


Abstract
Quantitative systems pharmacology (QSP), an emerging field that entails using modeling and computation to interpret, interrogate, and integrate drug effects spanning from the molecule to the whole organism to forecast treatment outcomes, is expected to enhance the efficiency of drug development. Since late 2017, the U.S. Food and Drug Administration has advocated the use of an analogous approach of model-informed drug development. This review focuses on issues pertaining to nanosized medicines (NP) and the potential utility of QSP to determine NP delivery and residence at extracellular or intracellular targets in vivo. The kinetic processes governing NP disposition and transport, interactions with biologic matrix components, binding and internalization in cells, and intracellular trafficking are determined, sometimes jointly, by NP properties (e.g., dimension, materials, surface charge and modifications, shape, and geometry) and target tissue properties (e.g., perfusion status, vessel pore size and wall thickness, vessel and cell density, composition of extracellular matrix, and void volume fraction). These various determinants, together with the heterogeneous tissue structures and microenvironment factors in solid tumors, lead to environment-, spatial-, and time-dependent changes in NP concentrations that are difficult to predict. Adding to the complexity is the recent discovery that NP surface-coating protein corona, whose composition depends on NP properties and which undergoes continuous evolution with time and local protein environments, is yet another unpredictable variable. Examples are provided to demonstrate the potential utility of QSP-based multiscale modeling to capture the physicochemical and biologic processes in equations to enable computational studies of the key kinetic processes in cancer treatments.
I. Introduction
Quantitative systems pharmacology (QSP) is an emerging field that is expected to advance pharmaceutical sciences and drug development. QSP entails using modeling and computation to interpret, interrogate, and integrate drug concentrations and effects on multiple scales spanning from the molecule to the whole organism to forecast treatment outcomes. The U.S. National Institutes of Health considers QSP a high priority for research and training program development (https://www.nigms.nih.gov/training/documents/systemspharmawpsorger2011.pdf). Its importance in the drug development field is highlighted by recent U.S. Food and Drug Administration (FDA) announcements stating that the agency advocates the use of an analogous approach, model-informed drug development, in New Drug Applications and Abbreviated New Drug Applications (https://wayback.archive-it.org/7993/20170403224108/https://www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AdvisoryCommitteeforPharmaceuticalScienceandClinicalPharmacology/ucm361585.htm; https://www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AdvisoryCommitteeforPharmaceuticalScienceandClinicalPharmacology/ucm535520.htm; https://www.fda.gov/Drugs/NewsEvents/ucm554182.htm; https://www.fda.gov/drugs/newsevents/ucm527823.htm) and by the agency’s implementation of a model-informed drug development pilot program in April 2018 (U.S. Food and Drug Administration, 2018).
This review focuses on the unique issues pertaining to nanosized medicines (NP) and the potential utility of QSP to determine the target site delivery and residence of NP. The emphasis is on solid tumors, which comprise heterogeneous tissue structures and microenvironmental factors that are known to affect drug/NP delivery and residence, drug/NP-biomatrix interactions, and tumor response to treatments. Some aspects of this review were presented at a public FDA workshop in October 2017 (Fang et al., 2018).
In this review, “NP” broadly refers to nanosized entities used in the treatment, diagnosis, monitoring, and control of biologic systems. This includes entities that fall under the FDA definitions of nanotechnology—that is, entities engineered to have a dimension of ∼1–100 nm or entities having a dimension of up to 1000 nm plus dimension-dependent physical or chemical properties or biologic effects (https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM588857.pdf). There are currently 12 FDA-approved parenteral NP products loaded with active pharmaceutical ingredients, including 7 liposomal preparations, 1 nanoparticle, 1 nanotube, and 3 lipid-drug complexes, with particle sizes between 45 and 150 nm (Table 1). Additional FDA-approved nanosized entities include 239 proteins and peptides (>2 kDa, typically >1 nm) (Usmani et al., 2017), 80 antibodies and derivatives (>40 kDa, >5 nm), and 4 antibody-drug conjugates (IgG-based, 149–160 kDa, ∼15 nm) (https://www.accessdata.fda.gov/scripts/cder/daf/). The FDA has published several guidance documents for industry related to nanotechnology products; the latest draft guidance was published on December 2017 (https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM588857.pdf).
FDA-approved parenteral NP products containing active pharmaceutical ingredients on the U.S. market as of October 2018
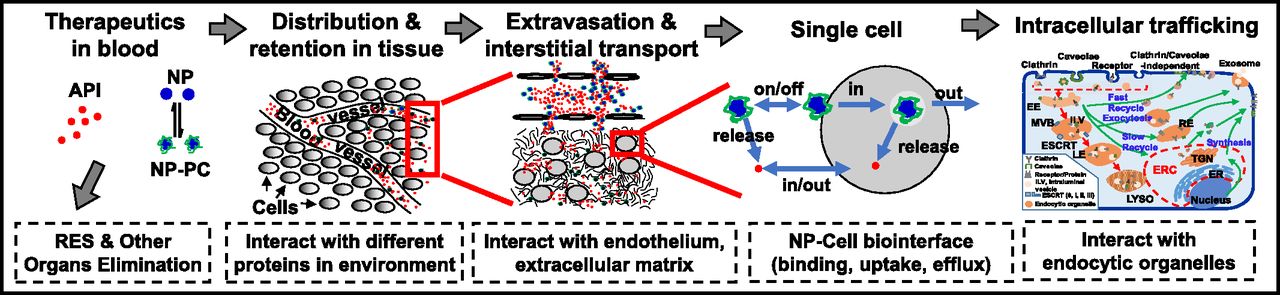
The efficacy of NP depends on the delivery and residence at the intended target site. Potential target sites are the tissue interstitium (e.g., diagnostics or therapeutics), cell membrane (e.g., antibodies), or intracellular compartments (e.g., DNA, antisense, RNA interference). Figure 1 shows the multiple processes involved in NP delivery from the injection site to the extracellular or intracellular target. As discussed below, there are many intersecting kinetic processes that ultimately determine the target site delivery of an NP and its cargo, and many of these processes are determined by NP properties as well as by tissue properties. In addition, some of these processes produce opposite outcomes; for example, a surface modification of an NP, such as pegylation, confers stealthiness to reduce clearance by the reticuloendothelial system (RES) but also reduces the cell membrane binding and subsequent endocytosis. The complexity of the system makes it a fertile ground for QSP research in which the physicochemical and biologic processes can be described in mathematical terms and models, so as to enable the use of computation to study how changes in individual NP or tissue properties, singly or in combination, affect the target site exposure.

Transport of NP from the injection site to the target site. After an intravenous injection, NP are distributed in blood and undergo the following processes: 1) interaction with proteins in blood to form NP-PC, 2) removal by phagocytic/RES entrapment or elimination by metabolism and excretion, 3) transport to organs and tissues via blood circulation, 4) extravasation into the tissue interstitium via transvascular diffusion or convection, 5) interaction with ECM components, 6) transport by interstitial diffusion and convection to reach individual cells, 7) interaction with cells (binding, internalization, efflux), and 8) intracellular trafficking and interaction with endocytic organelles. The drawing of NP, ECM components, and subcellular organelles is not to scale. Note that the proteins on the PC formed in blood may exchange with proteins in ECM (not depicted in the figure). See the text for more details on individual processes. API, active pharmaceutical ingredient; ER, endoplasmic reticulum; ERC, endocytic recycling compartment; ESCRT, endosomal sorting complexes required for transport; ILV, intraluminal vesicle; LYSO, lysosome; MVB, multivesicular body; TGN, trans-Golgi network. This figure is adapted from Li et al. (2012) and reprinted with permission.
The remaining sections of this review focus on the processes and determinants of NP exposure at target sites and the QSP-based approaches to depict and compute these dynamic and intersecting processes. Section II discusses the processes governing, and the factors affecting, the transport of NP and cargo from the injection site to the tumor interstitium. Section III discusses the interactions of NP with extracellular matrix (ECM) components and cells. Section IV discusses the NP internalization into cells and the intracellular NP trafficking through endocytic organelles. These three sections jointly describe the numerous determinants and barriers affecting drug/NP transport to the intended extracellular or intracellular target sites in tissues in general and solid tumors in particular. Readers are referred to our earlier reviews for more details and original citations on these aspects (Au et al., 2001, 2002, 2015, 2016; Jang et al., 2003; Lu et al., 2010, 2015; Wang et al., 2010, 2011; Li et al., 2012). Sections II, III, and IV also provide the equations describing the individual kinetic processes, whereas section V provides examples of applying these equations to compute the spatial-, environment- and time-dependent target site exposure of small molecule drugs and NP. Section VI discusses a unique NP property—that is, the formation and evolution of protein corona (PC). As of this writing, relatively little is known about PC except that it can affect NP transport and interactions with biologic matrices, and therefore is likely to play a role in the efficacy of nanomedicines. Section VII discusses the regulatory issues regarding NP therapeutics. Finally, section VIII summarizes the key information and discusses the potential application of QSP to unravel the understudied kinetic processes in cancer treatments.
II. Transport from Blood to the Tissue Interstitium
A. Fates of Nanosized Medicine in Blood
In blood, NP can undergo degradation (e.g., due to carrier instability), interact with blood components (e.g., interact with proteins to form PC, enter blood cells), undergo surface opsonization and subsequent entrapment by the mononuclear phagocytic system and RES, or be excreted by the kidneys (e.g., for small NP with a <6-nm diameter) (Nie, 2010). The remaining NP is transported via the blood circulation to the target organs and is extravasated by transvascular diffusion or convection to enter the tissue interstitium. NP cargo such as small molecule drugs can be released from NP within systemic blood or the tissue interstitium or intracellularly; the released cargo can interact with blood components, undergo elimination, or be transported to its intended targets.
B. Differences in Blood and Lymph Vasculatures in Normal and Tumor Tissues Lead to Differences in Nanosized Medicine Extravasation into the Tissue Interstitium
In humans, blood flow velocity and pressure in blood vessels decreases with vessel diameter (e.g., from ∼20 cm/s and ∼100 mmHg, respectively, in large arteries to ∼0.03 cm/s and ∼20 mmHg in small capillaries). Openings in the capillary wall range from small pores on endothelial cells to large openings due to discontinuous cell junctions and vary among organs (e.g., between 5 and 15 nm in most tissues, 60 nm in the bone marrow and lungs, 70–90 nm in the kidneys, ∼135 nm in hepatic sinusoid, and up to 5 µm in the terminal arterial capillary network of the splenic red pulp reticulum) (Sarin, 2010). Compared with normal tissues, tumor vasculature is typically disorganized, resulting in chaotic and tortuous paths and greater flow resistance. On the other hand, tumor blood vessels have much larger openings (100–780 nm) and are much leakier. In addition, larger tumors show heterogeneous blood vessel distribution, with a central avascular or necrotic region, an intermediate seminecrotic and poorly perfused region, and a stably perfused periphery. Normal tissues have functional lymphatic systems, whereas tumors typically do not. These differences in blood and lymph vasculatures can either promote or reduce NP delivery to tumors. For example, absence of interstitium-lymphatic fluid drainage in tumors increases the interstitial fluid pressure and thereby limits the transvascular and interstitial convective transport but also enhances the retention of NP. In addition, the larger tumor capillary pore/opening sizes enhance the ease of NP extravasation into the interstitial space. The spatial-dependent differences in blood vessel density and perfusion status within a tumor contribute to the uneven intratumoral drug/NP distribution.
Most of the knowledge regarding vasculatures and drug/NP delivery in tumors was obtained from preclinical models. Recent advances in intravital high-resolution imaging in tumors of human patients in the clinical setting reveal that 1) vessels are disorganized and tortuous, as observed in animal tumors; 2) ∼50% of tumor vessels are not open; and 3) vessel diameters are larger and vessel wall shear stress is lower than predicted from immunohistochemistry or preclinical intravital microscopy results (Fisher et al., 2016; Gabriel et al., 2018).
C. Transport within the Tissue Interstitium
The effects of NP properties, including size, surface charge or modifications, shape and geometry, and nanomaterials, on NP disposition and interactions with ECM/cells are summarized in Table 2. With respect to tissue properties, the fibrous proteins (e.g., collagen, elastin, fibronectin) and polysaccharides (e.g., hyaluronan, glycosaminoglycan) in ECM and tumor cells present physical resistance to interstitial diffusional and convective transport. The high interstitial fluid pressure, secondary to the absence of lymphatic drainage and accumulation of macromolecules and wastes, reduces the transvascular and interstitial convective transport. The smaller void volume fraction and, conversely, higher density of tumor cells or ECM components reduces the tissue porosity and NP diffusivity, whereas binding to ECM components or cells reduces the free NP fraction available for convective or diffusive transport (Au et al., 2001, 2002, 2015, 2016; Jang et al., 2003; Lu et al., 2010, 2015; Wang et al., 2010, 2011; Li et al., 2012; Abbiati and Au, 2018).
Effect of NP properties
D. Equation for Transvascular and Interstitial Transport
The transvascular and interstitial convective and diffusive transport processes are determined by dimension- and nanomaterial-dependent factors. These include 12 tissue properties [i.e., hydraulic conductivity of microvessel walls (Lpv), osmotic pressure in blood (πv) and in interstitial fluid (πint), protein reflection coefficient across the vascular wall (σp), pressure in blood vessels (Pv) and interstitial fluid (Pint), interstitial fluid flow velocity ( ), blood vessel surface area per unit of tissue volume (S/V), maximum NP binding sites per unit of tissue volume (Bmax), hydraulic conductivity of lymphatic vessel walls (Lpl), lymphatic vessel surface area per unit of tissue volume (Sl/V), and lymphatic vessel pressure (Pl)] and multiple factors that are jointly determined by both NP and tissue properties. For example, NP size relative to vessel pore size and vessel wall thickness determine the reflection coefficient σ and vascular diffusive permeability (Pd), and NP size relative to tissue void volume fraction as well as NP interactions with host organs and ECM or cells determine its interstitial diffusion coefficient (D). These physicochemical processes are captured in several well established equations, such as convective transport described by the combination of the Starling equation, Darcy’s law, and mass and momentum conservation laws and diffusive transport described by Fick’s First and Second laws (eqs. 1 and 2); the individual transport processes are denoted underneath the corresponding mathematic terms. Please see our earlier publications for the mathematic basis of these equations (Au et al., 2014).
), blood vessel surface area per unit of tissue volume (S/V), maximum NP binding sites per unit of tissue volume (Bmax), hydraulic conductivity of lymphatic vessel walls (Lpl), lymphatic vessel surface area per unit of tissue volume (Sl/V), and lymphatic vessel pressure (Pl)] and multiple factors that are jointly determined by both NP and tissue properties. For example, NP size relative to vessel pore size and vessel wall thickness determine the reflection coefficient σ and vascular diffusive permeability (Pd), and NP size relative to tissue void volume fraction as well as NP interactions with host organs and ECM or cells determine its interstitial diffusion coefficient (D). These physicochemical processes are captured in several well established equations, such as convective transport described by the combination of the Starling equation, Darcy’s law, and mass and momentum conservation laws and diffusive transport described by Fick’s First and Second laws (eqs. 1 and 2); the individual transport processes are denoted underneath the corresponding mathematic terms. Please see our earlier publications for the mathematic basis of these equations (Au et al., 2014).


Note that the partial differential equation (PDE), such as eq. 2, captures the spatiotemporal-dependent processes. Hence, spatial-dependent properties (e.g., differences in normal and tumor tissues) as well as the intratumoral heterogeneities (e.g., high vessel density in the tumor periphery and low density in the tumor center) can be accounted for by using model parameter values specific to individual spatial locations within the system. The PDE is solved using the computational finite element method (FEM), a numerical method for solving PDE-based engineering problems. In the FEM, the geometry of the system under study is virtually recreated using a computer-aided design tool, divided into smaller subdomain or finite elements, each represented by a set of simpler, spatial-independent element equations in place of the original PDE (Flynn et al., 1974; Macheras and Iliadis, 2006; Keener and Sneyd, 2009; Sharma, 2010; Müller and Kuttler, 2015). An example of using the FEM to solve the mass transport PDE, to depict the time- and spatial-dependent distribution of a smaller molecule drug in a solid tumor, is detailed elsewhere (Au et al., 2014).
The above transport models are applicable to an NP that is not bound to macromolecules. For NP that bind to tumor cells or ECM components, the concentration of the free or unbound NP available for diffusive transport needs to be adjusted (e.g., see eq. 3 in section III).
III. Interactions with the Extracellular Matrix and Cells
A. Nanosized Medicine Interactions with the Extracellular Matrix and Cells
NP may bind to cell membrane via specific binding (e.g., ligand-receptor interaction) or nonspecific binding driven by electrostatic interactions. The membrane binding facilitates the subsequent internalization. In general, positively charged NP bind more readily to the cell membrane compared with neutral or negatively charged NP (Table 2). After binding, NP enter cells via multiple mechanisms; the internalization involves an elaborated sequence of coordinated events (Khalil et al., 2006; Mathivanan et al., 2010; Huotari and Helenius, 2011). Among the micropinocytosis mechanisms that take up macromolecules or other chemical substances into cells by membrane invagination, caveolae-mediated endocytosis is used by smaller NP (∼50–100 nm diameter) and clathrin-mediated endocytosis by larger NP (∼100–150 nm) (Rejman et al., 2004; McMahon and Boucrot, 2011). Nucleation of clathrin domains, which cover about 0.5%–2% of the cell surface, promotes the assembly of clathrin triskelions, induces invagination of the membrane into clathrin-coated pits, and stabilizes the membrane curvature, resulting in clathrin-containing vesicles. The vesicle is detached from the membrane and released into the cytoplasm through the recruitment and action of dynamin, a membrane scission protein, at the neck of the budding vesicle. This is followed by disassembly of the clathrin coat and the clathrin triskelions are recycled back to the membrane. Caveolae are flask-shaped membrane invaginations that are 50–80 nm in size and lined with caveolin, a dimeric protein that binds cholesterol onto the cellular surface for intracellular trafficking. Caveolae-mediated endocytosis also uses dynamin as the scission protein. NP that use clathrin-mediated endocytosis for internalization frequently end up in the lysosomes, whereas NP that use caveolin-dependent endocytosis can sometimes escape lysosomal degradation. Clathrin- and caveolae-independent endocytoses are not well understood, but they take place in cells devoid of both clathrin and caveolae and are generally related to cholesterol-rich rafts with a diameter of 40−50 nm in the cell membrane; these small rafts can presumably be captured by and internalized within any endocytic vesicle (Doherty and McMahon, 2009; Behzadi et al., 2017; Kinnear et al., 2017). There are several other nonreceptor-mediated internalization mechanisms, including 1) fluid phase macropinocytosis, in which cytoskeleton rearrangement leads to cell membrane extensions that then fuse back onto the membrane to form a large vesicle (0.2–5 µm); 2) passive penetration of quantum dots (4 nm) into red blood cells as a result of increased membrane flexibility; and 3) disruption of membrane-mimicking lipid bilayers by organic and inorganic NP to cause formation of nanopores to enable NP penetration (Mercer and Helenius, 2009; Oh and Park, 2014; Behzadi et al., 2017). NP such as liposomes can also enter cells via fusion of the NP lipid-bilayer with the cell membrane, followed by release of NP cargo directly into cytosol (Lu et al., 2009; Schroeder et al., 2010). A recent example shows that the fusion of a nanoparticle-stabilized nanocapsule/small interfering RNA (siRNA) complex (∼180 nm) with the cell membrane in a cholesterol-dependent process represents an efficient approach for direct cytoplasmic delivery of siRNA (Jiang et al., 2015).
The binding and internalization of NP into cells depends on the NP concentrations and, in the event of receptor-mediated internalization, also the receptor availability and therefore follows first-order or saturable kinetics. The internalized NP may undergo exocytosis to re-enter the extracellular or interstitial fluid (Oh and Park, 2014; Behzadi et al., 2017).
B. Equation for Nanosized Medicine Interactions with Extracellular Matrix and Cells
Equation 3 shows the ordinary differential equations depicting concentration- and time-dependent extracellular binding, internalization, and exocytosis via a receptor-mediated mechanism.  (3)NPbound, NPEC, and NPIC are the respective concentration of membrane-bound, extracellular, and intracellular NP. These various NP concentrations can be experimentally measured and used to obtain the respective rate constant of NP binding to the cell membrane (kon,NP), NP dissociation from cell membrane binding sites (koff,NP), NP internalization (kin,NP), NP exocytosis (kexocytosis), and the maximum number of NP binding sites on the cell membrane (Bmax). ki is the first-order rate constant for an intracellular process such as degradation (e.g., in lysosomes; see section IV). Examples of the calculations are shown in our earlier publications (Gao et al., 2013; Wientjes et al., 2014; Wang et al., 2017).
(3)NPbound, NPEC, and NPIC are the respective concentration of membrane-bound, extracellular, and intracellular NP. These various NP concentrations can be experimentally measured and used to obtain the respective rate constant of NP binding to the cell membrane (kon,NP), NP dissociation from cell membrane binding sites (koff,NP), NP internalization (kin,NP), NP exocytosis (kexocytosis), and the maximum number of NP binding sites on the cell membrane (Bmax). ki is the first-order rate constant for an intracellular process such as degradation (e.g., in lysosomes; see section IV). Examples of the calculations are shown in our earlier publications (Gao et al., 2013; Wientjes et al., 2014; Wang et al., 2017).
For nonreceptor-mediated internalization, with the exception of passive diffusion that would follow a first-order concentration gradient-driven process, the other processes involve cytoskeleton or membrane changes that, to our knowledge, have not yet been described in mathematical terms.
IV. Intracellular Trafficking
A. Intracellular Processing of Nanosized Medicine
The far-right panel of Fig. 1 shows the schematic illustration of the known NP intracellular trafficking processes. The vesicles are internalized and become a part of early endosomes (EE). The two proposed models regarding EE organization are 1) fusion of vesicles (e.g., clathrin- or caveolin-containing vesicles) with each other to form the larger EE (Helenius et al., 1983) and 2) delivery of the vesicles to the preexisting EE (Oh and Park, 2014; Behzadi et al., 2017; Naslavsky and Caplan, 2018). Although it remains unclear which model is prevalent, the general consensus is that contents in EE are sorted into fast or slow recycling endosomes (REs), which return the contents (e.g., membrane receptor) to the cell membrane. EE can evolve into multivesicular bodies that proceed to either the acidified late endosomes (LEs; pH 5 to 6) and then to the enzyme-rich lysosomes (pH 4.5–5) or migrate to a pericellular location where their contents are released as exosomes. Exosomes are small membrane vesicles with an average diameter of between 30 and 100 nm (Raposo and Stoorvogel, 2013), and they can have diverse biologic functions (van Niel et al., 2018). For example, exosomes derived from cancer cells are involved in distal metastatic niche initiation (Melo et al., 2014; Costa-Silva et al., 2015), intercellular communications (e.g., during drug resistance development; Qu et al., 2016; Stone, 2016), and immune system modulation (Robbins and Morelli, 2014; Muller et al., 2016). We recently demonstrated that the exosome is an intercellular drug transfer mechanism with pharmacological consequences (Wang et al., 2017).
B. Equation for Nanosized Medicine Intracellular Trafficking
Figure 2 shows a kinetic model of intracellular trafficking, in which the transfer of the endocytosed NP among the intracellular organelles is described as a first-order catenary reaction based on the law of mass action. Equation 4 shows the general ordinary differential equation to depict the time-dependent changes in NP concentrations in an endocytic organelle (i.e., first-order input from an upstream compartment j into a destination compartment i and first-order output to its next downstream compartment z).
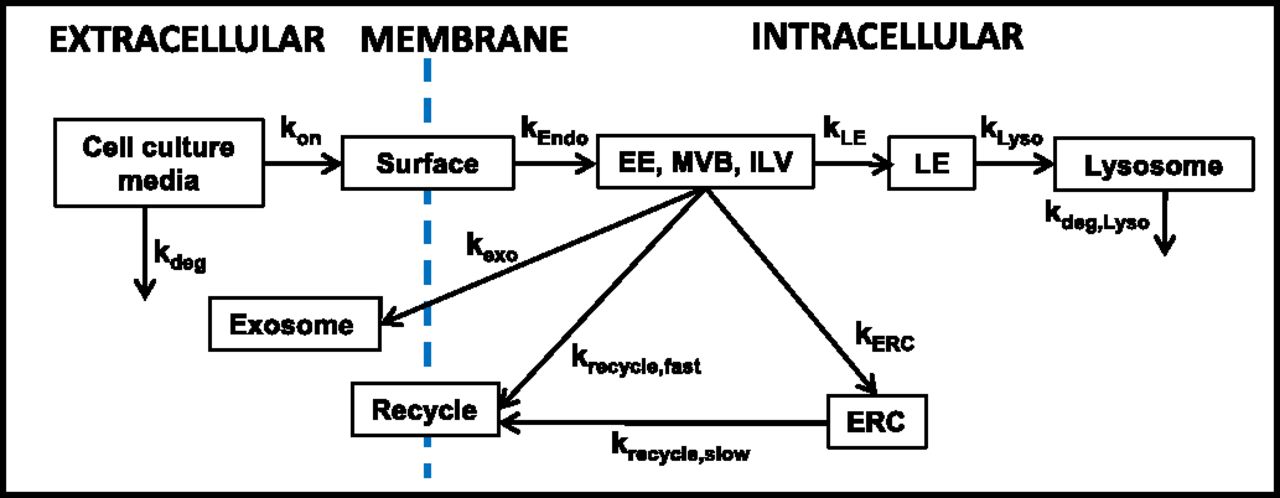
A kinetic model of intracellular trafficking. This kinetic model is based on the intracellular trafficking scheme shown in the far-right panel of Fig. 1. kx denotes the inter-compartmental transfer rate constant, with x being the destination compartment. ERC, endocytic recycling compartment; ILV, intraluminal vesicle; MVB, multivesicular body. This figure is adapted from Abbiati and Au (2018) and reprinted with permission.
 (4)The rate constants for transfer between endocytic compartments (e.g., EE to LE to RE) can be measured by following the appearance and disappearance of the NP (usually labeled with a fluorescent probe) in these organelles (detected by their respective markers; e.g., Rab5 for EE, Rab7 for LE, and Rab4/Rab11 for RE), where the colocalization of the NP and markers is monitored using laser confocal microscopy and quantitative imaging software such as Colocalizer Pro and ImageJ. An example of the intracellular trafficking and processing of an siRNA-loaded lipoplex is described elsewhere (Abbiati et al., 2017b).
(4)The rate constants for transfer between endocytic compartments (e.g., EE to LE to RE) can be measured by following the appearance and disappearance of the NP (usually labeled with a fluorescent probe) in these organelles (detected by their respective markers; e.g., Rab5 for EE, Rab7 for LE, and Rab4/Rab11 for RE), where the colocalization of the NP and markers is monitored using laser confocal microscopy and quantitative imaging software such as Colocalizer Pro and ImageJ. An example of the intracellular trafficking and processing of an siRNA-loaded lipoplex is described elsewhere (Abbiati et al., 2017b).
V. Examples of Applying Quantitative Systems Pharmacology to Estimate the Target Site Delivery and Residence of Small Molecule Drugs and Nanosized Medicines
A. Basis for Quantitative Systems Pharmacology–Based Multiscale Modeling and Computation
As discussed in sections II, III, and IV, the delivery and residence of NP to extracellular or intracellular targets involves multiple kinetic processes that are, in turn, controlled by numerous NP properties and tissue properties. In view of the system complexity, we advocate using QSP-based multiscale models to capture these various scale-, environment-, spatial-, time-, and concentration-dependent processes in order to compute how various NP properties and their changes affect the target site exposure. In these models, a tumor is viewed as a unit in which the intratumoral heterogeneities are represented in mathematical terms (e.g., high blood flow for a highly perfused tumor vs. low blood flow to the necrotic region, or large volume for a primary tumor vs. small volume for a micrometastasis). The numerical values of the respective model parameters are altered and simulations are performed to predict the magnitude of the effects of these diversities on spatiotemporal changes in the target site exposure of small molecule drugs and NP.
B. Examples of Using Multiscale Modeling and Computation to Determine Target Site Delivery and Residence
Three examples, including two from our group, that used multiscale modeling and the FEM to capture the diverse kinetic processes to calculate drug/NP exposure in tumors are discussed below.
The first example used modeling together with experimental data to evaluate whether liposomes can improve the delivery of doxorubicin to mouse and human tumors in vivo. The simulated results demonstrate that, due to differences in transvascular flux and drug/liposome deposition among mouse and human tumors, the typical tumor properties in mice would show greater drug levels for the liposomal formulation compared with the free drug, whereas human tumors would not show such benefit under multiple conditions (Hendriks et al., 2012).
The second example demonstrates the use of multiscale modeling to predict the spatiotemporal changes in concentrations of a small molecule drug within a tumor in vivo as functions of other readily measured parameters (in vitro drug-biomatrix interactions, in vivo drug disposition in the peritoneal fluid and whole organism). The whole organism model accounts for drug distribution to various organs and drug clearance from the body, whereas the tumor model accounts for intratumoral heterogeneities, spatial-dependent transport, and drug-biomatrix interactions. These models jointly enabled the prediction of drug concentrations as functions of time and spatial positions within a tumor. The model predictions show good agreement with the experimental results (e.g., 1% deviation for the area under the curve and 23% deviations for individual data points); these deviations are severalfold lower compared with the experimental variations between animals (Au et al., 2014).
The third example demonstrates the successful prediction of interstitial diffusive NP transport in a three-dimensional tumor cell spheroid system by a model that accounts for concentration gradient-dependent diffusive flux, depletion of the NP due to cell binding and internalization, and the effect of NP-PC. The model was used together with 1) in vitro NP-cell interaction parameters measured in avascular two-dimensional monolayer cell cultures plus 2) interstitial NP diffusivity calculated based on NP and tumor properties (NP size, tumor cell density, interstitial void volume fraction, or concentrations and diameters of ECM components) (Gao et al., 2013; Wientjes et al., 2014).
Other examples of modeling and computation based on PDE and FEM include investigations of tumor properties and pharmacokinetics, such as effects of vasculature normalization on tumor properties (Jain et al., 2007), interstitial fluid flow (Soltani and Chen, 2011), and effects of tumor shape and size on drug residence in tumors (Sefidgar et al., 2014).
Note that QSP model-based simulations in the above examples, as in other model-based simulations, employed a set of model parameter values and yielded single data points (i.e., no data variances), and hence are not suitable for typical statistical analyses that require such variance measurements.
VI. Interaction of Nanosized Medicine and Proteins to Form Protein Corona
A. Current Knowledge on Nanosized Medicine–Protein Corona
A unique feature of NP is the adsorption of proteins on its surface to form PC. This occurs among a wide variety of NP (e.g., metallic, metal oxide, carbon based, polymer coated, polymeric, quantum dots, liposomes) and many proteins in biologic milieu (e.g., apolipoprotein, complement protein, prothrombin, vitronectin, immunoglobulin, fibrinogen, serum albumin). PC was first reported in the mid-2000s and has since been proposed as an important determinant of NP disposition and functionality. Readers are referred to several reviews for more details and original citations (Lai et al., 2012; Monopoli et al., 2012; Treuel and Nienhaus, 2012; Walkey and Chan, 2012; Yang et al., 2013; Setyawati et al., 2015; Au et al., 2016; Barbero et al., 2017; Corbo et al., 2017; Ahsan et al., 2018).
Although protein-lipid interactions have been a major research field for several decades, the knowledge regarding NP-PC is relatively limited. For example, only 226 publications in 2017 contained both “nanoparticle” and “corona” as keywords. Most published studies have focused on analysis of PC protein composition and on NP interactions with single proteins (Huang et al., 2013; Winzen et al., 2015; Zhang et al., 2017; Raoufi et al., 2018). The latter results most likely are oversimplifications, since PC comprises multiple proteins and it is known that binding of one protein to an NP often induces conformational changes that affect the binding of other proteins (Selva Sharma and Ilanchelian, 2015; Vilanova et al., 2016; Raoufi et al., 2018). Two recent reviews suggest using molecular dynamic simulations to address the complexity of PC formation, in the hope of uncovering the major determinants under a wide variety of conditions and thereby improving the clinical translational of nanotechnologies (Ke et al., 2017; Mahmoudi, 2018).
B. Formation of Nanosized Medicine–Protein Corona
NP-PC formation depends on NP properties (material, surface properties, size, charge, shape, geometry, curvature), environment (ECM composition, pH, temperature, shear stress, ionic strength), and time (Strojan et al., 2017). High shear flow, by inducing conformational changes of proteins in plasminogen-rich PC, reduces NP binding to plasminogen receptors on cells compared with static shear flow (Jayaram et al., 2018). Surface modifications such as pegylation generally decrease PC formation, enabling the NP to avoid recognition and entrapment in the RES (Nuytten et al., 2010; Dobrovolskaia et al., 2014; Bargheer et al., 2015; Pelaz et al., 2015). Decreased NP size leads to higher surface curvature and reduced steric interactions among the pegylation molecules, thereby allowing more proteins to adsorb to the surface (Walkey et al., 2012). Lowering the NP surface positive charge reduces the electrostatic interaction with the negatively charged ECM components and thereby reduces PC formation (Mastorakos et al., 2015).
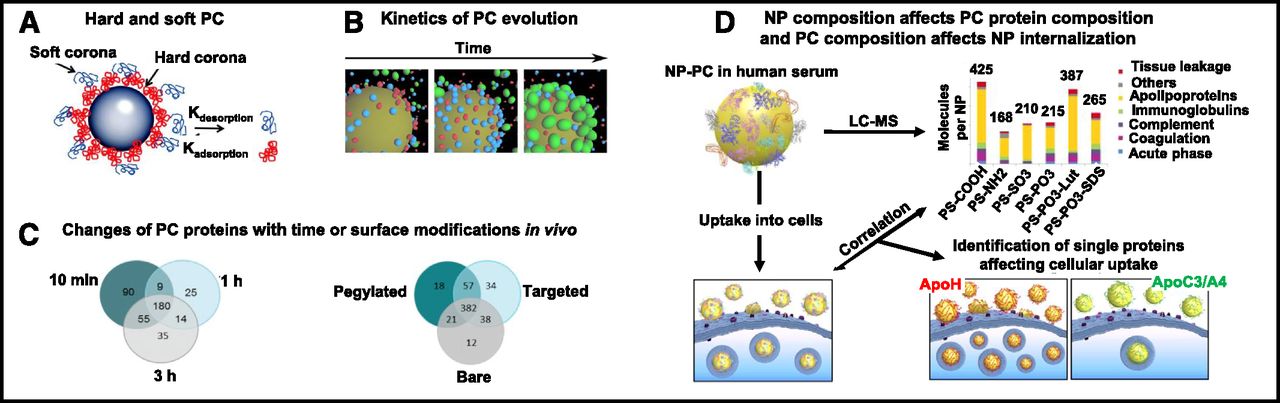
Figure 3A shows the multilayer PC. The first layer of high-affinity proteins on the NP surface (referred to as hard corona) is covered by a layer of low-affinity proteins (soft corona) (Rahman et al., 2013). Among the several thousand available proteins in human plasma, only a small fraction is present in the hard corona. Equally intriguing is that the proteins in the hard corona are not necessarily the most abundant proteins in plasma or those with the highest affinity for NP (Monopoli et al., 2012). PC formation, mediated by van der Waals forces and electrostatic interactions, occurs rapidly and is completed within minutes (Tenzer et al., 2013).

NP-PC. (A) Schematic illustration of hard and soft PC. The rate of protein adsorption and desorption determines their exchange time and lifetime in PC. The hard or soft PC is composed of multiple proteins. (B) Kinetics of evolution of proteins on PC. Colors indicate the following: red, albumin; blue, transferrin; and green, fibrinogen. (C) Venn diagrams of the numbers of unique proteins adsorbed onto pegylated liposomes recovered from blood of the CD1 mouse at 10 minutes, 1 hour, and 3 hours postinjection (left), and proteins adsorbed onto liposomes with different surface modifications (bare or nonpegylated, pegylated, conjugated with cell surface associated mucin 1 transmembrane glycoprotein antibody) recovered at 10 minutes postinjection (right). (D) NP composition affects PC protein compositions and PC composition affects NP uptake into cells. Analysis of NP-PC with LC-MS shows different PC protein compositions for different PS-COOH NP. Coating of NP by ApoA4 or ApoC3 reduces cellular uptake, whereas coating by ApoH increases uptake. Apo, apolipoprotein; LC, liquid chromatography; MS, mass spectrometry; MUC-1, cell surface associated mucin 1; PS-COOH, carboxy-functionalized polystyrene. Images in this figure were adapted from the following sources and are reprinted with permission: Rahman et al. (2013) (legend also) (A), Vilanova et al. (2016) (B), Hadjidemetriou et al. (2015, 2016) (C), and Ritz et al. (2015) (D).
C. Evolution of Nanosized Medicine–Protein Corona with Time In Vitro and In Vivo
PC evolves with time. Figure 3B shows an example of the evolution of PC in vitro, where the PC of a silica NP incubated with three proteins with increasing binding affinity (human serum albumin < transferrin < fibrinogen) changed from mostly albumin to mostly fibrinogen, indicating replacement of the low-affinity protein by the high-affinity protein over time (Vilanova et al., 2016).
Figure 3C shows two examples of the time-dependent evolution of PC in vivo, where NP was injected intravenously into CD1 mice (Hadjidemetriou et al., 2015, 2016). The left panel of Fig. 3C shows that both the number and the identity of PC proteins change with time, even though there is no change in the total protein amount (i.e., the number changed from 334 at 10 minutes to 228 at 1 hour and 284 at 3 hours, with only ∼50% of proteins present at all three times). The right panel of Fig. 3C similarly shows substantial changes in the PC proteins of liposomes with different surface modifications (bare or nonpegylated, pegylated, conjugated with cell surface associated mucin 1 transmembrane glycoprotein antibody) at 10 minutes.
D. Evolution of Nanosized Medicine–Protein Corona with Environment
The formation and stability of PC, due to the reversibility of noncovalent binding of proteins to the NP surface, depends on the concentrations and types of proteins available in the biologic milieu. Hence, the protein composition of PC evolves as NP translocates from one biologic compartment to another (e.g., from blood to tumor tissues or lymph) (Monteiro-Riviere et al., 2013; Riviere, 2013; Bonvin et al., 2017).
E. Effects of Nanosized Medicine–Protein Corona Formation and Evolution on Target Site Delivery and Residence
For NP comprising a nucleic acid–lipoplex or polyplex, interactions with proteins cause destabilization and release of nucleic acid (e.g., DNA and siRNA) (Buyens et al., 2008; Abbiati et al., 2017a). For NP other than a lipoplex or polyplex, the formation of PC has different effects, including an increased effective size (up to 150% for polystyrene and silica NP), a change of surface charge from positive to neutral or negative, increased opsonization, increased RES uptake, and pathobiological changes such as hemolysis and endothelial cell death (Tenzer et al., 2013; Au et al., 2016). PC on albumin-bound paclitaxel NP acts as a shield and reduces the drug release (Behzadi et al., 2014). In addition, the presence of PC proteins that are ligands to membrane receptors enhances the NP internalization (Fig. 3D) (Ritz et al., 2015). These changes in turn affect the NP transvascular and interstitial transport, internalization and intracellular processing, and consequently the target site exposure to NP. Similarly, evolution of PC protein compositions can be expected to cause spatial-, environment-, NP/protein concentration-, and time-dependent changes in NP disposition and functionality.
Interspecies differences in PC compositions have been shown to affect NP interactions with macrophages (Müller et al., 2018). Some proteins including complement C3 protein in the PC preformed in human plasma are diminished upon intravenous injection into mice, due to exchange of PC proteins in vivo (Chen et al., 2017).
Serum proteins in humans vary with age, ethnicity, and physiologic or disease state (Hanash, 2003; Conrads et al., 2004). Differences in PC among patients with and without cancer have been reported (Caputo et al., 2017).
In short, the formation and evolution of PC affects many aspects of NP disposition, transport, and interactions with host tissues or cells and therefore is likely to affect the target site exposure to NP.
F. Potential Utility of Quantitative Systems Pharmacology in Studying Nanosized Medicine–Protein Corona
In view of the large number of variables affecting NP-PC and their wide-ranging effects on multiple processes that determine the target site exposure, QSP would be a useful tool to interrogate the dynamic systems governing PC formation or evolution and their biologic consequences. However, many of the required qualitative and quantitative background information is currently unavailable, including data on the following: 1) effects of PC on the delivery, transport, residence, and internalization of NP; 2) exchange of PC proteins in different biologic matrices, as functions of time, concentrations of NP, and/or concentrations of proteins of interest; 3) effects of NP composition on PC formation and stability; 4) species differences; and 5) effects of physiologic and disease states. We propose that such background information is needed to improve the in vitro to in vivo and the animal to human translation of nanotechnologies.
VII. Unique Regulatory Issues Regarding Nanosized Medicine Products
Drug exposure at the target site is a critical determinant of treatment efficacy. FDA approval of generic drugs requires the generic product to be pharmaceutically equivalent to the reference listed drug (RLD) and to demonstrate bioequivalence (BE) when administered to patients under the conditions specified in the RLD product labeling. For BE, the FDA requires the confidence interval for the ratio of the averages (population geometric means) of measures for test and reference products to be within a limit, usually 80%–125% (https://www.fda.gov/downloads/drugs/guidances/ucm070244.pdf; https://www.fda.gov/downloads/Drugs/.../Guidances/UCM134846.pdf; https://www.fda.gov/downloads/drugs/guidances/ucm377465.pdf).
Since target site exposure data are usually not available, systemic exposure is often used as the surrogate measurement of BE, with the assumption that systemic BE equals target site BE. As discussed throughout this article, there are multiple dynamic processes and numerous factors affecting the delivery and residence of an NP and its cargo at the target site. Compounding this problem is the unpredictability due to the PC formation and the continuous exchanges of PC proteins with the proteins in the local environment. In other words, target site BE of NP products is achieved only when there is equivalence in each and every step involved in the delivery or removal of the NP from the injection site to the target site, including NP disposition and clearance on the whole organism level, transvascular and interstitial transport on the target organ level, internalization on the target cell level, interactions with subcellular organelles, release of cargo on each level (whole organism, organ, cell), interactions with proteins on each level, and interactions with ECM and intracellular endocytic organelles. Given this level of complexity, it is reasonable to expect that systemic BE of NP does not equal target site BE and, accordingly, the conventional systemic BE determination used to evaluate small molecular drugs does not apply to NP products.
A recent publication from the FDA outlines the paradigm for BE recommendation of parenteral NP products, which calls for formulation sameness and systemic BE (Zheng et al., 2017). However, formulation sameness may be difficult to achieve if the production of generic NP uses a different manufacturing process. On the other hand, regulatory decisions can be facilitated by 1) determining which NP properties will significantly alter the target site exposure and therefore cause systemic BE to not equal target site BE [i.e., the critical quality attributes or (CQAs)]; 2) determining, conversely, which NP properties are non-CQAs; 3) determining to what extent differences in CQAs and non-CQAs will (and will not) result in unacceptable changes in target site exposure rendering the generic NP to not have target site BE to RLD; and 4) determining the effects of PC on NP delivery and residence, including the effects of host-dependent factors (e.g., age, sex, ethnicity, physiologic and disease states) on PC formation and evolution in human subjects. In addition, QSP-based multiscale modeling represents a potentially useful tool in these endeavors, such as in assessing the quantitative relationship between individual NP properties (singly and in combinations) versus systemic and target site exposure to NP and cargo.
VIII. Conclusions and Perspectives
A major challenge in cancer nanotechnology is the inability of translating the successes in animals to humans (Prabhakar et al., 2013; Au et al., 2016). This could be a result of the many intersecting kinetic processes and the numerous variables causing scale-, environment-, and spatio-temporal–dependent changes in the target site exposure of NP. For example, the structural differences between animal and human tumors (e.g., larger size with greater stroma/ECM fraction in human tumors) would affect the NP transport. A better understanding of the interspecies differences in the tumor properties and the resulting differences in physicochemical processes such as transvascular and interstitial transport or NP-biomatrix interaction, together with QSP modeling to account for these differences, may facilitate the scale-up of animal data to forecast the likely outcomes in humans.
Second, although NP such as liposomes and polymeric NP have been studied for ≥40 years, the presence of PC was first recognized about 10 years ago. Because of its “newness,” relatively little information is available. We advocate additional research to obtain qualitative and quantitative data on the formation and evolution of PC. Such information can be used with modeling and computation to advance the knowledge on the effects of PC.
Third, the dimension- and material-dependent effects of NP, as they affect the target site pharmacokinetics and pharmacodynamics, present new challenges for regulatory decisions. This is in part because the established FDA guidelines to demonstrate BE for small molecule drugs are based on the assumption of kinetic equilibrium between the systemic circulation and the target site, which is likely not applicable to NP products.
Finally, this review outlines several QSP approaches to account for the time- and spatial-dependent effects of various factors on drug/NP delivery and residence in tumors. Another potential application of QSP models, once they have been established and validated, is to interrogate other difficult-to-measure kinetic processes, such as 1) pathobiological factors that are patient dependent, diverse in nature, and will for certain happen during the course of disease and treatments (e.g., disease progression leading to tumor size increase and/or micrometastasis, chemotherapy-induced apoptosis leading to changes in tumor cell density and interstitial void volume fraction, and antiangiogenic-induced changes in vasculatures and blood flow); and 2) environment- and time-dependent changes in ECM components or structures that would alter the NP transport and functionality. In short, QSP, although still in its infancy, represents a potentially useful tool to predict the NP delivery and residence at the therapeutic and toxicity targets or organs, and thereby improves in vitro to in vivo and preclinical to clinical translations and enables the prediction of the clinical safety profile and efficacy of NP products.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Au, Abbiati, Wientjes, Lu.
Footnotes
This work was supported in part by the University of Oklahoma [Mosier Chair of Pharmaceutical Sciences], the National Institutes of Health National Cancer Institute [Grants R01-CA158300, R01-CA163015, R43/R44-CA103133, R43/R44-CA107743, and R43-CA162448], the National Institutes of Health National Institute of Biomedical Imaging and Bioengineering [R01-EB015253], the National Institutes of Health National Institute of General Medical Sciences [R01-GM100487], and the National Institutes of Health National Center for Advancing Translational Sciences [R43-TR000356].
A portion of this work was previously presented at the following workshop: Au JLS (2017) Considerations for bioequivalence evaluation of nano-particulate/molecular medicine. U.S. Food and Drug Administration Public Workshop on Leveraging Quantitative Methods and Modeling to Modernize Generic Drug Development and Review; 2017 Oct 2–3; Silver Spring, MD.
Abbreviations
- BE
- bioequivalence
- CQA
- critical quality attribute
- ECM
- extracellular matrix
- EE
- early endosome
- FDA
- U.S. Food and Drug Administration
- FEM
- finite element method
- LE
- late endosome
- NP
- nanosized medicine
- PDE
- partial differential equation
- PC
- protein corona
- QSP
- quantitative systems pharmacology
- RE
- recycling endosome
- RES
- reticuloendothelial system
- RLD
- reference listed drug
- siRNA
- small interfering RNA
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics
References
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. Transport from Blood to the Tissue Interstitium
- III. Interactions with the Extracellular Matrix and Cells
- IV. Intracellular Trafficking
- V. Examples of Applying Quantitative Systems Pharmacology to Estimate the Target Site Delivery and Residence of Small Molecule Drugs and Nanosized Medicines
- VI. Interaction of Nanosized Medicine and Proteins to Form Protein Corona
- VII. Unique Regulatory Issues Regarding Nanosized Medicine Products
- VIII. Conclusions and Perspectives
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters