


Abstract
Reactive oxygen species (ROS) have been correlated with almost every human disease. Yet clinical exploitation of these hypotheses by pharmacological modulation of ROS has been scarce to nonexistent. Are ROS, thus, irrelevant for disease? No. One key misconception in the ROS field has been its consideration as a rather detrimental metabolic by-product of cell metabolism, and thus, any approach eliminating ROS to a certain tolerable level would be beneficial. We now know, instead, that ROS at every concentration, low or high, can serve many essential signaling and metabolic functions. This likely explains why systemic, nonspecific antioxidants have failed in the clinic, often with neutral and sometimes even detrimental outcomes. Recently, drug development has focused, instead, on identifying and selectively modulating ROS enzymatic sources that in a given constellation cause disease while leaving ROS physiologic signaling and metabolic functions intact. As sources, the family of NADPH oxidases stands out as the only enzyme family solely dedicated to ROS formation. Selectively targeting disease-relevant ROS-related proteins is already quite advanced, as evidenced by several phase II/III clinical trials and the first drugs having passed registration. The ROS field is expanding by including target enzymes and maturing to resemble more and more modern, big data–enhanced drug discovery and development, including network pharmacology. By defining a disease based on a distinct mechanism, in this case ROS dysregulation, and not by a symptom or phenotype anymore, ROS pharmacology is leaping forward from a clinical underperformer to a proof of concept within the new era of mechanism-based precision medicine.
Significance Statement Despite being correlated to almost every human disease, nearly no ROS modulator has been translated to the clinics yet. Here, we move far beyond the old-fashioned misconception of ROS as detrimental metabolic by-products and suggest 1) novel pharmacological targeting focused on selective modulation of ROS enzymatic sources, 2) mechanism-based redefinition of diseases, and 3) network pharmacology within the ROS field, altogether toward the new era of ROS pharmacology in precision medicine.
I. Introduction
For decades, the oxidative stress theory has placed ROS as the basis of numerous disease states (Ghezzi et al., 2017). Consequently, antioxidants have been broadly tested as therapeutic agents for neurodegenerative diseases (Schmidt et al., 2015), cancer (Nezis and Stenmark, 2012), brain ischemia (Lees et al., 2006), and endothelial dysfunction (Ashor et al., 2014), to name a few. So far, hardly any of them have reached the efficacy and or safety criteria sufficient for drug approval. There is only one approved antioxidant drug, edaravone, to treat a small subset (Abe et al., 2017) of patients with amyotrophic lateral sclerosis (Hardiman and van den Berg, 2017). From a clinical pharmacological perspective, the questions arise as to whether the oxidative stress theory is wrong, whether ROS are a therapeutic target at all, and whether the wrong diseases or wrong approaches have been chosen.
One fundamental theory of ROS biology that introduced a considerable bias for redox medicine has been the concept of whole-cell redox balance or homeostasis (Sies, 2019). Moreover, the notion that ROS are dangerous metabolic by-products to be controlled to a certain tolerable level to prevent oxidative stress remains as controversial theory. In fact, the existence of antioxidant enzymes such as superoxide dismutase (McCord and Fridovich, 1988) could be viewed in support of this bias. Many other second messengers, however, are also controlled by both synthesis (e.g., cyclases, kinases) and degradation (e.g., phosphodiesterases, phosphatases). This would argue both against the notion of ROS being primarily detrimental and against an ROS-specific cellular mechanism to ensure local steady-state levels of a signaling molecule. If ROS formation is dysregulated and triggers disease, this may happen only in a certain cellular compartment, whereas many other simultaneous ROS signaling events in different compartments may still be required for essential physiologic processes, such as cell differentiation and proliferation, immune response, vascular tone, and hormone synthesis (Altenhöfer et al., 2015). This would argue against a whole-cell redox status. Moreover, these latter physiologic roles of ROS can occur at both high and low concentrations, demonstrating a strictly quantitative criteria for ROS being physiologic or pathologic, and this may explain the recurrent failures in translating broadly acting antioxidants for clinical application (Steinhubl, 2008). Finally, disease-relevant ROS dysregulation may include the following: 1) the induction of ROS-forming enzymes and local overproduction of ROS; 2) translocation of ROS-forming enzymes to cellular sites where, physiologically, there is no ROS formation; 3) unphysiological ROS formation interfering with ROS-sensitive pathways; or 4) changes in the ROS formed, e.g., superoxide or peroxynitrite instead of hydrogen peroxide or nitric oxide (Dröge, 2002; Casas et al., 2015; Dao et al., 2015).
The novel and more clinically promising pharmacological approach to ROS reviewed here suggests selectively inhibiting only the disease-relevant ROS sources and leaving all others intact. To achieve this, disease relevance and target validation have to go beyond the concept of “small amounts of ROS beneficial or tolerable, large amounts detrimental” and no longer use terms such as “redox balance,” “redox gradient,” or “oxidative stress”; instead, the identification of a specific enzyme’s dysregulation as a causal disease mechanism is required (Ghezzi et al., 2017). Also, cases of secondary ROS formation as a by-product of many inflammatory reactions (e.g., NOX2 or NOS2) are most likely an epiphenomenon not suited as a therapeutic target. We thus strictly limit this review to clinical validations at the level of at least a phase II clinical trial or higher. This excludes, for now, some targets commonly considered in basic and preclinical research, such as 1) mitochondria and ROS-induced ROS release, which, in our view, may occur but are currently not realistic to be safely targeted pharmacologically in humans; or 2) other ROS sources for which hitherto no disease or therapeutic relevance has been shown.
Therefore, the ROS-forming and metabolizing enzymes considered for this review include NADPH oxidase (NOX) (Schramm et al., 2012; Rousset et al., 2015; Warren et al., 2017), NO synthase (NOS) (Li and Pagano, 2017), xanthine oxidoreductase (XOR) (Berry and Hare, 2004), monoamine oxidase (MAO) (Bortolato et al., 2008), and myeloperoxidase (MPO) (Malle et al., 2007). Additionally, we include proteins that are not generally considered by the ROS field. However, we view them as mechanistically related and therapeutically relevant (in particular, targets of ROS) if they are disease-relevant and amenable to pharmacological modulation, i.e., nuclear factor (erythroid-derived 2)-like 2 (NRF2) and its activators (Dao et al., 2015; Cuadrado et al., 2019) and the nitric oxide synthase (NOS)-soluble guanylate cyclase (sGC) signaling pair (Evgenov et al., 2006). Drugs targeting sGC (Sandner et al., 2019) do not modulate ROS formation directly but correct functional ROS-dependent dysregulation (Melichar et al., 2004).
Maturing the ROS field concerning successful drug discovery is confronted, however, with an even more significant challenge that the entire pharmaceutical industry and biomedicine field in general faces. Even for several drugs that are on the market, population-based studies fail to show patient-relevant benefits (Lee et al., 2012; Nosengo, 2016). Moreover, research covering drug approvals since the 1970s suggests that only a limited number of new drugs provide real advances over existing drugs; most studies put the proportion of real innovation at under 15% (Ebrahim, 2001). One of the factors contributing to this innovation roadblock is a conceptual knowledge gap applied to the whole field of medicine and our current definitions of diseases. Most diseases are defined based on a dominant symptom—indeed, mostly in one organ. However, they are often not defined based on a causal molecular mechanism, which for most common diseases, is not known. A drug, however, can only be effectively developed and applied in a precise manner if the molecular disease mechanism is known (Caldera et al., 2019; Choobdar et al., 2019).
As we are moving to molecular redefinitions of disease (Aguirre-Plans et al., 2018), we realize that common diseases are not defined by single causal proteins, unlike in highly penetrant monogenic diseases, but rather by a dysregulated signaling network as part of the cellular interactome (Sharma et al., 2015). Indeed, proteins associated with specific diseases are not randomly spread but tend to interact, forming connected subgraphs, i.e., disease modules (Menche et al., 2015). A given ROS source or ROS target may be part of one such disease module but not another. In fact, different modules may cause different or similar symptoms (phenotypes) (Ghiassian et al., 2016). Moreover, there are several ways of defining disease models. The Disease Module Identification DREAM Challenge was recently launched as an open competition to assess diverse identification methods (Choobdar et al., 2019). Although all models in the ROS field give almost identical results, we selected the first-neighbor approach obtained from validated protein-protein interactions (PPIs) within the experimentally assessed interactome (Zhang and Itan, 2019). These disease models were further pruned according to the subnetwork participation degree to correct for hub node proteins. Then, identification of disease models is, therefore, occurring because of the high number of interactions in the whole network. Thus, we present here the first draft of a network pharmacology approach to redox medicine based on clinically validated therapeutic ROS targets (seed genes) and associated construction of disease modules (Fig. 1).

A mechanotype approach to ROS-related diseases. Red shows validated ROS-related disease targets and their first neighbors (blue). A participation degree score was calculated for each protein by the ratio of total protein-protein interactions in the subnetwork to the total protein-protein interaction in the interactome. A cutoff of 0.25 was chosen for the participation degree score to filter non–cluster-relevant highly promiscuous proteins. These data suggest that ROS pathophysiology is split up into at least 10 distinct mechanotypes, which will become mechanistic disease definitions. Once metabolites are included, possible new module connections may appear, such as NOX4 linking into NOS1 signaling (Casas et al., 2019a).
This “ROSome” currently shows 10 distinct PPI modules. One module overlaps with cGMP signaling (ROS-cGMP, i.e., ROCG module). Moreover, modules may interact not only through PPI but also functionally through metabolites, as shown for the ROCG and the NOX4 module (Casas et al., 2019a), reducing the ROSome to nine functional modules. Therefore, we show here that different ROS sources belong to nonconnected modules and, thus, probably different diseases. Hence, we need to identify mechanism-related targets that will only be associated with some ROS-related disease phenotypes presenting specific ROS source-disease couples and no longer considering ROS as a general disease mechanism. Since all seed proteins are disease-relevant, these functional modules will be starting points for ROS-related target selection, diagnostics, and therapeutic prioritization as part of new mechanistic disease definitions. Thus, what we currently name a disease would be annotated as a phenotype or symptom of this “mechanotype” (Schmidt et al., 2018). Hence, we propose 1) novel pharmacological targeting focused on selective modulation of ROS enzymatic sources, 2) mechanism-based redefinition of diseases, and 3) implementation of network pharmacology approaches as the future of the ROS field in the new era of precision medicine.
II. ROS Revisited: From Oxidative Stress and Redox Biology to Redox Medicine
Based on the above, both the terms disease and redox medicine need to be renewed. One common bias has been a focus on the roles of ROS in disease rather than in physiology, thereby overlooking or neglecting many of their nonpathologic roles (Ghezzi et al., 2017). Physiologic ROS sources are expressed in different cell structures, i.e., mitochondria, peroxisomes, endoplasmic reticulum, lysosomes, and plasma membrane (Di Meo et al., 2016). Based on their distribution and colocalization, the activation of different ROS sources will result in differential subcellular superoxide (O2•), hydrogen peroxide (H2O2), hydroxyl, or [by interaction with nitric oxide (NO) or nitrite] peroxynitrite formation.
Physiologic ROS production is involved in several essential signaling functions regulating cell growth, necrosis, apoptosis, proliferation, and survival (Zuo et al., 2015); immune system functioning (Sena and Chandel, 2012); and mitocondrial metabolism. H2O2 alters protein function through post-translational modification (Finkel, 2012) of mitogen-activated protein kinase, phosphatase and tensin homolog (Tonks, 2005; Rojo et al., 2014), and others. Finally, autophagy in response to cellular stress involving ROS leads to programmed cell death (Scherz-Shouval and Elazar, 2011; Sena and Chandel, 2012; Pajares et al., 2017, 2018). However, ROS is among several other intracellular signaling transducers that sustain and control autophagy. Mitochondria-derived ROS indirectly activates AMP protein kinases, which are involved in several signaling mechanisms, e.g., glucose metabolism and inflammatory response. Indeed, AMP protein kinase inhibitors have been suggested as a possible pharmacotherapy for neuroprotection and cancer, although no compound reached the clinical stage (Viollet et al., 2010).
Immune cells destroy pathogens via the NOX2-mediated oxidative burst, a key mechanism for antiviral, antiparasitic, and antibacterial responses (Arsenijevic et al., 2000), and NOX-dependent regulation of toll-like receptor 4 (Ogier-Denis et al., 2008). In the nervous system, ROS regulate synaptic plasticity at the cellular level by modulating long-term potentiation as part of the learning process and memory formation and/or maintenance. Conversely, ROS dysregulation is involved in the impairment of memory in the aged brain in neurodegenerative diseases and dementia (Rojo et al., 2017, 2018). Moreover, NOX3 is essential for audition because of its role in otoconia formation, whereas different NO synthase isoforms play a role in blood pressure regulation, inflammation, and neuronal homeostasis. ROS toxifiers, e.g., MPO, use H2O2 to oxidize the halides I−, Br−, and Cl−, resulting in the subsequent hypopseudohalous acids with an extreme oxidative reactivity (Casas et al., 2015). ROS are therefore linked to several physiologic mechanisms, and if essential, their dysregulation of off-target inhibition may lead to pathology or drug side effects, respectively (Fig. 2).

Role of ROS in physiology and disease. ROS plays a crucial role in several physiologic mechanisms, i.e., angiogenesis, cell signaling, immune system, and memory formation. However, ROS have also been suggested to be part of several pathomechanisms because of a dysregulation of specific ROS sources that leads to unphysiological 1) regulation, 2) location, and 3) amount of ROS produced. Our approach is focused on inhibition of ROS formation through NOX, XO, MAO, NOS, and MPO. A parallel strategy focuses on the direct repair of ROS-induced damage through modulation of the cGMP-forming enzyme sGC using both activators [sGC activators (sGCas)] and stimulators (sGCs). ONOO−, peroxynitrite.
III. ROS Generators and Toxifiers in Disease
For several ROS sources and toxifiers, clear target validation has been demonstrated. For therapeutic application, this has to be evaluated in light of their physiologic function, possible side effects, and the resulting risk-benefit ratio. Thus, in the following sections, both of these aspects are discussed per target. In general, acute indications such as ischemic stroke (Kleinschnitz et al., 2010; Casas et al., 2017, 2019a,b) and neurotrauma (Stover et al., 2014) appear safer than those potentially requiring chronic therapy. Moreover, since most targets are embedded in multiprotein signaling modules (see above), multidrug approaches on the same module, i.e., network pharmacology (Casas et al., 2019a), with lower doses of each drug also seem more promising and safer than single-target, single-drug approaches.
A. Focus on Oxidases
1. NADPH Oxidase
Human NADPH oxidases are a family of seven transmembrane proteins, i.e., five mono-oxidases [NOX1, NOX2, NOX3, NOX4, and NOX5 (Wingler et al., 2011)] and two dual oxidases [DUOX1 and DUOX2 (van der Vliet et al., 2018)], each with specific tissue distribution and physiologic functions. Of these, NOX5 is the only one that is missing from the mouse and rat genome and requires knockin mouse models to be investigated in animal models, which has resulted in the fact that this isoform is preclinically understudied (Jha et al., 2017; Casas et al., 2019b; Touyz et al., 2019a,b). NOX1–4 catalytic units, but not NOX5, form a complex with the transmembrane protein p22phox and, further, with different cytosolic regulatory (e.g., p67phox or NOXA1; the small G protein Rac) and/or organizational protein partners (e.g., p47phox or NOXO1). Most NOX isoforms produce O2•−, whereas NOX4 primarily leads to H2O2 (Peng et al., 2019). NOX isoforms also differ regarding ROS generation kinetics (low and high output), abundance (low and high expression levels), subunit requirements, and modes of activation (basally active or inactive). Concerning regulation, NOX5 and the DUOXs stand out by being directly calcium-activated (Rigutto et al., 2009; Touyz et al., 2019b). However, unlike NOX5, DUOX produces H2O2 similar to NOX4; for details, see (Altenhöfer et al., 2015).
NOX1 is mainly expressed in the colon, where it appears to play a role in gut immune regulation (Schwerd et al., 2018). NOX2 was the first NOX to be identified, originally named phagocytic oxidase glycoprotein of 91 kDa, gp91phox, and is mainly expressed in leukocytes (eosinophils, neutrophils, macrophages). It represents a vital component of the phagocytic oxidative burst, and it has been suggested that it is involved in cell proliferation, differentiation, and survival; contractility; neuronal plasticity; and apoptosis (Diebold et al., 2015). Moreover, inflammatory cytokines signal through mitochondrial ROS production lead to the activation of the tumor necrotic factor receptor which promotes NF-kB inflammatory response. NLRP3-dependent inflammasomes are activated through ROS, promoting a quick inflammatory response (Tschopp, 2011). NOX3 appears to have the most restricted distribution, e.g., in the inner ear, where it is essential for the formation of otoconia, calcium carbonate crystals in the vestibule responsible for the detection of linear acceleration, gravity, and audition (Paffenholz et al., 2004; Kiss et al., 2006). A recent study, however, showed NOX3 overexpression in heart failure, suggesting new physiologic roles for this isoform (Bkaily et al., 2019). Conversely, NOX4 has the most ubiquitous distribution, including in the brain, endothelium, kidney, muscle, and adipose tissue. Therefore, NOX4 is constitutively active to preferentially produce H2O2, as opposed to all other NOXs, which produce superoxide. NOX4 plays a crucial role in cell differentiation, e.g., in preadipocytes and endothelial cells for angiogenesis (Schröder et al., 2009), as well as in neural precursor cells for hippocampal neurogenesis and memory formation (Choi et al., 2019; Yoshikawa et al., 2019). NOX5 is physiologically expressed in endothelium, spermatocytes, and lymphoid organs (Jagnandan et al., 2007). In mice, a triple NOX1-2-4 knockout is viable and lacks a distinct phenotype (Rezende et al., 2016). Thus, at least in mice under basal conditions, NOX1-2-4 appear to be neither essential for survival nor indispensable for basal physiology. This scenario changes, of course, upon infection or under stress condition (Odalys et al., 2015) or under stress conditions (Zhang et al., 2010). DUOX1 and 2 are expressed in the thyroid gland and are essential for thyroid hormone synthesis (Moreno et al., 2002).
Regarding validated pathophysiological roles, NOX enzymes are involved in several disease conditions primarily based on NOX dysregulation (Casas et al., 2015; Dao et al., 2015) (Table 1). Current drug developments for NOX inhibition focus on fibrotic and neurovascular disease indications, with NOX1, 4, and 5 as the main isoforms to be targeted (Casas et al., 2017; Jha et al., 2017; Teixeira et al., 2017). NOX1 mutations are also found in patients with inflammatory bowel diseases (Scherz-Shouval and Elazar, 2011; Hayes et al., 2015). Moreover, NOX1 appears to contribute to diabetic atherosclerosis (Di Marco et al., 2016) and retinopathies (Wilkinson-Berka et al., 2014). As a proinflammatory gene, it is not surprising that NOX2 has been suggested as a therapeutic target not only in inflammatory diseases, i.e., acute respiratory distress syndrome and chronic obstructive pulmonary disease, but also in muscle and neurodegenerative disorders such as Parkinson disease, amyotrophic lateral sclerosis, and schizophrenia, to name a few (Diebold et al., 2015). Moreover, the genetic deletion of NOX2 may reduce lung metastasis (Xu et al., 2016; van der Weyden et al., 2017). Because of its role in the inner ear, NOX3 could be a key target for hearing loss. NOX4-derived H2O2 leads to the pathologic fibrosis of liver (Lan et al., 2015) and lung (Carnesecchi et al., 2011) and blood-brain barrier breakdown and neurodegeneration upon ischemic stroke (Kleinschnitz et al., 2010; Casas et al., 2017, 2019a,b), but it is surprisingly protective in atherosclerosis (Gray et al., 2016). NOX5 might be a suitable target in cardiovascular pathologies (Touyz et al., 2019a) and stroke (Casas et al., 2019b). DUOX2 and DUOXA2 loss-of-function mutations lead to congenital hypothyroidism (Weber et al., 2013) and can be targeted for radiation-induced thyroid cancer (Ameziane-El-Hassani et al., 2016).
NOX isoforms: function and potential pathologic role
Concerning safety-benefit considerations, pharmacological inhibition of DUOX2 should most likely be circumvented to avoid hypothyroidism and bowel inflammation (Hayes et al., 2015). Also, the safety profile of inhibiting NOX2 is controversial based on its role in innate immunity (Sareila et al., 2011). A small residual activity, however, may be sufficient to confer antimicrobial activity (Diebold et al., 2015), although the risk of noninfectious complications similar to those in patients with chronic granulomatous disease is less clear (Henrickson et al., 2018). A potential concern regarding NOX1 and NOX4 inhibition is to enhance inflammation in the gut by inhibiting NOX1 and diminishing the potential protective mechanisms of NOX4 in vascular cells, thus promoting atherosclerosis (Schürmann et al., 2015; Gray et al., 2016) as well as enhancing the risks of kidney fibrosis (Nlandu Khodo et al., 2012) and liver cancer (Crosas-Molist et al., 2017). Acute indications such as ischemic stroke (Kleinschnitz et al., 2010; Casas et al., 2017) are likely to have, however, a low risk-benefit profile.
2. Nitric Oxide Synthases
NOSs are homodimeric NADPH binding flavoheme proteins additionally regulated by the redox-sensitive cofactor tetrahydrobiopterin (H4Bip), calmodulin, and several other modulatory interactions (Nedvetsky et al., 2002) to convert l-arginine (Schmidt et al., 1988) to NO. Three isoforms exist, and they were initially named according to their first observed cellular/tissue localization or expressional regulation, i.e., neuronal, inducible, and endothelial (i.e., NOS1, NOS2, and NOS3) (Schmidt et al., 1991; Förstermann et al., 1992; Chakrabarti et al., 2012; Liu et al., 2012; Caviedes et al., 2017). Qualitatively, three functional states need to be differentiated: 1) physiologic NO formation signaling via sGC into cGMP and PKG (Schmidt and Walter, 1994); 2) excessive NO formation leading to cellular and tissue damage (Kleinschnitz et al., 2016), e.g., in stroke; and 3) uncoupled states of NOS in which the activation of oxygen is not coupled to the oxidation of arginine but results in leakage of ROS, such as in endothelial dysfunction and hypertension (Li et al., 2015).
Several mechanisms have been suggested to cause NOS uncoupling, for which oxidation of H4Bip (tetrahydrobiopterin) to H2Bip (dihydrobiopterin) appears to be crucial (Schmidt et al., 1992; Bömmel et al., 1998). H2Bip leads to enzyme monomerization (Reif et al., 1999) and inhibition of activity (Kotsonis et al., 1999). In turn, supplementation with arginine (Schramm et al., 2002) and/or H4Bip in vitro (Kotsonis et al., 2000) or in vivo (Moens et al., 2011) recouples NOS and restores NO-cGMP signaling and function. An alternative approach to substituting H4Bip may be to facilitate its endogenous recycling via dihydrofolate reductase (Crabtree et al., 2009; Crabtree and Channon, 2011). Counterproductive competition of folic acid with H4Bip does not seem to occur. Another pathophysiologically relevant event is the accumulation of methylated arginine analogs, in particular asymmetric dimethylarginine (Antoniades et al., 2009), which inhibits NOS and appears to be an independent cardiovascular risk factor. This mechanism may be a rationale to clinically substitute l-arginine (Schramm et al., 2002) or l-citrulline (subsequently converted to l-arginine) to compete against asymmetric dimethylarginine–induced NOS inhibition, despite cellular l-arginine being sufficiently high to also thoroughly saturate NOS under disease conditions. Besides augmenting NO synthesis and subsequent cGMP formation, there may be situations in which NO levels are unphysiologically high, causing disease states, most likely in a cGMP-independent manner. These include inflammatory and hypoxic states. Attempts to exploit previous conditions clinically have all failed. Conversely, NOS inhibition using anti-pterins (Fröhlich et al., 1999; Kotsonis et al., 2001), such as ronopterin, in neurotrauma (Stover et al., 2014) and stroke (Kleinschnitz et al., 2016; Casas et al., 2019a) may be promising. Future pivotal clinical trials [No Synthase in Traumatic Brain Injury (NOSTRA) III, NCT02794168] to be finalized in 2020 will tell.
Concerning risk-benefit profile, NOS inhibition (chronic inhibition, in particular) could be detrimental for patients suffering from cardiovascular and renal diseases due to endothelial dysfunction, i.e., vasoconstriction, hypertension, atherosclerosis, and insulin resistance. Thus, acute indications seem to be more promising.
3. Xanthine Oxidase
Xanthine oxidoreductase is a terminal enzyme in the purine catabolic pathway that catalyzes the conversion of hypoxanthine to xanthine and then to uric acid (UA). XOR exists in two isoforms: xanthine oxidase (XO) and xanthine dehydrogenase (XDH). To note, XDH and XO are interconvertible, i.e., XDH can be converted into XO (Battelli et al., 1973). Thus, not only is XO an ROS-generating enzyme, but oxidative modification would convert residual XDH into XO to produce more ROS.
Physiologically, UA can directly activate the NACHT, LRR, and PYD domain–containing protein 3 inflammasome, resulting in toll-like receptor–independent production of interleukin (IL)-1β and IL-18 (Martinon et al., 2006). This is not only restricted to urate crystals but also an activity of soluble UA (Braga et al., 2017). Thus, UA adds a further element of complexity into possible ROS-independent activities of XO. Moreover, UA is not just a biomarker of XO activation, but it has also been indicated as a potent antioxidant (Ames, 1989).
The best-known pathologic role of XOR is independent of ROS in the accumulation of uric acid in gout. Text mining analysis presents ischemia-reperfusion injury (Granger et al., 1981) as the primary disease concept associated to XO (841 hits), followed by tumor development (422) and several cardiovascular diseases (CVD), including hypertension (169), diabetes (143), myocardial ischemia (125), myocardial infarction (101), atherosclerosis (118), coronary disease (115), and heart failure (111). XO can be activated in reperfusion because of the high availability of substrates. Angiotensin II can induce XO levels in aortic endothelial cells in vitro (Landmesser et al., 2007), which may be relevant in CVD states. For example, in patients with coronary artery disease, XO is activated along with NOX (Spiekermann et al., 2003), and plasma XO levels are associated with left ventricular hypertrophy in hypertension (Butts et al., 2019). Assuming that XO activation is a causal mechanism in disease, one might expect that UA would correlate with disease or disease severity. Despite all evidence of XO in CVD, the association of hyperuricemia and CVD has remained unclear and controversial (Feig et al., 2006). A positive correlation between UA levels and disease was observed for chronic kidney disease (Wang et al., 2018a), peripheral artery disease (Wang et al., 2018a), atrial fibrillation (Tamariz et al., 2014), and nonalcoholic fatty liver disease (Jaruvongvanich et al., 2017). In several other conditions, however, meta-analyses resulted in a negative correlation between UA and disease, including neurologic or psychiatric diseases such as Parkinson (Wei et al., 2018), Alzheimer (Mullan et al., 2018), depression (Bartoli et al., 2018), autism (Wen et al., 2017), and dementia (Khan et al., 2016). XO is also elevated in tissues and serum after viral or bacterial infection (Akaike et al., 1990). The induction of XO by pathogens is probably mediated by interferon (IFN), which acts as an XO inducer at the transcriptional level (Ghezzi et al., 1984). These findings may explain infections as risk factors for CVD.
For a risk-benefit profile of XO as a target, safety appears to be high based on the decade-long experience with antigout medication. It has been suggested that close monitoring should be taken into account in patients with impaired renal function (Jordan and Gresser, 2018). A recent study argues, however, against such a risk (Vargas-Santos et al., 2018).
4. Monoamine Oxidase
MAOs are flavin-containing enzymes located in the outer mitochondrial membrane. They catalyze the oxidative deamination of both endogenous and exogenous amines, especially modulating the metabolism of neurotransmitters and several drugs. During this process, MAOs generate a significant amount of H2O2 as a by-product and, indirectly, aldehyde and ammonia (Casas et al., 2015). MAOs exist in two isoforms, A and B, which differ in structure, substrate preference, inhibitor specificity, and tissue distribution (Binda et al., 2002; De Colibus et al., 2005; Finberg, 2014). MAO-dependent increases in H2O2 formation have been observed in isolated skeletal and cardiac myocytes (Kaludercic et al., 2010, 2014; Sorato et al., 2014; Deshwal et al., 2018); increased expression of MAO-B, but not MAO-A, has been observed in various other tissues upon aging, including brain (Fowler et al., 1997).
In mood disorder models related to stress and glucocorticoid administration, a decrease in neurotransmitter levels and the preferred utilization of serotonin and noradrenaline by MAO-A led to the development of MAO-A inhibitors as antidepressants. Clinical data are, however, not yet available. MAO-A–dependent ROS formation contributes to muscle dystrophy and other pathologies (Sorato et al., 2014), such us prostate tumorigenesis and cancer metastasis. Pharmacological or genetic inhibition of MAO-A reduced or even eliminated prostate tumor growth and metastasis in prostate cancer mouse models, whereas high MAO-A expression in cancer tissues correlated with worse clinical outcomes in patients (Wu et al., 2014a). These findings suggest that MAO-A has potential as a therapeutic target in cancer treatment. On the other hand, MAO-B is increased in Huntington, Alzheimer, and Parkinson diseases (Kumar et al., 2003; Gulyás et al., 2011; Krzysztoń-Russjan et al., 2013; Ooi et al., 2015), and inhibition of MAO-B is indicated in the treatment of Parkinson disease because of its efficacy in sparing endogenous dopamine (Bortolato and Shih, 2011; Fišar, 2016). Several reports suggest that, in addition to modulating neurotransmitter levels, MAO-dependent ROS generation promotes the development of neurodegenerative disorders by causing oxidative damage to neurons (Youdim et al., 2006; Bortolato et al., 2008). Additionally, MAO contributes to cardiovascular disease, including ischemia-reperfusion injury (Bianchi et al., 2005; Pchejetski et al., 2007; Carpi et al., 2009), pressure overload (Kaludercic et al., 2010, 2014), diabetic cardiomyopathy (Deshwal et al., 2018), postoperative atrial fibrillation (Anderson et al., 2014), and vascular dysfunction (Sturza et al., 2013). MAO activity is increased in the left and right ventricles from patients with ischemic heart disease (Manni et al., 2016). The mechanisms underlying MAO toxicity have mostly been attributed to excessive H2O2 and aldehyde generation, which leads to impaired mitochondria-endoplasmic reticulum cross talk and cardiomyocyte death (Kaludercic et al., 2014; Deshwal et al., 2018). Based on the known side effects of MAO inhibitors, their risk-benefit profile for novel severe indications appears to be beneficial.
B. Focus on Toxifiers
1. Myeloperoxidase
Myeloperoxidase is heme protein (Fiedler et al., 2000) that is located primarily in neutrophils and, to a lesser extent, in monocytes (Bainton et al., 1971) and uses H2O2 to oxidize the halides I−, Br−, and Cl− (but not F−) and the pseudohalide SCN− to give the corresponding hypohalous and hypopseudohalous acids, which have high oxidative reactivity (Jantschko et al., 2005). Besides hypo(pseudo)halous species (HOSCN), MPO produces other potent oxidative compounds, such as the nitrous radical (NO•) and peroxynitrite (Eiserich et al., 2002). The highly oxidative product, hypochlorous acid, has an essential role in immunity by causing oxidative damage to biomolecules of pathogens phagocytosed by neutrophils (Odell and Segal, 1988) and stimulating the release of inflammatory mediators. By oxidizing serotonin, MPO not only impairs physiologic functions of serotonin but also generates the neurotoxic compound tryptamine-4,5-dione (Ximenes et al., 2009). Besides, MPO-catalyzed oxidation of uric acid has been reported in patients with gout, in whom reactive metabolites from urate may contribute to the pathology of gout (Stamp et al., 2012) and may be implicated in several chronic inflammatory syndromes related to the central nervous system, cardiovascular system, respiratory system, and renal system, including glomerular and tubulointerstitial disorders (Klebanoff, 2005). MPO is involved in the development of atherosclerosis (Libby et al., 2011), which is related to the overexpression of the enzyme, and chloro-tyrosine (ClTyr) has been considered as a potential biomarker for CVD (see section VI. Biomarkers of ROS). MPO-dependent oxidative modification makes high-density lipoprotein dysfunctional, i.e., unable to contribute to cholesterol efflux (Smith, 2010). In gestational hypertension, MPO mediates endothelial dysfunction by directly consuming NO (Nicholls and Hazen, 2005; Rocha-Penha et al., 2017). However, MPO-deficient mice do not show enhanced endothelial function compared with wild-type mice, even when provoked with lipopolysaccharide (Golubinskaya et al., 2014). Moreover, high levels of MPO have been detected in brain tissues of patients with several neurodegenerative disorders (Gellhaar et al., 2017), such as Parkinson (Choi et al., 2005), Huntington (Choi et al., 2005), Alzheimer (Green et al., 2004), and multiple sclerosis (Nagra et al., 1997). However, mice lacking MPO are more susceptible to experimental autoimmune encephalomyelitis (Brennan et al., 2001). Studies on epileptic brains suggested that MPO is the key source of chlorination stress in epileptogenesis (Ray and Katyal, 2016). Patients with cystic fibrosis show high amounts of MPO in lung tissue. Moreover, the damaged tissues contain significant levels of 3-chlorotyrosine, 3,3′-dityrosine, and nitrotyrosine (van der Weyden et al., 2017). Finally, MPO correlates with several types of cancers, i.e., lung (Li et al., 2014), renal (Ramsaransing et al., 2003), esophageal (Inayama et al., 2007), epithelial ovarian (Saed et al., 2010), bladder (Hung et al., 2004), larynx (Cascorbi et al., 2000), and acute leukemia (Kim et al., 2012).
IV. Pharmacological Modulation of ROS
A. Inhibition of ROS Formation
1. NADPH Oxidase Inhibitors
Conventional methods used to measure NOX activity are prone to artifacts (Maghzal et al., 2012; Dao et al., 2020), and particular attention is required for assays used to measure H2O2 and O2•, such as L-012, luminol, or Amplex Red, because of their lack of specificity and further shortcomings (Dikalov and Harrison, 2014; Zielonka et al., 2014). Thus, methods to eliminate nonspecific hits are essential. This type of approach has indeed limited many reported NOX inhibitors, including apocynin, VAS2870, 2-acetylphenothiazine (ML-171), GKT136901.
Small-molecule NOX inhibitors are already well described in the literature (Jaquet et al., 2009; Altenhöfer et al., 2015). GSK2795039 was identified after a high-throughput screening performed by GlaxoSmithKline, which it had claimed in international patent application WO/2012/170752 or WO/2011/075559. Its IC50 is in the sub-micromolar range for NOX2, and it is inactive against other NOX isoforms. GSK2795039 is orally bioavailable, it can dose-dependently block NOX2 in a model of paw inflammation, and has a protective effect in the cerulein mouse model of pancreatitis. CPP11G and CPP11H are two recently discovered in silico–modeled NOX2 inhibitors that can improve endothelial cell inflammation and vessel dysfunction by abolishing tumor necrotic factor α-induced ROS production (Li et al., 2019). Thioridazine and several other phenothiazines, some of them already marketed for different indications (i.e., antipsychotic), are moderate inhibitors of NOX1–5 that are in the low micromolar range. Thioridazine is a useful pharmacological tool to inhibit NOX in vivo, as it was shown to mitigate O2− production in the spinal cord of a mouse model of amyotrophic lateral sclerosis (ALS). However, it did not show increased survival (Seredenina et al., 2016). Unfortunately, N-substituted phenothiazines are molecules with numerous described modes of action and, therefore, several off-target effects to be considered. Ewha-18278 (APX-115) inhibits NOX1, NOX2, and NOX4 in the low micromolar range. Ewha-18278 is orally available and shows therapeutic benefit in a mouse model of ovariectomy-induced osteoporosis (Joo et al., 2016) and mouse models of diabetic nephropathy (Cha et al., 2017).
NADPH oxidase inhibitors have attracted international patent submissions starting in 1990, when small companies and academia took the lead, whereas large pharmaceutical entities were mostly absent. The premiere patentee is the French company Genkyotex SA, which has built a pipeline of NOX inhibitors, although recently promising compounds have been patented by Actelion (MX2017016908) and Glenmark (WO2018203298). Although the earliest Genkyotex research had focused on the NOX2 isoform, the lead candidate GKT137831 (Setanaxib, but also recently abbreviated to GKT-831) is claimed as a NOX1/4 dual inhibitor. So far, it is one of the few NADPH oxidase inhibitors besides APX-115 (see below) that has reached the clinical trial stage. Apocynin, a methoxycatechol investigated by the Medical University of Lodz by inhalation in chronic congestive pulmonary disease and patients with asthma, is far from being NOX-specific and, thus, is not considered a NOX inhibitor in this context. GKT-831 is being developed for diabetic nephropathy in its first clinical efficacy studies, and in November 2015, an orphan drug designation was obtained in the United States and Europe for systemic sclerosis.
During the phase I program, this candidate exhibited a favorable pharmacokinetic profile that was dose-proportional over the 10- to 900-mg range. In the subsequent phase II trial (NCT02010242) in 155 patients with diabetic nephropathy and residual albuminuria, despite maximal inhibition of the renin-angiotensin-aldosterone system, the primary endpoint (reduction of albuminuria) was not reached. A phase II study with GKT-831 (400 mg once or twice daily for 24 weeks) in patients with primary biliary cholangitis (an orphan disease) started in June 2017 (NCT03226067) and met its primary and secondary interim efficacy endpoints in November 2018. Success in this indication could expand the clinical applications of GKT-831 to other liver fibrosis opportunities, such as nonalcoholic steatohepatitis.
In 2018, Genkyotex announced a phase II clinical trial with GKT-831 in patients with idiopathic pulmonary fibrosis, an indication that Genkyotex had initially chosen not to pursue. This indication follows another phase II trial in type 1 diabetes and kidney disease launched in Australia (Table 2). APX-115 is also clinically developed for the same indication, i.e., diabetic nephropathy (Lee et al., 2020), moving from phase I to II, which was originally announced for 2019 but has not yet been listed in clinicaltrials.gov.
Inhibition of ROS formation and toxification: translation to the clinics
Additional preclinical-stage compounds from the extensive Genkyotex NOX inhibitor portfolio, such as GKT136901, are currently under evaluation (Teixeira et al., 2017). Evidence in eye disease has been preclinically published in retinopathy of prematurity (Deliyanti and Wilkinson-Berka, 2015) but could very likely be extended to diabetic retinopathy. VAS2870, a NOX4 inhibitor from Vasopharm (claimed in WO/2005/111041), has recently been reported to inhibit the proliferation of retinal pigment epithelial cells after their epithelial-to-mesenchymal transition, a significant pathologic change involved in proliferative vitreoretinopathy (Yang et al., 2018). The Swedish company Glucox Biotech AB is also focused on NOX4 inhibitors and is reported to be looking into diabetes, Marfan syndrome, stroke, and cancer. GLX351322 (Anvari et al., 2015), GLX7013107, and GLX7013114 (GLX114) (Wang et al., 2018b) are compounds that are likely to be covered by the substantial intellectual property portfolio in Glucox’s name: WO/2016/133446, WO/2016/096720, WO/2014/064118, WO/2013/135803, WO/2008/008033, and WO/2003/087399. A remarkable range of less apparent indications for NOX inhibitors has been claimed by academia: cardiac arrhythmia (WO/2013/158782, University of California), muscular dystrophies (WO/2013/078261, University of Maryland), cataract and presbyopia (WO/2009/044294, Université de Genève), and a broad range of psychiatric disorders (WO/2009/052454, University of California). So far, it is not possible to estimate what clinical potential NOX inhibitors could have in these applications. Similarly, 1) the tetrahydroisoquinoline NOX2 inhibitors from Pittsburgh University (WO/2014/179592), 2) bi- and triaromatic compounds claimed by the University of Surrey (WO/2013/038136), and finally 3) quinazolines (WO/2013/181135, WO/2012/058211) and piperazines (WO/2012/173952) from Emory University merit significant attention (Fig. 3).
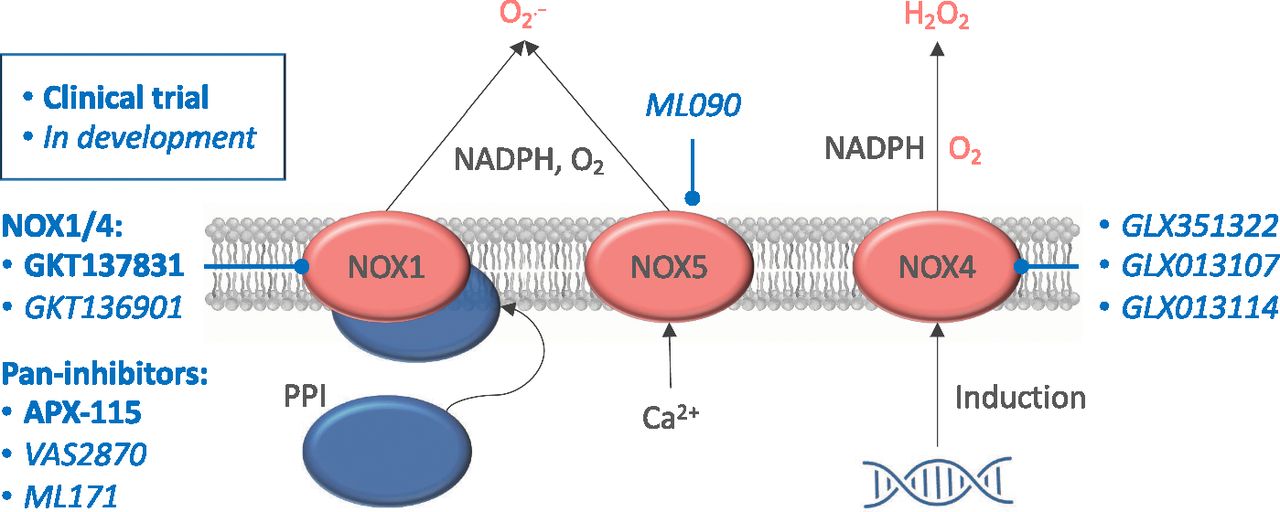
NADPH oxidase isoforms with clinical potential. NOX1 and 4 need a complete protein complex to be fully activated and generate superoxide (O2−; in the case of NOX1) and H2O2 (in the case of NOX4). NOX5 stands out by being directly calcium-activated and also producing superoxide. Under several pathophysiological conditions, all NADPH oxidase isoforms can be directly induced. Pan-inhibitors, dual NOX1/4 inhibitors, and specific NOX4 and NOX5 inhibitors are shown based on their current status, i.e., under clinical testing or preclinical development.
a. Limitations of NOX inhibitors
The observed protected role of NOX inhibition in several preclinical studies identifies NOX isoforms as a promising therapeutic target. However, the specificity of NOX inhibitors remains controversial. Widely used compounds such as diphenyleneiodonium chloride, ebselen, and VAS2870 inhibit all NOXs in similar micromolar or sub-micromolar ranges, compromising their usefulness as isoform-specific experimental tools. Moreover, apocynin, frequently used as a NOX2 inhibitor, resulted in complete inactivation of all NOXs, suggesting that its anti-inflammatory therapeutic properties cannot be attributed to NOX inhibition. Controversially, a recent report has shown that GKT137831 and its close analog, GKT136901, are indeed not active on NOX enzymes (Augsburger et al., 2019) but rather to an oxidant scavenging effect, as previously suggested (Schildknecht et al., 2014). However, several previous studies and in-house experimentation showed its NOX1/4 activity (Altenhöfer et al., 2015). Although the field is intensively criticized, highly specific compounds recently entered the pipeline. GSK2795039 (Hirano et al., 2015) remains the first NOX2 competitive inhibitor in which in vivo efficacy was thoroughly studied, although other compounds, i.e., CPP11G and CPP11H, are now in the early stages of development (Li et al., 2019). Moreover, GLX7013114 (Glucox, Sweden) has shown a NOX4-specific profile, whereas ML090 resulted in a potent NOX5 inhibitor (Casas et al., 2019b). An immediately applicable NOX inhibitor panel has been recently proposed that suggests ML171, VAS2870, M13, and ML090 as NOX1, 2, 4, and 5 inhibitors, respectively (at specific concentrations), allowing NOX-dependent target validation in scenarios in which using knockout/knockin animals is not possible (Dao et al., 2020). Thus, IC50 values of these compounds should be carefully considered for experimental design to ensure a suitable concentration range for both in vivo and in vitro studies. Despite recent advances, precise systematic evaluation using multiple redox test systems is still required to identify novel compounds to wisely target NOXs and their pathologic role.
2. NO Synthase Inhibitors
A large number of nitric oxide synthase inhibitors with various degrees of selectivity for NOS isoenzymes are available and claimed in patents. However, few have reached the clinical trial stage, and not all of these meet the criteria for pharmaceutical development ability. This applies primarily to the numerous derivatives of l-arginine, the physiologic NOS substrate:
NG-monomethyl-l-arginine (tilarginine; the archetypal, relatively nonselective NOS inhibitor, also endogenous),
NG-nitro-l-arginine methyl ester (inactive prodrug of NG-nitro-l-arginine) (Pfeiffer et al., 1996), and
other nitroarginine esters (l-NG-benzylarginine, l-NG-aminoarginine, iminoethylornithine).
Clinical trials have been conducted with most of these compounds. Many were conducted in stand-alone settings, e.g., tilarginine was tested in acute myocardial infarction complicated by refractory cardiogenic shock (the TRIUMPH trial, NCT00112281), which failed to reduce mortality (Alexander et al., 2007). Recently, l-arginine–derived NOS inhibitors have been employed as potentially synergistic adjuncts to other candidate compounds, such as tilarginine, to block blood flow in the gut to prevent glucagon-like peptide-2 release from gut lipid stores (ongoing study sponsored by the Toronto University Health Network, NCT03534661). Additionally, tilarginine is also used to treat cancers when combined with the anti–programmed death-1 monoclonal antibody pembrolizumab (Merck & Co.’s Keytruda), again as a blood flow disruptor (NCT03236935; phase Ib). Moreover, specific inducible NOS inhibitors, i.e., acetamidine derivatives, have been synthesized and evaluated that show high isoform specificity under sub-micromolar concentrations (Fantacuzzi et al., 2016). l-S-methylthiocitrulline is another amino acid derivative that is selective for neuronal NO synthase and has been reported in a phase II clinical study. However, stability issues and (more importantly) lack of patentability will prevent the further development of these compounds into drugs. The situation is different for 2-iminobiotin, a selective neuronal NOS and inducible NOS inhibitor that was originally patented by the Amsterdam University Medical Center for the treatment and prevention of perinatal asphyxia (WO/2001/074351) and is currently being developed by the Dutch company Neurophyxia B.V. The company has published an international patent application claiming the compound for cerebral hypoxia-ischemia and reperfusion injury (WO/2017/105237) and other pharmaceutical formulations (WO/2011/149349). The company has been recruiting patients for a phase II study to prevent hypoxic brain injury in patients with out-of-hospital cardiac arrest (NCT02836340) using an intravenous administration within 6 hours after the event. The current status of the trial is unknown.
The most advanced NOS inhibitor is Vasopharm Biotech GmbH’s ronopterin (VAS203; 4-aminotetrahydrobiopterine). It entered phase III development for moderate and severe closed traumatic brain injury in September 2016; recruitment for the 220-patient NOSTRA-III study (NCT02794168) was 50% complete in March 2018, with 35 European neurotrauma centers in Germany, Austria, France, United Kingdom, and Spain participating (Fig. 4). NOSTRA-III, which is due for completion in November 2019, aims to confirm the data from the phase IIa NOSTRA trial (NCT02012582), in which ronopterin produced a clinically significant two-point improvement in the extended Glasgow Outcomes Score—a clinically significant benefit (Stover et al., 2014). Vasopharm holds several patent applications on ronopterin and related pteridines (WO/2005/037286 and WO/2004/084906). With no drug treatment approved explicitly for neuroprotection after traumatic brain injury, considerable hope rests on this drug candidate (Table 2).

Therapeutic targeting of NO-sGC signaling in physiology and disease. Under physiologic conditions, NO produced by NOS1 and 3 activates the sGC, which results in cGMP formation and cell signaling regulation. However, dysregulation of this mechanism leads to increase NO and superoxide production through NOS1 and NOS3, respectively, resulting in nitrative damage. In oxidative stress conditions, the sGC heme can be either oxidized or removed (apo-sGC) and, therefore, is no longer an active enzyme. NOS inhibitors, sGC stimulators, and activators are shown based on their current status, i.e., already marketed, under clinical testing, or under preclinical development. L-NAME, l-arginine methyl ester; L-NMMA, L-NG-monomethyl arginine; SMTC, S-methyl-L-thiocitrulline; un-NOS3, uncouple endothelial NO synthase.
3. Xanthine Oxidoreductase Inhibitors
As mentioned earlier, the first XO inhibitor developed was allopurinol, currently used for the treatment of gout, followed by its derivative, oxypurinol. However, adverse events and the need for high doses (in the range of 300 mg) prompted research for novel XO inhibitors. Indeed, febuxostat has been approved by the Food and Drug Administration in 2009 and can be given at lower doses for hyperuricemia in adults suffering from gout, together with other drugs also being tested, i.e., topiroxostat. For this reason, many current trials test febuxostat rather than allopurinol. An earlier meta-analysis on congestive heart failure and other CVDs has shown an effect of allopurinol or oxypurinol on surrogate markers, including vascular function, as well as on malondialdehyde, a biomarker of excess ROS (Higgins et al., 2012). A recent trial with allopurinol in patients with chronic heart failure did not show an improvement in arterial stiffness, but it is important to note that these patients were not hyperuricemic (Alem et al., 2018). In August 2018, the University of Iowa completed the Xanthine Oxidase Inhibition in Renal Transplant Recipients trial (NCT01332799) to evaluate the effect of daily allopurinol on cardiovascular events and endothelial function in these vulnerable patients. This is in line with a small study conducted by the University of Dundee (NCT00688480) that investigated whether allopurinol can reduce both left ventricular hypertrophy and endothelial dysfunction in cardiovascular patients with renal dysfunction. Two earlier United States–based studies (NCT00181155, NCT00997542) had focused on heart failure. There are a few active clinical trials that focus on the effect of allopurinol as a suppressor of ROS generation: 1) XILO-FIST (NCT02122718), managed by the UK National Health System and the University of Glasgow, investigates whether allopurinol can reduce the formation of white matter hyperintensities, cardiac hypertrophy, and the onset of hypertonia after an ischemic stroke. The phase IV trial, which plans to randomize 464 patients, commenced in May 2014 and is estimated to last until September 2020 (Dawson et al., 2018). 2) University Hospital Tübingen, in collaboration with a dozen other European clinical centers, is recruiting for ALBINO. This phase III trial (NCT03162653), planned for 646 participants, investigates the drug’s efficacy and safety when administered immediately after birth to near-term infants with hypoxic-ischemic encephalopathy, in addition to standard hypothermic treatment. The estimated study completion date is December 2020. 3) A prospective randomized clinical trial with allopurinol versus topiroxostat (another XO inhibitor) in patients with chronic heart failure is currently ongoing in Japan (Sakuma et al., 2018). Nonpurine xanthine oxidase inhibitors have been developed to avoid the many side effects of allopurinol, but none has quite matched it in terms of efficacy. Of relevance are febuxostat (marketed as Uloric), which was discovered at the Japanese pharmaceutical company Teijin in 1998 and licensed to Takeda Pharmaceuticals and Ipsen, and topiroxostat (approval in Japan in 2013; sold as Topiloric and Uriadec). Patenting for xanthine oxidase inhibitors reveals low interest for new agents, which are almost exclusively focused on the treatment of gout (Fig. 5). Recent exceptions point toward applications in which ROS might be involved, including cancer (WO/2017/142091) and angina (WO/2010/136823). Thus, it seems highly unlikely that xanthine oxidase inhibitors would be developed into drugs that specifically address undesired ROS formation, not only because of side effects but also because of the possibility to interfere with purine metabolism (Table 2).
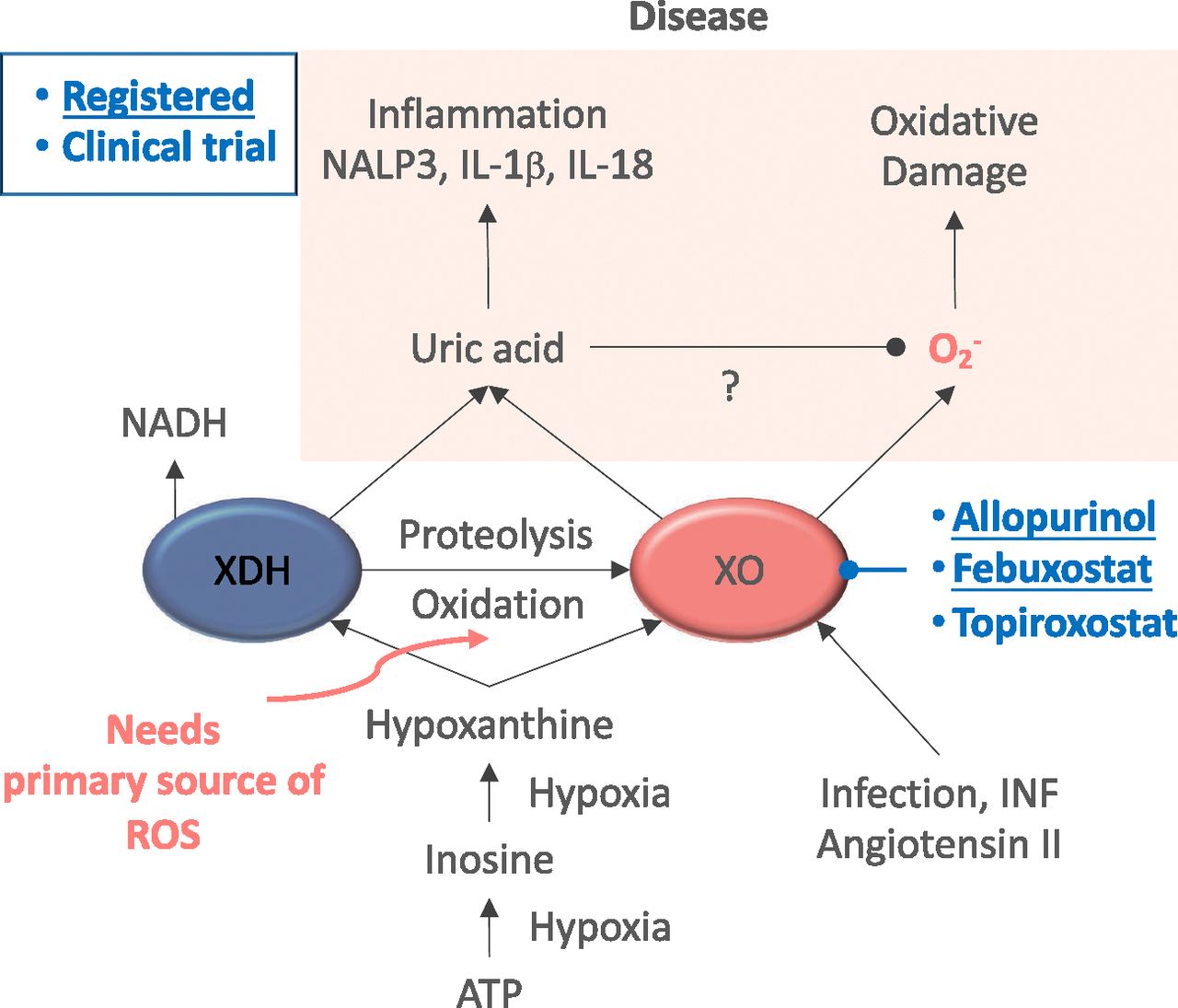
XO as a therapeutic target. In normoxic conditions, ATP levels remain stable, whereas under hypoxia, ATP is catabolized, leading to accumulation of purines, i.e., inosine and hypoxanthine. XDH under oxidative conditions gets oxidized, leading to XO. Later, XO catabolizes purines to uric acid while generating ROS. Uric acid can directly activate inflammatory pathways, resulting in toll-like receptor–independent production of IL-1β and IL-18, activation of NACHT, LRR and PYD domains-containing protein 3 (NALP3) and, therefore, inflammation and oxidative stress coursing with the disease. XO inhibitors are shown based on their current status, i.e., already marketed or under preclinical development.
4. Monoamine Oxidase Inhibitors
After the initial compounds, clorgyline and deprenyl, a wide array of MAO inhibitors have been developed, including the reversible inhibitor moclobemide for MAO-A. Reversible inhibition should be less prone to adverse effects and devoid of overdose toxicity (Lum and Stahl, 2012). However, the high degree of inhibition required for the pharmacological effects might be more challenging to achieve, since the administered inhibitor has to compete with endogenous substrates. The monoamine oxidase inhibitor that has been most intensely investigated for potential neuroprotective effects is rasagiline, an irreversible MAO-B inhibitor that has been approved as a symptomatic treatment of Parkinson disease. Efforts to prove an additional disease-modifying effect in this disease (NCT00256204) yielded ambiguous results, which caused the US Food and Drug Administration to refuse the corresponding application for an approved extension. Although rasagiline did seem to show indications of such a neuroprotective effect in ALS when added to the standard treatment riluzole (Ludolph et al., 2018), another trial found no effect on disease progression after 1 year of treatment (Statland et al., 2019). A phase IV clinical study investigating rasagiline for neuroprotection in retinal detachment (NCT02068625), suggested by data obtained with a mouse model (Eigeldinger-Berthou et al., 2012), was prematurely terminated on 31 December 2018 because of persistent recruitment difficulties. Its termination was reported in February 2019, although no further information regarding outcome has been provided yet (Table 2). Besides, multimodal compounds have been developed that combine the propargyl moiety of MAO inhibitors with compounds targeting other processes involved in neurodegenerative disorders. The best examples are ladostigil, which adds the anticholinesterase action of rivastigmine to rasagiline (Youdim et al., 2006). The latest MAO-B inhibitor released on the market is safinamide, a highly selective and reversible inhibitor of monoamine oxidase B. It is currently being used as an add-on therapy in patients with mid- to late-stage fluctuating Parkinson disease (Fabbri et al., 2015). The cardioprotective and antitumorigenic effects of MAO inhibitors are well established in animal models, so it is tempting to speculate whether the currently available compounds could be repurposed for the treatment of these pathologies. Notably, among the mitochondrial sources of ROS, MAO inhibition represents the only pharmacological approach devoid of effects on the vital functions of mitochondria related to energy conservation. This critical advantage is further enhanced by the clinical availability of MAO inhibitors, which is not the case with any other mitochondrial source of ROS. The availability of selective and reversible MAO inhibitors, currently used in patients with depression and neurodegenerative disorders and devoid of well known MAO inhibitor side effects, opens a possibility for the compounds to be considered as candidate drugs for the treatment of pathologies such as cardiovascular diseases or prostate cancer (Fig. 6).

Clinical perspective of monoamine oxidase inhibitors. MAO-A and MAO-B catalyze the oxidative deamination of endogenous and exogenous amines including several neurotransmitters, i.e., serotonin, norepinephrine, and dopamine, leading to H2O2 and aldehyde production. Dysregulation of MAOs under certain pathologic conditions results in MAO activity and, therefore, elevated availability of MAO subproducts, i.e., H2O2. MAO inhibitors are shown based on their current status, i.e., already marketed or under clinical development.
B. Inhibition of ROS Toxification
1. Myeloperoxidase Inhibitors
MPO contributes to many chronic inflammatory syndromes and aims to help treat, prevent, or at least stop the development of these diseases in high-risk patients (Malle et al., 2007; Lazarević-Pasti et al., 2015). According to the mechanism of inhibition, MPO inhibitors are divided into two main categories: 1) reversible inhibitors and 2) irreversible inhibitors (Soubhye et al., 2016).
a. Reversible inhibitors
Dietary flavonoids are useful for suppressing the lipid peroxidation of low-density lipoprotein occurring by MPO/nitrite or peroxynitrite (Schewe and Sies, 2005) and protecting the endothelial cell (Tian et al., 2017). Oral administration of (−)-epigallocatechin-3-gallate has been found to prevent colitis in mice and to be essential for mucoprotective effects (Nastase et al., 2016). Besides flavonoids, other natural compounds showed good activity against MPO, including ursolic acid, eugenol (Narasimhulu and Vardhan, 2015), and harmaline (Bensalem et al., 2014). Random and rational screening of non–anti-inflammatory drugs revealed that metoclopramide, hydralazine (Soubhye et al., 2017), and paroxetine (Soubhye et al., 2014) are potent MPO inhibitors. In vivo experiments have disclosed that different tryptamine compounds have an extensive therapeutic index as promising candidates for anti-inflammation. Use of N-acetyl lysyltyrosylcysteine amide (KYC) in mice with experimental autoimmune encephalomyelitis and middle cerebral artery occlusion can reduce the migration of myeloid cells. This inhibitor may be effective in the treatment of middle cerebral artery occlusion, reduce the formation of ClTyr and NO2Tyr, and consequently be useful for treating brain damage after stroke (Yu et al., 2016). In apolipoprotein E knockout mice that were given a high-fat diet, ferulic acid (INV-315) showed a reduction of atherogenic plaques and improvement of endothelial function, suggesting that the inhibition of MPO could be useful for treating atherosclerosis (Liu et al., 2012). None of these compounds have, however, been translated to the clinics so far.
b. Irreversible inhibitors
Although this is a niche mechanism, two of the major global players in the pharmaceutical industry, i.e., AstraZeneca and Pfizer, have embarked on the pharmacological development of these compounds. However, the efforts have never gained breadth. At least two candidates have been discontinued, and progress on the single remaining one stays slow. Only one compound, 4-aminobenzoic acid hydrazide, showed in vivo decreased development of aortic atherosclerosis (Békési et al., 2005) myeloid inflammation, tissue damage in experimental autoimmune encephalomyelitis, demyelinating diseases such as multiple sclerosis (MS) (Forghani et al., 2012), blood-brain barrier dysfunction (Üllen et al., 2013), and improved neurogenesis after ischemic stroke (Kim et al., 2016). AstraZeneca had advanced AZD3241 into phase II trials for Parkinson disease (NCT01527695, NCT01603069) and multiple system atrophy (NCT02388295; negative outcome). After discontinuing development in April 2017, the company released it for third-party development as a part of its Open Innovation initiative. AZD3241 has been replaced by AZD4831, which is in active development for heart failure with preserved ejection fraction (HFpEF). A phase I study (NCT03136991) in healthy males has been completed; the second one, in patients with HFpEF, is scheduled for completion at the end of 2020 (Table 2). Pfizer’s thiouracil derivative PF-06282999, an irreversible MPO inhibitor, entered clinical development for the treatment of acute coronary syndrome in June 2012. Pfizer announced its discontinuation in August 2017. Deduced from the most recent patenting activity, Bristol Myers Squibb Co. is now investing heavily in the discovery of MPO inhibitors (Fig. 7). The company has international patent applications for macrocyclic molecules (WO/2018/005336) and a broad range of triazolopyridines and triazolopyrimidines [WO/2017/160632, WO/2017/161145, WO/2017/040451, WO/2017/040450, WO/2017/04044, WO/2016/040419, WO/2016/040417 (Duclos et al., 2017)].
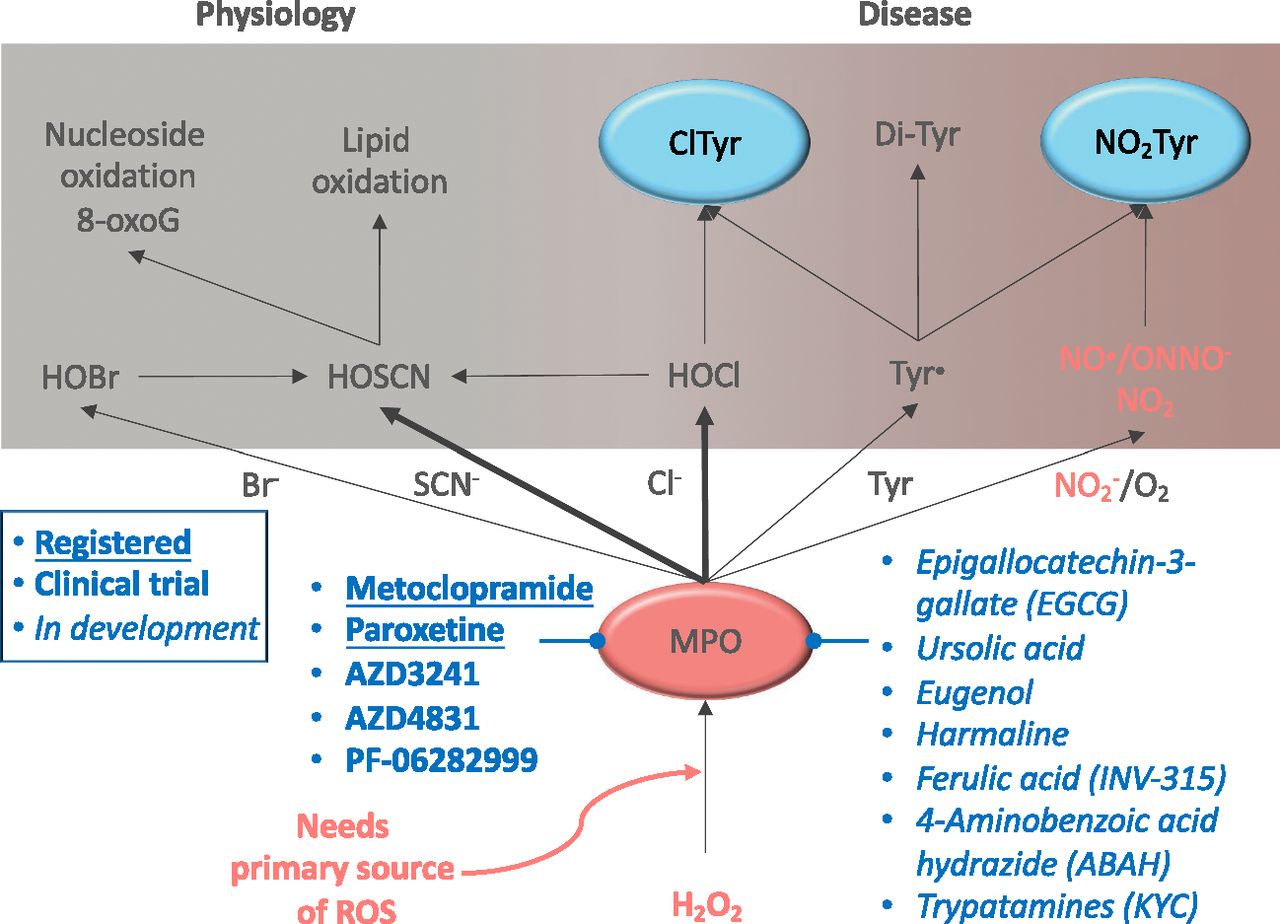
Role of myeloperoxidase in physiology and disease. MPO generates through H2O2 several reactive chlorinating (Cl−), bromating (Br−), and radical (SCN− NO2) species, which are later transformed into more reactive hypohalous acids [hypobromous acid (HOBr), hypothiocyanous acid (HOSCN), hypochlorous acid (HOCl)] and reactive nitric species (NO•/ONNO−). Thus, MPO actively contributes to nucleoside and lipid oxidation, together with protein (Tyr) nitration (NO2Tyr) and chlorination (ClTyr). Dysregulation of MPO results in a broad oxidative environment that contributes to several pathologic conditions. MPO inhibitors are shown based on their current status, i.e., already marketed, under clinical testing, or under preclinical development. 8-oxoG, 8-oxo-guanosine.
V. Targeting ROS Beyond Oxidases
A. Direct Antioxidants
Antioxidants have been used as nutraceuticals for the prevention of aging and disease or even as a therapeutic, often with little or no clinical control. The origin of the antioxidant therapy concept is tightly connected to the free radical theory of aging established by Harman (1956, 1972), which was later expanded to also include age-related diseases. According to this concept, free radicals, i.e., any molecular species capable of independent existence that contains an unpaired electron in an atomic orbital, are generated from several sources, including mitochondria, and cause progressive oxidant damage to macromolecules, such as DNA, that limit cell functionality. Consequently, antioxidants that reduce free radical reactions should protect against aging and disease. However, the existence of the large enzyme family of NADPH oxidases and DUOXs with the sole physiologic function to generate ROS clearly indicated that ROS have at least two qualities, and thus, antioxidants should be a double-edged sword.
In clinical trials, antioxidants have failed consistently to show benefits in humans (Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group, 1994; Marchioli, 1999; de Gaetano, 2001; Collins et al., 2002). Vitamin E has been studied in the context of cardiovascular disease in the CHAOS (Stephens et al., 1996) and HOPE (Hegele, 2000; Lonn et al., 2005) trials, in which it failed to improve cardiovascular mortality. Moreover, several meta-analyses focusing on the effect of vitamin E in CVD showed similar results, i.e., no clear evidence of a positive effect on either mortality or morbidity (Vivekananthan et al., 2003; Eidelman et al., 2004; Kris-Etherton et al., 2004; Shekelle et al., 2004). Similarly, no effect was detected in cancer (Bjelakovic et al., 2004) or other causes of mortality and quality of life (Bjelakovic et al., 2007).
N-acetylcysteine, which increases glutathione levels, and vitamin E promote the acceleration of tumor progression by reducing p53 expression in rodents. Similarly, supplements of vitamin E showed the same effect on p53 levels. Indeed, high doses of antioxidants in patients with diabetes were detrimental because of an indirect increase in blood glucose levels (Sarangarajan et al., 2017). Moreover, long-term supplementation also provides a risk for metabolic syndrome incidence. Vitamin E supplementation significantly increases prostate cancer risk and even provides evidence of increased hemorrhagic stroke risk. Moreover, chronic use of multivitamins without clinical control, especially lipid-soluble antioxidants at dosages above the upper safety limit, has been associated with increased health risks (Bjelakovic and Gluud, 2007). Moreover, during the past years, teratogenic effects were reported and associated with high intakes of vitamin A together with accelerated bone loss and risk of fracture due to increased osteoporosis.
Regarding vitamin C, epidemiologic studies initially suggested that low plasma levels correlate with increased inflammation and endothelial dysfunction (Wannamethee et al., 2006). In the EPIC study, a vitamin C–rich diet improved endothelium-dependent vasodilation and hemostasis (Sargeant et al., 2001). A later meta-analysis, however, failed to show benefit in reducing major cardiovascular events (Myung et al., 2013). Specific meta-analyses claimed a beneficial effect of vitamin C in breast cancer (Harris et al., 2014). Ultra–high-dose therapy with vitamin C may be beneficial in cancer through a pro- rather than an antioxidant effect. In colon cancer, the effects of vitamin C have been attributed to its oxidized form, dihydroascorbate (Chen et al., 2008; Yun et al., 2015). In either non–small-cell lung cancer or glioblastoma, the safety, not the efficacy, of high-dose vitamin C injections was tested (Schoenfeld et al., 2017). Vitamin C may help to alleviate some of the side effects of antitumor treatment (Carr et al., 2014), but conversely, other clinical trials had to be stopped because of severe side effects by vitamin C (Subbarayan et al., 2007). Thus, vitamin C fails to reduce all-cause mortality, and antioxidants do not exhibit beneficial effects in either cardiovascular or other diseases (Bjelakovic et al., 2012), whereas the antitumor, pro-oxidant effect of high-dose vitamin C dihydroascorbate is to some degree open, but far away from being recommended. Vitamin C has also been suggested to increase the risk of calcium oxalate kidney stones.
Coenzyme Q has been used in several trials with little or no benefit. Recently, the 2CARE study was conducted in a large cohort of patients with early Huntington disease. This study was stopped for futility, as it would be very unlikely (less than a 5% statistical probability) to see a benefit of coenzyme Q10 treatment over placebo (McGarry et al., 2017). Dietary polyphenols have also been tested, leading to another translational failure when positive data in animal models could not be replicated in humans (McDougall et al., 2005; Yang et al., 2011).
Some synthetic antioxidants also provided disappointing results. The spin-trapping synthetic antioxidant NXY-059 was used in two trials of patients with ischemic stroke (SAINTI and SAINTII). NXY-059 showed no effect on mortality and neurologic function (Lees et al., 2006; Shuaib et al., 2007). Several hypotheses have been proposed to explain the failure of most antioxidants in clinic practice, although the overall reason is still unknown:
Broadly interfering with ROS biology, which will inevitably also scavenge essential and physiologic ROS, is of general concern.
The concept that low ROS is beneficial or neutral and that high ROS is always detrimental is not sustainable.
There is no whole-body redox status, and even at a cellular level, there are independent compartments of ROS biology.
The molecular targets of the antioxidants are usually poorly defined, leading to a broad mechanism of action mainly focused on ROS scavenging with neither specific subcellular localization nor predefined enzymatic target (Cochemé and Murphy, 2010).
Absorption, distribution, metabolism, excretion, and bioavailability, i.e., pharmacokinetics and pharmacodynamics of specific antioxidants, may not have been analyzed sufficiently, and dosing may have been wrong or insufficient (Manach et al., 2005).
Inadequate trial duration may have resulted in the underestimation of any therapeutic effect. Chronic clinical trials might not last long enough to obtain positive outcomes, and post-trial follow-up may have had time and funding limitations (Steinhubl, 2008).
Nevertheless, most antioxidant therapies not only seem to be ineffective based on clinical evidence but are also associated with some risk of detrimental outcomes depending on the dose or pathology being treated (Salehi et al., 2018).
One exception to the above may be edaravone (3-methyl-l-phenyl-pyrazolin-5-one, MCI-186), which has been approved for stroke (Miyaji et al., 2015) and/or amyotrophic lateral sclerosis in some countries, including Japan (Radicut) and the United States (Radicava), but the market authorization application was withdrawn in Europe. Edaravone is considered to act as a free radical scavenger with neuroprotective effects both in in vitro and in vivo models of ROS-related neurodegenerative diseases, especially brain ischemia (Lei et al., 2014; Sun et al., 2015) and ALS (Xu et al., 2017). ROS have been considered as a critical player in ALS pathophysiology being involved in neuromotor degeneration and glial and endothelial dysfunctions. Indeed, edaravone, prevents motor neuron death (Nagase et al., 2016). Preclinical studies hypothesized that the neuroprotective effect of edaravone is mediated by plasma membrane stabilization (Wu et al., 2014b) via 1) scavenging hydroxyl radicals, 2) inhibiting lipid peroxidation, and 3) playing a potent antioxidant effect (Feng et al., 2011). Despite the recent approval for ALS treatment, there are still limitations, since the intravenous infusion is inconvenient for the patients and costly (Hardiman and van den Berg, 2017). Based on promising in vivo studies (Wu et al., 2014b), multiple clinical trials were conducted demonstrating an effect of edaravone in patients diagnosed with stroke (Otomo et al., 2003) and patients with acute cerebral large-vessel occlusion as a subanalysis of the RESCUE-Japan Registry (Miyaji et al., 2015). Several systematic reviews, however, found no evidence for edaravone’s efficacy (Lapchak, 2010), along with serious shortcomings in clinical trial design, randomization, patient numbers, inclusion and exclusion criteria, and outcome parameters (Feng et al., 2011). Edaravone has also been investigated in a pilot for the treatment of acute myocardial infarction (NCT00265239), ischemia-reperfusion injury in patients with kidney transplantation (NCT02644915), Parkinson disease, Alzheimer disease, atherosclerosis, retinal diseases, and diabetes mellitus. No clinical outcome data have been released (Ahmadinejad et al., 2017).
B. Indirect Antioxidants: NRF2 Activators
Considering the failure of most direct antioxidants in clinical studies, a new approach has been suggested focused on the activation of the endogenous redox homeostatic system. The advantage of this strategy is to achieve redox regulation in the tissues, cells, or organelles where it is required. Transcription factor nuclear factor (erythroid-derived 2)-like 2, the master regulator of the antioxidant response, is a very promising target, since it can be activated pharmacologically. NRF2 is a basic leucine zipper transcription factor that forms heterodimers and controls the basal and stress-inducible expression of over 250 genes through its binding to the enhancer antioxidant response element (Cuadrado et al., 2018, 2019). The most successful NRF2 activators are electrophile drugs that alter the structure of KEAP1 through the interaction with several redox-sensitive cysteines, including C151, C273, and C288. At least 30 recent patents for NRF2 modulators are indexed in the World Intellectual Property Organization. Although most of these compounds proved to be useful to some degree in preclinical proof-of-concept studies, only a few have entered clinical trials approved, and even fewer have reached phases II/III. The three most successful examples are sulforaphane, synthetic triterpenoids, and dimethyl fumarate. Sulforaphane is an isothiocyanate obtained from cruciferous plants, and it is already used so far in at least 32 clinical studies of chronic diseases with an oxidant and inflammatory component, including cancer, asthma, chronic kidney disease, type 2 diabetes, and cystic fibrosis (Houghton et al., 2013). In these studies, sulforaphane improved blood glucose, lipid profile, and molecular markers of ROS. Evgen Pharma Inc. has developed a new cyclodextrin complex formulation (Sulforadex) to increase its solubility and stability. Nowadays, this compound is under phase II clinical trial for the treatment of metastatic breast neoplasm and subarachnoid hemorrhage. Synthetic triterpenoids, derived from 2-cyano-3,12-dioxooleana-1,9(11)dien-28-oate, are the most potent activators of NRF2 and have been developed as antioxidant modulators of inflammation (Dinkova-Kostova et al., 2010). Despite excellent results in preclinical models, serious concerns were raised in the first clinical trial for diabetic nephropathy. Despite excellent results in preclinical models, serious concerns were raised in the first clinical trial for diabetic nephropathy since the therapeutic effects were not related to NRF2, but most likely to an off-target alteration of endothelin signaling (Chen et al., 2015). Reata Pharmaceuticals has developed a second generation of synthetic oleanane triterpenoid (RTA408). This compound is currently in phase II/III trials of patients with Friedreich ataxia, mitochondrial myopathies, immunooncology, and prevention of corneal endothelial cell loss after cataract surgery. The best therapeutic results for an NRF2 activator have been obtained so far with dimethyl fumarate (DMF), which is the only NRF2 activator that has been approved as a drug by the Food and Drug Administration and European Medicines Agency. Initially used for the treatment of psoriasis (Fumaderm), DMF is currently used as the first-line oral therapy for MS (Tedfidera) (Bomprezzi, 2015). NRF2 activation by DMF correlates with decreased levels of proinflammatory markers and macrophage infiltration in the spinal cord (Schimrigk et al., 2006), which is in line with phase II/III trials in which a significant reduction of MS lesions and annualized relapse was demonstrated (Fox et al., 2012) (Fig. 8; Table 3). However, uncontrolled Nrf2 activation can lead to deleterious effects due to the so-called reductive stress event, which is detrimental in muscle disorders and different cardiomyopathies (Pérez-Torres et al., 2017; Bellezza et al., 2018). A novel strategy for NRF2 activation is based on the development of PPI inhibitors. These small molecules interfere with the docking of NRF2 to KEAP1, hence preventing NRF2 degradation. Several NRF2/KEAP1 PPI inhibitors have been developed and are under patent protection, but further research is required to demonstrate that they are highly selective and that off-target effects are negligible (Cuadrado et al., 2019).

Clinical pharmacology of NRF2. The KEAP1 E3 ligase adapter is a homodimer that presents NRF2 to the cullin3 (CUL3)/RING-box protein (RBX) complex for ubiquitination and further proteasome degradation. This process restrains NRF2 protein levels. ROS (H2O2) and electrophiles alter KEAP1 and lead to a stalled KEAP1/NRF2/CUL3/RBX complex. Then, newly synthetized NRF2 accumulates in the nucleus, makes heterodimers with small transcription factor MAF (MAF) proteins, and activates target genes that contain the antioxidant response element (ARE). Clinically relevant electrophiles, such as sulforaphane, dimethyl fumarate, bardoxolone, or omaveloxone, disrupt the capacity of KEAP1 to target NRF2 for degradation and therefore increase NRF2 levels.
Targeting ROS beyond oxidases: translation to the clinics
C. Functional Repair of ROS-Induced Damage
All of the above approaches are aimed at reducing ROS levels, either by reducing their synthesis or toxification or by scavenging them. Functionally repairing ROS-induced damage is, however, in our view, a promising complementary strategy that is consistent with the identified ROS disease modules and, importantly, has already entered clinical trialing and practice.
1. sGC Activators
Physiologically, soluble guanylate cyclase mediates the generation of the second messenger cGMP upon stimulation by NO. NO-cGMP signaling regulates several physiologic mechanisms, including vasodilation, prevention of fibrosis, antithrombosis, anti-inflammation, and memory formation (Ding et al., 2004; Dasgupta et al., 2015). The effects of cGMP are mediated by a family of cGMP-dependent protein kinases (PKG) (Francis et al., 2010). In addition to its activation by cGMP, PKG has also been reported to be activated by oxidative modification, presumably by hydrogen peroxide, which can lead to vasorelaxation (Burgoyne et al., 2007) and angiogenesis (Burgoyne et al., 2015). Thus, ROS formation, on the one hand, interferes with endothelium-dependent relaxation (possibly via superoxide scavenging of NO) and, on the other hand, should induce vasorelaxation (by hydrogen peroxide acting as an endothelial hyperpolarizing factor). What is unclear is whether these apparently contradictory scenarios can coexist and what their pathophysiological or therapeutic relevance is.
Nonphysiologic ROS production can affect the NO-sGC-cGMP by direct chemical scavenging, resulting in redox state changes of the NO responder soluble guanylate cyclase or even, finally, in loss of its heme group. The nonactive resulting enzyme, called apo-sGC, is unable to respond to NO signaling, dramatically reducing the bioavailability of cGMP (Casas et al., 2015). The so-called sGC activators could functionally reverse this mechanism. These compounds bind to both the oxidase and heme-free forms of the enzyme, reactivating it as the common NO-stimulated sGC and preventing its proteasomal degradation (Meurer et al., 2009). sGC activations can replace the weakly bound oxidized heme in apo-sGC or also occupy the empty heme pocket after the loss of the heme group (Stasch et al., 2006), stabilizing the heme-free enzyme and preventing degradation (Dasgupta et al., 2015). The NO-sGC-cGMP signaling pathway is widely established as a potent cardiovascular target because of its role in hypertension and cardiovascular diseases. Therefore, sGC activators have been preclinically tested not only for cardiac hypertrophy (Costell et al., 2012), heart failure (Breitenstein et al., 2017), and ischemia-reperfusion injury (Nossaman et al., 2012) but also in other pathologies such as fibrosis (Sandner and Stasch, 2017) and diabetic nephropathy (Boustany-Kari et al., 2016) and stroke (Langhauser et al., 2018). After preclinical validation, some sGC activators have been translated to the clinical stage. Phase II clinical trials were initiated recently using cinaciguat (BAY 58-2667), which is a potent vasodilator (Dasgupta et al., 2015), as a treatment of acute heart failure (NCT01067859, NCT00559650) and heart decompensation (NCT01064037). Unfortunately, all trials for decompensated heart failure (COMPOSE 1, COMPOSE 2, and COMPOSE EARLY) were terminated because of systemic hypotension and difficulty enrolling patients. Another phase II clinical trial using a new sGC activator, ataciguat (HMR 1766), as a treatment of aortic valve calcification (NCT02481258) was completed in July 2018, with no results available so far (Fig. 4). Other indications, like neuropathic pain, have also been investigated (NCT00799656) in a phase II trial (Table 3).
2. sGC Stimulators
sGC stimulators can directly activate sGC, resulting in an increase of cGMP formation in pathologic conditions and preventing NO-dependent disorders (Nossaman et al., 2012). sGC stimulators bind to an allosteric binding site of Fe(II)heme-containing sGC, resulting in an NO-independent sGC stimulation. Therefore, contrary to sGC activators, stimulators do not work under the heme-free/oxidized form of sGC (Dao et al., 2015). Different sGC stimulators, i.e., BAY 41-8543 and BAY 41-2272, have been preclinically tested in animal models of pulmonary hypertension and show a decrease in systemic and pulmonary arterial pressure, which has been translated to the clinics (Nossaman et al., 2012). The clinical success of Bayer’s riociguat (Adempas) in pulmonary arterial hypertension prompted development efforts for repurposing sGC stimulators to other therapeutic indications, although their biologic effects are less directly related to ROS generation compared with the oxidases (Sandner et al., 2019). Leading these efforts is the collaboration between Merck Sharp & Dohme and Bayer, which are conducting Vericiguat Global Study in Subjects With Heart Failure With Reduced Ejection Fraction (VICTORIA) (NCT02861534), a large phase III clinical trial with vericiguat (BAY1021189/MK-1242-001) (Follmann et al., 2017) in over 4800 patients with HFpEF. It is scheduled for completion in 2019. When VICTORIA was announced in 2016, it was considered a surprise because vericiguat had produced a mixed performance in the preceding phase II SOCRATES-REDUCED trial. In fact, the candidate had failed to meet its primary endpoint of achieving a significant reduction in the biomarker N-terminal pro–B-type natriuretic peptide, although a subgroup given the highest dose, 10 mg, did show a benefit compared with placebo. SOCRATES-PRESERVED (NCT01951638), an earlier phase II trial in heart failure with preserved ejection fraction, is being followed up with VITALITY-HFpEF (NCT03547583), another phase II trial that is supposed to be completed in November 2019. An sGC stimulator would represent a promising mechanistic deviation from the current small-molecule therapeutic strategies for heart failure and could be especially important in HFpEF. However, development might be risky. Bayer has already discontinued cinaciguat (BAY 58-2667) for this indication. Indeed, Bayer has investigated combinations of its approved phosphodiesterase inhibitor vardenafil with nelociguat (desmethyl riociguat, BAY60-4552; WO/2003/095451) in phase II clinical trials for male erectile dysfunction. Highly interesting is Ironwood Pharmaceuticals’ olinciguat (IW-1701), an sGC stimulator in development for sickle cell disease. STRONG SCD (NCT03285178) is a phase II trial that investigates disease-specific symptoms and pharmacodynamic biomarkers over a 12-week treatment course; it is planned for completion in July 2019 (Fig. 4). Olinciguat is also in phase II for the esophageal sphincter disorder achalasia (Table 3).
VI. Biomarkers of ROS
Current drug therapy is imprecise (Schork, 2015), and since the 1950s, drug development has an ever-increasing risk to fail (Scannell et al., 2012). Even the most widely sold drugs in the United States only provide relative benefit in one in four patients treated. The number needed to treat (number of patients who would need to be treated with one specific drug to prevent one adverse outcome) remains extremely high in more than 90% of approved drugs (Schork, 2015). To move from here to precision medicine, patients need to be stratified based on the fact that they will respond with a high likelihood to a drug. Biomarker-based stratification of patients in different populations may help to target pharmacologically the different mechanisms affected in each patient. The National Institutes of Health Biomarkers Definitions Working Group defined a biomarker as “a characteristic that is objectively measured and evaluated as an indicator of normal biological process, pathogenic processes, or pharmacologic responses to a therapeutic intervention.” Moreover, clinically relevant biomarkers must have diagnostic or prognostic value or correlate with disease activity. Finally, the stability, feasibility, and cost-effectiveness are additional important criteria for clinical acceptance of a biomarker. In the ROS field, a wide range of biomarkers have already been tested, including in humans. However, none has entered clinical practice or any guideline.
A. Xanthine Oxidase and Myeloperoxidase
ROS-forming enzymes such as XO and MPO can be found circulating in blood, independent of the release mechanism. After administration of IFN or IFN inducers, XO activity increases in tissues as well as in plasma (Ghezzi et al., 1984). MPO is stored in granules of neutrophils and monocytes and is released during degranulation (Zhang et al., 2001). Clinical studies have previously used plasma or serum MPO levels as a potential prognostic biomarker of inflammation in cardiovascular disease (Vita et al., 2004), myocardial infarction (Cavusoglu et al., 2007; Khan et al., 2007), chronic obstructive pulmonary disease (Morrow et al., 2015), and cognitive impairment (Heringa et al., 2014). Another alternative is to measure MPO activity. 3-Chlorotyrosine, as well as chlorinated lipids, is a specific marker of MPO action highly elevated in low-density lipoproteins (Hazen and Heinecke, 1997). Several clinical studies have found pronounced 3-chlorotyrosine levels in cardiovascular (Cheng et al., 2008; Delporte et al., 2014) and rheumatoid arthritis (Wu and Pizzo, 2001; Stamp et al., 2012) in plasma or synovial fluid using high performance liquid chromatography or liquid chromatography–mass spectrometry. To date, there is no specific and reliable anti-ClTyr antibody available.
B. ROS-Dependent Protein Modification
Protein 3-nitrotyrosine (NO2Tyr), either within polypeptide sequences or as a free amino acid, is a marker of nitrative stress. At least two mechanisms for its formation exist: 1) chemically, in a diffusion-limited reaction between nitric oxide and superoxide and intermediate peroxynitrite; and 2) enzymatically, by heme peroxidases such as MPO or eosinophil peroxidase from nitrite and H2O2 (van der Vliet et al., 1997; Wu et al., 1999). In inflammatory disease states, NO2Tyr has been identified as a stable marker of nitrative stress (Herce-Pagliai et al., 1998). The analytical gold standard for NO2Tyr is mass spectrometry (Tsikas and Duncan, 2014), but this is not suitable for high-throughput analysis of clinical samples. Other quantitative approaches, such as ELISA (Pirro et al., 2007; Richardot et al., 2009), liquid chromatography (Shishehbor et al., 2003; Misko et al., 2013), or high performance liquid chromatography (Kaur and Halliwell, 1994), are currently explored to detect NO2Tyr clinically. However, extra effort is still needed in the field to further use NO2Tyr as a clinical biomarker, although the current data seem extremely promising. Clinical studies have already explored the potential of NO2Tyr as a marker of disease. It has been reported that NO2Tyr can estimate cerebrovascular stress after reperfusion in patients diagnosed with stroke (Bas et al., 2012). In addition, nitrated fibrinogen has also been detected in patient samples after ischemic stroke (Ill-Raga et al., 2015), and it has been suggested to be a cardiovascular disease risk factor (Heffron et al., 2009).
C. ROS-Dependent DNA/RNA Modification
DNA and RNA are also targets and biomarkers of ROS oxidation, with 8-oxo-2′-deoxyguanosine (8-oxodG) and 8-oxo-guanosine being the most common oxidative lesions, respectively. They are secreted into the urine and can be measured, usually by chromatography coupled with MS (Weimann et al., 2001). They provide information about systemic ROS and are lacking in high-ROS conditions in specific organs. Nevertheless, 8-oxodG and 8-oxo-guanosine are among the most relevant ROS biomarkers examined. In fact, recent systematic reviews and meta-analysis studies have validated and highlighted the role of 8-oxodG as a biomarker in, e.g., cardiovascular disease (Di Minno et al., 2016), chronic heart failure, and Parkinson disease (Wei et al., 2018).
The field of ROS biomarkers is constantly growing, and therefore, extensive wet laboratory validation studies are still required. We have highlighted some of the most ROS-relevant biomarkers, with a focus on stable oxidative modifications. However, there are plenty more worth mentioning—e.g., vasodilator-stimulated phosphoprotein phosphorylation levels in platelets are used in the clinic for drug monitoring. Although several methods have been discussed, antibody-based methods remain the most feasible in a point-of-care setting. Moreover, if disease definitions will become pathway modules, they need to be analyzed in cells or tissue. Since most proteins are present in most cells, most pathways may be assessable in circulating blood cells or platelets. One potential addition to this are recent developments in liquid biopsies and harvesting rare circulating cells such as tumor cells (Luecke et al., 2015) or endothelial cells (Farinacci et al., 2018).
VII. Summary
For decades, ROS have been proposed as a cause of aging and numerous diseases, primarily because of correlation with the detection of ROS or ROS biomarkers. Antioxidant therapy to scavenge ROS sounded like a logical intervention but failed for reasons that we now understand. The new therapeutic approaches reviewed here no longer focus on ROS but on classic pharmacological protein targets, i.e., 1) ROS sources, 2) ROS toxifiers, 3) activating the endogenous antioxidant defense systems, and 4) functionally repairing ROS-induced damage. They jointly represent the future of redox medicine, a field distinct of and synergistic with general ROS biology. Pathologic overexpression or overactivation of specific enzymatic sources of ROS can be selectively inhibited without affecting physiologic sources of ROS and physiologic signaling. With respect to isoforms of NOX, NOS, MAO, XO, and MPO, their inhibition has resulted in promising therapeutic approaches currently under development in late clinical stages, i.e., VAS203, in phase III for traumatic brain injury, and GKT-831, in phase III for diabetic nephropathy. XO and MAO inhibitors have been approved, although with different mechanisms in mind, but their clinical efficacy may still involve ROS aspects. In any case, the latter opens new clinical opportunities through repurposing strategies for mechanism-based ROS indications, e.g., XO inhibitors for congestive heart failure, MAO inhibitors for macula detachment, or NOX inhibitors for stroke. Activation of endogenous antioxidant defense systems such as the master redox regulator NRF2 may represent a viable alternative to the futile exogenous antioxidants, as NRF2 activators will physiologically modulate ROS and have indeed provided clinical success, e.g., in multiple sclerosis. These ROS-centric approaches are complemented by functionally repairing ROS-induced damage. The best example of this is soluble guanylate stimulators in pulmonary hypertension (riociguat) and heart failure (vericiguat) that sensitize sGC for lower NO levels to reach a physiologic cGMP signal. sGC activators are able to functionally repair damaged sGC in its apo-sGC state, e.g., for aortic valve calcification (ataciguat). The redefinition of diseases by causal molecular modules will lead to mechanism-based combination therapies, i.e., network pharmacology, and therapeutic synergy between many of the above drug principles if they act in the same module. Finally, mechanism-based validated biomarkers, once they become available, will enable 1) mechanism-based rather than phenotype-based patient stratification, 2) a higher success rate of subsequent interventional clinical trials, and 3) lower numbers needed to treat in clinical practice for ROS-related diseases as a role model for precision medicine.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Casas, Nogales, Mucke, Petraina, Cuadrado, Rojo, Ghezzi, Jaquet, Augsburger, Dufrasne, Soubhye, Deshwal, Di Sante, Kaludercic, Di Lisa, Schmidt.
Footnotes
This study was supported by the European Research Council Advanced Investigator Grant/RadMed [Grant 294683], Proof-of-Concept Grant/SAVEBRAIN [Grant 737586], and H2020 Project REPO-TRIAL [Grant 777111] (to H.H.H.W.S.) and short-term scientific missions by the COST Actions EU-ROS and Open-MultiMed, and Kootstra Talented Fellowship (Maastricht University) (to A.I.C.).
Abbreviations
- ALS
- amyotrophic lateral sclerosis
- ClTyr
- chloro-tyrosine
- CVD
- cardiovascular disease
- DMF
- dimethyl fumarate
- DUOX
- dual oxidase
- H4Bip
- tetrahydrobiopterin
- HFpEF
- heart failure with preserved ejection fraction
- H2O2
- hydrogen peroxide
- IFN
- interferon
- IL
- interleukin
- KEAP1
- Kelch-like ECH-associated protein 1
- MAO
- monoamine oxidase
- MPO
- myeloperoxidase
- MS
- multiple sclerosis
- NO
- nitric oxide
- NO2Tyr
- 3-nitrotyrosine
- NOS
- NO synthases
- NOSTRA
- No Synthase in Traumatic Brain Injury
- NOX
- NADPH oxidase
- NRF2
- nuclear factor (erythroid-derived 2)-like 2
- O2•
- superoxide
- 8-oxodG
- 8-oxo-2′-deoxyguanosine
- PKG
- protein kinase G
- PPI
- protein-protein interaction
- ROCG
- ROS-cGMP signaling network
- ROS
- reactive oxygen species
- sGC
- soluble guanylate cyclase
- UA
- uric acid
- XDH
- xanthine dehydrogenase
- XO
- Xanthine oxidase
- XOR
- xanthine oxidoreductase
- Copyright © 2020 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





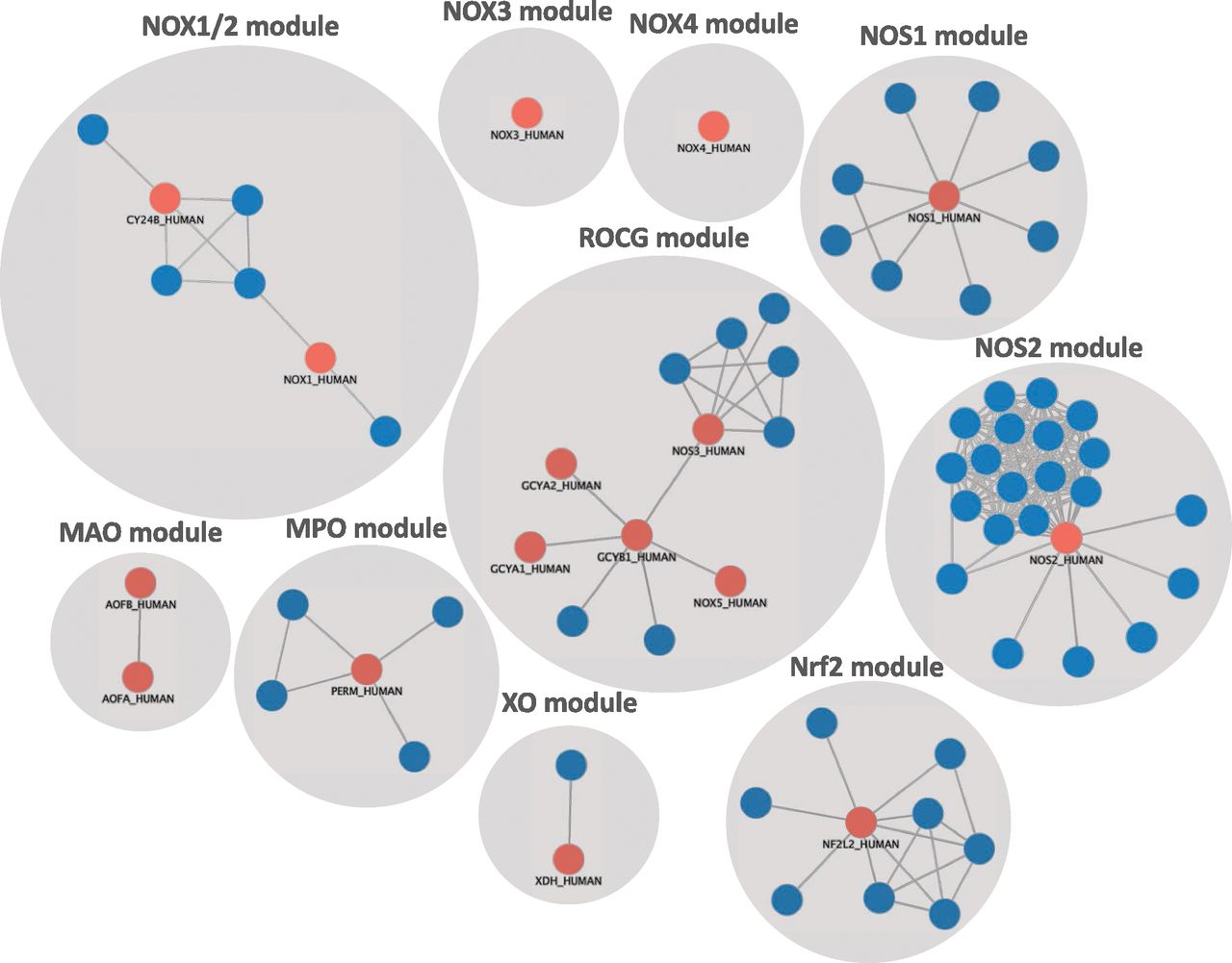{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
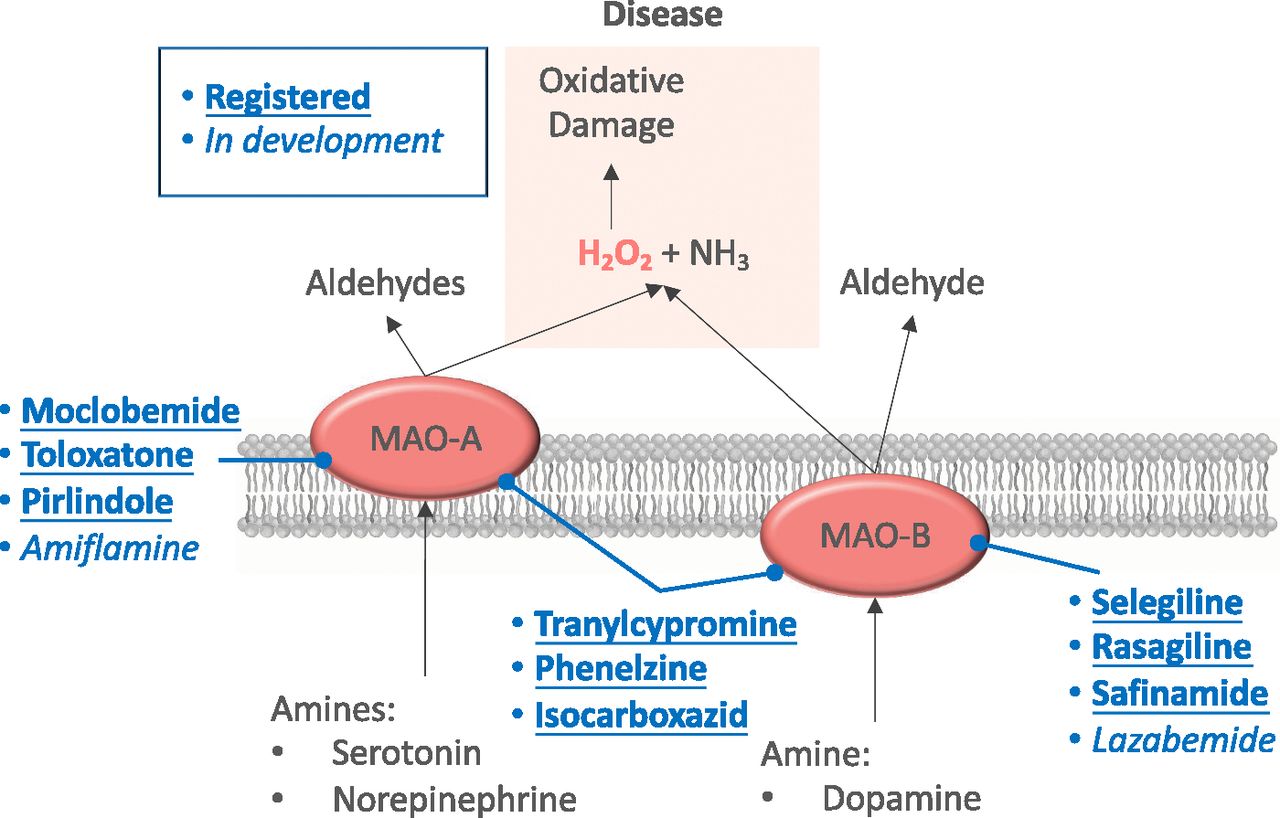{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. ROS Revisited: From Oxidative Stress and Redox Biology to Redox Medicine
- III. ROS Generators and Toxifiers in Disease
- IV. Pharmacological Modulation of ROS
- V. Targeting ROS Beyond Oxidases
- VI. Biomarkers of ROS
- VII. Summary
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters