluoxetine. Different mechanisms have been identified that can explain the efficacy of such combinations in clinical practice, such as 1) decreasing local inhibitory GABAergic tone in the dorsal raphe nucleus, through the antagonism of 5-HT6 receptors by olanzapine, leading to a potentiation of SSRI’s activity (Asaoka et al., 2015) and 2) increasing hippocampal neurogenesis and preventing BDNF decrease in stressed rats (Xu et al., 2006).
Beside the SGAs, there is evidence for the efficacy of augmentation with lithium in treatment-resistant MDD (Crossley and Bauer, 2007; Bauer et al., 2014; Nelson et al., 2014), and, accordingly, treatment guidelines consistently recommend this strategy (Bauer et al., 2017). Nevertheless, adjunctive lithium was less frequently prescribed in pharmacoepidemiologic surveys than augmentation with SGAs (Dold et al., 2016). Probably, the use of lithium in the clinical practice is limited by the requirement of continuous plasma level measurements to ensure the achievement of the therapeutic window and due to the anticipation of adverse effects (Nierenberg et al., 2006).
Paralleled by the enhancement of available evidence especially for the efficacy of SGA augmentation, pharmacoepidemiologic studies consistently found a substantial rise of SGA prescription in MDD over the last years (Mohamed et al., 2009; Konstantinidis et al., 2012; Gerhard et al., 2014). For instance, a significant increase of the proportion of patients with MDD receiving SGAs from 12.8% in 2000 to 28.3% in 2007 was found in a pharmacovigilance program analyzing 1826 inpatients in German-speaking countries (Konstantinidis et al., 2012). From a clinical viewpoint, augmentation of antidepressants with antipsychotic drugs is especially recommended for patients with MDD exhibiting psychotic features, whereas lithium should be preferably considered in patients with MDD displaying high risk for suicidal behavior (Kessing et al., 2005; Cipriani et al., 2013; Bauer et al., 2017; Dold and Kasper, 2017).
VII. Drug Discovery in Treatment-resistant Depression
A. Animal Models or Treatment-resistant Depression
The development of animal models in the field of psychiatry is very challenging because of the complex nature and the heterogeneity of these disorders. Therefore, although existing models may be able to reproduce specific features of the disease, they often show limited ability in mimicking the whole pathologic complexity, which is probably crucial for treatment resistance.
In a recent review, Willner and Belzung (2015) argued that among 18 potential models, the most promising are the Wistar Kyoto (WKY) and the congenital learned helplessness (cLH) rats, the CB1 (cannabinoid 1) receptor knockout, and organic cation transporter 2 null mutant mouse strains. The authors considered four criteria: increased stress responsiveness, decreased response to chronic antidepressant treatment, good response to novel antidepressant approaches, and the correspondence to a known clinical risk factor.
WKY rats were originally bred as normotensive control for the spontaneously hypertensive rat strain, but were later found to be characterized by heightened emotionality (Nam et al., 2014), enhanced response to repeated stress (Morilak et al., 2005), and increase propensity to develop learned helplessness (LH) (Belujon and Grace, 2014). Moreover, they are less responsive to antidepressants in the FST and less sensitive to serotonergic antidepressants (Lopez-Rubalcava and Lucki, 2000; Ivarsson et al., 2005). Notably, LH was normalized by ketamine in WKY rats, supporting the predictive validity of this model in the field of TRD (Belujon and Grace, 2014).
The cLH strain was generated by selecting animals that develop the most severe LH. After several breedings, cLH failed to escape without prior stress exposure (Vollmayr and Henn, 2001), they are anhedonic, and, different from noncongenital LH rats, their helpless behavior is not reversed by antidepressant treatment (Vollmayr et al., 2004; Sartorius et al., 2007; Vollmayr and Gass, 2013).
CB1 receptor knockout mice display increased CMS-induced anhedonia (Valverde and Torrens, 2012) and altered sensitivity to subchronic treatment with desipramine or paroxetine (Steiner et al., 2008), suggesting that they may represent a useful model for antidepressant resistance.
Organic cation transporter 2 null mutant mice show reduced levels of noradrenaline and serotonin, increased sensitivity to the effect of CMS (Courousse et al., 2015), and decreased response to chronic treatment with venlafaxine (Bacq et al., 2012).
A number of studies on treatment-resistant depression have also been carried out using other models, such as Flinders sensitive line (FSL) rats, CRCT1−/− mice, and adrenocorticotropic hormone administration (Kitamura et al., 2002; Iwai et al., 2013). FSL rats are one of the most studied “depressive” rat strains (Wegener et al., 2012). Although they were originally generated for cholinergic supersensitivity, they show a range of depression-like features, such as elevated immobility in the forced swim test (Overstreet and Wegener, 2013) and increased anhedonia in the CMS (Pucilowski et al., 1993). Recently, it was demonstrated that exposure of FSL to time-dependent sensitization exacerbated their depressive-like behavior (Brand and Harvey, 2017). Interestingly, although chronic treatment with imipramine, venlafaxine, or ketamine was ineffective in normalizing their behavioral defects as monotherapy, the combination of imipramine with either venlafaxine or ketamine resulted in significant anti-immobility effects and enhanced coping behaviors (Brand and Harvey, 2017).
CRTC1-deficient mouse line exhibits reduced dopamine and serotonin turnover and decreased expression of genes involved in neuroplasticity in the prefrontal cortex. Moreover, CRTC1−/− mice present several endophenotypes related to mood disorders, such as decreased sexual motivation, increased despair, anhedonia, and anxiety-like behavior (Breuillaud et al., 2012) as well as a blunted response to the antidepressants fluoxetine and imipramine in behavioral despair-related paradigms (Breuillaud et al., 2012; Meylan et al., 2016).
Chronic administration of adrenocorticotropic hormone in rats blocks the effects of antidepressants in the FST, alters monoamine response to stress, and downregulates HPA axis activity (Kitamura et al., 2002; Iwai et al., 2013; Walker et al., 2013a).
To summarize, unfortunately the animal models existing are few and, as mentioned above, show important limitations. Moreover, no animal model has been subjected to sequential application of different treatments a key requisite in human to be defined as TRD. Thus, there is still a large gap between clinical TRD and the possibility of effectively mimicking this condition in preclinical models.
B. Pharmacological Strategies in Treatment-resistant Depression
1. Glutamatergic System
Evidence demonstrating the crucial role of the glutamatergic system in the pathophysiology of depression led to the seminal observation that the blockade of the ionotropic NMDA receptor may result in a fast antidepressant response. Initial studies have demonstrated this effect at preclinical level; subsequently the rapid therapeutic efficacy of NMDA receptor antagonists was confirmed in patients with depression. In particular, a single intravenous infusion of ketamine, a noncompetitive NMDA receptor antagonist, at subanesthetic doses (0.5 mg/kg) produced a significant improvement of depressed symptoms in patients (Berman et al., 2000), an effect that has now been confirmed in a large number of clinical trials (Bobo et al., 2016). In addition, Zarate and coworkers (2006) demonstrated that ketamine was also effective in patients suffering from TRD, leading to a marked symptomatological improvement 2 hours after single ketamine infusion, which remained significant for almost 1 week (Zarate et al., 2006). The antidepressant efficacy of ketamine in treatment-resistant depression was confirmed by different studies (Ibrahim et al., 2011; Murrough et al., 2013; Papadimitropoulou et al., 2017).
The acute effects of ketamine have also inspired research to explore its potential as a life-saving therapy in TRD patients with an imminent risk of suicide (Price and Mathew, 2015). Beneficial effects on suicidal ideation have been obtained in randomized controlled trials where, in addition to a single subanesthetic dose of intravenous ketamine, thrice-weekly ketamine infusions over a 12-day period were administered [for a thorough review, see Price and Mathew (2015)].
It has to be noted that ketamine is a racemate that comprises R-(−)-ketamine (arketamine) and S-(+)-ketamine (esketamine) enantiomers. Esketamine has a threefold to fourfold higher affinity for N-methyl-d-aspartate (NMDA) receptors than arketamine, and its intravenous infusion has been effective in improving depression in TRD patients, with similar rates compared with the racemate (Paul et al., 2009; Singh et al., 2016). Further studies are ongoing to assess if alternative formulations of esketamine can be developed to avoid the inconvenience of intravenous infusion. A randomized double-blind phase II clinical trial assessed the efficacy and safety of intranasal esketamine compared with placebo in 67 adults with TRD. The results of this study show that intranasal esketamine (28–84 mg administered twice weekly) produced, after 1 week, a significant improvement of depressive symptoms, as assessed by the Montgomery-Åsberg Depression Rating Scale total score, with a significant ascending dose-response relationship and a sustained improvement also after 9 weeks adopting a lower frequency (weekly or every 2 weeks) of esketamine administration (Daly et al., 2018). Two Phase 3 clinical trials by Janssen Research & Development (Titusville, New Jersey) are currently ongoing to study the necessary frequency of dosing and duration of effect of intranasal esketamine (NCT02493868) as well as the long-term safety of this drug in patients with TRD (NCT02782104) (Daly et al., 2018).
Despite these important discoveries, the use of ketamine has many drawbacks, such as the development of transient psychotic/dissociative symptoms, abuse potential, and cognitive impairment and neurotoxicity, pointing to the need to develop drugs with similar properties but without undesirable effects. The safety profile of ketamine should be carefully considered, in particular when debating about its use in long-term studies for depression. Nevertheless, ketamine still remains a lead compound, which has stimulated the development of new antidepressant drugs with similar properties but lower propensity to produce undesirable effects.
Along this line of reasoning, a phase 2 clinical trial was conducted in patients suffering from TRD with CP-101,606 [traxoprodil (Pfizer; Groton, Connecticut)], an NMDA antagonist that is selective for the GluN2B subunit, which acts as negative modulator (Mott et al., 1998). The results of the study indicated that CP-101,606 was able to reduce both Montgomery-Asberg Depression Rating Scale (MADRS) and Hamilton Rating Scale for Depression scores and that 78% of CP-101,606-treated patients maintained the response status for 1 week and 32% for 30 days after the infusion (Preskorn et al., 2008). However, the clinical studies with CP-101,606 were discontinued at the end of 2010 due to potential cardiovascular toxicity of the compound.
Another NMDA-related compound with a promising efficacy in TRD is GLYX-13 [rapastinel (Allergan; Dublin, Ireland)], a partial agonist at the glycine site of the NMDA receptor with established antidepressant-like properties in several preclinical models of depression, including forced swim, learned helplessness, novelty-induced hypophagia (Burgdorf et al., 2013; Moskal et al., 2017). It was recently demonstrated that, when compared with ketamine, GLYX-13 may target a different population of glutamatergic NMDA receptors. However, similar to ketamine, it produces antidepressant effects by promoting exocytotic BDNF release with the consequent activation of TrkB-mTORC1 downstream signaling (Kato et al., 2017). Moreover, GLYX-13 is able to ameliorate the behavioral alterations in the social defeat stress model, although at a difference from R-ketamine; it was not able to correct stress-induced alterations of BDNF and synaptic proteins (Yang et al., 2016).
Interestingly, the results of a Phase 2 clinical trial demonstrated that a single intravenous dose of GLYX-13 was able to reduce the depressive symptoms in patients with treatment-resistant depression within 2 hours from the administration. Furthermore, the antidepressant action was lasting for 1 week and, different from ketamine, was not associated with psychotomimetic effects (Preskorn et al., 2015). Currently a Phase 3 randomized, double-blind, placebo-controlled, multicenter study of GLYX-13 as adjunctive therapy in major depressive disorder is ongoing. Specifically, the efficacy, safety, and tolerability of two doses of GLYX-13 will be evaluated in comparison with placebo adjunctive to antidepressant therapy in patients with major depressive disorder who have a partial response to antidepressant drugs (NCT02943564).
Moreover, apimostinel (NRX 1074), an orally bioavailable analog of GLYX-13 more potent as a glycine partial agonist, has also been developed (www.naurex.com/pipeline/nrx-1074). Up to now, its safety, tolerability, and pharmacokinetics have been evaluated in a Phase 1 study in normal, healthy volunteers orally treated with the drug (NCT02366364). A Phase 2 study to investigate the efficacy and safety of apimostinel following a single intravenous dose in subjects with major depressive disorder has been completed, although the results have not been disclosed yet (https://clinicaltrials.gov/ct2/show?term=NRX+1074&rank=2).
Another line of research is focused on glutamate metabotropic receptor, addressing the therapeutic potential for TRD of mGlu2/3 receptor blockade. These receptors, which are coupled to Gi/Go and inhibit glutamate release, are highly expressed in brain areas associated with emotion and cognition (Nicoletti et al., 2011). Preclinical studies have demonstrated that mGluR2/3 antagonists as well as negative allosteric modulators of these receptors exert relevant antidepressant effects in animal models of depression (Chaki, 2017). Interestingly mGluR2/3 antagonists can target mTOR signaling pathway (Dwyer et al., 2012) and enhance synaptic glutamate levels, leading to enhanced AMPA receptor transmission. Currently, two groups of mGlu2/3 receptor antagonists have been developed: the orthosteric mGlu2/3 receptor antagonists (MGS0039, LY341495, and LY3020371) that bind the orthosteric sites in the N terminus of the receptor and the negative allosteric modulators (RO4491533, RO1, and RO2) that bind to the allosteric sites (Chaki et al., 2013; Celanire et al., 2015).
The majority of the studies reported the effect of the orthosteric antagonists MGS0039 and LY341495, which displayed antidepressant properties not only in classic animal models of depression such as forced swimming, tail suspension, chronic unpredictable and chronic defeat stress (for review, see Chaki, 2017), but also in animal models not responding to classic antidepressants. For example, different from the tricyclic desipramine or the SSRI fluoxetine, orthosteric mGlu2/3 receptor antagonists were able to reverse the depressive-like phenotype in chronic corticosterone-treated mice (Ago et al., 2013), an effect also reported when the glucocorticoid was administered to rats (Koike et al., 2013). Another study reported that LY341495 showed antidepressant effects in the tail-suspension test performed on CD-1 mice, which appear relatively resistant to administration of the SSRI citalopram (Witkin et al., 2016).
At the moment, no published studies reported the potential antidepressant effects of mGlu2/3 receptor antagonists in patients with TRD. However, a Phase 2 clinical trial was conducted with RO4995819 (RG1578, Decoglurant) by Roche (Basel, Switzerland) to investigate the efficacy and safety of this compound versus placebo as an adjunctive therapy in patients with major depressive disorder having inadequate response to ongoing antidepressant treatment (NCT01457677). Despite the expectations, RO4995819 failed to demonstrate any significant antidepressant effects on primary or secondary outcomes, because no differences in MADRS total score, responses, and remission rates were observed between the active drugs and placebo (Umbricht et al., 2015). These disappointing results do not exclude the potential of mGlu2/3 receptor antagonists as antidepressants in TRD, considering that our knowledge on the role of mGlu2/3 receptor in depression is far from being complete. For example, it has been reported that, beside antagonists, selective mGlu2/3 receptor agonists may also exhibit antidepressant-like activity in animal models of depression (Matrisciano et al., 2008).
An alternative approach to mimic ketamine’s effects might be the use of mGluR5-selective antagonists or negative allosteric modulators of this receptor (Pałucha-Poniewiera and Pilc, 2016). Indeed, mGluR5s are functionally associated with NMDA receptors, and mGluR5 antagonists exert significant antidepressant effects in animal models of depression, possibly through a negative modulation of NMDA receptors (Chaki et al., 2013). Pharmacological blockade of mGlu5 receptors might therefore become another promising strategy for treatment of MDD. Accordingly, basimglurant, a negative allosteric modulator of mGluR5, has shown promising results in two clinical trials for TRD (NCT00809562, NCT01437657), in particular as adjunctive treatment to SSRIs or SNRIs (Quiroz et al., 2016).
2. Inflammatory System
As discussed above, activation of the immune system and neuroinflammation represent a primary event in the pathophysiology of TRD (Carvalho et al., 2013; Cattaneo et al., 2013; Kiraly et al., 2017). It is known that anti-inflammatory drugs such as nonsteroidal anti-inflammatory drugs can augment the clinical efficacy of monoaminergic antidepressants (Abbasi et al., 2012). Acetylsalicylic acid accelerates the antidepressant effect of fluoxetine in an animal model of depression (Brunello et al., 2006) and also shortens the onset of action of SSRI in non-responder patients with MDD (Mendlewicz et al., 2006).
Minocycline, a tetracyclic derivative with anti-inflammatory properties that inhibits microglial activation (Kohler et al., 2016), is currently being studied in a phase II clinical trial (NCT02456948) in patients with TRD as an adjunctive treatment to monoaminergic antidepressant monotherapy.
An alternative and innovative approach that is currently proposed for TRD is to selectively target the proinflammatory cytokines primary involved in TRD, such as TNF-α, IL-6, and IL-1β (Bortolato et al., 2015; Kappelmann et al., 2018).
IL-1β signaling plays a central role in the acquisition of the depression-like phenotype in stress-related animal models of depression (Maes et al., 2012). Stress-induced IL-1β release is driven by ATP-induced activation of the P2X purinoceptor 7 ion channel (P2X7) (Iwata et al., 2016). P2X7 is highly expressed in microglia and plays a key role in stress-induced neuroinflammation (Bhattacharya and Drevets, 2017). Along this line of reasoning, genetic deletion of P2X7 prevents the induction of a depressive-like phenotype in stress-related animal models of depression (Iwata et al., 2016). Therefore P2X7 may represent a novel therapeutic target for depression as also suggested by the observation that selective central nervous system-penetrable P2X7 antagonists reverse the anhedonic and depressogenic behaviors induced by chronic stress in animal models (Iwata et al., 2016). JNJ-54175446 is indeed a selective brain penetrant P2X7 antagonist that is currently being studied in a phase I single-arm, open-label trial (NCT03088644).
On a different note, it is important to say that, although clinical studies have examined the efficacy of cytokine antagonists for the treatment of depression, only one clinical trial has been designed using depression as primary outcome (Raison et al., 2013). These authors examined whether repeated intravenous administration of infliximab, a monoclonal antibody directed against TNF-α, was able to improve depressed mood in patients with TRD. While they found that infliximab did not exhibit a generalized efficacy in TRD, a post hoc analysis of this study has shown that infliximab exerted a clinically relevant antidepressant effect only in patients with TRD who exhibited elevated baseline levels of proinflammatory markers (based on hs-C-reactive protein >5 mg/l) (Raison et al., 2013). Future clinical trials with an adequate design and focused on patients with TRD with hs-C-reactive protein >5 mg/l and/or elevated TNF-α are needed to replicate these findings and to validate the efficacy of infliximab for TRD.
IL-6 has also been considered a new relevant pharmacological target in TRD (Zhou et al., 2017). Sirukumab (Janssen Biotech; Horsham, Pennsylvania) is a monoclonal antibody endowed with anti-inflammatory properties against IL-6 that has shown preliminary antidepressant efficacy in patients with rheumatoid arthritis (Sun et al., 2017). Sirukumab is a safe and well-tolerated drug, and improvement in depressive symptoms in patients with rheumatoid arthritis positively correlates with baseline soluble IL-6 receptor levels (Sun et al., 2017). According to the Biobehavioral Research Domain Criteria Matrix, it has been hypothesized that the symptoms of MDD most sensitive to sirukumab may be anhedonia and cognitive deterioration (Zhou et al., 2017). A phase II clinical trial is currently ongoing to evaluate the antidepressant efficacy of adjunctive subcutaneous sirukumab compared with placebo in patients with depression (NCT02473289). Along this line of reasoning, tocilizumab is a humanized monoclonal antibody against the IL-6 receptor currently studied in a phase II single-arm, open-label trial (NCT02660528) designed to evaluate the antidepressant effects of this drug in TRD patients.
3. Opioid System
In the last few years, different studies have clearly demonstrated the relevance of the opiodergic system as a novel pharmacological target in TRD. It is known that long-term opioid treatment of chronic pain can interfere with the outcome of depression treatment. In particular, long-term prescription of µ-opioid receptor agonists (for >90 days) is associated with new onset of TRD (Scherrer et al., 2016). On the contrary, the κ-opioid receptor (KOR) has emerged as a new pharmacological target for the treatment of TRD (Li et al., 2016b). KOR has a central role in reward system and in mediating the effects of chronic stress on dopamine release in the mesolimbic pathway (Abraham et al., 2018). It has been hypothesized that activation of KOR receptor by dynorphin leads to a reduction in dopamine release, thus producing anhedonia and depressive symptoms. Accordingly, KOR antagonists exert relevant antidepressant effects in animal models of depression (Taylor and Manzella, 2016).
Buprenorphine is a partial µ-opiod receptor agonist and KOR antagonist, which exerts clinically relevant antidepressant effects in TRD patients (Bodkin et al., 1995; Karp et al., 2014). Karp et al. (2014) found that buprenorphine acts as a rapid antidepressant drug when administered at an average dose of 0.4 mg/day in patients with TRD. Two different phase II clinical trials are currently evaluating the clinical efficacy of buprenorphine in TRD in older adults as an adjunct to venlafaxine (NCT 02181231) or as monotherapy in late-life treatment resistant depression (BUILD trial) (NCT01071538).
ALKS-5461 is a combination of buprenorphine and samidorphan, a μ-opioid receptor antagonist, that has been developed as sublingual tablets by Alkermes (Dublin, Ireland) as a potential treatment of TRD (Fava et al., 2016b). In a randomized, double-blinded, placebo-controlled phase 2 trial, the treatment with 2 mg/2 mg of buprenorphine/samidorphan provided significant antidepressant effects in MDD with a recent history of nonresponse to SSRIs or SNRIs (Fava et al., 2016b). This combination is safe, but with an increased occurrence of adverse events such as nausea, vomiting, dizziness, and headache. ALKS-5461 received fast track designation by the Food and Drug Administration for treatment-resistant depression in October 2013 (Li et al., 2016b). Two placebo-controlled MDD studies (also known as Forward-3 and Forward-4) were recently completed for ALKS-5461 with contrasting results, positive in Forward-3 and negative in Forward-4 due to higher positive response in the placebo-control group (Dhir, 2017). A phase III efficacy trial (Forward-5 study, NCT02218008) was recently concluded with 407 subjects recruited, where the 2/2 mg dose of ALKS-5461 yielded significant antidepressant outcomes compared with placebo (Rakesh et al., 2017). ALKS-5461 is currently being studied in an open-label, long-term (52 weeks) safety and tolerability trial (NCT02141399).
CERC-501 is a potent and selective κ-opioidergic receptor antagonist that does not interact with other opioidergic receptors (Rorick-Kehn et al., 2014). It was recently developed as an adjunctive treatment in patients with MDD (Dhir, 2017). The drug shows a good preclinical efficacy in animal models of depression (Rorick-Kehn et al., 2014). A phase I trial has demonstrated that CERC-501 has a good bioavailability in healthy volunteers, crosses the blood-brain barrier, and is also well tolerated (Lowe et al., 2014). Fava et al. (2016a) are currently examining the clinical efficacy of CERC-501 at two different doses (10 and 20 mg/day) in the so-called proof-of-concept phase II Trial of CERC-501 Augmentation of Antidepressant Therapy in TRD (RAPID KOR) (NCT01913535). An ongoing clinical trial called Fast-Fail Trials in Mood and Anxiety Spectrum Disorders (FASTMAS) will also examine the antidepressant effects of the drug with biologic markers (NCT02218736). Results from these two clinical studies are awaited and will be essential to validate the use of selective KOR antagonists as a novel pharmacological strategy in TRD.
4. Cholinergic System
Although a hyperactivation of the cholinergic system was been proposed a long time ago to contribute to the pathogenesis of MDD (Janowsky et al., 1972; Jeon et al., 2015), the interest for this system has been refueled recently, particularly with respect to the possibility of targeting muscarinic acetylcholine receptors (Jeon et al., 2015). A large number of preclinical and clinical studies have been conducted to examine the role of cholinergic system as a pharmacological target for the treatment of depression, although drugs acting on nicotinic acetylcholine receptors (nAChRs) have shown limited clinical efficacy in TRD if compared with recent clinical evidence of efficacy observed in patients with TRD with the muscarinic receptor antagonist scopolamine (Drevets et al., 2013).
A number of relatively small studies have provided support to the potential for cholinergic drugs targeting nicotinic receptors in TRD. Antagonism of α4β2 nicotinic acetylcholine receptors (nAChR) is known to exert antidepressant effects in preclinical models of depression (Philip et al., 2010). Accordingly, mecamylamine, an α4β2 nAChR antagonist, was effective as augmentation strategy for patients with SSRI-refractory MDD (George et al., 2008). CP-601,927, a partial agonist at the nicotinic acetylcholine receptor, has shown a relevant clinical efficacy only in patients with TRD with increased leptin levels (Fava et al., 2015), but further studies are needed to understand whether α4β2 nAChRs still represent a valid pharmacological target for the treatment of TRD.
The first evidence about the clinical efficacy of muscarinic receptor antagonists in TRD was obtained over three decades ago with biperiden, which induced a significant improvement in a small open-label study of inpatients with TRD (n = 10) (Kasper et al., 1981). The renewed interest in this field stems from the evidence that the muscarinic M1 and M2 receptor antagonist scopolamine (Witkin et al., 2014) elicits a rapid antidepressant response in patients with MDD (Furey and Drevets, 2006; Drevets and Furey, 2010). Interestingly scopolamine induces the mTOR pathway with a similar magnitude and timing of ketamine, leading to an increase in BDNF levels (Drevets et al., 2013). Two different studies have demonstrated that intravenous repeated infusions of a low dose of scopolamine (0.004 mg/kg) in patients with unipolar or bipolar depression significantly reduced symptoms of depression and anxiety a few days after the initial infusion (Furey and Drevets, 2006; Drevets and Furey, 2010). Placebo-adjusted remission rates were 56% and 45% for the initial and subsequent replication studies, respectively (Drevets et al., 2013). In the second study Drevets and Furey (2010) found that women were 33% more sensitive than men to the antidepressant effects of scopolamine. Recently Ellis et al. (2014) conducted a double-blind, placebo-controlled, crossover clinical trial to compare the antidepressant effects of scopolamine in patients with TRD and patients who were treatment naive with recurrent MDD or bipolar disorder. Scopolamine rapidly reduces symptoms in both treatment groups, with a greater improvement in patients who were treatment naive, but also with a sustained improvement in patients with TRD (Ellis et al., 2014). Khajavi et al. (2012) demonstrated, in a randomized double-blind, placebo-controlled study, the safe and efficacious use of oral scopolamine (1 mg/day) as an augmenting agent for moderate to severe MDD (as an adjunct to citalopram). Alternatives routes of administration of scopolamine, such as transdermal patch, are currently being studied (NCT00369915). Interestingly, a combination of intravenous scopolamine and ketamine for TRD is also currently being investigated in an open-label clinical study (NCT01613820).
5. Dopaminergic System
The role for dopamine transmission in depression is very complex and may depend upon different dopaminergic circuits in the brain. Reduced dopaminergic activity within the mesolimbic system contributes to anhedonia and apathy in severe depressive disorders (Leggio et al., 2013; Bech et al., 2015), although increased dopaminergic activity in the mesolimbic ventral tegmental area nucleus accumbens pathway has also been demonstrated in animal models of depression vulnerability with the possible involvement of BDNF signaling (Wook Koo et al., 2016).
One dopaminergic target that has been proposed for its potential in the treatment of depression is the dopamine D3 receptor, an autoreceptor (i.e., presynaptic receptor) highly expressed in the limbic system and involved in reward and cognitive functions (Sokoloff et al., 1990). D3 receptors exert inhibitory effects on dopamine impulse flow, dopamine synthesis, and dopamine release (Tepper et al., 1997). Recent studies suggest that antagonism on the D3 receptor can be considered a new pharmacological strategy for the treatment of neuropsychiatric disorders such as schizophrenia and MDD (Leggio et al., 2016). Selective D3 receptor antagonists have been proposed as a pharmacological tool for the treatment of depression (Leggio et al., 2013). As an example, buspirone is an anxiolytic drug that acts as a 5-HT1A partial agonist but also a potent antagonist for the D3 receptors (Bergman et al., 2013), which has been proposed as augmentation therapy in patients with MDD who do not respond to first-line therapy (Fleurence et al., 2009). Interestingly Appelberg et al. (2001) demonstrated that buspirone can exert clinically relevant antidepressant effects in patients with MDD not responsive to SSRIs and with initially high MADRS scores (>30), but larger studies are needed to validate the therapeutic potential of this drug in TRD.
Cariprazine is a new antipsychotic drug that acts as D2R and D3R partial agonist and 5-HT2B antagonist according to NbN classification, but with a high selectivity toward the D3 compared with the other receptors (De Deurwaerdere, 2016). Studies in animal models of depression, such as CMS, revealed potent antidepressant and antianhedonic-like activity of cariprazine (Papp et al., 2014). Interestingly the antianhedonic-like effects of cariprazine were not observed in D3-knockout mice, suggesting that these effects are mediated by dopamine D3 receptors (Duric et al., 2017). A first phase II randomized, double-blind, placebo-controlled trial (NCT01469377) demonstrated the efficacy and safety of cariprazine (2–4.5 mg/day) as adjunctive therapy in patients with MDD who have inadequate response to standard “monoaminergic” antidepressant therapy (Durgam et al., 2016). The results of two different phase III clinical trials, which were recently completed (NCT01838876) or are still ongoing (NCT01715805), will be essential to establish the clinical efficacy as well as the long-term safety and tolerability of cariprazine in TRD.
It is worth mentioning that pramipexole, a D3/D2 receptor agonist, is not only the most effective compound in the treatment of depression associated with Parkinson’s disease (Seppi et al., 2011), but it has been proposed as an augmentation strategy in TRD (Pae, 2014).
6. Neurotrophin Signaling
The development of new antidepressant able to rescue neurogenesis was recently explored in TRD. One possible approach might be to target the BDNF-TrkB signaling pathway (Zhang et al., 2016). NSI-189 is a benzylpiperizine-aminiopyridine compound developed by Neuralstem (Rockville, MD) which enhances neurogenesis both in the hippocampus as well as in the subventricular zone in mice (McIntyre et al., 2017). Interestingly this drug strongly upregulates neurotrophic factors (BDNF and stem cell factor) in rat hippocampal cells in tandem with increased neurogenesis in rat hippocampus (Tajiri et al., 2017), although the exact signaling pathways modulated by this drug remain to be established. Results from a recent proof-of-concept phase 1B, double-blind, randomized, placebo-controlled, multiple-dose study suggest that NSI-189 significantly reduces depressive symptoms and improves cognitive function in patients with MDD (Fava et al., 2016a). NSI-189 appears to be a safe drug with a multidomain profile able to improve cognitive function independently from its effects on affective symptoms (McIntyre et al., 2017). An ongoing phase II double-blind clinical study is now examining the efficacy of a 12-week treatment with NSI-189 (80 mg/day) in 220 patients with MDD (NCT02695472). Furthermore, a longitudinal observational cohort study is currently ongoing to evaluate the long-term safety of NS-189 and, most importantly, the durability of effect of this drug defined as the time until the start of a new antidepressant treatment (NCT02724735).
VIII. Nonpharmacological Approaches in Treatment-resistant Depression
Although our work is focused on pharmacological strategies for TRD, it must be kept in mind that, especially for patients with severe TRD, the use of nonpharmacological therapies may be recommended in combination with pharmacological treatment or even alone. The effectiveness of alternative approaches such as electroconvulsive therapy (ECT), transcranial magnetic stimulation, deep-brain stimulation, vagal nerve stimulation (Cusin and Dougherty, 2012; Carreno and Frazer, 2017), or even neurosurgical lesion procedures (Patel et al., 2013) have been evaluated in several studies, and they may represent valid therapeutic options. Nevertheless, it is important to point out that not all patients may benefit from these approaches, which are characterized by specific indications with respect to patient’s level of treatment resistance, response probability, severity of side effects, intolerance issues, and costs. For examples, among the neurostimulatory treatments, ECT is considered the most effective option in patients with TRD, with a response rate of almost 50% (Heijnen et al., 2010), but it also characterized by a high relapse rate after a successful course of therapy (Jelovac et al., 2013) and by cognitive side effects, which are instead less pronounced with transcranial magnetic stimulation (Ren et al., 2014).
In addition, psychiatrists may recommend psychotherapies (Trivedi et al., 2011) or even light-based therapies, exercise, and acupuncture (Qureshi and Al-Bedah, 2013), although clear evidence for the effectiveness of these approaches in TRD is very limited. The major benefits may be obtained with combinatory strategies. Indeed cognitive behavioral therapy has been reported as a valid option in combination with pharmacotherapy for primary care patients with TRD compared with usual care alone (Wiles et al., 2013; Morimoto, 2014).
IX. Conclusions
In summary, we provided a description of the main molecular mechanisms that may play a role in TRD. We suggest that the poor response to pharmacological intervention in patients with TRD may result from a limited impact of available treatments on key mechanisms that are altered in depressed subjects. These mechanisms, which include altered glutamatergic function, reduced neuroplasticity and brain trophism, enhanced inflammation, and dysregulation of the HPA axis, represent different aspects of a common background or may selectively contribute to different “dimensions” of the disease. As an example, fatigue has long been identified as a core depressive symptom known to be resistant to monoaminergic antidepressants (Ferguson et al., 2014) and strictly related to chronic inflammation in depression (Felger et al., 2016), suggesting that the ability to target inflammatory dysfunction with novel drugs may also lead to beneficial effects on specific disease features.
It is important to consider that there is a close link between these alterations that will ultimately affect global brain function. We believe that the ability to interfere with such network of changes represents a critical element for therapeutic response to pharmacological intervention. We propose a number of different approaches and discuss the potential for novel therapeutic strategies in TRD (as summarized in Fig. 1),

Hypothetical flowchart for the treatment of major depressive disorders (MDD). MDD (major depressive disorder) is a complex disease characterized by a plethora of molecular and functional alterations. The ability of “classical” antidepressant treatments (i.e., monoaminergic drugs) currently used in the clinic setting to modulate such dysfunctions will ultimately lead to remission or to the development of treatment-resistant depression (TRD). The condition of TRD can benefit from a number of well-established therapeutic strategies as well as from the possibility to target novel systems and pathways, which may allow restoring the normal brain function thus leading to remission.
Finally, sex differences also may represent a variable to take into account when developing new drugs and designing clinical trials. Indeed, despite major depressive disorders affecting women more than men (Wittchen et al., 2011; Whiteford et al., 2013) with a consequent greater antidepressant prescription in this sex, sex influence on antidepressant response are controversial. In particular, although several studies showed that women respond better to antidepressant than men, others suggested exactly the opposite and many studies detected no sex differences (Sramek et al., 2016). These contradictions may be explained by the enormous heterogeneity in the methodology used, from diagnostic nosology to the class of therapeutic agent, with differences also in the criteria employed for determining a significant response to drug treatment.
All in all, considering the complex nature of MDD and the vast heterogeneity of patients with depression, different treatments and their combinations should not only be able to restore normal function, but, more importantly, must promote resilience to reduce the long-term vulnerability to recurrent depressive episodes.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Caraci, Calabrese, Molteni, Bartova, Dold, Leggio, Fabbri, Mendlewicz, Racagni, Kasper, Riva, Drago.
Footnotes
↵1 M.A.R. and F.D. equally contributed to this work.
F.Car. received compensation as speaker/consultant from Eli Lilly, Lundbeck, Otzuka, Janssen-Cilag, Grunenthal and he has received research grants from Lundbeck. M.D. has received a travel grant from Janssen-Cilag. J.M. is a member of the Faculty of the Lundbeck International Neuroscience Foundation and of Advisory Board of Servier. G.R. has received consulting fees and/or honoraria from Indena, Servier, and Recordati. S.K. received grants/research support, consulting fees, and/or honoraria from Angelini, AOP Orphan Pharmaceuticals AG, AstraZeneca, Eli Lilly, Janssen, KRKA-Pharma, Lundbeck, Neuraxpharm, Pfizer, Pierre Fabre, Schwabe, and Servier. M.A.R. has received compensation as speaker/consultant from Lundbeck, Otzuka, Sumitomo Dainippon Pharma, and Sunovion, and he has received research grants from Lundbeck, Sumitomo Dainippon Pharma and Sunovion. F.Cal., R.M., L.B., G.M.L., C.F., and F.D. have no conflict of interest to declare.
Abbreviations
- AMPA
- alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- ATC
- anatomical-therapeutic-chemical
- BDNF
- brain-derived neurotrophic factor
- CB1
- cannabinoid 1
- cLH
- congenital learned helplessness
- CMS
- chronic mild stress
- CRCT1
- CREB-regulated transcription coactivator 1
- ECT
- electroconvulsive therapy
- eEF2
- eukaryotic elongation factor 2
- FSL
- Flinder sensitive line
- FST
- forced swim test
- GR
- glucocorticoid receptor
- GSRD
- European Group for the Study of Resistant Depression
- GWAS
- genome-wide association study
- HDAC
- histone deacetylases
- HNK
- (2R,6R)-hydroxynorketamine
- HPA
- hypothalamic-pituitary-adrenals
- 5-HT
- serotonin
- IL
- interleukin
- KCNK2/TREK1
- potassium channel subfamily K member 2
- KOR
- κ-opioid receptor
- LH
- learned helplessness
- MADRS
- Montgomery-Asberg Depression Rating Scale
- MDD
- major depressive disorder
- mGluR
- metabotropic glutamate receptor
- MIF
- macrophage inhibiting factor
- mTOR
- mammalian target of rapamycin
- nAChR
- nicotinic acetylcholine receptors
- NbN
- neuroscience-based nomenclature
- NET
- norepinephrine transporter
- NMDA
- N-methyl-d-aspartate
- SGA
- second generation antipsychotic
- SNRI
- serotonin and noradrenaline reuptake inhibitors
- SSRI
- selective serotonin reuptake inhibitors
- STAR*D
- Sequenced Treatment Alternatives to Relieve Depression Study
- TCA
- tricyclic antidepressants
- TDO
- tryptophan 2,3 dioxygenase
- TNF
- tumor necrosis factor
- TRD
- treatment-resistant depression
- TrkB
- tropomyosin receptor kinase B
- VPA
- valproic acid
- WKY
- Wistar Kyoto
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





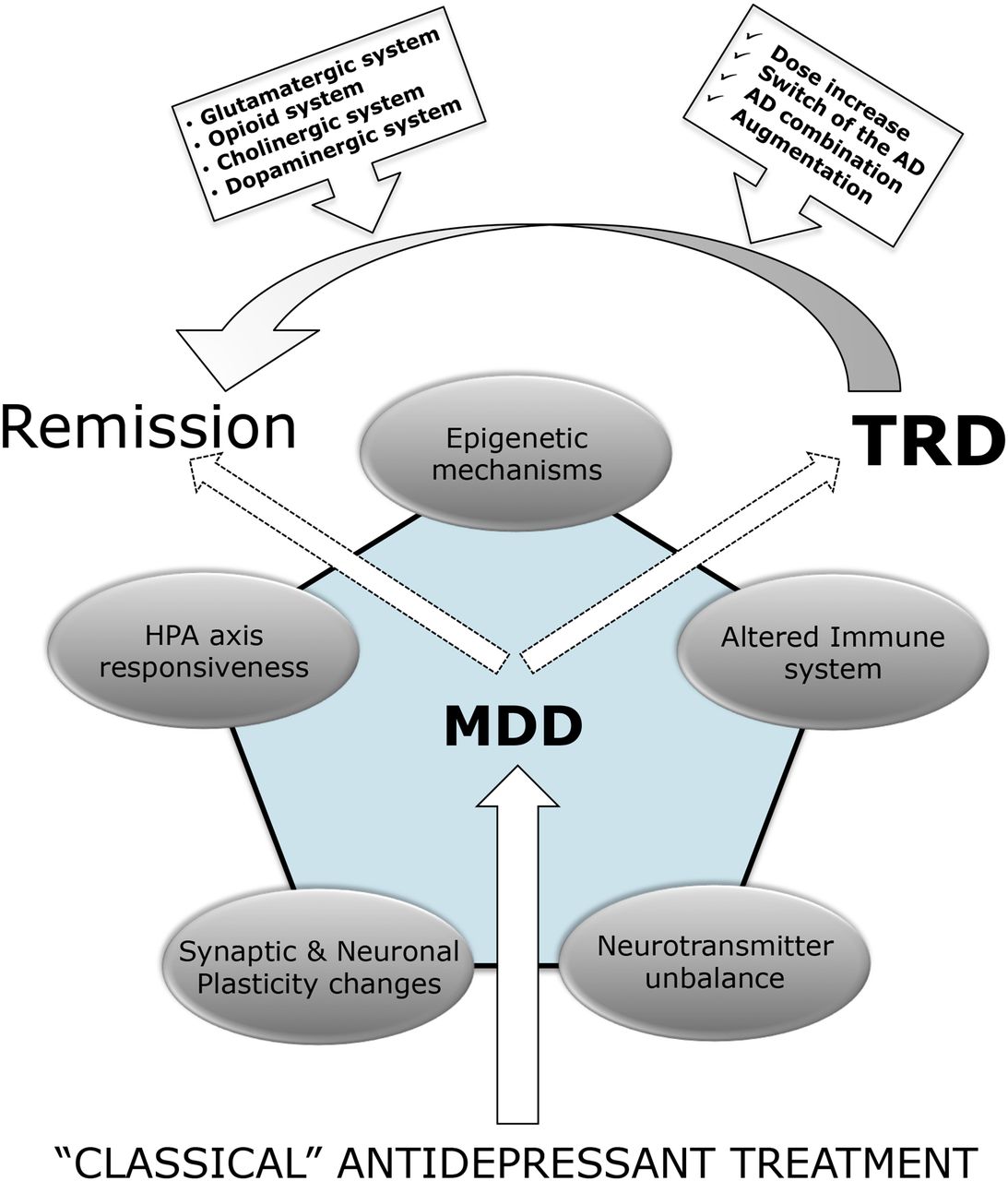{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. The New Neuroscience-Based Nomenclature and the Classification of Antidepressant Drugs: Implications for Drug Discovery and Clinical Practice
- III. Clinical Phenotypes of Treatment-resistant Depression
- IV. Genetics of Treatment-resistant Depression
- V. From the Neurobiology of Depression to Treatment Resistance
- VI. Current Therapeutic Strategies for Treatment-resistant Depression
- VII. Drug Discovery in Treatment-resistant Depression
- VIII. Nonpharmacological Approaches in Treatment-resistant Depression
- IX. Conclusions
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters