


Abstract
Imidazoline receptors historically referred to a family of nonadrenergic binding sites that recognize compounds with an imidazoline moiety, although this has proven to be an oversimplification. For example, none of the proposed endogenous ligands for imidazoline receptors contain an imidazoline moiety but they are diverse in their chemical structure. Three receptor subtypes (I1, I2, and I3) have been proposed and the understanding of each has seen differing progress over the decades. I1 receptors partially mediate the central hypotensive effects of clonidine-like drugs. Moxonidine and rilmenidine have better therapeutic profiles (fewer side effects) than clonidine as antihypertensive drugs, thought to be due to their higher I1/α2-adrenoceptor selectivity. Newer I1 receptor agonists such as LNP599 [3-chloro-2-methyl-phenyl)-(4-methyl-4,5-dihydro-3H-pyrrol-2-yl)-amine hydrochloride] have little to no activity on α2-adrenoceptors and demonstrate promising therapeutic potential for hypertension and metabolic syndrome. I2 receptors associate with several distinct proteins, but the identities of these proteins remain elusive. I2 receptor agonists have demonstrated various centrally mediated effects including antinociception and neuroprotection. A new I2 receptor agonist, CR4056 [2-phenyl-6-(1H-imidazol-1yl) quinazoline], demonstrated clear analgesic activity in a recently completed phase II clinical trial and holds great promise as a novel I2 receptor–based first-in-class nonopioid analgesic. The understanding of I3 receptors is relatively limited. Existing data suggest that I3 receptors may represent a binding site at the Kir6.2-subtype ATP-sensitive potassium channels in pancreatic β-cells and may be involved in insulin secretion. Despite the elusive nature of their molecular identities, recent progress on drug discovery targeting imidazoline receptors (I1 and I2) demonstrates the exciting potential of these compounds to elicit neuroprotection and to treat various disorders such as hypertension, metabolic syndrome, and chronic pain.
I. Introduction
Although the imidazoline receptor concept was proposed decades ago and the field has been consistently evolving, many pharmacologists have not yet heard about this receptor. This is not surprising because, to some extent, this concept has not received unanimous acceptance in the biomedical community. For example, in the 12th edition of Goodman & Gilman’s The Pharmacological Basis of Therapeutics (Brunton et al., 2011), there is only one sentence mentioning imidazoline receptors. In the fifth edition of the Guide to Receptors and Channels (Alexander et al., 2011), imidazoline receptors are only briefly described under the entry on α2-adrenoceptors. However, there has recently been renewed interest in increasing understanding and pharmacological study of this system due to exciting new developments regarding the therapeutic potential of imidazoline receptor ligands. This review provides a comprehensive update on the historical and current status of imidazoline receptor research and offers our perspective on future directions.
II. History of Imidazoline Receptors
The early history of imidazolines and their receptors is very closely linked to that of clonidine. Clonidine is also a product of the α2-adrenergic receptor history. The stories of imidazolines and clonidine have subsequently remained intertwined but each has developed separately over time.
Clonidine was designed and synthesized by Stähle (2000) in the 1960s. Clonidine contains an imidazoline ring—that is, a five-atom ring, two of which are nitrogen atoms. Clonidine was first code-named ST155 (Shaw et al., 1971). More specifically, clonidine is an aminoimidazoline because there is a nitrogen atom between the imidazoline ring and the phenyl ring (see Fig. 1) (Stähle, 2000). Thus, the word “imidazoline” comes directly from the chemical name of the series of which clonidine is a member. In very exceptional cases, some authors also refer to the iminoimidazolidine structure due to the delocalization of the double bond around the intercyclic nitrogen atom (Leclerc et al., 1980; Stähle, 2000). The compound obviously contracts the vascular vessels and would have been of interest in the treatment of rhinorrhea (Corboz et al., 2013). However, it was never developed or used as such, since clonidine was shown to reduce blood pressure and heart rate at the time of the first trials in humans (Nayler et al., 1966; McRaven et al., 1971; Stähle, 2000).

Chemical structure of clonidine.
A vasoconstrictive agent that lowers blood pressure was obviously not expected. Yet a drug that lowers blood pressure and simultaneously slows the heart rate was also very intriguing. The antihypertensive drugs known at that time reduced blood pressure because of their vasodilatory properties; peripheral vasodilators are known to usually accelerate cardiac rhythm through the baroreflex (Cohn et al., 2011). Therefore, clonidine constituted from the beginning some kind of double paradox. Credit goes to Kobinger who showed that the hypotensive effect of clonidine originates in the central nervous system (CNS) and that the same applies to its bradycardic effect (Kobinger, 1967; Kobinger and Walland, 1967, 1972a,b). In the initial study on this matter, Kobinger showed that central injections of very low doses of clonidine in the cisterna magna of cats (i.e., in the vicinity of the brainstem) produced hypotension and bradycardia (Kobinger, 1967; Scriabine et al., 1970). The dose used in that study was too low to induce any cardiovascular effect when injected intravenously. Thus, evidence for the central origin of the unexpected cardiovascular effects of clonidine was provided.
Clonidine, in fact, causes the vasoconstrictive effect for which it was conceived but only when administered intravenously. With this route of administration, the plasma concentration is sufficiently high for clonidine to activate the α-adrenergic receptors of the vascular wall, leading to a very transient hypertensive peak lasting less than 2 minutes (Shaw et al., 1971). When administered by any other route (e.g., oral, intramuscular, and a fortiori by direct intracerebral route), clonidine no longer provokes the hypertensive (vasoconstrictive) effect that is observed when administered intravenously. The hypotensive effect of clonidine is produced through sympathoinhibition. Direct recordings of electrical activity on sympathetic nerves, such as the renal and splanchnic nerves, confirmed this fact (Klupp et al., 1970; Dhasmana et al., 1972; Armstrong and Boura, 1973; Bralet and Rochette, 1973; Schmitt and Fénard, 1973a; van Zwieten, 1973).
Thus, the existence of a long-lasting hypotensive effect of central origin not preceded by any hypertensive phase has made clonidine an antihypertensive drug (McRaven et al., 1971). Clonidine was used with success in the 1970s to 1980s in the treatment of primary hypertension (Khan et al., 1970; Hoobler and Sagastume, 1971). At the time, clonidine represented great progress in the treatment of hypertension. Nevertheless, because of its side effects, interest in using clonidine for hypertension has waned and it has been largely replaced by newly marketed drugs that produce fewer side effects compared with clonidine (e.g., sedation, mouth dryness, sexual impotence, and rebound effects upon treatment cessation) (Delbarre and Schmitt, 1973; Cavero et al., 1977). Despite these adverse effects, clonidine is still widely used to treat symptoms due to the sympathetic activation observed during opioid withdrawal (Cottereau et al., 1979; Fantozzi et al., 1980; Gowing et al., 2017).
Because clonidine was designed as an α-adrenergic agonist, it was first tempting to propose that the stimulation of such receptors in the CNS could explain its hypotensive and bradycardic effects. Indeed, there are α-adrenergic receptors in the CNS. Schmitt was the first to describe that substances known for having α-adrenergic antagonist properties are able to prevent the hypotensive effect of clonidine (Delbarre and Schmitt, 1973; Bogaievsky et al., 1974). Of all α-adrenergic antagonists used at that time, Schmitt described the blocking properties of the clonidine cardiovascular effects by only two agents: yohimbine and piperoxan. It is interesting to note that the classic α-blockers of the time, such as phenoxybenzamine, were not mentioned by this Schmitt and Fénard (1973a). At the end of the 1970s, Starke et al. (1977) and Langer et al. (1977) described the existence of two α-adrenergic receptor subtype, namely the α1- and α2-subtypes, respectively. α2-Adrenoceptors were described to be mainly presynaptic in location and involved in the negative feedback control of the release of noradrenaline, the neurotransmitter from the orthosympathetic system, into the synapse (Starke and Endo, 1976; Langer et al., 1977; Miach et al., 1978). At that time, yohimbine and piperoxan were described as compounds selective for α2-adrenergic receptors (Bolme et al., 1974; Gold et al., 1978; Hunt et al., 1978; Drew et al., 1979; Guyenet and Cabot, 1981; Rouot et al., 1982). Binding studies also confirmed that clonidine had affinity for α2-adrenergic receptors (Greenberg et al., 1976; U’Prichard et al., 1977; Kapur et al., 1979; Rouot and Snyder, 1979).
It is interesting to note that initially the main adverse effects of clonidine and the first-generation derivatives of centrally acting antihypertensive drugs were attributed to the activation of α2-adrenergic receptors. This applies particularly to the sedative effects (Cavero and Roach, 1978; Drew et al., 1979). Nevertheless, a new idea germinated very rapidly—that is, to determine whether it was pharmacologically possible to distinguish the hypotensive effects from the unwanted side effects and to develop better tolerated centrally acting drugs for lowering blood pressure (i.e., having less or no sedative effect, the most common side effect of clonidine). During the 1970s to 1980s, numerous structural analogs of imidazolines were synthesized, both in pharmaceutical companies and in academic pharmacochemistry laboratories (Boudier et al., 1975; Hoefke et al., 1975; Rouot et al., 1976; Leclerc et al., 1980; Stähle et al., 1980). Most often, they were screened according to their ability to activate α2-adrenergic receptors, since the α2-adrenergic theory dictated the mechanism of the hypotensive action of imidazoline derivatives. The screening tests used were often binding assays relating to specific α2-adrenergic binding sites (Kapur et al., 1979; Leclerc et al., 1980; Carenzi et al., 1989). With hindsight, it can now easily be understood why the structural analogs have at best been shown to be hypotensive and sedative as was the lead product, clonidine. By following a different scenario, it was finally possible to discriminate, from a mechanistic point of view, between the favorable effects on blood pressure and the side effects such as sedation.
It must nevertheless be emphasized with this pharmacochemistry approach that it was possible to individualize a second generation of products called “hybrid” agonists. These latter compounds remained able to bind to both α2-adrenergic receptors and imidazoline receptors but with a lower affinity for α2-adrenergic receptors than that of clonidine, so that their selectivity for imidazoline receptors versus α2-adrenergic receptors was more favorable to I1 imidazoline receptors. These compounds, whose prototypes have been widely used in the treatment of hypertension, were moxonidine and rilmenidine (Gomez et al., 1991; Bousquet, 2001; Reid, 2001; Edwards et al., 2012). In agreement with the pharmacological concepts resulting from the above-mentioned research, these drugs have proven to be hypotensive but less sedative than the first-generation products such as clonidine (Bousquet, 2001).
To further study whether the hypotensive effects and side effects could be separated, the first step was to locate the site of action of clonidine and clonidine-like compounds within the CNS. Based on cross-section experiments, it was quickly concluded that the site(s) of action of clonidine was located within the brainstem, which is known to contain many structures involved in the autonomic regulations of cardiovascular functions (Schmitt and Fénard, 1973a; Trolin, 1975).
In the early 1970s, Bousquet and Guertzenstein (1973) reported that topical applications of a very low concentration of clonidine on a particular area of the ventral surface of the brainstem in cats lowered blood pressure. Beneath the surface of this rostroventral region of the medulla oblongata, there is a small nucleus containing sympathetic neurons, called the nucleus reticularis lateralis (NRL) (Bousquet et al., 1981; Bousquet and Feldman, 1987; Tibiriça et al., 1989, 1991, 1992). This nucleus acts as a vasopressor center, since its blockade by tetrodotoxin, which abolishes neuronal depolarization, leads to a fall in blood pressure (Bousquet et al., 1980). Studies using the microinjection technique (i.e., injection of microvolumes directly into the NRL region by a stereotaxic approach) confirmed that very low doses of clonidine reduce blood pressure (Bousquet et al., 1981; Gatti et al., 1988).
Other groups focused their attention on the nucleus tractus solitarii located in the dorsal part of the medulla oblongata, which acts as the first central relay of the baroreflex arc that has the role of a vasodepressive center (Schmitt and Fénard, 1972, 1973b; Reis et al., 1977; Rockhold and Caldwell, 1979, 1980; Howe, 1985). In very similar studies, it was shown that clonidine could also induce, at least partly, its hypotensive effect from this structure (Laubie et al., 1976; Lipski et al., 1976; Zandberg et al., 1979; Rockhold and Caldwell, 1980; Kubo and Misu, 1981; Vlahakos et al., 1985). However, there was a growing consensus that the NRL played a dominant role among the sites of action for clonidine (Antonaccio and Halley, 1977; Ernsberger and Haxhiu, 1997). From that moment on, studies concerning the pharmacological mechanism of action of clonidine-like compounds could be conducted directly at the site(s) of action of clonidine, leading to more accurate information.
It should be noted that during the same period, Ruffolo et al. (1979a,b,c, 1980a,b) published a series of articles, many of which described the differences in effects of substances bearing an imidazoline structure and others with a phenylethylamine structure on α-adrenergic receptors. This drew attention to the differences between imidazolines and phenylethylamines, in particular as far as their actions on α2-adrenergic receptors were concerned. In this context, a structure-activity relationship study was conducted by using the microinjection technique. In this study, α-methylnoradrenaline was used as a reference substance with a phenylethylamine structure and was highly selective for α2-adrenergic receptors. α-Methylnoradrenaline was not capable of inducing any hypotensive effect when it was directly injected into the NRL, whereas imidazoline compounds reduced blood pressure irrespective of their selectivity for α1- or α2-adrenergic receptors (Bousquet et al., 1984). It is on this basis that “sites preferring the imidazoline structure” were proposed for the first time (Bousquet et al., 1984). This initial study was followed by radioligand binding studies using membrane preparations from tissues collected in the rostro-ventrolateral region of the human medulla oblongata, which showed that about 80% of the specific [3H]clonidine high-affinity binding was not displaced by various catecholamines such as adrenaline, norepinephrine, and dopamine (Bricca et al., 1988, 1989, 1993, 1994; De Vos et al., 1994; Greney et al., 1994).
At the same time, Reis and colleagues performed similar experiments in the bovine brainstem and showed that 20%–30% of the [3H]para-aminoclonidine (PAC) high-affinity binding sites were also resistant to catecholamines (Meeley et al., 1986; Ernsberger et al., 1987). Biochemical confirmation of the existence of specific binding sites for the imidazoline compounds was therefore conclusive. These sites were defined as binding sites sensitive to imidazoline derivatives but insensitive to catecholamines. In fact, for the sake of semantic simplification, Donald Reis named them “imidazoline receptors” instead of “imidazoline-preferring receptors” (Ernsberger et al., 1987). Since then, the entire scientific community interested in these receptors has used this denomination consensually. The principle of this denomination has been modeled onto the benzodiazepine receptors, which also refers to the chemical structure of the compounds that bind to them and that act on them. As far as imidazolines and imidazoline-like compounds are concerned, specific high-affinity binding sites were associated with functions such as sympathetic inhibition and blood pressure reduction.
Together with some other properties such as stereospecificity, this association makes them genuine receptors (Laduron, 1988). Various types of experiments led to the subdivision of the imidazoline receptors into three receptor subtypes: I1, I2, and I3. Studies of functional pharmacology and/or binding led to this subclassification and the different aspects of these subtypes are discussed later in this review. Subsequently, clonidine was administered to engineered mice whose α2-adrenergic receptors were not functional. These experiments confirmed that clonidine can induce a hypotensive effect independently of any action on α2-adrenergic receptors (Bruban et al., 2001). However, as far as “hybrid” drugs are concerned, concomitantly targeting imidazoline receptors and α2-adrenergic receptors has a synergistic action on blood pressure (Bruban et al., 2002). Nevertheless, an exclusive action on the imidazoline receptors is enough to induce a hypotensive action (Bruban et al., 2002).
Given the fact that a hypotensive effect can be obtained by an exclusive action on imidazoline receptors and that the main adverse effects of clonidine are related to the activation of α-adrenergic receptors, it became conceptually possible to design drugs more selective for the imidazoline receptors and thus less active at the α2-adrenergic receptors. Structural analogs of clonidine devoid of any effect on α-adrenergic receptors are now available. Their potential therapeutic applications are currently being studied. Such compounds could represent a basis for the development of drugs to be used in hypertension and perhaps for other indications, as discussed later in this review (Fellmann et al., 2013a; Gasparik et al., 2015). Once the concept of imidazoline receptors was accepted by a large number of scientists interested in the potential therapeutic effects and uses of drugs targeting these receptors, they were quickly subdivided into three subtypes: I1, I2 and I3. This classification results from biochemical, pharmacological, and functional characterizations (Vauquelin et al., 1999; Morgan and Chan, 2001; Dardonville and Rozas, 2004; Li et al., 2015). This work will also describe the most recent prospects for I2 and I3 imidazoline receptors and their possible new clinical applications.
III. Endogenous Imidazoline Receptor Ligands
As a bona fide receptor, one line of research along the continued study and refinement of imidazoline receptors is to determine and characterize their endogenous ligands, an essential step for improving understanding of this new receptor system. Over the years, several substances have been put forward as endogenous ligands, including clonidine-displacing substance (CDS), harmane, imidazole-4-acetic acid-ribotide (IAA-RP), and agmatine.
A. Clonidine-Displacing Substance
One of the first articles to suggest the presence of an endogenous ligand was published in the 1980s by Atlas and Burstein (1984). Their work used an extract of bovine brain that when partially purified was able to displace [3H]clonidine binding in rat brain membranes. However, this [3H]clonidine binding was to α2-adrenoceptors, as further work showed that high-performance liquid chromatography (HPLC)–purified CDS extract would compete for [3H]yohimbine binding to α2-adrenoceptors (Atlas and Burstein, 1984) and also for [3H]rauwolscine binding to human platelets (Diamant et al., 1987). Other work demonstrated that the extract would displace labeled ligands from I1 and I2 receptors (Ernsberger et al., 1988; Coupry et al., 1990). Further work by Atlas and colleagues used plasma desorption mass spectrometry to determine that CDS had a mass of 587.8 Da and that it was not an amino acid or primary amine compound, as it was ninhydrin and fluorescamine negative in nature (Atlas et al., 1987; Atlas, 1994).
In that same decade, polyclonal antibodies were raised against PAC (Dontenwill et al., 1988) and binding of [3H]PAC to the antibody was inhibited by many imidazoline compounds but not by catecholamines. Moreover, [3H]PAC binding was inhibited by CDS in a concentration-dependent manner, demonstrating that CDS and these imidazolines were recognizing the same or a similar site on these antibodies (Dontenwill et al., 1988; Meeley et al., 1988). Another polyclonal antibody raised against idazoxan recognized CDS extracted from human serum and cerebrospinal fluid and led to the term “immunoreactive CDS” (Wang et al., 1993). Having a tool that recognizes immunoreactive CDS resulted in elegant studies detailing the presence of CDS in several tissues such as the brain, heart, small intestine, liver, kidney, adrenal gland, and serum (Meeley et al., 1992). It was apparent that this distribution was not similar to the distribution of agmatine in the body and that agmatine might not be the pharmacologically active component of CDS. That the active component of CDS remained a mystery for many years is not that unexpected, as different groups had variations in extraction and purification techniques and also the tissue used as a source of CDS. This mystery was set to be solved to some extent by a more rational approach to comparing CDS from different tissues and comprehensive spectroscopic methods. Initial work by Parker et al. (1999a,b) examined crude methanolic extracts of CDS from bovine lung, brain, and adrenal glands, which were partially purified by reverse-phase HPLC and then assayed for affinity at α2-adrenoceptors, I1 receptors, and I2 receptors. Each tissue proved to be a source of CDS. For bovine lung, fraction 21 collected via reverse-phase HPLC contained material that inhibited [3H]clonidine binding and corresponded to a peak of absorption at 276 nm (Parker et al., 1999a,b). In further work, Parker et al. (2000) were also able to extract CDS from NG108-15 cells that also inhibited [3H]clonidine binding. One component of CDS was identified as tryptophan, but although present, it was shown to be inactive at displacing [3H]clonidine binding or the selective I2 receptor ligand [3H]2-(2-benzofuranyl)-2-imidazoline (2-BFI) (Hudson et al., 1999b; Parker et al., 1999c). Finally, Parker et al. (2004) refined their techniques further and incorporated electrospray mass spectrometry and 1H NMR to further analyze purified bovine lung CDS. This resulted in the isolation and identification of the β-carbolines harmane and harmalan (Fig. 2) and revealed them to be biologically active components of CDS having nanomolar affinities at I1 and I2 receptors (Parker et al., 2004).

Chemical structures of candidate endogenous imidazoline receptor ligands harmane, IAA-RP, harmalan, and agmatine.
Although the study by Parker et al. (2004) provides interesting answers to the likely biologic activity of CDS and shows that harmane and harmalan are components of bovine lung CDS, it does not explain how or where these β-carbolines originated. It is known that β-carbolines are the condensation products of tryptamine, an indoleamine that has been shown to have moderate affinity for I2 receptors (Hudson et al., 1999b). It is likely that β-carbolines are formed in vivo via an enzymatically catalyzed Pictet–Spengler reaction (Rommelspacher et al., 1985; Susilo and Rommelspacher, 1987), although oxidation of tetrahydro-b-carbolines was shown to form harmane and norharmane is catalyzed by haem peroxidases (Herraiz and Galisteo, 2014), providing more evidence of endogenous production. Much research describes the pharmacology of β-carbolines, some of which will be discussed in the next section. Although β-carbolines are an active component of CDS, other active endogenous ligands have also been extracted from CDS (Li et al., 1994).
B. Harmane
As described above, harmane and harmalan were shown to be pharmacologically active components of CDS and also were shown to have affinity for I1 and I2 receptors (Husbands et al., 2001). However, harmane and several β-carbolines have a diverse pharmacology binding to several sites, including monoamine oxidase (MAO) as well as serotonin, dopamine, histamine, and benzodiazepine receptors (Glennon et al., 2000; Arib et al., 2010). Some β-carbolines, including harmane, are found in cooked foods and fermented drinks, and they may be associated with the pathogenesis of essential tremor and may also be involved in clinical conditions such as Parkinson disease and amnesia (Herraiz and Papavergou, 2004; Pfau and Skog, 2004; Laviţă et al., 2016). Despite harmane’s pharmacological diversity, it is an attractive prospect as an endogenous ligand for imidazoline receptors. At I1 receptors, harmane binds with nanomolar affinity, higher than that of agmatine (Hudson et al., 1999b; Husbands et al., 2001), and injection of harmane into the rat brainstem results in hypotension much like the injections of clonidine. Furthermore, these effects are blocked by the I1 receptor antagonist efaroxan, indicating a role for harmane in controlling blood pressure (Musgrave and Badoer, 2000). Like several I2 receptor ligands, harmane is a MAO inhibitor and this may represent a mechanism for modulating central monoamine levels (Glover et al., 1982; Lalies et al., 1999). This idea is supported by drug discrimination experiments performed in rats, in which animals trained to recognize 2-BFI and two MAOA inhibitors, moclobemide and RO41-1049 [N-(2-aminoethyl)-5-(3-fluorophenyl)-4-thiazolecarboxamide hydrochloride], fully substituted for 2-BFI, suggesting shared pharmacological mechanisms of action (MacInnes and Handley, 2002). The same study showed that several I2 receptor ligands, such as BU216 (3-[4,5-dihydroimidaz-2-yl]-quinoline hydrochloride), BU224 (2-[4,5-dihydroimidaz-2-yl]-quinoline hydrochloride), BU226 (2-[4,5-dihydroimidaz-2-yl]-isoquinoline hydrochloride), and LSL60101 (2-[2-benzofuranyl]-2-imidazole hydrochloride), and harmane were able to substitute for 2-BFI, whereas MAOB inhibitors did not. More support for an association of I2 receptors with MAOA was established using in vitro autoradiography in the rat brain (Anderson et al., 2006b). Analysis of the autoradiograms showed a highly significant correlation between the distribution of [3H]harmane and [3H]RO41-1049 binding in the CNS and also a significant correlation between the distribution and binding density of [3H]harmane– and [3H]2-BFI–labeled sites. The study did not rule out the possibility of a small population of I2 receptor binding not associated with MAOA distribution (Anderson et al., 2006b).
I2 receptor ligands have been shown to alleviate some behaviors associated with naloxone-precipitated morphine withdrawal in rats (Hudson et al., 1999a), and harmane is also able to mimic these affects presumably via I2 receptors (Aricioglu-Kartal et al., 2003). We now know that I2 receptors play a role in chronic pain, and harmane is reported to show antinociceptive activity comparable to I2 receptor–selective ligands (Aricioglu et al., 2003; Aglawe et al., 2014). Harmane also binds to I3 receptors located in the pancreas that are involved in the regulation of insulin secretion (Morgan et al., 2003). Although harmane binds to all classes of imidazoline receptor, it is interesting to note that extracts of rat CDS were also shown to stimulate insulin secretion (Chan et al., 1997). However, the ability of harmane to potentiate the glucose-dependent release of insulin from rat and human isolated islets did not entirely match the effects induced by the I3 receptor agonist efaroxan. Morgan et al. (2003) suggested that subtle differences in methodology could have been responsible for the differences seen or that, unlike efaroxan, harmane was able to access an intracellular pool of Ca2+ to mediate some of its effects.
In past studies, the availability of radioactive bioactive molecules has allowed investigation of their potential as putative neurotransmitters by studying their uptake and stimulated release from brain synaptosomes or tissue homogenates. With the availability of [3H]harmane, this approach was used to investigate the potential of harmane as the natural modulator or transmitter substance for imidazoline receptors (Abu Ghazaleh et al., 2015a). The uptake of [3H]harmane into rat cortical slices was examined and determined to be approximately 450 fmol/mg protein, greater than the uptake of [3H]dopamine and [3H]noradrenaline under the same conditions (260 ± 43.9 and 291.4 ± 56.1 fmol/mg protein, respectively). [3H]harmane uptake was not affected by monoamine reuptake blockers nor by I1 receptor or I2 receptor ligands. This apparent [3H]harmane uptake was not found to be Na+ dependent and was unaffected by blockade of the Na+-K+ ATPase pump with ouabain. Transiently elevating K+ stimulated the release of stored [3H]noradrenaline, [3H]serotonin, and [3H]dopamine in control superfusion experiments, whereas [3H]harmane release was unaffected (Abu Ghazaleh et al., 2015a). Taken together, the results did not support that, under the conditions employed, harmane was acting as a typical neurotransmitter. Work by the same group examined the effect of harmane on [3H]monoamine release from rat cortical tissue in vitro and found that harmane (100 μM) was able to enhance the K+ evoked release of [3H]serotonin but not that of K+ evoked release of [3H]noradrenaline or [3H]dopamine (Abu Ghazaleh et al., 2015b).
To date, the other β-carboline found in CDS (harmalan) has not been widely studied, so its status as a potential endogenous ligand for I receptors is unclear. Although harmane is found endogenously in many tissues, one has to be aware that exogenous sources are a confounder to some extent because β-carbolines are also present in foodstuffs and fermented beverages (Herraiz and Galisteo, 2003). Evidence demonstrates that harmane is an active constituent of CDS and affects blood pressure via I1 receptors, inhibits MAOA possibly via I2 receptors, and regulates insulin release via an I3 receptor–mediated process. Despite these attributes, attempts to demonstrate a neurotransmitter role for harmane have to date proved futile, at least in its radiolabeled form. One is reminded of the huge excitement when β-carbolines, extracted from human urine, were proposed as endogenous ligands for the benzodiazepine receptor and then the subsequent dismay when these substances were found to be an extraction artifact (Braestrup et al., 1980). However, the finding that harmane and several β-carbolines bind to imidazoline receptors with high affinity remains important and intriguing (Abu Ghazaleh et al., 2015c).
C. Imidazole-4-Acetic Acid-Ribotide
IAA-RP (Fig. 2) is derived by ribosylation of IAA in the brain (Prell et al., 2004). IAA, which is an agonist for GABAA receptors, has been shown to exist in the rat brain and to be ribosylated to form IAA-RP at micromolar levels in the brain (Prell et al., 2004).
Most of the observations to support IAA-RP as an endogenous ligand were comprehensively reported by Prell et al. (2004). They found that rat brain extracts contained 1.1 µg IAA-RP/g tissue, and neurons of the rostral ventrolateral medulla stained heavily for IAA-RP particularly on neuronal processes. IAA-RP displaced [3H]clonidine binding from adrenal medulla I1 receptors with a Ki value of 13 µM; IAA-RP stimulated [3H]arachidonic acid release from PC12 cells in a functional assay. Similarly, IAA-RP had high affinity for brainstem I1 receptors (Ki, 100 nM) determined using p-[125I]iodoclonidine as the radioligand. Under conditions in which the I1 binding component was masked, IAA-RP bound to presumed α2-adrenoceptors with a Ki of 210 µM. In a functional assay for I3 receptors, IAA-RP increased insulin secretion from rat and human pancreatic islet cells, an effect blocked by the I3 receptor antagonist KU-14R [2-(2-ethyl-2,3-dihydro-2-benzofuranyl)-1H-imidazole] (Prell et al., 2004). Using crude synaptosomic and vesicle-enriched rat brain P2 preparations, K+-evoked release of IAA-RP was shown to be Ca2+ dependent, thus demonstrating that IAA-RP has the characteristics of a neurotransmitter. One surprising finding of this study was that IAA-RP (100 nmol) microinjected into the rat brainstem elevated mean arterial blood pressure, and this effect was reversed by the I1 receptor agonist moxonidine. Overall, Prell et al. (2004) concluded that IAA-RP is a neurotransmitter and may also exhibit hormone-like activity in the periphery. More recently, IAA-RP immunoreactivity has been used to study the distribution of IAA-RP in the rat brain to demonstrate its neuronal location, particularly in structures such as the olfactory bulb, granule cells of the dentate gyrus, and superior colliculus, and it was noteworthy that the distribution of IAA-RP mirrored to some extent that of [3H]harmane (Anderson et al., 2006a; Friedrich et al., 2007). An electrophysiological study showed that IAA-RP elicits synaptic depression in rat hippocampal slice preparations in a concentration-dependent manner. This effect was blocked by efaroxan and inhibited to some extent by the I3 receptor antagonist KU-14R, leading Bozdagi et al. (2011) to speculate the involvement of an I3-like receptor as well as I1 receptors. Thus, IAA-RP is present in brain neurons, has functional effects including blood pressure modulation, and in the periphery stimulates release of insulin from the pancreas, making this substance a strong candidate as an endogenous ligand for I receptors.
D. Agmatine
The final candidate is agmatine (Fig. 2), a substance that has long been recognized as endogenous to the mammalian body and was proposed by Reis and colleagues as a ligand for I receptors (Li et al., 1994). Since then, an enormous amount of research has established agmatine as the front runner in terms of our understanding of a biologic modulator or transmitter substance for I receptors.
Agmatine is synthesized from l-arginine by arginine decarboxylase and hydrolyzed by agmatinase (Reis and Regunathan, 2000). In 2003, the entire proceedings of the fourth international symposium on agmatine and imidazoline systems were published as a single volume in memory of Donald Reis (Annals of the New York Academy of Sciences, Volume 1009). Agmatine was isolated from bovine brain CDS and determined to be present at 1.5–3.0 nmol/g tissue (Li et al., 1994). It soon became apparent that agmatine was a novel neurotransmitter in the brain, as its synthesis, uptake, vesicular storage, release by depolarization, and subsequent breakdown by agmatinase was demonstrated in the CNS (for review, see Reis and Regunathan, 2000). However, agmatine has a diverse pharmacology and the involvement of I receptors is not always evident (Piletz et al., 2013), particularly because some groups find agmatine to have low affinity for I1 and I2 receptors (Hudson et al., 1999b). For example, agmatine inhibits nitric oxide synthase (Regunathan and Piletz, 2003) and antagonizes N-methyl-d-aspartic acid (NMDA) receptors, which may explain the neuroprotective actions of agmatine in rodent models of stroke (Gilad et al., 1996) and glutamate-induced toxicity in cultured rat cerebellar granule cells (Olmos et al., 1999b). Several studies have detailed the effects of agmatine on nociception in rodent models of pain and showed that agmatine enhanced morphine-induced antinociception, an effect mediated through α2-adenoceptors and/or I receptors (Aricioglu et al., 2003; Bhalla et al., 2011). However, because agmatine is effective in chronic pain models versus acute models of nociception and selective I2 ligands such as 2-BFI, BU224, and CR4056 [2-phenyl-6-(1H-imidazol-1yl) quinazoline] are shown to have therapeutic potential in chronic pain models, I2 receptors rather than α2-adrenoceptors appear to be involved in nociception (Ferrari et al., 2011; Thorn et al., 2016a). In summary, many consider agmatine to be the endogenous ligand for I receptors despite its low affinity for these sites, and much published work favors agmatine over other putative endogenous substances such as IAA-RP or harmane (Piletz et al., 2013; Abu Ghazaleh et al., 2015c).
IV. Imidazoline Subtype 1 Receptors
A. Definition
Ernsberger et al. (1993) classified the imidazoline receptors and initiated the terminology “I1 and I2 imidazoline sites.” At that time, conclusions were mainly based on data from specific binding experiments using [3H]clonidine and [3H]idazoxan (Michel and Ernsberger, 1992; Ernsberger et al., 1993). According to this definition, the specific binding sites labeled with tritiated clonidine but resistant to catecholamines (i.e., nonadrenergic receptors) were designated as I1 imidazoline receptors by convention.
B. Specific Binding Properties, Selective Ligands, and Tissue and Subcellular Localization
1. Selective Ligands
Clonidine has been shown to specifically bind to nonadrenergic receptors, particularly non–α2-adrenergic receptors, in cell membrane preparations taken from the rostro-ventrolateral part of the brainstem, which contains the main site of the hypotensive action of clonidine (see section II). These clonidine-labeled binding sites themselves are sensitive to other imidazoline compounds or even imidazoles. These imidazoline sensitive binding sites, which are insensitive to catecholamines and even to histamine, are different from those that are labeled by another imidazoline (namely, tritiated idazoxan).
Indeed, idazoxan has a much lower affinity than clonidine for clonidine-labeled sites (Michel and Insel, 1989; Brown et al., 1990; Coupry et al., 1990; Wikberg et al., 1991). These experiments analyzing in detail the binding of tritiated clonidine and that of tritiated idazoxan led to the classification of I1 receptors as having high affinity for tritiated clonidine, whereas I2 receptors are those sites with high affinity for tritiated idazoxan. Clonidine and idazoxan are therefore the two historical markers of the two main classes of imidazoline receptors. Subsequently, many other substances have been used to further investigate specific imidazoline binding. When studied with selective ligands, the specific binding of imidazolines to nonadrenergic receptors was shown to be saturable, reversible, and of high affinity (Greney et al., 1994). 125I-labeled para-iodoclonidine was used in a number of studies to characterize the specific binding to I1 receptors (Ernsberger et al., 1993). Radiolabeled idazoxan has also been used in attempts to purify the I1 receptor by chromatography (Greney et al., 1997). Ernsberger et al. (1993) showed that there is no correlation between affinities of imidazoline compounds for α2-adrenergic receptors and their hypotensive effects in vivo.
Because imidazoline binding sites are responsible for the hypotensive effects of imidazoline substances and of their close derivatives, these specific binding sites can be considered as authentic functional receptors (Gomez et al., 1991; Ernsberger et al., 1993; Bousquet, 2001). Efaroxan is an imidazoline drug that antagonizes the functional effects of clonidine, including its hypotensive effects. This compound does not significantly compete with α2-adrenergic receptors (Ernsberger and Haxhiu, 1997); thus, the definition of the I1 imidazoline receptors has somewhat improved. It is now a receptor that is sensitive to clonidine and antagonized by efaroxan (Ernsberger and Haxhiu, 1997).
Compound RX82-1002 [2-(2,3-dihydro-2-methoxy-1,4-benzodioxin-2-yl)-4,5-dihydro-1H-imidazole hydrochloride] has also been used to mask α2-adrenergic receptors in various studies relating to the specific binding of [3H]clonidine. As long as very selective ligands of I1 receptors were not available, it was necessary to follow a strategy of masking α2-adrenergic receptors to study the I1 receptors. For example, RX82-1002 was used for this purpose (Bruban et al., 2001). The first ligands thus developed were a series of aminopyrrolines whose first prototypes were LNP509 [cis-/trans-dicyclopropylmethyl-(4,5-dimethyl-4,5-dihydro-3H-pyrrol-2-yl)-amine], LNP906 [2-(5-azido-2-chloro-4-iodo-phenylamino)-5-methyl-pyrroline], LNP911 [(2-(2-chloro-4-iodo-phenylamino)-5-methyl-pyrroline], and LNP599 [3-chloro-2-methyl-phenyl)-(4-methyl-4,5-dihydro-3H-pyrrol-2-yl)-amine hydrochloride] (Fig. 3). The most studied was LNP599 because of its pharmacodynamic and pharmacokinetic characteristics, which could make it a lead compound for the development of new therapeutics in the cardiovascular or metabolic field (Gasparik et al., 2015). Details will be provided later in this section.

Chemical structures of aminopyrrolines of interest LNP911, LNP906, LNP509, and LNP599.
2. Tissue Distribution
When the binding experiments were carried out with mixed ligands (i.e., capable of binding to both α2-adrenergic receptors and imidazoline receptors), it was necessary to follow the strategies of avoiding or eliminating the binding to α2-adrenergic receptors so to focus only on binding to I1 imidazoline receptors. In fact, two strategies have been used: namely, the masking of α2-adrenergic receptors by a specific and selective ligand of these receptors, such as α-methyl-noradrenaline or rauwolscine, or using membrane preparations from cells that do not express α2-adrenergic receptors but only I1 imidazoline receptors, as is the case of chromaffin cells of the adrenal gland (PC12 line). Thus, it has been demonstrated that moxonidine, a second-generation ligand, was more selective for I1 than for α2-adrenergic receptors compared with clonidine (Regunathan et al., 1991b; Wang et al., 1992).
I1 binding sites were found in the bovine brainstem (Ernsberger et al., 1987, 1988; Bricca et al., 1989) and also in the human, rabbit, and rat brain (Bricca et al., 1989, 1993). Similar observations were made by Kamisaki et al. (1990) in rat brains and by Ernsberger et al. (1987) in neurons taken from the ventrolateral portion of the bovine brainstem.
Although most of the imidazoline receptor autoradiography experiments published to date have been devoted to I2 receptors, some studies have used I1 ligands. King et al. (1995a,b,c) used [3H]idazoxan, [3H]para-aminoclonidine, and [3H]rilmenidine to describe sites corresponding to the definition of specific I1 binding sites in the kidney and brain of rats. De Vos et al. (1994) used [3H]idazoxan to differentiate imidazoline receptors from α2-adrenergic receptors in the human CNS (De Vos et al., 1994). Using [3H]para-aminoclonidine and [3H]idazoxan, MacKinnon et al. (1993) showed the existence of I1 binding sites in the rat kidney and also I2 binding sites under α2-adrenergic receptor masking conditions. MacKinnon et al. (1993) found some differences, notably in the receptor density that was lower in the kidney than in the human and bovine brain. In a study of membrane preparations from proximal tubular cells of rabbit using [3H]idazoxan, specific I1 binding sites were found in this particular region of the kidney (Gargalidis-Moudanos and Parini, 1995). Escribá et al. (1994) achieved immunodetection of imidazoline receptors in the rat brain and the human brain in particular.
Imidazoline binding sites were also found in the rat kidney (Ernsberger et al., 1990), in human platelets (Piletz et al., 1991), in chromaffin cells of the adrenal gland (Separovic et al., 1996), as well as in cat and rabbit carotid sinuses (Kou et al., 1991). Piletz and Sletten (1993) characterized I1 imidazoline receptors on human and rabbit platelets. They showed in binding experiments in human platelets that in addition to the classic α2-adrenergic binding of this ligand, the [3H]PAC radioligand also binds to nonadrenergic sites corresponding to the definition of I1 imidazoline receptors (Piletz et al., 1991). In addition, specific I1 binding sites have been described in the dog prostate gland (Felsen et al., 1994).
There are no detectable I1 receptors in cardiovascular tissues, including the heart; thus far, only I2 receptors have been found. However, Ernsberger et al. (1998) showed, with para-iodoclonidine, that there are I1 receptors in the carotid bodies. Nevertheless, using the binding technique, I1 imidazoline receptors were shown to be present in the atria and ventricles of rat hearts (El-Ayoubi et al., 2002, 2004). Several teams have shown the existence of imidazoline binding sites in adipocytes of different species, including the hamster and rat (MacKinnon et al., 1989; Langin et al., 1990; Fellmann et al., 2013b; Weiss et al., 2015). Weiss et al. (2015) demonstrated the existence of specific I1 binding sites of imidazolines in hepatic cell lines, and Molderings et al. (1995) also found fairly abundant amounts in the rat stomach. In summary, the expression of the specific I1 binding sites is rather ubiquitous and concerns mainly the CNS but also the digestive and endocrine system, cardiovascular system, and adipose tissue.
3. Subcellular Distribution
In a study performed on membrane preparations from neurons taken from the ventrolateral part of the brainstem by a discontinuous sucrose density gradient, Ernsberger and Shen (1997) showed that I1 receptors were predominantly on nonmitochondrial membranes. This localization differentiates them clearly from I2 receptors (Ernsberger and Shen, 1997). Heemskerk et al. (1998) subsequently showed that high-affinity I1 receptors in bovine brain tissue are particularly expressed by synaptic membranes, and most likely from presynaptic terminals. Although another team observed a partial mitochondrial localization of I1 receptors, it nevertheless confirmed that the highest expression levels were in the plasma membrane fractions in the rat cerebral cortex (Hosseini et al., 1998). Keller and García-Sevilla (2015) used immunodetection to localize I1 receptors in the membrane fractions of mouse and human brains. As can be seen, subcellular localization studies are relatively few, but they all converge toward a major localization on the plasma membranes for the expression of these receptors, a location very different from that of the I2 receptors, which will be detailed later in this review.
4. Second-Generation Imidazoline Subtype 1 Receptor Ligands
To achieve the second-generation central antihypertensive drugs moxonidine and rilmenidine, various substitutions were made on the aromatic part of the imidazoline molecule in the case of moxonidine and on the oxazoline structure instead of an authentic imidazoline structure in the case of rilmenidine (Bricca et al., 1989).
The antihypertensive moxonidine is a second-generation I1 receptor–selective drug, with a 10- to 700-fold greater affinity for I1 receptors than for α2-adrenergic receptors (Ernsberger et al., 1993). Similarly, rilmenidine was developed for the same purposes and is, like moxonidine, used as an antihypertensive drug with fewer adverse effects, particularly sedation (Reid, 2001).
Tritium-labeled moxonidine and rilmenidine have also been used as markers for I1 receptors in various experimental studies (King et al., 1992, 1993, 1995c, 1998). The development of imidazoline-like drugs, which were more selective for I1 receptors than for α2-adrenergic receptors, demonstrated that modifications of the chemical structure could improve this selectivity. Then, new pharmacochemistry projects were developed to further improve this selectivity through structure-activity relationship analysis. Thus, a chemical series of pyrroline compounds has been exploited and tested for biologic activities and also for its specific binding properties.
In this pyrroline series, LNP509, which is a dicyclopropyl-methyl-pyrrol-amine, was the first to have no detectable affinity for α2-adrenergic receptors but was still able to lower blood pressure after central administration (Schann et al., 2001). This validated the concept that substances with neither activity nor affinity for α2-adrenergic receptors were nevertheless capable of lowering blood pressure by sympathetic inhibition. Another structural modification also led to the development of substances that are highly selective for I1 receptors and induce hypotensive activity even after intravenous administration. These are methylated derivatives on the heterocyclic group; the prototype in this case was LNP630 [(2,6-dichloro-phenyl)-(4-methyl-4,5-dihydro-1H-imidazol-2-yl)-amine] (Schann et al., 2012). From this ligand, compounds were formulated to try to develop substances that could lead to new centrally acting antihypertensive drugs that were better tolerated than first-generation drugs. Thus, new molecules of the 2-aryl-imino-pyrrolidine family have been proposed. In this series, the LNP599 molecule has a nanomolar affinity for I1 receptors, has no detectable affinity or activity for the α2-adrenergic receptor, and decreases blood pressure at relatively low doses regardless of the route of administration. This molecule serves as a prototype for development of new drugs to treat hypertension or the metabolic syndrome. This perspective will be detailed later in this review (Gasparik et al., 2015).
5. Selective Imidazoline Subtype 1 Receptor Ligands with High Affinity as Pharmacological Probes
For the purposes of biochemical investigations concerning imidazoline receptors, the development of molecules with a very high affinity and selectivity for the I1 receptors has been a priority. In the pyrroline series, LNP911 was the first molecule that exhibited very high affinity for I1 receptors and a very high selectivity for I1 receptors over α2-adrenergic receptors (Greney et al., 2002). Subsequently, a photoactivatable function has been added in the LNP906 structure to irreversibly label the I1 receptor with high affinity. LNP906 has also been shown to be an I1 receptor antagonist. These two substances, LNP911 and LNP906, have been used to study the I1 receptors (Greney et al., 2002; Urosevic et al., 2004). The entire range of compounds needed to study the target receptors is now available, and they are also useful to explore hypotheses for the development of new drugs.
6. Receptor–Receptor Interactions
As early as the mid-1990s, Hieble and Ruffolo (1995) suggested the existence of possible interactions between I1 receptors and α2-adrenergic receptors and wondered about the possible effects of multiple interactions between receptors and between agonists and antagonists on both types of receptors. Greney et al. (2000) used different cell lines, one expressing only α2-adrenergic receptors (HT29 cells), a second line expressing only I1 receptors (PC12 cells), and a third one expressing both of them (NG108-15 cells). These authors showed that both receptors are individualized entities, but they can couple to the same cAMP transduction pathway. Thus, interactions between the I1 receptors and the α2-adrenergic receptors may occur at least at the level of transduction pathways that they have in common (Greney et al., 2000). Shortly afterward, Bruban et al. (2002) showed that a α2-adrenergic agonist and a selective I1 receptor agonist, devoid of any affinity for α2-adrenergic receptors, synergistically lower blood pressure, suggesting functional interaction(s) between the two types of receptors. In this work, LNP509 was the I1 receptor agonist used, whereas α-methyl-noradrenaline was the reference α2-adrenergic agonist (Bruban et al., 2002). Chen et al. (2003) performed a study on Chinese hamster ovary cells expressing human α2-adrenergic receptors transfected with cDNA encoding human imidazoline receptor antisera-selected (IRAS) protein (a candidate protein of the I1 receptor). In these modified cells expressing both types of receptors, Chen et al. (2003) also showed an interaction between the two types of receptors. In summary, although the effects of molecular mechanisms beyond the interactions between I1 receptors and α2-adrenergic receptors are not yet fully elucidated, this interaction is presently strongly supported by experimental data.
Nevertheless, additional receptor–receptor interactions have also been proposed. A cannabinoid agonist, WIN52212-2 [(R)-(+)-[2,3-dihydro-5-methyl-3-[(4-morpholino)methyl]pyrrolo-[1,2,3-de]-1,4-benzoxazin-6-yl](1-naphthyl)methanone; phenyzoline,4,5-dihidro-2-(2-phenylethyl)-1H-imidazole], and agmatine used as an imidazoline receptor agonist could reduce central temperature by a synergistic mechanism in rats (Rawls et al., 2006). A similar synergistic interaction between agmatine and cannabinoid agonists was observed for their antinociceptive effects in the hot plate test (Aggarwal et al., 2009). Also in the field of antinociceptive effects, Stone et al. (2007) showed that there is a possible synergy between I1 receptors and opioids at the spinal level based on the selective I1 receptor agonist, diethyl-phenyl-amino-imidazoline, ST91 [2-(2,6-diethylphenylamino)-2-imidazoline hydrochloride] (Stone et al., 2007). Boxwalla et al. (2010) showed that an endothelin receptor antagonist is able to potentiate the antinociceptive effects of clonidine, indicating a negative interaction between endothelin receptors and imidazoline receptors. Chan and Morgan (1998) showed that two potent σ receptor agonists were able to increase insulin secretion from on islets of Langerhans isolated from rats. They studied the possible interaction between σ receptors and the imidazoline receptors (possibly of the I1 receptor) by using an I1 receptor antagonist efaroxan, and they suggested the existence of complex interactions of these two types of receptors at the pancreatic level (Chan and Morgan, 1998). Experiments were performed in rats using selective NMDA and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/kainate receptor antagonists and the I1 receptor agonists clonidine and moxonidine (Wang et al., 2007). The glutamate receptor antagonists abolished the hypotensive actions of clonidine and even its actions on heart rate. Wang et al. (2007) therefore suggested the existence of an interaction between the imidazoline receptors and the glutamate receptors.
7. Transduction Mechanisms
As expected, the hypothesis of a coupling of the I1 receptors with G proteins has been the subject of the greatest number of investigations. Regunathan et al. (1991a) explored this hypothesis using adrenal chromaffin cells of bovine origin. Clonidine did not alter basal or forskolin-stimulated cAMP production, nor did it alter the hydrolysis of basal, guanosine 5′-3-O-(thio)triphosphate–stimulated, or carbachol-stimulated phosphoinositide hydrolysis. In contrast, clonidine increased cGMP production and calcium uptake. Regunathan et al. (1991a) concluded from this seminal study that I1 receptors did not appear to be coupled to a second messenger system involving classic G proteins. Other studies quantified the densities of different G proteins in platelets of patients with depression to analyze possible associations with the expression of imidazoline receptors (García-Sevilla et al., 1996). Positive correlations were found between the immunoreactivity of I1 receptors and either Gαq/11, Gαi2, or Gβ proteins. The correlation with Gαq/11 protein suggested that I1 receptors could be coupled to a phosphoinositide pathway at least in platelets (García-Sevilla et al., 1996). Thus, a transduction mechanism in the platelets of patients with depression (i.e., the phosphoinositide pathway) that does not exist in chromaffin cells has been described. The question of a possible coupling mechanism of I1 receptors with G proteins was reanalyzed by Takada et al. (1997). These authors were interested in a possible involvement of G proteins sensitive to pertussis toxin, and they analyzed the effects of the toxin on the antiarrhythmic actions of various agonists and antagonists for either α2-adrenergic receptors or selective for the I1 receptors in a rat model of arrhythmias induced by the halothane-adrenaline mixture. They observed that the arrhythmogenic effect of adrenaline was prevented by rilmenidine and that this action was completely blocked by pretreatment with pertussis toxin. The same applied for dexmedetomidine, a selective α2-adrenergic agonist, suggesting that both types of receptors could be functionally coupled to G proteins sensitive to pertussis toxin (Takada et al., 1997). Greney et al. (2002) also took up this question and showed that a selective I1 receptor agonist, benazoline, reduced cAMP levels stimulated by forskolin in rat pheochromocytoma (PC12) cells, but this effect was insensitive to pertussis toxin. It therefore seems that at least for these G proteins sensitive to pertussis toxin, the situation may be variable depending on the tissues or cells studied. In addition, two teams focused their attention on the possible effects of selective I1 receptor agonists on phosphatidylcholine-selective phospholipase C activity in PC12 cells, which do not express α2-adrenergic receptors, and in cells from the rostro-ventrolateral region of the medulla oblongata. Thus, Separovic et al. (1997) showed that moxonidine caused an accumulation of diacylglycerides and released tritiated phosphocholine in cells previously labeled with tritiated clonidine. These effects were blocked by a PC-PLC inhibitor. These results demonstrated the involvement of PC-PLC in the effects resulting from the stimulation of I1 receptors (Separovic et al., 1997). This hypothesis was echoed by Zhang et al. (2001) who observed that activation of phosphatidylphospholipase C by I1 receptors in the PC12 cell line could result in secondary phosphorylation of a mitogen-activated protein kinase (MAPK). Similar results were later described with a protein considered as a candidate for the I1 receptor (i.e., IRAS protein) (Zhang and Abdel-Rahman, 2005). The fact that this transduction mechanism involves PC-PLC and extracellular signal-regulated kinase (ERK) provides support for the similarity between this IRAS and the I1 receptor and immunohistochemistry confirms that the major site of hypotensive action of rilmenidine was in the medulla oblongata. It should be noted that in these experiments, the I1-selective antagonist efaroxan antagonized the effect induced by rilmenidine. This study showed that, as in PC12 cells, ERK1/2 MAPK (p42/44) appears to be involved in effects mediated by the activation of I1 receptors in brain tissue (Zhang and Abdel-Rahman, 2005). Edwards et al. (2001) used moxonidine as an I1 receptor agonist in PC12 cells and showed that ERK and c-Jun N-terminal kinase were activated by the treatment and could therefore play a role in the signaling pathways coupled to I1 receptors. Edwards et al. (2001) suggested that these receptors may also play a role in cell growth.
The same team also showed that the activation of I1 receptors can abolish the nerve growth factor–activated signaling pathway by increasing the levels of a specific phosphatase through ERK dephosphorylation (Edwards and Ernsberger, 2003). Yamanaka et al. (2010) investigated the possible involvement of the phosphatidylinositol 3-kinase/Akt signaling pathway in the antiarrhythmic effects of the centrally administered I1 receptor agonist, rilmenidine. The results of this study showed that the pertussis toxin–sensitive G protein, phosphatidylinositol 3-kinase/Akt GSK3β, is coupled with I1 receptors (Yamanaka et al., 2010). Weiss et al. (2015) showed that a ligand selective for I1 receptors, LNP509, was able to increase the phosphorylation of 5′ adenosine monophosphate-activated protein kinase (AMPK) in hepatic cells. AMPK is involved in cellular energy homeostasis. In case of low cellular energy, AMPK increases glucose and fatty acid uptake and oxidation. This activation of the AMPK pathway could explain, at least partially, the favorable effect of I1 receptor activation on insulin sensitivity (Weiss et al., 2015). In a study of the effects of I1 receptor activation by moxonidine on the development of hepatic fibrosis, Zhang et al. (2017) showed that I1 receptor activation negatively regulates the course of hepatic fibrosis in an Nrf2-dependent pathway. Indeed, both in vivo and in vitro, moxonidine activated Nrf2 signaling, whereas knockout or knockdown of Nrf2 enhanced the antifibrotic and anti-inflammatory effects of moxonidine (Zhang et al., 2017).
Based on a fairly wide range of experimental models involving various species, tissues, and cell lines, it appears that I1 receptors may couple to several transduction mechanisms involving G proteins, the cAMP pathway, phospholipase C–selective phosphatidylcholine, MAPKs (ERK1/2 and c-Jun N-terminal kinase), mitogen-activated protein kinase phosphatase 2 phosphatase, and even nitric oxide. I1 receptors have been described in various peripheral tissues, including the liver, kidney, and adipose tissue. Now, there is evidence in favor of intracellular functions associated with these receptors, and I1 receptor–selective ligands, at least those that are sufficiently lipophilic to cross the plasma membrane, can target these cytoplasmic receptors. Thus, there is presently experimental evidence that, in addition to transmembrane I1 receptors, intracellular I1 receptors exist in various peripheral tissues and may be associated with metabolic functions.
8. Attempts to Clone Imidazoline Subtype 1 Receptors
Polyclonal antibodies have been developed in rabbits that specifically label a 70-kDa protein that binds I1 receptor–selective ligands. This protein, capable of binding labeled idazoxan, was purified from solubilized bovine chromaffin cell membranes by affinity chromatography (Escribá et al., 1994). With these antibodies, a cDNA clone isolated from a human hippocampus cDNA library encoded transcripts of 6 and 9.5 kb. The 6-kb mRNA was enriched particularly in the brain and endocrine glands compared with other tissues (Piletz et al., 2000), and in situ hybridization showed enrichment of this mRNA in neurons of rat hippocampus and also in the cerebellar cortex. The protein encoded by this cDNA has been proposed as the I1 receptors (Piletz et al., 2000). Piletz et al. (2000) also showed that the IRAS-selected cDNA-1 (IRAS-1) encodes a 267-kDa protein that was immunologically consistent with the I1 receptor protein, and significant correlation was observed between total IRAS mRNA and Bmax values (I1 receptor density in different rat tissues). However, the amino acid sequence of IRAS is different from that of the transmembrane I1 receptors. Mouse IRAS was identified with nisharin (Alahari et al., 2000; Sun et al., 2007), which is an intracellular protein that plays a role in regulating cell structure. The same group reported that nisharin is involved in the brainstem control of blood pressure (Maziveyi and Alahari, 2015). The fact that this protein has an intracellular localization is quite consistent with the described actions of imidazoline-like compounds on transduction pathways involving AMPK or Nrf2 (see above). Zhang and Abdel-Rahman (2008) also showed that inhibition of nisharin expression decreases the hypotensive effect of rilmenidine.
C. Cellular Effects
Here only the main effects that were not addressed in section IV.B.7 and that may contribute to effects observed in vivo or may be of interest in the future will be mentioned.
1. Imidazoline Subtype 1 Receptors and Apoptosis, Cell Viability, Growth, and Proliferation
The effects of a selective I1 receptor agonist on apoptosis in PC12 cells appears to be complex and depends on the experimental model of apoptosis. Thus, benazoline had a facilitating effect on apoptosis in the serum deprivation model, whereas it had a protective action in the model of induction of apoptosis by tumor necrosis factor α (TNF-α). In both cases, the use of a selective I1 receptor antagonist confirmed that these effects involved I1 receptors (Dupuy et al., 2004). Molderings et al. (2007) demonstrated in PC12 cells that moxonidine and also agmatine have an antiproliferative effect linked to their action on I1 receptors, but they also involve receptors of the S1P family. McLean et al. (2014) confirmed that another selective I1 receptor agonist, S43126 (2-[40-methoxyphenyl]-4,5-dihydro-1H-imidazole), has an antiproliferative effect on PC12 cells. Using cardiomyocytes from rat neonates, the selective I1 receptor agonist moxonidine protects against cell death induced by starvation or by interleukin-1b (Aceros et al., 2014). In summary, it appears that the activation of I1 receptors most often has a protective effect on cell viability.
2. Imidazoline Subtype 1 Receptors and Insulin and Adiponectin
Using insulin-secreting Min6 β-cells Tesfai et al. (2012) showed that I1 receptor activation with a selective I1 receptor ligand, S43126, had a glucose-dependent insulinotropic action; this effect was prevented by efaroxan, a selective I1 receptor antagonist. Weiss et al. (2015) showed that activation of I1 receptors on tissues targeted by insulin (i.e., liver and adipose tissues) also led to an improvement in insulin sensitivity. LNP599, a compound highly selective for I1 receptors, stimulates the secretion of adiponectin by 3T3-L1 adipocyte cultures (Fellmann et al., 2013a; Weiss et al., 2015). In addition to the work just mentioned, Yang et al. (2012) observed on these same cells that a selective I1 receptor agonist can increase the expression of farnesoid X nuclear receptors and thus improve hepatic steatosis. This effect appears to be due to an increase in intracellular calcium and an increase in p38 phosphorylation. An improvement in hepatic steatosis was also observed in vivo in mice treated with rilmenidine (Yang et al., 2012).
I1 receptor activation negatively regulates the progression of fibrosis through a Nrf2-dependent pathway in hepatic stellate cells. This hepatic I1-dependent antifibrotic action was also observed in vivo in mice (Zhang et al., 2017). In human platelets, when α2-adrenergic receptors are blocked by yohimbine, the addition of dexmedetomidine (a α2-adrenergic agonist/I1 receptor agonist) was actually found to suppress ADP-induced platelet aggregation, and this effect was blocked by efaroxan, an I1 receptor–selective antagonist. The specific activation of I1 receptors thus leads to an interesting platelet antiaggregation effect (Kawamoto et al., 2015).
3. Imidazoline Subtype 1 Receptors and Neurons
In rats, immunohistochemistry showed an increase in ERK1/2 (p42/44) MAPK in the ventrolateral part of the brainstem associated with the hypotensive effect of rilmenidine and this effect was abolished by efaroxan (Zhang and Abdel-Rahman, 2005). In a whole-cell patch-clamp study performed on rat dorsal striatum slices, moxonidine significantly decreased GABAA receptor–mediated inhibitory postsynaptic currents through the involvement of presynaptic I1 receptors (Tanabe et al., 2006). In organotypic cultures of mouse hippocampal slices, dexmedetomidine had a postconditioning effect dependent on I1 receptors (Dahmani et al., 2010).
Once again, the cellular effects on neurons implicating I1 receptors appear complex and depend much on the localization within the CNS.
D. Imidazoline Subtype 1 Receptors and In Vivo Effects
Here we will only discuss the effects that have been repeatedly documented in the scientific literature. Obviously, the cardiovascular and metabolic effects largely dominate the picture.
1. Effects on Blood Pressure and Heart Rate
Effects on blood pressure and heart rate were analyzed in detail in section I. It is accepted that all I1 receptor agonists that are sufficiently lipophilic to cross the blood–brain barrier reduce arterial pressure and heart rate in all mammalian species tested, including humans, effects that are mediated by a central sympathoinhibitory action. According to Mahmoudi et al. (2018), I1 receptor agonists may have interesting clinical applications in hypertensive patients with intracerebral hemorrhage by reducing post-intracerebral brain injury. This hypothesis is based on the fact that not only do these drugs allow the control of hypertension, but they also have a set of potential effects (e.g., anti-inflammatory, antiedematous, anti-inflammatory, and antiapoptotic effects) that can circumvent posthemorrhagic complications (Mahmoudi et al., 2018).
2. Antiarrhythmic Effects
Cardiac antiarrhythmic effects of I1 receptor agonists have been observed in several species, in several experimental models, and with various I1-selective drugs. Thus, Leprán and Papp (1994) observed a protective effect of moxonidine against arrhythmias induced by myocardial ischemia in rats; this beneficial effect extended even to the reperfusion period. Rilmenidine has a protective effect against bicuculline-induced cardiac arrhythmias in rabbits (Roegel et al., 1996, 1998). Rilmenidine and dexmedetomidine have protective effects against adrenaline-induced arrhythmias in halothane anesthetized dogs (Kamibayashi et al., 1995; Mammoto et al., 1995, 1996). In the same experimental model in rats, moxonidine and rilmenidine are also clearly protective (Kagawa et al., 2005; Yamanaka et al., 2010). Similarly, moxonidine is protective against ouabain-induced arrhythmias in guinea pigs (Mest et al., 1995). In a model of neurogenic arrhythmias induced by electrical stimulation of the posterior hypothalamus, moxonidine also exerts a significant protective effect in rabbits (Poisson et al., 2000). In summary, the antiarrhythmic properties associated with the activation of I1 receptors are amply documented.
3. Effects in Congestive Heart Failure
In a clinical study of 32 patients with class III congestive heart failure according to the New York Heart Association classification, moxonidine was administered in single doses of 0.4 or 0.6 mg per patient compared with placebo. Moxonidine caused a modest vasodilator effect accompanied by a significant reduction in systemic and pulmonary arterial pressures as well as a reduction in the plasma concentration of noradrenaline (Swedberg et al., 2000). Unfortunately, this effect of moxonidine was not confirmed in the MOXCON clinical trial, which involved chronic treatment of patients with fairly severe heart failure. In this trial, the sympathetic activity was probably inhibited too much in patients who, at this advanced stage, require sympathetic activity (Cohn et al., 2003; Pocock et al., 2004). In the hamster model of cardiomyopathy, moxonidine improved cardiac performance by both central and direct cardiac sympathoinhibitory actions (Stabile et al., 2011). The same team showed that moxonidine prevents left ventricular hypertrophy and cardiac remodeling in hypertensive rats and in hamsters with cardiomyopathy (Mukaddam-Daher, 2012). Therefore, despite interesting experimental observations, moxonidine did not display efficacy in patients with congestive heart failure. Two possible contributors to this finding involve too intense sympathoinhibition with respect to the disease severity in the patients tested and the insufficient selectivity of the test drug, moxonidine, for the I1 receptors.
4. Effects on Glucose and Lipid Metabolism and Interest in Metabolic Syndrome
The group of symptoms consisting of arterial hypertension and at least two of the following abnormalities (i.e., insulin resistance, hyperlipidemia, obesity, or at least overweight) form what is now called the metabolic syndrome (Fellmann et al., 2013a). This syndrome is known to be associated with high cardiovascular risk in patients (Mongraw-Chaffin et al., 2018).
In rats with fructose-induced insulin resistance, moxonidine completely prevented the development of insulin resistance, hyperinsulinemia, and hypertension (Ernsberger et al., 1999). Spontaneously hypertensive obese (SHROB) rats have a faK mutation of leptin receptors that naturally attenuates them. SHROB rats exhibit hypertension, glucose intolerance, and insulin resistance. In SHROB rats, chronic treatment with moxonidine reduced blood pressure and improved glucose intolerance. It also decreased fasting insulin and free fatty acids (Ernsberger et al., 1999). Koletsky et al. (2003) also achieved similar results with rilmenidine in SHROB rats, with a reduction in triglycerides and cholesterol (Velliquette et al., 2006; Niu et al., 2011). In another model of metabolic syndrome (high-fat diet–induced obesity), clonidine and rilmenidine had similar beneficial blood pressure and metabolic effects that were accompanied by a reversal of microvascular rarefaction in skeletal muscles and in the heart (Nascimento et al., 2016). It is interesting to note that under acute conditions, moxonidine and rilmenidine cause hyperglycemia, but less than that induced by clonidine in the same experimental conditions. The acute effects were observed in normal rats, fructose-fed rats, and SHROB rats (Rösen et al., 1997; Velliquette and Ernsberger, 2003). This acute effect is apparently related to a α2-adrenergic activity, whereas the beneficial action of the chronic treatments is the consequence of the activation of I1 receptors.
Since I1 receptor agonists not only lower blood pressure but improve glucose intolerance, insulin resistance, and hyperlipidemia, it made sense to think of the metabolic syndrome as a possible new application for such compounds. As can be seen above, at least in the case of glucose intolerance, it appears that the action on α2-adrenergic receptors of I1/α2-adrenoceptor mixed agonists is likely to limit their favorable effects. Making drugs highly selective for I1 receptors would be an interesting challenge to test the hypothesis according to which such selectivity would improve the therapeutic efficacy in metabolic syndrome but would also have a better tolerance profile. In a pharmacochemistry study, Gasparik et al. (2015) designed and synthesized a series of aminopyrrolines that were highly selective for I1 receptors to the extent that some of them lacked any detectable affinity and activity at α2-adrenergic receptors. A lead compound LNP599 was selected on the basis of these pharmacological properties but also for its favorable lipophilicity to allow passage through the blood–brain barrier (Gasparik et al., 2015). In an experimental model of metabolic syndrome in rats (i.e., rats with spontaneously hypertensive heart failure), this compound had favorable effects on blood pressure because of its sympathoinhibitory activity as well as favorable effects on insulin resistance, glucose tolerance, and lipid profile, and it also stabilized body weight. The metabolic effects of LNP599 were associated with the stimulation of adiponectin secretion (Fellmann et al., 2013a). A specific study showed that in addition to sympathetic inhibition and stimulation of adiponectin secretion by adipocytes, the I1-selective agent LNP599 improves insulin sensitivity in the liver (Weiss et al., 2015).
In conclusion of this section on the pharmacology of I1 receptors and their ligands, the recent development of very selective pharmacological agents suggests the possibility of developing new centrally acting drugs with fewer side effects compared with the historical reference drugs such as clonidine, moxonidine, and rilmenidine (see section II). A summary of the pharmacological effects mediated via I1 receptors is provided in Table 1. It is also pharmacologically legitimate to think about new indications of such new drugs: the metabolic syndrome already appears as an interesting target.
Summary of the pharmacological effects mediated by imidazoline receptors
V. Imidazoline Subtype 2 Receptors
A. Definition, Distribution, and Composition
As discussed above, I2 imidazoline receptors are defined as the nonadrenergic binding sites that bind [3H]-idazoxan with high affinity and bind [3H]-p-aminoclonidine and [3H]-clonidine at substantially lower affinity (Regunathan and Reis, 1996). Largely identified by ligand binding, I2 receptors are found in many organs, tissues, and cell types, including but not limited to the brain, kidney, liver, colon, placenta, urethra, prostate, adrenal medulla, carotid bodies, astrocytes, glial cells, platelets, pancreatic cells, and vascular smooth muscle cells (Regunathan and Reis, 1996).
Unlike I1 receptors, which are considered a homogenous entity, the I2 receptors are highly heterogenous. Largely through binding studies, it was initially proposed to differentiate I2 receptors into I2A and I2B subtypes based on their binding affinity to the drug amiloride (Diamant et al., 1992). This seemed consistent with drug competition curve studies, which typically showed biphasic competitor binding curves for a number of I2 ligands (Miralles et al., 1993). However, functional evidence supporting such a classification is lacking. Studies using a polyclonal antiserum against idazoxan-binding proteins found four different protein bands, which slightly differed between rat and rabbit brain tissues (rat brains: approximately 30, 45, 66, and 85 kDa; rabbit brains: approximately 30, 57, 66, and 85 kDa) (Olmos et al., 1999a). Repeated treatment with I2 receptor ligands such as BU224 and tracizoline, also known as LSL61122 (2-styryl-4,5-dihydro-lH-imidazole) and valldemossine, significantly reduced the 30-, 45-, and 66-kDa protein levels in the mouse brain, whereas treatment with idazoxan, the purported I2 receptor antagonist, increased these levels (Keller and García-Sevilla, 2015), demonstrating the biochemical correlates of I2 receptor ligands and these proteins. Using the prototypical I2 receptor ligand 2-BFI to generate an affinity column, other studies successfully isolated and sequenced one of the proteins, which was identified as the brain creatine kinase (Kimura et al., 2009). These investigators demonstrated that brain creatine kinase binds [3H]2-BFI, and this binding was attenuated by BU224 or the irreversible I2 receptor ligand BU99006. Thus, the identity of the approximately 45-kDa protein band as reported earlier (Olmos et al., 1999a) has likely been solved. However, the identities of other protein bands remain a mystery. Furthermore, the functional correlates between various reported pharmacological effects of I2 receptor ligands and brain creatine kinase remains elusive.
At the subcellular level, I2 receptors are primarily located at the outer membrane of mitochondria and are thought to be novel allosteric binding sites of MAOA and MAOB (Tesson et al., 1991, 1995; Parini et al., 1996). However, MAO likely only accounts for a portion of the total I2 receptor binding, as substantial binding remains in MAO knockout mice (Remaury et al., 2000; Anderson et al., 2006b). Consistent with this observation, brain creatine kinase is apparently unrelated to MAO (Kimura et al., 2009).
In summary, the current consensus is that the I2 receptors represent a group of heterogenous proteins that I2 receptor ligands such as idazoxan and 2-BFI recognize; some of these binding sites are related to MAO and others are not. Therefore, it is important to note that referring to “I2 receptors” in the literature does not refer to a specified molecular identity. Rather, it refers to several different and potentially biologically diverse protein molecules. This clarification proves to be useful in interpreting many recent functional results.
B. Synthetic Imidazoline Subtype 2 Receptor Ligands
Because idazoxan was initially used to characterize I2 receptors but it also binds to adrenoceptors, medicinal chemistry efforts have been actively developing selective I2 receptor ligands with low I1 and α2-adrenogic receptor binding activities. The chemical structures of some commonly used I2 receptor ligands are given in Fig. 4. A large library of selective I2 receptor ligands have been developed over the years and the effort continues. For example, a recent study describes a new family of (2-imidazolin-4-yl) phosphonates that have high affinity for I2 receptors (Abás et al., 2017). Among those compounds, several such as 2-BFI, BU224, phenyzoline, and CR4056 have been commonly used in previous studies to explore I2 receptor pharmacology and have become valuable research tools. Among these synthetic ligands, two are worthy of discussion because both have moved from preclinical research to human studies.
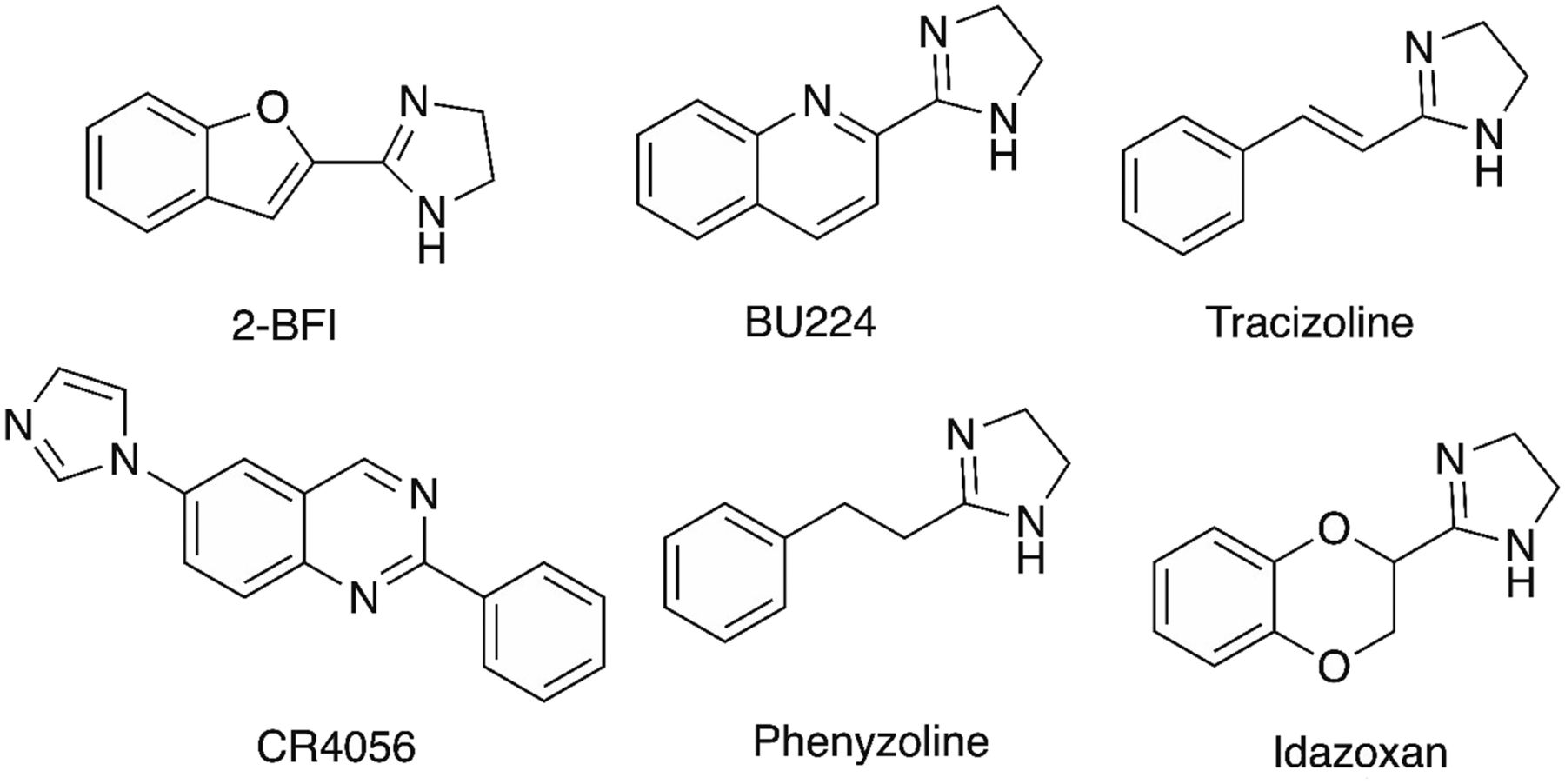
Chemical structures of I2 receptor ligands 2-BFI, BU224, tracizoline, CR4056, phenyzoline, and idazoxan.
11C-BU99008 is a new radiolabeled I2 receptor ligand that has been used in positron emission tomography (PET) studies with human volunteers to demonstrate the distribution of these receptors in the brain (Tyacke et al., 2018). In the future, this PET ligand may make great inroads in expanding our understanding of I2 receptors in neurodegenerative disorders. In fact, 11C-BU99008 was developed with the knowledge that the I2 receptor density changes in several psychiatric conditions particularly where there is marked gliosis. Therefore, this PET ligand could not only be used to confirm and monitor neurodegenerative disease, but it would also detect glioblastomas. The authors of this study had previously established that 11C-BU99008 has low affinity for MAOB and high affinity and selectivity for I2 receptors (Parker et al., 2014). Nonetheless, in their current study, volunteers were cotreated with the irreversible MAO inhibitor isocarboxazid, which showed that the signal in the human brain from 11C-BU99008 was unaffected by MAO inhibition. In contrast, pretreatment of volunteers with an oral dose of idazoxan (20 and 80 mg) showed marked attenuation of the 11C-BU99008 signal across all brain regions (Fig. 5). Although this initial study represents PET scans from a small number of volunteers, the clarity of the 11C-BU99008 signal in the human brain is remarkable, with structures such as the striatum, thalamus, amygdala, and other key areas clearly visible (Fig. 5). To quote the authors’ conclusions, “This new clinical imaging tool has paved the way for more in-depth clinical investigations into the role of I2 binding sites in disease states and in the development of potential therapies” (Tyacke et al., 2018, p. 1602).

PET imaging of I2 receptors in healthy volunteers treated with the radioligand 11C-BU99008. Data adapted from Tyacke et al. (2018) with permission.
Another significant advancement in the field is the development of CR4056 (Fig. 4) which has shown positive results in one phase II clinical trial for the treatment of chronic pain related to knee osteoarthritis (Rovati et al., 2017) (see below for further details).
C. Neuropharmacology of Imidazoline Subtype 2 Receptor Ligands
In recent years, an increasing number of studies have used selective I2 receptor ligands in various functional assays and have identified several increasingly promising therapeutic uses of I2 receptor agonists. In addition, the study of these compounds also provides novel insights into understanding the nature of I2 receptors. Interestingly, although I2 receptors have a wide distribution, most studies related to I2 receptor functions focus on the CNS. This probably is not surprising given the fact that earlier correlational studies using human tissue consistently report that the expression of I2 receptors was significantly altered in several neuropsychiatric diseases (García-Sevilla et al., 1999). This section will review four well characterized in vivo effects of I2 receptor ligands: analgesic, discriminative stimulus, neuroprotective, and hypothermic effects. These pharmacological effects are also summarized in Table 1.
1. Pain
One of the best studied pharmacological effects of I2 receptor ligands is their potential analgesic effects, which have been demonstrated with various I2 receptor ligands across multiple preclinical pain models.
The overall findings for I2 receptor agonists support the notion that these compounds are not very effective for acute nociception in thermal stimulus-induced pain models. Earlier studies found that I2 receptor ligands, including 2-BFI, tracizoline, phenyzoline, and LSL60101, do not produce significant antinociception in rodent models of acute nociception such as radiant tail flick and hot plate assays (Boronat et al., 1998; Sánchez-Blázquez et al., 2000; Gentili et al., 2006). More recent studies from Li’s group are consistent with these findings. 2-BFI and phenyzoline only produced mild antinociception, whereas other selective I2 receptor ligands such as BU224 and S22687 (5-[2-methyl phenoxy methyl]-1,3-oxazolin-2-yl) amine) had no effect in a warm water tail flick assay (Thorn et al., 2011; Sampson et al., 2012).
I2 receptor agonists are generally efficacious for chemical stimulus-induced pain. For example, in one study, 2-BFI, BU224, and morphine were found equally effective in decreasing the writhing response in a writhing test (Li et al., 2011). In another study employing intraplantar injection of capsaicin-induced mechanical allodynia, CR4056, which as mentioned above is a structurally novel I2 receptor agonist, completely reversed capsaicin-induced neurogenic/inflammatory allodynia, and the effect was dose-dependently prevented by idazoxan (Ferrari et al., 2011). In the formalin test (a widely used chemical stimulation-induced persistent pain model), the rodents demonstrate nocifensive behaviors that typically include two phases: an early phase I that is primarily neurogenic and a later phase II that is primarily inflammatory (Hunskaar and Hole, 1987). 2-BFI, BU224, and CR4056 all dose-dependently reduced the flinching response during phase II (Thorn et al., 2016b).
Overall, several studies have consistently shown that I2 receptor agonists are effective in various animal models of chronic pain. Thus, the selective I2 receptor agonists 2-BFI, BU224, and tracizoline all significantly reduced the mechanical allodynia and thermal hyperalgesia in a complete Freund’s adjuvant (CFA)–induced inflammatory pain model (Li et al., 2014). 2-BFI and phenyzoline also both attenuated mechanical allodynia in a rat peripheral neuropathic pain model [chronic constriction injury (CCI)] (Li et al., 2014; Thorn et al., 2017). The antiallodynic effects of 2-BFI and phenyzoline were attenuated by idazoxan, confirming an I2 receptor–mediated mechanism (Li et al., 2014; Thorn et al., 2015; Siemian et al., 2016a). In a series of studies, CR4056 demonstrated a robust antihyperalgesic effect in several different rat models of chronic pain. CR4056 reduced mechanical allodynia in CFA-treated rats (Ferrari et al., 2011), attenuated mechanical hyperalgesia in diabetes-induced neuropathic pain (Ferrari et al., 2011), and reduced neuropathic pain induced by chronic treatment with the chemotherapeutic agent bortezomib (Meregalli et al., 2012). Interestingly, CR4056 was also effective in a rat model of fibromyalgia (Ferrari et al., 2011). When acidic saline (pH 4, 150 μl) is injected into the right gastrocnemius muscle, rats demonstrate a persistent mechanical allodynia condition, which is thought to mimic human chronic pain syndromes such as fibromyalgia (Nielsen et al., 2004). CR4056 significantly reversed acidic saline-induced mechanical allodynia (Ferrari et al., 2011). In another study, CR4056 was fully effective in reversing mechanical allodynia in a postoperative pain model, and this effect was blocked by idazoxan (Lanza et al., 2014). In the same study, no sex difference was found for CR4056-induced antinociception (Lanza et al., 2014). Together, these studies convincingly show that I2 receptor agonists are able to attenuate various chronic painful conditions and may represent a novel class of analgesics with a broad spectrum of analgesic activity. These findings are significant because many chronic pain conditions respond poorly to existing pharmacotherapies; as such, I2 receptor agonists could provide urgently needed treatments for certain complex chronic pain conditions. Indeed, a phase II multisite, randomized placebo-controlled clinical trial on CR4056 in a group of 213 patients with knee osteoarthritis was recently completed (Rovati et al., 2017). In this study, CR4056 at oral doses of 100 or 200 mg twice daily produced significant analgesic activity in a short 2-week study. Not only were the doses well tolerated and no serious adverse events were noted, but interestingly the analgesic effect was more prominent in patients with obesity (Rovati et al., 2017). This is very exciting progress, and larger long-term trials are planned based on these results. In addition, the potential clinical indications have been expanded to other painful conditions including an ongoing phase II clinical trial studying the analgesic effect of CR4056 in patients with postoperative dental pain. If CR4056 is eventually approved, this would be an analgesic with a completely novel mechanism of action and the first-in-class drug to treat chronic pain based on I2 receptor pharmacology.
The existing evidence supports that I2 receptor agonists are most effective for the management of chronic pain and persistent pain. Because chronic pain is long-lasting and pharmacotherapies of chronic pain require repeated drug use, the possibility of the development of analgesic tolerance has to be carefully considered. The available data suggest that such a possibility is quite low for I2 receptor agonists. For example, daily treatment with 2-BFI or CR4056 at the dose that produced the maximal antihyperalgesic effect for at least 1 week failed to produce observable tolerance in CFA-treated rats (Li et al., 2014). In another study using the chemotherapeutic agent-induced neuropathic pain model, daily treatment with a CR4056 dose (6 mg/kg) that completely prevented mechanical allodynia for 3 weeks did not produce observable antinociceptive tolerance (Meregalli et al., 2012). Using a much more aggressive treatment regimen (twice-daily treatment for 19 days), phenyzoline at the dose that produced near maximal antinociceptive effect only produced a slight antinociceptive tolerance in CFA-treated and CCI rats (Thorn et al., 2017). In contrast, oxycodone produced dramatic antinociceptive tolerance under similar treatment conditions (Thorn et al., 2017). In summary, this accumulating evidence suggests that the possibility for developing tolerance to the antinociceptive effects for I2 receptor agonists is relatively low as long as the doses used are close to therapeutically relevant doses.
Given the complexity and diversity of painful conditions, it is unrealistic to expect one analgesic drug to treat all pain. Combination therapy could offer a more effective approach for pain control. This strategy may be able to achieve better analgesia and produce fewer adverse effects due to the smaller doses needed. Several studies have examined the antinociceptive interactions between I2 receptor ligands and other analgesic agents. One study reported that 2-BFI shifted the opioid antinociceptive dose-effect curves leftward and markedly enhanced the antinociceptive effects of morphine and tramadol (Thorn et al., 2011). Although I2 receptor agonists are not effective for acute pain, combining them with opioids to treat acute pain could substantially reduce the opioid dose needed, which is obviously clinically beneficial. Similar results were found in a writhing test in which the combination of morphine and 2-BFI or BU224 produced synergistic antinociception (Li et al., 2011). The interaction between 2-BFI and opioids is partially determined by the efficacy of the opioids at the µ-opioid receptors. For example, although both the high-efficacy opioid fentanyl and the moderate-efficacy opioids morphine and oxycodone produced additive antinociceptive interactions with 2-BFI in CFA-treated rats (Li et al., 2014; Thorn et al., 2015), lower-efficacy opioids such as buprenorphine and NAQ (17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-[(3′-isoquinolyl) acetamido] morphine) produced synergistic interactions with 2-BFI in the same pain model (Siemian et al., 2016b). The interaction also seems to depend on which I2 receptor agonist is used. For example, phenyzoline produced synergistic antinociceptive interactions with oxycodone in CFA-treated rats (Thorn et al., 2015), whereas CR4056 produced synergistic interactions with morphine in models of capsaicin-induced neurogenic pain (Ferrari et al., 2011) and postoperative pain (Lanza et al., 2014). Together, these studies strongly suggest that I2 receptor agonists and opioids produce overall preferable antinociceptive interactions under several different painful conditions and may be a viable combination therapy strategy to treat pain. Several factors such as the drugs used (both I2 receptor ligands and opioids) and the pain conditions are important determinants of drug–drug interactions.
Besides the favorable antinociceptive interactions, increasing evidence also suggests that I2 receptor agonists can actually reduce some adverse effects related to prolonged opioid use. Chronic opioid use often leads to significant adverse effects such as constipation, dependence, and increased propensity to addiction. Two studies examined the concern of opioid-induced physical dependence. In one study, BU224 reduced some naltrexone-precipitated observable withdrawal signs in morphine-dependent rats (Hudson et al., 1999a). In a more systematic study, naltrexone treatment disrupted food-maintained operant responding in rats treated chronically with morphine (Thorn et al., 2016b). 2-BFI treatment reduced the development of tolerance to morphine and attenuated naltrexone-precipitated body weight loss (Thorn et al., 2016b). I2 receptor agonists (2-BFI, BU224, and CR4056) also significantly reduced the development of antinociceptive tolerance to repeated morphine treatment (Thorn et al., 2016b). Combined, these results suggest that I2 receptor agonists may decrease some adverse effects related to opioid use.
2. Discriminative Stimulus Effects
Drug discrimination is a powerful behavioral pharmacological procedure to study the in vivo mechanism of action of a novel compound. Over half a century’s research has convincingly demonstrated that most psychoactive drugs can be distinguished by various species (from mice to humans) and be classified based on their discriminative stimulus effects, and such classifications correlate highly with a specific cellular mechanism of action (Schuster and Johanson, 1988). For example, if a group of rats are trained to recognize a drug with a known mechanism of action as a discriminative stimulus, drugs with the same or similar (overlapping) mechanisms of action typically are able to elicit responding on the same operandum as the training drug (i.e., the test drug “substitutes” for the training drug), consistent with their shared pharmacological mechanism(s) of action.
Several groups have successfully used drug discrimination procedures to study the discriminative stimulus effects of various imidazoline I2 receptor agonists and characterized their pharmacological specificity (Jordan et al., 1996; MacInnes and Handley, 2002; Qiu et al., 2014a,b, 2015). In an early study, rats were trained to discriminate the prototypical I2 receptor agonist 2-BFI (6 mg/kg, i.p.) from saline (Jordan et al., 1996). Rats readily learned the discrimination and substitution studies showed that idazoxan and the α2-adrenoceptor antagonist ethoxy idazoxan fully substituted for 2-BFI. The MAO inhibitors moclobemide and pargyline also fully substituted for 2-BFI, suggesting that MAO inhibition plays an essential role in mediating the discriminative stimulus effects of 2-BFI (Jordan et al., 1996). In a follow-up study by the same group, several known I2 receptor ligands were tested: BU224 and BU226 fully substituted, whereas BU216, LSL60101, and LSL60125 (2-[6-methoxybenzofuran-2-yl] imidazole hydrochloride) partially substituted for 2-BFI (MacInnes and Handley, 2002). The reversible MAOA inhibitors, moclobemide and RO41-1049, and the naturally occurring MAOA inhibitor β-carbolines (harmane, norharmane, and harmaline) all exhibited significant and dose-dependent substitution for 2-BFI (MacInnes and Handley, 2002). It is interesting that the reversible MAO-B inhibitors lazabemide and RO16-1649 were not able to produce 2-BFI–like discriminative stimulus effects, suggesting that the activity of MAOA but not MAO-B is important in mediating the discriminative stimulus effects of 2-BFI (MacInnes and Handley, 2002). MAOA is a limiting factor in monoamine synthesis and is critical in monoaminergic biology. Because I2 receptor agonists have been shown to increase extracellular monoamine levels in the brain (Hudson et al., 1999a), it is therefore reasonable to assume that the modulation of monoaminergic activity may be involved in the discriminative stimulus effects of I2 receptor agonists such as 2-BFI (but see section V.D). Indeed, the monoamine-releasing drugs amphetamine and fenfluramine both dose-dependently substituted for 2-BFI, whereas reuptake inhibitors for norepinephrine (desipramine, reboxetine) and serotonin (clomipramine, citalopram) all only partially substituted for 2-BFI (MacInnes and Handley, 2003). These results suggest that the noradrenergic and serotonergic mechanisms are important in 2-BFI discrimination.
In an attempt to expand the existing knowledge, we trained rats to discriminate several other I2 receptor agonists (Qiu et al., 2014a,b, 2015). Rats can readily learn to discriminate 5.6 mg/kg BU224 (i.p.) from saline. Other I2 receptor ligands, including phenyzoline, tracizoline, CR4056, RS45041 [4-chloro-2-(imidazolin-2-yl)isoindoline hydrochloride], and idazoxan, as well as the MAOA inhibitor harmane, all showed full substitution for BU224 (Qiu et al., 2015). Using a newer I2 receptor agonist phenyzoline (32 mg/kg) as the discriminative cue, RS45041, CR4056, phenyzoline, tracizoline, and harmane all showed full substitution (Qiu et al., 2015). Finally, in rats discriminating CR4056 (10 mg/kg, i.p.) from its vehicle, tracizoline, phenyzoline, RS45041, and idazoxan all fully substituted for CR4056, whereas 2-BFI partially substituted and BU224 failed to substitute for CR4056 (Qiu et al., 2014a). Interestingly, harmane failed to produce a significant CR4056-like discriminative stimulus effect under this situation (Qiu et al., 2014a). In summary, these studies strongly suggest that most of the selective I2 receptor ligands studied thus far share similar pharmacological mechanisms of action that presumably are an essential component of I2 receptors, which mediate the characteristic interoceptive cue of the I2 receptor agonists.
Among these findings, substitution tests with idazoxan emerge as a surprising outlier. In nearly all pain studies, idazoxan reliably antagonizes the effects of many I2 receptor agonists (Ferrari et al., 2011; Li et al., 2011, 2014; Lanza et al., 2014) and it has long been considered the only known I2 receptor antagonist. However, in all of the drug discrimination studies, idazoxan fully substituted for the training I2 receptor agonists. This is surprising because it essentially shows that idazoxan acts as an agonist in one assay (drug discrimination) but as an antagonist in a different assay (antinociception) when interacting with the same I2 receptor ligands (e.g., 2-BFI, CR4056). The only reasonable explanation for these results seems to be that there exists more than one “I2 receptor” and that idazoxan has different pharmacological properties at each of them. This is reasonable, given that I2 receptors have long been known to actually include several proteins (Olmos et al., 1999a). Thus, idazoxan could act like an agonist at one of these I2 receptors (e.g., to produce discriminative stimulus effects) and like an antagonist at another (different) I2 receptor (e.g., for antinociception). This notion is sufficient to explain the apparently discordant in vivo results as discussed previously (Qiu et al., 2014a, 2015; Thorn et al., 2016a) and is further supported by two recent findings. First, 2-BFI and BU224 led to seizures in mice at higher doses, an effect that was not attenuated by idazoxan (Min et al., 2013). Second, idazoxan did not block the 2-BFI– and CR4056–induced antinociceptive effect in phase II of the formalin test (Thorn et al., 2016b). Thus, it is logical to propose at least two functionally different I2 receptors: one is idazoxan sensitive and another is idazoxan insensitive (Thorn et al., 2016b).
3. Neuroprotection
It was known over 2 decades ago that idazoxan could against protect neuronal damage in a rat model of transient global forebrain ischemia (Gustafson et al., 1989, 1990). This neuroprotection may be partially explained by a small hypothermic effect induced by idazoxan (Craven and Conway, 1997). However, a later study failed to support these findings. In a rat focal cerebral hypoxia-ischemia model, idazoxan was found not only to be ineffective but to worsen the brain damage induced by hypoxia (Antier et al., 1999). This discrepancy is difficult to reconcile due to the descriptive nature of the studies.
Recent studies showed more consistent results using a selective I2 receptor ligand 2-BFI. 2-BFI was found to exert significant neuroprotection in a rat model of cerebral ischemia (middle cerebral artery occlusion model) (Han et al., 2009, 2010, 2012). Several cellular and molecular mechanisms appear to be involved in 2-BFI–induced neuroprotection. For example, NMDA receptor–mediated currents can be selectively blocked by the endogenous imidazoline receptor ligand agmatine in rat hippocampal neurons; agmatine did not block AMPA or kainate-induced currents (Yang and Reis, 1999). Moreover, imidazoline receptor drugs and the candidate IRAS/nischarin (Piletz et al., 1999; Sun et al., 2007) have been associated with antiapoptotic and/or cytoprotective functions, because they were shown to reduce the levels of proapoptotic proteins and to protect cells from cell death induced by noxious stimuli (Choi et al., 2002; Dontenwill et al., 2003; Garau et al., 2013). The NMDA receptor is a glutamate receptor subtype with many available competitive and noncompetitive antagonists. NMDA receptor activation by endogenous glutamate or analogs such as NMDA opens the receptor cation channel, allowing Ca2+ influx and an increase in the intracellular calcium concentration (Ca2+i). Prolonged receptor activation by increasing Ca2+i may activate potentially damaging Ca2+-dependent enzymes. Noncompetitive NMDA receptor antagonists such as MK-801 (dizocilpine), ketamine, and phencyclidine block Ca2+ entry by binding to a site within or at the mouth of the cation channel (MacDonald and Nowak, 1990). 2-BFI and idazoxan both produced transient and reversible inhibition of intracellular calcium influx through NMDA receptors (Jiang et al., 2010). Whereas 2-BFI inhibits NMDA-stimulated currents, it did not affect AMPA-stimulated currents (Han et al., 2013). Interaction of I2 receptor ligands with NMDA receptors was previously reported (Olmos et al., 1996, 1999b), and these results support modulation of NMDA receptor activity by I2 receptor ligands. These mechanisms were used to explain the neuroprotective effects of 2-BFI and idazoxan in a glutamate toxicity test using cortical neurons in vitro (Jiang et al., 2010; Han et al., 2013). In the cerebral ischemia model, 2-BFI reduced terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling–positive cells, preserved the integrity of subcellular structures, and significantly increased the Bcl-2 expression level, further confirming neuroprotection at the cellular and molecular levels (Han et al., 2010). To further extend the molecular observations from 2-BFI to other I2 receptor ligands, a recent study examined the effects of acute and chronic treatment with four different I2 receptor ligands (2-BFI, tracizoline, BU224, and LSL60101) on a battery of molecules within the canonical apoptotic pathways (Garau et al., 2013).
In many in vitro and in vivo models, NMDA receptor blockade may provide a molecular basis for the neuroprotective actions of imidazoline receptor ligands. However, imidazoline receptor ligands also interact (in the micromolar range) with the MAO enzymes (e.g., see Parini et al., 1996; Ozaita et al., 1997; Ferrari et al., 2011) and various cation channels [reviewed in Olmos et al. (1999a)]. These compounds also block ATP-sensitive K+ channels in pancreatic β-cells and rat insulinoma cells, which leads to the stimulation of insulin release (Jonas et al., 1992; Olmos et al., 1994; Berdeu et al., 1997; Proks and Ashcroft, 1997). Imidazoline receptor ligands can also inhibit the acetylcholine-induced secretion of catecholamines in adrenal chromaffin cells by blocking nicotinic acetylcholine receptors (Musgrave et al., 1995). I2 receptor ligands also interact with the 5-HT3 receptor channel in N1E-115 cells, inhibiting the veratridine-induced influx of guanidinium into these cells (Molderings et al., 1996). Moreover, in vitro interactions with red cell Gardos channels (Coupry et al., 1996) and rat brain NMDA receptors (Olmos et al., 1996) have also been reported for these compounds [see Olmos et al. (1999b)].
Although effects of I2 receptor ligands on the variety of potential mechanisms as discussed above may be useful to interpret the neuroprotective effects of certain I2 receptor ligands, 2-BFI was the only selective I2 receptor ligand that was ever tested in in vivo functional studies that involve a well characterized animal model and generalization to other I2 receptor ligands has not been attempted. This is problematic because marked inconsistencies exist in the effects of the I2 receptor ligands on biochemical events, and it is essentially impossible to link any of those changes to a specific I2 receptor component. In addition, the effects are also only correlational, and it is unknown whether the molecules are actually on the I2 receptor signaling pathway.
4. Body Temperature
Imidazoline I2 receptor ligands can reduce body temperature, and this effect is mediated by I2 receptors as shown by pharmacological antagonism studies (Thorn et al., 2012). Indeed, the hypothermic effect was significantly reversed by the prototypical I2 receptor antagonist idazoxan, but not by the I1 receptor antagonist efaroxan or the α2-adrenoceptor antagonist yohimbine (Thorn et al., 2012). In addition, this is a general effect that is shared by all I2 receptor ligands that have been studied in rats and is highly dose and time dependent. This simple and straightforward in vivo assay (hypothermia) is increasingly used as a preliminary study of newly synthesized I2 receptor ligands and is particularly useful when combined with specific receptor antagonists (Abás et al., 2017). As discussed above, because body temperature reduction can produce significant neuroprotection under certain conditions such as in cerebral ischemia (Craven and Conway, 1997), the observed I2 receptor agonist-induced neuroprotection could be partially attributable to drug-induced hypothermia (Craven and Conway, 1997).
D. Pharmacological and Neurobiological Mechanisms
Although the potential mechanisms discussed above on the neuroprotective actions of I2 receptor agonists are intriguing and helpful, evidence increasingly suggests that these mechanisms are unlikely to account for the in vivo pharmacological effects of I2 receptor agonists in pain and drug discrimination. For example, unlike 2-BFI, LSL60101 treatment did not change the apoptotic biomarkers (Garau et al., 2013), but it enhanced morphine antinociception similar to 2-BFI and substituted for 2-BFI in rats trained to discriminate 2-BFI (Sánchez-Blázquez et al., 2000; MacInnes and Handley, 2002). Moreover, although tracizoline showed a different pattern in manipulating the canonical apoptotic pathways (Garau et al., 2013) than 2-BFI, the two compounds showed strikingly similar behavioral effects: they both produced antihyperalgesia, reduced body temperature, and demonstrated symmetrical substitution in drug discrimination (Thorn et al., 2012; Li et al., 2014; Qiu et al., 2015).
Existing evidence strongly suggests that I2 receptor agonists are able to modulate the monoaminergic system possibly via inhibition of MAOA activity. Several I2 receptor agonists such as 2-BFI and BU224 and the novel I2 receptor agonist CR4056 have been shown to inhibit human recombinant MAOA activity in a concentration-dependent manner, and in vivo these drugs caused marked increases in norepinephrine and serotonin content in the rat cerebral cortex and lumbar spinal cord (Ferrari et al., 2011; see also Jones et al., 2007). Li’s group has examined the involvement of the monoaminergic system in 2-BFI– and CR4056–induced antinociception in two chronic pain rat models: CFA-induced inflammatory pain and CCI-induced neuropathic pain (Siemian et al., 2018). 2-BFI induced dose-dependent antiallodynia in CCI rats; pretreatment with the selective serotonin reuptake inhibitor fluoxetine or the norepinephrine reuptake inhibitor desipramine dose-dependently and significantly enhanced the effects of 2-BFI and shifted the 2-BFI dose-effect curve leftward (Siemian et al., 2018). The dopamine reuptake inhibitor GBR12909 failed to alter the antiallodynic effect of 2-BFI under the same condition. To further explore the involvement of serotonin and norepinephrine in 2-BFI–induced antinociception, we used a combination of para-chlorophenylalanine and fenfluramine to deplete endogenous serotonin and used DSP-4 [N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine], a neurotoxin selective for noradrenergic neurons, to deplete norepinephrine. This treatment protocol dramatically attenuated the antinociceptive effect of 2-BFI (Siemian et al., 2018). These results clearly demonstrate that serotonin and norepinephrine, but not dopamine, are important in 2-BFI–induced antinociception. Because both serotonin and norepinephrine include multiple subtype receptors, the role of some receptors that reportedly are involved in pain processing was studied in I2 receptor agonist-induced antinociception. Thus, for both 2-BFI– and CR4056–induced antinociception in CCI rats, the selective 5-HT1A receptor antagonist WAY100135 [(S)-N-tert-butyl-3-(4-(2-methoxyphenyl)-piperazin-1-yl)-2-phenylpropanamide], the 5-HT2A receptor antagonist MDL100907 [(R)-(+)-α-(2,3-dimethoxyphenyl)-1-[2-(4-fluorophenyl)ethyl]-4-piperinemethanol], and the α1-adrenergic receptor antagonist WB4101 [2-(2,6-dimethoxyphenoxyethyl)aminomethyl-1,4-benzodioxane hydrochloride], but not other monoamine receptor antagonists for 5-HT2C, adrenergic α2, dopamine D1, and D2 receptors, dose-dependently attenuated the effects of 2-BFI and CR4056. These results suggest that several monoaminergic receptors, including 5-HT1A receptors, 5-HT2A receptors, and adrenergic α1 receptors, are critically involved in the I2 receptor agonist-induced antinociceptive effects. These results are consistent with the findings that I2 receptor agonists inhibit MAOA activity and increase the central monoamine levels, and they support the central role of the monoaminergic system in mediating I2 receptor agonist-induced antinociception (Siemian et al., 2018).
However, none of the monoaminergic ligands altered 2-BFI–induced hypothermia nor 2-BFI–induced discriminative stimulus effects (Siemian et al., 2018). These results strongly suggest that I2 receptor agonists produce antinociceptive effects and discriminative stimulus effects via different pharmacological mechanisms. Results from another study support this notion. Siemian et al. (2017) examined the role of intracellular Ca2+ signaling in 2-BFI–induced antinociception. The l-type Ca2+ channel blockers verapamil and nimodipine, the calmodulin antagonist W-7 [N-(6-aminohexyl)-5-chloro-1-naphthalenesulfonamide hydrochloride], and the internal Ca2+ release inhibitor ryanodine all attenuated the antinociceptive effects of 2-BFI. This effect is somewhat specific to 2-BFI, as the same treatment did not alter the antinociceptive effect of oxycodone and acetaminophen. In contrast, in rats reliably discriminating 5.6 mg/kg 2-BFI from saline, verapamil, nimodipine, and W-7 did not alter the discriminative stimulus effects of 2-BFI. Again, these results strongly suggest that I2 receptor agonist-induced antinociception and some other behavioral effects may not be mediated by the same mechanisms. Given the crucial role of internal Ca2+ in mediating neurotransmitter exocytosis, it is not surprising that the inhibition of internal Ca2+ signaling by blocking a key step of the chain reaction disrupts the release of monoamines and the subsequent pharmacological effects of I2 receptor agonists. A schematic figure detailing the proposed mechanistic pathways of I2 receptor agonist-induced antinociception is provided in Fig. 6.

Two mechanisms underlying imidazoline I2 receptor agonist-induced analgesia. I2 receptor agonists can increase the synaptic monoamine (5-HT and norepinephrine) level via MAO inhibition, which can subsequently activate the individual monoaminergic receptors. I2 receptor agonists can also inhibit central glial activity, attenuate the secretion of proinflammatory cytokines, and then attenuate the neural activity of the second-order pain-projecting pathways. 5-HT, serotonin; AR, adrenergic receptor; I2R, I2 receptor.
Immunocompetent cells in the CNS, including microglia and astrocytes, have become increasingly recognized as essential players in pain pathophysiology. Glial cells express I2 receptors and several studies have shown that I2 receptor agonists can modulate glial activity. For example, in mice with brain and spinal cord injury induced by experimental autoimmune encephalomyelitis, repeated 2-BFI administration reduced spinal microglial activation, reduced levels of cytokines such as interferon-γ and TNF-α in the blood and spinal cord, and improved symptom severity scores (Li et al., 2012; Zhu et al., 2015). In an effort to evaluate whether I2 receptor agonists produce antinociception by directly modulating glial activities, Siemian et al. (2018) examined the glial reactivity in the dorsal horn of the spinal cord and the TNF-α levels in CCI rats. CCI injury led to robust microglial and astrocyte activity in the dorsal horn of the spinal cord, which was significantly reduced by repeated 2-BFI treatment at the dose that significantly reduced the mechanical allodynia in the same rats. The TNF-α level in the lumbar spinal cord tissue was significantly higher in CCI rats than sham rats, and this increase was significantly attenuated by 2-BFI treatment. To provide direct evidence that 2-BFI acts directly on the glial cells, in vitro mouse cortical astrocyte cultures were used and 2-BFI treatment was found to markedly reduce LPS-stimulated astrocyte activation and reduced TNF-α levels. Together, these results provide direct evidence that 2-BFI could directly suppress glial activity in CCI rats, which contributes to the observed antinociceptive activities induced by 2-BFI. This potential mechanism of action of I2 receptor agonists is depicted in Fig. 6.
VI. Imidazoline Subtype 3 Receptors
I3 receptors represent a putative binding site that is different from I1/I2 receptors. The nature and the functional characterization of this receptor remains elusive and little progress has been made in the past 2 decades. A brief review of this receptor will be provided here primarily based on the earlier literature (Eglen et al., 1998; Morgan and Chan, 2001).
It has long been recognized that phentolamine, an imidazoline, and other imidazoline compounds administered to human subjects results in an alteration in glycemic control (Cerasi et al., 1969). Later studies established that these compounds may influence the insulin secretory activity of pancreatic β-cells and that imidazoline binding sites exist on β-cells (Bousquet et al., 1984; Coupry et al., 1987; Ernsberger et al., 1987). Indeed, using an antiserum that is often used to identify I2 receptors, pancreatic islet tissue was found to contain protein bands that are consistent with I2 receptors (Morgan and Chan, 2001). The existence of I1 receptors is, however, controversial. Using pancreatic β-cells as the bioassay, some I2 receptor agonists were able to regulate insulin secretion, whereas others did not (Morgan et al., 1999). For example, 2-BFI is an efficient potentiator of glucose-induced insulin secretion in these cells at high concentrations (high micromoles), whereas its typical concentrations for interacting with I2 receptors are much lower (low nanomoles). On the other hand, other I2 receptor ligands (e.g., idazoxan, RS45041-190) do not stimulate insulin release nor do they antagonize the insulin secretory effect of other imidazoline compounds on β-cells (Chan et al., 1994; Berdeu et al., 1995). These results suggest that 2-BFI and related compounds may not directly modulate β-cell insulin secretion via I2 receptors.
One proposed model to explain how imidazoline compounds alter insulin secretion states that these compounds bind to a site associated with (or present on) the ATP-sensitive potassium (KATP) channel, thereby causing a reduction in the rate of potassium efflux. This in turn leads to an increase in the membrane potential (toward a less negative value) and culminates in membrane depolarization with the subsequent gating of voltage-sensitive Ca2+ channels and the triggering of insulin secretion (Morgan and Chan, 2001). This model is based primarily on the findings that imidazoline compounds such as phentolamine can alter the permeability of the β-cell plasma membrane to both ion flux measurements and by electrophysiological analysis of membrane current flow (Chan et al., 1991; Dunne, 1991). Patch-clamp recording revealed that imidazoline compounds can reduce the rate of potassium efflux through KATP channels. Clearly, this mechanism is drastically different from the conventional recognition of I1 and I2 imidazoline receptors and has been suggested to involve I3 receptors.
The β-cell KATP channel is an octomeric complex composed of two subunits: Kir6.2 and sulfonylurea receptor 1 (SUR1). Earlier studies showed that imidazoline compounds retain their activity as KATP channel blockers when truncated Kir6.2 is expressed in the absence of SUR1, suggesting that SUR1 is not required for imidazoline compounds to regulate the ionic conductance of Kir6.2 and that the I3 receptor is located within Kir6.2 itself (Ishida-Takahashi et al., 1996; Proks and Ashcroft, 1997). Subsequent biochemical evidence supports this notion (Monks et al., 1999). Thus, I3 receptors represent a binding site that is located on the Kir6.2-subunit of KATP channels.
Given that imidazoline compounds are able to modulate insulin secretion via I3 receptors, this receptor may have the potential to be a novel drug target for the development of new antidiabetic drugs. Unfortunately, little progress has been made in this regard. This is due in large part to the fact that how imidazoline compounds stimulate insulin secretion remains to be fully elucidated. Whether the I3 receptor is a viable drug target awaits much more detailed mechanistic investigations.
VII. Summary and Conclusions
Since the last systematic review on the topic of imidazoline receptors and their endogenous ligands (Regunathan and Reis, 1996), the field of imidazoline receptors has seen uneven progress: some quite dramatic and some much less so. The endogenous imidazoline receptor ligands remain an enigma and multiple candidates have been studied and discussed. The past 2 decades also saw a boom in the research on the purported endogenous imidazoline receptor ligand agmatine, including human trials to demonstrate the safety and potential therapeutic benefits for certain pain conditions (Keynan et al., 2010; Gilad and Gilad, 2014). The physiologic and pharmacological effects of agmatine continue to be studied and expanded to new arenas, including learning and memory (Moretti et al., 2014), pain (Li and Zhang, 2011), opioid dependence (Wu et al., 2008), and depression (Freitas et al., 2016). Sufficient preclinical evidence supports promising clinical translations, and the recent call for more translational studies with agmatine is a good reflection of the maturity of this field (Piletz et al., 2013).
Until recently, moxonidine and rilmenidine were considered representative I1 receptor agonists that modulate blood pressure. However, both compounds only have marginal I1/α2-receptor selectivity (approximately 10-fold) and their clinical efficacy cannot be solely attributed to I1 receptor activity. In this regard, novel compounds such as LNP599 demonstrate extraordinary I1 receptor selectivity and exciting therapeutic efficacy in hypertension and the metabolic syndrome (Fellmann et al., 2013a). These new developments may pave the way toward a newer generation of pharmacotherapy against hypertension and related complications.
For many years, one major impediment for I2 receptor research was the lack of well characterized in vivo functionalities that can be related to this receptor with confidence. The most prominent development in this field is the mounting and unequivocal findings that I2 receptor agonists are effective analgesics. This is endorsed by the recent successful phase II clinical trial of CR4056, a selective I2 receptor agonist (Rovati et al., 2017). For the first time, it seems that an I2 receptor–based analgesic could become a reality. This would prompt further research to explore other therapeutic potentials of I2 receptor agonists, such as neuroprotection. Using 11C-BU99008 as a ligand for imaging I2 receptors in the human brain in real time, researchers can now study these receptors in various neurodegenerative states (Tyacke et al., 2018).
In contrast, there is really not much progress on I3 receptor pharmacology and the research on this receptor remains in its infancy. Further research should remain focused on deciphering the nature of this enigmatic receptor and on developing new and selective I3 receptor ligands to facilitate more extensive pharmacological studies.
In summary, despite several decades of efforts, pharmacologists remain puzzled by the nature of the so-called “imidazoline binding sites.” The continued endeavor to decipher the molecular identities of these receptors remains important. However, exciting progress has been made from a different perspective: translational pharmacology. With the new developments in I1 and I2 receptor pharmacology and their synthetic ligands, now may be the prime time to focus more on imidazoline receptor drug discovery and translating preclinical findings to novel pharmacotherapies to benefit millions of patients suffering from hypertension, metabolic syndrome, chronic pain, and stroke, as imidazoline receptor agonists show the most promising therapeutic potential for these conditions. Looking forward, it is not too radical to predict that imidazoline receptor–based novel therapies are on the horizon.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Bousquet, Hudson, García-Sevilla, Li.
Footnotes
P.B. and A.H. contributed equally to this work.
J.A.G.-S. and J.-X.L. contributed equally to this work.
Abbreviations
- 2-BFI
- 2-(2-benzofuranyl)-2-imidazoline
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- AMPK
- 5′ adenosine monophosphate-activated protein kinase
- BU216
- 3-[4,5-dihydroimidaz-2-yl]-quinoline hydrochloride
- BU224
- 2-[4,5-dihydroimidaz-2-yl]-quinoline hydrochloride
- BU226
- 2-[4,5-dihydroimidaz-2-yl]-isoquinoline hydrochloride
- CCI
- chronic constriction injury
- CDS
- clonidine-displacing substance
- CFA
- complete Freund’s adjuvant
- CNS
- central nervous system
- CR4056
- 2-phenyl-6-(1H-imidazol-1yl) quinazoline
- DSP-4
- N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine
- ERK
- extracellular signal-regulated kinase
- HPLC
- high-performance liquid chromatography
- IAA-RP
- imidazole-4-acetic acid-ribotide
- IRAS
- imidazoline receptor antisera-selected
- KATP
- ATP-sensitive potassium
- KU14-R
- 2-(2-ethyl-2,3-dihydro-2-benzofuranyl)-1H-imidazole
- LNP509
- cis-/trans-dicyclopropylmethyl-(4,5-dimethyl-4,5-dihydro-3H-pyrrol-2-yl)-amine
- LNP599
- 3-chloro-2-methyl-phenyl)-(4-methyl-4,5-dihydro-3H-pyrrol-2-yl)-amine hydrochloride
- LNP630
- (2,6-dichloro-phenyl)-(4-methyl-4,5-dihydro-1H-imidazol-2-yl)-amine
- LNP906
- 2-(5-azido-2-chloro-4-iodo-phenylamino)-5-methyl-pyrroline
- LNP911
- (2-(2-chloro-4-iodo-phenylamino)-5-methyl-pyrroline
- LSL60101
- 2-[2-benzofuranyl]-2-imidazole hydrochloride
- LSL60125
- 2-[6-methoxybenzofuran-2-yl] imidazole hydrochloride
- LSL61122 (tracizoline)
- 2-styryl-4,5-dihydro-lH-imidazole
- MAO
- monoamine oxidase
- MAPK
- mitogen-activated protein kinase
- MDL100907
- (R)-(+)-α-(2,3-dimethoxyphenyl)-1-[2-(4-fluorophenyl)ethyl]-4-piperinemethanol
- NAQ
- 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-[(3′-isoquinolyl) acetamido] morphine
- NMDA
- N-methyl-d-aspartic acid
- NRL
- nucleus reticularis lateralis
- PAC
- para-aminoclonidine
- PC-PLC
- phosphatidylcholine-specific phospholipase C
- PET
- positron emission tomography
- phenyzoline
- 4,5-dihidro-2-(2-phenylethyl)-1H-imidazole
- RO41-1049
- N-(2-aminoethyl)-5-(3-fluorophenyl)-4-thiazolecarboxamide hydrochloride
- RS45041
- 4-chloro-2-(imidazolin-2-yl)isoindoline hydrochloride
- RX82-1002
- 2-(2,3-dihydro-2-methoxy-1,4-benzodioxin-2-yl)-4,5-dihydro-1H-imidazole hydrochloride
- S22687
- 5-[2-methyl phenoxy methyl]-1,3-oxazolin-2-yl) amine
- S43126
- 2-[40-methoxyphenyl]-4,5-dihydro-1H-imidazole
- SHROB
- spontaneously hypertensive obese
- ST91
- 2-(2,6-diethylphenylamino)-2-imidazoline hydrochloride
- SUR1
- sulfonylurea receptor 1
- TNF-α
- tumor necrosis factor α
- W-7
- N-(6-aminohexyl)-5-chloro-1-naphthalenesulfonamide hydrochloride
- WAY100135
- (S)-N-tert-butyl-3-(4-(2-methoxyphenyl)-piperazin-1-yl)-2-phenylpropanamide
- WB4101
- 2-(2,6-dimethoxyphenoxyethyl)aminomethyl-1,4-benzodioxane hydrochloride
- WIN52212-2
- (R)-(+)-[2,3-dihydro-5-methyl-3-[(4-morpholino)methyl]pyrrolo-[1,2,3-de]-1,4-benzoxazin-6-yl](1-naphthyl)methanone
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}