


Abstract
Recent studies have strived to find an association between rapid antidepressant effects and a specific subset of pharmacological targets and molecular pathways. Here, we propose a broader hypothesis of encoding, consolidation, and renormalization in depression (ENCORE-D), which suggests that, fundamentally, rapid and sustained antidepressant effects rely on intrinsic homeostatic mechanisms evoked as a response to the acute pharmacological or physiologic effects triggered by the treatment. We review evidence that supports the notion that various treatments with a rapid onset of action, such as ketamine, electroconvulsive therapy, and sleep deprivation, share the ability to acutely excite cortical networks, which increases synaptic potentiation, alters patterns of functional connectivity, and ameliorates depressive symptoms. We proceed to examine how the initial effects are short-lived and, as such, require both consolidation during wake and maintenance throughout sleep to remain sustained. Here, we incorporate elements from the synaptic homeostasis hypothesis and theorize that the fundamental mechanisms of synaptic plasticity and sleep, particularly the homeostatic emergence of slow-wave electroencephalogram activity and the renormalization of synaptic strength, are at the center of sustained antidepressant effects. We conclude by discussing the various implications of the ENCORE-D hypothesis and offer several considerations for future experimental and clinical research.
Significance Statement Proposed molecular perspectives of rapid antidepressant effects fail to appreciate the temporal distribution of the effects of ketamine on cortical excitation and plasticity as well as the prolonged influence on depressive symptoms. The encoding, consolidation, and renormalization in depression hypothesis proposes that the lasting clinical effects can be best explained by adaptive functional and structural alterations in neural circuitries set in motion in response to the acute pharmacological effects of ketamine (i.e., changes evoked during the engagement of receptor targets such as N-methyl-D-aspartate receptors) or other putative rapid-acting antidepressants. The present hypothesis opens a completely new avenue for conceptualizing and targeting brain mechanisms that are important for antidepressant effects wherein sleep and synaptic homeostasis are at the center stage.
I. Introduction
Major depression is a highly prevalent and disabling psychiatric illness. Common symptoms include depressed mood, anhedonia, and cognitive dysfunction, and the patient’s emotional state is frequently dominated by negative thinking and low self-esteem. Recurrent, self-reflected, and uncontrollable rumination is one of the hallmarks of disease phenomenology (Nolen-Hoeksema et al., 2008; Hamilton et al., 2015; Whisman et al., 2020). Furthermore, depression is associated with negative biases in cognition, which predispose depressed individuals to exhibit a better recall for negative information compared with healthy subjects (Harrington et al., 2017). Patients suffering from depression may avoid social interaction and linger in despair and suicidal thoughts, often with tragic consequences.
The etiology of depression is poorly understood but involves complex neurobiological and neurodevelopmental underpinnings. Depression is not the result of any specific pathology affecting only single-cell types, brain areas, or neural networks but is characterized by structural and functional alterations of a more complex cortico-limbic circuitry (Price and Drevets, 2012). Moreover, although the illness may manifest suddenly, the underlying pathology takes time to develop and depends on both environmental and genetic factors. Nevertheless, certain treatments, most notably subanesthetic ketamine, can ameliorate the core symptoms of depression and suicidality within just a few hours. These remarkable effects of ketamine have been intimately connected with its ability to increase neurotrophic signaling and synaptogenesis along with the facilitation of synaptic plasticity in brain areas implicated in the pathophysiology of depression (Duman and Aghajanian, 2012; Duman et al., 2016; Rantamäki and Yalcin, 2016). Indeed, interest in manipulating some of the molecular mechanisms of synaptic plasticity directly with novel drugs is increasing. Synaptic plasticity, however, is an activity-dependent property of neural networks to change, (Changeux and Danchin, 1976; Thoenen, 1995; Hensch, 2005; Park and Poo, 2013) and its control cannot be outsourced for a specific molecular entity or any antidepressant treatment of that matter (Castrén, 2005, 2013; Castrén and Rantamäki, 2010).
Synaptic plasticity is proportionally high during early life, when neural networks are developed and fine-tuned (Hensch, 2005). During these periods, information from environmental interaction is encoded into stabilizing synapses and networks at a high rate. This activity-dependent interplay is thought to shape the structure and function of these networks for later life. However, animal studies suggest that dendritic spines of the cerebral cortex eventually become highly stable, with a majority lasting throughout life (Grutzendler et al., 2002; Yang et al., 2009). While stability increases, plasticity is not entirely lost. This is best exemplified by the lifelong ability to learn. Learning is dependent on functional changes in neuronal circuitry, which can be modified via alterations to the number (formation and elimination), morphology, and strength of synaptic contacts (Trachtenberg et al., 2002). Animal studies suggest that high-frequency stimulation-induced long-term potentiation (LTP; cellular model of learning) increases the formation of dendritic spines in the developing brain, whereas synapses may preferentially increase in size in older animals (Lang et al., 2004; Bailey et al., 2015). The main pharmacological targets of ketamine, that is, glutamatergic N-methyl-D-aspartate receptors (NMDARs) and α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptors (AMPARs), are crucial components for neural activity and synaptic plasticity. Mechanistic studies investigating ketamine’s antidepressant actions have focused on these receptor ion channels and their downstream effectors (Zanos and Gould, 2018), whereas more global mechanisms contributing to synaptic plasticity have received less attention.
A single subanesthetic dose of ketamine produces antidepressant effects within hours, and the effects are most prominent 24 hours after the treatment. The antidepressant effects of ketamine may continue even up to a week or two. We hypothesize that these lasting clinical effects of ketamine can be best explained by adaptive functional and structural alterations in neural circuitries set in motion in response to the acute pharmacological effects of ketamine (i.e., changes evoked during the engagement of receptor targets such as NMDARs). Most importantly, the timescale of the effects set forth by ketamine suggests that fundamental mechanisms associated with synaptic homeostasis and sleep play key roles in sustaining the antidepressant effects. Here, we build on the synaptic homeostasis hypothesis (SHY) of sleep (Tononi and Cirelli, 2003) and propose the hypothesis of encoding, consolidation, and renormalization in depression (ENCORE-D), which provides novel perspectives for understanding and examining the complex neurobiology around rapid-acting antidepressants.
II. Synaptic Homeostasis Hypothesis as the Foundation for Encoding, Consolidation, and Renormalization in Depression
Accumulating evidence suggests that sleep is essential for long-lasting changes in synaptic function, plasticity, and learning (Maquet, 2001; Stickgold, 2005; Tononi and Cirelli, 2014). Tononi and Cirelli (2003) have described a mechanism for the regulation of synaptic plasticity during wake and sleep, known as SHY (Fig. 1). It proposes that the restoration of synaptic homeostasis is the fundamental function of sleep. This homeostasis is challenged during waking by increases in synaptic potentiation and by immense synaptogenesis during early development (Tononi and Cirelli, 2014). Accordingly, during waking, the brain is active and in an optimal state for learning, which is encoded into long-lasting changes in the strength, number, and wiring of neuronal connections, fueled by molecular cascades and plasticity-related proteins such as brain-derived neurotrophic factor (BDNF) and activity-regulated cytoskeleton-associated protein (Arc) (Bramham et al., 2008; Shepherd and Bear, 2011). In contrast, during sleep, our brains become disconnected from the external world. The state of disconnection associated with stages of deep sleep is ideal for the systematic renormalization of synaptic strength because the brain is not influenced by ongoing sensory stimuli (Tononi and Cirelli, 2019). SHY further proposes that synaptic downscaling during deep sleep is crucial for preserving synaptic strength at a sustainable level. It allows for further synaptic plasticity to take place during the next day while preventing the metabolic costs associated with excessive potentiation and excitability during waking. In other words, “sleep is the price the brain pays for plasticity” (Tononi and Cirelli, 2014).

Synaptic homeostasis hypothesis (SHY) (see Tononi and Cirelli, 2003, 2014). During waking, cortical synapses become potentiated proportionally to their activity, resulting in a net increase in synaptic strength. Synaptic potentiation during waking is connected to a homeostatic increase in slow-wave activity (SWA) during non–rapid eye movement (NREM) sleep, during which the systematic normalization of synaptic strength (synaptic downscaling and renormalization) takes place. Larger and most potentiated synapses may remain unscaled. Furthermore, weak connections may be eliminated, whereas the relative strength of the remaining connections is preserved. This mechanism allows further synaptic plasticity to take place during the next day while preventing the metabolic costs associated with excessive potentiation and excitability during waking.
SHY indicates that synaptic potentiation (e.g., evoked by neural activity) during waking is connected to the homeostatic increase of slow-wave sleep (SWS) during consecutive non–rapid eye movement (NREM) sleep. This can be measured in electroencephalogram (EEG) recordings as ∼0.5–4 Hz slow-wave activity (SWA) and is proposed to reflect the strength of cortical synapses (Tononi and Cirelli, 2003). Indeed, a motor-learning task evokes the homeostatic increase of SWA during subsequent sleep in the cortical area representative of the given action during sleep along with improvement in task performance in humans (Huber et al., 2004). Moreover, selective SWA disruption in the motor cortex during post-training sleep impairs subsequent motor performance (Fattinger et al., 2017), whereas facilitation of SWA may aid the consolidation of declarative memories (Marshall et al., 2006; Lu et al., 2018).
During SWS, cortical and thalamic neurons oscillate between up and down states, characterized by the tendency to fire and to be silent, respectively (Tononi and Cirelli, 2019). This slow and synchronized rhythmic activity has been proposed as the basis for sleep-dependent synaptic down-selection, along with some other forms of neural activity, such as hippocampal sharp wave-ripples (Norimoto et al., 2018). Throughout the course of SWS, the cortex proposedly undergoes a process of synaptic renormalization to produce a global yet specific downscaling of synaptic strength. Larger and most potentiated synapses appear most likely to remain unscaled compared with smaller and less potentiated synapses (de Vivo et al., 2017). As further posited by SHY, synaptic connections that are most active during sleep are preferentially spared (Tononi and Cirelli, 2019). Such protection from downscaling has been evaluated in computer simulations (Hashmi et al., 2013; Nere et al., 2013) and has recently gained support from an animal study, in which urethane was used to reproduce the up and down states of NREM sleep combined with state-specific optogenetic stimulations (González-Rueda et al., 2018).
The states of wakefulness and learning are accompanied by LTP-like changes and increases in synaptic density and neuronal complexity in the brains of animals subjected to manipulations such as an enriched environment or whisker stimulation (Moser et al., 1997; Kolb et al., 1998; Knott et al., 2002; Tononi and Cirelli, 2014). In line with SHY, studies have demonstrated that synapse size and the number of AMPARs in synaptic sites decrease during sleep (Vyazovskiy et al., 2008; de Vivo et al., 2017; Diering et al., 2017). These changes occur along with decreases in cortical firing rates and synchrony (Vyazovskiy et al., 2009) and in the frequency and amplitude of miniature excitatory postsynaptic currents (Liu et al., 2010). Moreover, studies investigating the evoked responses from transcranial magnetic stimulation (TMS) in humans suggest that cortical synaptic plasticity is restored after sleep but impaired following sleep deprivation (Kuhn et al., 2016). Time spent awake also results in a net increase in cortical spines in mice, whereas sleep results in spine loss (Maret et al., 2011; Yang and Gan, 2012). These changes may not be, however, limited to the cortex, as a recent study suggests that sleep-dependent synaptic renormalization may also occur in the hippocampus (Spano et al., 2019).
Sleep is likely involved in several mechanisms beyond synaptic renormalization proposed by SHY. Indeed, sleep is often considered to be a key stage for memory consolidation (Diekelmann and Born, 2010). For example, the active consolidation hypothesis proposes that memory traces encoded during waking are reactivated and consolidated during sleep (Diekelmann and Born, 2010). These processes have been mainly investigated in the hippocampus, where reactivation of neural ensemble activity occurs during sleep; however, such reactivations may also occur in concert or independently in other brain areas (Marshall and Born, 2007). Moreover, differences in sleep-dependent processing likely exist between various types of memory traces. These hypotheses may thus represent various characteristics of wake- and sleep-dependent plasticity that are not mutually exclusive.
III. The Neurobiological Premise of Major Depression
Major depression can emerge at almost any age; however, it is most prevalent in adults aged 18–64 years, with a median age of onset in the 20s (Kessler et al., 2003, 2012). Notably, this is around the period when prefrontal cortical development and synaptic density eventually stabilize toward the adult range (Chugani et al., 1987; Huttenlocher and Dabholkar, 1997; Petanjek et al., 2011; Tamnes et al., 2017). Although depression is often diagnosed in adulthood, a vast amount of evidence indicates that it has developmental components in many cases. Already in 1649, René Descartes referenced the involvement of what is now called an implicit (i.e., nondeclarative) memory by describing how “[an aversive childhood experience may] remain imprinted on the child’s brain to the end of his life [without] any memory remaining of it afterwards” (Schacter, 1987). Indeed, the incorporation of early negative memory traces and experiences may contribute to dysfunctional network architecture and altered implicit cognition, which is known to be highly relevant for depressive disorders and several associated behavioral characteristics, such as negativity (Gaddy and Ingram, 2014) and suicidal thinking (Nock et al., 2010). Such alterations are most likely to develop during sensitive periods and are difficult to remodel thereafter. Animal studies have demonstrated that even slight changes in the organization of neuronal networks during sensitive periods contribute to functional alterations that persist throughout life (Greenhill et al., 2015). These developmental windows exist for various brain functions and networks, including those engaged in higher-order cognitive functions and emotional processing (Silbereis et al., 2016). Thus, early life adversity-driven structural alterations may be important in the onset of depressive disorders later in life, an idea supported by numerous studies (Newman et al., 1996; Pine et al., 1999; Hammen et al., 2000; Heim and Nemeroff, 2001; Dougherty et al., 2004; Cohen et al., 2006; Kitayama et al., 2006; Kessler et al., 2010; Teicher et al., 2014; Li et al., 2016c; Bandoli et al., 2017; Ellis et al., 2017; Tyborowska et al., 2018; Martins-Monteverde et al., 2019; Ohashi et al., 2019). Notably, severe forms of early life stress, adversity, and maltreatment are associated with the most difficult treatment-resistant forms of depression (Bernet and Stein, 1999; Nanni et al., 2012; Williams et al., 2016).
Depression is recognized not to result from any specific pathology affecting only single-cell types or areas but is characterized by altered functioning of a more complex cortico-limbic circuitry. The medial prefrontal cortex, particularly the subgenual cingulate cortex (SCC), along with connecting subcortical structures, such as the hippocampus (Videbech and Ravnkilde, 2004; Kempton et al., 2011; Santos et al., 2018), the amygdala, and the nucleus accumbens, are affected (Ressler and Mayberg, 2007; Heshmati and Russo, 2015; Akil et al., 2018). Hyperactive hypothalamus-pituitary adrenal axis and the overproduction of stress hormones, decreased neurotrophic support, and reduced neuronal plasticity are thought to underlie some of the atrophic changes associated with depression (Lee et al., 2002; Duman and Monteggia, 2006; Castrén et al., 2007; Liu et al., 2017). These changes commonly manifest as a decreased volume of cortical and limbic areas in patients suffering from depression (Lorenzetti et al., 2009; Kempton et al., 2011; Schmaal et al., 2016, 2017), and they are also supported by both human imaging and postmortem studies showing reductions in cortical neurons and synapses (Rajkowska et al., 1999; Kang et al., 2012; Holmes et al., 2019). The extent of these changes is associated with the duration and severity of the disorder. Studies in animal models have demonstrated that exposure to chronic stress results in similar atrophic alterations (Duman and Aghajanian, 2012).
Dysregulation of connectivity within cortical and limbic networks is also evident in depressive disorders. Notably, although major depression is commonly associated with reduced neural activity, synaptic plasticity, and even atrophy in several regions, other areas and networks may exhibit increased activity or hyperconnectivity, which contributes to the overall dysregulation of information processing and symptomatology such as rumination. The triple network model proposes that the default mode network (DMN), salience network (SN), and central executive network (CEN) are at the core of several psychopathologies (Menon, 2011). In particular, depression is associated with increased DMN connectivity (Greicius et al., 2007; Hamilton et al., 2015), whereas connectivity or activity within SN and CEN are suggested to be reduced (Menon, 2011). It has been suggested that a switch between internally directed cognition of the DMN and externally directed cognition of the CEN is modulated by SN (Sridharan et al., 2008; Goulden et al., 2014).
Dysregulation of the DMN may be of pivotal importance in depression, as it is associated with self-referential thinking, remembering the past, future planning, and the evaluation of survival cues (Buckner et al., 2008). Moreover, abnormal DMN activity and functional connectivity correlate with the severity of depression and rumination (Greicius et al., 2007; Sheline et al., 2010; Berman et al., 2011). Importantly, the activity of the DMN is underpinned by patterns of hyperconnectivity between the DMN and other regions related to symptoms of depression. For example, Hamilton et al. (2015) have proposed that the primary dysfunction contributing to depressive rumination is not overactivity of the DMN per se but rather the level of connectivity with the SCC, which demonstrates increased activity (Mayberg et al., 1999). Deep-brain stimulation of the SCC white matter tract has been shown to reverse symptoms in treatment-resistant depressive patients along with the normalization of SCC activity (Mayberg et al., 2005); however, a large multisite randomized sham-controlled trial failed to reproduce statistically significant antidepressant efficacy (Holtzheimer et al., 2017). Future functional imaging and elaborate deep-brain stimulation studies that involve (multi)targeting key brain areas and networks will not only yield further important insights into the effectiveness of these treatments but also elucidate the underlying pathophysiology of depression.
Several pathophysiological paths may dictate the emergence of depression and the associated structural and functional alterations. Apart from depression being born out of early-life predisposition, late-onset depression may also rely on mechanisms involving implicit cognitive processes and networks (Beck, 2008; Gotlib and Joormann, 2010; Rock et al., 2014; Rayner et al., 2016), which confer vulnerability to stress and strengthen negative thought patterns through continued conscious and nonconscious mental exercise. This may lead to changes in neural networks determined by activity-dependent synaptic plasticity, measurable as changes in synaptic strength and altered neural network balance in favor of the hyperactive circuits. Conversely, persistent hyperactivity can also facilitate readjustments in functional and structural connectivity through mechanisms of homeostatic plasticity (Fauth and Tetzlaff, 2016). Over the course of worsening of depression, global levels of synaptic potentiation may decrease because general patterns of activity are “dominated” by only a subset of networks. Several other circuits and networks concurrently demonstrate dysregulated activity and connectivity, resulting in long-term depression–like plasticity, which ultimately culminates in reversible structural atrophy. This may be evidenced by the lowest levels of synaptic density in several brain areas in patients with the highest levels of depression measured indirectly using a radioligand for synaptic vesicle glycoprotein 2A (Holmes et al., 2019). A vicious cycle could be created by the skewed balance between hypo- and hyperactive networks. For example, hyperactive depressogenic networks may repeatedly reach a saturation of synaptic plasticity, which consistently builds their relative strength during waking and maintains it over the renormalization of synaptic strength during SWS. Similar processes leading to alterations in synaptic connectivity are likely shared by many other psychiatric disorders, such as schizophrenia and post-traumatic stress disorder (Krystal et al., 2017a,b).
The processes leading to the depressed state are likely further fueled by stress; inflammation (Vogelzangs et al., 2012); cognitive, emotional, and genetic factors (Joormann and Siemer, 2011; Dunn et al., 2015); sleep problems (Nutt et al., 2008); and other disposing factors, such as lack of gratifying daily exercise and social communication. Overall, depression is associated with reduced physical activity patterns along with dysregulation of sleep and circadian rhythm (Nutt et al., 2008; Hasler et al., 2010; Burton et al., 2013). Conversely, sleep disturbances are significantly associated with an increased risk of depression (Baglioni et al., 2011; Li et al., 2016b) and suicidality (Malik et al., 2014; Bernert et al., 2015). Moreover, altered sleep architecture is particularly associated with affective disorders (Benca et al., 1992). Depressed patients commonly exhibit disturbances in sleep continuity, a shortening of rapid eye movement (REM) latency, increases in REM density, and increased total REM sleep time, although these changes are not specific for affective disorders (Riemann et al., 2020). Notably, REM sleep plays an important role in emotional processing (Tempesta et al., 2018), and renormalization of amygdala activity is suggested to be one of the physiologic roles of REM, a process likely disrupted in patients suffering from anxiety (Van Der Helm et al., 2011). Furthermore, depression is often associated with reduced SWS and a lower delta sleep ratio, which is indicative of a smaller decrease in SWA occurring from the first NREM episode to the last (Kupfer et al., 1990; Armitage, 2007). Most importantly, some of these sleep-related abnormalities are present even when patients are in remission (Rush et al., 1986), and they are associated with an increased risk of relapse (Modell et al., 2002).
IV. Examining Rapid Antidepressant Effects through Encoding, Consolidation, and Renormalization in Depression
Several attempts have been made to pinpoint the precise mechanistic basis underlying ketamine’s antidepressant effects. Ketamine’s abilities to boost, at subanesthetic doses, glutamatergic firing and to increase AMPAR function are among the key areas of interest (Zanos and Gould, 2018; Zanos et al., 2018b). These ketamine-induced changes are thought to normalize dendritic spine loss and synaptic plasticity within the prefrontal cortex and hippocampus through molecular mechanisms involving, but not limited to, BDNF (Liu et al., 2012; Lepack et al., 2014) and its primary target tropomyosin receptor kinase B (TrkB) (Yang et al., 2015), mitogen-activated protein kinase (MAPK) (Réus et al., 2014), mammalian target of rapamycin (mTOR) (Li et al., 2010), and glycogen synthase kinase 3β (GSK3β) (Beurel et al., 2011) (for reviews see Duman and Aghajanian, 2012; Duman et al., 2016; Rantamäki and Yalcin, 2016; Rantamäki, 2019) (Fig. 2). The following questions then arise: how does ketamine master these mechanisms, and how do they lead both to rapid and sustained antidepressant effects and to the rescue of abnormal functional connectivity and plasticity?

General mechanistic principles underlying rapid antidepressant effects. (A) Evoked glutamate bursts (e.g., through disinhibition of inhibitory interneurons) and AMPAR-mediated facilitation of BDNF signaling and synaptogenesis within the medial prefrontal cortex are considered important for the rapid antidepressant effects of ketamine. (B) Ketamine is readily distributed in the body after systemic administration; however, it undergoes rapid elimination and metabolism. The antidepressant effects of ketamine become most evident when its psychotropic actions and acute pharmacological effects fade out, and the therapeutic effects may last for days or weeks. ENCORE-D proposes that the key to understanding the full spectrum of neurobiological changes triggered by ketamine is in the adaptations triggered within the brain in response to a pharmacological “challenge.”
According to the prevailing view, ketamine has almost an ingenious ability to regulate a chain of molecular events connected with synaptic plasticity. This has led some researchers to suggest that mere regulation of any of these effectors leads to rapid and sustained antidepressant responses. Although this idea has turned out to be promising in some rodent studies (Shirayama et al., 2002; Chen et al., 2012; Fukuda et al., 2016; Hasegawa et al., 2019), it essentially ignores how these molecular events are part of activity-dependent synaptic plasticity (Changeux and Danchin, 1976; Thoenen, 1995; Hensch, 2005; Park and Poo, 2013) (see Introduction). Moreover, such molecular perspectives fail to appreciate the temporal distribution of the effects of ketamine on cortical excitation and plasticity as well as the prolonged influence on depressive symptoms (Fig. 2). The acute pharmacological effects of ketamine are short-lived, lasting only a few hours, though many neurobiological alterations become evident long after the immediate engagement of target receptors such as NMDARs. Notably, emerging preclinical data indicate that specific metabolic byproducts of ketamine, namely, hydroxynorketamines (HNKs; especially 2R,6R-HNK), bring about rapid antidepressant effects in rodents, thus suggesting that the gradual buildup of these metabolites take part in ketamine’s rapid and sustained antidepressant effects (Zanos et al., 2016). HNKs do not share the pharmacological profile of ketamine (Zanos et al., 2016; Lumsden et al., 2019) and thereby challenge the original idea that NMDARs inhibition plays an essential role in (rapid) antidepressant effects (Trullas and Skolnick, 1990; Skolnick et al., 1996). This topic is subject to active ongoing work and debate in the preclinical domain, while patient studies are still underway (Collingridge et al., 2017; Zanos et al., 2018a; Hashimoto, 2019). However, a recent study did find a correlation between plasma 2R,6R-HNK levels and the antidepressant and antisuicidal effects of ketamine in patients (Grunebaum et al., 2019), but higher rather than lower levels correlated with less clinical improvement.
Instead of solely giving the credit to ketamine or to some of its metabolites, or to any particular molecular event for that matter, ENCORE-D proposes that ketamine’s ability to transiently excite cortical neurocircuits triggers the intrinsic mechanisms of synaptic plasticity (Waltereit and Weller, 2003; Peineau et al., 2008; Hoeffer and Klann, 2010; Lu et al., 2014) and homeostasis upon drug withdrawal (i.e., after the disengagement from target receptors), leading to facilitated encoding of activity-dependent changes and increases in synaptic strength. These synaptic changes are then consolidated during subsequent steps that involve changes in transcription and protein synthesis. In accordance with SHY, these activity-dependent alterations may reach their final state through the global renormalization of synaptic weights during subsequent sleep, resulting in long-term changes in circuit activity and functional connectivity. Fundamental mechanisms of sleep, and particularly SWA, are at the core of this process, which results in the engraving of what can be thought of as a pharmacologically induced “memory.” This is not a memory trace in a strictly explicit sense. It is instead recapitulated as the sum of synaptic changes that the pharmacological effect sets in motion and that are subsequently consolidated and sustained throughout sleep. These alterations ultimately affect the patterns of neural activity and the flow of implicit cognitive processes, promoting thought patterns free of depression. As will be discussed, such a hypothesis may be relevant not only for ketamine but also for other manipulations capable of eliciting rapid and sustained antidepressant effects. We now overview evidence supporting the key steps of the ENCORE-D hypothesis (Fig. 3).

A simplified model of the ENCORE-D hypothesis. (A) During the development of depression, susceptible neuronal networks become gradually hyper- (red) or hypoactive (blue) under predisposing environmental conditions, and they retain that state at the expense of optimal network functionality. This may manifest as, for example, uncontrollable self-focused rumination, depressive thoughts, anhedonia, and cognitive dysfunctions. Rapid-acting antidepressants (i.e., ketamine represented in the figure) have excitatory effects that are reflected in the activity of local circuits, leading to increases in cortical synaptic strength and the re-emergence of global functional connectivity patterns. This breaks the recursive cycle of rumination and allows for the immediate relief of depressive symptoms. (B) Increased excitatory tone and synaptic strength are reflected in the homeostatic emergence of waking slow-wave activity (SWA) after the acute pharmacological or physiologic effects of the said intervention have ceased. This phase is associated with the activation of several pathways implicated in synaptic plasticity and protein synthesis, and it contributes to the subacute consolidation of synaptic change. In subsequent slow-wave sleep (SWS), SWA is increased proportionally to the increase in cortical synaptic strength, and the global renormalization of synaptic strength takes place. During this period of renormalization, the increased activity of previously strengthened circuits offers protection from synaptic downscaling. Therefore, groups of synapses may maintain their relative potentiation, whereas others are relatively depotentiated. However, if no further excitatory stimulus or other therapy is applied, susceptible neuronal networks may again gravitate toward their depressogenic state over the course of several sleep-wake cycles. Once depressogenic patterns of activity are re-established, symptoms of depression re-emerge.
A. Encoding Activity into Synaptic Change
1. Cortical Excitation
Though competing explanations for the rapid antidepressant effects of ketamine have been proposed, most converge on the idea that subanesthetic (i.e., antidepressant) doses of ketamine lead to widespread cortical excitation and increases in energy metabolism (Breier et al., 1997; Lu et al., 2008; Li et al., 2016a; Abdallah et al., 2018a) (Fig. 2). These effects have been suggested to occur through the inhibition of NMDARs present on GABAergic interneurons, which leads to decreased inhibition of excitatory pyramidal neurons and increased glutamatergic signaling (Homayoun and Moghaddam, 2007), as demonstrated in animals (Moghaddam et al., 1997; Chowdhury et al., 2017) and humans (Abdallah et al., 2018a). The excitatory effects of ketamine are dose-dependent, as anesthetic doses decrease glutamate activity (Moghaddam et al., 1997), and “markers of excitation,” such as the phosphorylation of p44/42-MAPK, are differentially regulated by low and high doses of ketamine in rodents (Li et al., 2010; Kohtala et al., 2019b). The rapid antidepressant effects of ketamine have been proposed to be dependent on this glutamatergic excitation and the regulation AMPARs, as blocking these channels abolishes antidepressant-like behavioral responses in rodents (Maeng et al., 2008; Koike et al., 2011; Koike and Chaki, 2014).
The altered “balance of inhibition and excitation” produced by ketamine is thought to be displayed as increased high-frequency gamma oscillations on a cortical EEG. The effects of ketamine on gamma power are dose- and area-specific; furthermore, they are most prominent during the peak of pharmacological effects when the target receptors are preferentially occupied (Maksimow et al., 2006; Kohtala et al., 2019a,b), but they remain upregulated several hours after drug administration in patients (Nugent et al., 2019a). Moreover, patients who have lower baseline gamma power and experience larger increases in response to ketamine also report better antidepressant responses (Nugent et al., 2019a). In another study, ketamine responders displayed a difference in peak gamma response to a somatosensory stimulus when compared with nonresponders, further suggesting that altered excitation and synaptic potentiation are important first steps for ketamine’s antidepressant effects (Nugent et al., 2019b).
Global cortical excitation is perhaps even more obvious with many of the other treatments possessing rapid antidepressant potential. Electroconvulsive therapy (ECT) remains as one of the most effective interventions for treatment-resistant depression. Though series of consecutive treatments are typically required for therapeutic effects of ECT, some studies have shown antidepressant effects already after the first treatment episodes (Rich, 1984; Fligelman et al., 2016). The alternating current induced by ECT essentially forces activity into groups of cortical neurons (Fink, 2014). Neurons along the traveling current, dictated by electrode placement, begin firing simultaneously, which results in the propagation and generalization of epileptiform activity. Similar to ECT, pharmacological convulsants such as flurothyl have been successfully used to treat depression in the past. In animals, seizures induced by electroconvulsive shock (ECS; an animal model of ECT) have been suggested to facilitate glutamate release and NMDAR activation (Reid and Stewart, 1997). Notably, findings related to ECS have provided important evidence for the upregulation of BDNF following seizures (Nibuya et al., 1995). These findings formed the basis for the neurotrophin hypothesis of depression, which for the first time suggested that neurotrophic support is an important feature in the pathophysiology of depression and antidepressant actions (Duman et al., 1997).
Sleep deprivation, another nonpharmacological treatment of depression and an important tool for examining SHY in relation to sleep (see above), also increases cortical excitation (Huber et al., 2013; Meisel et al., 2015; Ly et al., 2016) and extracellular glutamate levels (Dash et al., 2009). In addition, several other experimental rapid-acting depression treatments, such as the antimuscarinic agent scopolamine (Furey and Drevets, 2006; Voleti et al., 2013; Chowdhury et al., 2017; see also Park et al., 2019) and psychedelic drugs (Scruggs et al., 2003; Muschamp et al., 2004; Vollenweider and Kometer, 2010; Kometer et al., 2013; Carhart-Harris et al., 2016, 2018), acutely increase cortical excitation and glutamate activity despite obvious pharmacological differences. We recently reported that nitrous oxide (“laughing gas”), an NMDAR-blocking anesthetic possessing rapid antidepressant potential (Nagele et al., 2015), increases markers of cortical excitation, including the expression of immediate early genes (IEGs) such as c-Fos (Fos proto-oncogene), Arc, and Homer1 (homer protein homolog 1) (Kohtala et al., 2019b). Whether other anesthetics with antidepressant potential, such as isoflurane (Langer et al., 1995; Weeks et al., 2013; Antila et al., 2017) or propofol (Mickey et al., 2018), also regulate cortical excitation in a similar manner remains to be investigated. Several reports have, however, highlighted the paradoxical excitation that frequently takes place during general anesthesia, characterized by a pattern of burst-suppression in the cortical EEG (Kroeger and Amzica, 2007; Ferron et al., 2009). It could be that the variable therapeutic outcomes of general anesthesia reported in depressed patients are related to the unpredictable nature of such treatments on cortical excitation.
2. Synaptic Potentiation and Spinogenesis
Neural communication in neocortical circuits is adjusted in coordination with experience. Changes can be made by altering the strength of synaptic connections or through the growth and retraction of dendritic spines and axonal boutons (Holtmaat and Svoboda, 2009). Synaptic potentiation, synaptic strength, and spine size correlate with one another, with stronger spines being larger in size and capable of more effective neurotransmission. Indeed, studies using caged glutamate released at individual spines have demonstrated increased LTP taking place along with increases in spine size (Matsuzaki et al., 2004).
Changes in the phosphorylation and trafficking of AMPARs constitute the early phase of LTP (E-LTP) formation (Esteban et al., 2003; Yang et al., 2008), which is the starting point for activity-induced synaptic alteration. This change is set in motion by an increased Ca2+ flow through NMDARs, which leads to the activation of calcium-dependent enzymes such as CaMKII (De Koninck and Schulman, 1998). CaMKII regulates several targets, including AMPARs (Incontro et al., 2018), p44/42-MAPK (Zhu et al., 2002), and BDNF-TrkB signaling (Harward et al., 2016), and its activity is associated with the control of spine size and synaptic strength (Lee et al., 2009; Pi et al., 2010). Among other things, spine size and synaptic strength are regulated by the constant dynamic motion of AMPARs to and from the postsynaptic membrane (Derkach et al., 2007). The activation of NMDARs and the following Ca2+ influx is thought to play a role in the phosphorylation, lateral diffusion, and incorporation of GluR1 subunit containing AMPARs from extrasynaptic sites to the active postsynaptic densities (PSDs). Moreover, small GTPases rat sarcoma protein and Ras-related protein have been found to control AMPAR subunit trafficking and synaptic potentiation through mechanisms requiring MAPK activation (Zhu et al., 2002). Increased activation and expression of GluR1 subunits in the synaptic membrane contributes to enhanced channel conductance and function, which are important for E-LTP expression (Hayashi et al., 2000; Derkach et al., 2007; Kristensen et al., 2011), whereas subsequent accumulation of GluR2-containing AMPARs may contribute to maintaining LTP. Concomitantly, activity-induced signaling cascades, including MAPK, reach the nucleus and activate transcription factors (Barco et al., 2002). This leads to changes in the expression of IEGs, such as c-Fos (Fos proto-oncogene), which orchestrates further transcriptional events. IEG mRNAs, which are encoding proteins that support the functions of synapses and dendrites, are transported to the synapse for local protein synthesis (e.g., Bdnf, Homer-1a, Arc) (Flavell and Greenberg, 2008; Lisman et al., 2018). These processes, involving changes in transcription and translation, are generally deemed to contribute to the late phase of LTP (L-LTP) (Derkach et al., 2007; Costa-Mattioli et al., 2009).
Ketamine has been shown to regulate CaMKII, eukaryotic elongation factor 2 kinase (Adaikkan et al., 2018), GSK3β (Beurel et al., 2016), and the phosphorylation of GluR1 subunits (Zhang et al., 2016), altering the expression, composition, and trafficking of AMPARs to the postsynaptic membrane. The blocking of AMPARs abolishes the ketamine-induced BDNF release and phosphorylation of MAPK in primary neuronal cultures, suggesting that these processes are dependent on increased glutamatergic signaling (Lepack et al., 2016). Ketamine also induces several transcriptional changes in IEGs, including altered Homer1 expression (de Bartolomeis et al., 2013; Ficek et al., 2016). These IEGs are translated and accumulate in the PSD, which refers to complex scaffolds of proteins located in close proximity to the postsynaptic membrane in dendritic spines. Proteins such as Homer1, Arc, and PSD-95 are important components of the PSD and contribute to glutamatergic signaling and synaptic function. Changes in Homer1a expression are particularly interesting because it has been associated not only with the homeostatic regulation of sleep (Maret et al., 2007; Mackiewicz et al., 2008) but also with the antidepressant-like effects of ketamine, sleep deprivation, and ECS in rodents (Conti et al., 2007; Serchov et al., 2015). A recent study demonstrating that systemic administration of cell-permeable Homer1a recapitulates the behavioral responses of ketamine in rodents provides a further link between Homer1a and rapid antidepressant responses (Holz et al., 2019).
The administration of ECSs also produces prominent synaptic potentiation in rodents. Studies have shown that brief electroconvulsive seizures essentially reduce the degree to which further LTP can be induced in the dentate gyrus of anesthetized rats. Indeed, analyses of the EPSPs (excitatory postsynaptic potentials) and population spike size suggest that LTP induction had already occurred after ECS induced seizure activity, thus hampering further potentiation attempts (Stewart and Reid, 1993; Stewart et al., 1994). These results are also supported by a study in which hippocampal EPSP characteristics were monitored throughout a series of ECS treatments. The increase in EPSP slope developed gradually over the course of the first five seizures, whereas a single seizure did not induce a significant change (Stewart et al., 1994). The saturation of LTP-like plasticity and synaptic strength appears to resist further potentiation attempts, whereas weaker synapses are more likely to gain in strength (Abraham et al., 2001; Abraham, 2008). Similar to ECS, sleep deprivation is thought to saturate LTP-like plasticity in humans (Kuhn et al., 2016) and rats (Campbell et al., 2002; Vyazovskiy et al., 2008). Ample evidence demonstrates the upregulation of IEGs in response to both ECS and sleep deprivation (Cirelli and Tononi, 2000).
Apart from synaptic potentiation, the growth of dendritic spines and their retraction offer powerful ways to modulate circuit activity. In some cortical areas, alterations in the density of spines and synapses have been demonstrated to occur after exposure to different training paradigms, enriched environments (Greenough et al., 1985; Moser et al., 1994, 1997; Kolb et al., 2008), and sensory stimulation (Knott et al., 2002; Trachtenberg et al., 2002; Holtmaat et al., 2006). On the other hand, chronic stress decreases spine density in areas implicated in depression, such as the prefrontal cortex and hippocampus; this finding is commonly observed in animal models of depression (Qiao et al., 2016). Though the structures of dendritic and axonal arbors remain relatively stable in adulthood, even subtle changes in the growth or retraction of boutons and dendritic spines may affect functional and behavioral outcomes. Several factors are thought to be important for the structural plasticity of dendritic spines, including presynaptic activity (Engert and Bonhoeffer, 1999; Maletic-Savatic et al., 1999), glutamate release (Richards et al., 2005), neurotrophins (Tanaka et al., 2008), mTOR signaling (Lipton and Sahin, 2014), CaMKII (Glazewski et al., 2000), and GSK3β (Cuesto et al., 2015; Cymerman et al., 2015; Ochs et al., 2015), among others.
Furthermore, animal studies investigating the effects of ketamine on spine synapse alterations have found that a single subanesthetic dose of ketamine increases the synthesis of synaptic proteins and the number of dendritic spines in the medial prefrontal cortex (Li et al., 2010; Moda-Sava et al., 2019). In the chronic unpredictable stress model of depression, ketamine is able to rapidly reverse the loss of spines along with the amelioration of depressive behavior, an effect that is blocked by the administration of rapamycin, which is an mTOR antagonist (Li et al., 2011). In addition to ketamine, ECS has been shown to rescue deficits in spine morphology induced by the stress hormone corticosterone in the mouse cortex along with the induction of activity-dependent synthesis of BDNF (Maynard et al., 2018). A similar rescue of stress-induced dendritic changes after ECS treatment has been shown in the hippocampus of rats (Hageman et al., 2008; Kaastrup Müller et al., 2015). However, in a recent study, the increase in prefrontal cortical spines was not required for the rapid effects of ketamine on animal behavior or circuit function (Moda-Sava et al., 2019), thus suggesting that alterations in synaptic strength and circuit function may be responsible for the fast onset of antidepressant effects, whereas spinogenesis contributes to the sustained action. Upcoming clinical studies will be important to elucidate the role of synaptogenesis and underlying mechanisms such as mTOR in rapid and sustained antidepressant effects.
3. Acute Changes in Circuit Function
Current network models of brain function suggest that functionality is generated through the activation neural ensembles consisting of groups of neurons (Yuste, 2015). ENCORE-D proposes that rapid antidepressant effects are set forth by increased cortical excitation, activation of neural ensembles, and the resulting synaptic potentiation during stimulation or pharmacological action. These changes rapidly alter the computational processes in cortical circuits, ultimately modulating patterns of functional connectivity and the emergence of behavioral states. Such reorganization may also contribute to the acute increase in prefrontal functional connectivity reported in depressed patients treated with ketamine (Abdallah et al., 2018b). Indeed, recent studies suggest that the optogenetic activation of groups of neurons in the visual cortex can generate neural ensembles that spontaneously recur in rodents (Carrillo-Reid et al., 2016) and that learning can rapidly engender memory engrams in the human neocortex (Brodt et al., 2018).
As indicated by ENCORE-D, the local and more global rearrangements in neural communication result in the rapid amelioration of depressive thought patterns. Once implicit cognitive patterns are restored, the underlying neural ensembles and networks begin to gather synaptic strength in competition with prevailing depressogenic network activity. In other words, the stimulus-facilitated activation of previously “dormant” circuits may rapidly switch the balance toward more unconstrained patterns of local and global activity. Notably, ketamine has been shown to reverse stress-induced changes in the ensemble activity of prefrontal cortical projection neurons accompanied by antidepressant-like behavioral changes in rodents (Moda-Sava et al., 2019).
Changes in functional connectivity are also associated with the antidepressant effects of ECT and neuromodulatory treatments. For example, a study by Argyelan et al. (2016) used resting-state functional magnetic resonance imaging to measure the fractional amplitude of low-frequency fluctuation and found that depressed patients had higher baseline activity and connectivity in the SCC, with higher values predicting a better response to ECT. This increased activity was reduced after a single ECT treatment and completely normalized at the end of the treatment course. On the other hand, Liu et al. (2015) reported an increase in the local activity and connectivity of the SCC in response to ECT. Moreover, Perrin et al. (2012) investigated the effects of an ECT treatment course on global functional connectivity and found a decrease localized to a limited area within the left dorsolateral prefrontal cortex (DLPFC) in severely depressed patients. A recent study by Leaver et al. (2018) proposed that pretreatment connectivity measures of the DLPFC and SCC predict ECT treatment outcomes. Several studies have also reported functional connectivity changes in response to repetitive TMS (rTMS) in depressed patients (Ge et al., 2017; Kito et al., 2017; Richieri et al., 2017; Du et al., 2018; Iwabuchi et al., 2019). Positive responses to rTMS have been associated with reduced connectivity of the SCC to the DMN (Philip et al., 2018), along with reduced activity of the SCC and connectivity with the DLPFC (Hadas et al., 2019), supporting the hypothesis of altered connectivity in these areas underlying depressive symptoms (Hamilton et al., 2015). Moreover, sleep-deprived healthy participants and depressed patients demonstrate alterations in functional connectivity patterns (Bosch et al., 2013; Chen et al., 2018; Kong et al., 2018). In particular, after sleep deprivation, depressed patients demonstrate reduced connectivity between the ACC and DMN (Bosch et al., 2013).
In synthesis of the discussed studies, ENCORE-D proposes that rapid-acting antidepressants share the property of eliciting pronounced cortical excitation and synaptic potentiation, thus promoting altered encoding of information and changes in network dynamics. The increase in excitatory tone effectively leads to acute alterations in functional connectivity, as reported with ketamine, sleep deprivation, ECT, and rTMS. Areas and networks implicated in treatment responses include, but are not limited to, the ACC and SCC, the DLPFC, and the DMN. However, future studies are likely to offer better perspectives on which specific brain regions and networks are most relevant for antidepressant action. As suggested by ENCORE-D, the switch in connectivity entails the disruption of depressogenic network dominance and the reinstatement of normal patterns of connectivity and activity, evidenced as a decrease in rigid patterns of thought, such as depressive rumination, which is a core symptom of depressive disorders. These changes, however, are short-lived, and as such, they require consolidation to remain sustained.
B. Subacute Consolidation of Synaptic Change
1. Protein Synthesis and Synaptic Consolidation
Synaptic consolidation generally refers to the postencoding process, which stabilizes stimulus-induced activity into a more stable (“storable”) form. This is assumed to occur within a short time from the induction of a memory trace, being finalized in synaptic alterations that confer resistance to amnesic agents. In animals, the administration of protein synthesis inhibitors does not affect short-term improvement (i.e., short-term memory) in task performance while disrupting long-term memory (Davis and Squire, 1984). The activation of pathways important for protein synthesis during memory consolidation suggests an intertwined relationship between memory and translation (Costa-Mattioli and Sonenberg, 2008). Indeed, de novo protein synthesis is required for the consolidation of induced LTP, which is the basis for the formation of lasting memories (Goelet et al., 1986; Cammalleri et al., 2003; Bekinschtein et al., 2007). Moreover, the induction of L-LTP through electrical or pharmacological stimulation is thought to require changes in gene expression along with the local translation of mRNAs in dendrites (Kelleher et al., 2004; Sutton and Schuman, 2006; Costa-Mattioli and Sonenberg, 2008). The blocking of either the transcription steps or the translation inhibits the formation of lasting synaptic change.
The translation of new protein products is suggested to contribute to both the stabilization of activity-induced changes in synaptic strength and the structural plasticity of dendritic spines (Vanderklish and Edelman, 2002; Miniaci et al., 2008; Tanaka et al., 2008). Moreover, a mechanism of synaptic tagging has been proposed to explain how short-term memories transform into long-term memories (Frey and Morris, 1997). In synaptic tagging, a stimulus establishes molecular “tags” in active synapses that can then become further potentiated in L-LTP by associating with newly synthesized plasticity-related proteins. Kinases triggered during E-LTP are thought to contribute to L-LTP, such as MAPK (Thomas and Huganir, 2004), CaMKII (Ma et al., 2015), protein kinase B (Pen et al., 2016), phosphoinositide 3-kinase (Asrar et al., 2009), and protein kinase C (Jalil et al., 2015).
Activation of mTOR and MAPK, which initiate protein synthesis through eukaryotic translation initiation factors 4E and 4E-binding protein 1, are among the multiple pathways involved in translation regulation for plasticity and memory (Hay and Sonenberg, 2004; Amorim et al., 2018). Key components of these pathways are MAPK-interacting serine/threonine-protein kinase 1 and 2. The disruption of this translation mechanism has been shown to result in depression-like behavior in mice (Aguilar-Valles et al., 2018). Importantly, several studies associate the increased phosphorylation TrkB, MAPK, mTOR, and GSK3β with the antidepressant-like effects of ketamine in rodents (Li et al., 2010; Beurel et al., 2011; Lepack et al., 2016; Sun et al., 2016). A seminal study by Li et al. (2010) demonstrated that a subanesthetic dose of ketamine induced phosphorylation changes in mTOR, p70S6K, eukaryotic translation initiation factor 4E-binding protein 1, p44/42-MAPK, and protein kinase B within 30 minutes, whereas synaptic proteins Arc, Synapsin I, PSD95, and GluR1 were upregulated hours later. The blocking of mTOR by rapamycin also blocks ketamine-induced antidepressant-like effects and accelerated fear extinction responses (Li et al., 2010; Girgenti et al., 2017), suggesting that protein synthesis is important for the consolidation of these effects. Similarly, the blocking of MAPK signaling diminishes ketamine’s antidepressant-like effects (Réus et al., 2014). Moreover, studies have suggested that the activation of mTOR and protein synthesis are linked with ketamine’s ability to increase the formation of dendritic spines in the prefrontal cortex (Li et al., 2010; Moda-Sava et al., 2019). This effect likely relies on pharmacologically induced excitation, as electrophysiological experiments have shown increased spine formation to occur rapidly within hours of an LTP-inducing stimulus (Engert and Bonhoeffer, 1999; Maletic-Savatic et al., 1999; Toni et al., 1999; Tang et al., 2002).
Among other targets implicated in synaptic consolidation and ketamine’s effects is GSK3β, a promiscuous kinase with multiple functions and targets (Li and Jope, 2010). Its activity can be blocked by increased phosphorylation at the serine-9 residue, which has been shown to take place in LTP induction (Peineau et al., 2007) and ketamine administration (Beurel et al., 2011). This inactivation following LTP disrupts the induction of long-term depression in synapses for up to an hour, possibly by altering AMPAR trafficking, and it has been suggested to maintain information encoded during LTP for subsequent consolidation (Peineau et al., 2007). The inhibition of GSK3β has been proposed to be necessary for the rapid antidepressant-like effects of ketamine observed in mice (Beurel et al., 2011), with coadministered lithium (an unspecific GSK3 inhibitor) leading to additive effects (Liu et al., 2013). However, a recent clinical trial reported that lithium did not increase or prolong the effects of ketamine in depressed patients (Costi et al., 2019).
Notably, many studies have focused on either immediate or relatively sustained molecular changes induced by ketamine, measured during the acute pharmacological effects of the drug or during the day following treatment. These approaches have generally not accounted for the temporal gradients of molecular changes that may take place during acute pharmacological effects of ketamine and thereafter (t1/2 ∼10–15 minutes in mice) (Maxwell et al., 2006). To this end, we investigated the time-dependent effects of nitrous oxide and flurothyl on these molecular responses, as the exceptional pharmacokinetics of these compounds allows for the differentiation of acute effects from effects emerging immediately after. Nitrous oxide produced no regulation in the phosphorylation of TrkB, GSK3β, and p70S6K (downstream of mTOR) during gas administration (i.e., during NMDAR blockade); instead, changes took place gradually after the gas flow was terminated (Kohtala et al., 2019b). Similarly, these signaling mechanisms remained unaltered immediately during a flurothyl-induced seizure but, again, appeared gradually during the postictal period. These results suggest that pathways involved in synaptic plasticity and protein synthesis are differentially regulated during acute excitatory effects and periods following thereafter. Detailed time- and dose-dependent studies of ketamine on these pathways must, however, be carried out.
2. Homeostatic Emergence of Wake Slow-Wave Activity
SWA, characterized by ∼0.5–4 Hz high amplitude oscillations in the EEG, occurs predominantly during physiologic deep NREM sleep; however, it also occurs under states of sedation and anesthesia and, in some cases, locally during prolonged waking (Nir et al., 2017). Several studies have suggested that rapid-acting antidepressants share common features of regulating SWA during sleep. Notably, the pronounced upregulation of SWA can also occur during quiet waking (Sachdev et al., 2015). Though increased waking SWA has previously been considered to be mainly a pathologic EEG signature, it has also been suggested to serve as a proxy for cortical plasticity (Assenza and Di Lazzaro, 2015). The increases in waking SWA appear to follow similar principles as the upregulation of SWS, with high levels of neuronal activity resulting in increases in subsequent EEG slowing. For example, the arousal-promoting effects of subanesthetic ketamine are countered by increases in the intensity of SWA after the acute pharmacological effects have dissipated and again during sleep in rats (Feinberg and Campbell, 1993, 1995). Similarly, albeit more pronounced and long-lasting, increases in SWA take place following administration with MK-801 (dizocilpine; a potent and durable NMDAR antagonist) (Campbell and Feinberg, 1996a,b), sharing features with the SWS rebound caused by sleep deprivation (Campbell and Feinberg, 1999). Increases in waking SWA have also been noted following the cessation of nitrous oxide administration in humans (Henrie et al., 1961; Williams et al., 1984; Foster and Liley, 2011).
The emergence of rebound SWA is not limited to pharmacological agents that possess antidepressant properties. Postictal increases in SWA after ECT are widely documented, and other convulsive therapies, such as flurothyl, share similar features (Chusid and Pacella, 1952; Fink and Kahn, 1957; Chatrian and Petersen, 1960; Kriss et al., 1978; Silfverskiöld et al., 1987; Sackeim et al., 1996; Perera et al., 2004). Notably, pronounced postictal EEG slowing has been associated with rapid antidepressant responses (Folkerts, 1996) and clinical improvement after ECT (Nobler et al., 1993; Suppes et al., 1996). As already noted, though a single seizure is usually insufficient to produce marked symptomatic relief in depression, some patients have responded already to a single ECT (Rich, 1984; Fligelman et al., 2016). In further support of the idea of the homeostatic emergence of waking SWA in response to neural activity, a clinical study using cortical intermittent theta burst stimulation found increases in waking delta frequency power following the stimulation (Assenza et al., 2015).
We have recently observed an increase in waking SWA following the withdrawal of nitrous oxide, after subanesthetic ketamine, and as a rapid response to flurothyl-induced seizures in mice (Kohtala et al., 2019b; Kohtala et al., unpublished data). This phenomenon is best exemplified by nitrous oxide, which evokes SWA within 5–15 minutes after gas cessation when the drug has been essentially eliminated (exhaled unchanged within minutes) from the body (Kohtala et al., 2019b). The period of nitrous-oxide–induced waking SWA, along with coinciding increases in the phosphorylation of TrkB, GSK3β, and p70S6K, continues long after drug withdrawal (Kohtala et al., 2019b) and overlaps with a probable time period for changes involved in synaptic consolidation. Whether subanesthetic ketamine regulates this molecular cascade similarly during homeostatic SWA remains to be investigated.
The MAPK pathway is a possible candidate involved in the homeostatic upregulation of SWA, as increases in the phosphorylation of p44/42-MAPK occur during the acute excitatory effects of nitrous oxide, subanesthetic ketamine (Kohtala et al., 2019b), and ECS/flurothyl (Hansen et al., 2007; Rosenholm M., et al. unpublished data). Conversely, large decreases in MAPK phosphorylation occur during SWA induced by sedative or anesthetic agents and during the homeostatic regulation of waking SWA (Kohtala et al., 2016, 2019b). Intriguingly, the MAPK pathway has been shown to regulate sleep duration through activity-induced gene expression during wakefulness, with p44/42-MAPK deletion or inhibition significantly increasing the duration of wakefulness in mice (Mikhail et al., 2017). Though the function of this phenomenon remains almost completely unstudied, ENCORE-D suggests that the period dominated by waking SWA in response to rapid-acting antidepressants represents a physiologically meaningful step for the subacute consolidation of activity-induced synaptic changes, involving alterations in both protein synthesis and energy metabolism, resembling deep or local sleep. Notably, a recent study conducted in zebrafish demonstrates the homeostatic emergence of a sleep-like state immediately following acute administration of pharmacological agents that prominently increase neuronal activation (Reichert et al., 2019).
In summary of this section, ENCORE-D proposes that the acutely emerging homeostatic increase in waking SWA after cortical excitation, which coincides with the activation of several pathways involved in synaptic plasticity and protein synthesis, contributes to the subacute consolidation of synaptic change. Moreover, the homeostatic processes that are activated in response to excitation may share common mechanisms with the emergence of waking SWA after cortical stimulation or increased sleep SWA after TMS or sleep deprivation. Future studies that specifically investigate the electrophysiological, molecular, and cellular changes occurring immediately after rapid-acting antidepressant treatments, as well as their functional and clinical significance, will be of great importance to test this hypothesis.
C. Renormalization and Consolidation of Synaptic Strength during Sleep
1. Slow Waves and Rapid Antidepressant Responses
Sleep plays an extremely important role in learning and memory consolidation (Abel et al., 2013). During different stages of sleep, rich patterns of neural activity take turns in periods of REM and NREM sleep throughout the cortex. Neurons previously activated during the day are recruited by patterns of reactivation, which are thought to contribute to the consolidation of previously encoded information. Increasing knowledge of the mechanisms of sleep suggests that complex interactions between different brain regions, emerging neural activity, and the phases of sleep act in conjunction to modulate learning and memory. For understanding the basic idea of ENCORE-D, we now focus on examining mechanisms associated with SWS from the perspective of SHY (Tononi and Cirelli, 2003). However, it is important to recognize that several different mechanisms throughout the complex patterns of sleep periods may be ultimately involved.
According to SHY, synaptic potentiation during active waking is connected to the homeostatic increase in SWA during sleep. In other words, the higher the level of cortical synaptic potentiation, the higher the intensity of slow waves during subsequent sleep. This connection between preceding neural activity and the emergence of subsequent sleep SWA has been demonstrated in numerous studies. For example, SWS is increased after sleep deprivation (i.e., recovery sleep) (Cajochen et al., 1999; Huber et al., 2000) and after cortical TMS (Huber et al., 2007). Local increases in SWA emerge after sensory or motor stimuli in experience-related brain areas during NREM sleep (Kattler et al., 1994; Huber et al., 2004; Mascetti et al., 2013), whereas arm immobilization reduces SWA in the sensorimotor area (Huber et al., 2006) and dark rearing in the visual cortex (Miyamoto et al., 2003). Moreover, several studies have investigated the effects of sleep manipulations for the consolidation of nondeclarative memories (Stickgold, 2005; Miyamoto et al., 2017). For example, boosting sleep spindles enhances finger-sequence tapping task performance (Lustenberger et al., 2016), and the perturbation of sleep slow waves in the motor cortex decreases motor performance (Fattinger et al., 2017). Notably, SWS also correlates with cerebral protein synthesis in monkeys (Nakanishi et al., 1997) and rodents (Ramm and Smith, 1990) and has been suggested to contribute to the formation of dendritic spines after learning (Yang et al., 2014).
During stages of deep sleep, cycles of SWS and synaptic renormalization take place (Tononi and Cirelli, 2003). Based on the previous activity of the synapses and the neural networks they are part of, synapses may lose potentiation, remain unchanged, or, in some cases, gain strength (Fig. 1). This renormalization of synaptic strength during sleep contributes to adjusting the baseline of neural activity for the subsequent waking. The ENCORE-D hypothesis proposes that the sleep following ketamine and other rapid-acting antidepressant treatments is important for the sustained consolidation of synaptic changes and the full realization of the antidepressant effects through the renormalization of synaptic weights. Moreover, ENCORE-D proposes that in depression, the balance between waking neural activity and synaptic renormalization in sleep is compromised. Depressed patients spend significant amounts of time in rumination, whereas complex cognitive tasks, or even routine daily activities, may seem insurmountable. This is perhaps also evidenced in decreased patterns of motor activity during the day, whereas increases in activity take place during the night (Burton et al., 2013). Rapid-acting antidepressants administered during waking cause rapid changes in synaptic strength and circuit function, which are subacutely consolidated in the following hours. During subsequent sleep, a period of further consolidation, along with the reactivation of neural activity and the renormalization of synaptic strength, takes place, thereby allowing homeostatic processes to readjust network function in a sustained manner. Under these circumstances, previously hypoactive but now pharmacologically potentiated neural networks can conserve some of their gained strength throughout SWS and the accompanying synaptic renormalization.
In support of ENCORE-D, clinical evidence suggests that the most robust antidepressant responses to subanesthetic ketamine become evident during the following day (i.e., after one night’s sleep) (Berman et al., 2000; Zarate et al., 2006a; Lapidus et al., 2014; Phillips et al., 2019). Studies have also demonstrated the normalization of global functional connectivity patterns when measured 24 hours after treatment (Abdallah et al., 2017a,b) and decreases in suicidal cognition within the same time frame (Price et al., 2014, 2009; Wilkinson et al., 2018). Likewise, antisuicidal responses in depressed patients have been associated with reductions in wakefulness the night following ketamine treatments (Vande Voort et al., 2017). Moreover, a recent double-blind placebo-controlled crossover functional magnetic resonance imaging imaging study demonstrated the normalization of connectivity between the insula and DMN in depressed patients when compared with healthy controls 2 days after ketamine administration (Evans et al., 2018). The authors have highlighted the role of the insula in emotional information processing and in modulating the switch between the CEN and DMN, which may be involved in the improvement of depressive symptoms.
The most direct evidence supporting the importance of SWA in rapid-antidepressant action originates from a clinical study that demonstrated increases in sleep SWA after subanesthetic ketamine treatments and further showed that this increase correlated with antidepressant efficacy (Duncan et al., 2013a). Moreover, a low baseline delta ratio has been demonstrated to predict a better mood response to ketamine the day following treatment (Duncan et al., 2013b). These studies suggest that ketamine’s sustained effects on mood may be related to subsequent SWS through the accrual of synaptic potentiation during wakefulness and by the modulation of synaptic renormalization during sleep. In addition, ECT-induced increases in SWS (as well as increased REM sleep duration and a decrease in REM density) have been reported to occur in remitting patients but not in nonremitters (Göder et al., 2016), suggesting that the regulation of brain activity during sleep is implicated in antidepressant responses to ECT.
Several mechanisms have been proposed to be potentially involved in the processes that lead to the global yet selective renormalization of synaptic strength during sleep as proposed by SHY (reviewed by Tononi and Cirelli (2019). Different rules could apply in the renormalization of synaptic strength, several of which have been tested in computer simulations (Hashmi et al., 2013; Nere et al., 2013). One of these is a “protection from depression” mechanism, which suggests that when a neuron fires strongly during sleep, because of the prominent coactivation of its different inputs, its synapses maintain their strength throughout sleep. The end result is a relative increase of strength in these protected synapses in relation to other synapses that are more likely to be downscaled. This idea is supported by a recent in vivo study, which demonstrated a similar effect during urethane anesthesia that mimics the up and down states of neural activity during NREM sleep (González-Rueda et al., 2018). Another recent study has lent support to the function of the up states of SWA in synaptic down-selection by studying neuroprosthetic learning in an experiment in which the activity of neurons in the motor cortex were paired with the control of a feeding tube (Gulati et al., 2017). The authors observed that after sleep, a slight increase occurred in the peak firing of the direct units in causal control of the task activity, whereas the firing of most of the indirect units were markedly reduced. The lack of firing during sleep was found to predict synaptic down-selection. Furthermore, when closed-loop optogenetic inhibition was used to reduce firing during the on states of slow oscillations, sleep-induced learning improvements were prevented, and the decrease in indirect unit activity did not occur. In addition to slow-wave oscillations, hippocampal sharp wave-ripples may not only promote synaptic weakening but also allow for previously activated hippocampal place cells to maintain their firing rate throughout sleep (Norimoto et al., 2018).
Apart from patterns of neural activity, several plasticity-related proteins may be important for the renormalization of synaptic strength during sleep. Among potential candidates are Homer1a and Arc, which are known to increase during extended waking and excitatory neuronal activity (Cirelli and Tononi, 2000; Diering et al., 2017). Though the overall levels of Homer1a in the cortex are higher during wake than in sleep, the amount of Homer1a in the PSDs increases in sleep and acts to replace longer isoforms of Homer (Diering et al., 2017). This effectively inactivates the signaling complex with metabotropic glutamate receptors, facilitates the endocytosis of AMPARs from the cell membrane, and results in the downscaling of synaptic strength. Indeed, the dysfunctional regulation of Homer1a may be involved in the development of network imbalances, as suggested by ENCORE-D. For example, a genome-wide association study, combined with neuroimaging, found a single-nucleotide polymorphism of Homer1 to exert significant influence over prefrontal cortical activity during cognitive and motivational processes (Rietschel et al., 2010). Furthermore, in the context of synaptic tagging, Arc has been proposed to act as one of the tags for synaptic depression by promoting the endocytosis of AMPARs (Chowdhury et al., 2006). Arc has also been suggested to accumulate, particularly in synapses that have not been prominently activated, resulting in decreased synaptic strength (Okuno et al., 2012). At this time, though the role of altered TrkB, GSK3β, p70S6K, and MAPK signaling during SWS remains unknown, it is plausible they are involved in processes of consolidation or renormalization throughout sleep.
2. Sustained Antidepressant Effects
A significant yet scarcely studied issue of ketamine treatments is the gradual relapse of depressive symptoms, which typically already occur a few days after the treatment. ENCORE-D proposes that if no further excitatory stimulus or other therapy is applied following the initial treatment, then susceptible neuronal networks may begin to regravitate toward an unfavorable, depressogenic state. This occurs during several subsequent wake-sleep cycles, effectively resulting in the loss of a treatment-induced increase in the relative potentiation of nondepressogenic networks. Depressive thought patterns again emerge along with a more “rigid” functional connectivity. Indeed, Evans et al. (2018) reported that the connectivity changes induced by ketamine in depressed patients were reversed after 10 days, in line with the duration of ketamine’s antidepressant effects and in alignment with a viable timeframe for multiple cycles of renormalization to occur. Moreover, repeated ketamine infusions have cumulative effects in treating depression, and patients that relapse between treatments still respond to new infusions (Phillips et al., 2019). This suggests that such changes can be reestablished with further treatments.
Following these same principles, ENCORE-D proposes that therapeutic sleep deprivation also increases cortical excitation through the accrual of synaptic potentiation or other mechanisms of altered excitability. Similar to the rapid effects of ketamine, sleep deprivation results in a decrease in depressive symptoms in approximately half of patients (Wu and Bunney, 1990). This decrease in symptoms builds up throughout the sleepless night and is maximal the following day. However, contrary to ketamine, the effects of sleep deprivation are typically highly transient, with a large majority of patients relapsing after a night of sleep or even after taking a short nap. ENCORE-D proposes that this difference in sustainability is because of differences in both the consolidation of excitation-induced synaptic change and the renormalization during treatment-evoked SWS. Although both treatments lead to changes in neural activity and patterns of connectivity (i.e., rapid antidepressant effects), the changes induced by sleep deprivation may not be sufficiently consolidated or protected from subsequent renormalization but are instead lost in time, like tears in rain. The mechanistic basis for such a difference remains unknown; however, it is well acknowledged that sleep deprivation results in impairments of learning and memory while decreasing long-lasting forms of synaptic plasticity (for reviews, see Havekes et al., 2012; Abel et al., 2013). These detrimental changes may occur through the reduced expression of genes that are important in regulating translation for memory encoding and consolidation. Moreover, both ketamine and sleep deprivation increase glutamate release, whereas ketamine may also block postsynaptic and extrasynaptic NMDARs, which contribute differentially to processes such as the activation of the mTOR pathway, the activity of transcription factors, and BDNF synthesis (Sutton et al., 2007; Autry et al., 2011; Nosyreva et al., 2013; Miller et al., 2016). Here, the metabolites of ketamine, such as HNKs, may also play an important role.
Unlike subanesthetic ketamine, ECT is commonly associated with cognitive dysfunction, including impaired learning and memory (Nuninga et al., 2018). The major differences in the extent, duration, and selectivity of the excitatory activity produced by rapid antidepressant treatments may ultimately determine their functional consequences and the onset of the antidepressant effects. Though ECT-induced seizures are highly pronounced and global forms of excitatory activity, ketamine drives cortical excitation in a subtler way, essentially facilitating more physiologic levels of neural activity. Moreover, excitation may involve particular (micro)circuits, depending on the properties of the treatment or the pharmacology of the drug. Different drugs and treatments are thus likely to have varying effects on a network level. Notably, ECT, once the traveling current and neural activation generalizes into a global seizure, remains in no way selective regarding which neuronal populations are activated; it thus effectively disregards the physiologic patterns of neural communication. The processes of encoding, consolidating, and subsequently renormalizing synaptic strength during SWA may consequently also be highly disorganized, which can have not only therapeutic but also detrimental effects on memory and cognitive functions.
V. Discussion of Encoding, Consolidation, and Renormalization in Depression and Its Implications for Antidepressant Treatments
We and others have previously proposed models for integrating mechanisms of synaptic plasticity into antidepressant actions (Castrén, 2005, 2013; Castrén and Rantamäki, 2010; Leuchter et al., 2015; Harmer et al., 2017). According to the network hypothesis of antidepressant action, long-term treatment with conventional antidepressants such as fluoxetine produce a heightened state of cortical plasticity that allows for rewiring of synaptic connections in the adult brain. Importantly, such rewiring is not considered to be orchestrated by the drugs themselves but by the network activity in collaboration with environmental stimuli. Rapid-acting antidepressants alleviate depression within hours, indicating that their mode of action is fundamentally different from conventional antidepressants, although the treatments trigger several shared molecular pathways (Rantamäki et al., 2007, 2011; Rantamäki, 2019). Moreover, in line with the network hypothesis, the effects of rapid-acting antidepressants can hardly be explained by basic pharmacological principles. For example, the antidepressant effects of ketamine become most evident when its psychotropic actions (Berman et al., 2000) and acute pharmacological effects on NMDARs fade, and the therapeutic effects may last for days or weeks. Such dynamic and long-lasting effects of a pharmacologically short-acting drug can be best explained by adaptive functional and structural alterations in neural circuitries, involving physiologic mechanisms of sleep and plasticity.
Building on aspects of the network hypothesis of antidepressant action and SHY (Tononi and Cirelli, 2003), ENCORE-D proposes that to understand rapid antidepressant effects, we should pay attention to the physiologic homeostatic adaptations triggered within the brain in response to pharmacological (i.e., NMDAR blockade) or physiologic challenges (e.g., convulsions, sleep deprivation) and its consecutive release (see Workman et al., 2018) (Fig. 4). In this framework, fundamental mechanisms of synaptic plasticity and sleep are at the center of attention. In short, ENCORE-D suggests that altered synaptic encoding takes place during treatment-induced neural excitation and network activity, which is subsequently consolidated during periods that overlap with the emergence of waking SWA in the following hours. Finally, the synaptic and network alterations set forth by the treatment reach a more stable form during different stages of sleep, when synaptic renormalization, and likely many other processes related to consolidation and reactivation of neural activity patterns, takes place. The hypothesis proposes that during these phases, rapid-acting antidepressants trigger important yet temporally distinct mechanisms for the emergence of sustained alterations in neural networks.

Potential implications of the ENCORE-D hypothesis for the treatment of depression. Rapid and sustained antidepressant effects critically rely on homeostatic adaptations (e.g., slow EEG activity during waking and sleep) triggered within the brain in response to the intervention in question (e.g., ketamine). The ability of the intervention to evoke such adaptations depends on several baseline variables (cortical excitability and sleep, genetics, medication) and specific aspects related to the dosing of the treatment (e.g., administration route, circadian time of administration, pharmacokinetics and thus duration of acute pharmacological effects, and external factors). Subjective experiences during the treatment, along with supportive psychotherapy and mechanisms of sleep, may influence the sustainability of the clinical outcome.
As posited by ENCORE-D, the different temporal phases constitute mechanisms that work in conjunction to achieve alterations in network function. Indeed, agents that facilitate SWA more directly and without preceding cortical excitation do not bring about therapeutic effects despite activating some of the molecular cascades implicated in synaptic plasticity and antidepressant responses, such as TrkB signaling and the inhibition of GSK3β (Kohtala et al., 2019b). Though these molecular events are important for the mechanisms underlying antidepressant effects, relevant neural activity is required to harness these molecular machineries into producing meaningful activity-dependent alterations in synaptic structure and function. As demonstrated by several animal experiments, disrupting either glutamatergic neurotransmission (Zanos et al., 2016) or these molecular mechanisms (Li et al., 2010; Beurel et al., 2011; Pochwat et al., 2017) results in diminished antidepressant-like responses because without activity, the machinery does not have a purpose, and without the machinery, activity cannot elicit sustainable change. Further studies are required to elucidate how the molecular and functional alterations directly activated by sedative-anesthetic drugs differ from those that occur during the homeostatic emergence of waking SWA or subsequent SWS in response to cortical excitation. Thus, ENCORE-D predicts that efforts to directly and specifically target individual components of complex molecular pathways are unlikely to lead to antidepressant outcomes in patients. This hypothesis is, however, built on studies of treatments possessing rapid antidepressant effects currently in the clinical domain, and it aims to explain their effects through a common neurobiological framework. This does not rule out that rapid and sustained antidepressant effects might also be achieved via mechanisms not aligning with this hypothesis, such as by using novel treatments not yet proven in the clinical setting. With the current enthusiasm about novel antidepressant developments, the principles of ENCORE-D will need to be re-examined and expanded as new findings emerge.
It is also important to recognize that merely increasing SWS may not be beneficial for treating depression. On the contrary, some preliminary studies have found that the reduction of SWS is associated with modest improvements in depressed mood (Landsness et al., 2011) or negative affect (Cheng et al., 2015). From the perspective of ENCORE-D, the dysfunctional renormalization of synaptic strength (i.e., reduced downscaling in depressogenic or facilitated downscaling in nondepressogenic areas) may constitute one characteristic that maintains depressive information processing. One possibility is that reducing SWS in depressed patients could allow nondepressogenic areas to also maintain higher levels of synaptic potentiation throughout sleep, resulting in a small restoration of network balance. However, for more robust antidepressant responses such as those produced by ketamine and perhaps ECT, likely determined by the path of the initial electrical current (Leaver et al., 2018), synaptic activation and potentiation in specific circuits, along with putative protection from renormalization during SWS, may be required. Moreover, focusing merely on alterations in the amount of SWS is misleading because changes in its temporal expression may be equally important. For example, several studies have suggested that the delta sleep ratio may be indicative of clinical outcomes in the treatment of depression (Kupfer et al., 1990; Nissen et al., 2001; Duncan et al., 2013b; Lotrich and Germain, 2015), with lower values associated with a higher risk of relapse (Kupfer et al., 1990). A recent study demonstrated that a lower delta sleep ratio predicted mood disturbance in depressed individuals who were subjected to a 3-hour sleep-delay challenge, highlighting the distinction between the accumulation of SWS and its dissipation in regulating emotional functioning (Goldschmied et al., 2019). Importantly, it must be emphasized that the role of SWS, or delta sleep ratio, in either rapid or sustained antidepressant effects of ketamine remains to be thoroughly investigated. To further unravel these mechanisms, experiments in which the process of SWA/SWS accumulation, along with its emergence during both wake and subsequent sleep are manipulated, are essential.
One intriguing aspect related to the study of novel treatments of depression is ketamine’s ability to produce rapid antidepressant responses, whereas several other drugs targeting glutamatergic neurotransmission have failed in patient studies (Ibrahim et al., 2012; Quiroz et al., 2016; Sanacora et al., 2017; Henter et al., 2018). It is tempting to speculate that the pharmacokinetics (Fig. 4) and receptor-binding properties of ketamine allow durable yet not too extensive cortical excitation to take place, especially when compared with less or more potent or long-lasting NMDAR antagonists. For example, while both ketamine and MK-801 (dizocilpine) are noncompetitive inhibitors of NMDAR ion channels, the channel-trapping capability is lower for ketamine (Zanos et al., 2018a). Moreover, memantine, which is a low affinity, voltage-dependent, uncompetitive NMDAR antagonist (Gilling et al., 2009), has not demonstrated robust antidepressant effects in clinical trials (Zarate et al., 2006b; Kishi et al., 2017). Though ketamine’s acute effects last for a few hours (Clements and Nimmo, 1981; Mathew and Zarate, 2016), memantine has a half-life of several days in humans (Matsunaga et al., 2018) as well as differential effects on NMDA receptor desensitization (Glasgow et al., 2017). Notably, one of the proposed mechanisms of action for the use of memantine in the treatment of neurodegenerative diseases is its neuroprotective properties against glutamatergic excitotoxicity (Parsons et al., 1999), whereas the antidepressant effects of subanesthetic ketamine have been directly linked to an acute increase in glutamate bursting (Moghaddam et al., 1997). These properties may explain why ketamine is unique in provoking relatively rapid homeostatic adaptations, and they encourage the search for novel, rapid-acting antidepressants among short-acting agents that influence glutamate bursting.
Several other features may also explain the unique effects of ketamine. Notably, a recent animal study suggests that the gradual metabolism of ketamine and the appearance of specific metabolites, preferentially 2R,6R-HNK, account for the therapeutic lag, and they mediate antidepressant effects through a mechanism not involving NMDAR blocking (Zanos et al., 2016). It is plausible that these ketamine metabolites may directly increase neural excitation and engage mechanisms proposed by ENCORE-D, as evidenced by increased gamma oscillations in some preclinical studies (Zanos et al., 2016, 2019). However, it currently remains unknown whether these metabolites have clinical efficacy by themselves or if they contribute to augmenting or sustaining ketamine’s effects. These questions will hopefully be answered by upcoming clinical trials and animal experiments.
An interesting line of research employing motor-activity monitoring in depressed patients has associated the rapid antidepressant effects of ketamine with the regulation of circadian timekeeping (Duncan et al., 2017, 2018). These studies suggest that parameters of circadian timekeeping may be important in uncovering the mechanisms of both rapid and sustained responses to ketamine as well as the identification of patients who are most likely to benefit from the treatment. In a model proposed by Duncan et al. (2017), depressed patients exhibit diminished interactions of sleep homeostatic and circadian mechanisms, leading to the promotion of a depressed mood. Treatment with ketamine increases plasticity, SWS, and sleep quality while ameliorating depressive symptoms. At the same time, ketamine modulates circadian timing and output. This results in both weakened interaction of homeostatic and circadian mechanisms and an acute reduction of the circadian mood component. The authors suggest that in sustained responses, the interaction between homeostatic and circadian is strengthened through reciprocal activation, which facilitates a more functionally relevant interaction and greater temporal organization of the transcriptome. Conversely, in relapse, the interaction between homeostatic and circadian returns to its weakened state. As such, this theoretical and experimental approach is highly important for recognizing and integrating motor and circadian aspects into the study of rapid-acting antidepressants, some of which may turn out to be highly relevant in the context of ENCORE-D as well.
Apart from obvious pharmacodynamic and kinetic differences, varying doses, dosing paradigms, and routes of administration may be major contributors to the effects of different rapid antidepressant drugs (Fig. 4). For ketamine, subanesthetic doses produce increases in glutamatergic neurotransmission, whereas higher anesthetic doses may suppress neural activity (Moghaddam et al., 1997; Chowdhury et al., 2017; Abdallah et al., 2018a). Subanesthetic and anesthetic doses also differ in the molecular pathways that are acutely activated (Kohtala et al., 2019a). Nonetheless, it remains to be investigated whether higher anesthetic doses may also reach excitatory concentrations once the majority of the drug is metabolized. Such postanesthesia reactions are well known by anesthesiologists and are often referred to as emergence phenomena, which may manifest as, for example, agitation, confusion, and hallucinations (Marland et al., 2013). Importantly, this phenomenon is by no means limited to ketamine; it can occur upon awakening from general anesthesia conducted with various drugs. Altogether, general anesthetics can facilitate paradoxical excitation of the cortex when the drug concentrations are low (Voss et al., 2008), an effect that may hold unprecedented potential to trigger rapid antidepressant effects.
It is likely that no optimal dosage or intensity of treatment exists that would be beneficial for every patient. In this regard, volatile and gaseous anesthetics hold great potential, either as a sole agent or in combination with another short-acting NMDAR antagonist, to titrate the dosing almost in real time to elicit sufficient excitation. It is important to keep in mind, however, that these effects are not restricted to NMDAR antagonists, as the manipulation of several other receptors and physiologic mechanisms may efficiently produce similar neurobiological consequences. Excitatory drugs and treatments unarguably do not contain any inherent information that would effectively target the underlying pathology of depression. Instead, ENCORE-D proposes that they exert their beneficial effects by unbalancing the current state of neural and network homeostasis, which allows innate neurobiological mechanisms to adequately adjust. In this context, neurophysiological measures such as the emergence of increased rebound SWA and subsequent SWS may turn out to be particularly useful in determining optimal dosing and treatment paradigms for bringing about a remedying state in a personalized manner (Fig. 4).
New avenues of research may also arise from understanding rapid-acting antidepressants from the perspective of memory functions. For example, a stimulus may have no immediate effect on synaptic strength but can modulate the subsequent expression of plasticity, a phenomenon known as metaplasticity (Young and Nguyen, 2005). Indeed, investigations into spacing LTP induction in an intermittent manner have suggested that dividing training over time produces stronger and longer-lasting memories than a single bout of intensive learning (Lynch et al., 2013; Wang et al., 2014). Whether such physiologic mechanisms hold relevance for developing more effective rapid-acting antidepressant treatments (i.e., intermittent administration paradigms) remains to be investigated. In this context, treatments such as nitrous oxide, short-acting ketamine analogs (Dimitrov et al., 2019), and theta burst stimulation may be particularly useful for several consecutive intermittent periods of administration in a single session (Fig. 4). For these types of novel treatment paradigms, conventional drug-development principles that aim for stable, steady-state concentrations and long half-lives may need to be discarded.
One neglected aspect in the research and treatment of brain disorders is related to the variability in the state of the brain. The mechanisms implicated in ENCORE-D provide the basis for the idea that the baseline state of the underlying neural networks may influence rapid antidepressant outcomes (Fig. 4). For example, ketamine reportedly has distinct electrophysiological, as measured by magnetoencephalography, and behavioral effects when administered to depressed or healthy subjects (Nugent et al., 2019a). Though depressed patients exhibit rapid improvements in their symptoms, healthy controls may even display increases in depressive symptoms for up to a day after ketamine administration. A possible explanation offered by ENCORE-D is that unless dysfunctional connectivity is present, ketamine may negatively influence network homeostasis. In support of the idea that baseline conditions affect the outcome of ketamine treatment, mice subjected to the chronic social defeat model of depression have been found to elicit glutamate functional hyperconnectivity and altered responses to ketamine when compared with naïve mice (McGirr et al., 2017).
Following the idea of variability in brain states, emphasis should also be placed on studies that address whether the timing of treatment contributes to its clinical effects (Ruben et al., 2019) (Fig. 4). Treatments given during the early morning could produce different outcomes than those in the evening, as cortical excitability is regulated by circadian rhythmicity (Ly et al., 2016) as well as wake and sleep (Kuhn et al., 2016). To the best of our knowledge, the impact of timing has not been addressed with ketamine, or other rapid-acting antidepressants, although such experiments have been planned (Zhuo et al., 2019). These principles may also prove to be crucial for the refinement of the basic research of psychiatric disorders in general. Laboratory rodents, such as mice and rats, are widely used in biomedical research to understand the pathologic processes underlying depression and to study and develop antidepressants. Though many important discoveries have been made, the translation of preclinical observations into novel treatments is scarce. ENCORE-D urges us to consider that a part of this translational gap is explained by our ignorance of the most fundamental aspects of animal physiology: the circadian rhythm and sleep. Most rodent species used for biomedical research are nocturnal. Although the contrast between active, wakeful, and vigilant states during light and dark periods in rodents is not as stark as in humans, (nocturnal) mice and rats are clearly more active during the dark period and sleep in bouts predominantly during the day. Yet, and solely because of the convenience of the experimenter, a vast majority of rodent studies are conducted during the inactive period. A recent observation of prominent circadian variation in ketamine metabolism (Martinez-Lozano Sinues et al., 2017) suggests that the timing of administration may significantly contribute to the neurobiological effects set forth by ketamine and its metabolites.
The mechanisms proposed by ENCORE-D may also be important for the antidepressant actions of psychedelic drugs, which have recently gained renewed interest in psychiatry. Psilocybin, a prodrug of the 5-HT2A agonist psilocin, has shown promise in a range of psychiatric conditions, including treatment-resistant depression. Studies suggest that, unlike ketamine, these drugs may have positive effects lasting for months (Carhart-Harris et al., 2016), although proper placebo-controlled randomized clinical trials are still lacking. Interestingly, the expression of 5-HT2A receptors is especially enriched in the high-level association regions, such as those within the DMN (Beliveau et al., 2017). Compared with ketamine, the effects of psilocin may thus be more focused to this hub network, which is dysregulated in depressive states (Carhart-Harris and Friston, 2019). Molecular mechanisms suggested to underlie the effects of other rapid-acting antidepressants, such as TrkB and mTOR signaling, are also associated with psychedelics and their ability to promote structural and functional plasticity (Ly et al., 2018). An important distinction between the therapeutic use of ketamine and classic psychedelics comes from the therapeutic context (Fig. 4). Psychedelic therapy uses psychedelic drugs to promote peak inner experiences during a session together with a therapist. It may turn out that the extraordinarily long-lasting effects of psychedelic therapy arise when the person and his or her experiences are the focus of the treatment. Salient stimuli, for example, a powerful psychologic experience, combined with a pharmacologically facilitated state may be expected to produce the most persistent changes in memory and cognitive patterns. In this context, examining the principles of ENCORE-D together with previously proposed theoretical perspectives, such as the entropic brain hypothesis (Carhart-Harris et al., 2014) and the relaxed beliefs under psychedelics framework (Carhart-Harris and Friston, 2019), may turn out to be particularly fruitful. These emerging perspectives also encourage further research into both ketamine-assisted psychotherapy (Dore et al., 2019) and the impact of subjective experience on the therapeutic outcome.
ENCORE-D also provides a plausible neurobiological basis for the rapid and sustained antidepressant effects. For example, sleep deprivation is a relatively efficient rapid-acting antidepressant (efficient increase in activity/encoding); however, its therapeutic effects are seldom sustained (dysfunctional consolidation and/or renormalization). On the other hand, ketamine may sufficiently allow both phases to take place. According to one recent hypothesis (Wolf et al., 2016), sleep deprivation extends the period when cortical neurons gather synaptic strength or excitability. This allows synapses to reach an optimal zone for LTP inducibility (Normann et al., 2007), which compensates for the attenuated associative synaptic plasticity in depression and leads to the remediation of depressive symptoms. This hypothesis does not exclude the principles of ENCORE-D, which proposes that the increase in cortical excitability induced by sleep deprivation (and other putative rapid-acting treatments) is important for altering patterns of brain activity and subsequent synaptic changes. However, though alterations in LTP inducibility may exist in some brain areas of depressed patients, ENCORE-D suggests that the observed decrease in cortical excitability is mainly representative of the pathophysiological process itself, meaning that neural activity is more concentrated on select depressogenic networks wherein synaptic plasticity may even be facilitated (Nissen et al., 2010). This skewed balance leads other networks to exhibit a relative decrease in activity over the course of disease progression, which continues to worsen in a vicious cycle as depressive symptoms increase.
Finally, the advantage of ENCORE-D is that it does not depend on any synaptic pathology, molecular pathway, or deficit that affects global synaptic plasticity; instead, it relies on mechanisms of activity-dependent network tuning in several brain networks associated with major depression. Though many aspects of the hypothesis remain conceptual and speculative, and, at this stage, require further testing, this perspective provides a plausible framework for explaining how depression may arise from early adverse psychologic experiences and/or later cognitive behaviors over time. Moreover, ENCORE-D provides a mechanistic framework for a variety of pharmacological and physiologic treatments capable of eliciting rapid antidepressant effects, and it suggests that understanding the shared mechanisms between these treatments may provide fundamental insights into both rapid antidepressant action and physiologic brain function.
Acknowledgments
We thank Heikki Tanila, Okko Alitalo, and Chiara Cirelli for discussions and advice. Okko Alitalo is also thanked for preparing the final figures.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Rantamäki, Kohtala.
Footnotes
This work has been supported by the Academy of Finland (T.R.) [Grants 276333, 305195, 312664], the Business Finland (T.R.), the Brain and Mind Doctoral Program (S.K.), the Orion Research Foundation (S.K.), and the Emil Aaltonen foundation (S.K.). T.R. and S.K. are listed as coinventors on a patent application, wherein new tools enabling the development of rapid-acting antidepressants and the efficacy monitors thereof are disclosed based on the basic principles of ENCORE-D. T.R. and S.K. have assigned their patent rights to the University of Helsinki but will share a percentage of any royalties that may be received by the University of Helsinki.
Abbreviations
- AMPAR
- α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor
- Arc
- activity-regulated cytoskeleton-associated protein
- BDNF
- brain-derived neurotrophic factor
- CaMKII
- Ca2+/calmodulin-dependent protein kinase II
- CEN
- central executive network
- DLPFC
- dorsolateral prefrontal cortex
- DMN
- default mode network
- ECS
- electroconvulsive shock
- ECT
- electroconvulsive therapy
- EEG
- electroencephalogram
- E-LTP
- early phase LTP
- ENCORE-D
- encoding, consolidation, and renormalization in depression
- EPSP
- excitatory postsynaptic potential
- GluR1/2
- glutamate AMPA receptor subunits 1 and 2
- GSK3β
- glycogen synthase kinase 3β
- HNK
- hydroxynorketamine
- IEG
- immediate early gene
- L-LTP
- late phase LTP
- LTP
- long-term potentiation
- MAPK
- mitogen-activated protein kinase
- mTOR
- mammalian target of rapamycin
- NMDAR
- N-methyl-D-aspartate receptor
- NREM
- non–rapid eye movement
- PSD
- postsynaptic density
- REM
- rapid eye movement
- rTMS
- repetitive TMS
- SCC
- subgenual cingulate cortex
- SHY
- synaptic homeostasis hypothesis
- SN
- salience network
- SWA
- slow-wave activity
- SWS
- slow-wave sleep
- TMS
- transcranial magnetic stimulation
- TrkB
- tropomyosin receptor kinase B
- Copyright © 2020 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





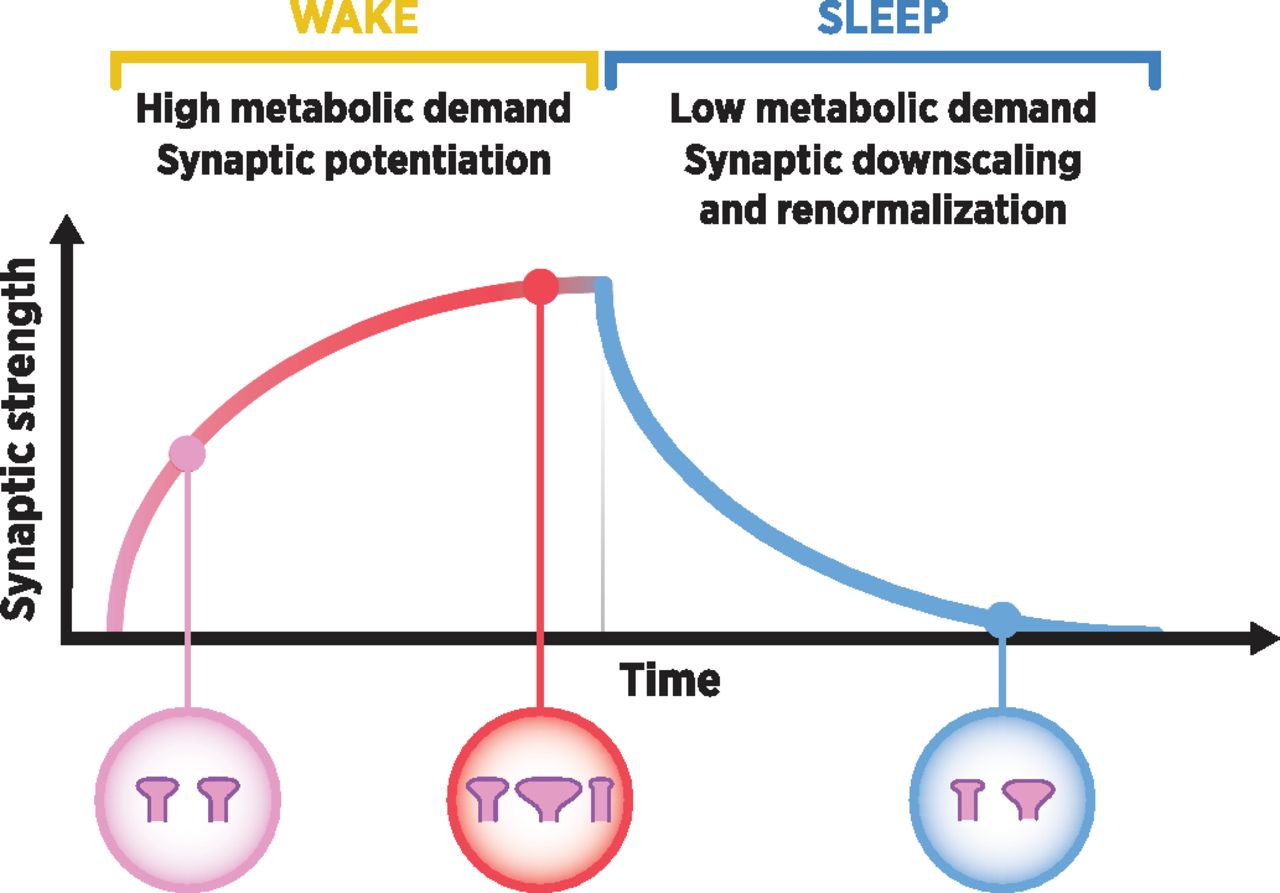{kind=link}
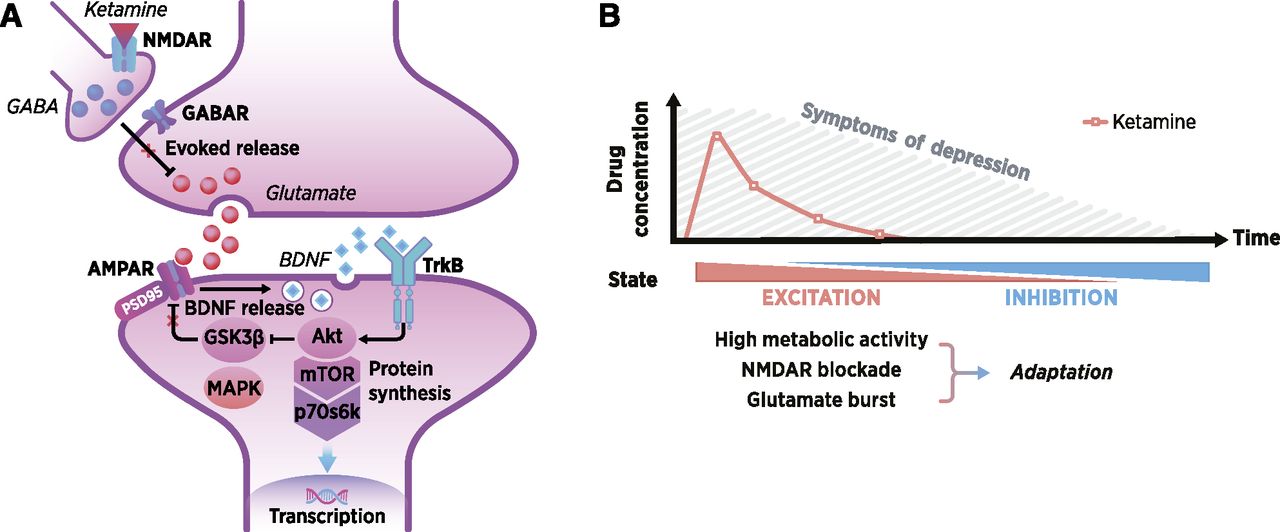{kind=link}
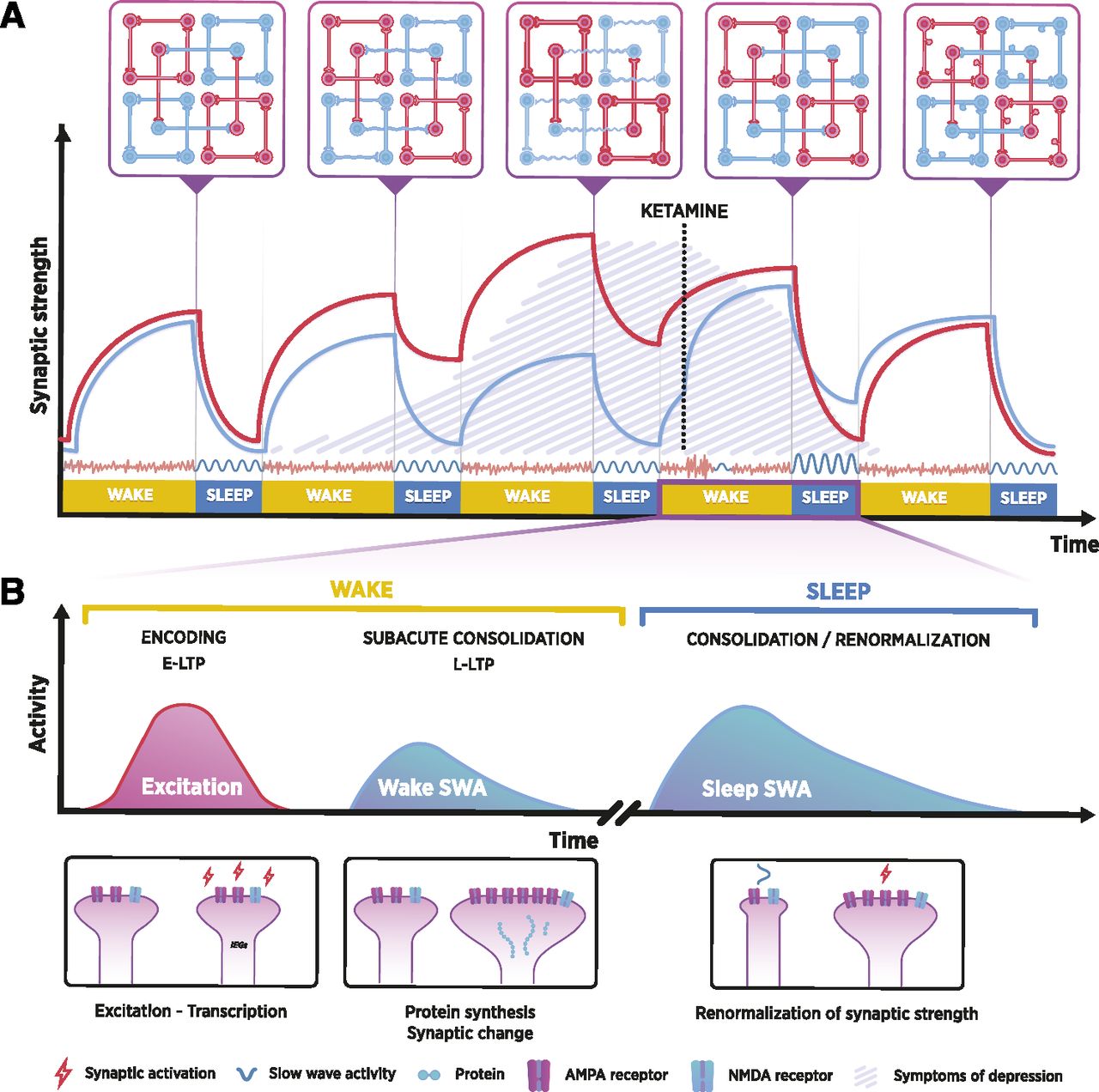{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. Synaptic Homeostasis Hypothesis as the Foundation for Encoding, Consolidation, and Renormalization in Depression
- III. The Neurobiological Premise of Major Depression
- IV. Examining Rapid Antidepressant Effects through Encoding, Consolidation, and Renormalization in Depression
- V. Discussion of Encoding, Consolidation, and Renormalization in Depression and Its Implications for Antidepressant Treatments
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters