


Article Figures & Data
Figures
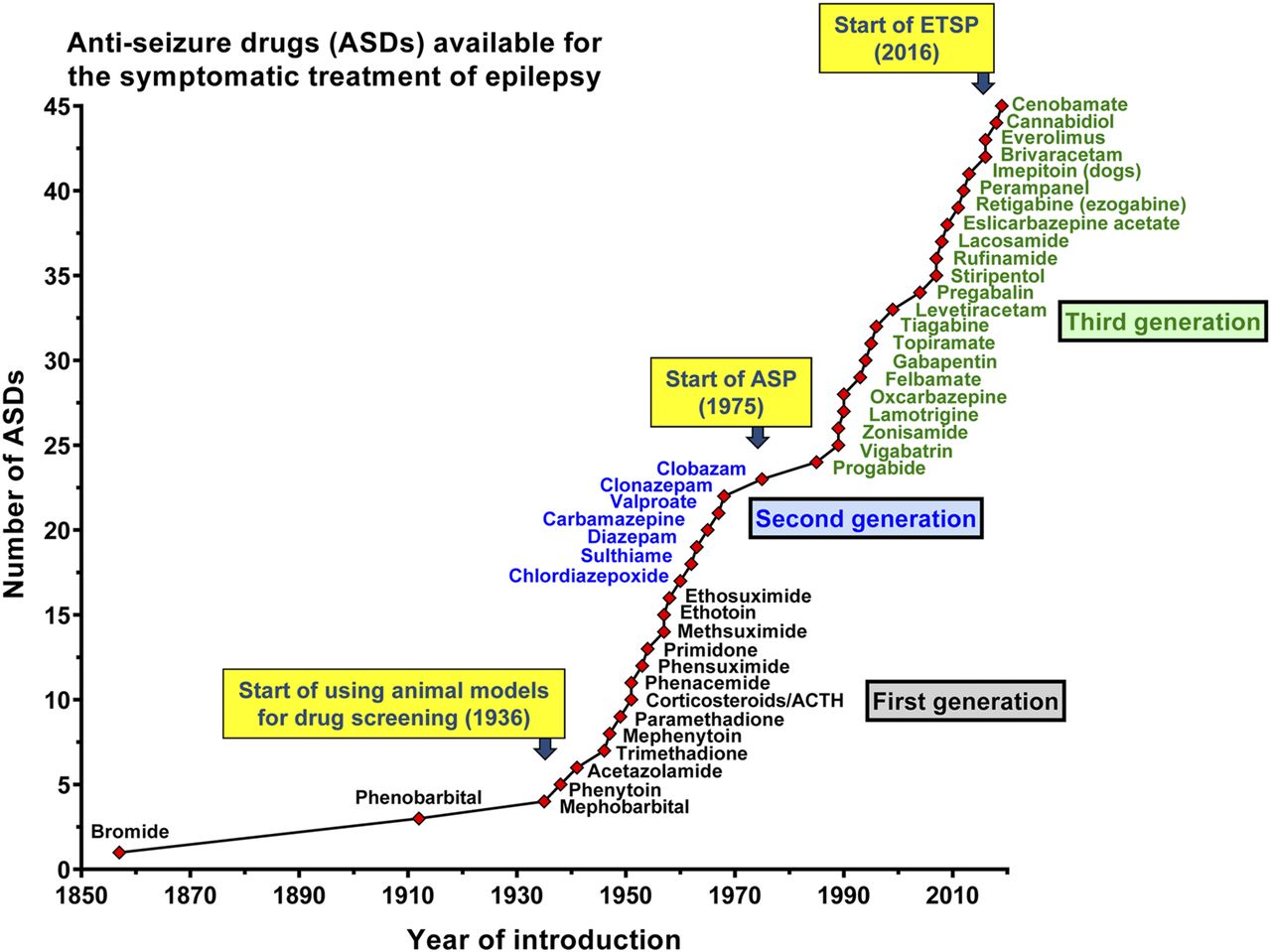
- Fig. 1.
Introduction of antiseizure drugs (ASDs) to the market from 1853 to 2019. Licensing varied from country to country. We give here the year of first licensing or the first mention of clinical use in a country of Europe, the United States, or Japan. We have not included all derivatives of listed ASDs nor ASDs used solely for treatment of status epilepticus. The first generation of ASDs, entering the market from 1857 to 1958, includes potassium bromide, phenobarbital, and a variety of drugs that were mainly derived by modification of the barbiturate structure, including phenytoin, primidone, trimethadione, and ethosuximide. The second-generation ASDs, including carbamazepine, valproate, and the benzodiazepines, which were introduced between 1960 and 1975, differed chemically from the barbiturates. The era of the third-generation ASDs started in the 1980s with “rational” (target-based) developments such as progabide, vigabatrin, and tiagabine, i.e., drugs that were designed to selectively target a mechanism that was thought to be critical for the occurrence of epileptic seizures. The figure also illustrates the impact of preclinical seizure models on ASD development. The use of seizure models for drug screening started with the experiments performed by Merritt and Putnam in the 1930s, who used an electroshock seizure model in cats, leading to the discovery of phenytoin. Subsequently, the electroshock model was adapted to rodents and, together with chemical seizure models, used for drug screening in diverse laboratories, leading to discovery of various additional ASDs. In 1975, the NIH/NINDS ASP was established in the United States as part of a larger Antiepileptic Drug Development program to promote industry interest in ASD development. Since its start, the seizure tests have been performed at a contract facility based at the University of Utah, using three rodent models, i.e., the maximal electroshock seizure (MES) test, the pentylenetetrazole (PTZ) seizure test, and the rotarod test for assessing neurotoxicity. Later, other seizure models were added. The seizure tests were performed on a blinded and confidential basis and at no cost to the ASP participants, thus providing opportunities for researchers from academia and industry in the United States and abroad to submit compounds for screening in a battery of well established rodent seizure models. Approximately 32,000 compounds from more than 600 participants from 38 countries have been screened by this program, and the ASP has contributed to bringing nine currently available ASDs to market since 1990 (Kehne et al., 2017). More recently (2016), the ASP has been renamed Epilepsy Therapy Screening Program (ETSP) with the refocused mission to identify novel agents that will help address the considerable remaining unmet medical needs in epilepsy, particularly ASD-resistant seizures (Kehne et al., 2017). Figure modified from Löscher and Schmidt (2011). For further details, see text and Löscher et al. (2013a).
- Fig. 2.
Pharmacoresistant epilepsy workflow for the Epilepsy Therapy Screening Program (ETSP). The figure has been provided by John Kehne and slightly modified for consistency with the text of this review. For details, see text and Kehne et al. (2017) and Wilcox et al. (2020).
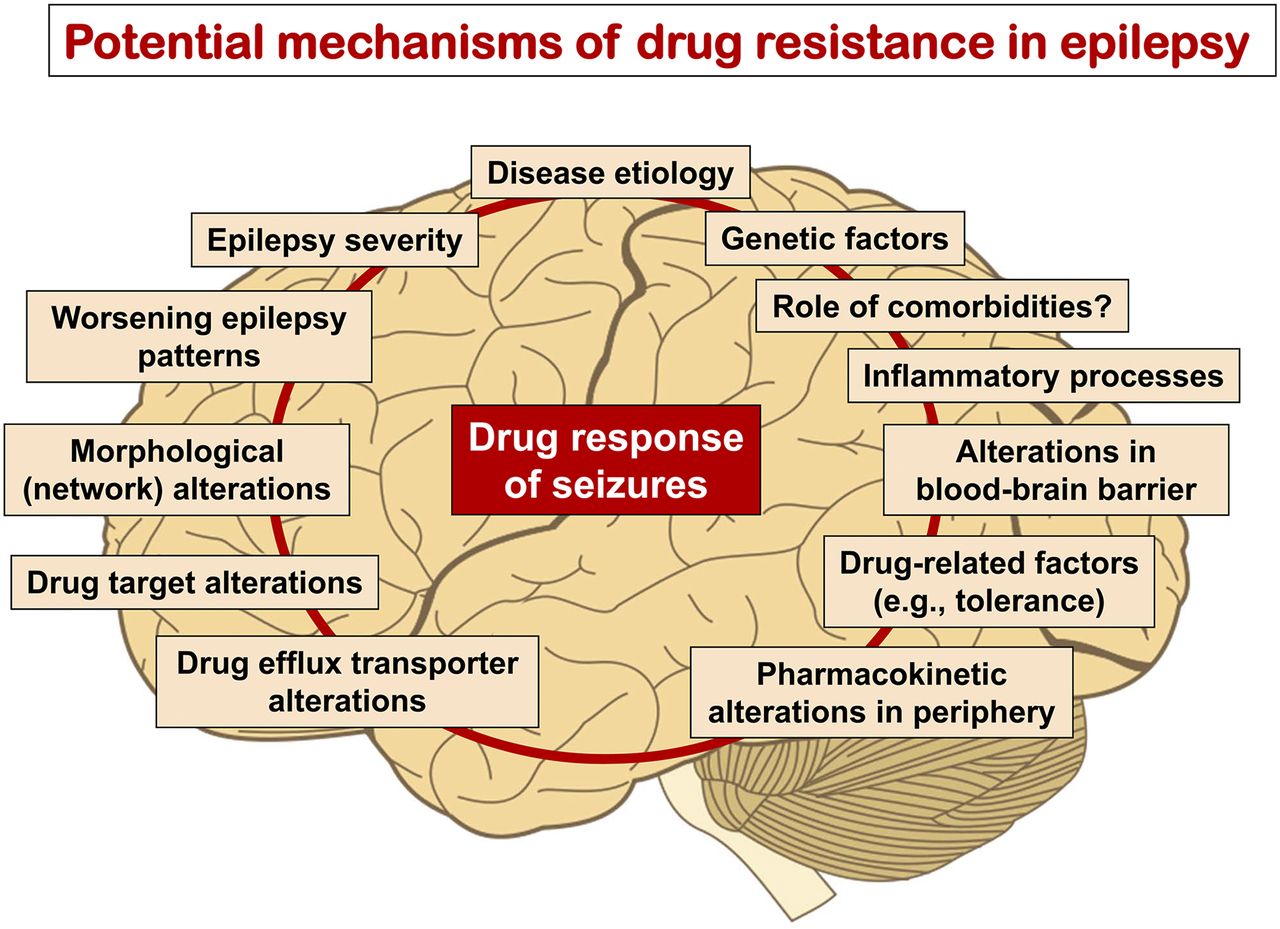
- Fig. 3.
Various potential mechanisms of ASD resistance or factors predicting poor outcome have been implicated in patients with epilepsy and animal models of medically resistant seizures, indicating that intrinsic or acquired resistance to ASDs is a multifactorial phenomenon. Based on these findings, a number of hypotheses of ASD resistance, including the target, transporter, network, intrinsic severity, and genetic variant hypotheses, have been suggested (see text). These hypotheses are not mutually exclusive but may be relevant for the same patient, thus complicating any strategy to counteract or reverse pharmacoresistance. Modified from Löscher et al. (2013a).
- Fig. 4.
Differences between ASD responders and nonresponders in two animal models of DRE. For comparison, alterations associated with ASD resistance in patients are shown. Those alterations that occur both in the models and in patients are highlighted by the colored boxes. For details, see Löscher (2011), Löscher et al. (2013a), and Löscher (2016).
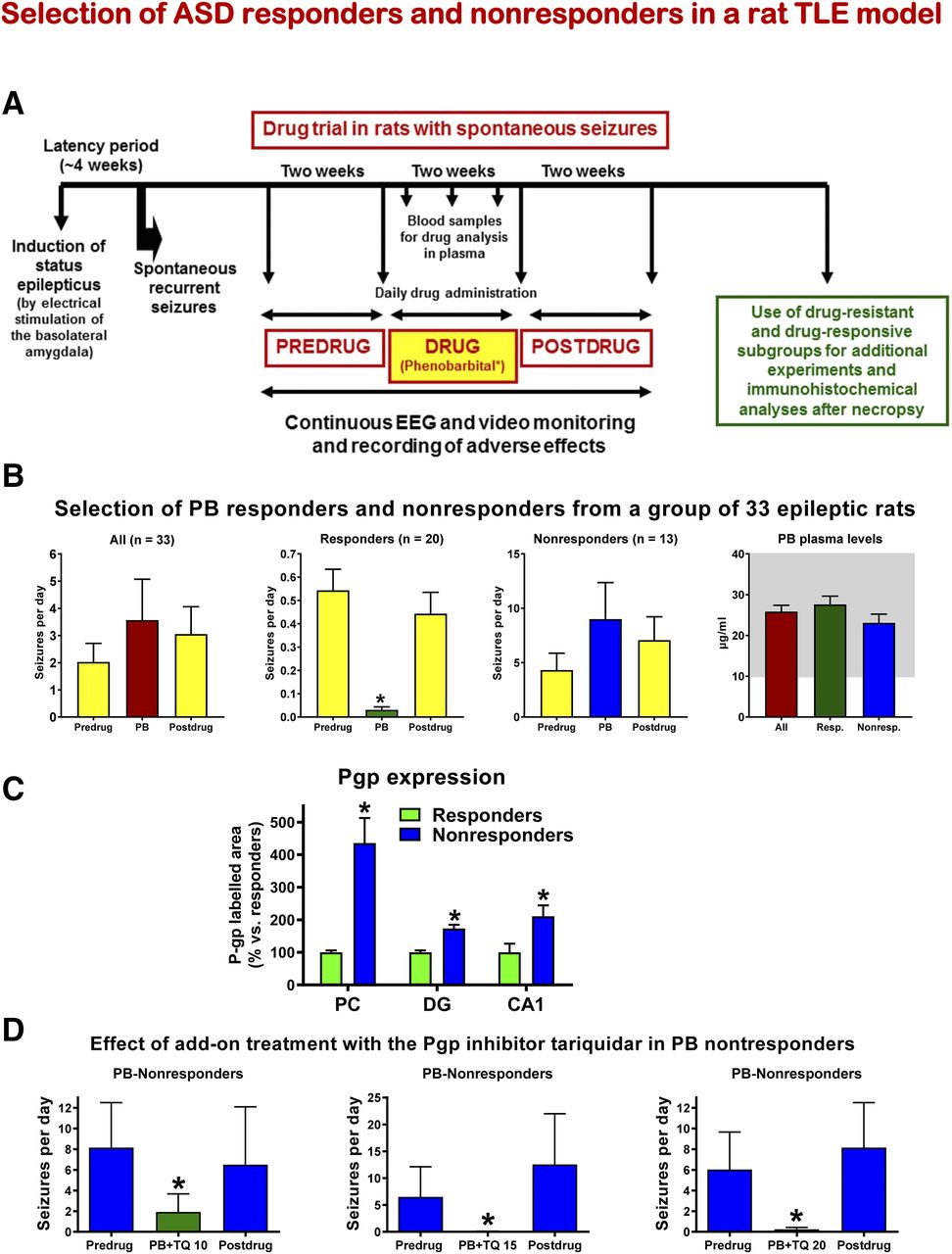
- Fig. 5.
Selection and characterization of ASD responders and nonresponders by phenobarbital in a rat model of TLE in which spontaneous recurrent seizures (SRS) develop following sustained electrical stimulation of the basolateral amygdala. (A) Schematic illustration of selection of drug-resistant and drug-responsive epileptic rats by prolonged administration of phenobarbital. (B) Effect of phenobarbital (PB) on SRS. About 5 months after the electrically induced SE, SRS were recorded over a period of 2 weeks before onset of PB treatment (predrug control), followed by drug treatment of 2 weeks and then a 2-week postdrug control period. All data are shown as means ± S.E.M. The graphs in (B) show 1) average seizure data from 33 epileptic rats from three prospective experiments, 2) respective data from 20 responders, 3) data from 13 nonresponders, and 4) average plasma concentration of PB from the blood samples taken at the end of the treatment period. The shaded area indicates the therapeutic plasma concentration range of PB. In the responder group, PB significantly suppressed SRS compared with the pre- and postdrug periods (*P < 0.001). Note the higher average frequency of SRS in nonresponders versus responders. (C) Pgp expression in brain capillary endothelial cells of responders and nonresponders. Significant differences are indicated by asterisk (*P < 0.05). (D) Coadministration of PB and the Pgp inhibitor tariquidar lead to a significant (*P < 0.05) suppression of SRS in PB nonresponders. Three different doses of tariquidar (10, 15, and 20 mg/kg) were used, demonstrating a dose-dependent effect. Data are from Brandt et al. (2004), Volk and Löscher (2005), Brandt et al. (2006), Bethmann et al. (2007), and Brandt and Löscher (2014).
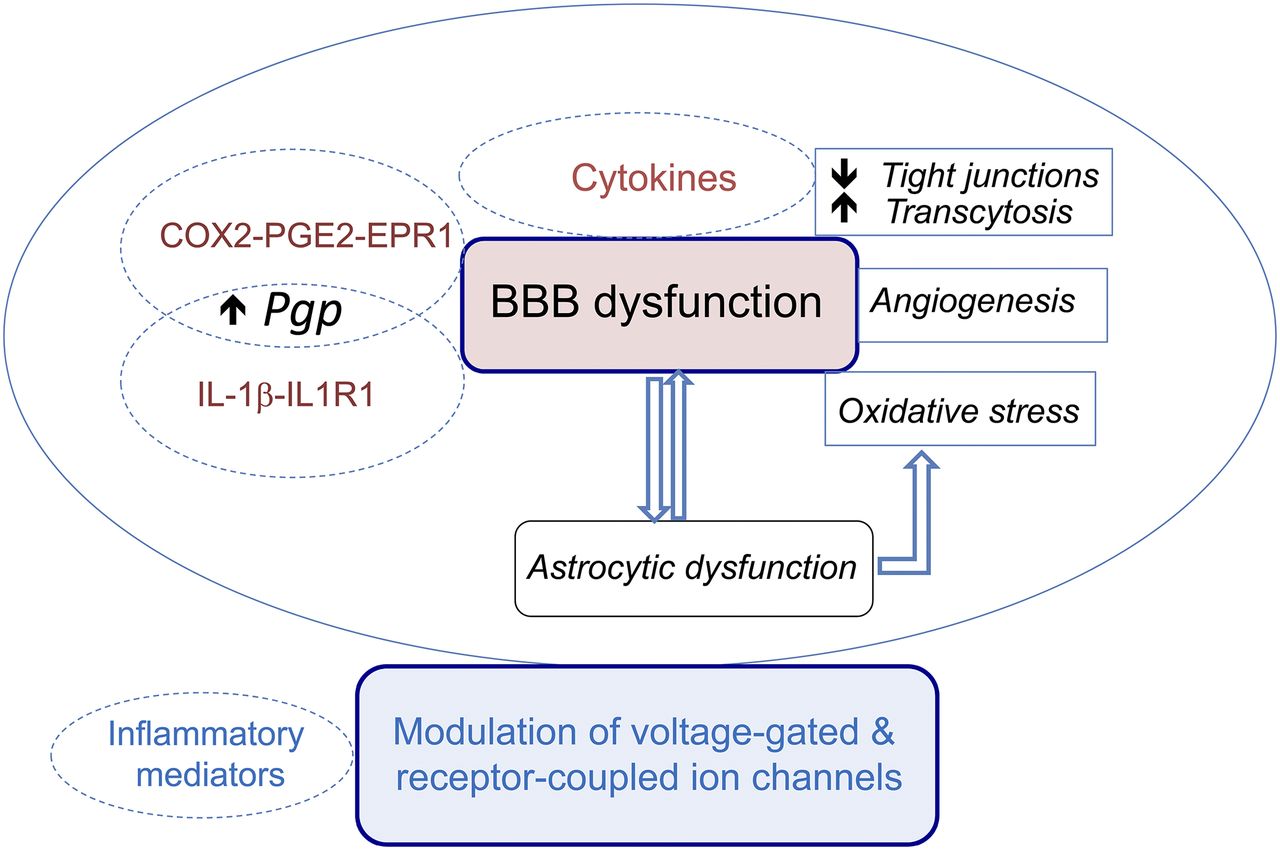
- Fig. 6.
Schematic representation of the evidence-based pathologic links between inflammatory mediators and mechanisms of drug resistance. Inflammatory mediators (including but not limited to cytokines) may contribute to drug-resistant seizures mainly by three (nonmutually exclusive) pathways: 1) the induction of BBB dysfunction by promoting breakdown of tight junctions or inducing transocytosis, aberrant angiogenesis generating “leaky” vessels, and oxidative stress. The inflammatory phenotype of astrocytes is pivotal for these actions to take place, and reciprocally, BBB permeability changes may promote the expression of inflammatory molecules in astrocytes. This vicious cycle contributes to recurrent seizures, cell loss, and maladaptive neuronal network plasticity, therefore contributing to increase the “intrinsic severity” of the disease. Morever, BBB dysfunction will enhance albumin brain extravasation into the brain parenchyma and potentially increase the “buffering” effect of albumin binding to drugs, thus decreasing functionally relevant unbound drug levels at brain target sites. 2) Another mechanism is the induction of Pgp in endothelial cells, and likely in perivascular astrocytes, by specific inflammatory pathways involving COX2-PGE2-EP1R and the IL-1beta-IL-1R1 axis, thus contributing to the transporter hypothesis of drug resistance. 3) Inflammatory mediators can also induce post-translational modifications in voltage-gated and receptor-operated ion channels resulting in less responsive ASD targets, which may contribute to the pharmacodynamic (target) hypothesis of drug resistance. Details and references are reported in the main text.





Tables
- TABLE 1
Molecular targets of clinically used ASDs
Adapted from Rogawski and Löscher (2004), Rogawski et al. (2016), and Sills and Rogawski (2020)
Molecular target ASDs that act on target Voltage-gated ion channels Voltage-gated sodium channels Phenytoin, fosphenytoin,a carbamazepine, oxcarbazepine,b eslicarbazepine acetate,c lamotrigine, lacosamide; possibly topiramate, zonisamide, rufinamide Voltage-gated calcium channels (T-type) Ethosuximide Voltage-gated potassium channels (Kv7) Retigabine (ezogabine) GABA-mediated inhibition GABAA receptors Phenobarbital, primidone, stiripentol, benzodiazepines, (including diazepam, lorazepam, midazolam and clonazepam), possibly topiramate, felbamate, retigabine (ezogabine) GAT1 GABA transporter Tiagabine GABA transaminase Vigabatrin Glutamic acid decarboxylase Possibly valproate, gabapentin, pregabalin Presynaptic release machinery SV2A Levetiracetam, brivaracetam α2δ subunit of calcium channels Gabapentin, pregabalin Ionotropic glutamate receptors AMPA receptor Perampanel Carbonic anhydratase inhibition Acetazolamide, topiramate, zonisamide, possibly lacosamide Disease-specific mTORC1 signalingd Everolimus Lysosomal enzyme replacemente Cerliponase alfa (recombinant tripeptidyl peptidase 1) Mixed/unknown Valproate, felbamate, topiramate, zonisamide, rufinamide, adrenocorticotrophin (ACTH), cannabidiol, cenobamate AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazole-propionate.
↵a Fosphenytoin is a prodrug for phenytoin.
↵b Oxcarbazepine serves largely as a prodrug for licarbazepine, mainly S-licarbazepine.
↵c Eslicarbarbazepine acetate is a prodrug for S-licarbazepine.
↵d In patients with epilepsy because of tuberous sclerosis complex (TSC).
↵e In patients with epilepsy because of neuronal ceroid lipofuscinosis type 2.
Seizure or epilepsy model Species Mode of seizure or epilepsy induction Chronic model Development of spontaneous seizures Selection of responders and nonresponders reported Throughput 6-Hz seizure model Mouse Transcorneal electrical stimulation No No n.a. High 6-Hz seizure model Rat Transcorneal electrical stimulation No No n.a. High Allylglycine-induced seizures Mouse Intraperitoneal administration of chemoconvulsant No No n.a. High Allylglycine-induced seizures Zebrafish larvae Bath application of chemoconvulsant No No n.a. Very high 6-Hz kindling Mouse Transcorneal electrical stimulation Yes No No Intermediate Lamotrigine-resistant kindled animals Rat Repeated electrical stimulation of the amygdala Yes No No Intermediate Lamotrigine-resistant kindled animals Mouse Repeated electrical stimulation of the amygdala Yes No No Intermediate Intrahippocampal kainate model Mouse Intracerebral injection of kainate Yes Yes Yes Intermediate Post-traumatic seizures Rat Fluid percussion injury Yes Yes No Low Cortical dysplasia model Rat In utero exposure to methylazoxymethanol acetate plus kainate exposure Yes No No Low Dravet models Mice Genetic modulation Yes Yes No Low NMDA model of epileptic spasms Rat (immature) Intraperitoneal administration of chemoconvulsant No No No High Multiple-hit model of infantile spasms Rat PN3 unilateral i.c.v. doxorubicin and intracortical lipopolysaccharide plus PN5 intraperitoneal p-chlorophenylalanine (→ increases spasm frequency) Yes Yes No Low Phenytoin-selected kindled animals Rat Repeated electrical stimulation of the amygdala Yes No Yes Low Phenobarbital-selected animals with spontaneous seizures Rat Prolonged electrical stimulation of amygdala induction of a status epilepticus Yes Yes Yes Low Canine patients with DRE Dog Natural disease (structural or idiopathic according to IVETF guidelines) Yes Yes Yes (based on clinical response) Very low i.c.v., intracerebroventricular; IVETF, International Veterinary Epilepsy Task Force; n.a., not applicable; PN, postnatal day.
- TABLE 3
Proof-of-concept of drug resistance hypotheses
As suggested by Sisodiya (2003), at least four criteria must be satisfied for a proposed drug-resistance mechanism of epilepsy to be accepted; the mechanism must 1) be detectable in epileptogenic brain tissue, 2) have appropriate functionality, 3) be active in drug resistance (and not be an epiphenomenon), and 4) drug resistance should be affected when the mechanism is overcome. These criteria are based on the famous Koch’s postulates, which were originally proposed by Robert Koch in 1890 to establish a causal relationship between a bacterium and a disease.
Drug-resistance hypothesis in epilepsy Detectable in brain (or peripheral) tissues of nonresponders Appropriate functionality Active in ASD resistance Resistance reversed when mechanism is overcome Target hypothesis + (rat) + (rat) ? (rat) ? (rat) + (human) + (human) ? (human) ? (human) Transporter hypothesis + (rat) + (rat) + (rat) + (rat) + (human) + (human) + (human) ? (human) Pharmacokinetic hypothesis - (rat) - (rat) ? (rat) ? (rat) + (human) ? (human) ? (human) ? (human) Neural network hypothesis + (rat) ? (rat) ? (rat) ? (rat) + (human) ? (human) ? (human) + (human) Intrinsic severity hypothesis + (rat)a ? (rat) ? (rat) ? (rat) + (human)a ? (human) ? (human) ? (human) Gene variant hypothesis + (rat) + (rat) ? (rat) ? (rat) + (human) + (human) +/? (human) +/? (human) Epigenetic hypothesis + (rat/mouse) +/? (rat/mouse) +/? (rat/mouse) +/? (rat/mouse) + (human) ? (human) ? (human) ? (human) Neuroinflammation/blood-brain barrier + (rat, mouse) + (rat) + (rat) + (rat) + (human) ? (human) ? (human) ? (human) ↵a Increased seizure frequency/density compared with ASD responders.
- TABLE 4
Potential etiology-specific drugs (“precision medicine”) that are currently used or discussed for treatment of severe pediatric-onset epilepsies
Drugs are listed according to mutated genes. For a source of references, please refer to Wang et al. (2017) and Mesraoua et al. (2019). Note that mutations of the same gene may result in different clinical phenotypes, as recently shown for KCNQ2 mutation, in which the majority of patients have loss-of-function mutations but a small percentage have gain-of-function mutations associated with a different phenotype (Demarest and Brooks-Kayal, 2018). Except for everolimus in TSC-associated focal epilepsy and for cannabidiol and fenfluramine in Dravet syndrome, none of the treatments listed in this table have been validated in randomized controlled trials in patients with the indicated mutations, and for some of these treatments, evidence for efficacy is speculative or controversial, with most entries being anecdotal or in fact not “precision” (see comments). Clinicians should not consider this table as constituting support for treatment with these agents.
Mutated gene Gene name Encoded protein function Type of epilepsy Potentially beneficial therapy Comments CHRNA4 Cholinergic receptor nicotinic alpha 4 subunit Nicotinergic acetylcholine receptor Nocturnal frontal lobe epilepsy Zonisamide, acetazolamide, and nicotine patches Zonisamide and acetazolamide are not really “precision.” Nicotinergic agents are theoretically of possible use, but none have been proven to be of value currently GRIN2A Glutamate ionotropic receptor N-methyl-D-aspartate (NMDA) type subunit 2A Glutamate (NMDA) receptor Focal epilepsy and speech disorder with or without mental retardation Memantine Has been proposed on the basis of two studies only, none published since 2015 KCNQ2 Potassium voltage-gated channel subfamily Q member 2 Potassium channel Benign familial neonatal seizures or, in infancy and childhood, EIEE Retigabine/ezogabine Has in vitro evidence to support its use in gain-of-function mutants, but prospective controlled trials are still lacking KCNT1 Potassium sodium-activated channel subfamily T member 1 Potassium channel EIEE Quinidine The evidence is equivocal, with many negative reports after the initial reports of benefit PCDH19 Protocadherin 19 Cell adhesion molecule EIEE Potassium bromide, clobazam Only anecdotal evidence. Better rationale for hormonal treatment with allopregnanolone. PLCB1 Phospholipase C beta 1 Enzyme EIEE Inositol Not any evidence for this in humans PRRT2 Proline-rich transmembrane protein 2 Unclassified Benign familial infantile seizures Carbamazepine, oxcarbazepine Not really precision (mechanism-based) treatments SCN1A Sodium voltage-gated channel alpha subunit 1 Voltage-gated sodium channel Dravet syndrome GABAergic drugs, fenfluramine, cannabidiol Fenfluramine and cannabidiol cannot be considered precision (mechanism-based) treatments SCN2A Sodium voltage-gated channel alpha subunit 2 Voltage-gated sodium channel Benign familial infantile seizures or EIEE High levels of phenytoin; levetiracetam Not yet clear whether levetiracetam can be considered precision (mechanism-based) treatment SCN2A Sodium voltage-gated channel alpha subunit 2 Voltage-gated sodium channel EIEE, status epilepticus Lidocaine, acetazolamide Evidence for precision (mechanism-based) treatment status limited SCN8A Sodium voltage-gated channel alpha subunit 8 Voltage-gated sodium channel Benign familial infantile seizures or EIEE High levels of phenytoin or carbamazepine; amitriptyline, nilvadipine, carvedilol Based on one study for one mutation in SCN8A SLC2A1 Solute carrier family 2 member 1 Transporter Idiopathic generalized epilepsy Ketogenic diet Bypasses the pathophysiology to provide an alternative energy supply to the brain STXBP1 Syntaxin-binding protein 1 Membrane trafficking EIEE Levetiracetam, folinic acid, vigabatrin Only anecdotal evidence TSC1 and 2 TSC (tuberous sclerosis complex) subunits 1 and 2 Unclassified; mutations lead to increased activity of mTOR Tuberous sclerosis Everolimus A precision treatment with support from clinical trials and licensed for particular uses in tuberous sclerosis complex EIEE, early infantile epileptic encephalopathy.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}