


Abstract
The calcium-sensing receptor (CaSR) is a class C G protein–coupled receptor that responds to multiple endogenous agonists and allosteric modulators, including divalent and trivalent cations, L-amino acids, γ-glutamyl peptides, polyamines, polycationic peptides, and protons. The CaSR plays a critical role in extracellular calcium (Ca2+o) homeostasis, as demonstrated by the many naturally occurring mutations in the CaSR or its signaling partners that cause Ca2+o homeostasis disorders. However, CaSR tissue expression in mammals is broad and includes tissues unrelated to Ca2+o homeostasis, in which it, for example, regulates the secretion of digestive hormones, airway constriction, cardiovascular effects, cellular differentiation, and proliferation. Thus, although the CaSR is targeted clinically by the positive allosteric modulators (PAMs) cinacalcet, evocalcet, and etelcalcetide in hyperparathyroidism, it is also a putative therapeutic target in diabetes, asthma, cardiovascular disease, and cancer. The CaSR is somewhat unique in possessing multiple ligand binding sites, including at least five putative sites for the “orthosteric” agonist Ca2+o, an allosteric site for endogenous L-amino acids, two further allosteric sites for small molecules and the peptide PAM, etelcalcetide, and additional sites for other cations and anions. The CaSR is promiscuous in its G protein–coupling preferences, and signals via Gq/11, Gi/o, potentially G12/13, and even Gs in some cell types. Not surprisingly, the CaSR is subject to biased agonism, in which distinct ligands preferentially stimulate a subset of the CaSR’s possible signaling responses, to the exclusion of others. The CaSR thus serves as a model receptor to study natural bias and allostery.
Significance Statement The calcium-sensing receptor (CaSR) is a complex G protein–coupled receptor that possesses multiple orthosteric and allosteric binding sites, is subject to biased signaling via several different G proteins, and has numerous (patho)physiological roles. Understanding the complexities of CaSR structure, function, and biology will aid future drug discovery efforts seeking to target this receptor for a diversity of diseases. This review summarizes what is known to date regarding key structural, pharmacological, and physiological features of the CaSR.
I. Introduction
A. Identification and Cloning of the Calcium-Sensing Receptor
Ca2+ is an essential ion, both intracellularly and extracellularly, in mammals. Intracellular Ca2+ (Ca2+i) is maintained at approximately 100 nM but rises to low micromolar concentrations upon membrane or endoplasmic reticulum Ca2+ channel opening, thus serving as an important second messenger (Brini et al., 2013). Ca2+ also functions as a key first messenger via activation of the calcium-sensing receptor (CaSR) (Alexander, et al., 2017; Bikle et al., 2019), which plays a pivotal role in tightly regulating ionized (free) extracellular calcium (Ca2+o). In human plasma, total calcium (referred to herein as calcium to signify ionized and nonionized calcium) levels are maintained between 2.1 and 2.6 mM, of which roughly half is in an ionized form (Brini et al., 2013).
In the mid 1980s, there was significant interest in the mechanisms regulating parathyroid hormone (PTH) release from the parathyroid glands. It was consequently shown that elevated Ca2+o increased Ca2+i levels and decreased PTH release (LeBoff et al., 1985; Nemeth et al., 1986). In the following years, elevated Ca2+o was demonstrated to increase inositol phosphate (IP) and decrease cAMP levels, which led to the suggestion of a cell surface calcium-sensing G protein–coupled receptor (GPCR) (Nemeth and Scarpa, 1986, 1987; Brown et al., 1987a; Chen et al., 1989). Further evidence for the receptor was provided via activation of Ca2+-sensitive Cl− channels in Xenopus oocytes injected with mRNA isolated from bovine parathyroid cells (Racke et al., 1993), which subsequently led to expression cloning of the bovine CaSR (Brown et al., 1993). In isolated parathyroid cells, the cloned bovine CaSR was activated (in rank order of potency) by gadolinium (Gd3+) > neomycin > Ca2+o > magnesium (Mg2+) and signaled through elevation of Ca2+i, providing strong evidence of the cloned receptor being the long-sought CaSR (Brown et al., 1993).
Analyses of the cloned receptor sequence revealed a 1085 amino acid–long protein consisting of a large amino-terminal extracellular domain (ECD) of 613 amino acids comprised of a “Venus flytrap” (VFT) domain, which closes upon activation much like the VFT plant, and a cysteine-rich domain, a 7-transmembrane (7TM) domain of 250 amino acids and an intracellular carboxy terminus of 222 amino acids (Brown et al., 1993). The analyses also revealed that the CaSR was homologous to the metabotropic glutamate receptors, which were later shown to form the class C GPCRs together with GABAB, taste type 1; GPRC6A; and a handful of orphan receptors (Wellendorph and Bräuner-Osborne, 2009). The structurally conserved class C GPCR VFT domain is homologous to bacterial periplasmic binding proteins, and thus it has been predicted that class C GPCRs arose from fusion of the GPCR 7TM with a periplasmic binding protein (O’Hara et al., 1993). Nucleic acid hybridization techniques quickly led to cloning of the human (Garrett et al., 1995a), rat (Riccardi et al., 1995; Ruat et al., 1995), rabbit (Butters et al., 1997), chicken (Diaz et al., 1997), and shark (Nearing et al., 2002) CaSR orthologs, and genome data base mining subsequently suggested that the CaSR is evolutionarily conserved in flies and worms (Bjarnadóttir et al., 2005).
B. General Gene Structure
The human CASR gene has been mapped to chromosome 3q13.3-21 by fluorescence in situ hybridization (Janicic et al., 1995) and linkage analyses (Chou et al., 1992). The human CaSR is encoded by seven exons, of which exons 2-6 encode the ECD, and exon 7 encodes the 7TM and intracellular carboxy terminus (Pollak et al., 1993; Pearce et al., 1995). Two different 5′-untranslated promoter regions, termed exon 1A and exon 1B, have been identified in humans (Chikatsu et al., 2000), and both splice with the same site in exon 2. As recently reviewed (Hendy and Canaff, 2016), the promoters, and thus CaSR expression, are regulated by cis-elements responding to 1,25-dihydroxyvitamin D [1,25(OH)2D], proinflammatory cytokines, and the transcription factor glial cells missing-2 (GCM2s).
Tissue-specific splice variants lacking exon 3 (Bradbury et al., 1998) and exon 5 (Oda et al., 1998) have been reported, but their function (if any) remains elusive. The exon 5 splice variant is of particular interest because it is functional in growth plate chondrocytes (Rodriguez et al., 2005) despite being nonfunctional when recombinantly expressed in HEK293 and CHO cells. These latter findings led to an initial underestimation of the role of the CaSR in bone development because the original exon 5 knockout mouse (Ho et al., 1995) displayed a mild bone phenotype compared with a more severe phenotype in the exon 7 knockout mouse model (Chang et al., 2008).
C. Tissue Distribution
mRNA probes and antibodies have revealed that the CaSR is widely expressed both in tissues directly involved in controlling systemic Ca2+o homeostasis as well as in tissues with other functions. As detailed in the section, V. (Patho)physiology of the Calcium-Sensing Receptor and Its Ligands, the plasma calcium level is mainly regulated via actions on the parathyroid gland (PTH release), thyroid gland (calcitonin release, although calcitonin in humans is less important than in rodents), and kidney (production of 1,25(OH)2D3 and regulation of ion excretion), but other tissues, such as the bone (release of skeletal Ca2+o) and small intestine (Ca2+o absorption), also play a role both via direct CaSR activation and via PTH, calcitonin, and 1,25(OH)2D3 (Brown and MacLeod, 2001; Brown, 2013; Lee et al., 2019). In addition, the CaSR is expressed in a range of tissues not involved in systemic Ca2+o homeostasis, such as the keratinocytes of the skin (VE. Calcium-Sensing Receptor in Keratinocytes), colon (VF. Calcium-Sensing Receptor in the Gastrointestinal Tract), pancreas (VG. Calcium-Sensing Receptor in the Pancreas), mammary glands (VH. Calcium-Sensing Receptor in Mammary Glands), airway smooth muscle and epithelium (VI. Calcium-Sensing Receptor in Airway Smooth Muscle and Epithelium), vascular smooth muscle and endothelium (VJ. Calcium-Sensing Receptor in the Vasculature), and the brain (VK. Calcium-Sensing Receptor in the Brain and Nervous System), in which the CaSR regulates a range of (patho)physiological functions.
D. Signal Transduction Pathways
The principal CaSR signaling pathways are shown in Fig. 1. The CaSR primarily elicits its functions by coupling to the Gi/o and Gq/11 families of heterotrimeric G proteins to activate intracellular signaling pathways that inhibit PTH synthesis and release from parathyroid cells (A. Calcium-Sensing Receptor in the Parathyroid Glands). CaSR activation of Gi/o proteins leads to inhibition of the cAMP-synthesizing enzyme, adenylate cyclase, causing a decrease in intracellular cAMP levels (Chang et al., 1998; Kifor et al., 2001). CaSR coupling to Gq/11 is usually considered the primary signaling pathway, which activates phospholipase C (PLC)-β to hydrolyze phosphatidylinositol 4,5-bisphosphate to the second messengers, IP3 and diacylglycerol (Brown et al., 1993; Chang et al., 1998). IP3 triggers release of Ca2+i from intracellular stores, such as the endoplasmic reticulum, and diacylglycerol alone or in combination with Ca2+i activates protein kinase C (PKC). Cytosolic phospholipase A2, which is the rate-limiting enzyme in arachidonic acid metabolism, is also activated by the CaSR-mediated Gq/11 pathway through calmodulin and the Ca2+/calmodulin-dependent protein kinase II (Handlogten et al., 2001).

Key CaSR-signaling pathways. The CaSR primarily couples to Gq/11 and Gi/o proteins to mediate many of its physiological responses, including PTH release. The CaSR may also couple to G12/13, but the physiological relevance of this is unknown; therefore, G12/13 is semitransparent in the figure. AC, adenylate cyclase; ADIS, agonist-driven insertional signaling; Akt, protein kinase B; β-Arr, β-arrestin; CaM, calmodulin; DAG, diacylglycerol; EGFR, epidermal growth factor receptor; ER, endoplasmic reticulum; PI3K, phosphatidylinositol 3-kinase; PI4K, phosphatidylinositol 4-kinase; PLA2, phospholipase A2; PLD, phospholipase D.
The importance of the Gq/11 pathway in CaSR physiology has been demonstrated by the similarities between selective parathyroid knockout of the genes encoding Gαq (Gnaq) and Gα11 (Gna11) in mice, which results in a phenotype with almost all the features of Casr germline knockout mice (Wettschureck et al., 2007). Similarly, human CASR and GNA11 loss- or gain-of-function mutations cause familial hypocalciuric hypercalcemia (FHH) types 1 (CASR) and 2 (GNA11) or autosomal dominant hypocalcemia (ADH) types 1 (CASR) or 2 (GNA11), respectively (Pollak et al., 1993, 1994; Nesbit et al., 2013a) (VI. Calcium-Sensing Receptor–Related Genetic Diseases and Therapeutic Interventions).
Studies of CaSR coupling to G12/13 are limited because of a lack of inhibitors and suitable functional readouts. However, the CaSR activates phospholipase D in Madin-Darby canine kidney cells through a Gq/11- and Gi/o-independent pathway involving activation of the Rho family of small GTPases, most likely via G12/13 coupling (Huang et al., 2004). The G12/13 pathway is also likely to be the Gq/11- and Gi/o-independent pathway that activates the phosphatidylinositol 4-kinase responsible for the first step in inositol biosynthesis through Rho (Huang et al., 2002). However, CaSR can activate RhoA by a Gq/11 pathway in HEK293 cells (Pi et al., 2002) and phospholipase D by a PKC-dependent mechanism likely mediated by Gq/11 in HEK293 cells and parathyroid cells (Kifor et al., 1997), so it remains unclear whether CaSR also couples to G12/13 in these cells.
CaSR coupling to Gs and the consequent increase in intracellular cAMP levels activates PKA and stimulates PTH-related protein (PTHrP) release in immortalized and malignant breast cells and in the AtT-20 pituitary tumor-derived cell line (Mamillapalli et al., 2008; Mamillapalli and Wysolmerski, 2010) (VH. Calcium-Sensing Receptor in Mammary Glands). Stimulation of cAMP production is not observed in HEK293 cells recombinantly expressing the CaSR (Thomsen et al., 2012a), and the molecular mechanism for the switch in G protein preference in breast cancer and AtT-20 cells remains unknown.
The CaSR activates several mitogen-activated protein kinase (MAPK) cascades, including extracellular signal-regulated kinase (ERK) 1/2, p38 MAPK, and c-Jun N-terminal kinase to regulate PTHrP release, proliferation, and other functions (MacLeod et al., 2003; Tfelt-Hansen et al., 2003; Chattopadhyay et al., 2004). ERK1/2 is activated by phosphorylation (pERK1/2) through multiple CaSR-mediated pathways, including parallel G protein–dependent pathways involving either Gq/11 and PKC or Gi/o and epidermal growth factor receptor transactivation (Kifor et al., 2001; MacLeod et al., 2004; Thomsen et al., 2012a). Ras and phosphatidylinositol 3-kinase are also involved in ERK1/2 activation by the CaSR (Hobson et al., 2003), but it is unclear whether this pathway overlaps with the Gq/11- or Gi/o-dependent pathways. The CaSR can also activate ERK1/2 through a β-arrestin–dependent and G protein–independent pathway (Thomsen et al., 2012a). Furthermore, an Arg6803.32Gly [numbering shown in superscript after residue numbers throughout this manuscript is based on Ballesteros-Weinstein numbering assigned in Ballesteros and Weinstein (1995) for class A GPCRs and in Dore et al. (2014) for class C GPCRs] CaSR mutation associated with ADH1 selectively increases β-arrestin–dependent ERK1/2 activation, in which the mutation is predicted to disrupt an extracellular salt bridge between Arg6803.32 and Glu767 in the second extracellular loop (ECL) (Gorvin et al., 2018a).
In some cell types, the CaSR stimulates opening of L-type voltage-gated Ca2+ channels (Fajtova et al., 1991; McGehee et al., 1997; Muff et al., 1988) and nonselective cation channels, including transient receptor potential cation channels (Ye et al., 1996; El Hiani et al., 2006; Meng et al., 2014), although the pathways that couple the CaSR to ion channels are poorly defined.
II. Agonists and Allosteric Modulators
A. Endogenous and Exogenous Agonists
1. Polyvalent Cations
The CaSR is now well-known for its ability to sense fluctuations in Ca2+o. CaSR radioligand-binding assays to quantify the affinity of Ca2+o and other agonists have to date not been possible because of low agonist affinity, a lack of suitable radioligands, and complexities in quantifying agonist binding to multiple binding sites. However, spectroscopic studies indicate Ca2+o binds to the VFT with an affinity in the range of 3.0–5.0 mM (Zhang et al., 2014b). These findings are supported by the use of an operational model of agonism for receptors with multiple agonist binding sites, in which Ca2+o affinity at the full-length CaSR was 1.1–1.3 mM (Gregory et al., 2020). The low millimolar Ca2+o affinity is consistent with Ca2+o potency in healthy human subjects, in which Ca2+o suppresses PTH secretion with an approximate IC50 of 1.2 mM (which is also the approximate free Ca2+o concentration in human serum) (Brown, 1991; Ramirez et al., 1993), whereas in cultured parathyroid cells, the Ca2+o IC50 for PTH release is closer to 1 mM (Brown, 1983, 1991). The Ca2+o-PTH relationship is characterized by a Hill coefficient greater than unity (Brown, 1983, 1991; Ramirez et al., 1993). This is because multiple Ca2+o ions bind to the CaSR in a positively cooperative manner, allowing the CaSR to respond to minute changes in Ca2+o concentrations that span less than 100 µM (Brown, 1983, 1991; Ramirez et al., 1993). Thus, although Ca2+o is considered the primary endogenous and therefore orthosteric agonist of the CaSR, strictly speaking it is an allosteric modulator of its own activity.
In addition to Ca2+o, the CaSR is activated by many other polyvalent cations, including Mg2+, zinc, manganese, ferrous iron, strontium (Sr2+), barium, cadmium, cobalt, nickel, lead, terbium, Gd3+, europium, and yttrium (Brown et al., 1990; Ruat et al., 1996; Handlogten et al., 2000). Trivalent cations are generally more potent than divalent cations, of which Ca2+o and Mg2+ are the most physiologically relevant. The role of non-Ca2+o cations in CaSR-mediated (patho)physiology is unknown. Agonists that mimic the actions of Ca2+o at the CaSR have traditionally been called type I calcimimetics.
Although much larger and structurally more complex than the small cations described above, polyamines are CaSR agonists. Polyamines are found in all eukaryotes, with spermine, spermidine, and their diamine precursor putrescine the most abundant in mammals. Polyamines are synthesized ubiquitously in the body and are also ingested in the diet and secreted by intestinal bacteria. Although polyamines activate the CaSR in the absence of Ca2+o, there is some evidence they also potentiate the potency of Ca2+o (Quinn et al., 1997). Spermine is the most potent CaSR agonist, followed by spermidine and then putrescine (Quinn et al., 1997). Spermine IC50 for suppression of PTH release from cultured bovine parathyroid cells is ∼200 µM (Quinn et al., 1997). Blood polyamine concentrations in healthy humans are ∼5–10 µM (Casti et al., 1982; Soda et al., 2009), concentrations that are likely sufficient to activate the CaSR in tissues where receptor density is high. In the lung, polyamines and other polycations stimulate CaSR-mediated airway contraction (Yarova et al., 2015) (described in VI. Calcium-Sensing Receptor in Airway Smooth Muscle and Epithelium). Intriguingly, other overlapping functions of the CaSR and polyamines exist, including promotion of osteoblast, keratinocyte, vascular smooth muscle cell, and gastrointestinal epithelial cell differentiation and proliferation (Riccardi and Kemp, 2012; Leach et al., 2014; Miller-Fleming et al., 2015). Thus, polyamines may contribute to multiple (patho)physiological processes mediated by the CaSR.
Not surprisingly, additional positively charged molecules activate the CaSR, including poly-L-arginine, protamine, and aminoglycoside antibiotics, including neomycin, tobramycin, and gentamicin (McLarnon and Riccardi, 2002). Poly-L-arginine is a mimetic of eosinophil major basic protein released to activate mast cells, neutrophils, basophils, and macrophages in asthma and other allergic diseases. Ca2+i mobilization in CaSR-HEK293 cells stimulated by the related eosinophil cationic protein was completely absent in untransfected HEK293 cells and was blocked by structurally distinct CaSR inhibitors, demonstrating a CaSR-dependent signaling mechanism (Yarova et al., 2015).
B. Endogenous and Exogenous Allosteric Modulators
Allosteric modulators bind to sites that are topographically distinct from the orthosteric binding site and act to either potentiate [positive allosteric modulators (PAMs)], inhibit [negative allosteric modulators (NAMs)], or have no effect on (neutral allosteric ligands) the binding or efficacy of the orthosteric agonist. Allosteric modulators may also be agonists (or inverse agonists) in the absence of orthosteric agonists and can simultaneously act as agonists and PAMs (PAM-agonists). CaSR PAMs have been termed type II calcimimetics and CaSR NAMs calcilytics.
1. L-Amino Acids
L-amino acids are endogenous CaSR activators that are generally recognized as PAMs. Thus, L-amino acids have no activity in the absence of Ca2+o or another cationic activator, such as Gd3+ or spermine, but potentiate CaSR-mediated responses in the presence of submaximal concentrations of cationic activators (Conigrave et al., 2000). In a Ca2+i mobilization assay performed in CaSR-HEK293 cells, the magnitude of Ca2+o potentiation mediated by 10 mM amino acids followed the rank order L-Phe, L-Trp, L-histidine > L-alanine > L-serine, L-proline, L-glutamic acid > L-aspartic acid (but not L-lysine, L-arginine, L-leucine, and L-isoleucine) (Conigrave et al., 2000). Similarly, in human parathyroid cells in culture, aromatic amino acids, such as L-Trp and L-Phe, were the most potent L-amino acid CaSR activators in Ca2+i mobilization assays (Conigrave et al., 2004). Thus, the CaSR, like a number of other class C GPCRs, is a promiscuous sensor of L-amino acids (Conigrave and Hampson, 2006, 2010; Smajilovic et al., 2014).
As would be expected for a positive binding interaction, L-amino acids and Ca2+o markedly enhance the CaSR's sensitivity to one another in a reciprocal manner (Conigrave et al., 2000). Based on observations of Ca2+i mobilization and PTH secretion assays in vitro, amino acids support normal physiological Ca2+o sensitivity and thus underpin the physiological Ca2+o concentration set point for the parathyroid at around 1.1–1.2 mM (Conigrave et al., 2004).
Recent crystal structures of the CaSR’s VFT (Zhang et al., 2016) and entire extracellular (Geng et al., 2016) domains as well as mutational studies suggest that L-amino acids and analogs might be better viewed as coagonists of the receptor rather than PAMs (see III. Receptor Structure). As detailed later, L-amino acids display pronounced biased signaling properties (IIC. Biased Agonism and Biased Allosteric Modulation), and L-amino acid signaling appears to be attenuated by PKC-mediated phosphorylation of Thr888 in the C-terminal tail of CaSR (IVA. Phosphorylation and Dephosphorylation).
2. γ-Glutamyl Peptides
Wang et al. (2006) demonstrated that the γ-glutamyl peptide, glutathione, is a potent activator of the CaSR and of another class C GPCR, the fish 5.24 receptor. Subsequently, various natural and synthetic analogs of glutathione were found to activate the CaSR in the presence of threshold Ca2+o concentrations in a similar manner to L-amino acids. A receptor double mutant (Thr145Ala + Ser170Thr) exhibits similar impairments of function when exposed to either L-amino acids (Mun et al., 2005) or the glutathione analog, S-methylglutathione (Broadhead et al., 2011), suggesting overlapping binding sites. Interestingly, γ-glutamyl peptides active at the CaSR are also potent activators of kokumi taste (Ohsu et al., 2010; Amino et al., 2016).
3. pH
Large supraphysiological changes in buffer pH alter the potencies of Ca2+o and Mg2+ at the CaSR (Quinn et al., 2004). In the blood, pH rarely varies by more than 0.2 U; however, this represents a change in H+ concentration of ∼58%. Such acidosis can occur in advanced chronic kidney disease (CKD), which has relevance to the CaSR (see V. (Patho)physiology of the Calcium-Sensing Receptor and Its Ligands). Interestingly, altering buffer pH from 7.4 to just 7.2 or 7.6 elicits significant attenuation or enhancement of CaSR signaling, respectively, as observed in both HEK293 cells and bovine parathyroid cells (Campion et al., 2015). The site of H+ action is unknown, although it is not apparently mediated via the CaSR’s extracellular histidine residues (Campion et al., 2015). Crucially, pathophysiological changes in pH elicit significant changes in PTH secretion from isolated human parathyroid cells (Campion et al., 2015). This indicates the potential clinical relevance of altered acid or base balance in CaSR-modulated mineral metabolism.
4. Phosphate
Crystallization of the CaSR ECD has revealed up to four anion binding sites (Geng et al., 2016) (see III. Receptor Structure), and a recent study has revealed that phosphate inhibits the CaSR directly and in a noncompetitive manner (Centeno et al., 2019). This phosphate effect is more substantial than can be explained by buffering of free Ca2+o ions, and mutation of Arg62 inhibits the phosphate action. Exposure of human and murine parathyroid cells to pathophysiological phosphate concentrations induces rapid and reversible PTH secretion indicative of a receptor-mediated action (Centeno et al., 2019). Similarly, other anions, such as sulfate (SO42−), act as inhibitors of the CaSR (Geng et al., 2016) potentially also acting via Arg62 (Centeno et al., 2019).
5. Osmolarity
High sodium chloride (NaCl) concentrations are inhibitory for the CaSR, such that concomitant Ca2+o concentration-response curves are right-shifted, whereas lowering the NaCl concentration raises the potency of Ca2+o for the CaSR (Quinn et al., 1998). Accordingly, in dispersed bovine parathyroid cells, raising extracellular osmolarity with either NaCl or sucrose elicits rapid (within minutes) and substantial PTH secretion, an effect that cannot be suppressed by raising Ca2+o concentrations (Chen et al., 1987). Although this means that the CaSR could represent an ionic strength sensor where it is expressed in, for example, the renal tubules or the subfornical organ of the brain, there is little evidence to date that the CaSR is a substantive contributor to mammalian osmoregulation. Indeed, Na+ is a well-known negative allosteric modulator of multiple class A GPCRs, in which it binds in a conserved 7TM domain pocket. Therefore, allosteric modulation of GPCRs, at least by Na+, is likely a general phenomenon. Nonetheless, some severe gain-of-function clinical CaSR mutations (see VI. Calcium-Sensing Receptor–Related Genetic Diseases and Therapeutic Interventions) can elicit a Bartter-like salt-wasting syndrome, whereas loss-of-function CaSR mutations can enhance the natriuretic response to loop diuretics indicative of mild Na+ retention (Huang and Miller, 2010; Tyler Miller, 2013).
6. Small-Molecule Allosteric Modulators
A detailed review on the discovery and development of CaSR small-molecule drugs has recently been published (Nemeth et al., 2018). Therefore, for the purposes of this review, the focus will be on small molecules for which detailed pharmacological or clinical data are available. To date, all CaSR small-molecule binding sites have been localized to the 7TM domain and/or ECLs (Petrel et al., 2003, 2004; Miedlich et al., 2004; Bu et al., 2008; Leach et al., 2016). These sites are distinct from the predominant Ca2+o-, L-amino acid, or γ-glutamyl binding sites in the ECD (see III. Receptor Structure), and thus all small-molecule CaSR drugs identified so far are allosteric.
For the majority of small-molecule PAMs and NAMs, pharmacological characterization has been based on their ability to potentiate or inhibit a single concentration of Ca2+o, usually in a Ca2+i mobilization or IP accumulation assay (see Table 1). This approach provides a measure of modulator potency, which is a composite value of affinity, cooperativity (the magnitude and direction of modulator potentiation or inhibition of the orthosteric agonist), and efficacy (i.e., agonism or inverse agonism). Although potency measurements facilitate drug comparisons in a series when in vitro assays are performed under identical conditions, they can be misleading when different assay conditions are employed (e.g., different orthosteric agonist concentrations, different signaling outputs) (Gregory et al., 2018). Therefore, more recent work has quantified PAM and NAM affinity, cooperativity, and efficacy values as separate parameters using an operational model of allosterism or an allosteric ternary complex model (Davey et al., 2012; Leach et al., 2013, 2016; Cook et al., 2015; Diepenhorst et al., 2018; Gregory et al., 2018, 2020).
Representative CaSR agonists or endogenous and small-molecule allosteric modulators and their pharmacological properties
7. Small-Molecule Positive Allosteric Modulators
The structural and chemical diversity of small-molecule CaSR PAMs is relatively limited, with few distinct series discovered. Two chemically and structurally related small-molecule PAMs, cinacalcet and evocalcet (Table 1), are clinically approved. Cinacalcet is FDA-approved for the treatment of primary hyperparathyroidism in patients who cannot undergo parathyroidectomy, and for hypercalcemia in adults with parathyroid carcinoma. Cinacalcet is also FDA-approved for secondary hyperparathyroidism in patients on renal replacement therapy, and has been used off-label to treat naturally occurring loss-of-function mutations in the CaSR or its signaling partners that cause disorders of Ca2+o and PTH homeostasis (described in VI. Calcium-Sensing Receptor–Related Genetic Diseases and Therapeutic Interventions). Cinacalcet was the first GPCR allosteric modulator to be approved for clinical use in 2004. Evocalcet was approved in Japan in 2018 for the treatment of secondary hyperparathyroidism in patients on dialysis. Cinacalcet and evocalcet potentiate Ca2+o activity at the CaSR, thus left-shifting the Ca2+o-PTH concentration-response relationship in the body. This means lower Ca2+o concentrations are required to suppress PTH release, thus normalizing elevated serum PTH levels. However, both cinacalcet and evocalcet carry a risk of hypocalcemia in patients that limits their clinical utility (Fukagawa et al., 2018), presumably in part from potentiation of the CaSR in the kidney and enhanced CaSR-mediated calcitonin secretion from thyroid parafollicular C cells (see V. (Patho)physiology of the Calcium-Sensing Receptor and Its Ligands). Furthermore, cinacalcet and evocalcet are associated with adverse gastrointestinal side effects, including nausea and vomiting, which may occur via the CaSR expressed in the gastrointestinal tract. In rats and humans, however, evocalcet appears to have reduced actions in the gastrointestinal tract in comparison with cinacalcet (Fukagawa et al., 2018; Kawata et al., 2018).
Cinacalcet and evocalcet belong to the arylalkylamine family of PAMs derived from the nonselective calcium channel blocker, fendiline. A number of structurally related arylalkylamine PAMs have been identified, including NPS R-467 and NPS R-568 (the precursors to the discovery of cinacalcet), calindol, and calcimimetic B (Table 1). The activity of these PAMs is highly dependent upon their stereoselectivity, in which the R-configuration of the methyl between the aromatic and secondary nitrogen is more active than the S-configuration (Nemeth et al., 2018). Although NPS R-568, cinacalcet, and calindol exhibit similar affinity and cooperativity values when measured in a Ca2+i mobilization assay (Davey et al., 2012; Cook et al., 2015; Leach et al., 2016; Diepenhorst et al., 2018; Keller et al., 2018), R,R-calcimimetic B has a roughly 10-fold higher affinity but comparable cooperativity (Cook et al., 2015). Although concentrations of cinacalcet that exceed 1 µM weakly activate the CaSR in the absence of divalent cations (Nemeth et al., 2018), suggesting it is a “PAM agonist,” arylalkylamine PAMs demonstrate negligible agonism at concentrations that robustly potentiate CaSR activity (Cook et al., 2015; Keller et al., 2018). In contrast, R,R-calcimimetic B is a PAM and a partial agonist at micromolar concentrations (Cook et al., 2015). Arylalkylamine PAMs also exhibit pronounced positive interactions with L-amino acids (Zhang et al., 2002a) and glutathione (Broadhead et al., 2011).
A benzothiazole series of CaSR PAMs that is structurally and chemically distinct from the arylalkylamines has been discovered. These PAMs include the small benzothiazole, AC265347 (Table 1), which has been characterized in detail. AC265347 has comparable affinity and cooperativity to cinacalcet when measured in a Ca2+i mobilization assay (Cook et al., 2015; Leach et al., 2016; Diepenhorst et al., 2018), and similar to the arylalkylamine PAMs, AC265347 is a PAM agonist, although AC265347 is more potent and efficacious as an agonist than the arylalkylamines (Cook et al., 2015). Although AC265347 has not been tested in humans, in healthy rats, AC265347 suppressed serum PTH levels with greater potency than cinacalcet and demonstrated a lower propensity to cause hypocalcemia (Ma et al., 2011).
Trisubstituted urea compounds have been identified as another potent class of CaSR PAMs (Temal et al., 2013) (Table 1). Benzothiazole trisubstituted urea (BTU) compound 13 (Deprez et al., 2013) is the best characterized of this series. BTU compound 13 has similar affinity and cooperativity to cinacalcet at the CaSR in a Ca2+i mobilization assay (Cook et al., 2015; Diepenhorst et al., 2018). Much like AC265347, BTU compound 13 suppressed PTH levels in a rat model of CKD while avoiding significant hypocalcemia (Deprez et al., 2013).
8. Peptide Positive Allosteric Modulator, Etelcalcetide
In 2017, a novel CaSR PAM, etelcalcetide (chemical name N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-argininamide disulfide with L-cysteine hydrochloride), was FDA-approved for the treatment of secondary hyperparathyroidism in patients with CKD on dialysis. Etelcalcetide is administered intravenously at the end of dialysis. Similar to cinacalcet and evocalcet, etelcalcetide is associated with adverse gastrointestinal side effects and hypocalcemia (Hamano et al., 2017).
Etelcalcetide is comprised of seven D-amino acids linked via a disulfide bond to L-cysteine. Not surprisingly, given it is the only peptide CaSR PAM identified, etelcalcetide has a unique mode of PAM action in comparison to small-molecule PAMs that involves binding to the CaSR via disulfide bond formation (Alexander et al., 2015) (see III. Receptor Structure). Although etelcalcetide has been classified as a PAM agonist, assays used to discern agonism contained 0.5 mM MgCl2; therefore, it is currently uncertain whether observed etelcalcetide efficacy for stimulation of IP1 accumulation in the absence of Ca2+o is true agonism or potentiation of Mg2+ (Walter et al., 2013). The affinity and cooperativity of etelcalcetide at the CaSR has not been quantified, but its potency for potentiation of 1.2 mM Ca2+o in an HEK293 IP1 accumulation assay was 25 µM (Walter et al., 2013).
9. Small-Molecule Negative Allosteric Modulators
Due to the role of the CaSR in regulation of PTH secretion, there was significant interest in the development of CaSR NAMs that could stimulate PTH release. Intermittent and transient increases in serum PTH levels enhance the formation of new bone via the differentiation and proliferation of bone-forming osteoblasts. This is evidenced by clinical use of recombinant PTH1-34 injections to promote bone formation in osteoporosis. However, if PTH levels remain elevated, PTH stimulates the differentiation and proliferation of bone-resorbing osteoclasts, resulting in bone breakdown (Dobnig and Turner, 1997).
Although several pharmaceutical companies have embarked on CaSR NAM discovery programs, similar to CaSR PAMs, there is fairly limited structural and chemical diversity in the NAM scaffolds identified to date. NPS 2143 (Table 1) was one of the first CaSR NAMs to be discovered (Gowen et al., 2000) and is structurally and chemically related to cinacalcet and other arylalkylamines. Like CaSR PAMs, NAMs have generally been evaluated for their potency to inhibit a single Ca2+o (usually EC80) concentration. Nevertheless, more recent studies have employed an operational model of allosterism to quantify NPS 2143 activity and have indicated that NPS 2143 binds at the CaSR with micromolar to submicromolar affinity depending on the assay (Table 1) (Davey et al., 2012; Leach et al., 2013, 2016; Gregory et al., 2020). Importantly, NPS 2143 is a partial NAM at the CaSR, meaning that it does not fully inhibit Ca2+o-mediated signaling (Cook et al., 2015; Leach et al., 2016; Gregory et al., 2018).
In rats, NPS 2143 stimulated the release of PTH, resulting in an increase in bone turnover markers, but it did not promote the formation of new bone (Gowen et al., 2000). The lack of new bone formation was hypothesized to be due to the prolonged, rather than transient, PTH release in response to NPS 2143, resulting in both bone formation and resorption. Efforts to develop shorter-acting CaSR NAMs based on the structure of NPS 2143 led to the discovery of ronacaleret (Fitzpatrick et al., 2011a,b, 2012) and JTT-305/MK-5442 (Shinagawa et al., 2011) (Table 1). However, in rats, JTT-305/MK-5442 did not increase bone mass and density (Fisher et al., 2012), whereas in human clinical trials, both ronacaleret and JTT-305/MK-5442 lacked efficacy in treating postmenopausal osteoporosis (Fitzpatrick et al., 2011a,b, 2012; Halse et al., 2014).
Further efforts to identify additional CaSR NAMs that may prove successful in treating osteoporosis led to the discovery of four chemically distinct NAM series exemplified by the quinazolinones ATF936 and AXT914 (Gerspacher et al., 2010), the pyridine Bristol Myers Squibb (BMS) compound 1 (Arey et al., 2005), a series of 3H-quinazoline-4-ones and 3H-pyrimidine-4-ones (Shcherbakova et al., 2005; Didiuk et al., 2009), and benzimidazoles (Gerspacher et al., 2010). A recent study revealed that the affinity of ATF936 was 17-fold higher than that of ronacaleret, and ATF936 also demonstrated higher negative cooperativity (Josephs et al., 2019) (Table 1). However, despite findings that the quinazolinone NAMs may be superior to ronacaleret in terms of desirable drug properties, when AXT914 was evaluated for its effects on bone turnover in humans, the trial was terminated early because of a lack of effect on bone turnover markers and a propensity to cause hypercalcemia (John et al., 2014).
After the failure of three different NAMs in human clinical trials of osteoporosis, the development of CaSR NAMs diminished. However, there has been recent interest in repurposing these NAMs for the treatment of Ca2+o homeostasis disorders caused by gain-of-function mutations in the CaSR or its interactors (described in VI. Calcium-Sensing Receptor–Related Genetic Diseases and Therapeutic Interventions). Indeed, the NAM, NPSP795 (SHP635), has recently undergone clinical testing for its therapeutic potential in the treatment of ADH1 (Roberts et al., 2019).
10. Calhex 231: A Mixed Positive Allosteric Modulator and Negative Allosteric Modulator
Although the arylalkylamine, calhex 231, was originally classified as a NAM based on its ability to inhibit an EC100 Ca2+o concentration (Kessler et al., 2006), a recent study has revealed that calhex 231 is both a PAM and a NAM (Gregory et al., 2018). This novel mode-switching mechanism is due to allostery across the CaSR dimer, wherein calhex 231 acts as a PAM when it occupies a single protomer in the dimer and a NAM when bound to both protomers. Mixed PAM and NAM activity was observed in HEK293 cells stably expressing the CaSR and in primary cultures of human parathyroid cells, demonstrating that mode-switching may occur under physiological conditions (Gregory et al., 2018). Because several CaSR NAMs have been characterized based on their ability to modulate only a single Ca2+o concentration, it is unclear at present whether other CaSR allosteric modulators also exhibit mixed PAM and NAM activity. However, whereas other CaSR NAMs were identified from high throughput screens of large compound libraries, calhex 231 originated from a PAM scaffold (Kessler et al., 2004, 2006), which likely contributes to its mixed PAM and NAM activity.
C. Biased Agonism and Biased Allosteric Modulation
Given that the CaSR responds to a diverse array of different ligands, it is unsurprising that the CaSR is subject to biased agonism and biased modulation. Biased agonism is the phenomenon by which distinct ligands stabilize preferred GPCR signaling states, with each state having the potential to stimulate or inhibit discrete subsets of the full repertoire of intracellular signaling pathways that couple to a given receptor (Kenakin and Christopoulos, 2013). This is in contrast to the earlier dogma that all agonists activate the same subsets of GPCR signaling pathways to greater (e.g., full agonists) or lesser (e.g., partial agonists) extents. Similarly, biased modulation arises when an allosteric ligand differentially modulates different agonist-mediated signaling pathways.
For instance, in CaSR-HEK293 cells, Ca2+o preferentially mediates stimulation of Ca2+i mobilization over pERK1/2, whereas spermine preferentially activates pERK1/2 (Thomsen et al., 2012a). Similarly, L-amino acids activate Ca2+i mobilization and ERK phosphorylation (Lee et al., 2007) and also inhibit cAMP synthesis. However, they are inactive in stimulating phosphatidylinositol-PLC and various other signaling events, including Rho-dependent actin stress fiber formation (Davies et al., 2006) and cAMP responsive element-binding protein phosphorylation (Avlani et al., 2013), and appear to promote Ca2+i mobilization via a G12/13/transient receptor potential cation 1–dependent Ca2+o influx pathway (Rey et al., 2005, 2006).
Evidence from patients with FHH suggests CaSR bias may arise in part from spatial and temporal CaSR-signaling patterns. Loss-of-function germline mutations of the adaptor-related protein complex-2 (AP2)-S1 gene, which encodes the sigma subunit of the heterotetrameric AP2σ, cause FHH3 (Nesbit et al., 2013b; Hannan et al., 2015a). AP2σ forms part of the heterotetrameric AP2 that plays a critical role in clathrin-mediated endocytosis. AP2σ mutations increase CaSR cell surface expression yet reduce CaSR signaling because CaSR residency time in clathrin-coated pits is increased, consequently impairing CaSR Gq/11 signaling from endosomes (Gorvin et al., 2018c). In contrast, Gi/o-mediated signaling is less sensitive to AP2σ mutations. Thus, whereas the plasma membrane localized CaSR signals via Gq/11 and Gi/o, endosomal CaSRs signal predominantly via Gq/11 (Gorvin et al., 2018c).
It must be noted that many of the studies reporting differential CaSR-mediated pathway activation have not been performed in a systematic manner using identical conditions across assays (e.g., buffers, duration of agonist stimulation, etc.) or the same cellular background. Furthermore, bias has not been quantified in these studies. Therefore, it remains to be definitively proven whether biased agonism is truly operative at the CaSR or whether previous observations were due to observational bias (e.g., different assay conditions, different cell types) or system bias (e.g., the relative efficiency with which the receptor couples to different pathways).
Nonetheless, small-molecule allosteric modulators do appear to exhibit true biased modulation at the CaSR. Evidence of biased modulation comes from reversals in the magnitude of cooperativity in different pathways between distinct PAMs or NAMs or from differences in PAM or NAM affinity for receptor states that couple to different signal transducers. For instance, although cinacalcet and NPS 2143 preferentially potentiate or inhibit, respectively, Ca2+o-mediated Ca2+i mobilization over pERK1/2, AC265347 and R,R-calcimimetic B show reversed bias for CaSR-mediated pERK1/2 over Ca2+i mobilization (Cook et al., 2015; Leach et al., 2016; Diepenhorst et al., 2018). Similarly, AC265347, NPS R-568, and calindol, but not cinacalcet or R,R-calcimimetic B, have a higher functional affinity (i.e., an affinity quantified in a functional assay using an operational model of allosterism (Leach et al., 2007)) for the CaSR state that signals to IP1 accumulation versus Ca2+i mobilization (Cook et al., 2015; Diepenhorst et al., 2018), whereas cinacalcet, NPS R-568, and NPS 2143 all have a higher functional affinity for the CaSR state that couples to membrane ruffling (Davey et al., 2012).
Evidence for small-molecule PAM and NAM bias also comes from pharmacochaperone studies, which reveal that although cinacalcet, AC265347, and BTU compound 13 are all PAMs in multiple CaSR-mediated signaling assays, only cinacalcet positively modulates the trafficking of an endosomally-trapped, naturally occurring mutant CaSR, rescuing its cell surface expression back to levels comparable to wild-type CaSR (Leach et al., 2013; Cook et al., 2015; Diepenhorst et al., 2018). In contrast, although NPS 2143 is a NAM of CaSR signaling, it is a PAM of loss-of-expression mutant receptor trafficking (Leach et al., 2013). This is in contrast to the actions of NPS 2143 at the wild-type CaSR, wherein it reduces CaSR surface expression (Huang and Breitwieser, 2007), suggesting naturally occurring mutations (which cause Ca2+o homeostasis disorders; see VI. Calcium-Sensing Receptor–Related Genetic Diseases and Therapeutic Interventions) may engender bias in CaSR function. Indeed, Ca2+o-mediated bias toward Ca2+i mobilization is abolished by some naturally occurring mutations (Leach et al., 2012).
Although the physiological relevance of biased agonism and biased modulation at the CaSR is not at present known, differences in the propensity of CaSR PAMs to cause hypocalcemia could be linked to this phenomenon. For instance, as already mentioned, R,R-calcimimetic B and AC265347 are effective suppressors of PTH release. However, in comparison with cinacalcet, R,R-calcimimetic B and AC265347 demonstrate reduced propensity to cause hypocalcemia in rats successfully treated for severe hyperparathyroidism induced by CKD (R,R-calcimimetic B) or in normal rats (AC265347). The reduced incidence of hypocalcemia with R,R-calcimimetic B and AC265347 is presumably linked, in part, to their lower potency and efficacy for the stimulation of calcitonin secretion versus suppression of PTH release (Henley et al., 2011; Ma et al., 2011). Importantly, although suppression of PTH release has been associated with pERK1/2, calcitonin release is independent of pERK1/2 in rat medullary thyroid carcinoma cells (Thomsen et al., 2012b). This highlights differences in the coupling specificity of the CaSR in distinct tissues and is consistent with observations that when compared with cinacalcet, AC265347 and R,R-calcimimetic B show reversed bias for CaSR-mediated pERK1/2 over Ca2+i mobilization.
Another apparent difference between CaSR PAMs points toward putative clinical advantages for cinacalcet. The CaSR agonist Sr2+ reduces the differentiation of bone-resorbing osteoclasts (Bonnelye et al., 2008) and stimulates osteoclast apoptosis (Hurtel-Lemaire et al., 2009) (described in V. (Patho)physiology of the Calcium-Sensing Receptor and Its Ligands). In cultured osteoclasts differentiated from human CD14+ monocytes, although cinacalcet potentiated Sr2+-mediated tartrate-resistant acid phosphatase expression (a marker of osteoclast activity) and robustly inhibited osteoclast-mediated hydroxyapartite artificial bone resorption, AC265347 and BTU compound 13 were without effect in these two assays (Diepenhorst et al., 2018). Although it is not clear whether differences in the biased profile of AC265347 and BTU compound 13 versus cinacalcet are responsible for their distinct PAM activities in osteoclasts, it is interesting that only cinacalcet, and not AC265347 or BTU compound 13, can pharmacochaperone loss-of-function mutant CaSRs potentially via differential stabilization of different conformations of the CaSR. A more detailed understanding of the signaling and trafficking pathways that couple the CaSR to its many physiological responses will aid our understanding of why the CaSR responds to so many endogenous activators and may facilitate the development of biased compounds with improved, tissue-specific effects.
In addition to bias engendered by small-molecule allosteric modulators, CaSR autoantibodies that cause acquired hypocalciuric hypercalcemia can act as biased allosteric modulators. Biased autoantibodies directed against the CaSR VFT can potentiate IP accumulation while inhibiting pERK1/2 generation (Makita et al., 2007; Makita and Iiri, 2014), whereas others inhibit pERK1/2 generation but have no effect on IP accumulation (Pallais et al., 2011). Importantly, cinacalcet corrected the severe hypercalcemia associated with acquired hypocalciuric hypercalcemia caused by a biased autoantibody (Makita et al., 2019). Taken together, these findings once again highlight how bias and allostery are key features of CaSR (patho)physiology and drug actions.
III. Receptor Structure
To date, the complete structure of the CaSR has not been determined. Current CaSR structural knowledge comes from the inactive (Geng et al., 2016) and active (Geng et al., 2016; Zhang et al., 2016) crystal structures of the CaSR ECD in isolation, from mutagenesis studies and homology modeling of the 7TM based on the crystal structures of the metabotropic glutamate receptors (mGluRs) 1 and 5 7TMs (Dore et al., 2014; Christopher et al., 2015, 2019), and from comparisons with the low resolution cryogenic electron microscopy (cryo-EM) structure of mGlu5 (Koehl et al., 2019).
The CaSR is an obligate homodimer (Romano et al., 1996; Bai et al., 1998a; Ward et al., 1998; Ray et al., 1999; Zhang et al., 2001; Pidasheva et al., 2006), with each protomer comprised of an extracellular VFT domain (amino acids 20–542) and a cysteine-rich (CR) domain (9 Cys residues within amino acids 542–612) that links the VFT to the prototypical GPCR 7TM domain (amino acids 613–862) (Fig. 2). The 7TM domain is followed by a long intracellular tail (amino acids 863–1078), which is predicted to be largely unstructured but is important for trafficking and phosphorylation (Bai et al., 1998b; Chang et al., 2001; Stepanchick et al., 2010; Zhuang et al., 2012).

Structural conformation of the CaSR. (A) Model of the CaSR based on homology with full-length mGlu5 (PDB 6N51). The CaSR (cartoon ribbon) comprises an extracellular VFT domain composed of lobe 1 (LB1, dark blue) and lobe 2 (LB2, teal) and a CR domain (yellow) anchored to the 7TM (orange). (B) Inactive ECD monomer (PDB 5K5T). The bilobed VFT adopts an open conformation revealing a conserved binding cleft between the two lobes. (C) Inactive ECD dimer (left, front view; right, side view). The CR domains of the inactive ECDs are separated. (D) Active ECD monomer (PDB 5K5S). Upon activation, the bilobed VFT closes the amino acid–binding site, narrowing the cleft. (E) Active ECD dimer (left, front view; right, side view). Upon activation, each protomer (orange and yellow) is drawn closer together.
A. Calcium-Sensing Receptor Extracellular Domain
1. Structural Overview of the Calcium-Sensing Receptor Extracellular Domain
The VFT extends outside the cell and is comprised of two lobe subdomains (lobe 1 and 2; Fig. 2), with each lobe forming part of a ligand binding cleft. In other class C GPCRs, this cleft forms the orthosteric binding pocket (Kunishima et al., 2000; Tsuchiya et al., 2002; Muto et al., 2007). However, in the CaSR, it is an allosteric or coagonist binding site for L-amino acids, with Ca2+o and other cations binding elsewhere.
Two recent VFT crystal structures confirm that the CaSR VFT forms a dimer, with each CaSR protomer orientated next to each other as mirror images (Fig. 2). The dimer orientation of the extracellular domain is similar to that reported for other class C GPCRs, including mGluRs (Kunishima et al., 2000; Tsuchiya et al., 2002; Muto et al., 2007) and the GABAB receptor (Geng et al., 2012, 2013). In the inactive state, the two VFT lobes adopt an open conformation [buried surface area of 740 Å2, calculated using methods described in Krissinel and Henrick (2007)], and the interdomain cleft is empty. In contrast, the active-state structures adopt a closed conformation and a resulting increase in the buried surface area to just over 1000 Å2 between the VFT lobes (Fig. 2). Upon VFT closure, the interdomain cleft interface rotates 29°, mediated by interactions between the two lobes of the VFT (Geng et al., 2016).
The crystal structure of the CaSR VFT plus the CR domains shows an 83-Å distance between the CR domains when the CaSR VFT is in the open (inactive) conformation, which is reduced to 23 Å once the VFT is closed (active; Fig. 2D) (Geng et al., 2016). This change is consistent with other X-ray structures of class C ECDs (Muto et al., 2007; Chappell et al., 2016), likely driving a similar reorientation of the 7TM domains as seen in the mGlu5 cryo-EM structure a "transition-state" that is partially active but not coupled to G proteins (Koehl et al., 2019). This reorientation is sustained by the rigid CR domain and its nine Cys residues, which form five covalent disulfide bonds: four within the CR domain and one that anchors the CR domain to lobe 2 of the VFT. Consequently, mutation of the Cys residues compromises this rigidity, impacting significantly on receptor function (Fan et al., 1998).
2. Amino Acid and γ-Glutamyl Peptide Binding Site
Although Ca2+o has long been considered the orthosteric agonist for the CaSR, Ca2+o does not occupy the conserved cleft that forms the orthosteric binding site in other class C GPCRs. Both mutagenesis (Zhang et al., 2002b, 2014a; Mun et al., 2004, 2005) and, more recently, the crystal structures of the VFT have revealed that L-amino acids bind the conserved cleft (between lobe 1 and 2), similar to L-Glu binding in the mGluRs (Wellendorph and Bräuner-Osborne, 2009). Thus, L-amino acids and analogs might be better viewed as coagonists rather than PAMs.
The binding of L-Trp (Geng et al., 2016) or the tryptophan derivative, L-1,2,3,4-tetrahydronorharman-3-carboxylic acid (TNCA) (Zhang et al., 2016), stabilizes closing of the bilobed domains through hydrogen bonding and hydrophobic interactions with the receptor. Mutational analysis of residues in the conserved interdomain cleft support binding of L-Trp here (Zhang et al., 2002b, 2014a; Mun et al., 2004, 2005). Interestingly, residues in the conserved cleft are also important for Ca2+o activation of the CaSR (Bräuner-Osborne et al., 1999; Kunishima et al., 2000; Tsuchiya et al., 2002; Muto et al., 2007; Geng et al., 2013; Jacobsen et al., 2017), suggesting that L-amino acids are required for Ca2+o activation in line with a classification as coagonists. However, these mutational studies have not accounted for mutation-induced changes in receptor expression, therefore the mutation-induced signaling impairments may be due to reduced receptor expression and consequent reductions in apparent agonist efficacy.
Receptor contacts with L-Trp or TNCA are predominantly through backbone interactions, and the fact that these interactions are largely not L-Trp- or TNCA-specific means other amino acids could be accommodated within this pocket, explaining the L-amino acid promiscuity of the CaSR (see IIB. Endogenous and Exogenous Allosteric Modulators). Interestingly, TNCA was not included as a constituent of the crystallization conditions. This highlights not only the diversity of ligands that can bind and activate the CaSR but also suggests that TNCA has such high affinity for the CaSR that it is difficult to remove during the purification process.
γ-Glutamyl peptides are also potent CaSR PAMs that can promote Ca2+o-dependent Ca2+i mobilization, suppress intracellular cAMP levels, and inhibit PTH secretion from normal parathyroid cells (see IIB. Endogenous and Exogenous Allosteric Modulators) (Broadhead et al., 2011). This activity is lost when Thr145 and Ser170 located in the interdomain cleft are mutated to Ala, indicating that the γ-glutamyl peptides likely share the same binding site as the amino acids (Mun et al., 2005; Broadhead et al., 2011).
3. Cation Binding Sites
In both crystal structures of the CaSR VFT domain, cation binding sites were identified, but these sites differed in their number, location (with the exception of cation binding site 1), and the cation that was bound to each site (Fig. 3).

Binding sites within the CaSR crystal structures. ECD conformations of the (A) active (PDB 5K5S) and (B) inactive (PDB 5K5S) calcium-bound structures and the VFT conformations of the (C) active Mg2+-bound (PDB 5FBK) and (D) active Mg2+- and Gd3+-bound (PDB 5FBH) structures. Crystal structures are shown as cartoon ribbon within the transparent molecular surface and colored as in Fig. 2. Hydrogen bond interactions (dashed lines) of calcium (red spheres), Mg2+ (green spheres), and Gd3+ (yellow spheres) with key residues or water molecules (black spheres) are shown for each proposed binding site.
Anomalous difference mapping indicated four Ca2+o binding sites in the VFT structure solved by Geng et al. (2016) (Fig. 3). In lobe 1 of the active (L-Trp bound and closed) VFT conformation, backbone carbonyl oxygen atoms of Ile81, Ser84, Leu87, and Leu88 coordinate Ca2+o binding at cation binding site 1 (PDB: 5K5S). There was no Ca2+o coordinated at cation binding site 1 in the inactive structure (PDB: 5K5T), even though this site is not significantly different in the active versus inactive structures (Geng et al., 2016). As such, it is possible that Ca2+o, which was used at a lower concentration in the crystallization conditions for the inactive structure, could bind to this site without the need for the VFT domain to be closed.
Cation binding site 2 is located adjacent to the L-Trp binding site above the interdomain cleft in lobe 1 of the VFT. Cation binding site 2 is occupied by Ca2+o in both the inactive and active structures, in which Ca2+o is coordinated by the hydroxyl group of Thr100 in both states and by the carbonyl of Asn102 via a water molecule in the active structure. Thr145 also lines cation binding site 2 and forms part of the L-Trp binding cleft in the active state (Geng et al., 2016).
The hydroxyl groups of Ser302 and Ser303 coordinate cation binding site 3, either directly or indirectly through water molecules, at the edge of the interdomain cleft of lobe 2. The closing of lobe 1 and lobe 2 of the VFT is facilitated by Ca2+o stabilization of a conformation that permits an interdomain hydrogen bond interaction between lobe 1 residue Arg66 and lobe 2 residue Ser301 (Geng et al., 2016).
Finally, upon agonist binding, cation binding site 4 forms part of the homodimer interface bridging the lobe 2 domain of one subunit and the CR domain of the second subunit. Three interfacial residues, the carboxylate group of Asp234, and carbonyl oxygen of Glu231 and Gly557, coordinate Ca2+o binding to site 4 (Geng et al., 2016).
The anomalous difference map intensities varied at each of the Ca2+o binding sites, where intensity was ranked as Ca2+o binding site 1 = 2 > 3 > 4. The lower anomalous signal for Ca2+o in sites 3 and 4 indicates incomplete occupancy or higher flexibility at these positions in the crystal lattice. The authors suggested the lower signal reflects a lower Ca2+o affinity at these sites. In support of a lower Ca2+o binding affinity for cation binding site 4, the authors proposed that Ca2+o binding at site 4 stabilizes the active homodimer conformation, and thus the site is occupied only at elevated concentrations required for receptor activation (Geng et al., 2016).
In contrast to the structures by Geng et al. (2016), Zhang et al. (2016) identified two cation binding sites in their active VFT structures. Electron density and geometric restraints were used to identify Mg2+ occupying these cation binding sites, one of which overlapped with cation binding site 1 in the structure by Geng et al. (2016). However, in contrast to the unoccupied cation binding site 1 in the inactive structure by Geng et al. (2016), cation binding site 1 was occupied by Mg2+ in the inactive structure by Zhang et al. (2016). The Mg2+ is coordinated by Ser84 and backbone interactions with Ile81, Ile87, and Leu88, in addition to two water molecules. This site is similarly occupied by a Mg2+ cation in the rat mGlu1 VFT structure (Kunishima et al., 2000).
The second Mg2+ binding site (cation-binding site 5) is located at the dimerization interface of lobe 2 and is coordinated through Ser240 and four water molecules (Zhang et al., 2016). The highly conserved residues Glu228 and Glu231 from one protomer and Glu241 from the other protomer surround this site.
Anomalous difference maps identified a Gd3+ binding site (cation binding site 6) coordinated by Glu232, Glu228, and Glu229 adjacent to cation binding site 5 on the lobe 2 dimerization interface (PDB: 5FBN) (Zhang et al., 2016). The Glu228Ile and the double mutant Glu228Ile/Glu229Ile have previously been shown to reduce Mg2+-induced Ca2+i mobilization; therefore, other cations could bind here (Huang et al., 2009).
The crystal structures of the ECD suggest that Ca2+o and other cations play a role in: 1) local stabilization of the CaSR ECD; and 2) activation of the receptor via stabilization of the homodimer through cation binding at sites 4-6 (Jensen et al., 2002; Geng et al., 2016; Zhang et al., 2016). It is unknown whether Ca2+o alone can activate the receptor or whether it requires the presence of the cleft-binding ligands. Although Geng et al. (2016) obtained an active (closed) structure in the absence of amino acids, an unidentified continuous stretch of density in the conserved interdomain cleft was observed, which could be attributed to an endogenous ligand or a ligand acquired during the crystallization process. If ligands that bind the conserved interdomain cleft are difficult to remove during crystallography studies, it is likely that these same ligands are present during in vitro assays that measure CaSR activation. Furthermore, cations identified in the crystal structures could be artifacts of the crystallization conditions and merely stabilize the crystal contacts required for structure determination. Although mutagenesis was used to corroborate the observed cation binding sites (Geng et al., 2016; Zhang et al., 2016), these mutational studies neither accounted for mutation-induced changes in receptor expression nor quantified changes in cation affinity and efficacy. Therefore, a reduction in cation binding upon mutation of these sites has not been validated. Furthermore, analysis of Ca2+o-binding proteins to predict the CaSR’s Ca2+o sites, coupled with mutagenesis and spectroscopic techniques to validate the predictions, confirmed multiple VFT Ca2+o binding sites, but they differed to those identified in the VFT crystal structures (Huang et al., 2007, 2009; Kirberger et al., 2008; Wang et al., 2009, 2010; Zhao et al., 2012). Moreover, analyses of a “headless” CaSR, in which the ECD has been removed, has shown that Ca2+o can also activate the CaSR via binding sites in the 7TM domain (see IIIB. Calcium-Sensing Receptor 7-Transmembrane Domain) (Ray and Northup, 2002; Leach et al., 2016). Accordingly, the cooperative binding of Ca2+o at multiple binding sites likely maximizes the CaSR’s ability to respond to Ca2+o over a narrow physiological range. Additional active-state structures, biophysical studies, and mutagenesis work are required to fully understand how these sites interact.
Site-directed mutagenesis and functional studies show that Ca2+o and L-amino acids potentiate each other’s activity in a positively cooperative manner (Conigrave et al., 2000; Zhang et al., 2002b, 2014a,b; Mun et al., 2005). Under physiological conditions, L-amino acids potentiate Ca2+o potency for evoking intracellular responses (Conigrave et al., 2007), and mutating residues important for L-amino acid binding eliminated L-Phe potentiation of Ca2+i mobilization (Conigrave et al., 2000; Zhang et al., 2002a). The ability of Ca2+o and L-amino acids to cooperatively activate the CaSR was further demonstrated using saturation transfer difference NMR (Zhang et al., 2014b). Using saturation transfer difference NMR, L-Phe was estimated to bind to the CaSR with an affinity of ∼10 mM in the absence of Ca2+o, whereas in the presence of Ca2+o, L-Phe affinity was increased. Similarly, and as expected for reciprocal cooperativity, the binding affinity of Ca2+o in the presence of 10 mM L-Phe was increased. Therefore, dual binding of Ca2+o and amino acids enhances the sensitivity of the CaSR to changes in concentrations of these ligands.
4. Anion Binding Sites
A total of four anion binding sites in the inactive and active extracellular domain structures were identified based on electron density and crystallization conditions (Geng et al., 2016). Anion binding sites 1-3 are located above the interdomain cleft in lobe 1, and anion site 4 is located in lobe 2. Although SO42− and PO43− anions were modeled into these structures, it is possible other anions may be present. These anions act to stabilize the local conformation of the receptor in the crystal structure because, in the absence of PO43− in the inactive crystal structure, several binding site residue side chains are disordered. In the inactive structure, anions were bound at sites 1-3, whereas in the active structure, only sites 2 and 4 were occupied. In the crystal structures, anions may have stabilized the CaSR to aid crystallization. However, like all GPCRs, the CaSR can sample multiple conformations not captured in these crystal structures. Thus, under physiological conditions, anions may act to stabilize intermediate CaSR states.
5. Etelcalcetide Binding Site
The polypeptide allosteric modulator etelcalcetide binds to a distinct site in the CaSR’s VFT domain and requires a covalent S-S bond formed directly with the CaSR VFT to retain activity (Alexander et al., 2015). This interaction occurs when the free Cys482, which is located at the back of VFT lobe 1 near the hinge loops, exchanges with a L-Cys disulphide bound to a D-Cys in the etelcalcetide D-amino acid peptide sequence. Despite this covalent linkage, the interaction appears transient, and the effect of etelcalcetide on plasma PTH levels rapidly diminishes immediately after withdrawal of intravenous injection (Alexander et al., 2015). It is not known how etelcalcetide binding potentiates CaSR activity at a structural level; therefore, further structural and mutagenesis studies are needed to determine the conformational changes stabilized by etelcalcetide that mediate its PAM activity.
B. Calcium-Sensing Receptor 7-Transmembrane Domain
1. Structural Basis of Calcium-Sensing Receptor 7-Transmembrane Activation
The only full-length class C GPCR structure is of mGlu5, which was determined using cryo-EM. The full-length mGlu5 structure shows how the inactive (or open) VFT receptor complex disrupts the interface between the 7TM domains, whereas the activated (closed) complex forces a reorientation of the 7TM domains, fostering an interface between the top of TM6 and TM7 (Koehl et al., 2019). Without a comparable structure available for the CaSR, similar conformational changes driving CaSR activation can only be hypothesized. Nevertheless, there is significant structural and functional data that are available for the CaSR 7TM that is important for understanding its activity.
Like all GPCRs, the CaSR’s 7TM helices are joined by intracellular loops (ICLs) 1-3, which are important for effector coupling, and ECLs 1-3, in which ECL2 and ECL3 contain a number of residues important for receptor activation (Leach et al., 2012; Goolam et al., 2014). Structural and biochemical data for other GPCR classes show that receptor activation involves an outward movement of TM5 and TM6 to permit G protein coupling and signal transduction. 7TM movements are driven by a number of conserved amino acid sequences important for receptor activation, which are known as switch motifs. How this process may happen in the CaSR is discussed in this section.
Although the CaSR responds to a diverse array of stimuli through its VFT, the VFT is not required for the receptor to respond to Ca2+o. The CaSR 7TM domain alone signals in response to Ca2+o, albeit with lower potency and a significant reduction in the Ca2+o Hill coefficient (Ray and Northup, 2002; Leach et al., 2016). This indicates that the CaSR 7TM also contains one or more orthosteric binding sites. Regrettably, no structures of the CaSR 7TM have been determined experimentally. However, sequence comparisons between the CaSR and mGlu1 or mGlu5 reveal that putative switch motifs important for receptor activation are shared throughout the 7TMs of class C GPCRs, guiding our understanding of CaSR activation.
With the lack of a CaSR 7TM domain structure, the CaSR 7TM has been the subject of extensive mutagenesis and structure-function studies in an attempt to understand this domain. Guided by naturally occurring and engineered mutations and sequence homology with other GPCRs, residues important for Ca2+o activity, allosteric modulation, biased agonism, and biased modulation have been identified (Leach et al., 2013, 2014; Goolam et al., 2014; Cook et al., 2015). Indeed, the putative Ca2+o binding site within the 7TM has been predicted using this approach, in which Ca2+o is hypothesized to mediate an interaction network between Glu767ECL2 and Glu8377.32 (Leach et al., 2016).
The mGlu1 and mGlu5 X-ray structures revealed an ionic lock formed between Lys3.50 (Lys6983.50 in the CaSR) and Glu6.35 (Glu8036.35 in the CaSR) (Dore et al., 2014; Christopher et al., 2015, 2019). These ionic lock residues are conserved across class C GPCRs and this “switch motif” is believed to stabilize the inactive conformation of the class C 7TM domain in the absence of agonist (Dore et al., 2014). Furthermore, a conserved sequence in class A GPCRs important for their activation called the “toggle” switch motif (protein sequence: FxxCWxP6.50) is replaced by a “wl switch motif” (protein sequence: W6.50L6.51) in class C GPCRs (Trzaskowski et al., 2012). Although the wl switch motif differs markedly in sequence from the class A toggle switch motif, most notably by its lack of Pro6.50 to induce a characteristic kink in TM6 (Lagerström and Schiöth, 2008), Trp6.50 in the class C GPCR wl motif (Trp8186.50 in the CaSR) is in an identical position to Trp6.48 in the class A GPCR FxxCWxP6.50 motif (Trzaskowski et al., 2012; Dore et al., 2014). Rotation of the Trp6.48 side chain is a central feature of the toggle switch motif during class A GPCR activation. Molecular dynamic simulations suggest a similar rotation of Trp6.50 may occur in mGluR2 upon activation (Perez-Benito et al., 2017), whereas the mGlu5 crystal structures demonstrate that Trp6.50 can alternate between two distinct rotomers when bound to different NAMs, indicating it differentially orientates upon binding of different ligands (Dore et al., 2014; Christopher et al., 2015, 2019). Thus, it is hypothesized that Trp6.50 in class C GPCRs fulfills an equivalent toggle switch function to Trp6.48 in class A GPCRs (Trzaskowski et al., 2012; Dore et al., 2014). Finally, the CaSR and other class C GPCRs contain a P7.56KxY motif, which is believed to perform an analogous role to the NP7.50xxY(x)5/6F motif (wherein F sits five or six residues away from the Y) in class A GPCRs. The NP7.50xxY(x)5/6F motif undergoes significant rearrangement during activation (Fritze et al., 2003; Katritch et al., 2013; Dore et al., 2014). Nevertheless, without high resolution structures of the CaSR and with only inactive mGlu1 and mGlu5 7TM structures available, it is difficult to confidently determine any importance of these motifs to CaSR activation and effector coupling.
2. Small-Molecule Allosteric Modulator Binding Sites
The CaSR 7TM contains allosteric binding sites for small-molecule allosteric modulators (Ray and Northup, 2002; Petrel et al., 2003, 2004; Miedlich et al., 2004; Hu et al., 2006; Bu et al., 2008; Gerspacher et al., 2010; Leach et al., 2016; Gregory et al., 2018; Keller et al., 2018; Josephs et al., 2019). These sites have been established by mutagenesis studies that examined changes in modulator potency or affinity coupled with homology modeling to understand the context of this mutagenesis data. Initial homology modeling was based on the solved X-ray crystallography structures of class A GPCRs (Miedlich et al., 2004; Hu et al., 2006; Bu et al., 2008; Gerspacher et al., 2010), but this was later extended to modeling based on the NAM-bound 7TM structures of mGlu1 and mGlu5 (Leach et al., 2016; Gregory et al., 2018; Keller et al., 2018; Josephs et al., 2019).
Mutagenesis and homology modeling has established that the CaSR 7TM domain contains an extended allosteric binding pocket formed by Phe6682.56, Arg6803.32, Phe6843.36, Phe6883.40, Glu767ECL2, Leu7765.43, Trp8186.50, Phe8216.53, Tyr8256.56, Val833ECL3, Ser834ECL3, Glu8377.32, Ala8407.35, Ile8417.36, and Ala8447.39 (Leach et al., 2016). This extended pocket overlaps with the allosteric and orthosteric binding sites in biogenic amine class A GPCRs (Kruse et al., 2013) and contains multiple binding sites. For instance, arylalkylamine PAMs and NAMs, such as cinacalcet and NPS 2143, are predicted to form direct salt-bridge interactions with Glu8377.32 at the top of the extended binding pocket supported by substitutions of Glu8377.32 with uncharged or positively charged amino acids, which abolish or significantly reduce arylalkylamine activity (Miedlich et al., 2004; Bu et al., 2008; Leach et al., 2016; Jacobsen et al., 2017; Gregory et al., 2018; Keller et al., 2018; Josephs et al., 2019). AC265347 is believed to bind lower in the allosteric pocket because it lacks the capacity to interact with Glu8377.32 (Leach et al., 2016). Although ATF936 is predicted to bind in a comparable position to the arylalkylamines, mutation of Glu8377.32 has no effect on ATF936 potency or affinity; therefore, some of its binding interactions with the CaSR differ to the arylalkylamines (Gerspacher et al., 2010; Josephs et al., 2019).
Excitingly, the established 7TM allosteric pocket is unlikely to be the only binding site for small-molecule allosteric modulators. The CaSR NAM, BMS compound 1, does not appear to use this binding site because it interacts in a noncompetitive manner with NPS 2143 and is largely unaffected by many of the 7TM mutations that reduce the affinity of other CaSR NAMs (Arey et al., 2005; Josephs et al., 2019). Thus, there remains scope for allosteric modulator binding to multiple sites in the CaSR 7TM.
3. Structural Basis of Small-Molecule Allosteric Modulator Cooperativity, Efficacy, and Bias
Fitting an operational model of agonism or allosterism to functional CaSR data has revealed structural features important for allosteric cooperativity, agonism, and bias. For the PAMs cinacalcet, NPS R-568, and AC265347, mutations Glu767ECL2Ala, Val8176.49Ala, or Ala8447.37Val all reduced the cooperativity of these PAMs (Leach et al., 2016; Keller et al., 2018). However, substantial differences between PAMs have also been described. For instance, although mutation of Phe6883.40Ala, Tyr8256.57Ala, or Leu8487.43Ala reduced the cooperativity of the two arylalkylamine PAMs, cinacalcet and NPS R-568, mutation of Ala6151.42Val or Lys831ECL3Ala only reduced the cooperativity of cinacalcet. Furthermore, mutation of Trp8186.50Ala, which is part of the wl motif discussed above, increased cooperativity of cinacalcet but had no significant effect on NPS R-568 cooperativity. Although structurally and pharmacologically similar, the divergent residues mediating cinacalcet or NPS R-568 cooperativity demonstrate how subtle differences in chemical scaffolds can stabilize distinct structural conformations of the CaSR 7TM domain (Leach et al., 2016; Keller et al., 2018).
The PAM agonist, AC265347, demonstrated further differences from cinacalcet and NPS R-568. For instance, unlike cinacalcet and NPS R-568, mutations Tyr8256.57Ala or Leu8487.43Ala had no effect on AC265347 cooperativity, whereas mutation of Phe6883.40Ala altered AC265347 cooperativity (Leach et al., 2016; Keller et al., 2018). Interestingly, AC265347 biased modulation of pERK1/2 versus Ca2+i mobilization was altered by the mutations Leu7765.43Ala or Trp8186.50Ala. Here, these two mutations increased or decreased AC265347 cooperativity in pERK1/2 assays without altering cooperativity in Ca2+i mobilization assays, providing some insight into 7TM residues that specifically mediated CaSR signaling toward a specific signaling pathway (Cook et al., 2015; Leach et al., 2016). Furthermore, allosteric agonism mediated by AC265347 has different requirements to Ca2+o agonism. Although mutation of Leu7765.43Ala or V8176.49Ala reduced efficacy of both AC265347 and Ca2+o, mutations Phe6843.36Ala or Phe6883.40Ala decreased AC265347 efficacy without altering the efficacy or affinity of Ca2+o (Leach et al., 2016; Keller et al., 2018).
Similar to residues that transmit cooperativity mediated by PAMs, distinct amino acids transmit negative cooperativity mediated by different NAMs. For instance, of the residues analyzed to date, only the mutation Leu7765.43Ala significantly altered NPS 2143 cooperativity (Leach et al., 2016). In contrast, a number of mutations that had no effect on NPS 2143 cooperativity increased or decreased ATF936 cooperativity, including Glu767ECL2Ala, Trp8186.50Ala, and Ile8417.36Ala (Josephs et al., 2019). Other NAMs were sensitive to different mutations (Josephs et al., 2019). Further analysis of additional 7TM mutations will help to unravel cooperativity networks that drive global and ligand-specific allosteric effects.
C. Calcium-Sensing Receptor Dimerization
Like all class C GPCRs, CaSR dimerization is a key feature governing receptor function. The dominant interaction underpinning the CaSR dimer is two covalent disulfide bonds formed at the top of lobe 1 of the VFT domains between Cys129 and Cys131 (Ray et al., 1999). However, the CaSR is not dependent on the disulfide links for activity, as is evidenced by mutation of these residues to Ser, which does not alter surface expression or Ca2+o potency in vitro (Fan et al., 1998; Zhang et al., 2001).
Dimerization influences allosteric modulation at the CaSR. For instance, negative allosteric modulators must bind both protomers to block signaling, whereas PAMs only need occupy one protomer to exert their full modulatory effect (Hauache et al., 2000; Jacobsen et al., 2017; Gregory et al., 2018). This feature likely reflects agonist-mediated signal transmission through the CaSR, which occurs across the dimer rather than propagating through a single protomer (Hauache et al., 2000). Consequently, transactivation across the dimer can result in unique pharmacology for CaSR allosteric modulators. An example is calhex 231, which shows positive allosteric activity when bound to the allosteric site in only one protomer but shows negative allosteric activity when occupying both the allosteric sites of the dimer (Gregory et al., 2018).
Immunoprecipitation data have demonstrated that the CaSR forms heterodimers in vitro with mGlu1/5 or the GABAB receptor, with heterodimers detected in bovine and mouse brain lysates, respectively (Gama et al., 2001; Chang et al., 2007). On the other hand, fluorescence resonance energy transfer studies have revealed that the CaSR does not heterodimerize with its closest receptor homolog, the GPRC6A receptor (Jacobsen et al., 2017). Heterodimerization may facilitate the varied functional roles of the CaSR in different tissues, particularly in the brain, wherein the expression of the GABAB receptor regulates CaSR expression and vice versa (discussed in VK. Calcium-Sensing Receptor in the Brain and Nervous System).
D. Calcium-Sensing Receptor Glycosylation
The CaSR VFT domain contains 11 potential N-linked glycosylation sites; however, not all of these sites have been experimentally verified. The CaSR is glycosylated in the endoplasmic reticulum with mannose (immature) carbohydrate prior to mature complex glycosylation processing in the Golgi. Disruption of at least three glycosylation sites can impair receptor processing and cell surface expression (Ray et al., 1998). Eight glycosylation sites (Asn90, Asn130, Asn261, Asn287, Asn446, Asn468, Asn488, and Asn541) have been experimentally validated, whereas questions remain over the three remaining sites (Asn386, Asn400, and Asn594). Notably, Asn594 was glycosylated in the solved X-ray crystal structure, whereas Asn386 was mutated to Gln to prevent glycosylation and aid crystallization. However, it is unclear whether this observation at a truncated CaSR sample reflects the glycosylation arrangement of the full-length CaSR. Importantly, the functional role of glycosylation beyond controlling surface expression needs further investigation.
IV. CaSR Regulation
A. Phosphorylation and Dephosphorylation
PKC-mediated phosphorylation of the CaSR provides a rapid and quickly reversible mechanism for inhibiting receptor activity. Indeed, treatment of parathyroid cells with PKC-activating phorbol esters overcomes the inhibitory effect of Ca2+o on PTH release (Brown et al., 1984; Nemeth et al., 1986). When first cloned, the CaSR was predicted to contain five PKC consensus motifs, although 54 serine and threonine residues reside in the receptor’s C-terminal tail and ICLs (Garrett et al., 1995a). However, the key inhibitory phosphorylation site is Thr888 in the C-terminal tail (Bai et al., 1998b). Thr888 is most likely phosphorylated by PKCα (Young et al., 2014) and dephosphorylated by a calyculin A–sensitive protein phosphatase (McCormick et al., 2010).
The functional importance of inhibitory Thr888 phosphorylation is most apparent with the clinical mutant, Thr888Met, which cannot be phosphorylated. In vitro Thr888Met is a gain-of-function mutant, whereas it suppresses PTH secretion in vivo, resulting clinically in ADH (see VIC. Autosomal Dominant Hypocalcemia and Bartter Syndrome Type V) (Lazarus et al., 2011). Therefore, CaSR phosphorylation contributes significantly to CaSR activity in vivo and thus to the overall control of PTH secretion and Ca2+o homeostasis [reviewed in Conigrave and Ward (2013)].
The nonphosphorylatable mutant, Thr888Val, also produced a significant gain of function, which was not further enhanced by comutating the other four predicted PKC sites (Bai et al., 1998b). However, PKC inhibition at the wild-type CaSR resulted in a greater gain of function than produced at the Thr888Val mutant, thus it appeared likely that another unknown site may be phosphorylated in tandem with Thr888. However, the identity of this site has remained elusive. In mGlu5, the key PKC phosphorylation site, Ser839 (Kim et al., 2005), aligns not with Thr888 in the CaSR but with Ser875, a residue not originally predicted to be phosphorylated by PKC (Garrett et al., 1995a). Intriguingly, current data indicate removal of this putative phosphorylation site from the CaSR (Ser875Ala) also produces a gain of function, similar to that of Thr888Ala, whereas a phosphomimetic mutation at this site (Ser875Asp) produces a loss of function (Binmahfouz et al., 2019). The double Ser875Ala plus Thr888Ala mutant exhibits a greater gain of function than Ser875Ala alone, and concomitant PKC inhibition exerts no further signal enhancement. Thus, Ser875 is most likely the second major inhibitory PKC site in the CaSR (Binmahfouz et al., 2019).
Ca2+o induces biphasic concentration-dependent phosphorylation of Thr888 in CaSR-HEK cells, with 0.5-2.5 mM Ca2+o eliciting increased Thr888 phosphorylation after 10 minutes, whereas 2.5-5 mM Ca2+o decreases phosphorylation apparently by activating a calyculin A-sensitive protein phosphatase (McCormick et al., 2010). The decrease in Thr888 phosphorylation mediated by 2.5-5 mM Ca2+o occurs at the same Ca2+o concentrations that elicit sustained, as opposed to oscillatory, Ca2+i mobilization. Consistent with this, the Thr888Ala mutant is not only gain-of-function but also exhibits less oscillatory and more sustained Ca2+i mobilization, as does the wild-type CaSR when cotreated with a PKC inhibitor (Davies et al., 2007). Furthermore, PKC-dependent phosphorylation of Thr888 attenuates L-amino acid–dependent signaling in a manner similar to its effect on Ca2+o (Bai et al., 1998b; McCormick et al., 2010). Since PKC-mediated Thr888 phosphorylation is thus a critical regulator of CaSR function, differential CaSR phosphorylation could provide a mechanism to permit biased signaling in different cells or in response to various agonists.
CaSR signaling is also modulated by the GPCR kinases (GRKs). Specifically, overexpression of GRK2 and GRK3 decreases CaSR-induced IP formation in a HEK-derived cell line by >70% (Lorenz et al., 2007). Mutating GRK2 so that it could no longer bind Gq overcame the inhibitory effect of GRK2 on CaSR signaling, indicating that GRK2 inhibition of CaSR signaling might be caused by sequestering of Gq rather than by phosphorylation of the CaSR. Overexpression of either β-arrestin 1 or β-arrestin 2 partly inhibits CaSR-induced IP production, and this effect was abolished by deleting all five of the predicted PKC sites as identified by Bai et al. (1998b) (Lorenz et al., 2007).
B. Internalization and Agonist-Driven Insertional Signaling
In heterologous expression systems, the CaSR undergoes constitutive internalization (Reyes-Ibarra et al., 2007; Gorvin et al., 2018c; Mos et al., 2019) and agonist-induced internalization (Lorenz et al., 2007; Zhuang et al., 2012; Nesbit et al., 2013b). Furthermore, CaSR internalization is increased by the CaSR PAM, NPS R-568, and agonist-induced but not constitutive internalization is inhibited by the NAM, NPS 2143 (Mos et al., 2019). GPCR internalization usually involves desensitization by kinase phosphorylation and subsequent β-arrestin binding followed by recruitment to clathrin-coated pits by β-arrestin and the clathrin-binding AP2 heterotetramer (Hanyaloglu and von Zastrow, 2008). As described above, phosphorylation by PKC and GRKs, or β-arrestin recruitment, is involved in CaSR desensitization (Pi et al., 2005b; Lorenz et al., 2007). Similarly, agonist-induced CaSR internalization requires β-arrestin, which is in contrast with the GABAB and mGluRs, which function independently of β-arrestin (Pin and Bettler, 2016). However, constitutive and agonist-induced CaSR internalization is largely independent of Gq/11 and Gi/o in HEK293 cells (Mos et al., 2019), thus indicating that G protein–mediated activation of PKC is not involved in this cell line. Studies of the internalization mechanisms of endogenously expressed CaSRs in nonrecombinant cells are still lacking because of the technical difficulty of performing such experiments.
The CaSR is also predicted to couple directly to AP2σ through a dileucine motif in the CaSR C-terminal tail (Nesbit et al., 2013b). Similar to loss-of-function mutations in the CASR or GNA11 genes (Pollak et al., 1993; Nesbit et al., 2013a), mutations that disrupt the CaSR interaction with AP2σ reduce the sensitivity of CaSR-expressing cells to Ca2+o (Nesbit et al., 2013b). Similarly, germline mutations of the AP2S1 gene that lead to alteration of Arg15 in AP2σ cause FHH3 (Nesbit et al., 2013b), which is clinically the most severe of the three FHH types (Hannan et al., 2015a) (VI. Calcium-Sensing Receptor–Related Genetic Diseases and Therapeutic Interventions). AP2σ Arg-15 mutations inhibit CaSR internalization (Nesbit et al., 2013b; Gorvin et al., 2018c), and the functional similarity between loss-of-function mutations in the CASR, GNA11, and AP2S1 genes shows a close relationship between internalization and CaSR signaling. This relationship could be explained by reduced resensitization and/or intracellular signaling when internalization is inhibited (Reyes-Ibarra et al., 2007; Zhuang et al., 2012; Gorvin et al., 2018c).
After internalization, GPCRs are either resensitized and recycled to the cell surface or degraded (Hanyaloglu and von Zastrow, 2008). Cell surface expression of CaSR is constant under basal conditions (Reyes-Ibarra et al., 2007; Zhuang et al., 2012), which means constitutively internalized receptors are replaced. In heterologous cells, internalized CaSR is recycled through Rab11a-dependent slow-recycling endosomes (Reyes-Ibarra et al., 2007) to be sorted to lysosomes for degradation (Grant et al., 2011; Zhuang et al., 2012). The CaSR’s C-terminal tail is involved in postendocytic sorting, as deletion of residues 920–970 increased cell surface expression and reduced colocalization with a lysosomal marker (Zhuang et al., 2012). Similarly, overexpression of associated molecule with the SH3 domain of signal-transducing adapter molecule, which interacts with the CaSR C-terminal tail, downregulated cell surface CaSR (Herrera-Vigenor et al., 2006; Reyes-Ibarra et al., 2007).
The interaction of 14-3-3 proteins with an arginine-rich motif in the CaSR C-terminal tail partly retains an intracellular CaSR pool (regulated by Ser899 phosphorylation) (Stepanchick et al., 2010; Grant et al., 2011, 2015), but the CaSR is upregulated at the cell surface upon agonist stimulation via a Gq/11-dependent mechanism called agonist-driven insertional signaling (Grant et al., 2011; Gorvin et al., 2018c). This process involves rapid mobilization of the intracellular pool of receptors to the cell surface and initiation of receptor synthesis to support prolonged upregulation (Grant et al., 2011, 2015). The rapid increase in cell surface receptors is proposed to support the high sensitivity of the CaSR to increases in Ca2+o.
V. (Patho)physiology of the CaSR and Its Ligands
A. Calcium-Sensing Receptor in the Parathyroid Glands
CaSR expression appears during parathyroid development in response to key parathyroid-determining genes, including GCM2 (encoding Gcmb) and SHH (encoding the inhibitory controller, Sonic Hedgehog) (Grevellec et al., 2011). Consistent with a direct connection between Gcmb and CaSR expression, GCM2 control elements have been identified in the CASR promoters (Canaff et al., 2009), and short hairpin RNA directed against GCM2 in parathyroid cell cultures suppressed the protein levels of Gcmb and the CaSR (Mizobuchi et al., 2009).
The CaSR’s nonredundant roles in Ca2+o metabolism have been clearly established by the hypercalcemic disorders, neonatal severe hyperparathyroidism (NSHPT) and FHH, and the hypocalcemic disorders, ADH and Bartter syndrome type V. These are discussed together with animal models of loss- and gain-of-function mutations of the CaSR in VI. Calcium-Sensing Receptor–Related Genetic Diseases and Therapeutic Interventions.
The CaSR negatively controls parathyroid function by suppressing acute PTH secretion primarily from chief cells [review: (Conigrave, 2016)], inhibiting cell proliferation and thus cell number and gland size (Fan et al., 2018), and reducing PTH gene transcription [review: (Chen and Goodman, 2004)]. It also activates the local synthesis, particularly in parathyroid oxyphil cells, of 1,25(OH)2D3 (Ritter et al., 2012), a recognized inhibitor of PTH synthesis. The CaSR’s effects on cell proliferation are particularly noticeable in the context of primary hyperparathyroidism (e.g., due to adenomatous disease) or hyperplasia in the context of CKD. Interestingly, in parathyroid adenoma and CKD, cellular CaSR expression is reduced (Kifor et al., 1996). Nonetheless, in patients with CKD and in rat models of secondary hyperparathyroidism, sustained treatment with cinacalcet suppresses parathyroid gland size as well as serum PTH levels (Colloton et al., 2005; Yamada et al., 2015). Similarly, exposure of parathyroid cells to cinacalcet in vitro suppresses proliferation and promotes apoptosis (Tatsumi et al., 2013).
The parathyroid CaSR continuously monitors the Ca2+o concentration as well as various other stimuli that affect CaSR function, including the plasma levels of L-amino acids (Conigrave et al., 2004), pH (Campion et al., 2015), ionic strength (Quinn et al., 1998), and, perhaps, locally generated polyamines (Quinn et al., 1997) (see II. Agonists and Allosteric Modulators). CaSR activity in the parathyroid glands is resistant to desensitization, in part because of efficient receptor recycling as well as a large intracellular receptor pool that undergoes a high rate of trafficking from the endoplasmic reticulum and Golgi to the plasma membrane [reviews: (Breitwieser, 2013; Ray, 2015)]. Whether agonist-driven insertional signaling operates in parathyroid glands is unknown, but the CaSR interacts with a signaling assembly dependent on caveolin-1 (Kifor et al., 1998) and undergoes AP2σ-regulated endocytosis (Nesbit et al., 2013b; Gorvin et al., 2018c).
1. Parathyroid Hormone Secretion Control
The set point for the CaSR’s half-maximal inhibitory effect on PTH secretion lies at the lower limit of the normal free Ca2+o concentration range (1.1–1.3 mM). In this way, the disinhibited parathyroid provides the body’s primary defense against hypocalcemia (Ca2+o < 1.1 mM). However, the parathyroid CaSR does not provide the primary defense against hypercalcemia, which is mediated by CaSRs in the renal cortical thick ascending limbs (TALs) of Henle’s loop, which accelerate Ca2+o excretion (Kantham et al., 2009; Loupy et al., 2012). Furthermore, as Ca2+o increases, its inhibitory effect on PTH secretion suppresses bone resorption.
Two distinct paradigms for CaSR-mediated inhibition of PTH secretion have been identified at the cellular level: 1) stimulation of Gi/o proteins, which oppose cAMP-dependent increases in PTH secretion mediated by Gs-coupled receptors, for example, adrenaline (β-adrenergic receptors 1 and 2), dopamine, histamine, and prostanoid receptors [review: (Conigrave, 2016)]; and 2) inhibition of endogenous PTH secretion mechanisms that occur in the absence of exogenous activators, at least in part via stimulation of Gq/11 proteins. Endogenous PTH secretion mechanisms may depend upon the intrinsic production of activators for parathyroid secretion [review: (Conigrave, 2016)] or may represent true constitutive secretion (Muresan and MacGregor, 1994).
2. Calcium-Sensing Receptor Structure and Function in the Parathyroid
The primary protein form adopted by the CaSR in parathyroid cells is a disulphide-linked homodimer (Kifor et al., 2003) similar to that observed when the CaSR is expressed in HEK293 cells (Bai et al., 1998a). However, it may also form heterodimers with some other receptors, for example, GABAB receptors, as reported for growth plate chondrocytes (Cheng et al., 2007), with unknown consequences for parathyroid cell signaling and function (see IIIC. Calcium-Sensing Receptor Dimerization).
The CaSR in the parathyroid couples to various signaling pathways as it does when expressed heterologously in HEK293 cells and in other cell types [review: (Conigrave and Ward, 2013)]. As described above, in parathyroid cells the CaSR couples to multiple heterotrimeric G proteins, including, most notably, Gi/o and Gq/11. Of these, Gi/o supports Ca2+o-mediated suppression of PTH secretion stimulated by Gs-coupled receptors [e.g., for dopamine (Brown et al., 1990)] but not intrinsic PTH secretion (Brown et al., 1992). Of apparently greater importance, Gq/11 signaling is absolutely required for CaSR-mediated control of PTH secretion. Thus, mice that are global null for Gα11 or have conditional deletion of Gαq in the parathyroid exhibit mild-moderate hyperparathyroidism. Interestingly, however, crossbreeding to generate mice that are both global null for Gα11 and parathyroid null for Gαq results in severe neonatal hyperparathyroidism (Wettschureck et al., 2007) that is comparable to that seen in human neonates with homozygous or compound heterozygous loss-of-function CaSR mutations (Pollak et al., 1993). Consistent with these observations, loss-of-function mutations of Gα11, which only partially impair signaling, have been linked to a variant form of FHH in humans, now known as FHH2, and gain-of-function mutations of Gα11 have been linked to a variant form of ADH, now known as ADH2 (Nesbit et al., 2013a). The Arg60Cys and Ile62Val gain-of-function mutations in Gα11 also induce ADH2 in mice, in which treatment with the NAM, NPS 2143, or the specific Gq/11 inhibitor, YM-254890, increases PTH and Ca2+o concentrations (Gorvin et al., 2017; Roszko et al., 2017). These findings demonstrate the critical importance of Gq/11 in control of PTH synthesis and/or secretion. CaSR signaling in the parathyroid is also negatively regulated by the GTPase activator RGS5, and overexpression of RGS5 in the parathyroid induces hyperparathyroidism in mice (Koh et al., 2011). Whether RGS5 has a preference for either Gq or G11 in parathyroid cells is unknown. Interestingly, studies in other tissues suggest that RGS5 preferentially suppresses the function of Gq with little or no effect on G11 (Ladds et al., 2007). Whether this might support a parathyroid-based preference for CaSR-mediated activation of G11 rather than Gq is unknown.
The mechanism by which the CaSR controls PTH secretion downstream of Gq/11 is surprisingly ill-defined. Contributing factors appear to include phosphatidylinositol-PLC, which is robustly activated by Ca2+o stimulation in parathyroid cells (Brown et al., 1987a; Shoback et al., 1988); Ca2+i signals whose frequency and amplitude are dependent on the phosphorylation status of Thr888 (McCormick et al., 2010; Lazarus et al., 2011); and the MAPK, ERK1/2 (Corbetta et al., 2002). Evidence has also been presented for a convergent signaling pathway mediated by α-klotho and the CaSR on PTH synthesis and parathyroid hyperplasia downstream of FGF receptors (Fan et al., 2018). In other work, parathyroid Na+/K+-ATPase activity has been implicated in the control mechanism (Brown et al., 1987b; Imura et al., 2007). Whether this might operate via changes in cell volume or intracellular ion concentrations is unclear; changes in Ca2+i concentration appear to have been excluded (Brown et al., 1987b).
B. Calcium-Sensing Receptor in the Thyroid Gland
In the thyroid the CaSR is expressed at high levels in a relatively small subpopulation of cells, the parafollicular C cells (Garrett et al., 1995b). In C cells, the CaSR acts to promote secretion of the peptide hormone calcitonin. Evidence that the CaSR stimulates calcitonin secretion is supported by studies in CaSR knockout mice, in which plasma calcitonin levels were suppressed (Fudge and Kovacs, 2004; Kantham et al., 2009). Thus, elevated Ca2+o stimulates calcitonin release, which, in turn, lowers the plasma calcium level, primarily by suppressing bone resorption. Both the CaSR and calcitonin (or calcitonin gene–related peptide) genes are under inhibitory regulation by thyroid transcription factor-1 in C cells, and CaSR activation promotes calcitonin synthesis, at least in part by suppressing the levels of thyroid transcription factor-1 (Suzuki et al., 1998). CaSR coupling to G proteins in C cells is similar to its coupling in parathyroid cells and various other cell types, and the C-cell CaSR thereby activates plasma membrane phospholipases and cellular protein kinases (McGehee et al., 1997). Activation of the CaSR also stimulates acute elevations in Ca2+i in various C-cell models. In some cell types, this occurs via Ca2+o entry through plasma membrane L-type voltage-gated Ca2+ channels (Muff et al., 1988; Fajtova et al., 1991; McGehee et al., 1997) and, in others, via Ca2+i mobilization (Freichel et al., 1996). Recently, the amino acid–activated CaSR was shown to stimulate calcitonin release from human C cells (Mun et al., 2019). Despite these insights, the molecular mechanisms by which the CaSR stimulates calcitonin secretion are largely unknown.
C. Calcium-Sensing Receptor in the Kidney
CaSR expression in the kidney is one of the highest in the body, and the renal CaSR plays a major role in the regulation of renal function in both a hormone-dependent and independent fashion [see Riccardi and Valenti (2016) and references therein]. A large body of functional, molecular, and genetic evidence indicates that the kidney CaSR plays a crucial role in mineral ion homeostasis. Indeed, the CaSR is widely expressed along the nephron at both the apical and basolateral sides of kidney cells, and thereby it is uniquely poised to monitor both urine and plasma and alter the final ultrafiltrate composition accordingly (Riccardi et al., 1998; Graca et al., 2016). Urinary calcium excretion mirrors serum calcium levels and is directly proportional to the filtered calcium load (Brown, 1991). Within the kidney, the TAL of Henle’s loop is the main site for active divalent cation movement, mostly via the paracellular route, and is coupled to NaCl reabsorption (Friedman, 1998). The latter occurs through a concerted action of an apical Na+:K+:2Cl− cotransporter, NKCC2, and this is followed by basolateral exit via the voltage-gated Cl− channel, chloride channel Kb, and the Na+:K+:ATPase. Overall, NaCl movement generates a favorable transepithelial electrochemical gradient for positively charged ions to move from the urine toward the basolateral side. In concert, the tight junctional proteins, claudins 14, 16, and 19, establish a divalent cation-selective permeable route, thereby allowing Ca2+o (and Mg2+) reabsorption (Gong and Hou, 2014). The TAL has the highest CaSR expression, and here the CaSR is expressed basolaterally (Riccardi et al., 1998). In the event of hypercalcemia, CaSR activation dampens Ca2+o reabsorption in two ways: firstly, it inhibits NaCl reabsorption, hence the driving force for divalent cation movement; secondly, it directly reduces Ca2+o and Mg2+ junctional permeability through its actions on claudin 14 by activating microRNA-9 and -374 (Gong and Hou, 2014). If the hypercalcemic stimulus persists, hypercalciuria can occur with excess urinary calcium excretion in the terminal collecting duct.
In the presence of hypovolemia, the antidiuretic hormone, vasopressin, promotes water reabsorption through the insertion of aquaporin-2 water channels into the lumen of inner medullary collecting duct cells. However, excessive water reabsorption could lead to suprasaturating urinary calcium concentrations and attendant pathologic kidney stone formation, which could severely impair renal function. The CaSR is expressed at the luminal side of inner medullary collecting duct cells, where it monitors Ca2+o concentration in the urine (Sands et al., 1997). Thus, CaSR activation inhibits the tubular response to vasopressin by limiting the number of apical aquaporin-2 water-channel insertions (Procino et al., 2012). In addition, CaSR activation stimulates the activity of the proton pump, V-ATPase, thereby evoking urine acidification and reducing the risk of precipitation (Renkema et al., 2009).
Further, the kidney proximal tubule is a major site of PTH action that promotes a phosphaturia by inhibiting the activity of the Na+:Pi cotransporters, Npt2a, and Npt2c (Murer et al., 2001). Excess phosphate in the urine could also exacerbate the risk of calcium-phosphorus stone formation by the distal nephron. In the proximal tubule, a luminal CaSR blunts the phosphaturic action of PTH and promotes acid secretion via stimulation of the Na+:H+ exchanger, Na+:H+ exchanger 3 (Capasso et al., 2013). Thus, by monitoring both urine and plasma composition, together with the integration of inputs deriving from urinary phosphate content, concentration, and acidification, the renal CaSR accomplishes divalent cation homeostasis while minimizing the risk of developing nephrolithiasis and nephrocalcinosis, which could arise as a consequence of enhanced urinary calcium excretion by the TAL (Hebert et al., 1997). The corollary is that altered CaSR expression or function due to CaSR mutations leads to FHH1, ADH1, or Bartter syndrome type V (see VI. Calcium-Sensing Receptor–Related Genetic Diseases and Therapeutic Interventions). In all circumstances, the aberrant calciuria is not the consequence of an impairment of renal function but rather the result of altered Ca2+o sensing by the CaSR in the parathyroid glands and the kidney. In the context of CKD, hyperphosphatemia caused by decreased renal phosphate excretion and acidosis may both elicit CaSR underactivation, leading to secondary hyperparathyroidism. Therefore, similarly to the parathyroid CaSR, the kidney CaSR is a drug target and, indeed, pharmacological CaSR PAMs are employed to rectify abnormal Ca2+o sensing by the kidney (Riccardi and Valenti, 2016). Furthermore, the use of NAMs for the treatment of nephrolithiasis and nephrocalcinosis could also be postulated (Riccardi and Valenti, 2016). Finally, it should be noted that CaSR PAMs increase urinary calcium excretion by means of their actions on both the parathyroid and kidney CaSR, and indeed, cinacalcet promotes calciuria in patients with secondary hyperparathyroidism, but this occurs in the absence of an increase in urine output (Courbebaisse et al., 2012). Given the clinical use of PAMs, the impact of their long-term use on urine production, acidification, and concentration, particularly in the context of kidney stone formation, remains to be fully understood (Riccardi and Valenti, 2016).
D. Calcium-Sensing Receptor in the Bone
The CaSR is expressed by several types of bone cells, including osteoblasts, osteocytes, osteoclasts, and some chondrocytes (Santa Maria et al., 2016). Although some controversies exist, there is good evidence that Ca2+o and the CaSR contribute to skeletal development and maintenance (Chang et al., 2008; Goltzman and Hendy, 2015; Hannan et al., 2018a) and that bone CaSRs may even contribute to overall Ca2+o homeostasis (Al-Dujaili et al., 2016). Elucidation of the CaSR’s roles in skeletal tissue was historically complicated by models that examined global deletion of the Casr gene (Kos et al., 2003). Global Casr deletion has numerous effects, partly through large alterations in PTH secretion and changes in serum calcium and phosphate concentrations (Hannan et al., 2018a), thus it is difficult to elucidate tissue-specific CaSR effects. To further complicate matters, early Casr knockouts involved deletion of Casr exon 5, which results in mice encoding a nonfunctional CaSR lacking a portion of its extracellular domain (Kos et al., 2003). When Casr exon 5–deleted mice were crossed with mice that had a deletion of Gcm2 (which results in no parathyroid gland development) or the Pth gene, the skeletal abnormalities seen in the global Casr knockout mice were largely abolished (Kos et al., 2003; Tu et al., 2003). Furthermore, studies of the Casr exon 5–deleted mice revealed an alternatively spliced Casr transcript in the growth plate and other organs, such as skin, that could compensate for the absence of full-length CaSR in cartilage and bone (Rodriguez et al., 2005). Nonetheless, alternative Casr knockout models and bone-specific Casr deletion has confirmed that the CaSR is critical to bone development and maintenance, as described below.
1. Osteoblast Calcium-Sensing Receptors
Perhaps the clearest evidence for a role of the CaSR in skeletal development and maintenance comes from studies in which exon 7 of the Casr gene was deleted during different stages of osteoblast differentiation. Casr exon 7 deletion removes most of the 7TM and C-terminal tail, resulting in a nonfunctional receptor (Chang et al., 2008; Dvorak-Ewell et al., 2011). Casr exon 7 deletion was achieved by Cre-recombinase in osteoblasts under the control of the 2.3-kb Col(I) α1 subunit promoter [2.3Col(I)-Cre], which is expressed in early- and late-stage cells of the osteoblast lineage (Chang et al., 2008); an α1(I) collagen promoter [Col 3.6-Cre], which is expressed throughout cells of the osteoblastic lineage (Dvorak-Ewell et al., 2011); or the osterix promoter, which is expressed in early osteoblasts (Chang et al., 2008). In studies using the collagen-Cre promoters, heterozygous Casr knockout mice grew relatively normally (Chang et al., 2008; Dvorak-Ewell et al., 2011). In contrast, homozygous Casr knockout using any of the Cre promoters resulted in severe bone defects (Chang et al., 2008; Dvorak-Ewell et al., 2011). There was marked reduction in the size of the knockout mice and their skeletons evident as early as 3 days after birth, and at day 20 the weight of the Casr knockout mice was only 30% that of controls (Chang et al., 2008; Dvorak-Ewell et al., 2011). Their skeletons were severely undermineralized, even in the skull, as well as in the vertebrae and long bones (Chang et al., 2008; Dvorak-Ewell et al., 2011). There was a marked reduction in bone volume in both the trabecular and cortical bones (Chang et al., 2008). Most of these mice died with multiple fractures by 3 to 4 weeks after birth (Chang et al., 2008; Dvorak-Ewell et al., 2011). Osteoblasts from Casr knockout mice were poorly differentiated, with both early and late differentiation markers markedly reduced, compared with controls (Chang et al., 2008; Dvorak-Ewell et al., 2011). mRNA levels of the local growth factor, insulin-like growth factor-1, were also substantially decreased, as were those of factors supporting cell survival, such as B-cell lymphoma 2 (Bcl-2) and Bcl-2L1 (Chang et al., 2008). In contrast, mRNA for IL-10, an inducer of apoptosis in many cell types, was increased, along with evidence of augmented apoptotic osteoblast and osteocyte numbers in sections from bone (Chang et al., 2008; Dvorak-Ewell et al., 2011). mRNA-encoding genes that inhibit mineralization, such as osteopontin, ankylosis protein, and nucleotide pyrophosphatase/phosphodiesterase 1 (NPP1), showed increased expression in the knockouts (Dvorak-Ewell et al., 2011). In addition to impaired osteoblastic differentiation and activity, deletion of Casr in early and late osteoblasts led to increased expression of mRNA for the bone resorption-promoting protein, receptor activator of nuclear factor-κB ligand, together with a doubling of osteoclast numbers and activity, with bone loss in trabecular and cortical bone (Chang et al., 2008; Dvorak-Ewell et al., 2011). Whether these effects of osteoblast Casr knockout are entirely specific is not clear, since transplantation of vertebrae from 10-day-old wild-type or homozygous Casr knockout mice into athymice mice resulted in no differences in the volume or composition of transplanted bones, as assessed by microCT or histomorphometry after 4 weeks (Al-Dujaili et al., 2016). Furthermore, bones of transgenic mice that expressed a constitutively active mutant CaSR in late osteoblasts under the control of the osteocalcin promoter (Dvorak et al., 2007) also showed increased receptor activator of nuclear factor-κB ligand expression and increased osteoclast activity, with resultant bone loss over the lifespan of the mice, but only in trabecular and not cortical bone (Dvorak et al., 2007). Other osteoblastic markers and function were largely unaffected, except for a slight decrease in bone-forming activity indicated by a small drop in mineral apposition rate (Dvorak et al., 2007). It is difficult to explain these observations in a comprehensive model of the CaSR’s role in bone, despite attempts to propose age-related differences in the interactions between the CaSR and PTH/PTH1R in bone or the presence of mild hyperparathyroidism in the CaSR knockout models, but not in the mutant constitutively active CaSR mice (Dvorak et al., 2007; Goltzman and Hendy, 2015). Under conditions of expression of a constitutively active CaSR, however, normal feedback mechanisms would not function. The existing evidence indicates that Ca2+o and the CaSR, together with PTH/PTHrP and PTH1R, interact with one another in whole animals in ways that cannot easily be predicted (Goltzman and Hendy, 2015; Santa Maria et al., 2016; Yang and Wang, 2018).
2. Osteoclast Calcium-Sensing Receptors
Local regulatory pathways relevant to Ca2+o and the CaSR are likely to involve osteoclasts, which, along with bone marrow monocytes and macrophages, express the CaSR (House et al., 1997; Kameda et al., 1998; Diepenhorst et al., 2018). Activation of the CaSR in these cells with high concentrations of Ca2+o or Sr2+ S inhibited osteoclast maturation and secretion of acid phosphatase, which is critical for bone resorption, and increased apoptosis of mature osteoclasts, all of which would suppress bone resorption (Zaidi et al., 1991; Kameda et al., 1998; Kanatani et al., 1999; Mentaverri et al., 2006; Diepenhorst et al., 2018). Although high Ca2+o concentrations were required to activate these osteoclast responses, this might be relevant in vivo, since an acid environment, as present in resorption pits, increases Ca2+o potency at the CaSR (Quinn et al., 2004), and the Ca2+o concentration in bone resorption pits can be as high as 40 mM (Silver et al., 1988). There is some recent evidence that cinacalcet can inhibit the actions of osteoclasts (Diepenhorst et al., 2018), raising the possibility of CaSR activation in osteoclasts as a potential antiresorptive strategy in osteoporosis. However, another study found no effect of cinacalcet on osteoclast-mediated resorption (Shalhoub et al., 2003).
3. Osteoblast and Osteoclast Calcium-Sensing Receptors as Therapeutic Targets
Given the negative effects of CaSR deletion on bone mass and bone cell survival (Chang et al., 2008; Dvorak-Ewell et al., 2011; Santa Maria et al., 2016), it follows that there would be interest in targeting the CaSR in osteoblasts/osteocytes for a bone anabolic effect (Marie, 2010; Goltzman and Hendy, 2015; Diepenhorst et al., 2018). Indeed, there is evidence that Sr2+, which displays higher potency than Ca2+o in osteoblasts (Brennan et al., 2009), increased bone mineral density and reduced fractures in the clinic (Reginster et al., 2005). Other receptors, including GPRC6A, may also mediate the effects of Sr2+ (Pi et al., 2005a; Rybchyn et al., 2009). Unfortunately, reported cardiovascular side effects of Sr2+ ranelate (marketed as Protelos/Osseor) narrowed the potential patient population so that this agent was withdrawn from the market. Nevertheless, preclinical studies showed Sr2+ reduced bone-resorbing signals and increased bone cell anabolism and survival under stress (Bonnelye et al., 2008; Brennan et al., 2009; Rybchyn et al., 2011). They also reported that Sr2+ stimulated the important bone anabolic Wnt pathway downstream of the CaSR and Akt phosphorylation in osteoblasts (Rybchyn et al., 2011). CaSR-dependent activation of the Wnt pathway in bone cells was in turn dependent on the formation of a complex involving CaSR, Homer1 (a long isoform of this scaffold protein), and mechanistic target of rapamycin complex-2, which phosphorylates Akt on Serine 475 (Rybchyn et al., 2019). These observations provide proof of principle that selective activation of the CaSR in osteoblasts might be a suitable strategy for osteoporosis therapies, either alone or in combination with other anabolic agents, such as intermittent PTH. Intermittent PTH has anabolic effects on bone but also stimulates osteoclast activity. Given that CaSR activation in osteoclasts suppresses bone resorption, as discussed above, and has anabolic effects on bone, the use of CaSR PAMs in conjunction with intermittent PTH may reduce the likelihood of hypercalcemia and enhance the bone anabolic effects of intermittent PTH. Indeed, administration of the PAM, NPS R-568, in combination with intermittent PTH in mice reduced blood Ca2+o concentrations, increased trabecular bone, and increased cortical bone strength compared with intermittent PTH alone (Santa Maria et al., 2016). The effect of intermittent PTH on trabecular bone volume as a fraction of total bone volume was slightly but significantly blunted in mice in which the Casr gene was deleted in early and late osteoblasts (Al-Dujaili et al., 2016).
4. Chondrocyte Calcium-Sensing Receptors
High levels of CaSR protein are present in hypertrophic chondrocytes in the growth plate of long bones (Santa Maria et al., 2016). When mice with the loxP sites flanking exon 7 of the Casr were crossed with mice expressing the Cre transgene under the control of the type II collagen α1 subunit [Col(II)] promoter [Col(II)-Cre], which targets growth plate chondrocytes and other types of chondrocytes, they all died in utero at around E13 (Chang et al., 2008). Whether this was due to interference in heart valve development is unclear. When the Col(II)-Cre promoter was modified to a tamoxifen-inducible variant and 4-hydroxytamoxifen was given at E18-19, the resultant growth plate chondrocyte targeted Casr knockout and produced small, undermineralized skeletons, with expansion and reduced mineralization of the hypertrophic zone of the growth plate (Chang et al., 2008). Gene expression analysis confirmed reduced expression of terminal differentiation markers and reduced expression of insulin-like growth factor-1 and its receptor (Chang et al., 2008).
The CaSR is also expressed in articular cartilage chondrocytes, with increased expression reported in chondrocytes from osteoarthritic joints (Burton et al., 2005). Increased expression of the CaSR was also reported in cartilage endplate chondrocytes adjacent to degenerated intervertebral discs from human subjects along with high total calcium concentrations and low water content (Grant et al., 2016). Treatment of cartilage endplate chondrocytes in vitro with increasing Ca2+o resulted in lower accumulation of collagens I and II and aggrecan, whereas catabolic enzymes were increased, an effect that was abrogated by knockdown of the CASR (Grant et al., 2016). The authors proposed a role for increased Ca2+o and the CaSR in intervertebral disc degeneration (Grant et al., 2016). In a dental malocclusion model affecting the temporomandibular joint in rats, increased expression of CaSR in articular chondrocytes and in the endoplasmic reticulum of these cells was also observed (Zhang et al., 2019). These provided some evidence to support a role for endoplasmic reticulum expressed CaSR, as opposed to cell membrane CaSR in chondrocytes under stress (Zhang et al., 2019). Increased whole-cell CaSR and increased endoplasmic reticulum CaSR were also observed in vitro using articular cartilage chondrocytes exposed to shear stress (Zhang et al., 2019). Shear stress resulted in increased expression of chondrocyte terminal differentiation markers, such as alkaline phosphatase, osteocalcin, and matrix metalloprotease-13, which contributes to cartilage degradation. Critically, local CaSR knockdown or the use of the NAM, NPS 2143, reduced the shear stress-induced increases in terminal differentiation markers in chondrocytes in culture and reduced the severity of osteoarthritis in the temporomandibular joint of a rat model of dental malocclusion (Zhang et al., 2019). In contrast, injection of the PAM, cinacalcet, into the temporomandibular joint of these rats promoted thinning and loss of articular cartilage (Zhang et al., 2019). These studies in chondrocytes raise the possibility that the chondrocyte CaSR is a potential therapeutic target for prevention or management of joint degeneration.
E. Calcium-Sensing Receptor in Keratinocytes
The CaSR is highly expressed in keratinocytes, the main epidermal cell type. Moreover, an ionic calcium gradient exists in the epidermis, which increases from the basal proliferative layer to reach a maximum in the stratum granulosum, wherein the keratinocytes are well-differentiated, decreasing again in the relatively water-deficient lipid-containing cells of the stratum corneum (Menon et al., 1985; Celli et al., 2010). The epidermal calcium gradient and the CaSR are critically important for various epidermal functions, including keratinocyte differentiation, water and xenobiotic barrier function, and wound healing (Tu and Bikle, 2013; Hannan et al., 2018a). Interestingly, the epidermal calcium gradient is predominantly present in intracellular organelles of keratinocytes, such as the endoplasmic reticulum and Golgi, although an extracellular gradient makes some contribution to the gradient (Celli et al., 2010).
Keratinocytes cultured in low-calcium media (<0.07 mM) proliferate well. Raising the Ca2+o concentration above 0.1 mM promotes differentiation, as indicated by the appearance of E-cadherin/catenin complexes (adherens junctions) and desmosomes, upregulation of keratins 1 and 10, stratification of cells, and then formation of cornified envelope precursors (Braga et al., 1995). Disruption of the permeability barrier of the skin by tape stripping disrupts the epidermal calcium gradient, resulting in disorganization of the normally differentiated cell layers (Menon et al., 1994). When the calcium gradient is re-established over the next day or so, the permeability barrier also recovers. Skin diseases, such as psoriasis, characterized by abnormal barrier function also exhibit a loss of the calcium gradient (Menon and Elias, 1991).
CaSR expression increases in upper layers of the epidermis with the increase in differentiation, with high expression in the stratum granulosum but weak expression in the corneocytes (Komuves et al., 2002). There is some expression of the CaSR on the plasma membrane of keratinocytes, but its predominant localization in these cells is intracellular and in the cytoplasm around the nucleus (Komuves et al., 2002). This perinuclear localization is also seen, although not to the same extent, in rodent osteoblasts and chondrocytes (Chang et al., 1999). It is likely that Ca2+o signals to the keratinocyte via the plasma membrane CaSR in a manner similar to that of more classic calcium targets, such as the parathyroid or kidney. The function of the intracellular CaSR is unclear at this time. Knockdown or inactivation of the CaSR in keratinocytes abrogates calcium-induced inhibition of proliferation and stimulation of differentiation of these cells (Tu et al., 2008). Not surprisingly, in mice in which there had been knockdown of the CaSR in the epidermis, skin barrier function was disrupted with impaired differentiation of keratinocytes, and these problems were exacerbated by a low-calcium diet (Tu et al., 2012). Keratinocytes from these epidermal Casr−/− mice had blunted Ca2+i mobilization in response to Ca2+o, decreased Ca2+i pools, defective cell-cell adhesion, and reduced expression of differentiation markers (Tu et al., 2012).
In contrast, mice engineered to constitutively overexpress the CaSR in basal keratinocytes displayed enhanced keratinocyte differentiation and barrier formation during development as well as accelerated hair growth at birth (Turksen and Troy, 2003). There was hypertrophy of the suprabasal keratinocyte layers with increased expression of early and late differentiation markers together with upregulation of epidermal growth factor and noncanonical Wnt-signaling pathways (Turksen and Troy, 2003).
In the epidermis, there are interactions between the CaSR and the vitamin D system in skin. The active vitamin D hormone, 1,25(OH)2D, increases the calcium response in keratinocytes (Ratnam et al., 1999). Deletion of the epidermal CaSR reduces expression of both the vitamin D receptor (VDR) and CYP27B1, the enzyme that produces 1,25(OH)2D from 25-hydroxyvitamin D (Tu et al., 2012). It is likely that these effects on the vitamin D system contribute to impaired differentiation of the epidermis in these mice and reduced function of the innate immune system (Schauber et al., 2007). Moreover, 1,25(OH)2D increases transcription of the CASR (Canaff and Hendy, 2002).
It has previously been reported that wound healing is impaired in mice with epidermal deletion of the VDR (Oda et al., 2017). Very low dietary calcium or deletion of the Casr gene exacerbates this impairment in wound healing in epidermal VDR-deficient mice (Oda et al., 2017). There is a robust increase in Ca2+ in the bed of wounds within minutes of injury (Jungman et al., 2012), rapid increase in Ca2+i in cells near the site of the wound, and spreading to surrounding cells (Tsutsumi et al., 2013), with all of these indicating that the CaSR may play an important role in wound healing. The CaSR is coexpressed with E-cadherin at the cell membranes of migratory keratinocytes. Blockade of either the CaSR or E-cadherin inhibited keratinocyte proliferation and migration after wound induction (Tu et al., 2019). Accordingly, the PAM, NPS R-568, accelerated wound healing in normal mice, potentially pointing to the epidermal CaSR as a therapeutic target to enhance repair of skin wound (Tu et al., 2019).
Mice with epidermal knockout of the VDR are more susceptible to UV- or chemically induced skin tumor formation (Zinser et al., 2002; Ellison et al., 2008). Neither the epidermal VDR knockout mice nor mice with epidermal Casr knockout develop skin tumors spontaneously, but mice null for both epidermal Vdr and Casr are reported to spontaneously develop squamous cell carcinomas (Bikle et al., 2015). In keratinocytes, stimulation of Wnt signaling results in β-catenin translocation to the nucleus and subsequent transcriptional activity, which may be important in skin tumorigenesis (Wei et al., 2007; Youssef et al., 2012). The VDR appears to suppress this β-catenin transcriptional activity in skin (Wei et al., 2007), in part by helping to keep β-catenin at the cell membrane as part of the E-cadherin/catenin complex (adherens junctions). As noted earlier, the CaSR is also important for the development of the E-cadherin/catenin complex, which helps to retain β-catenin at the cell membrane by promoting wound healing and differentiation of the skin cells (Oda et al., 2017) and inhibiting nuclear translocation and associated protumorigenic activities of β-catenin (Wei et al., 2007). This is in direct contrast with osteoblasts, wherein activation of the CaSR predominantly promotes β-catenin stabilization, subsequent β-catenin translocation to the nucleus, and increased transcriptional activity (Rybchyn et al., 2011). Some preliminary data indicate that both CaSR PAMs and NAMs enhance DNA repair after UV damage in cultured keratinocytes (Yang et al., 2016), although the mechanism and why PAMs and NAMs have a similar effect are unclear. How this observation fits with observed effects of CaSR knockdown in mice remains to be examined.
F. Calcium-Sensing Receptor in the Gastrointestinal Tract
The CaSR is expressed along the entire gastrointestinal tract in parietal and G cells of stomach gastric glands (Ray et al., 1997; Busque et al., 2005; Feng et al., 2010; Engelstoft et al., 2013), epithelial and enteroendocrine cells of the small and large intestine (Liou et al., 2011; Wang et al., 2011; Cheng et al., 2014; Alamshah et al., 2017), and neurons of the submucosal and myenteric plexuses of the enteric nervous system (ENS) (Geibel et al., 2006; Cheng, 2012; Tang et al., 2018). In the gastrointestinal tract, the CaSR functions as a nutrient sensor, binding not only Ca2+, Mg2+, and other cations but also L-amino acids and dipeptides and polypeptides (e.g., glutamyl dipeptides, poly-L-lysine). The CaSR is involved in regulation of gastric acid and hormone secretion, nutrient absorption, intestinal fluid homeostasis, energy metabolism, cellular differentiation and proliferation, motility and enteric nerve activity, maintenance of gut microbiota, immune homeostasis, and intestinal inflammation (Dufner et al., 2005; Ceglia et al., 2009; Geibel and Hebert, 2009; Feng et al., 2010; Brennan et al., 2014; Cheng et al., 2014; Tang et al., 2016b, 2018; Alamshah et al., 2017; Sun et al., 2018).
The CaSR responds to alterations in nutrient levels by regulating hormone secretion from enteroendocrine cells (Geibel and Hebert, 2009; Liou et al., 2011; Wang et al., 2011; Alamshah et al., 2017; Liu et al., 2018). In global Casr knockout mice, gastric G cell number was significantly reduced, suggesting the CaSR regulates G cell growth. Further, in wild-type but not knockout mice, NPS 2143 inhibited gastrin secretion after gavage of Ca2+o, L-Phe, or cinacalcet (Feng et al., 2010). In rat whole-stomach preparations, ex vivo exposure to Ca2+o increased acid production in parietal cells by enhancing H+-K+-ATPase activity. These effects were potentiated by L- but not D-amino acids, implicating the CaSR (Busque et al., 2005). The function of the recently identified acid secretory protein, vacuolar H+-ATPase, in parietal cells is also dependent on CaSR activity (Kitay et al., 2018).
The amino acid–stimulated CaSR may influence appetite and satiety via stimulatory effects on satiety hormones, cholecystokinin (CCK), glucagon-like peptide 1 (GLP-1) and protein YY (PYY) (Alamshah et al., 2017), and/or inhibitory effects on the release of the appetite-stimulating hormone ghrelin (Engelstoft et al., 2013). In the mouse enteroendocrine cell line, STC-1, L-Phe increased PYY and GLP-1 secretion, an effect inhibited by NPS 2143, suggesting involvement of the CaSR (Alamshah et al., 2017). In a transgenic mouse model expressing a CCK promoter–driven enhanced GFP, the CaSR was enriched in CCK-producing duodenal I cells, in which L-Phe and cinacalcet induced Ca2+i changes and stimulated CCK in the presence of Ca2+o (Liou et al., 2011). L-Phe- and Trp-stimulated CCK secretion was inhibited by calhex 231 (Wang et al., 2011). In intestinal L-cells, the CaSR was involved in peptone-stimulated GLP-1 release (Pais et al., 2016), whereas in a ghrelinoma cell line, the CaSR partially mediated the L-Phe, L-Ala, and peptone-induced secretion of octanoyl ghrelin (Vancleef et al., 2015). In swine duodenum, ex vivo L-Trp perfusion induced secretion of CCK and glucose-dependent insulinotropic peptide and upregulated CaSR expression. This effect was inhibited by NPS 2143 (Zhao et al., 2018). To conclude that the Trp-induced gut-hormone secretion is mediated by the CaSR, further proof is needed. In mice and rats, L-Phe reduced short-time food intake and plasma ghrelin release, affecting the appetite of the animals (Alamshah et al., 2017). In minks, CaSR-mediated secretion of CCK and PYY led to emesis (Wu et al., 2017). These findings might explain why cinacalcet and other CaSR PAMs cause gastrointestinal side effects (Block et al., 2017).
Specific knockout of intestinal epithelial cell Casr leads to epithelial cell hyperproliferation and changes in intestinal crypt structure driven by β-catenin signaling (Rey et al., 2012). Mice additionally have decreased intestinal transepithelial resistance and reduced levels of colonic tight-junction proteins, suggesting that the epithelial CaSR maintains intestinal barrier function (Cheng et al., 2014). The inadequate epithelial barrier function was associated with lower amounts of beneficial Lactobacilli bacteria and more Deferribacteraceae bacteria, which are linked to colitis (Cheng et al., 2014). This intestinal dysbiosis has been associated with more severe proinflammatory responses in the intestinal epithelium-specific Casr null mice compared with wild-type controls (Owen et al., 2016).
The amino acid-stimulated CaSR has recently been found to suppress intestinal inflammation in inflammatory bowel disease and other settings [reviews: (Owen et al., 2016; Sun et al., 2018)]. Inflammatory cytokine expression, including IL-1R, was higher in the distal colons of the CaSR knockout mice, in addition to a marked increase in nuclear factor-κB–dependent genes (Cheng et al., 2014). These mice developed more severe colitis with delayed recovery than the CaSR-expressing littermates when challenged with dextrane sulfate sodium (DSS) (Cheng et al., 2014). In a mouse model of colitis, poly-L-lysine (commonly used as a food preservative) and glutamyl dipeptides reduced DSS-induced inflammation, whereas intravenous administration of NPS 2143 inhibited this effect (Mine and Zhang, 2015; Zhang et al., 2015). Dietary supplementation of Trp, L-Phe, and Tyr also reduced the expression of intestinal inflammatory markers in piglets after short-term induction of inflammation by lipopolysaccharides (Liu et al., 2018). However, a recent study found that the CaSR PAMs, cinacalcet and GSK3004774 (an intestine-specific modulator), did not reduce the inflammatory effects of DSS, whereas NPS 2143 ameliorated the DSS-induced symptoms and reduced immune cell infiltration (Elajnaf et al., 2019).
The anti-inflammatory effect of the CaSR was also shown in vitro in cell lines. In a colonic myofibroblast cell line, activation of the CaSR inhibited tumor necrosis factor-α secretion (Kelly et al., 2011) and increased expression of bone morphogenetic protein-2, which is a promoter of colonic epithelial barrier maturation (Peiris et al., 2007). In colon cancer cell lines, amino acids and dipeptides inhibited proinflammatory cytokine secretion, and the effect was reversed by NPS 2143 (Mine and Zhang, 2015; Zhang et al., 2015). Inflammatory cytokines, such as tumor necrosis factor-α, IL-1β, and IL-6, increased the expression of the CaSR at the mRNA and protein level in some colon cancer cell lines (Fetahu et al., 2014), which could be a defense mechanism against inflammation in the intestines.
Bicarbonate (HCO3−) secretion in the colon is fine-tuned by the CaSR (Tang et al., 2015). However, it seems that the neurogenic secretory responses in the intestinal epithelium are mediated mainly by the CaSR expressed in the ENS and not in the epithelium (Geibel et al., 2006; Cheng, 2012; Tang et al., 2018). Because abnormalities of the ENS affect the severity of intestinal inflammation and contribute to the pathogenesis of inflammatory bowel disease (Margolis et al., 2011), the CaSR could be a potential therapeutic target.
Increased dietary intake of calcium reduces the risk of several cancers. The inverse correlation between calcium intake and risk of colorectal cancer has been known for decades, although the mechanisms driving the protective effect of calcium were not clear. There is some evidence that the CaSR is one of the central mediators of the antitumorigenic effects of calcium and acts as a tumor suppressor (Kállay et al., 1997, 2003; Yang et al., 2018). In colon cancer cells, activation of the CaSR increased differentiation and reduced proliferation, epithelial-to-mesenchymal transition, and expression of stem cell markers (Aggarwal et al., 2015, 2017). The signaling pathways involved in these processes still need to be determined. Interestingly, in the upper intestinal tract, it seems that the CaSR functions as an oncogene, as it promoted gastric cancer cell proliferation (Xie et al., 2017).
G. Calcium-Sensing Receptor in the Pancreas
The CaSR is expressed in pancreatic acinar cells (Bruce et al., 1999), which promote digestion via nutrient-stimulated release of digestive enzymes and fluid. The CaSR is also expressed in the pancreatic islets on glucagon-secreting α cells and insulin-secreting β cells (Babinsky et al., 2017). Thus, the CaSR may influence not only protein metabolism but also carbohydrate and fat metabolism.
Ca2+o is critical for pancreatic islet function and acts via voltage-gated Ca2+ channels to trigger the exocytosis of insulin- and glucagon-containing secretory granules from β- and α-cells, respectively (Rorsman et al., 2012). Ca2+o also activates the pancreatic islet CaSR, with ex vivo and in vitro studies demonstrating a role for the CaSR in mediating islet hormone secretion. Thus, stimulation of isolated human islets and an insulin-secreting mouse cell line (MIN6) with the PAM, NPS R-568, potentiated Ca2+o-mediated insulin secretion (Gray et al., 2006), whereas knockdown of the CaSR through RNA interference diminished glucose-induced insulin secretion in MIN6 cells that were cultured as pseudoislets (Kitsou-Mylona et al., 2008). Studies involving MIN6 pseudoislets have also revealed CaSR-stimulated insulin secretion to be mediated by PLC and the MAPK pathway (Gray et al., 2006). Furthermore, CaSR-mediated MAPK activation in MIN6 cells induces β-cell proliferation, thus highlighting a potential role for the CaSR in the regulation of β-cell mass (Kitsou-Mylona et al., 2008). The CaSR also upregulates the expression of E-cadherin in MIN6 cells, which is associated with increased adherence between neighboring β-cells (Hills et al., 2012). Thus, the CaSR may facilitate cell-to-cell communication within an individual pancreatic islet to coordinate insulin secretion from β-cells (Hodgkin et al., 2008). In addition, the level of islet CaSR expression correlates with insulin secretion from isolated wild-type mouse pancreatic islets (Oh et al., 2016). Studies in wild-type mice have shown that islet CaSR expression increases with age, which may compensate for the insulin resistance in aged mice by increasing insulin secretion (Oh et al., 2016). Transient stimulation of isolated human islets with Ca2+o and NPS R-568 also promoted glucagon secretion, thereby indicating a role for the CaSR in α-cells (Gray et al., 2006). The intestinal CaSR, which is activated by dietary amino acids and peptides, may also influence pancreatic islet function by regulating the secretion of incretin hormones. In support of this, studies involving isolated mouse intestine have shown that the CaSR is expressed in GLP-1-secreting L-cells and also that oligopeptides enhance GLP-1 secretion by activation of the CaSR (Diakogiannaki et al., 2013).
The role of the CaSR in systemic glucose homeostasis has been investigated in studies involving human subjects. An association study reported a common coding-region CASR gene variant to be an independent determinant of plasma glucose concentrations in renal transplant recipients (Babinsky et al., 2015). However, another study involving patients with FHH1 (caused by germline loss-of-function CASR mutations) did not reveal any alterations in insulin secretion or glucose tolerance (Wolf et al., 2014). The effect of altered CaSR function on glucose tolerance has also been evaluated in an ADH1 mouse model, which is referred to as Nuclear flecks (Nuf) because the mutant mouse was initially identified to have nuclear cataracts (Babinsky et al., 2017). Nuf mice, which harbor a germline gain-of-function CaSR mutation (Leu723ICL2Gln) causing hypocalcemia, have impaired glucose tolerance and hypoinsulinemia in association with reductions in pancreatic islet mass and β-cell proliferation (Babinsky et al., 2017). Nuf mice also lack glucose-mediated suppression of glucagon secretion, which was associated with an increase in α-cell proliferation and an impairment of α-cell membrane depolarization (Babinsky et al., 2017). Administration of the NAM, ronacalceret, ameliorated the hypocalcemia and glucose intolerance of Nuf mice, and these findings highlight the potential utility of targeted CaSR compounds for modulating glucose metabolism (Babinsky et al., 2017).
H. Calcium-Sensing Receptor in Mammary Glands
The CaSR is expressed in breast epithelial cells, in which its main role is to fine tune maternal Ca2+ metabolism by balancing Ca2+o mobilization and usage: It ensures the supply of Ca2+ for milk while protecting against maternal hypocalcemia (Cheng et al., 1998; VanHouten et al., 2004, 2007; Kim and Wysolmerski, 2016). The expression of the CaSR is increased during lactation when it regulates Ca2+o transport into milk. In parallel, it inhibits synthesis of PTHrP by coupling with Gi to inhibit adenylyl cyclase activity and cAMP production (VanHouten et al., 2004). During milk production, the CaSR enables the lactating breast to participate in the regulation of systemic Ca2+o and bone metabolism. VanHouten and Wysolmerski (2013) suggested a negative feedback between systemic Ca2+o delivered to the lactating breast and PTHrP synthesis and secretion by mammary epithelial cells. When the mother’s serum calcium level is adequate, the CaSR in breast epithelial cells stimulates calcium secretion into milk, but reduces Ca2+o usage when the mother’s calcium supply becomes limited (VanHouten and Wysolmerski, 2013). In mammary epithelial cells, the CaSR regulates Ca2+o transport by altering the activity of the plasma membrane Ca2+ ATPase 2; however, the detailed molecular mechanism is not yet known (VanHouten et al., 2007; VanHouten and Wysolmerski, 2007).
The CaSR is expressed also in the neoplastic mammary gland. In contrast with normal breast cells, in breast cancer cells the CaSR stimulates PTHrP secretion. This is possible because the CaSR switches coupling from Gi/o to Gs, leading to stimulation of cAMP and PTHrP synthesis (Mamillapalli et al., 2008). The higher PTHrP levels, secreted because of activation of the CaSR, inhibit the cell cycle inhibitor p27kip1 and the apoptosis-inducing factor, stimulating cell proliferation and reducing apoptosis (Kim et al., 2016). Moreover, PTHrP is an activator of osteoclasts and often stimulates osteolytic bone destruction when secreted from cancer cells that metastasize to bone (Wysolmerski, 2012).
The CaSR is highly expressed by metastatic breast cancer cells and potentiates their osteolytic ability, promoting a more aggressive behavior. In vitro, the CaSR promoted breast cancer cell migration only in cells capable of forming bone metastases (e.g., MDA-MB-231, MCF-7) but not in BT474 cells that have no bone-metastatic potential, even though CaSR levels were similar in all cell types (Saidak et al., 2009). It has been shown recently that breast cancer cells overexpressing the wild-type CaSR injected intratibially into BALB/c-Nude mice led to osteolytic lesions through an epiregulin-mediated mechanism (Boudot et al., 2017). The oncogenic potential of the CaSR in breast cancer cells was also suggested by the fact that activation of the CaSR by NPS R-568 or Ca2+o increased secretion of proangiogenic and chemotactic cytokines and growth factors from the highly invasive MDA-MB-231 breast cancer cells (Hernández-Bedolla et al., 2015). Another group, however, found that activating the CaSR with Ca2+o induced sensitivity of MCF-7 and MDA-MB-435 cells to the chemotherapeutic drug paclitaxel and reduced malignant behavior. The paclitaxel-resistant cells expressed no CaSR (Liu et al., 2009). This group suggested a positive link between the tumor suppressive functions of BRCA1 and the CaSR (Promkan et al., 2011).
I. Calcium-Sensing Receptor in Airway Smooth Muscle and Epithelium
Asthma is characterized by airway hyperresponsiveness, inflammation, and remodeling of the conducting airways. A number of mechanisms, many driven by inflammation, have been hypothesized to contribute to airway hyperresponsiveness and/or remodeling. Among these, local increases of polycations are seen in the airways of patients who are asthmatic (Kurosawa et al., 1992) and, vice versa, increased inflammation increases the local concentration of polycations. Furthermore, the polycations, eosinophil cationic protein and major basic protein, are markers for asthma severity and stability. Elevated arginase activity increases the consumption of L-arginine to enhance production of the polycations, spermine, spermidine, and putrescine (North et al., 2013). Indeed, arginase inhibitors have been proposed to have therapeutic potential for allergic asthma (van den Berg et al., 2018). Recent evidence suggests that the CaSR is expressed in the airway epithelium, smooth muscle, and inflammatory cells and that polycations act at the CaSR and are directly implicated in the pathogenesis of asthma (Yarova et al., 2015). Yarova et al. (2015) have also shown that inhaled CaSR NAMs delivered topically reverse-airway hyperresponsiveness, inflammation, and remodeling in in vivo models of allergic asthma and other inflammatory lung diseases, such as chronic obstructive pulmonary disease. Inhaled NAMs also show efficacy in nonallergic asthma, which is often associated with poor response to steroids, and for which currently there is no treatment (Riccardi unpublished observations). Four CaSR NAMs have been studied in humans; NPSP795, ronacaleret, AXT914, and JTT-305 (IIB. Endogenous and Exogenous Allosteric Modulators, Table 1), which could be repurposed, via the inhaled route, as novel asthma treatments. Crucially, delivery of CaSR NAMs directly to the lung does not significantly affect serum calcium levels up to 24 hours post-treatment, suggesting absence of any significant systemic overspill and possible effects on whole-body mineral ion homeostasis in vivo. Thus, CaSR NAMs could provide a new therapeutic approach to treating inflammatory lung disease in humans.
J. Calcium-Sensing Receptor in the Vasculature
The CaSR is expressed in the intima, media, and adventitia of the blood vessels in endothelial, smooth muscle cells and in the perivascular neurons. Although consumption of dietary calcium reduces blood pressure (Nakamura et al., 2019; Rietsema et al., 2019), direct actions of Ca2+o on isolated blood vessels have yielded contrasting effects, with both relaxation and constriction reported (Bohr, 1963). Furthermore, the molecular mechanisms underlying these actions are elusive. Studies carried out over the last two decades indicate that the CaSR could mediate at least some of the effects of Ca2+o on vascular function, with opposing effects in the endothelium and smooth muscle cell layers of the blood vessels. Specifically, CaSR activation by the CaSR PAM, cinacalcet, in the vascular endothelium leads to hyperpolarization and attendant nitric oxide release and vasodilatation (Smajilovic et al., 2007). In contrast, studies of Casr gene ablation in the vascular smooth muscle cells show that activation of the CaSR in these cells leads to contraction, as evidenced by the fact that Casr knockout mice exhibit impaired vascular reactivity, hypotension, and reduced contractile response to Ca2+o (Schepelmann et al., 2016). Thus, the CaSR sets blood vessel tone by integrating prorelaxing (endothelium-mediated) actions with procontractile (smooth muscle–mediated) effects (Schepelmann et al., 2016). Therefore, altered CaSR expression within either the endothelium or smooth muscle could account for the abnormal vascular reactivity seen in advanced CKD or in type 2 diabetes. Indeed, systemic administration of the CaSR PAM, NPS R-568, initially evokes an increase in blood pressure in control and in uremic rats (a model for advanced CKD), which is followed by a reduction in blood pressure, but only in uremic animals (Odenwald et al., 2006), suggesting partial loss of the CaSR-dependent contractile component of the vasculature.
However, there is some controversy regarding the role of the CaSR in regulating blood pressure. In ex vivo studies in rat mesenteric arteries, relaxant responses to cinacalcet and calindol were not blocked by calhex 231 (Thakore and Ho, 2011), which at the time was believed to be a CaSR NAM (but has now been shown to have mixed PAM and NAM activity, see II.B. Endogenous and Exogenous Allosteric Modulators). Nonetheless, Ca2+o influx into these vessels stimulated by the α1 adrenergic receptor agonist, methoxamine, was inhibited, not potentiated, by cinacalcet and calindol, as were contractions in response to an L-type calcium channel activator (Thakore and Ho, 2011). Given that arylalkylamine PAMs are structurally related to the nonselective calcium channel blocker, fendiline, and have low affinity for calcium channels, the relaxing effects of CaSR PAMs in arteries may in part arise from off-target calcium channel effects. Further support for a non-CaSR-mediated effect of PAMs in the vasculature comes from findings that, although the S-enantiomers of arylalkylamine PAMs have little activity at the CaSR, the effects of NPS R-568 on vascular tone, blood pressure, and heart rate are not stereoselective and only occur at concentrations in excess of those required to inhibit PTH secretion (Nakagawa et al., 2009).
End-stage CKD is associated with impaired mineral ion metabolism, which can lead to pathologic vascular calcification of the medial layer of the blood vessel, left ventricular hypertrophy, and increased cardiovascular mortality (Locatelli et al., 2002). CaSR expression is significantly reduced in the medial layer of calcifying blood vessels and is completely absent in areas of extensive medial calcification, suggesting an involvement of the CaSR in the vascular calcification process (Alam et al., 2009). Human and bovine vascular smooth muscle cells exposed to Ca2+ and phosphate concentrations mimicking those seen during pathologic CKD exhibit marked calcification in vitro, an effect that is exacerbated by CaSR downregulation and that is reversed by the CaSR PAM, NPS R-568 (Alam et al., 2009). In addition, NPS R-568 reduces blood pressure and ameliorates cardiac remodeling in animal models of advanced CKD in vivo (Ogata et al., 2003). Taken together, these results suggest that loss of CaSR expression by the medial layer of the blood vessels in advanced CKD leads to vascular calcification and that CaSR PAMs might be vasculo-protective by directly restoring normal CaSR expression levels within the vasculature. However, CaSR PAMs reduce systemic levels of serum Pi and PTH through their actions on the parathyroid CaSR, and parathyroidectomy suppresses vascular calcification (Kawata et al., 2008); therefore, PAM-mediated reduction of vascular calcification may be dependent on activation of parathyroid CaSRs. Although in vitro and in vivo studies support a direct role for the vascular CaSR in protecting vascular function, human observational studies of clinical evaluation of the CaSR PAM, cinacalcet, assessed by the Evaluation of Cinacalcet Hydrochloride Therapy to Lower Cardiovascular Events randomized controlled trial failed to reach its endpoints (reduction of all-cause and cardiovascular mortality in patients with advanced CKD) (Chertow et al., 2012). However, a recent Bayesian meta-analysis combined with a systematic literature review concluded that once subject ages and high drop-out rates throughout the trial are accounted for, cinacalcet treatment does reduce mortality rates in patients with secondary hyperparathyroidism on hemodialysis (Lozano-Ortega et al., 2018). Therefore, further clinical studies are needed to fully evaluate the effects of CaSR PAMs on cardiovascular and all-cause mortality in patients with advanced CKD.
Finally, it should be pointed out that the CaSR is also expressed in arterial smooth muscle cells of the pulmonary vasculature, wherein receptor activation leads to pulmonary vasoconstriction and proliferation. Here, CaSR NAMs prevent the development and progression of pulmonary hypertension in mouse and rat models in vivo (Tang et al., 2016a). Thus, targeting the CaSR in the pulmonary arteries with inhaled NAMs might provide a novel treatment of patients with idiopathic pulmonary hypertension.
K. Calcium-Sensing Receptor in the Brain and Nervous System
For a comprehensive review of all evidence for CaSR function in the brain, readers are directed to a recent review (Giudice et al., 2019).
Although the role of the CaSR in human brain requires validation, the CaSR is expressed throughout the rat brain, with particular abundance in the hippocampus, striatum, cerebellum, pituitary, and olfactory bulb (Ruat et al., 1995). However, CaSR expression can change with developmental age (Vizard et al., 2008), which supports a role for the CaSR in brain development. For instance, rat CaSR expression increases in fetal oligodendrocyte precursor cells and postnatal immature oligodendrocytes during myelination of nerve axons, but expression declines in mature oligodendrocytes (Chattopadhyay et al., 1998, 2008; Ferry et al., 2000).
Although hyperparathyroidism and consequent early lethality resulting from global Casr ablation preclude determination of the role of the CaSR in brain development, concomitant Casr and PTH ablation prevents hyperparathyroidism, and mice survive to adulthood (Kos et al., 2003). In brains of Casr−/−/Pth−/− mice, neuron and glial cell differentiation markers were reduced after birth, whereas differentiation of neural stem cells from Casr−/− mice was delayed (Liu et al., 2013). These mice also had reduced numbers of gonadotropin-releasing hormone-positive neurons in the hypothalamus. These findings suggest a role for the CaSR in neuron and glial cell differentiation.
To elucidate region-specific CaSR brain functions, hippocampus-specific Casr ablation 3 weeks postbirth has been undertaken (Kim et al., 2014). Although mice did not display an obvious phenotype under normal conditions, they were protected from hippocampal neuronal damage in response to ischemia-induced injury, which mimics injury sustained during cardiac arrest or stroke. In line with these findings, hypoxia increases CaSR expression in rat hippocampal neurons in vivo and in vitro (Bai et al., 2015), but neuroprotection from ischemia is blocked when the related class C GPCR, GABABR1, is also ablated (Kim et al., 2014). This may be explained by the discover of an increase in CaSR expression in hippocampal neurons in culture upon suppression of GABABR1 levels (Chang et al., 2007), which is also observed in cortical neurons of mice who have experienced controlled cortical impact as a model for traumatic brain injury (Kim et al., 2013). In support of a role for the CaSR in the hippocampus, in rat hippocampal neurons from wild-type but not Casr−/− mice, CaSR activation opens nonselective cation channels (Ye et al., 1997b). Similarly, transfection of a dominant negative Arg185Gln mutant CaSR into pyramidal neurons of hippocampal brain slice cultures resulted in significantly shorter and less complex dendritic branching (Vizard et al., 2008). Taken together, these studies suggest that CaSR NAMs could be neuroprotective.
In addition to neuroprotective effects upon brain injury, inhibition of brain CaSRs may afford neuroprotection in Alzheimer disease. The first evidence for a possible role of the CaSR in Alzheimer disease came from a study that demonstrated activation of nonselective cation channels in cultured hippocampal pyramidal neurons from wild-type rats and mice but not from Casr−/− mice (Ye et al., 1997a). Although additional studies have since suggested β-amyloid proteins activate the CaSR (Conley et al., 2009; Dal Pra et al., 2014), these findings warrant further validation. Nonetheless, CaSR expression is increased in the hippocampus of an Alzheimer disease mouse model (Gardenal et al., 2017), and there is a positive association between CaSR SNPs and Alzheimer disease, although this is only in patients who do not harbor the Alzheimer risk allele encoding apolipoprotein E4 (Conley et al., 2009). Furthermore, in human cortical astrocytes and neurons in culture, neurotoxic β-amyloid25–35 stimulates full-length β-amyloid42 secretion, an effect that is blocked by the CaSR NAM, NPS 2143 (Armato et al., 2013; Chiarini et al., 2017b). NPS 2143 also blocked β-amyloid25–35–mediated GSK-3β activation and subsequent phosphorylation of τ in cultured human astrocytes (Chiarini et al., 2017a).
The CaSR has also been implicated in the etiology of neuroblastomas, tumors originating from precursor nerve cells of the sympathetic nervous system. Approximately 98% of neuroblastomas are associated with spontaneous mutations in a variety of genes (Aygun, 2018). Analysis of mRNA from neuroblastoma tumors indicates that although the CaSR is expressed in benign differentiated tumors, epigenetic hypermethylation of the CASR P2 promoter region silences CASR transcription in some aggressive neuroblastomas (de Torres et al., 2009; Casalà et al., 2013). Similarly, two noncoding CASR SNPs (rs7652579 and rs1501899) that reduce CaSR expression are present in homozygous or heterozygous form in 58% of neuroblastoma tumors but in only 47% of the general population (Masvidal et al., 2017). In a subset of ganglioneuromas, CASR expression was absent in four out of six tumors harboring rs7652579 and rs1501899. However, neuroblastoma patients with rs7652579 and rs1501899 SNPs did not have poorer outcomes or survival (Masvidal et al., 2017). In contrast, neuroblastomas with a haplotype SNP in the CASR gene-coding region were associated with poorer outcomes, including increased risk of death (Masvidal et al., 2013). Importantly, cinacalcet reduced neuroblastoma tumor growth in immunocompromised mice carrying neuroblastoma xenografts by inducing endoplasmic reticulum stress, tumor differentiation, and fibrosis as well as upregulation of cancer-testis antigens (Rodríguez-Hernández et al., 2016).
Finally, approximately 40% of patients harboring ADH1 gain-of-function CASR mutations present with seizures (Gorvin, 2019). Although this could be associated with the consequent reduction in serum calcium concentrations, a gain-of-expression CaSR mutation, Arg898Gln, was identified in a patient with idiopathic epilepsy who did not have low serum concentrations of calcium or PTH (Kapoor et al., 2008). These findings suggest a possible role for CaSR in neurotransmission, which is supported by numerous in vitro studies suggesting the CaSR regulates synaptic transmission and neuronal activity via activation of nonselective cation channels on presynaptic terminals [reviewed in Jones and Smith (2016)].
VI. Calcium-Sensing Receptor–Related Genetic Diseases and Therapeutic Interventions
A. Loss- and Gain-of-Function Mutations in the Calcium-Sensing Receptor and Its Signaling Partners
Alterations in CaSR signaling, which lead to derangements of mineral homeostasis, can result from loss-of-function germline mutations of the CASR gene on chromosome 3q21.1, which cause FHH1 and NSHPT, or gain-of-function germline CASR mutations, which lead to ADH1 and Bartter syndrome type V (Fig. 4; Table 2) (Hannan et al., 2012; Hannan and Thakker, 2013). In addition, loss- and gain-of-function germline mutations of the GNA11 gene on chromosome 19p13.3, which encodes Gα11, are associated with FHH2 and ADH2, respectively (Table 2) (Nesbit et al., 2013a; Hannan et al., 2016). Furthermore, loss-of-function germline mutations of the AP2S1 gene on chromosome 19q13.3, which encodes AP2σ, cause FHH3 (Table 2) (Nesbit et al., 2013b; Hannan et al., 2015a).

CaSR snakeplot with residues linked to loss- and gain-of-function germline mutations. Snakeplot of the CaSR showing the location of the ECD, 7TM, ICLs, ECLs, and carboxy terminus. Sites of loss- and gain-of-function germline mutations causing FHH1/NSHPT (red), ADH1/Bartter syndrome type V (green), or both FHH1/NSHPT and ADH1/Bartter syndrome type V (yellow), respectively. Snakeplot generated by GPCRdb (Munk et al., 2016) with data from the Human Gene Mutation Database (Stenson et al., 2012). C-term, C terminal; N-term, N terminal.
Calcitropic disorders caused by germline CASR, GNA11, and AP2S1 mutations
B. Familial Hypocalciuric Hypercalcemia and Neonatal Severe Hyperparathyroidism
FHH is a genetically heterogeneous autosomal dominant disorder characterized by lifelong nonprogressive elevations of serum calcium concentrations, mild hypermagnesemia, normal or mildly raised serum PTH concentrations, and low urinary calcium excretion (Table 2) (Hannan and Thakker, 2013). FHH1 (OMIM 145980) accounts for ∼65% of all FHH cases and is usually an asymptomatic disorder. It has been associated with >150 different CASR mutations (Hannan et al., 2018a). The majority (>85%) of these loss-of-function CASR mutations are heterozygous missense substitutions, which are predominantly located in the VFT of the CaSR ECD and also in the 7TM (Hannan et al., 2012). These FHH1-associated missense mutations cause a loss of function by diminishing the signaling responses of CaSR-expressing cells (Leach et al., 2012) or by reducing CaSR anterograde trafficking and cell surface expression (Huang and Breitwieser, 2007; White et al., 2009). In addition, FHH1-causing missense mutations may induce biased agonism by switching from a wild-type CaSR that preferentially increases Ca2+i mobilization to mutant receptors that demonstrate equal preference for Ca2+i and MAPK pathways or that preferentially act via MAPK (Leach et al., 2012, 2013; Leach and Gregory, 2017). Between 10% and 15% of FHH1 cases are caused by heterozygous deletion, insertion, nonsense, and splice-site mutations that lead to nonsense-mediated decay of mRNA and CaSR haploinsufficiency or truncate the CaSR protein (Hannan et al., 2012). The offspring of two parents with FHH1 can harbor biallelic loss-of-function CASR mutations that cause NSHPT (OMIM 239200), which is associated with marked hyperparathyroidism that leads to hypercalcemia and bone demineralization causing fractures and respiratory distress (Hannan and Thakker, 2013). Occasionally, biallelic loss-of-function CASR mutations can lead to primary hyperparathyroidism, which presents in adulthood (Table 2) (Hannan et al., 2010). Furthermore, some heterozygous mutations (e.g., Arg185Gln) can cause NSHPT due to dominant negative effects on the wild-type CaSR (Bai et al., 1997).
FHH2 (OMIM 145981) is the least common form of FHH and has been reported in four probands to date (Nesbit et al., 2013a; Gorvin et al., 2016, 2018b). FHH2 appears to have a mild clinical presentation with serum-adjusted total calcium concentrations usually between 2.55 and 2.80 mM (normal range 2.10–2.55 mM). Urinary calcium excretion may be normal or low (Table 2) (Nesbit et al., 2013a; Gorvin et al., 2016, 2018b). The GNA11 mutations reported in FHH2 probands consist of three missense substitutions (Thr54Met, Leu135Gln, Phe220Ser) and an in-frame isoleucine deletion (Ile200del) (Nesbit et al., 2013a; Gorvin et al., 2016, 2018b). All of these mutations impair CaSR-signaling responses and are located within key domains of the Gα11 protein (Nesbit et al., 2013a; Gorvin et al., 2016, 2018b). Thus, the Ile200del and Phe220Ser mutations are located within the Gα11 GTPase domain and are predicted to diminish the interaction of Gα11 with the CaSR or PLC, respectively (Nesbit et al., 2013a; Gorvin et al., 2018b). In contrast, the Leu135Gln mutation is situated within the PLC-interacting portion of the Gα11 helical domain, and the Thr54Met mutation is located at the interface between the helical and GTPase domains and may potentially affect GTP binding (Nesbit et al., 2013a; Gorvin et al., 2016).
FHH3 (OMIM 600740) has been reported in >60 FHH probands and has a more marked clinical phenotype than FHH1. Thus, FHH3 is associated with significant elevations of serum calcium and magnesium and also a significantly reduced urinary calcium excretion compared with FHH1 (Table 2) (Hannan et al., 2015a; Vargas-Poussou et al., 2016). In addition, hypercalcemic symptoms, low bone mineral density, and alterations in cognitive function have been described in some patients with FHH3 (McMurtry et al., 1992; Hannan et al., 2015a). Nearly all FHH3 cases are caused by a missense mutation of the AP2σ Arg15 residue (Arg15Cys, Arg15His, or Arg15Leu) (Fujisawa et al., 2013; Nesbit et al., 2013b; Hendy et al., 2014; Hannan et al., 2015a; Howles et al., 2016; Vargas-Poussou et al., 2016; Hovden et al., 2017). In addition, a genotype-phenotype correlation has been observed at the AP2σ Arg15 residue with the Arg15Leu mutation being associated with significant increases in serum calcium and an earlier age of presentation compared with patients harboring the Arg15Cys or Arg15His AP2σ mutations (Hannan et al., 2015a; Hovden et al., 2017). The AP2σ subunit forms part of the heterotetrameric AP2 complex, which is involved in clathrin-mediated endocytosis (Kelly et al., 2008), and the FHH3-causing AP2σ Arg15 mutations have been shown to reduce CaSR endocytosis and impair endosomal signaling from the internalized CaSR (Gorvin et al., 2018c). However, given the role of AP2 in clathrin-mediated endocytosis, it remains to be established whether phenotypic observations, such as cognitive deficits in FHH3, are attributable to CaSR dysregulation or potentially due to alterations in the endocytosis of other plasma membrane proteins.
C. Autosomal Dominant Hypocalcemia and Bartter Syndrome Type V
ADH is comprised of two genetically distinct variants, designated ADH1 and 2, which are caused by germline gain-of-function mutations of the CaSR and Gα11, respectively (Table 2) (Hannan et al., 2016). ADH1 (OMIM 601198) is characterized by mild-to-moderate hypocalcemia in association with mild hypomagnesemia, hyperphosphatemia, and serum PTH concentrations that are usually detectable but within the lower half of the reference range (Nesbit et al., 2013a). Patients with ADH1 have significantly increased urinary calcium excretion compared with patients with hypoparathyroid (Yamamoto et al., 2000), and ∼10% of patients with ADH1 have an absolute hypercalciuria (Nesbit et al., 2013a). Some patients with ADH1 may have ectopic calcifications and/or elevations in bone mineral density (Pearce et al., 1996), and patients with a severe gain-of-function CaSR mutation may also develop a Bartter syndrome (referred to as Bartter syndrome type V) (Table 2), which is characterized by renal salt wasting leading to volume depletion, hyper-reninemic hyperaldosteronism, and hypokalemic alkalosis (Watanabe et al., 2002). Over 90 different ADH1-causing CaSR mutations have been reported (Hannan and Thakker, 2013; Hannan et al., 2016), and around 95% of these are heterozygous missense substitutions, whereas ∼5% are in-frame or frameshift insertion or deletion mutations (Hannan et al., 2012). ADH1 mutations cluster within the second loop of the VFT domain (residues 116–136), which contributes to the dimeric CaSR interface (Geng et al., 2016) (Fig. 4). A second ADH1 mutational hotspot is located in a region that encompasses transmembrane helices 6 and 7 and the intervening third ECL of the CaSR (residues 819–837) (Hannan et al., 2016). This transmembrane region may participate in a network of interactions with other transmembrane helices (Dore et al., 2014), thereby causing the CaSR to adopt an inactive conformational state.
ADH2 (OMIM 615361) (Table 2) has been reported in seven probands (Mannstadt et al., 2013; Nesbit et al., 2013a; Li et al., 2014a; Piret et al., 2016; Tenhola et al., 2016). Patients with ADH2 generally have mild-to-moderate hypocalcemia, in keeping with the serum biochemical phenotype of ADH1 (Hannan et al., 2016). However, ADH2 is associated with a milder urinary phenotype, with significantly reduced urinary calcium excretion compared with ADH1 (Li et al., 2014a). Moreover, short stature caused by postnatal growth insufficiency has been reported in two ADH2 kindreds (Li et al., 2014a; Tenhola et al., 2016). ADH2-causing mutations all comprise missense substitutions (Arg60Cys, Arg60Leu, Arg181Gln, Ser211Trp, Val340Met, and Phe341Leu), which enhance CaSR-mediated signaling responses, consistent with a gain of function (Nesbit et al., 2013a; Li et al., 2014a; Piret et al., 2016). ADH2-causing mutations cluster at the interface between the Gα11 helical and GTPase domains (Piret et al., 2016) and may enhance the exchange of GDP and GTP, thereby leading to G protein activation. ADH2 mutations also affect the C-terminal portion of the Gα11 protein, which facilitates G protein–GPCR coupling (Piret et al., 2016).
D. Animal Models of Genetic Diseases
Mouse models for FHH, NSHPT, and ADH have been generated using gene knockout and knock-in techniques and also by using chemical mutagenesis (Piret and Thakker, 2011).
1. Familial Hypocalciuric Hypercalcemia/Neonatal Severe Hyperparathyroidism Mouse Models
A mouse model lacking the CaSR was generated by replacing part of exon 5 with a neomycin resistance gene (Ho et al., 1995). Mice harboring this germline heterozygous CaSR deletion (Casr+/−) had mild hypercalcemia and hypocalciuria, similar to patients with FHH1, whereas mice with a homozygous CaSR deletion (Casr−/−) had a phenotype resembling NSHPT with parathyroid hyperplasia, severe hypercalcemia, bone abnormalities, and retarded growth (Ho et al., 1995). The Casr−/− mice died within the first 30 days of life (Ho et al., 1995), which was attributed to severe hyperparathyroidism. In support of this, correction of the hyperparathyroidism by the additional germline ablation of the Pth or Gcm2 genes rescued the early lethality and bone demineralization in Casr−/− mice (Kos et al., 2003; Tu et al., 2003). The importance of the parathyroid CaSR in the pathogenesis of NSHPT has been further highlighted by mice harboring a parathyroid-specific ablation of the CaSR, which developed severe hypercalcemia and hyperparathyroidism (Chang et al., 2008; Fan et al., 2018). In contrast, mice with a kidney-specific ablation of the CaSR do not have alterations in serum calcium or PTH but are hypocalciuric, and these findings support an independent role of the kidney CaSR in the regulation of urinary calcium excretion (Toka et al., 2012).
A mouse model for FHH2 has been generated by chemical mutagenesis using the N-ethyl-N-nitrosourea–alkylating agent (Howles et al., 2017). The mutant mice harbor a germline loss-of-function Gna11 mutation, Asp195Gly (D195G) (Howles et al., 2017). Heterozygous (Gna11+/195G) mice have mild hypercalcemia and normal plasma PTH concentrations (Howles et al., 2017). Homozygous (Gna11195G/195G) mice have significantly increased plasma calcium and PTH concentrations compared with Gna11+/195G and wild-type mice (Howles et al., 2017). However, Gna11195G/195G mice do not have growth retardation, bone demineralization, or early lethality to suggest an NSHPT phenotype (Howles et al., 2017). Thus, these studies indicate that the loss-of-function D195G Gα11 mutation is associated with a mild calcitropic phenotype. Furthermore, the Gna11+/195G and Gna11195G/195G mice have no alterations in urinary calcium excretion (Howles et al., 2017), which suggests that Gα11 may not play a major role in the renal handling of calcium.
2. Autosomal Dominant Hypocalcemia Mouse Models
Three different ADH1 mouse models have been reported (Hough et al., 2004; Dong et al., 2015). Nuf mice (described in V.G. Calcium-Sensing Receptor in the Pancreas) weregenerated by chemical mutagenesis using the isopropyl methane sulfonate–alkylating agent (Hough et al., 2004). Heterozygous and homozygous Nuf mice have hypocalcemia, hyperphosphatemia, reduced plasma PTH concentrations, and ectopic calcifications caused by a germline gain-of-function CaSR mutation, Leu723Gln (Hough et al., 2004). Two knock-in mouse models, which harbor ADH1-causing germline Cys129Ser or Ala843Glu gain-of-function CaSR mutations, have also been generated (Dong et al., 2015). Homozygous mutant knock-in mice exhibited embryonic or perinatal lethality, whereas heterozygous knock-in mice have hypocalcemia, hyperphosphatemia, reduced plasma PTH, hypercalciuria, and renal calcifications, consistent with the phenotype of patients with ADH1 (Dong et al., 2015).
Two mouse models for ADH2 have been described (Gorvin et al., 2017; Roszko et al., 2017). One mouse model, which is known as Dark skin 7, was generated by N-ethyl-N-nitrosourea chemical mutagenesis (Gorvin et al., 2017) and harbors a germline gain-of-function Gα11 mutation, Ile62Val, whereas the other ADH2 mouse model was generated by CRISPR-Cas9 gene editing and harbors a human ADH2-causing germline Gα11 mutation, Arg60Cys (Roszko et al., 2017). Both of these ADH2 mouse models have hypocalcemia, hyperphosphatemia, reduced plasma PTH, and normocalciuria in association with increased skin pigmentation (Gorvin et al., 2017; Roszko et al., 2017).
E. Therapeutic Interventions—Successes and Failures
CaSR PAMs represent a targeted therapy for symptomatic forms of FHH (Hannan et al., 2018b) and potentiate the signaling responses of cells expressing FHH-associated CaSR, Gα11, or AP2σ mutant proteins in vitro (Table 3) (Rus et al., 2008; Leach et al., 2013; Babinsky et al., 2016; Howles et al., 2016; Gorvin et al., 2018b). Furthermore, cinacalcet treatment is effective at decreasing serum calcium concentrations in patients with FHH1 and has been reported to improve hypercalcemic symptoms occasionally associated with FHH1, such as anorexia, polydipsia, and constipation (Table 3) (Alon and VandeVoorde, 2010; Rasmussen et al., 2011; Sethi et al., 2017). However, the response of NSHPT to cinacalcet is variable and appears to depend on the underlying CASR mutation. Indeed, cinacalcet rectifies the hypercalcemia and hyperparathyroidism in patients with NSHPT harboring a heterozygous Arg185Gln CaSR mutation (Reh et al., 2011; Gannon et al., 2014; Fisher et al., 2015) but is less effective for patients with NSHPT with biallelic truncating CASR mutations (Table 3) (García Soblechero et al., 2013; Atay et al., 2014), which would be a consequence of the truncated mutant receptor being unable to bind cinacalcet or couple with intracellular signaling proteins (Hannan et al., 2018b). Cinacalcet has also rectified the hypercalcemia in a mouse model for FHH2 (Howles et al., 2017) and ameliorated the hypercalcemia in a patient with symptomatic FHH2 (Table 3) (Gorvin et al., 2018b). Furthermore, cinacalcet is an effective therapy for symptomatic hypercalcemia caused by all three types of FHH3-causing Arg15 AP2σ mutations (Table 3) (Howles et al., 2016). However, hypocalcemic symptoms have occurred in a cinacalcet-treated child affected by FHH3 and the chromosome 22q11.2 deletion syndrome (Tenhola et al., 2015). Thus, long-term surveillance is required to detect hypocalcemia in cinacalcet-treated patients with FHH (Howles et al., 2016).
Summary of key studies assessing effectiveness of PAMs and NAMs for FHH, NSHPT, and ADH
Adapted from Hannan FM, Olesen MK, Thakker RV. Calcimimetic and calcilytic therapies for inherited disorders of the calcium-sensing receptor–signaling pathway. Br J Pharmacol (2018) 175:4083–4094.
CaSR NAMs have been evaluated as a potential targeted therapy for ADH. In vitro studies have demonstrated that NPS 2143 normalizes the signaling responses associated with gain-of-function CASR and GNA11 mutations, which cause ADH1 and ADH2, respectively (Table 3) (Letz et al., 2010; Leach et al., 2013; Hannan et al., 2015b; Babinsky et al., 2016). However, NPS 2143 is less effective for gain-of-function mutations causing Bartter syndrome type V (Letz et al., 2010; Leach et al., 2013). In contrast, the quinazolinone-derived NAMs rectify gain-of-function CASR mutations that cause Bartter syndrome V (Table 3) (Letz et al., 2014). CaSR NAMs have also been characterized in vivo, and single-dose studies have demonstrated that NPS 2143 significantly increases circulating concentrations of calcium and PTH in ADH1 and ADH2 mouse models (Table 3) (Hannan et al., 2015b; Gorvin et al., 2017; Roszko et al., 2017). In addition, repetitive dosing studies have shown that the NAM, JTT-305/MK-5442, prevents the occurrence of nephrocalcinosis in mouse models of ADH1 (Dong et al., 2015). Furthermore, the NAM, NPSP795, has been administered to five ADH1 patients in a phase IIa clinical trial and increased plasma PTH concentrations and reduced urinary calcium excretion (Table 3) (Roberts et al., 2019). However, circulating calcium concentrations were not altered in these patients, and the optimal dosing regimen of NPSP795 for ADH remains to be established.
VII. Conclusions and Perspective
The CaSR is a highly complex GPCR, as evidenced by its widespread tissue expression and varied physiological roles, its capacity to respond to multiple stimuli that act via numerous binding sites, and the ability of different stimuli to bias CaSR signaling toward distinct subsets of G protein–dependent and independent signaling pathways. The existence of multiple allosterically linked binding sites for endogenous CaSR ligands demonstrates how allostery is fundamental to CaSR activity. It is therefore unsurprising that the CaSR was the first GPCR for which an allosteric therapeutic, cinacalcet, was FDA-approved. The clinical success of cinacalcet in treating various forms of hyperparathyroidism highlights the potential of targeting the CaSR with allosteric drugs. Given the many fundamental roles of the CaSR, the CaSR is a putative therapeutic target for numerous diseases beyond Ca2+o homeostasis, including asthma, diabetes, and cancer. Thus, drug discovery efforts at the CaSR will no doubt continue.
In addition to the aforementioned CaSR (patho)physiology, ongoing research is expanding the known roles of this receptor. Analysis of CASR SNPs supports associations between CaSR expression or activity and the risk of kidney stones (Vezzoli et al., 2011), vascular calcification (Babinsky et al., 2015), breast cancer (Li et al., 2014b; Wang et al., 2017), psoriasis (Zuo et al., 2015), and serum glucose concentrations (Babinsky et al., 2015). Furthermore, the sensitivity of the CaSR to amino acids and other stimuli raises the possibility that Ca2+o is not solely responsible for CaSR-mediated Ca2+o homeostasis. Indeed, high dietary protein intake modestly increases bone density at some sites and reduces hospital stay after fracture (Dawson-Hughes, 2003; Shams-White et al., 2017). In contrast, low-protein diets induce secondary hyperparathyroidism (Kerstetter et al., 2000; Dubois-Ferrière et al., 2011) and acute increases in L-amino acids suppress PTH secretion and potentiate Ca2+o-mediated Ca2+i mobilization in human parathyroid cells (Conigrave et al., 2004). Thus, the CaSR could couple protein metabolism to changes in Ca2+o homeostasis (Conigrave et al., 2002, 2008).
Although novel analytical methods, such as the operational model of allosterism, have facilitated quantification of CaSR drug actions, CaSR drug discovery still suffers from limited tools to directly probe drug binding (e.g., commercially available radioligands or fluorescently labeled ligands) and from the lack of 7TM and full-length CaSR structures for structure-based drug discovery. Furthermore, given the critical importance of Ca2+o homeostasis to human health, novel drugs that target the CaSR outside the parathyroid glands and kidney must have limited on-target effects in these tissues (e.g., by delivery of the drug regiospecifically to the targeted tissue). Alternatively, biased signaling has the potential to revolutionize our ability to target GPCRs in a tissue-specific manner by directing receptor signaling toward desirable pathways that mediate therapeutic effects at the expense of pathways linked to unwanted effects. Furthermore, major advances in GPCR structural biology resulting in 7TM and full-length structures of the class C GPCRs, mGlu1, and mGlu5 (Dore et al., 2014; Wu et al., 2014; Koehl et al., 2019) provide confidence for forthcoming CaSR structural biology efforts. Thus, the future holds much promise for the design of novel drugs that target the CaSR.
Acknowledgments
This article was written as a result of updating the International Union of Basic and Clinical Pharmacology–British Pharmacological Society (IUPHAR-BPS) data base by the CaSR Nomenclature Subcommittee for the International Union of Pharmacology and colleagues.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Leach, Hannan, Josephs, Keller, Møller, Ward, Kallay, Mason, Thakker, Riccardi, Conigrave, Bräuner-Osborne.
Footnotes
The authors acknowledge funding from the Lundbeck Foundation (T.C.M. and H.B.-O.), the Marie Sklodowska-Curie Actions of the European Union’s Horizon 2020 research and innovation programme under REA Grant Agreements 675228 (H.B.-O., F.M.H., D.T.W., E.K., R.V.T., and D.R.) and 797497 (T.C.M. and H.B.-O.), the Australian Research Council (Future Fellowship FT160100075 and Discovery Project DP170104228, to K.L.), the Australian National Health and Medical Research Council (Project grant APP1138891, to K.L., A.D.C., and R.S.M.), the CASS Foundation (grant 8613, to A.N.K., K.L.), the Wellcome Trust (Investigator Award 106995/Z/15/Z, to R.V.T.), and the National Institute for Health Research (Senior Investigator Award NF-SI-0514-10091, to R.V.T.).
Abbreviations
- ADH
- autosomal dominant hypocalcemia
- AP2
- adaptor-related protein complex-2
- BMS
- Bristol Myers Squibb
- BTU
- benzothiazole trisubstituted urea
- Ca2+i
- intracellular calcium
- Ca2+o
- extracellular calcium
- CaSR
- calcium-sensing receptor
- CCK
- cholecystokinin
- CKD
- chronic kidney disease
- Col
- collagen
- CR
- cysteine-rich
- cryo-EM
- cryogenic electron microscopy
- DSS
- dextrane sulfate sodium
- EC
- effective concentration
- ECD
- extracellular domain
- ECL
- extracellular loop
- ENS
- enteric nervous system
- ERK
- extracellular signal-regulated kinase
- FDA
- Food and Drug Administration
- FHH
- familial hypocalciuric hypercalcemia
- GCM2
- glial cell missing-2
- GLP-1
- glucagon-like peptide 1
- GNA
- guanine nucleotide-binding protein α
- GPCR
- G protein–coupled receptor
- GRK
- GPCR kinase
- HEK
- human embryonic kidney
- ICL
- intracellular loop
- IL
- interleukin
- IP
- inositol phosphate
- MAPK
- mitogen-activated protein kinase
- mGluR
- metabotropic glutamate receptor
- NAM
- negative allosteric modulator
- NPS
- Natural Product Services
- NSHPT
- neonatal severe hyperparathyroidism
- 1,25(OH)2D
- 1,25-dihydroxyvitamin D
- OMIM
- Online Mendelian Inheritance of Man
- p-
- phosphorylated
- PAM
- positive allosteric modulator
- PDB
- Protein Data Base
- PKC
- protein kinase C
- PLC
- phospholipase C
- PTH
- parathyroid hormone
- PTHrP
- PTH-related protein
- PYY
- protein YY
- SNP
- single-nucleotide polymorphism
- TAL
- thick ascending limb
- 7TM
- 7-transmembrane
- TNCA
- L-1,2,3,4-tetrahydronorharman-3-carboxylic acid
- VDR
- vitamin D receptor
- VFT
- Venus flytrap
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





{kind=link}
{kind=link}
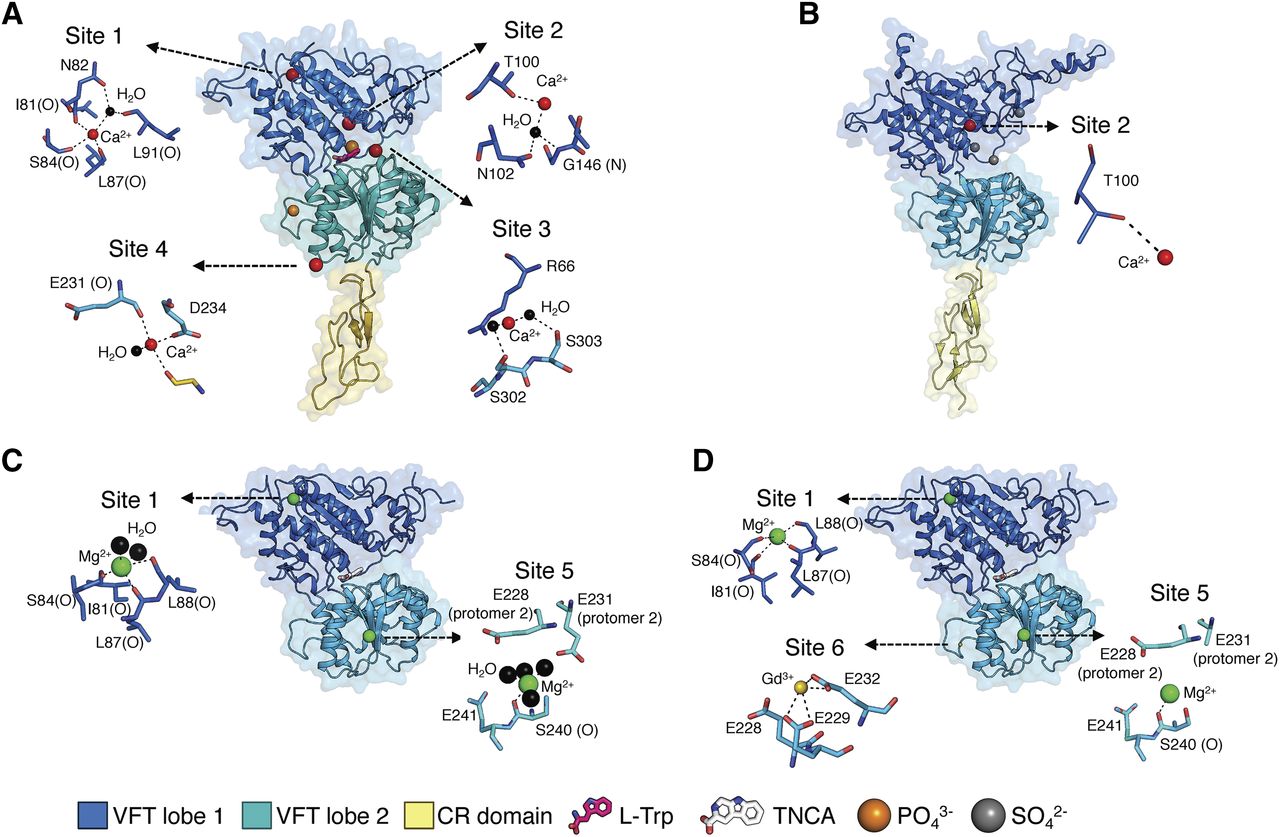{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. Agonists and Allosteric Modulators
- III. Receptor Structure
- IV. CaSR Regulation
- V. (Patho)physiology of the CaSR and Its Ligands
- VI. Calcium-Sensing Receptor–Related Genetic Diseases and Therapeutic Interventions
- VII. Conclusions and Perspective
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters