


Abstract
Ketamine, a racemic mixture consisting of (S)- and (R)-ketamine, has been in clinical use since 1970. Although best characterized for its dissociative anesthetic properties, ketamine also exerts analgesic, anti-inflammatory, and antidepressant actions. We provide a comprehensive review of these therapeutic uses, emphasizing drug dose, route of administration, and the time course of these effects. Dissociative, psychotomimetic, cognitive, and peripheral side effects associated with short-term or prolonged exposure, as well as recreational ketamine use, are also discussed. We further describe ketamine’s pharmacokinetics, including its rapid and extensive metabolism to norketamine, dehydronorketamine, hydroxyketamine, and hydroxynorketamine (HNK) metabolites. Whereas the anesthetic and analgesic properties of ketamine are generally attributed to direct ketamine-induced inhibition of N-methyl-D-aspartate receptors, other putative lower-affinity pharmacological targets of ketamine include, but are not limited to, γ-amynobutyric acid (GABA), dopamine, serotonin, sigma, opioid, and cholinergic receptors, as well as voltage-gated sodium and hyperpolarization-activated cyclic nucleotide-gated channels. We examine the evidence supporting the relevance of these targets of ketamine and its metabolites to the clinical effects of the drug. Ketamine metabolites may have broader clinical relevance than was previously considered, given that HNK metabolites have antidepressant efficacy in preclinical studies. Overall, pharmacological target deconvolution of ketamine and its metabolites will provide insight critical to the development of new pharmacotherapies that possess the desirable clinical effects of ketamine, but limit undesirable side effects.
I. Introduction
(R,S)-Ketamine (hereafter referred to as ketamine) is a phenylcyclohexylamine derivative (mol. wt. = 237.73) consisting of its two optical enantiomers, (S)- and (R)-ketamine (Adams et al., 1978). It became commercially available for human use in 1970 as a rapid-acting i.v. anesthetic (Dundee et al., 1970). Ketamine was derived from phencyclidine (PCP) with the aim of lessening the serious psychotomimetic/psychodysleptic side effects and abuse potential of the parent drug, which was subsequently removed from the market in 1978 (Mion and Villevieille, 2013). However, ketamine still induces dissociative effects (Domino et al., 1965; Erdemir et al., 1970; Oye et al., 1992; Krystal et al., 1994; Bowdle et al., 1998; Newcomer et al., 1999; Lahti et al., 2001; Pomarol-Clotet et al., 2006) and has abuse potential (Siegel, 1978; Reich and Silvay, 1989; Dalgarno and Shewan, 1996; Stewart, 2001; Morgan and Curran, 2012), although to a lesser extent than PCP. Despite these side effects, ketamine has proven to be a desirable drug due to its short half-life and lack of clinically-significant respiratory depression (Clements et al., 1982; Gorlin et al., 2016). In addition to its well-characterized anesthetic action in adults, children, and obstetric patients, ketamine possesses analgesic effects (Weisman, 1971), anti-inflammatory effects (Roytblat et al., 1998), and antidepressant activity (Berman et al., 2000; Zarate et al., 2006; also see Wolff and Winstock, 2006).
A. Clinical Therapeutic Effects
1. Anesthetic
Ketamine induces general, dissociative anesthesia in animals (McCarthy et al., 1965; Chen et al., 1966; Bree et al., 1967) and humans (Domino et al., 1965; Corssen and Domino, 1966; Virtue et al., 1967; Miyasaka and Domino, 1968; Domino, 2010). Moreover, ketamine is also used as an adjunct to local anesthetics in veterinary practice and in humans (Green et al., 1981; Bion, 1984; Gomez de Segura et al., 1998; Hawksworth and Serpell, 1998; Kathirvel et al., 2000).
Dissociative anesthesia—a form of anesthesia that lacks complete unconsciousness but is characterized by catatonia, catalepsy, and amnesia—is achieved in humans at ketamine doses ranging from 1 to 2 mg/kg administered i.v. (bolus) or 4–11 mg/kg administered i.m. (Sage and Laird, 1972; Sussman, 1974; Dachs and Innes, 1997; Weber et al., 2004; Green et al., 2011; Gao et al., 2016). Peak ketamine plasma concentrations of approximately 1200–2400 ng/ml, or 5–10 μM, are necessary to induce dissociative anesthesia (Little et al., 1972; Idvall et al., 1979; Grant et al., 1983).
The average steady-state plasma concentration necessary to achieve anesthesia with ketamine was reported to be 2200 ng/ml, or 9.3 μM (Idvall et al., 1979). Oral (500 mg; Craven, 2007) or intrarectal (8–15 mg/kg; Idvall et al., 1983; Malaquin, 1984; Malinovsky et al., 1996) administration of ketamine are sufficient to induce sedation and/or general anesthesia in humans.
Awakening from ketamine-induced anesthesia occurs at plasma concentrations ranging from 640 to 1100 ng/ml or 2.7–4.7 μM (Idvall et al., 1979; Reich and Silvay, 1989). White et al. (1985) showed that administration of the racemic mixture of ketamine (5- to 7-minute i.v. infusion of 50 mg/min for a total dose of 275 ± 25 mg) induced general anesthesia in five healthy adult volunteers, as indicated by the absence of an eyelid reflex. Upon termination of the infusion, it took approximately 11 ± 3 minutes for the volunteers to open their eyes (1900–3300 ng/ml or 8.0–14 μM serum concentration), and approximately 45 ± 10 minutes for them to fully regain proper orientation of self, place, and time (3.78–4.62 μM serum concentration; White et al., 1985).
Intranasal (S)-ketamine at the doses of 3–9 mg/kg induces sedation in patients (Tsze et al., 2012). As an anesthetic for humans, (S)-ketamine is reported to be twice as potent as the racemic mixture and approximately three times more potent than (R)-ketamine (White et al., 1985; Schüttler et al., 1987; Himmelseher and Pfenninger, 1998). Specifically, the total i.v. dose required for the induction of anesthesia is 275 ± 25 mg for racemic ketamine, 140 ± 21 mg for (S)-ketamine, and 429 ± 37 mg for (R)-ketamine (White et al., 1985). Time needed to regain complete orientation of self, place, and time following a 5- to 7-minute i.v. administration of (S)-ketamine (25 mg/min; total dose: 140 ± 21 mg) or (R)-ketamine (75 mg/min; total dose: 429 ± 37 mg) was 21 ± 2 minutes (500–900 ng/ml or 2.1–3.8 μM serum concentration) and 18 ± 3 minutes (2200–3200 ng/ml or 9.3–13 μM serum concentration), respectively (White et al., 1985). These data indicate that the (S)-ketamine isomer is a more potent anesthetic compared with (R)-ketamine, given that a threefold higher dose of (R)-ketamine is required to elicit a comparable level of sedation. Additionally, the serum concentration of (R)-ketamine that caused half-maximal median frequency decrease (IC50) in electroencephalographic oscillations was measured to be 2000 ± 500 ng/ml (8.0 ± 2.0 µM), versus 1800 ± 500 ng/ml (7.6 ± 2.0 µM) for the racemic drug and 800 ± 400 ng/ml (3.4 ± 1.7 µM) for the (S)-ketamine isomer (Schüttler et al., 1987).
2. Analgesic
An early report of the analgesic effects of ketamine was provided by Weisman (1971), who observed these effects in pediatric ophthalmologic procedures (Weisman, 1971). Ketamine is described to provide a form of analgesia quantitatively and qualitatively similar to opioids, but with less respiratory depressive effects, as was reported in pediatric patients treated for fractures (Kennedy et al., 1998), burns (see McGuinness et al., 2011), or in cases of traumatic amputation (Bonanno, 2002). When administered i.v. or i.m., ketamine’s analgesic effects are associated with plasma concentrations ranging between 70 and 160 ng/ml, or approximately 0.29–0.67 μM (Clements and Nimmo, 1981; Grant et al., 1981; Clements et al., 1982; Flood and Krasowski, 2000).
Intravenous ketamine is used as an analgesic to reduce chronic and acute postoperative pain (Laskowski et al., 2011). Adequate analgesia is achieved at subanesthetic doses of ketamine, as low as 0.15–0.25 mg/kg, when administered i.v. (Roytblat et al., 1993; Backonja et al., 1994; Eide et al., 1994), or 0.5–1 mg/kg when administered i.m. to patients following acute trauma (Hirlinger and Dick, 1984). In addition, ketamine’s antinociceptive and analgesic effects have been observed when ketamine is administered as follows: 1) orally at the dose of 0.5 mg/kg twice per day for 15 days (as adjuvant to morphine; Lauretti et al., 1999) or at the single dose of 2 mg/kg (Marchetti et al., 2015); 2) intranasally at a dose ranging from 10 to 50 mg twice per day (Carr et al., 2004); 3) transdermally at the dose of 25 mg released throughout a 24-hour period (Azevedo et al., 2000); 4) s.c. at a dose ranging from 0.05 to 0.15 mg/kg per hour for 7 days (Eide et al., 1995); and 5) rectally at the dose of 10 mg/kg (Tanaka et al., 2000). Following oral dosing, lower ketamine concentrations in the blood may be required to achieve analgesia compared with the other routes of administration (maximum concentration, Cmax = 45 ± 10 ng/ml or 0.19 ± 0.04 μM; Grant et al., 1981). Continuous infusion of a subanesthetic dose of ketamine (titrated from 10 to 40 mg/h; maintained for 5 days) has been demonstrated to be effective in improving pain in patients suffering from complex regional pain syndrome (CRPS), resulting in plasma concentrations of both (S)- and (R)-ketamine ranging between 200 and 225 ng/ml (0.84–0.95 μM; Goldberg et al., 2010; Moaddel et al., 2010).
The use of intranasal (S)-ketamine as an analgesic may be of particular relevance in prehospital settings, where i.v. administration is difficult, and in cases where acute administration for injuries is required, because it reduces pain scores within 5 minutes following administration (Johansson et al., 2013). Similar to their differential anesthetic effects, there is evidence supporting that (S)-ketamine is a more potent analgesic drug compared with racemic ketamine and (R)-ketamine in humans, although (S)-ketamine also produces more side effects (Oye et al., 1992; Mathisen et al., 1995).
3. Antidepressant
Evidence of ketamine’s antidepressant actions dates back to the 1970s. In preclinical studies, ketamine was found to exert effects similar to those observed following administration of classic antidepressant drugs (i.e., tricyclic antidepressants and monoamine oxidase inhibitors) in rodents (Sofia and Harakal, 1975). In particular, oral administration of ketamine to mice reversed reserpine-induced hyperthermia at the dose of 40 mg/kg and prevented tetrabenazine-induced ptosis with an ED50 of 27.6 mg/kg (Sofia and Harakal, 1975), which are phenotypes reversed by classical antidepressants (Delini-Stula, 1980). Early evidence of ketamine’s possible antidepressant properties in humans was described in 1973 by Khorramzadeh and Lotfy (1973), who reported that i.v. ketamine at the subanesthetic doses of 0.2–1.0 mg/kg (i.v. bolus) resulted in emotional discharge and facilitation of psychotherapy in a cohort of 100 psychiatric inpatients. However, the precise depression symptoms that were improved with ketamine were not well delineated in the context of modern diagnostic criteria and therapeutic definitions. In this study, ketamine was in fact referred to as a general abreactive agent (Khorramzadeh and Lotfy, 1973).
The first placebo-controlled study suggesting ketamine has antidepressant actions was reported in 2000. Based on the results reported in that study, an i.v. 40-minute infusion of 0.5 mg/kg ketamine induced a robust and rapid antidepressant response in patients suffering from depression compared with placebo (Berman et al., 2000). This finding was subsequently replicated in a double-blind, placebo-controlled, randomized clinical trial involving patients suffering from treatment-refractory major depression (Zarate et al., 2006). In particular, Zarate et al. (2006) demonstrated that ketamine exerts an antidepressant effect that becomes evident within 2 hours postinfusion, and lasts for an average of 7 days in patients who have failed to respond to at least two prior classical antidepressant medications. Several other clinical trials have replicated these findings in patients suffering from treatment-refractory depression (e.g., Murrough et al., 2013a; Lapidus et al., 2014). To address the functional unblinding of treatment status due to the dissociative effects of ketamine, which occur even at low subanesthetic doses, Murrough et al. (2013a) used a psychoactive placebo (i.e., midazolam) and demonstrated a higher response rate for the patients who received ketamine (64%) compared with those who received midazolam (28%). Ketamine is also reported to exert antidepressant actions in patients suffering from bipolar depression (Diazgranados et al., 2010a; Zarate et al., 2012b). (S)-ketamine has been shown effective as an antidepressant administered both via i.v. and intranasal routes (Singh et al., 2016a; Daly et al., 2018; Canuso et al., 2018). Additional studies have shown that ketamine reduces suicidal ideation (Price et al., 2009; DiazGranados et al., 2010b; Ballard et al., 2014) and decreases anhedonia (Lally et al., 2014, 2015; Ballard et al., 2017) in patients suffering from major depression. Intranasal (S)-ketamine also decreased suicidal ideation in patients suffering from depression (Canuso et al., 2018).
The most commonly used subanesthetic antidepressant dose of ketamine (0.5 mg/kg; 40-minute infusion) results in a maximal plasma concentration (Cmax) of ∼185 ng/ml or ∼0.78 μM ketamine, as calculated from the results of Zarate et al. (2012a). Nevertheless, there is some evidence for antidepressant responses achieved at doses as low as 0.1 mg/kg (5-minute i.v. infusion or i.m. injection), resulting in ketamine Cmax of ∼75 ng/ml (0.32 μM—estimated) as reported in a small pilot (n = 15) double-blind, placebo-controlled crossover study in patients suffering from treatment-resistant depression (Loo et al., 2016). Although this study indicated that lower doses of ketamine, which produce fewer side effects, could be effective in the treatment of depression, this finding awaits replication in a larger study.
4. Anti-Inflammatory
Inflammation is a critical homeostatic mechanism used by the body to fight infections and to heal tissue injuries (Selye, 1976; Hirsiger et al., 2012). Inflammatory reactions are triggered once immune cells of the innate immune system become activated, whether by invading pathogens or tissue damage. Release of proinflammatory cytokines by these cells then activate members of the adaptive immune system to initiate an inflammatory response (Newton and Dixit, 2012).
Ketamine administration during or prior to surgical operations has been used for a more favorable postoperative outcome, primarily due to its actions to reduce the production of excess proinflammatory cytokines. Anti-inflammatory actions (i.e., reduction of proinflammatory cytokines) of preoperative subanesthetic doses of 0.15–0.25 mg/kg (single i.v. bolus) ketamine were described in humans (Roytblat et al., 1998; Beilin et al., 2007; Russabrov et al., 2008). Ketamine was shown to inhibit immune reaction-induced proinflammatory cytokine production, including nuclear factor κB, and to decrease blood levels of tumor necrosis factor-α, interleukin 6 (IL-6), C-reactive protein, and/or inducible nitric oxide synthase (Larsen et al., 1998; Kawasaki et al., 1999, 2001; Lankveld et al., 2005; Beilin et al., 2007; Loix et al., 2011; De Kock et al., 2013). The ability of ketamine to reduce proinflammatory cytokine levels may be of clinical relevance, given that elevated IL-6 levels have been associated with poor postoperative outcomes (Oka et al., 1992; Hennein et al., 1994; Cremer et al., 1996). However, this possibility awaits systematic investigation.
In addition to its effects on the proinflammatory cytokines, ketamine dose dependently reduces inflammation-induced nitric oxide production (Shimaoka et al., 1996; Li et al., 1997; Yang et al., 2005). The anti-inflammatory effects of ketamine have been observed when the drug was administered prior to, and following an immune stimulation, indicating that ketamine may be able to prevent exacerbation of inflammation, and also reduce existing inflammation (Loix et al., 2011). There is evidence that ketamine can alleviate postoperative trauma-induced hyperalgesia by modulating the inflammatory response, which is beneficial in the context of chronic postoperative pain (Stubhaug et al., 1997; De Kock et al., 2001; Suzuki et al., 2006; Remerand et al., 2009).
Ketamine has also been shown to correct abnormal inflammatory bone markers in major depressive disorder. In particular, a 40-minute i.v. infusion of ketamine (0.5 mg/kg) increased levels of the osteoprotegerin receptor activator of nuclear factor κB ligand and osteopontin—predictive markers of bone inflammation—in patients with major depressive disorder, but had no effect in healthy controls (Kadriu et al., 2017). Moreover, serum levels of the proinflammatory cytokines tumor necrosis factor-α, interferon γ, and interleukin 2, 5, and 10 were unaltered following a 40-minute i.v. subanesthetic infusion of ketamine (0.5 mg/kg) in patients suffering from depression, whereas levels of the anti-inflammatory cytokine IL-6 were reported to increase 230 minutes postketamine infusion (Park et al., 2017). However, this effect of ketamine on IL-6 levels was not associated with the antidepressant actions of the drug (Park et al., 2017). It is possible that the infusion itself led to an acute stress-related increase in IL-6 levels, given that this has been observed following saline infusion as well (Cho et al., 2009). Overall, these findings indicate that the anti-inflammatory actions of ketamine occur primarily in the presence of immunostimulation, whereas the drug does not exert any effects on cytokine balance in the absence of an inflammatory reaction (Loix et al., 2011). Thus, ketamine may act as an immunomodulator, and not as an immunosuppressive agent, which is of particular importance because ketamine is commonly administered during the induction of anesthesia, prior to surgery.
Relevant doses and plasma concentrations of ketamine used for clinical therapeutic effects are listed in Table 1.
Relevant doses and plasma concentrations of ketamine for its clinical use and side effects in humans
B. Side Effects
1. Psychoactive Effects
a. Dissociative and psychotomimetic effects
Ketamine dose dependently exerts broad influences on consciousness and perception, with some patients reporting dissociative and extracorporeal sensations (out-of-body experiences/illusions) when recovering from ketamine-induced anesthesia (Garfield et al., 1972; White et al., 1980, 1982). Whereas these effects of ketamine established the drug as a dissociative anesthetic (Domino et al., 1965), the same effects have been noted following subanesthetic doses as well (e.g., Krystal et al., 1994).
The most common psychoactive effects reported after a single subanesthetic i.v. administration of ketamine include dissociation (distortions in visual, auditory, or somatosensory stimuli, or alterations in the perception of self or time), positive psychotomimetic effects (conceptual disorganization, hallucinations, suspiciousness, unusual thought content), and negative psychotomimetic effects (blunted affect, emotional withdrawal, motor retardation). These effects were reported in both randomized controlled studies (e.g., Malhotra et al., 1996; Anand et al., 2000; Berman et al., 2000; Hetem et al., 2000; Abel et al., 2003; Zarate et al., 2006; Diazgranados et al., 2010a; Zarate et al., 2012b; Murrough et al., 2013b, 2015; Downey et al., 2016; Hu et al., 2016; Li et al., 2016) and nonrandomized or open label studies (e.g., Phelps et al., 2009; Mathew et al., 2010; Valentine et al., 2011; Ibrahim et al., 2012; Ionescu et al., 2015). For instance, a randomized, double-blind, placebo-controlled study by Krystal et al. (1994) showed that a 40-minute i.v. infusion of the subanesthetic dose of 0.5 mg/kg ketamine (resulting Cmax estimated to be ∼100–250 ng/ml or 0.42–1.1 µM) leads to perceptual aberrations that are consistent with dissociative states, as well as positive and negative psychotomimetic symptoms. These effects emerged within 10 minutes of the beginning of ketamine infusion and subsided within 40 minutes of treatment termination. In contrast, little to no psychoactive effects were observed at the dose of 0.1 mg/kg (resulting in ∼25–50 ng/ml or 0.1–0.2 µM plasma ketamine concentration; Krystal et al., 1994). Ketamine (0.3 mg/kg bolus; Cmax = ∼120 ng/ml or 0.5 µM) has also been shown to exacerbate psychotic symptoms in patients suffering from schizophrenia (Lahti et al., 2001). Similarly, Malhotra et al. (1997) also reported that ketamine increased psychotic symptoms in patients suffering from schizophrenia when given as a single i.v. bolus of 0.12 mg/kg, followed by a 60-minute infusion of 0.65 mg/kg (total dose 0.77 mg/kg).
Experiencing illusions and alterations in hearing, vision, and proprioception has been attributed to the actions of (S)-ketamine (Oye et al., 1992; Mathisen et al., 1995; Vollenweider et al., 1997), whereas feelings of relaxation were associated with the actions of (R)-ketamine (Vollenweider et al., 1997). In particular, at equimolar doses producing average plasma ketamine levels of 379 ± 71 ng/mg (i.e., 1.59 ± 0.30 μM) and 389 ± 74 ng/mg (i.e., 1.64 ± 0.31 μM) for (S)- and (R)-ketamine, respectively, the (S)-ketamine enantiomer caused acute psychotic reactions at a mean plasma ketamine level of 539 ng/ml (i.e., 2.27 μM), whereas (R)-ketamine was not associated with these psychotomimetic actions. In contrast, (R)-ketamine administration induced a feeling of “well-being” and a beneficial effect on mood as measured by the Eigenschaftsworterliste (EWL) mood rating scale (Vollenweider et al., 1997). A clinical study conducted by Mathisen et al. (1995) showed that 56% of patients who suffered from orofacial pain and were treated with (S)-ketamine (0.45 mg/kg, i.m.; serum Cmax = ∼120 ng/ml or 0.5 µM) experienced illusions, whereas only 22% of those treated with (R)-ketamine experienced illusions, even though a higher dose of (R)-ketamine was used (1.8 mg/kg, i.m.; serum Cmax = ∼590 ng/ml or 2.5 µM). In this study, the prevalence of illusions among patients treated with (S)-ketamine was comparable to that observed among patients treated with (R,S)-ketamine at a dose of 0.9 mg/kg, i.m.; serum Cmax = ∼297 ng/ml or 1.25 µM (Mathisen et al., 1995). Alterations in hearing were reported in 78%, 67%, and 57% of patients treated with (S)-, (R)-, and (R,S)-ketamine, respectively, whereas blurred vision was reported by 100%, 78%, and 85% of patients receiving (S)-, (R)-, and (R,S)-ketamine, respectively. Additionally, treatment with (S)-ketamine led to proprioceptive disturbances in 100% of patients, as compared with 56% and 71% of patients receiving either (R)- or (R,S)-ketamine, respectively (Mathisen et al., 1995). Although 43% of patients treated with (R,S)-ketamine reported dreams and hallucinations, neither effect was reported by patients treated with either (S)- or (R)-ketamine (Mathisen et al., 1995).
A study conducted in healthy volunteers showed no differences in the postanesthetic effects of (S)- (140 ± 21 mg), (R)- (429 ± 37 mg), or (R,S)-ketamine (275 ± 25 mg) in their propensity to elicit floating sensations (average 67% of individuals), diplopia (double vision; 60%), or dizziness (47%; White et al., 1985). These effects occurred at higher plasma concentrations of (R)-ketamine compared with (S)- and (R,S)-ketamine (White et al., 1985; Mathisen et al., 1995).
b. Memory and cognitive impairment
In addition to the dissociative and psychotomimetic symptoms, several studies have identified unfavorable effects of subanesthetic administration of ketamine on cognition (also see Ke et al., 2018). Studies have reported that ketamine decreases mental sharpness (Mathew et al., 2010), concentration (Pfenninger et al., 2002), recall and recognition (Malhotra et al., 1996), as well as explicit (episodic and semantic) and implicit (procedural) forms of memory (Harris et al., 1975; Ghoneim et al., 1985; Newcomer et al., 1999; Morgan et al., 2004; Honey et al., 2005; Driesen et al., 2013) either during or shortly after administration (for dosing details, see Table 1).
Vigilance, verbal fluency, and delayed recall are also impaired during/immediately following a 40-minute i.v. infusion of 0.5 mg/kg ketamine (resulting in plasma Cmax estimated to be ∼100–250 ng/ml or 0.42–1.1 µM); these effects subside shortly after termination of the infusion (Krystal et al., 1994). Global cognitive function and immediate recall appear to remain intact during ketamine infusion (Krystal et al., 1994). Based on results obtained from cross-sectional studies, long-term ketamine abuse is also associated with cognitive impairments (Morgan and Curran, 2012; Zhang et al., 2018; Morgan et al., 2004). However, the nature of these studies makes it difficult to fully control for the impact of other comorbid or environmental factors (Morgan and Curran, 2012; Zhang et al., 2018).
c. Abuse
Whereas the acute psychotropic effects of ketamine may cause discomfort for some individuals (Domino et al., 1965), its dissociative properties have made it desirable for recreational use (Siegel, 1978; Stewart, 2001). However, some users may experience increased agitation or anxiety/panic attacks (Siegel, 1978; Jansen, 2000; Weiner et al., 2000; Arditti et al., 2002). Within 10 minutes following initiation of a 40-minute i.v. infusion of a subanesthetic dose of 0.5 mg/kg ketamine (resulting in plasma Cmax estimated to be ∼100–250 ng/ml or 0.42–1.1 µM), healthy subjects reported feelings of being “high” (i.e., subjectively comparable to that of alcohol intoxication; Krystal et al., 1994). A lower ketamine dose of 0.1 mg/kg (resulting in plasma Cmax = ∼25–50 ng/ml or 0.1–0.2 µM) induced a mild euphoria (i.e., buzzing) feeling (Krystal et al., 1994).
Although controlled studies addressing the abuse potential of ketamine are lacking, valuable information about both the acute and chronic effects of ketamine has been derived from reports of recreational use (see Corazza et al., 2013). In general, doses used for recreational ketamine intake may range between 1 and 2 mg/kg (i.v.), 50 and 150 mg (i.m.), 100 and 500 mg (oral), or 30 and 400 mg (intranasal insufflation; Siegel, 1978; Dalgarno and Shewan, 1996; Jansen, 2000; Arditti et al., 2002; Wolff and Winstock, 2006; Bokor and Anderson, 2014). Although the effects of specific doses used for recreational use cannot be directly determined due to a lack of controlled studies assessing these, users report that lower doses induce mild stimulatory, dissociative, and hallucinogenic effects, whereas higher doses yield psychotomimetic symptoms and separation from reality (Stewart, 2001; Wolff and Winstock, 2006).
The most common route of recreational administration is nasal insufflation, with an onset of feeling “high” ranging between 5 and 10 minutes, and lasting between 40 and 75 minutes (Dalgarno and Shewan, 1996; Stewart, 2001; Wolff and Winstock, 2006). At peak levels of intake, users report that ketamine induces a highly dissociative experience marked by an altered state of consciousness and sensory detachment (colloquially referred to as the k-hole), which some describe as being comparable to a near-death experience (Jansen, 1989; Stewart, 2001; Wolff and Winstock, 2006; Bokor and Anderson, 2014).
At plasma concentrations ranging from 50 to 200 ng/ml (0.21–0.84 µM; Bowdle et al., 1998), ketamine dose dependently enhances sensory perception (i.e., intensity of sound), emotional connectedness, feelings of unreality, and out-of-body experiences, and may be associated with visual hallucinations, altered perceptions of self and time, and floating sensations (Hansen et al., 1988; Bowdle et al., 1998; Jansen, 2000; Muetzelfeldt et al., 2008; Wilkins et al., 2012). Undesired effects reported by illicit users include dizziness, blurred vision, slurred speech, vomiting, palpitations, and chest pain (Siegel, 1978; Dalgarno and Shewan, 1996; Weiner et al., 2000; Muetzelfeldt et al., 2008); see section on peripheral effects below. It has been hypothesized that diminished tactile and musculoskeletal sensations caused by ketamine lead to feelings of weightlessness or detachment from oneself, which may contribute to extracorporeal sensations (Collier, 1972; White et al., 1982). Additionally, long-term use of ketamine may lead to flashbacks, attentional and other cognitive dysfunctions, and decreased sociability, but continued use is reinforced by the other psychotropic effects (Siegel, 1978; Jansen, 2000; Zhang et al., 2018). Despite its reinforcing properties, instances of ketamine dependence are relatively scarce (Bobo and Miller, 2002; Lim, 2003; Blier et al., 2012), but have been reported (Morgan and Curran, 2012). There is also evidence to suggest that repeated use of ketamine may lead to drug tolerance (Dalgarno and Shewan, 1996; Jansen and Darracot-Cankovic, 2001; Pal et al., 2002).
2. Direct and Indirect Peripheral Effects
At subanesthetic doses (∼0.5 mg/kg administered i.v. over 40 minutes), ketamine can lead to vestibular perturbations, including dizziness (Wan et al., 2015) and nausea/vomiting (Ghoneim et al., 1985; Krystal et al., 1994; Morgan et al., 2004). Ketamine’s actions on the sympathetic nervous system (Traber and Wilson, 1969; Traber et al., 1970) are associated with broad cardiovascular outcomes (e.g., tachycardia, hypertension, palpitations) evident in both clinical (0.5–1.0 mg/kg i.v.; Strayer and Nelson, 2008; Murrough et al., 2013b) and recreational settings (100–200 mg i.m. or s.c.; Weiner et al., 2000). Although generally considered clinically insignificant, mild respiratory depression is reported at doses ranging from 0.39 to 3.0 mg/kg (Domino et al., 1965; Idvall et al., 1979; Bourke et al., 1987). Additionally, hemodynamic effects (i.e., arterial pressure and heart rate) have not been found to vary significantly among (S)-, (R)-, and (R,S)-ketamine (White et al., 1985), although at least one study suggests that (S)-ketamine specifically contributes to (R,S)-ketamine’s cardiovascular effects, such as increased blood pressure (Geisslinger et al., 1993). Overall, a recent retrospective analysis in individuals who received 684 i.v. ketamine infusions (0.5 mg/kg over 40 minutes) reported that alterations in blood pressure are modest, well tolerated, and clinically insignificant (Riva-Posse et al., 2018).
Ocular effects (e.g., nystagmus, diplopia, dilation) are reported in recreational contexts (Weiner et al., 2000; Stewart, 2001), as well as clinically, at subanesthetic doses of ketamine (e.g., 0.25 mg/kg i.v.; Backonja et al., 1994; Krystal et al., 1994). Some ocular effects (i.e., blurred vision) have been primarily associated with (S)-ketamine (Mathisen et al., 1995). Additionally, musculoskeletal effects (e.g., myoclonus, twitching, spasms, ataxia, fasciculation) have been noted in cases of ketamine abuse (Corssen and Domino, 1966; Felser and Orban, 1982; Wolff and Winstock, 2006; Bokor and Anderson, 2014).
Prolonged recreational use of ketamine is associated with urological complications that include dysuria, increased frequency and urgency of urination, incontinence, pain, hematuria, and ulcerative cystitis (Shahani et al., 2007; Chu et al., 2008; Tsai et al., 2009; Meng et al., 2013; Skeldon and Goldenberg, 2014). It has been suggested that ketamine may have a direct detrimental impact on the interstitial cells of the bladder, since cystoscopy has shown erythema, edema, and epithelial inflammation in long-term ketamine users (Shahani et al., 2007; Chu et al., 2008). Moreover, computer tomography revealed marked bladder wall thickening, mucosal enhancement, and perivesical inflammation associated with recreational ketamine use (Mason et al., 2010). There is at least one case report of subanesthetic ketamine (0.1 mg/kg per hour i.v. administration for 12 hours), being associated with urinary urgency and incontinence (Vickers et al., 2017).
3. Long-Term Effects
Given that ketamine’s maintenance of therapeutic efficacy often requires repeated administration of the drug (e.g., Blier et al., 2012; Segmiller et al., 2013; Szymkowicz et al., 2013), it is important to consider the side effects that may be uniquely associated with chronic ketamine exposure. The effects resulting from long-term ketamine treatment are either poorly defined or scarcely reported (reviewed by Short et al., 2018). To date, repeated ketamine abuse has been most consistently associated with long-lasting memory-related deficits (Morgan et al., 2006; Morgan and Curran, 2012; Zhang et al., 2018). Deaths caused by ketamine overdose, in the absence of multidrug intoxication, are very rare (Gill and Stajic, 2000; Jansen, 2000), although accidental deaths caused by falls from heights, extreme hypothermia, or car accidents involving individuals using ketamine have been reported (Gill and Stajic, 2000; Jansen, 2000; Jansen and Darracot-Cankovic, 2001).
Overall, there is no report, to our knowledge, involving a lethal dose of ketamine in humans. Nevertheless, in rats, intravenous administration of (R,S)-ketamine and (S)-ketamine at the dose of 40 mg/kg induced significant lethality; whereas, all animals that received (R)-ketamine at the same dose survived (Marietta et al., 1977). Although these findings indicate that caution should be taken when using ketamine treatment long-term, there is evidence that repeated administration of subanesthetic doses of ketamine may have beneficial long-term effects. For instance, repeated subanesthetic ketamine has been shown to improve clinical outcomes for treatment-resistant depression (Rasmussen et al., 2013; Loo et al., 2016; Cusin et al., 2017). Repeated ketamine administration has also been associated with attenuation of the acute ketamine-induced dissociation, derealization, and dizziness over time (Grott Zanicotti et al., 2013; Singh et al., 2016b). Nevertheless, dissociative and psychotomimetic effects have been observed in randomized controlled studies examining the effects of repeated i.v. (Lai et al., 2014; Loo et al., 2016; Singh et al., 2016b), i.m. (Loo et al., 2016), and s.c. (Loo et al., 2016; George et al., 2017) subanesthetic ketamine exposure.
Most, if not all, side effects of ketamine are dose dependent, transient, and self-resolving (Wan et al., 2015; Kishimoto et al., 2016; Loo et al., 2016). However, to more fully assess ketamine’s therapeutic utility across clinical contexts, future studies should aim to systematically assess the safety and efficacy of either acute or chronic ketamine treatment, in terms of both short- and long-term outcomes.
4. Neurotoxicity
With emerging indications requiring repeated ketamine administration (e.g., antidepressant actions), there are concerns of more profound untoward effects of treatment, including the induction of Olney lesions. First reported in 1989, Olney lesions are characterized by vacuoles occurring in the cytoplasmic compartment of selected neuronal populations, where lysis of mitochondria was reported (Olney et al., 1989, 1991). These neuronal vacuolation events occur primarily in the posterior cingulate and retrosplenial cortices following administration of N-methyl-D-aspartate receptor (NMDAR) antagonists (e.g., PCP, MK-801, and ketamine) in rats (Olney et al., 1989; Fix et al., 1993; Carliss et al., 2007). At low doses, vacuolation appears to reverse within 24 hours of administration, suggesting that permanent cell damage does not occur when noncompetitive (PCP, MK-801, ketamine, and dextrorphan) or competitive (CPP, CGS 19755, and CGP 37849) NMDAR antagonists are used at clinically relevant doses (Olney et al., 1989; Allen and Iversen, 1990; Hargreaves et al., 1994). However, there remains a possibility that high doses (or perhaps repeated administration at low doses) of NMDAR antagonists, such as ketamine, could lead to selective irreversible damage. For instance, preclinical studies in rats have shown that administration of a high dose of MK-801 (i.e., 5 mg/kg) leads to necrosis in a small subset of neurons (Allen and Iversen, 1990; Auer, 1996; Kuroda et al., 2015)—an effect that was associated with an age-dependent increase in mortality rate (Auer, 1996). Additionally, studies in nonhuman primates have reported that repeated daily ketamine administration (1 mg/kg per day, i.v.): 1) reduced white matter integrity in fronto-thalamo-temporal connections as assessed by diffusion tensor imaging following a 3-month treatment (Li et al., 2017), and 2) increased cell death in the prefrontal cortex as assessed by terminal deoxynucleotidyl transferase-mediated digoxigenin-deoxyuridine nick-end labeling staining of brain sections obtained from animals treated for 6 months (Sun et al., 2014).
Vacuolation was found to occur following administration of high (i.e., 40–60 mg/kg, s.c.) but not low-to-moderate doses (5–20 mg/kg s.c.) of ketamine in rats (Olney et al., 1989; Jevtovic-Todorovic et al., 2001). Notably, these doses are much higher than the doses required for the analgesic, anti-inflammatory, or antidepressant actions of the drug. Therefore, the relevance of Olney lesions to human repeated ketamine use is controversial and difficult to assess. One magnetic resonance imaging study reported that recreational ketamine users (total time of ketamine use: 0.5–12 years) presented with cortical atrophy in the frontal, parietal, and occipital lobes, and that measurable atrophies were associated with initiation of drug use occurring 2–4 years prior (Wang et al., 2013). In addition, another study in recreational users (total time of ketamine use: 1–10.5 years) reported a loss of frontal cortical white matter microstructure integrity that was correlated with total lifetime ketamine use (Liao et al., 2010).
Yeung et al. (2010) reported the presence of hyperphosphorylated tau (microtubule associated protein)-positive cells in the prefrontal and entorhinal cortices of nonhuman primates and mice receiving daily administrations of ketamine (1 mg/kg, i.v. bolus for monkeys and 30 mg/kg, i.p. injections for mice) across a period of 3–6 months. Tau hyperphosphorylation has been associated with the memory decline observed in Alzheimer’s disease patients (Augustinack et al., 2002; Huang and Jiang, 2009), possibly indicating a mechanism underlying memory impairment following ketamine use (see Memory and cognitive impairment section). Moreover, chronic intermittent administration of (S)-ketamine resulted in a loss of parvalbumin immunoreactivity in the hippocampus and prefrontal cortex of mice (Yang et al., 2016), consistent with findings in animal models of psychosis and schizophrenia (Lodge et al., 2009; Gonzalez-Burgos et al., 2015). In line with the lower potency of (R)-ketamine to inhibit the NMDARs compared with the (S)-ketamine enantiomer (see section N-Methyl-D-Aspartate Receptors), chronic intermittent administration of (R)-ketamine, unlike that of (S)-ketamine (both administered at 10 mg/kg i.p., once per week for a total period of 8 weeks in mice), resulted in no loss of parvalbumin immunoreactivity (Yang et al., 2016). Overall, considering the expanding applications of ketamine, it will be critical to further define the long-term effects of chronic ketamine use.
Relevant doses and plasma concentrations of ketamine that result in untoward side effects in humans are listed in Table 1.
II. Pharmacokinetics
A. Metabolism
Ketamine undergoes extensive metabolism (Fig. 1), initially via nitrogen demethylation to norketamine, a reaction that is catalyzed primarily by the cytochrome P450 liver enzymes CYP2B6 and CYP3A4 (Kharasch and Labroo, 1992; Yanagihara et al., 2001; Hijazi and Boulieu, 2002; Portmann et al., 2010; Mossner et al., 2011; Desta et al., 2012; Rao et al., 2016). The demethylation of ketamine occurs in a stereoselective manner, as CYP3A4 demethylates the (S)-ketamine enantiomer more rapidly than the (R)-ketamine enantiomer, whereas CYP2B6 demethylates both enantiomers of ketamine with near equal efficiency (Portmann et al., 2010). The individual variability in the metabolism of ketamine (Hijazi and Boulieu, 2002; Cheng et al., 2007; Desta et al., 2012) has been attributed, in part, to differences in the expression of P450 enzymes (Shimada et al., 1994; Hijazi and Boulieu, 2002).

Major metabolic pathways. In the predominant metabolic pathway, racemic ketamine [(R,S)-KET] is initially metabolized to norketamine [(R,S)-norKET], by either CYP2B6 or CYP3A4. Subsequently, norketamine can be further metabolized to form DHNK or the HNKs. Hydroxylation of norketamine at the six position by CYP2A6 results in (2R,6S;2S,6S)-hydroxynorketamine [(2R,6R;2S,6S)-HNK]. Alternatively, CYP2B6 or CYP2A6 can hydroxylate norketamine at the four position, resulting in the 4-hydroxy isomers. In the third case, CYP2B6 can hydroxylate norketamine at the five position, resulting in (2R,5S;2S,5R)-HNK and (2R,5R;2S,5S)-HNK. (R,S)-DHNK can result either from direct dehydrogenation from norketamine via CYP2B6 or via dehydration from either diastereomer of the 5-hydroxynorketamines via a nonbiologically catalyzed process.
Following demethylation of ketamine to norketamine, norketamine is further metabolized to the hydroxynorketamines (HNKs) and dehydronorketamine (DHNK) (Fig. 1). Early studies noted that the HNKs are formed through the hydroxylation of the cyclohexyl ring of norketamine at various locations (Adams et al., 1981). Several of these HNK metabolites have been detected in humans following ketamine infusion, with (2R,6R;2S,6S)-HNK and (2S,6R;2R,6S)-HNK being the predominant circulating HNKs in plasma (Moaddel et al., 2010; Zarate et al., 2012a). Metabolism to (2R,6R;2S,6S)-HNK is primarily carried out by CYP2A6 and CYP2B6 (Moaddel et al., 2010; Desta et al., 2012). These enzymes are also responsible for the formation of the (2S,4S;2R,4R)- and (2S,5S;2R,5R)-HNKs. CYP3A4 and CYP3A5 are the principal enzymes identified to catalyze the conversion of norketamine to (2S,4R;2R,4S)-HNK, whereas CYP2B6 is predominantly responsible for the catalysis of the conversion of norketamine to (2S,5R;2R,5S)-HNK (Desta et al., 2012). The other secondary metabolite is DHNK (Chang and Glazko, 1972; Adams et al., 1981). DHNK is directly formed from norketamine primarily via the actions of the CYP2B6 enzyme, or from 5-HNK via a nonenzymatic dehydration event (Adams et al., 1981; Bolze and Boulieu, 1998; Turfus et al., 2009; Portmann et al., 2010; Desta et al., 2012).
In addition to the major metabolic pathways of ketamine, there are several other pathways that have also been studied (Fig. 2). One of these pathways is the direct hydroxylation of ketamine to 6-hydroxyketamine (HK) (Woolf and Adams, 1987; Desta et al., 2012). Metabolism of ketamine to (2R,6R;2S,6S)-HK is primarily catalyzed by CYP2A6, whereas (2S,6R;2R,6S)-HK production is catalyzed by the CYP3A4 and CYP3A5 enzymes (Desta et al., 2012). The formation of (2R,6R;2S,6S)-HK is associated with greater hydroxylation of (S)-ketamine relative to (R)-ketamine, suggesting this reaction is enantioselective (Desta et al., 2012). The (2R,6R;2S,6S)-HK metabolite is readily demethylated via CYP2B6 to the corresponding HNK (Desta et al., 2012). However, analogous demethylation from the (2S,6R;2R,6S)-HK metabolite is reported to occur very slowly, with a modest contribution from CYP3A5. In addition to 6-HKs, evidence for the production of the 4-HK metabolite has been reported (Adams et al., 1981; Moaddel et al., 2010; Desta et al., 2012; Zarate et al., 2012a). Whereas hydroxylation of the phenyl ring was initially ruled out as being part of the metabolism of ketamine, more recent studies have provided evidence for the formation of such hydroxyphenyl ketamine metabolites via the actions of the CYP2C9 [primarily for the (R)-ketamine enantiomer] and flavin-containing mono-oxygenase enzymes [primarily for the (S)-ketamine enantiomer; Desta et al., 2012]. Lastly, phenolic isomers of HNKs have also been observed, potentially resulting from the hydroxylation of norketamine (Turfus et al., 2009).

Minor metabolic pathways. Although the majority of ketamine is metabolized via the major metabolic pathways (Fig. 1), there are several minor metabolic pathways, which provide unique, albeit low abundance, ketamine metabolites. The aryl ring of ketamine can be directly hydroxylated by flavin-containing mono-oxygenase enzymes or CYP2C9 to provide hydroxyphenyl-ketamine (hydroxyphenyl-KET). 4-Hydroxyketamine has also been observed; however, the metabolic enzymes responsible for this are currently unknown. CYP3A5 can directly hydroxylate ketamine at the six position to provide (2R,6S;2S,6R)-HK. Demethylation of (2R,6S;2S,6R)-HK with CYP3A5 provides (2R,6S;2S,6R)-HNK. CYP2A6 can also directly hydroxylate ketamine to provide (2R,6R;2S,6S)-HK, which is then transformed to (2R,6R;2S,6S)-HNK. Finally, norketamine can be hydroxylated via an unknown enzyme directly on the aryl rich to provide hydroxyphenyl-norketamine (hydroxyphenyl-norKET).
A population pharmacokinetic model was constructed for ketamine and its metabolites in patients suffering from treatment-resistant bipolar depression in a study that identified norketamine, DHNK, and (2R,6R;2S,6S)-HNK as the major circulating metabolites in plasma following a single 40-minute i.v. infusion of ketamine (0.5 mg/kg) (Zhao et al., 2012). These were also the major metabolites identified in plasma of patients suffering from unipolar or bipolar depression (Zarate et al., 2012a) or CRPS (Moaddel et al., 2010) and treated with ketamine. Specifically, norketamine, DHNK, and (2R,6R;2S,6S)-HNK were detected in the plasma of patients suffering from treatment-resistant unipolar and bipolar depression as early as 40 minutes after the end of i.v. ketamine administration (0.5 mg/kg delivered during a single 40-minute infusion; Zarate et al., 2012a; Zhao et al., 2012). The average time for metabolites to reach peak plasma concentration was estimated to be approximately 1.33 hours for both (R)- and (S)-norketamine and 3.83 hours for (R)-DHNK, (S)-DHNK, and (2R,6R;2S,6S)-HNK (Zhao et al., 2012). In plasma samples from these patients, the ratios of (S)- to (R)-ketamine, (S)- to (R)-norketamine, and (S)- to (R)-DHNK were 0.84, 1.0, and 0.67, respectively, during a 40- to 230-minute postinfusion period (Zhao et al., 2012). Similar to these findings, i.v. administration of ketamine (2 mg/kg) in surgical patients resulted in a plasma ratio of (S)- to (R)-ketamine of 0.91 (Geisslinger et al., 1993). Additionally, CRPS patients receiving continuous i.v. ketamine infusion at a dose of 40 mg/h over a total period of 5 days had plasma ratios of (S)- to (R)-ketamine, (S)- to (R)-norketamine, and (S)- to (R)-DHNK of 0.77, 0.71, and 0.71, respectively (Moaddel et al., 2010).
Following a 40-minute i.v. ketamine infusion at 0.5 mg/kg in patients diagnosed with treatment-resistant major depressive disorder, peak plasma concentrations were 204.13 ± 101.46 ng/ml or 0.86 ± 0.43 μM for ketamine (at 40 minutes), 73.54 ± 31.86 ng/ml or 0.33 ± 0.14 μM for norketamine (at 80 minutes), 13.27 ± 6.92 ng/ml or 0.06 ± 0.03 μM for DHNK (at 110 minutes), and 23.19 ± 11.88 ng/ml or 0.097 ± 0.05 μM for (2R,6R;2S,6S)-HNK (at 230 minutes) (Zarate et al., 2012a); see Table 2. In patients with treatment-resistant bipolar depression, peak plasma concentrations were 177.23 ± 53.8 ng/ml or 0.75 ± 0.23 μM for ketamine (at 40 minutes), 69.96 ± 19.98 ng/ml or 0.31 ± 0.09 μM for norketamine (at 80 minutes), 50.5 ± 27.44 ng/ml or 0.23 ± 0.12 μM for DHNK (at 110 minutes), and 37.59 ± 14.23 or 0.16 ± 0.06 μM for (2R,6R;2S,6S)-HNK (Zarate et al., 2012a; Table 2). In a patient with CRPS receiving chronic ketamine treatment (infusion beginning at 10 mg/h, titrated to 40 mg/h, and lasting 5 consecutive days), significant plasma levels of several HNK metabolites were detected, with (2R,6R;2S,6S)- and (2R,6R;2S,6S)-HNKs being the major metabolites present in samples obtained on day 3 (Moaddel et al., 2010).
Pharmacokinetic comparison for (2R,6R) and (2S,6S)-HNK in humans and rodents
Values represent mean ± S.D.
In a study conducted by Cohen et al. (1973), brain concentrations of ketamine metabolites were measured following tail vein administration of ketamine (20 mg/kg) in rats. These authors showed that both ketamine and norketamine rapidly accumulated in the brain with peak concentrations achieved within 1 minute of administration (Cohen et al., 1973). Subsequently, it was demonstrated that (2R,6R;2S,6S)-HNK also accumulates in brain tissue shortly after dosing (Leung and Baillie, 1986; Paul et al., 2014; Moaddel et al., 2015b). Intravenous tail vein injection (2-minute infusion) of 20 mg/kg (S)- or (R)-ketamine to rats resulted in higher brain levels of (2S,6S)-HNK relative to (2R,6R)-HNK, respectively, with maximal concentrations of 769 ± 133 ng/g or 3.21 ± 0.55 μmol/kg at 20 minutes for (2S,6S)-HNK and 274 ± 47 ng/g or 1.14 ± 0.20 μmol/kg at 10 minutes for (2R,6R)-HNK (Moaddel et al., 2015b; Table 2). It was hypothesized that the difference was due to a passive uptake process of these metabolites into the brain (Moaddel et al., 2015b). A ∼1:1 ratio for the plasma:brain levels of the corresponding (2R,6R;2S,6S)-HNK was observed, indicating that blood-brain barrier penetration or the central nervous system transport process was not mediated by an enantioselective process (Leung and Baillie, 1986; Moaddel et al., 2015b). Importantly, no in situ metabolism was observed when ketamine was incubated with rat brain microsomes (S9 fraction) (Moaddel et al., 2015b). Likewise, ketamine metabolites were below detectable levels in the brain of mice following in vivo intracerebroventricular administration of ketamine (P.Z., R.M., J.N.H., T.D.G., unpublished data), a finding that suggests that local ketamine metabolism does not occur in the brain.
In mice, norketamine, DHNK, and (2R,6R;2S,6S)-HNK metabolites were detected in plasma within 10 minutes of i.p. administration of 10 mg/kg ketamine (Can et al., 2016; Zanos et al., 2016). The maximum plasma concentrations were 561.89 ± 86.09 ng/ml or 2.36 ± 0.18 μM at 10 minutes for ketamine, 1098.89 ± 216.89 ng/ml or 4.91 ± 0.97 μM at 10 minutes for norketamine, 83.92 ± 53.63 ng/ml or 0.38 ± 0.24 μM at 30 minutes for DHNK, and 674.59 ± 278.23 ng/ml or 2.81 ± 1.16 μM at 10 minutes for (2R,6R;2S,6S)-HNK (Zanos et al., 2016), as summarized in Table 2. In the brain, ketamine (1162.34 ± 202.05 ng/g or 4.89 ± 0.85 μmol/kg tissue), norketamine (450.94 ± 199.7 ng/g or 2.02 ± 0.89 μmol/kg tissue), and (2R,6R;2S,6S)-HNK (498.35 ± 50.99 ng/g or 2.08 ± 0.21 μmol/kg tissue) were detected within 10 minutes of ketamine administration (Zanos et al., 2016). The maximum brain concentration of ketamine was 51.66% higher than the corresponding plasma concentration, whereas the brain tissue concentrations of norketamine and (2R,6R;2S,6S)-HNK were 58.96% and 26.13% lower than the corresponding maximum plasma concentrations, respectively (Zanos et al., 2016). Levels of DHNK in brain tissue were below the limits of quantification, consistent with the findings that DHNK partitions into red blood cells (Moaddel et al., 2016) and has poor penetration of the blood-brain barrier (Can et al., 2016).
B. Absorption
Ketamine is administered to humans via multiple routes, including i.v., i.m., oral, intranasal, epidural, and intrarectal (Malinovsky et al., 1996; Andrade, 2017b). The most typical route of administration is via i.v. infusion, which rapidly attains maximum plasma concentrations (e.g., Clements et al., 1982; Weber et al., 2004). Intramuscular administration, which is used in emergency cases of uncooperative patients, neonates, and children, has high bioavailability of 93%, with peak plasma concentrations achieved within 5–30 minutes of administration (e.g., Clements et al., 1982); however, a population pharmacokinetic analysis reported a much lower bioavailability following i.m. administration of ketamine in children (41%; Hornik et al., 2013). In contrast, oral bioavailability of ketamine is limited to 16%–29%, with peak concentration levels of the drug occurring within 20–120 minutes (Grant et al., 1981; Clements et al., 1982; Sekerci et al., 1996; Chong et al., 2009; Rolan et al., 2014; Karch and Drummer, 2015), due to extensive first-pass hepatic metabolism (e.g., Kharasch and Labroo, 1992; Yanagihara et al., 2003). Oral bioavailability of (S)-ketamine was calculated to be 8%–11% (Peltoniemi et al., 2012; Fanta et al., 2015), consistent with the greater first-pass metabolism of (S)-ketamine relative to (R,S)-ketamine. Intranasal and intrarectal ketamine bioavailability is 45%–50% and 25%–30%, respectively (Malinovsky et al., 1996; Yanagihara et al., 2003). Intranasal administration is considered an attractive alternative to the i.v. administration of ketamine because it is less invasive, results in rapid systemic absorption, and is not subject to first-pass hepatic metabolism (Malinovsky et al., 1996).
Following oral administration of (2S,6S)-HNK in rats (20 mg/kg), maximum plasma concentrations were reached at 0.4 ± 0.1 hour. Oral bioavailability of (2S,6S)-HNK was estimated to be 46.3% in rats (Moaddel et al., 2015b). In mice, the oral bioavailability of (2R,6R)-HNK is estimated to be approximately 50% at the dose of 50 mg/kg (P.Z., R.M., J.N.H., T.D.G., unpublished data). The oral bioavailability of other ketamine metabolites remains to be determined.
C. Distribution
Ketamine is rapidly distributed into highly perfused tissues, including the brain, and has a plasma protein binding between 10% and 50% (Wieber et al., 1975; Dayton et al., 1983; Sinner and Graf, 2008; Peltoniemi et al., 2012, 2016; Karch and Drummer, 2015), resulting in a large steady-state volume of distribution (Vd = 3–5 l/kg; Karch and Drummer, 2015). A single i.v. bolus administration of an anesthetic dose of racemic ketamine in humans (2 mg/kg) leads to equal plasma concentrations of (S)-ketamine and (R)-ketamine 1 minute postadministration (Cmax = ∼1800 ng/ml or 7.6 µM—estimated from Geisslinger et al., 1993). However, i.v. (bolus) administration of 1 mg/kg (S)-ketamine resulted in a higher plasma concentration of the drug 1 minute postinfusion (Cmax = ∼2600 ng/ml: 11 µM—estimated from Geisslinger et al., 1993). These results are particularly important when comparing the outcomes of (S)-ketamine with those of the racemic ketamine or (R)-ketamine, because lower doses of (S)-ketamine are required to produce similar or greater ketamine concentrations in the plasma (e.g., White et al., 1985; Mathisen et al., 1995). Notably, there is no interconversion between (S)- and (R)-ketamine, because administration of (S)-ketamine does not result in the formation of (R)-ketamine in vivo, and vice versa (Geisslinger et al., 1993; Ihmsen et al., 2001). Plasma of patients suffering from treatment-resistant bipolar depression, who were treated with a 40-minute i.v. infusion of 0.5 mg/kg (R,S)-ketamine, had a ratio of (S)- to (R)-ketamine of 0.84 (Zhao et al., 2012), with peak ketamine concentrations of 177.23 ± 53.8 ng/ml or 0.75 ± 0.23 μM (Zarate et al., 2012a).
In mice, administration of subanesthetic doses of either (S)- or (R)-ketamine (10 mg/kg, i.p.) resulted in similar brain levels of both drugs [area under the curve (AUC)last = 483.1 hours.ng/ml or 2.03 hours.μmol/kg versus 591.9 hours.ng/ml or 2.48 hours.μmol/kg, respectively], with peak levels being Cmax = 1743 ± 560.6 ng/g or 7.33 ± 2.36 μmol/kg for (S)-ketamine and 1886 ± 459.6 ng/ml or 7.93 ± 1.93 for (R)-ketamine at 10 minutes postinjection (Zanos et al., 2016). Similarly, there were no differences in (S)-ketamine (Cmax = 2732 ± 535 ng/ml or 11.49 ± 2.25 μM) and (R)-ketamine (Cmax = 3430 ± 400 ng/ml or 14.4 ± 1.68 μM) levels in the plasma of rats 10 minutes following an i.v. administration of 20 mg/kg of each of these enantiomers (Moaddel et al., 2015b).
Direct i.p. administration of (2R,6R)-HNK and (2S,6S)-HNK in mice results in a 1:1 ratio between circulating plasma and brain tissue concentrations (Table 2), with higher total levels (AUClast) of (2S,6S)-HNK observed in plasma and brain tissue compared with (2R,6R)-HNK (brain: 7.55 versus 3.05 h.μmol/kg; plasma: 11.60 versus 3.22 h.μM; Zanos et al., 2016; Table 2). Following i.v. administration of (2S,6S)-HNK in rats (20 mg/kg), total drug exposure was calculated as AUClast = 28,981 ± 6162 h.ng/ml or 120.91 ± 25.71 h.μM, with a volume of distribution Vd = 7.35 ± 0.74 l/kg (Moaddel et al., 2015b). Following oral administration of (2S,6S)-HNK to rats (20 mg/kg), total drug exposure was AUClast = 10,120 ± 1313 h.ng/ml or 42.22 ± 5.48 h.μM (Moaddel et al., 2015b).
D. Elimination
Although plasma levels of ketamine are below detectable limits within 1 day following an i.v. antidepressant dose of ketamine (0.5 mg/kg administered over a 40-minute infusion), circulating levels of DHNK and (2R,6R;2S,6S)-HNK were observed for up to 3 days after ketamine infusion in patients diagnosed with bipolar depression (Zhao et al., 2012) or treatment-resistant major depression (Zarate et al., 2012a). Norketamine and ketamine were detectable for up to 14 and 11 days, respectively, in the urine of children who received anesthetic doses of ketamine, with reported concentrations of 0.1–1442 ng/ml (or 0.0004–0.031 μΜ) for norketamine and 2–1204 ng/ml (or 0.008–5.06 μM) for ketamine (Adamowicz and Kala, 2005).
In adult humans, ketamine has a high rate of clearance and a short elimination half-life (2–4 hours; Clements et al., 1982; White et al., 1985; Domino, 2010). White et al. (1985) also demonstrated a short elimination half-life (155–158 minutes) for both (S)-ketamine and (R)-ketamine. Elimination of ketamine is primarily performed by the kidneys, with low levels excreted as ketamine (2%), norketamine (2%), and DHNK (16%) (Haas and Harper, 1992; Lin and Lua, 2004; Adamowicz and Kala, 2005; Karch and Drummer, 2015; Dinis-Oliveira, 2017). The majority of the drug (∼80%) is excreted as the glucuronic acid-labile conjugates of HK and HNK (Dinis-Oliveira, 2017), which are eliminated in urine and bile (Chang and Glazko, 1974).
In adult humans, terminal plasma half-life and the clearance rates of ketamine do not significantly differ between i.v. (half-life = 186 minutes; total body clearance = 19.1 ml/min per kilogram) and intramuscular (half-life = 155 minute; total body clearance = 23.2 ml/min per kilogram) routes of administration (Clements et al., 1982). However, there is evidence that repeated administration of ketamine prolongs its elimination time. For example, Adamowicz and Kala (2005) reported that, among three instances of single i.v. infusions of ketamine during a 2-year period (doses ranged from 0.75 to 1.59 mg/kg), the elimination of ketamine was slowed from 2 days following the first infusion to 5 days after the second, and 11 days following the third. Elimination of norketamine remained constant (i.e., 5 days after each infusion; Adamowicz and Kala, 2005).
When compared with adults, ketamine is eliminated approximately twice as fast in children (Haas and Harper, 1992). This is in accordance with evidence supporting a longer duration of anesthesia in adults relative to children following i.m. administration of 6 mg/kg ketamine (Grant et al., 1981, 1983; Akin et al., 2005). Moreover, a negative correlation between age and ketamine dose per body weight required for anesthesia was reported in children (Lockhart and Nelson, 1974). These differences might be due to differences in the enzymatic metabolism of ketamine in children, as compared with adults (Edginton et al., 2006).
In humans, (S)-ketamine has a slightly longer elimination half-life than racemic ketamine [∼5 hours for (S)-ketamine versus 2–4 hours for racemic ketamine; Hagelberg et al., 2010; Peltoniemi et al., 2012], and its systemic clearance is faster when administered alone than when administered in the racemic mixture [26.3 ± 3.5 ml/kg per minute for (S)-ketamine versus 14.8 ± 1.7 ml/kg per minute when administered as the racemic ketamine; Ihmsen et al., 2001]. This may suggest an inhibition of (S)-ketamine’s clearance by the (R)-ketamine enantiomer when the racemic mixture is administered (Kharasch and Labroo, 1992). Such inhibition could contribute to the prolonged awakening time in patients receiving racemic ketamine relative to those receiving (S)-ketamine (White et al., 1985). We note that systemic clearance of (R)-ketamine following racemic ketamine administration is 13.8 ± 1.3 ml/kg per minute, which is similar to the (S)-ketamine enantiomer (14.8 ± 1.7 ml/kg per minute; Ihmsen et al., 2001).
Following i.v. administration of (2S,6S)-HNK in rats (20 mg/kg), the clearance rate was calculated to be 704 ± 139 ml/kg per hour, with an elimination half-life of 8.0 ± 4.0 hours. Oral administration of this metabolite resulted in an elimination half-life of 3.8 ± 0.6 hours (Moaddel et al., 2015b).
Overall, it is important to note that there are important species differences in regard to half-life values, AUCs, Cmax, and clearance rates of ketamine and its metabolites (see Table 2; also see Zarate et al., 2012a; Zanos et al., 2017c). This should be taken into consideration when comparing the behavioral actions of specific dose regimens for ketamine and its metabolites in mice, rats, and humans. Nevertheless, the brain levels of ketamine and its metabolites following administration of ketamine in humans are not known, and, therefore, direct comparisons are not straightforward.
III. Pharmacodynamics of Ketamine and Its Metabolites
As aforementioned, ketamine is a NMDAR antagonist, and ketamine’s well-characterized analgesic and anesthetic effects are primarily attributed to NMDAR inhibition (Franks and Lieb, 1994). However, ketamine’s pharmacological targets are not limited to NMDARs. It has been reported that ketamine interacts with several other receptors and ion channels, including dopamine, serotonin, sigma, opioid, and cholinergic receptors, as well as hyperpolarization-activated cyclic nucleotide-gated (HCN) channels. Ketamine typically has a lower affinity (higher inhibitory constant—Ki—values) for these receptors and channels compared with NMDARs, and independent laboratories have not validated many of the reported findings.
Early pharmacodynamic studies of (R,S)-ketamine were conducted in rats and examined the anesthetic effects of the parent compound and its two principal metabolites, (R,S)-norketamine and (2R,6R;2S,6S)-HNK (Leung and Baillie, 1986). The results demonstrated that a 40 mg/kg i.v. bolus administration of (R,S)-ketamine and (R,S)-norketamine produced anesthetic actions and increased spontaneous locomotor activity during the postanesthetic recovery phase, whereas (2R,6R;2S,6S)-HNK (same dose) had no anesthetic or hyperlocomotor effects. As a result, (2R,6R;2S,6S)-HNK was described as an inactive metabolite, and the majority of the pharmacodynamic assessments were carried out with only (R,S)-ketamine and (R,S)-norketamine. However, it has been recently demonstrated that ketamine’s HNK metabolites are biologically active (Moaddel et al., 2013; Singh et al., 2013, 2015, 2016c; Paul et al., 2014; Zanos et al., 2016; Cavalleri et al., 2017; Yao et al., 2017; Wray et al., 2018). The (2S,6S)- and (2R,6R)-HNK metabolites have been shown to exert antidepressant-relevant behavioral responses in rodents (Zanos et al., 2016; Pham et al., 2017a, but see Shirayama and Hashimoto, 2018, as well as Yang et al., 2017). Consistent with the more potent antidepressant actions of (R)-ketamine compared with the (S)-ketamine enantiomer, (2R,6R)-HNK was shown to be a more potent antidepressant than (2S,6S)-HNK in several animal tests (Zanos et al., 2016).
A. N-Methyl-D-Aspartate Receptors
Historically, the primary recognized receptor target of ketamine is the NMDAR, in which ketamine acts as a noncompetitive open-channel blocker (Lodge et al., 1982; Anis et al., 1983; MacDonald et al., 1987). NMDARs are glutamatergic ion channels made of different combinations of four subunits encoded by one of seven genes: GluN1, GluN2A–D, and GluN3A–B (Vyklicky et al., 2014). NMDARs are highly permeable to calcium ions, which can trigger the activation of a number of intracellular pathways in neurons and glial cells. At resting state, NMDAR channels are tonically blocked by magnesium (Mg2+). Efficient receptor activation requires the following: 1) membrane depolarization, which displaces the Mg2+ block, and 2) binding of both glutamate and the coactivator glycine and/or D-serine (Paoletti et al., 2013).
Ketamine was initially characterized as a NMDAR antagonist by David Lodge and colleagues (Lodge et al., 1982; Anis et al., 1983), a finding that was subsequently confirmed by other investigators (Harrison and Simmonds, 1985; Thomson et al., 1985). Ketamine binds to the allosteric phencyclidine (PCP) site that is located within the channel pore of the NMDAR, and thus it blocks the receptor noncompetitively (Kohrs and Durieux, 1998; Mion and Villevieille, 2013). Ketamine has a relatively high (∼86%) trapping capability (binding within the ion channel pore following closure of the channel) to block NMDARs, via binding to the same site as PCP (>98% trapping) and MK-801 (100% trapping; Huettner and Bean, 1988; Lerma et al., 1991; MacDonald et al., 1991; Jahr, 1992; Orser et al., 1997). The binding affinity of ketamine to the PCP binding site has been reported to be between 0.18 and 3.1 μM in the presence of Mg2+ (Table 3; Wong et al., 1986, 1988; MacDonald et al., 1987; Kornhuber et al., 1989; Reynolds and Miller, 1989; Sharif et al., 1991; Bresink et al., 1995; Lynch et al., 1995; Parsons et al., 1995; Kapur and Seeman, 2001, 2002; Sun and Wessinger, 2004; Seeman et al., 2005; Gilling et al., 2009; Moaddel et al., 2013; Bonifazi et al., 2015; Wallach et al., 2016; Kang et al., 2017; Morris et al., 2017).
Molecular targets of ketamine and its metabolites
Values represent mean ± S.E., unless otherwise indicated.
NMDAR blockade is thought to underlie the dissociative anesthetic and amnesic effects of ketamine, as well as the antidepressant, analgesic, and altered psychotomimetic effects induced by the drug (White et al., 1980; Oye et al., 1992; Yeung et al., 2010; Li et al., 2010, Autry et al., 2011; Miller et al., 2014). Ketamine-induced cognitive deficits are also hypothesized to be due to NMDAR inhibition (Shaffer et al., 2014). (S)-ketamine has an approximately fourfold higher affinity/potency for the PCP site of the NMDAR compared with the (R)-isomer, and twice that of the racemic mixture [(S)-ketamine: Ki = 0.3–0.69 μM; (R)-ketamine: Ki = 1.4–2.57 μM; and (R,S)-ketamine: Ki = 0.18–3.1 μM, in the presence of extracellular Mg2+ (Ebert et al., 1997; Kohrs and Durieux, 1998; Moaddel et al., 2013; Zanos et al., 2016; Temme et al., 2018)]. The effects of (S)-ketamine and (R)-ketamine were also assessed on NMDA receptor-activated cation currents of whole-cell voltage-clamped cultured rat hippocampal neurons (Zeilhofer et al., 1992). These authors showed that both enantiomers exhibited voltage- and use-dependent blockades of NMDAR currents, with (S)-ketamine being about twice as potent compared with (R)-ketamine (IC50 = 0.80 versus 1.53 μM, respectively; Zeilhofer et al., 1992). Moreover, (S)-ketamine has 2.5–3 times higher potency to inhibit NMDA-evoked currents in cat dorsal horn neurons compared with the (R)-ketamine enantiomer (Lodge et al., 1982). This higher affinity/potency of the (S)-ketamine isomer is hypothesized to explain why (S)-ketamine is a more potent anesthetic than (R,S)-ketamine (Yamakura and Shimoji, 1999). Consistent with these stereospecific differential potencies to inhibit the NMDAR by ketamine's isomers, the ED50 value for induction of hypnosis (loss of righting reflex) was lower for (S)-ketamine and (R,S)-ketamine (3.5 and 5.6 mg/kg, respectively) compared with (R)-ketamine (10.3 mg/kg; Marietta et al., 1977). Similarly, Ryder et al. (1978) showed that (S)-ketamine is an ∼3 times more potent analgesic, 1.5 times more potent hypnotic (loss of righting reflex) and 1.8 times more potent locomotor stimulant agent compared with (R)-ketamine. In particular, the median effective analgesic (s.c.) doses were found to be 6.5, 3.7 and 11 mg/kg for (R,S)-ketamine, (S)-ketamine and (R)-ketamine, respectively (Ryder et al., 1978). The median hypnotic doses for (R,S)-ketamine, (S)-ketamine and (R)-ketamine were calculated to be 45, 38 and 56 mg/kg, respectively (Ryder et al., 1978). In addition, (S)-ketamine (25 mg/kg, s.c.) induced a more profound disruption in sensorimotor gating compared with the (R)-ketamine (25 mg/kg, s.c.) enantiomer in the rat pre-pulse inhibition paradigm, although (R)-ketamine also showed a subtle effect in this study compared with the control-treated rats (Littlewood et al., 2006). In agreement with this finding, Yang et al. (2015) showed disruption of sensorimotor gating and hyperlocomotion to only occur from administration of (S)-ketamine, but not (R)-ketamine in mice. Subanesthetic concentrations of ketamine (40-minute i.v. infusion; 0.5 mg/kg), which exert antidepressant actions in patients suffering from major depression (Zarate et al., 2012a), resulted in a maximum of 31% ± 18% NMDAR occupancy (Shaffer et al., 2014). This occupancy is similar to the NMDAR occupancy estimated (32% ± 6% maximum; Shaffer et al., 2014) following an antidepressant-relevant dose of ketamine in rats (10 mg/kg, i.p.; Yeung et al., 2010). Nevertheless, (R)-ketamine was reported to be a more potent and longer-lasting antidepressant compared with the (S)-ketamine enantiomer in several rodent models (Zhang et al., 2014; Yang et al., 2015; Zanos et al., 2016; Fukumoto et al., 2017), when using a 30-fold dose range (Zanos et al., 2016). There do not appear to be differences in brain exposure of the two enantiomers (Zanos et al., 2016; Fukumoto et al., 2017), thus challenging the NMDAR inhibition hypothesis as the sole mediator of the antidepressant actions of ketamine.
In membrane fractions of postmortem human brain homogenates, IC50 values for [3H]MK-801 displacement by (S)- and (R)-ketamine were reported to be 1.6–1.9 and 7.2–10 μM, respectively, in the presence of extracellular Mg2+ (Oye et al., 1992). Similarly, in rat cortical tissue (S)-ketamine inhibited NMDA (10 μΜ)-evoked currents with an IC50 of 0.9 ± 1.4 μM, whereas (R)-ketamine was a less potent inhibitor with an IC50 of 3.0 ± 1.4 μM (Ebert et al., 1997). Whole-cell patch-clamp electrophysiological recordings obtained from human embryonic kidney (HEK)293T cells transfected with different NMDAR subunits revealed that, in the absence of extracellular Mg2+, ketamine inhibits the NMDARs containing GluN1/GluN2A (IC50 = 0.33 ± 0.01 μM) and GluN1/GluN2B (IC50 = 0.31 ± 0.02 μM) subunit compositions with a modestly higher potency than GluN1/GluN2C (IC50 = 0.51 ± 0.01 μM) and GluN1/GluN2D (IC50 = 0.83 ± 0.02 μM) subunits (Kotermanski and Johnson, 2009). In contrast, in the presence of physiologic levels of Mg2+ (1 mM), ketamine blocks NMDAR containing GluN1/GluN2C (IC50 = 1.18 ± 0.0 μM) and GluN1/GluN2D (IC50 = 2.95 ± 0.02 μM) subunits, with a higher potency than the GluN1/GluN2A (IC50 = 5.35 ± 0.34 μM) and GluN1/GluN2B (IC50 = 5.08 ± 0.02 μM) subunits (Kotermanski and Johnson, 2009). Nevertheless, Yamakura et al. (1993) failed to identify differences in ketamine-induced inhibition of the different NMDAR receptor subunits in Xenopus oocytes injected with subunit-specific mRNAs synthesized in vitro. These findings highlight a lack of clarity on any differential effects of ketamine on NMDAR subtypes composed of different subunits.
Studies have shown that (S)-ketamine inhibits NMDARs composed of GluN1/GluN2C (IC50 = 1.11 μM) and GluN1/GluN2D (IC50 = 1.50 μM) with higher potency than those composed of GluN1/GluN2A (IC50 = 16.10 μM) in the presence of 2 mM Mg2+ (Dravid et al., 2007). (S)-ketamine’s potency to inhibit GluN1/GluN2B (IC50 = 1.55 μM) is reported to be similar to its potency to inhibit GluN1/GluN2C- and GluN1/GluN2D-containing NMDARs in the presence of 2 mM Mg2+ (Dravid et al., 2007). These findings indicate that any preferential potency of ketamine is likely not the result of higher affinity of ketamine to bind to the GluN2C-NMDARs per se, but may be due to differential capacity for Mg2+ binding, or interactions between the drug and Mg2+ within the channel (Kotermanski and Johnson, 2009; Kotermanski et al., 2009). Thus, ketamine may differentially block specific NMDAR subtypes in the brain depending upon local Mg2+ concentrations. In support of this concept, in the absence of Mg2+, ketamine blocks GluN2B-containing NMDARs with a higher potency compared with the NMDARs containing other GluN2 subunits, as measured using recombinant NMDAR GluN2A–D subunits expressed in Xenopus oocytes (Dravid et al., 2007).
In the presence of extracellular Mg2+, ketamine’s N-demethylated metabolite, norketamine, also inhibits the NMDAR. (S)-norketamine has a reported Ki of 1.70–2.25 μM for NMDARs in the spinal cord and the cerebral cortex, whereas (R)-norketamine has an approximately eight times lower binding affinity (Ki = 13.0–26.46 μM; Ebert et al., 1997; Moaddel et al., 2013); also see Table 3. In accordance with these findings, (S)-norketamine (IC50 = 3.0 ± 0.8 μM) more potently inhibited NMDA (10 μΜ)-evoked currents than (R)-norketamine (IC50 = 39 ± 1.4 μM) in rat cerebral cortical neurons (Ebert et al., 1997). Therefore, because NMDAR inhibition was considered the primary mechanism of action of ketamine, the clinical effects of the drug were initially attributed to ketamine and norketamine (Leung and Baillie, 1986; Hirota and Lambert, 2011; Singh et al., 2014).
DHNK and HNK metabolites display weak or no ability to displace [3H]MK-801 binding to NMDARs. (R)-DHNK has lower affinity than (S)-DHNK (59.7–74.6 and 39.0–42.0 μM, respectively) for displacing [3H]MK-801 binding to the NMDAR (Moaddel et al., 2013; Morris et al., 2017). (2S,6S)-HNK has a Ki = 10.4–21.0 μM for displacing [3H]MK-801 binding, whereas (2R,6R)-HNK does not bind to the NMDAR-PCP site with appreciable affinity (Ki > 100 μM; Moaddel et al., 2013; Morris et al., 2017). In addition, at concentrations up to 10 μM, neither (2S,6S)-HNK nor (2R,6R)-HNK functionally inhibit NMDA-evoked currents in rat hippocampal interneurons (Zanos et al., 2016). Lack of functional NMDAR inhibition by (2R,6R)-HNK at 10 μM was also reported by Suzuki et al. (2017). At a higher concentration (50 μM), (2R,6R)-HNK moderately (∼40%) inhibited NMDAR-mediated miniature excitatory postsynaptic currents recorded from cultured hippocampal neurons in the absence of Mg2+. This finding supported the contention that, at concentrations higher than those relevant to antidepressant treatment and in the absence of Mg2+, (2R,6R)-HNK might functionally inhibit NMDARs (Suzuki et al., 2017; Zanos et al., 2017a). Notably, at the same concentration (50 μM) and under the same experimental conditions, ketamine induced >90% inhibition of NMDAR-mediated miniature excitatory postsynaptic currents recorded from hippocampal neurons (Suzuki et al., 2017). (2R,6S)-, (2S,6R)-, (2R,5R)-, (2S,5S)-, (2S,5S)-, (2R,5S)-, (2S,5R)-, (2R,4S)-, (2S,4R)-, (2R,4R)-, and (2S,4S)-HNKs do not have significant affinity to displace [3H]MK-801 binding (Ki > 100 μM; Morris et al., 2017).
There is also evidence that, by reducing extracellular levels of D-serine, ketamine’s enantiomers and its metabolites may indirectly decrease the activation of NMDARs (Singh et al., 2013). D-serine, an endogenous NMDAR coagonist that binds to the glycineB site, is required for activation of the NMDAR complex (Paoletti et al., 2013) and is produced by enzymatic L-serine enantioconversion catalyzed by serine racemase (Wolosker et al., 2008). Incubation of PC-12 cells with increasing concentrations of (S)- and (R)-ketamine exerted differential effects on the intracellular and extracellular D-serine levels. Specifically, application of (S)-ketamine was associated with increased intracellular D-serine (EC50 = 0.82 ± 0.29 μM) and decreased extracellular levels of D-serine (IC50 = 0.82 ± 0.29 μM; Singh et al., 2015). In contrast, (R)-ketamine decreased both intracellular (IC50 = 0.94 ± 0.16 μM) and extracellular levels of D-serine (IC50 = 0.70 ± 0.10 μM; Singh et al., 2015; Table 3). Similar findings were observed using 1321N1 cells and primary hippocampal and cortical neuronal cells. Singh et al. (2015) also demonstrated that inhibition of the amino acid transporter, ASCT2, resulted in qualitatively similar effects to those induced by (S)-ketamine on D-serine levels. In addition, coincubation with sub-saturating concentrations of an ASCT2 inhibitor and (S)-ketamine resulted in an additive effect in both PC-12 cells and primary neuronal cells in regard to D-serine levels, indicating that the effects of (S)-ketamine might be due to an inhibition of the amino acid transporter systems.
The differential effects of ketamine’s enantiomers on D-serine levels might contribute to their differential behavioral effects. Indeed, whereas (S)-ketamine is a more potent anesthetic and analgesic drug (Marietta et al., 1977; White et al., 1985) than (R)-ketamine, (R)-ketamine is a more potent and longer-lasting antidepressant than (S)-ketamine in several animal tests (Zhang et al., 2014; Yang et al., 2015; Zanos et al., 2016; Fukumoto et al., 2017). In fact, D-serine plays a role in synaptic plasticity (Henneberger et al., 2010), and baseline plasma D-serine levels are negatively correlated with ketamine treatment response in patients suffering from major depression (Moaddel et al., 2015a), indicating a possible role of D-serine levels in the antidepressant responses of ketamine (also see Hashimoto, 2014). In vivo, sub-chronic (14-day) administration of ketamine to rats was shown to reduce serine racemase mRNA levels in the forebrain (Watanabe et al., 2010). However, a single administration of ketamine at the dose of 50 mg/kg resulted in an enhancement of serine racemase mRNA levels in the striatum, hippocampus, and cortex of rats (Takeyama et al., 2006), an effect that is predicted to induce an increase rather than a decrease in D-serine levels. Indeed, a single administration of (R)-ketamine (10 mg/kg, i.p.) slightly, but significantly increased cortical D-serine/L-serine ratio in mice (Ma et al., 2017). Therefore, further in vivo confirmation of the effects of ketamine and its enantiomers on D-serine levels is warranted.
DHNK has also been shown to modify D-serine levels. Singh et al. (2013) demonstrated that incubation of PC-12 and 1321N1 cells with 5–90 nM DHNK decreased the relative intracellular D-serine concentrations. Because DHNK is not produced in the brain and does not cross the blood-brain barrier in ketamine-treated rodents (Can et al., 2016; Moaddel et al., 2016), the behavioral relevance of this metabolite’s actions on D-serine levels is not clear (Zanos et al., 2016).
HNKs are also capable of reducing intracellular D-serine concentrations in PC-12 cells, with (2S,6S)-HNK being more potent than (2R,6R)-HNK (IC50s are reported to be 0.18 ± 0.04 and 0.68 ± 0.09 nM, respectively; Singh et al., 2016c). It is possible that the HNK-induced reduction of intracellular D-serine levels may also result in a reduction of extracellular levels of this amino acid. However, modulation of extracellular levels of D-serine may not be an important determinant of the antidepressant effects of HNKs, because at least in mice, (2R,6R)-HNK exerts more potent antidepressant actions than (2S,6S)-HNK (Zanos et al., 2016) and DHNK (Sałat et al., 2015). In addition, electrophysiological studies failed to identify any inhibitory effects of antidepressant-relevant concentrations of these metabolites on NMDAR function (Zanos et al., 2016, 2017b; Suzuki et al., 2017), as decreased extracellular D-serine levels would predict. Moreover, acute D-serine administration induces ketamine-like antidepressant behavioral and biochemical responses in rats (Wei et al., 2017), further complicating the possible functional role of decreased extracellular D-serine levels following ketamine administration. Further verification, possibly using human brain-derived cells or measuring extracellular D-serine levels in vivo following administration of ketamine and/or its metabolites, would be informative in determining the functional relevance of these results.
B. Hyperpolarization-Activated Cyclic Nucleotide-Gated Channels
HCN channels are voltage-gated cation channels (HCN1–HCN4; Luthi and McCormick, 1998; Wahl-Schott and Biel, 2009). Activation of these channels by membrane hyperpolarization is facilitated by cyclic nucleotides, including cAMP. In the central nervous system, HCN channels play a major role in controlling neuronal excitability, synaptic activity, and rhythmic oscillations (Shah, 2014).
There is a report of subunit-specific inhibitory effects of ketamine (EC50 = 8.2–15.6 μM) on HCN1–HCN2 heteromeric channels and hyperpolarization-activated pacemaker currents (Ih; Chen et al., 2009). This may be relevant to the anesthetic actions of ketamine, as ketamine-induced anesthesia was significantly suppressed in HCN knockout mice (Chen et al., 2009). Additionally, (S)-ketamine was found to be more potent at inhibiting these channels (EC50 = 4.1–7.4 μM) compared with racemic ketamine (EC50 = 8.2–15.6 μM; Chen et al., 2009), concordant with the greater anesthetic potency of (S)-ketamine. Indeed, it has been hypothesized that NMDAR inhibition is not the sole mechanism underlying the anesthetic properties of ketamine (Petrenko et al., 2014). Further studies are required to substantiate the exact role of HCN channel inhibition in this regard, and to replicate these findings.
In addition to a possible role in the anesthetic properties of ketamine, HCN1 channel inhibition may have a role in ketamine’s antidepressant actions because reduced HCN1 activity in the hippocampus has been associated with antidepressant effects in rodents (Lewis et al., 2011; Kim et al., 2012; Han et al., 2017). Of interest, mice lacking the HCN1 gene did not manifest ketamine-induced reductions in immobility time in the forced-swim test following chronic oral corticosterone treatment (Li et al., 2014). Furthermore, following ketamine administration, these mice did not show increased sucrose preference or decreased latency to feed in the novelty-suppressed feeding test (Zhang et al., 2016). Unfortunately, these results cannot be unambiguously interpreted as evidence that HCN1 mediates the antidepressant effects of ketamine because HCN1 deletion by itself induced baseline behavioral changes compatible with reduced depressive-like behavior (e.g., decreased immobility in the forced-swim test). In addition, Zhang et al. (2016) suggested that reduction of HCN1 function by ketamine was secondary to inhibition of presynaptic NMDARs. The authors did not test the hypothesis that direct inhibition of HCN1 by ketamine, as was suggested by Petrenko et al. (2014), accounts for its antidepressant effects. There are currently no published data on the activity of ketamine’s metabolites on the function of HCN1 channels or the involvement of these channels on the behavioral effects of these metabolites.
C. GABA Uptake and GABA Receptors
The primary inhibitory neurotransmitter, GABA, activates both the ionotropic GABA receptor subtypes A and C (GABAA and GABAC) and the metabotropic GABA receptor subtype B (GABAB) in the brain (Jacob et al., 2008). Electrophysiological studies have revealed that high concentrations of ketamine potentiate GABAergic inhibitory postsynaptic currents in neurons of guinea pig olfactory cortical slices (300 μM; Scholfield, 1980) and of rat hippocampal slices (500 μM; Gage and Robertson, 1985). At high concentrations, ketamine potentiates GABA-activated GABAA receptors ectopically expressed in Xenopus oocytes (365 μM; Lin et al., 1992) and HEK293 cells (>500 μM; EC50 = 1.2 mM; Flood and Krasowski, 2000; but see Anis et al., 1983). There is also evidence for an inhibitory effect of ketamine on GABA uptake (Ki = 6.2 ± 1.1; IC50 = 50 μM), as assessed by the [3H]GABA-binding assay in striatal synaptosomes of rats (Mantz et al., 1995), indicating that ketamine might cause an increase in extracellular GABA levels. Indeed, it was shown that intramuscular administration of ketamine to rats increases GABA content in the brain and in synaptosomal-enriched fractions of the brain (Wood and Hertz, 1980). However, the effect of ketamine on GABA uptake was not found at clinically relevant concentrations in mice (IC50 > 1000 μM; Wood and Hertz, 1980).
The functional relevance of the actions of ketamine on GABAA receptors is not clear because ketamine concentrations required to modify the activity of these receptors are much higher than those achieved following ketamine administration in clinical settings for anesthetic, analgesic, anti-inflammatory, and antidepressant effects. Nevertheless, there is preclinical evidence for both agonist and antagonist properties of ketamine at GABAA receptors. For example, peripheral administration of subthreshold doses of ketamine (0.1 mg/kg, i.p.) combined with the GABAA receptor agonist muscimol (0.1 mg/kg, i.p.) induced a synergistic antidepressant behavioral response in the acute (30 minutes postinjection) forced-swim test in mice (Rosa et al., 2016). In contrast, direct infusion of muscimol into the infralimbic prefrontal cortex abolished the sustained (24 hours postinjection) antidepressant behavioral effects of ketamine in rats (Fuchikami et al., 2015), suggesting that the in vivo interaction between ketamine and GABAA receptors might be brain region specific. In addition, ketamine-associated delirium occurring at anesthetic doses (1–2 mg/kg, i.v. infusion) is effectively minimized via pretreatment with benzodiazepines (positive allosteric modulators of GABAA receptors; Dundee and Lilburn, 1978; Perumal et al., 2015). In contrast, Irifune et al. (2000) showed that both muscimol and the benzodiazepine receptor agonist diazepam augment ketamine-induced anesthesia, whereas the GABAA receptor antagonist bicuculline antagonizes ketamine-induced anesthesia in mice. Finally, at subanesthetic doses, ketamine does not bind to GABAA receptors in the human brain, as assessed by positron emission tomography (PET) scan imaging (Salmi et al., 2005), and does not alter GABAA receptor function at anesthetic-relevant concentrations (i.e., 10 μM) in HEK293 cells in vitro (Flood and Krasowski, 2000). These findings indicate that, at least at subanesthetic doses, ketamine itself might only indirectly affect GABAA receptor activity to exert any relevant behavioral effects. Indeed, it is postulated that ketamine-induced dissociation/psychotomimetic effects are due to NMDAR blockade on GABAergic inhibitory interneurons, an action that is presumed to disinhibit excitatory neurotransmission via decreased GABA release, and to consequently reduce activation of the GABAA receptors in glutamatergic synapses (Moghaddam et al., 1997; Farber et al., 1998; Homayoun and Moghaddam, 2007; Hare et al., 2017; Wohleb et al., 2017).
D. Cholinergic Receptors
Ketamine is reported to bind to both muscarinic and nicotinic acetylcholine receptors (mAChRs and nAChRs, respectively). To date, five subtypes of mAChRs have been identified (M1–M5). These are metabotropic receptors that signal primarily, although not exclusively, through Gαi/o (M1, M3, and M5) or Gαq (Eglen, 2005). Binding of ketamine to the mAChR subtypes M1, M2, and M3 has been described. In particular, assessment of [3H]quinuclidinyl benzilate displacement revealed that (S)-ketamine has ∼twofold higher affinity than (R)-ketamine for mAChRs (Hustveit et al., 1995). In a subsequent study, Hirota et al. (2002) demonstrated that, with Ki values of approximately 45, 294, and 246 μM, respectively, ketamine displaced [3H]N-methyl scopolamine binding to M1, M2, and M3 mAChRs ectopically expressed in Chinese hamster ovary (CHO) cells. However, the authors reported that ketamine had no significant effect on basal or methacholine-induced Ca2+ signals in M1-expressing CHO cells (Hirota et al., 2002). In contrast, Durieux (1995) reported that, at clinically relevant concentrations, ketamine inhibited M1 mAChR activation in Xenopus oocytes (IC50 = 5.7 μM). The apparently discrepant results could be accounted for by the fact that receptors ectopically expressed in CHO cells and oocytes can have differential sensitivity (e.g., McIntyre et al., 2001). These findings suggest the need for additional studies to identify the exact effects of ketamine on mAChRs and the functional relevance of these effects.
In contrast to the metabotropic mAChRs, nAChRs are ionotropic receptors, which are nonselective cation channels activated by the neurotransmitter acetylcholine. These receptors are composed of five subunits. To date, 10 α (α1–α10) and four β (β1–β4) nAChR subunits have been cloned, and different combinations of these subunits give rise to a number of functional nAChR subtypes, which are expressed in neuronal and non-neuronal cells and have specific pharmacological, functional, and kinetic properties (Albuquerque et al., 2009).
Ketamine is reported to act as a noncompetitive, open-channel blocker of the α7, α4β2, α4β4, and α3β4 nAChR subtypes (Flood and Krasowski, 2000; Yamakura et al., 2000; Coates and Flood, 2001; Jozwiak et al., 2002; Pereira et al., 2002). Ketamine concentration dependently blocked acetylcholine (1 mM)-induced activation of α4β4 nAChRs ectopically expressed in Xenopus oocytes (IC50 = 0.24 ± 0.03 μM), with complete inhibition being achieved with 10 μM ketamine (Flood and Krasowski, 2000). In addition, with IC50 values of 20 and 50 μM, respectively, ketamine inhibited acetylcholine (1 mM)-induced activation of α7 and α4β2 nAChRs ectopically expressed in oocytes (Coates and Flood, 2001). Moreover, Moaddel et al. (2013) showed that ketamine inhibits nicotine-induced α3β4 nAChR activation (IC50 = 3.1 μM). Because ketamine concentrations up to ∼10 μM are within the clinically relevant range, some nAChR subtypes may underlie the effects of ketamine in vivo.
Moaddel et al. (2013) also demonstrated that at 100 nM (R,S)-DHNK reduced the amplitude of acetylcholine-induced whole-cell currents in KXα7R1 cells that ectopically express α7 nAChRs by approximately 60%. At the same concentration, the metabolites (2S,6S)-HNK, (2R,6R)-HNK, and (R,S)-norketamine also reduced the amplitude of acetylcholine-induced α7 nAChR currents by approximately 54%, 51%, and 45%, respectively (Moaddel et al., 2013). In this study, the authors reported that (R,S)-DHNK was not acting as a channel blocker at α7 nAChRs, because its inhibitory effect was voltage independent. Moreover, (R,S)-DHNK did not bind at the agonist-binding site of the α7 nAChRs, because it did not displace α-bungarotoxin binding, suggesting that this metabolite might be acting as a negative allosteric modulator (Moaddel et al., 2013).
The α7 nAChR antagonist activity of ketamine metabolites may have implications for the antidepressant action of ketamine. It is noteworthy in this context that blockade of α7 nAChRs results in antidepressant effects in rodent models (Mineur and Picciotto, 2010; Philip et al., 2010). Thus, modulation of α7 nAChR activity could also be one of the underlying mechanisms involved in ketamine’s antidepressant actions, possibly through its metabolites. In support of this hypothesis, nAChR antagonists are aleady in clinical trials for the treatment of depression (Mineur and Picciotto, 2010; Philip et al., 2010).
Other nAChR subtypes are also sensitive to inhibition by therapeutically relevant concentrations of ketamine metabolites. Specifically, in KXα3β4R2 cells stably expressing rat α3β4 nAChRs, ketamine and (R,S)-norketamine were reported to inhibit the α3β4 nAChR with IC50 values of approximately 3.1 and 9.1 μM, respectively, whereas DHNK, (2S,6S)-, or (2R,6R)-HNK did not have any significant effect on these receptors (IC50 > 200 μM; Moaddel et al., 2013). Therefore, one cannot rule out the possibility that α3β4 nAChRs also contribute to the pharmacological effects of ketamine.
E. Monoaminergic Receptors and Transporters
Dopamine (DA) and serotonin [5-hydroxytryptamine (5-HT)] receptors are metabotropic receptors, with the exception of the 5-HT receptor subtype 3, which is ionotropic. Five different subtypes of DA receptors (D1–5R) and seven subtypes of 5-HT receptors (5-HT1–7R) have been characterized in the central nervous system (De Felice, 2017). Neurotransmitter transporters regulate DA and 5-HT uptake across the cellular/intracellular membranes and play a key role in the regulation of dopaminergic and serotoninergic neurotransmission.
Although there is some evidence that ketamine may act on DA receptors and transporters (see Table 3), there is also conflicting evidence indicating that ketamine does not directly alter dopaminergic signaling; also see Kokkinou et al. (2018). Ketamine was reported to have high affinity (0.06–1.0 μM) for D2 receptors (D2Rs; Kapur and Seeman, 2002; Seeman et al., 2005) and to act as a partial agonist at these receptors (EC50 = 0.9 ± 0.4 μM). This partial agonist activity at D2Rs was suggested to contribute to the psychotomimetic effects of ketamine (Kapur and Seeman, 2002; Seeman et al., 2005). This is further supported by the finding that a ketamine-induced decrease in D2R binding is significantly correlated with schizophrenia-related symptoms in humans, as measured by PET scanning (Breier et al., 1998).
There are several published studies reporting ketamine-induced decreases in striatal D2R binding (an indirect measure of DA release) in humans. Specifically, subanesthetic doses of ketamine (i.v. bolus of ketamine; 0.12 mg/kg), followed by an i.v. infusion of 0.65 mg/kg ketamine over 60 minutes (0.29–0.45 μM; Breier et al., 1998), 0.5 mg/kg ketamine over 20 minutes (Smith et al., 1998), or 0.014 mg/kg per minute (S)-ketamine for 90 minutes (Vollenweider et al., 2000) decreased striatal D2R binding. However, subsequent studies have failed to replicate these findings using single-photon emission computed tomography/PET techniques. In particular, when ketamine was administered as an i.v. bolus (0.12 mg/kg) followed by a constant 70-minute i.v. infusion (0.65 mg/kg per hour), resulting in an average plasma concentration of 0.59 ± 0.22 μM in healthy individuals, there was no change in D2R availability in striatal regions of the brain (Kegeles et al., 2002). Similarly, i.v. infusion of ketamine (66-minute infusion yielding a stable plasma ketamine concentration of 1.23 ± 0.12 μM) in healthy male volunteers did not alter striatal D2R binding (Aalto et al., 2002). In accordance with the lack of direct effects of subanesthetic doses of ketamine on the dopamine D2R, psychotomimetic effects of a 0.23 mg/kg bolus or 0.65 mg/kg per hour infusion of ketamine were not blocked by administration of a D2R antagonist in humans (Krystal and D’Souza, 2001). In addition, ketamine was not found to modify electrically evoked accumbal DA release measured in real-time using fast-scan cyclic voltammetry in mice (Can et al., 2016). In contrast to the previously mentioned studies, it was also reported that ketamine lacks functional agonist and antagonist activity on all of the DA receptor subtypes at concentrations up to 10 μM (Can et al., 2016).
Inhibition of the DA transporter by ketamine in HEK293 cells has also been reported (Ki = 62.9 μM; Nishimura et al., 1998). However, this effect did not occur at concentrations up to 10 μM (Can et al., 2016). Because this effect was only observed at relatively high concentrations, its functional/clinical relevance remains to be determined.
Ketamine was also reported to bind to 5-HT2 receptors with an affinity of 15 ± 5 μM (Kapur and Seeman, 2002). This finding might be relevant to the analgesic effects of the drug, because the 5-HT2B/2C receptor antagonist methysergide inhibited the analgesic effects of ketamine in rats (Crisp et al., 1991), implicating serotonergic signaling in the mechanisms of ketamine analgesia. Moreover, ketamine administration induced an increase in extracellular serotonin (5-HT) levels in the prefrontal cortex and dorsal raphe nucleus of mice (Pham et al., 2017b).
In nonhuman primates, ketamine administration was shown to significantly increase accumbal and ventral pallidum 5-HT1B receptor binding, an effect that was blocked by pretreatment with the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor antagonist 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX; Yamanaka et al., 2014). This finding may suggest that this effect of ketamine is involved in its antidepressant actions because AMPA receptor activation is a convergent mechanism of ketamine’s antidepressant actions, and NBQX administration abolishes ketamine’s effects in several animal tests of antidepressant efficacy (Maeng et al., 2008; Autry et al., 2011; Koike et al., 2011; Walker et al., 2013; Fukumoto et al., 2014; Koike and Chaki, 2014; Zhou et al., 2014; Yang et al., 2015; Zanos et al., 2016; Zanos et al., 2018b,c). Antagonist actions of ketamine on 5-HT3 receptors have been reported, but these occur at higher than clinically relevant concentrations (Ki > 90 μM; IC50 > 100 μΜ; Appadu and Lambert, 1996; Yamakura et al., 2000; Ho and Flood, 2004).
Several studies suggest that increased 5-HT levels are necessary for the antidepressant-like effects of ketamine in rodents. For instance, pharmacological treatments that reduce 5-HT levels in the brain abolished ketamine’s antidepressant behavioral effects in the forced-swim test (du Jardin et al., 2016; Pham et al., 2017b) and in the novelty-suppressed feeding test (Fukumoto et al., 2014) in rodents. In addition, higher extracellular 5-HT levels were positively correlated with ketamine’s antidepressant activity in the forced-swim test in mice (Pham et al., 2017b). Whether these effects are due to a direct action of ketamine on 5-HT receptors is not clear and needs further investigation. Nevertheless, there are also conflicting data on the effects of ketamine on 5-HT receptors, because even at very high concentrations (1 mM) ketamine only slightly altered [3H]5-HT or [3H]spiroperidol binding to 5-HT1 or 5-HT2 receptors, respectively (Martin et al., 1982). It is thus possible that ketamine may interact with serotonin uptake as opposed to directly binding to serotonin receptors. Indeed, administration of antidepressant doses of ketamine (1.5 mg/kg; 40-minute infusion) to nonhuman primates reduced serotonin transporter (SERT) activity (Yamamoto et al., 2013), an effect that was hypothesized to reflect direct binding of ketamine to SERTs to regulate 5-HT reuptake. However, in vitro work indicated that ketamine inhibits SERTs at concentrations ranging from 75 (Azzaro and Smith, 1977) to 162 μM (Nishimura et al., 1998), which are not only above the antidepressant-relevant concentrations, but also well above the clinical anesthetic concentrations of ketamine. At antidepressant-relevant concentrations, ketamine does not have an agonist or antagonist effect on SERTs (Can et al., 2016). Therefore, in vivo evidence of ketamine-induced inhibition of serotonin reuptake (Martin et al., 1982) could be attributed to indirect interactions of ketamine with the serotonergic system, at least at subanesthetic concentrations.
Finally, although there is some evidence that ketamine acts as an uptake inhibitor at norepinephrine transporters (NETs; Ki = 66.8 μM; Nishimura et al., 1998), the inhibition constant indicates that ketamine would not modulate NET function at clinically relevant concentrations (up to 10 μΜ). There is also a noted lack of functional activity of clinically relevant concentrations of ketamine (up to 10 μΜ) on NETs (Can et al., 2016).
To date, there is only one published study assessing the effect of ketamine’s metabolites on monoaminergic receptors and transporters. At concentrations up to 10 μM there was no agonist or antagonist activity of (S)-norketamine, (R)-norketamine, (S)-dehydronorketamine, (R)-dehydronorketamine, (2S,6S)-HNK, or (2R,6R)-HNK on D1, D2, D3, D4, or D5 receptors; DA transporter; NET; or SERT (Can et al., 2016). Although these findings suggest that direct effects of ketamine metabolites on DA receptors or monoaminergic transporters do not account for the antidepressant actions of ketamine, it cannot be ruled out that, at higher concentrations, ketamine’s metabolites may interact with and directly or indirectly modify the activity of these receptors and transporters.
F. Opioid Receptors
Opioid receptors are expressed throughout the central nervous system as well as in peripheral tissues (Trescot et al., 2008). These receptors are G protein–coupled receptors and are classified into three subtypes (μ-, δ-, and κ-opioid receptors) (Kieffer and Gaveriaux-Ruff, 2002). A primary function of opioid receptor activation is inhibiting the transmission of nociceptive stimuli, resulting in analgesia (Trescot et al., 2008; Stein, 2016).
Ketamine was reported to activate human recombinant μ-, κ-, and δ-opioid receptors expressed in CHO cells (Ki = 42.1, 28.1, and 272 μΜ, respectively; Hirota et al., 1999). (S)-ketamine was shown to have higher affinity compared with (R)-ketamine for binding to opioid receptors. In particular, (S)-ketamine’s affinities range from 11 to 29 μΜ for the μ-, 25–28 μΜ for the κ-, and 130–205 μΜ for the δ-opioid receptors (Hustveit et al., 1995; Hirota et al., 1999; Nemeth et al., 2010). In contrast, (R)-ketamine has affinities ranging from 28 to 84 μΜ for the μ-, 60–100 μΜ for the κ-, and 130–286 μΜ for the δ-opioid receptors (Hustveit et al., 1995; Hirota et al., 1999). Similar to these findings, (S)-ketamine (IC50 = 23.0 ± 1.2 μM) showed higher potency to bind and displace the non-specific opioid ligand [3H]dihydromorphine compared with the racemic ketamine (IC50 = 16.3 ± 7.4 μM) and (R)-ketamine (IC50 = 45.5 ± 3.2 μM; Finck and Ngai, 1982).
The actions of ketamine at opioid receptors are hypothesized to be involved in its analgesic effects (Finck et al., 1988; Pacheco Dda et al., 2014), and these findings are consistent with the higher potency of (S)-ketamine in measures of antinociception compared with (R)-ketamine (White et al., 1980; Oye et al., 1992; Mathisen et al., 1995). However, the exact roles of opioid receptors in mediating these effects are unclear.
Although the analgesic actions of ketamine are blocked by intracerebroventricular administration (i.e., direct brain exposure) of μ- and δ-, but not κ-opioid receptor antagonists in mice (Pacheco Dda et al., 2014), global opioid receptor inhibition achieved by systemic administration of naloxone does not prevent ketamine-induced analgesia in humans (Mikkelsen et al., 1999). These seemingly opposing findings indicate that ketamine might induce analgesia via an indirect interaction with the opioid system, or may exert an opioid receptor subtype-specific action. In support of such a subtype-specific effect, in vivo evidence indicates that ketamine might be a κ-opioid receptor agonist and a μ-opioid receptor antagonist. Specifically, ketamine microinjection in the periaqueductal gray region, which contains μ- but not κ-opioid receptors, did not exert antinociceptive effects, but blocked the effects of the μ-opioid receptor agonist, morphine, in the same region (Smith et al., 1985). This could explain the failure of naloxone to block ketamine-induced analgesia and supports a subtype-specific role of opioid receptors in ketamine-induced analgesia. However, it should be noted that these findings are in opposition of the results of Pacheco Dda et al. (2014), as discussed above. Furthermore, it has been reported that subeffective doses of ketamine only partially antagonize the cataleptic actions of morphine in rats (Hance et al., 1989); however, a combination of subeffective doses of ketamine and morphine induced catalepsy in the same experimental settings (Hance et al., 1989). Altogether, these findings implicate the opioid system in ketamine’s analgesic effects, but further studies are needed to clarify the exact mechanisms. Interactions between ketamine and the opioid system may be more relevant in chronic pain, in which ketamine reduces opioid tolerance. In particular, via acting on the downstream extracellular signal-regulated kinase 1/2 pathway, ketamine (10 μΜ) abolished μ-opioid receptor-induced desensitization in vitro (Gupta et al., 2011).
Activation of κ-opioid receptors by ketamine (EC50 = 29 μΜ) was reported to be involved in ketamine’s effects on attention and visual perception in rats assessed in the five-choice serial reaction time task (Nemeth et al., 2010). Pretreatment with a selective κ-opioid receptor antagonist blocked the ability of ketamine (20 mg/kg) to impair attention and visual perception (Nemeth et al., 2010). In contrast, in healthy volunteers, inhibition of opioid receptors by naloxone augmented the dissociative and cognitive deficit effects induced by a subthreshold administration of ketamine (1-minute 0.23 mg/kg bolus, followed by a 60-minute 0.58 mg/kg ketamine infusion; Krystal et al., 2006), possibly indicating an antagonist activity of low doses of ketamine on the opioid receptors in vivo. Nevertheless, further work is needed to establish the exact role of opioid receptor modulation in mediating the side effects of ketamine. There are currently no published studies assessing the effects of ketamine’s metabolites on opioid receptors. However, there is recent evidence indicating that (2R,6R;2S,6S)-HNK (at 10 and/or 30 mg/kg, s.c.) does not possess antinociceptive properties and does not alter opioid (morphine) tolerance in rats (Lilius et al., 2018a), indicating a possible lack of interactions between this metabolite and the opioid receptor system. Notably, norketamine, similar to ketamine, attenuates tolerance to morphine (Lilius et al., 2018b).
G. Sigma Receptors
Another potential site of action of ketamine is the sigma receptor. Sigma receptors are classified into two different subtypes, sigma I and II receptors (σ1R and σ2R, respectively; Bowen et al., 1989). Although a third subtype has also been suggested to exist (σ3R), it has not been fully defined (Myers et al., 1994).
As illustrated in Table 3, ketamine binding has been described at both σ1R and σ2R (Smith et al., 1987; Hustveit et al., 1995; Robson et al., 2012). The first evidence of direct binding of ketamine to sigma receptors came from Klepstad et al. (1990), who demonstrated that (R)-ketamine has a potency of IC50 = ∼15 μM to bind to sigma receptors, whereas (S)-ketamine has an IC50 of ∼100 μM. Although these findings were qualitative and not absolutely quantitative due to the different brain regions used for the binding studies, Hustveit et al. (1995) confirmed these initial findings by showing that (R)-ketamine has an affinity of 19 μM at sigma receptors, whereas (S)-ketamine shows ∼15-fold lower binding affinity for these receptors (131 μM). Robson et al. (2012) reported that (R,S)-ketamine has a preferential affinity for the σ2R (Ki = 26.3 μΜ) compared with the σ1R (Ki = 139.6 μΜ). In support of such binding in vivo, studies in nonhuman primates showed competition binding between ketamine and [11C]SA5845, a sigma receptor PET tracer (Kortekaas et al., 2008).
Sigma receptors are promising targets for antidepressant treatment (Fishback et al., 2010), and activation of these receptors induces antidepressant behavioral responses in animals (Matsuno et al., 1996; Skuza and Rogoz, 2002; Wang et al., 2007; Lucas et al., 2008) and humans (Pande et al., 1999). As a result it could be postulated that the action of ketamine on sigma receptors is involved in the mechanisms underlying its antidepressant responses, consistent with (R)-ketamine’s higher binding affinity for these receptors, and its more potent antidepressant effects compared with (S)-ketamine in rodent models (Zhang et al., 2014; Yang et al., 2015; Zanos et al., 2016; Fukumoto et al., 2017).
Although administration of σ1R and σ2R antagonists did not attenuate the antidepressant behavioral effects of ketamine in mice (Robson et al., 2012), administration of a σ1R-selective antagonist blocked the potentiating effects of ketamine on nerve growth factor–induced neurite outgrowth, and thus, modulation of neuroplasticity-related pathways (Robson et al., 2012) that are involved in the antidepressant effects of the drug (see Kavalali and Monteggia (2012), Gerhard et al. (2016)). These findings may indicate that ketamine’s actions on sigma receptors could be involved in the neuroplasticity-related effects of the drug, and thus indirectly involved in its antidepressant actions. There is currently no evidence of activity of ketamine’s metabolites on sigma receptors.
H. Voltage-Gated Sodium Channels
Voltage-gated ion channels are among the first identified ion channels and are involved in the generation of action potentials (Hodgkin and Huxley, 1952). Local anesthetics typically induce concentration-dependent inhibition of sodium channel activity, via binding to sites within the channel pore (Becker and Reed, 2012). Whereas ketamine has been shown to act as a local anesthetic in veterinary practice and in human patients (Bion, 1984; Gomez de Segura et al., 1998; Hawksworth and Serpell, 1998; Kathirvel et al., 2000), there is currently conflicting evidence regarding the effects of ketamine on voltage-gated sodium channel activity.
In isolated guinea pig ventricular myocytes, ketamine at concentrations ranging from 30 to 300 μΜ induced a concentration-, but not use-dependent tonic inhibition of sodium channel currents (16%–36% inhibition; Hara et al., 1998b). In contrast, both tonic (IC50 = 866.2 ± 34.7 μΜ) and phasic (IC50 = 314.8 μΜ) blockade of sodium channels was induced by ketamine in rat dorsal root ganglion neurons (Zhou and Zhao, 2000). In addition, ketamine was reported to block voltage-gated sodium channels in Xenopus oocytes (tonic inhibition: IC50 = 800 μΜ; phasic inhibition: IC50 = 2.3 mM; Wagner et al., 2001). Moreover, although Benoit (1995) demonstrated no inhibitory effect of ketamine on nodal sodium- channel currents of myelinated nerve fibers, others have reported up to 71.1% blockade of sodium channel conductance by ketamine (ED50 = 1.1 mM; Frenkel and Urban, 1992). A study assessing the effects of (S)- and (R)-ketamine on the activity of sodium channels showed that (S)-ketamine inhibits voltage-gated sodium channels with an apparent potency of 240 ± 60 and 59 ± 10 μΜ in neuronal and skeletal muscle isoforms, respectively. The potency of (R)-ketamine to block sodium channels was lower (IC50 = 333 ± 93 and 181 ± 49 μΜ in neuronal and skeletal muscle isoforms, respectively; Haeseler et al., 2003) than that of (S)-ketamine. Similarly, Schnoebel et al. (2005) showed stereoselective tonic block of voltage-gated sodium currents with (S)-ketamine being more potent than (R)-ketamine (IC50 = 128 and 269 μΜ, respectively). These findings predict that (S)-ketamine is a more effective local anesthetic than (R)-ketamine.
These discrepancies in the effects of ketamine on the activity of sodium channels might be due to differences in the cell populations and experimental procedures used. Overall, these findings support an inhibitory effect of ketamine on sodium channels, which is a characteristic of local anesthetics (Becker and Reed, 2012). These effects occur at concentrations well above circulating ketamine levels relevant for general anesthesia, but could possibly be relevant to local ketamine anesthesia.
I. L-Type Voltage-Dependent Calcium Channels
The L-type voltage-dependent calcium channel (VDCC) family consists of four different members referred to as Cav1.1–Cav1.4 (Catterall, 2011). Cav1.2 is the main LTCC expressed in the mammalian brain (Bhat et al., 2012). Antagonism of L-type VDCCs by ketamine has been reported over a wide range of concentrations. In porcine tracheal smooth muscle cells, for example, ketamine blocked VDCCs with an IC50 of 1 mM (Yamakage et al., 1995). In rabbit portal vein smooth cells, 1 mM ketamine completely inhibited VDCC currents (Yamazaki et al., 1992). Finally, in bullfrog single atrial cells, ketamine inhibited VDCC currents with an IC50 of 9.2 μΜ (Hatakeyama et al., 2001). This effect was not use-dependent, and ketamine did not act as an open channel blocker. Nonuse–dependent tonic inhibition of VDCCs was also reported in isolated guinea pig ventricular myocytes, in which 30–300 μΜ induced a 26%–53% inhibition (Hara et al., 1998b). The discrepancies in the reported concentrations at which ketamine inhibits VDCCs may be due to differences in species, cell type, or experimental preparations (e.g., differences in bath solutions).
Despite evidence of VDCC inhibition by ketamine, an in vitro study showed that AMPA receptor activation and subsequent increases in brain-derived neurotrophic factor release and mechanistic target of rapamycin complex 1 activation—actions that are believed to underlie ketamine’s antidepressant properties—require VDCC activation (Jourdi et al., 2009). Furthermore, ketamine’s antidepressant effects in mice are blocked by pretreatment with L-type calcium channel antagonists (Lepack et al., 2014). These findings are not in line with the inhibitory activity of ketamine at these channels, as previously described. However, it should be noted that these studies were performed in cells derived from peripheral tissues (e.g., heart, trachea) and that effects of ketamine on VDCCs in brain-derived cells at antidepressant-relevant concentrations have not been reported to our knowledge. In addition to the possible role of ketamine-induced inhibition of calcium channels in mediating the antidepressant actions of the drug, blockade of calcium channels has been also hypothesized to be involved in the psychoactive/psychotomimetic effects of ketamine (Lisek et al., 2016).
IV. Conclusions
Ketamine has been in clinical use as an anesthetic since the 1970s. However, novel use indications (e.g., chronic pain and depression) and mechanisms of action (e.g., HNK metabolites) are still emerging. Its use as an anesthetic, analgesic, anti-inflammatory, and antidepressant drug has reignited research to understand the neurobiological mechanisms underlying these effects of ketamine, as well as its metabolites, and several molecular and cellular targets have been identified to date.
Ketamine is typically used clinically (and in preclinical research) as the racemic mixture, (R-S)-ketamine. It is extensively and rapidly metabolized in vivo, resulting in the formation of norketamine and HKs, followed by production of the secondary metabolites, DHNK and the HNKs. Although ketamine, norketamine, and HNKs readily cross the blood-brain barrier, DHNK does not appear to reach pharmacologically active brain concentrations, at least in the mouse brain (Can et al., 2016). Ketamine, and to a lesser extent, norketamine, are noncompetitive antagonists of the NMDAR ion channel. DHNK and HNKs, however, appear to have much lower potency to inhibit the NMDAR, if at all (Morris et al., 2017; Suzuki et al., 2017; Zanos et al., 2017a).
In addition to the NMDAR, we reviewed findings supporting ketamine’s actions at a number of receptors and ion channels, including DA, 5-HT, opioid, cholinergic, sigma, and GABAA receptors, as well as monoamine transporters and HCN, sodium, and L-type VDCCs. Ketamine’s well-characterized anesthetic effects are primarily attributed to NMDAR inhibition. However, there is evidence that HCN channel inhibition might also be involved in the anesthetic properties of ketamine. Similarly, evidence implicates NMDAR inhibition in the analgesic actions of ketamine, with the possibility of opioid receptors also playing a role.
A. (R)- and (S)-Ketamine as Antidepressants
Much attention has focused upon the NMDAR as the primary pharmacological target responsible for ketamine’s antidepressant actions. However, in contrast to this prediction, clinical studies have suggested no—or only modest—antidepressant efficacy of some alternative NMDAR antagonists. To date, these drugs lack the robust, rapid, or sustained antidepressant effectiveness of ketamine, and some (e.g., memantine) have been proven clinically ineffective in multiple studies (Newport et al., 2015). Moreover, Hashimoto and colleagues first reported superior and longer-lasting antidepressant actions of (R)-ketamine compared with (S)-ketamine in rodent models (Zhang et al., 2014; Yang et al., 2015; Fukumoto et al., 2017). These findings were also supported by Zanos et al. (2016), who showed that (S)-ketamine’s antidepressant behavioral effects only become apparent at higher doses compared with (R)-ketamine. The increased potency of (R)-ketamine does not seem to be related to a U-shaped dose response of the drugs, as it has been shown more potent compared with (S)-ketamine with up to a 30-fold range of doses in multiple mouse tests of antidepressant efficacy (Zanos et al., 2016; Fukumoto et al., 2017). Administration of equal doses of (R)- and (S)-ketamine did not yield different levels of these enantiomers in the brains of rodents (Zanos et al., 2016; Fukumoto et al., 2017), indicating that increased antidepressant potency of (R)-ketamine in rodent models is not due to greater brain levels of the drug. These preclinical rodent data indicate that (R)-ketamine is a more potent antidepressant compared with the (S)-ketamine enantiomer (Zhang et al., 2014; Yang et al., 2015; Zanos et al., 2016; Fukumoto et al., 2017), suggesting that it is unlikely that ketamine exerts its antidepressant actions solely via inhibition of the NMDAR (Zanos et al., 2018a). Nevertheless, we note that preclinical rodent studies have also indicated rapid-acting antidepressant behavioral actions of (S)-ketamine in mice (Zhang et al., 2014; Yang et al., 2015; Zanos et al., 2016; Fukumoto et al., 2017).
The finding that ketamine exerts rapid antidepressant actions, within hours of administration, has focused extensive research efforts on understanding this phenomenon. This finding and elucidation of the relevant mechanisms involved have the potential to revolutionize the treatment of depression, considering that currently approved antidepressants take weeks or even months to exert their full antidepressant effects (Rush et al., 2006), and many patients suffering from major depressive disorders are resistant to classic antidepressant pharmacotherapies. Similar to racemic ketamine, clinical human studies in depressed patients have indicated that a 40-minute, i.v. infusion of (S)-ketamine exerts rapid antidepressant actions (within 2 hours of administration; Singh et al., 2016a). In addition, there are reported dose-dependent antidepressant actions of intranasally administered (S)-ketamine (dose of 28–84 mg, twice per week for a total of 2 weeks) in treatment-resistant depressed patients under oral classic antidepressant treatment (Daly et al., 2018). (S)-ketamine is currently in phase III clinical trials as an antidepressant (Andrade, 2017a).
If in human depression, as is the case in mouse models, (R)-ketamine has superior potency to (S)-ketamine, this would have advantages considering its fewer side effects due to less potent inhibition of the NMDAR (see section N-Methyl-D-Aspartate Receptors). However, there are currently no published clinical studies assessing the antidepressant efficacy of (R)-ketamine in depressed patients. Furthermore, there are no published clinical studies directly comparing the antidepressant actions of the (S)- and (R)-ketamine enantiomers, or comparing the actions of either enantiomer to the racemic mixture.
B. Utility of Ketamine’s Hydroxynorketamine Metabolites as Drug Treatments
It was reported that metabolism of ketamine is necessary for its full antidepressant actions in mice (Zanos et al., 2016). Specific HNK metabolites of ketamine, (2S,6S)-HNK, and (2R,6R)-HNK, derived from the metabolism of (S)-ketamine and (R)-ketamine, respectively, do not bind to or functionally inhibit the NMDAR at antidepressant-relevant concentrations (Morris et al., 2017; Suzuki et al., 2017; Zanos et al., 2017a), but do exert antidepressant behavioral effects similar to those observed following administration of ketamine itself (Zanos et al., 2016). These findings further challenge the NMDAR inhibition hypothesis of ketamine’s antidepressant actions. In addition, (2R,6R)-HNK exerts antidepressant effects without the sensory dissociation, ataxia, and abuse liability of ketamine in animal tests (Zanos et al., 2016). Indeed, the psychoactive side effects of ketamine, including dissociation and changes in sensory perception, as well as its abuse potential, have been attributed to the NMDAR-inhibition effects of ketamine (Shaffer et al., 2014).
The recent findings that ketamine metabolites are involved in the antidepressant actions of ketamine suggest the possible use of these metabolites in the treatment of depression and open new paths for investigating their role in other brain disorders. As we discussed earlier, HNK metabolites may contribute to the clinical effects of subanesthetic doses of ketamine, perhaps due to their direct actions on nAChRs (Moaddel et al., 2013), indirect actions on D-serine (Singh et al., 2013, 2016c), or other targets that have not yet been identified. This work may provide a framework for a novel ketamine metabolite paradigm, which posits clinically relevant effects dependent upon metabolic conversion of ketamine, but it does not involve NMDAR inhibition (Singh et al., 2014). Nevertheless, future preclinical studies are needed to support the contention that NMDAR inhibition is not required for the effectiveness of ketamine’s metabolites as fast-acting antidepressants and to identify the underlying mechanism of action of these metabolites. It also remains to be investigated whether ketamine metabolites have a role in the anti-inflammatory or analgesic actions of ketamine.
C. Future Directions
Identification of the targets responsible for the different behavioral effects of ketamine, and potentially its metabolites, is critical for the development of novel pharmacotherapies that will lack the side effects of ketamine, including psychotomimetic effects, changes in sensory perception, and abuse potential. In addition to the recently completed studies in preclinical depression models (Zanos et al., 2016), studies of ketamine metabolites in animal models of pain, inflammation, depression, and suicidality (see Gould et al., 2017) are essential to better understand their therapeutic potential. Expanded clinical use of racemic ketamine, (S)-ketamine, (R)-ketamine, and potentially key metabolites [i.e., (2R,6R)-HNK] represents an important opportunity to define new therapies for unmet medical conditions and to better define pharmacology–phenotype relationships. Notably, clinical exploration of these agents is feasible given the historical safety knowledge surrounding racemic and enantio-pure forms of ketamine (and thus its metabolites) when given acutely. As such, investments in alternate routes of administration, dosing strategies, and drug combination assessments are not overly burdensome. Furthermore, the breadth of potential indications coupled to ketamine’s multiple pharmacological targets offers an unprecedented window of insight into new mechanisms for future therapeutic interventions. Given these factors, it is clear that the comprehensive understanding of ketamine and ketamine metabolite pharmacology presents invaluable opportunities in both basic and translational research and clinical care.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Zanos, Moaddel, Morris, Riggs, Highland, Georgiou, Pereira, Albuquerque, Thomas, Zarate, Gould.
Footnotes
This work is supported by a National Institutes of Health (NIH) Grant MH107615 and a Harrington Discovery Institute Scholar-Innovator Grant to T.D.G., a National Association for Research on Schizophrenia and Depression Young Investigator Award to P.Z., and the National Institute of Aging (R.M.), National Institute of Mental Health (C.A.Z.), and National Center for Advancing Translational Sciences (C.J.T.) NIH intramural research programs.
The authors declare competing financial interests: R.M. and C.A.Z. are listed as coinventors on a patent for the use of (2R,6R)-hydroxynorketamine, (S)-dehydronorketamine, and other stereoisomeric dehydro- and hydroxylated metabolites of (R,S)-ketamine in the treatment of depression and neuropathic pain. P.Z., R.M., P.J.M., C.J.T., C.A.Z., and T.D.G. are listed as coinventors on a patent application for the use of (2R,6R)-hydroxynorketamine and (2S,6S)-hydroxynorketamine in the treatment of depression, anxiety, anhedonia, suicidal ideation, and post-traumatic stress disorders. R.M., P.J.M., C.A.Z., and C.J.T. have assigned their patent rights to the U.S. government but will share a percentage of any royalties that may be received by the government. P.Z. and T.D.G. have assigned their patent rights to the University of Maryland, Baltimore, but will share a percentage of any royalties that may be received by the University of Maryland, Baltimore. All other authors declare no competing interests.
Abbreviations
- σ1R
- sigma I receptor
- σ2R
- sigma II receptor
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- AUC
- area under the curve
- CHO
- Chinese hamster ovary
- CRPS
- complex regional pain syndrome
- D2R
- D2 receptor
- DA
- dopamine
- DHNK
- dehydronorketamine
- HCN
- hyperpolarization-activated cyclic nucleotide-gated channel
- HEK
- human embryonic kidney
- HK
- hydroxyketamine
- HNK
- hydroxynorketamine
- 5-HT
- serotonin
- IL-6
- interleukin 6
- mAChR
- muscarinic acetylcholine receptor
- nAChR
- nicotinic acetylcholine receptor
- NET
- norepinephrine transporter
- NMDAR
- N-methyl-D-aspartate receptor
- PCP
- phencyclidine
- PET
- positron emission tomography
- SERT
- serotonin transporter
- VDCC
- voltage-dependent calcium channel
- U.S. Government work not protected by U.S. copyright
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵





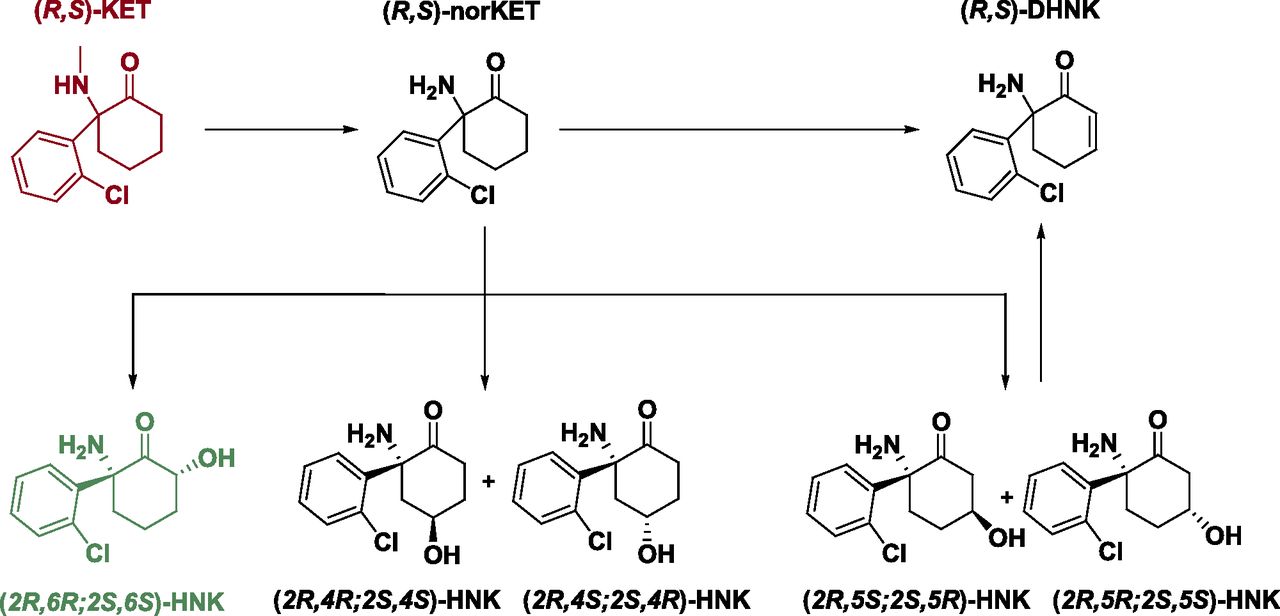{kind=link}
{kind=link}