


Visual Overview

Abstract
Post-translational modifications of cellular substrates with ubiquitin and ubiquitin-like proteins (UBLs), including ubiquitin, SUMOs, and neural precursor cell–expressed developmentally downregulated protein 8, play a central role in regulating many aspects of cell biology. The UBL conjugation cascade is initiated by a family of ATP-dependent enzymes termed E1 activating enzymes and executed by the downstream E2-conjugating enzymes and E3 ligases. Despite their druggability and their key position at the apex of the cascade, pharmacologic modulation of E1s with potent and selective drugs has remained elusive until 2009. Among the eight E1 enzymes identified so far, those initiating ubiquitylation (UBA1), SUMOylation (SAE), and neddylation (NAE) are the most characterized and are implicated in various aspects of cancer biology. To date, over 40 inhibitors have been reported to target UBA1, SAE, and NAE, including the NAE inhibitor pevonedistat, evaluated in more than 30 clinical trials. In this Review, we discuss E1 enzymes, the rationale for their therapeutic targeting in cancer, and their different inhibitors, with emphasis on the pharmacologic properties of adenosine sulfamates and their unique mechanism of action, termed substrate-assisted inhibition. Moreover, we highlight other less-characterized E1s—UBA6, UBA7, UBA4, UBA5, and autophagy-related protein 7—and the opportunities for targeting these enzymes in cancer.
Significance Statement The clinical successes of proteasome inhibitors in cancer therapy and the emerging resistance to these agents have prompted the exploration of other signaling nodes in the ubiquitin-proteasome system including E1 enzymes. Therefore, it is crucial to understand the biology of different E1 enzymes, their roles in cancer, and how to translate this knowledge into novel therapeutic strategies with potential implications in cancer treatment.
I. Ubiquitin-Like Protein Conjugation System
Ubiquitin is an 8.5-kDa, 76–amino acid polypeptide that serves as a post-translational modifier of cellular substrates (Bedford et al., 2011). Similar to other post-translational modifications, the process of reversible enzymatic attachment of ubiquitin to proteins is termed ubiquitylation (also known as ubiquitination). This post-translational modification is involved in modulating turnover, function, interaction, and/or localization of cellular proteins and therefore regulates a wide range of biologic processes (Rape, 2018). These include, among other functions, protein homeostasis, cell cycle regulation, DNA repair, transcriptional regulation, and endocytosis (Haglund et al., 2003; Ciechanover, 2005; Dhananjayan et al., 2005; Huang and D’Andrea, 2006; Gilberto and Peter, 2017). Given its indispensable biologic roles, ubiquitin is evolutionarily conserved among eukaryotes and, as the name suggests, is ubiquitously expressed in most tissues (Goldstein et al., 1975).
In eukaryotes, a group of proteins collectively known as ubiquitin-like proteins (UBLs) share sequence homology and have a similar three-dimensional structure to ubiquitin (Hochstrasser, 2009; van der Veen and Ploegh, 2012). There are more than a dozen UBLs, which are classified into eight families, including neural precursor cell-expressed developmentally downregulated protein 8 (NEDD8), small ubiquitin-like modifier (SUMO), human leukocyte antigen F-associated transcript 10 (FAT10/ubiquitin D), interferon-stimulated gene (ISG) 15, autophagy-related protein (ATG) 8, ATG12, ubiquitin-fold modifier 1 (UFM1), and ubiquitin-related modifier 1 (URM1) protein families. These UBLs, known as type I UBLs, are similarly involved in post-translational modifications that modulate a wide variety of cellular processes (Cappadocia and Lima, 2018). In contrast, type II UBLs are not conjugated to cellular substrates but rather exist as a part of proteins with multiple domains, and these include ubiquitin-like 5 (UBL5)/HTLV-I U5RE-binding protein 1 (HUB1) and fau and its ubiquitin-like domain (FUBI) (Rossman et al., 2003; Oka et al., 2014). Here, our focus will be on ubiquitin and type I UBLs, which we will simply refer to as UBLs.
Post-translational modifications with UBLs are brought about by a sequential enzymatic cascade that involves three classes of enzymes: E1 activating enzymes, E2 conjugating enzymes, and E3 ligases (Nalepa et al., 2006) (Fig. 1). The function of E1 enzymes is to activate UBLs in an ATP-dependent multistep enzymatic reaction whereby the UBL becomes attached to the catalytic cysteine residue of the E1 enzyme via a high-energy thioester bond [symbolized by a tilde (∼)], forming a UBL-loaded E1 enzyme (E1∼UBL). The E1-bound UBL is then transferred from the catalytic cysteine of the E1 enzyme to the catalytic cysteine of a cognate E2 enzyme via a transthiolation reaction, forming a UBL-loaded E2 enzyme (E2∼UBL) (Cappadocia and Lima, 2018). In concert with their cognate E3 ligases, the UBL is conjugated to cellular proteins and/or phospholipids by the E2 enzyme, forming a UBL-modified substrate usually via an isopeptide linkage with a lysine residue (Schulman and Harper, 2009). E3 ligases may function as either adaptors that bring E2s in close proximity to cellular substrates for UBL conjugation (represented by U-box E3s and RING finger E3s) or UBL acceptors that form thioester intermediates with UBLs for subsequent transfer to their substrates (represented by homologous to the E6-AP carboxyl terminus [HECT] domain E3s) (Nakayama and Nakayama, 2006). In the human proteome, there exist eight known E1s, >40 E2s, and >600 E3s, with the E3 ligases controlling substrate specificity (Clague et al., 2015). As UBL conjugation is a reversible process, the UBL signal can be removed by another set of enzymes, known collectively as deubiquitylases (DUBs) and UBL-specific proteases (ULPs), which catalyze proteolytic cleavage of these post-translational modifications from their substrates. There exist more than 100 DUBs and ULPs in the human proteome (Clague et al., 2015; Ronau et al., 2016). A subset of ubiquitylated proteins, specifically those tagged with K48- and K11-linked polyubiquitin chains, are identified by the proteasome, deubiquitylated, and degraded into peptides to maintain protein homeostasis in the cell (Swatek and Komander, 2016). The cellular machinery orchestrating ubiquitin-dependent proteasomal degradation is collectively known as the ubiquitin-proteasome system (UPS) (Nalepa et al., 2006). Apart from degradative ubiquitylation, conjugation with other forms of ubiquitin (e.g., K63-linked polyubiquitin chains and monoubiquitin) and UBLs serves nondegradative functions in many aspects of cellular signaling (Bedford et al., 2011; Swatek and Komander, 2016).
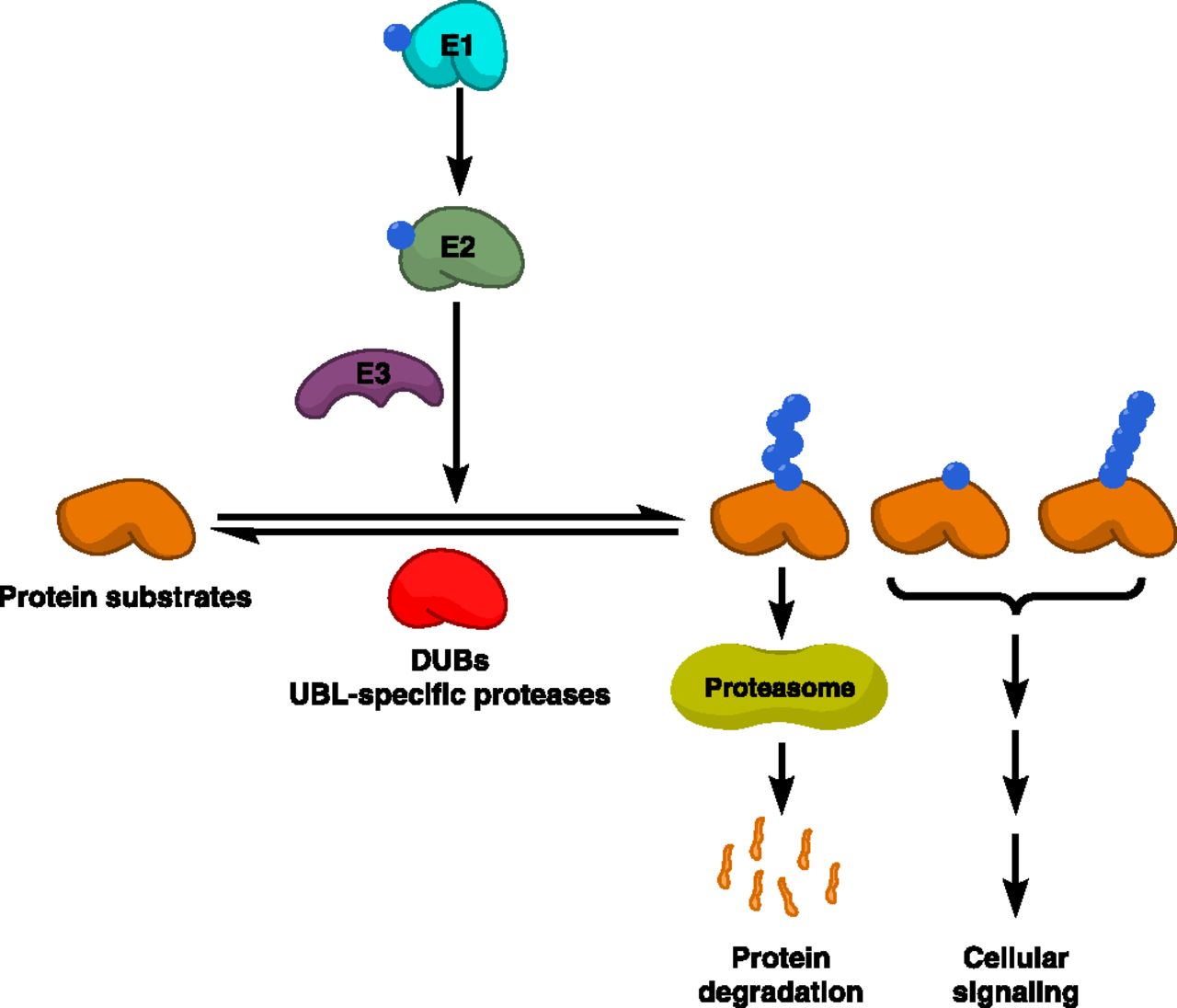
Ubiquitin and UBL conjugation system. The UBL conjugation system comprises three enzyme classes that act sequentially to catalyze UBL conjugation: UBL-activating enzymes (E1), UBL-conjugating enzymes (E2), and UBL E3 ligases. Protein substrates are conjugated with different forms of UBLs. Proteins conjugated with Lys48 (K48)- or K11-linked polyubiquitin chains are recognized by the proteasome, the major proteolytic machinery in the cell that degrades such proteins into smaller peptides. Other forms of UBL conjugation are involved in various cellular signaling pathways. UBL conjugation is reversed by other classes of enzymes, including DUBs, which deconjugate ubiquitin signals, and ULPs, which deconjugate other UBL signals.
Historically, drug discovery within the ubiquitin and UBL systems started with two parallel programs targeting the proteasome and E3 ligases. Despite the nonselective nature of proteasome inhibition, this program has led to the discovery of clinically useful drugs (the prototype of which is bortezomib), whereas the E3 ligase program is making slower progress (Mattern et al., 2014). Moreover, researchers have explored the therapeutic targeting of other signaling nodes in these systems including E1 enzymes that lie at the apex of the UBL conjugation cascade and thereby control degradative as well as nondegradative signaling processes in the cell (Schulman and Harper, 2009).
In this Review, we highlight different E1 enzymes, their biologic roles, and pathobiologic alterations, particularly in the context of cancer. In addition, we discuss in more detail the biochemical mechanisms of UBL activation and therapeutic strategies known so far to target different E1 enzymes with a focus on mechanism-based E1 inhibitors that have been advanced to clinical trials.
II. E1 Enzymes
A. Members of the E1 Enzyme Class
In the human proteome, eight E1 enzymes are known to activate UBLs (Schulman and Harper, 2009; Clague et al., 2015). These include ubiquitin-like modifier–activating enzyme (UBA) 1, NEDD8-activating enzyme (NAE), SUMO-activating enzyme (SAE), UBA6, UBA7, UBA4 (MOCS3), UBA5, and autophagy-related protein (ATG) 7. Based on their structural and biochemical properties, E1s have been subdivided into canonical (UBA1, NAE, SAE, UBA6, UBA7) and noncanonical (UBA4, UBA5, and ATG7) E1s (Schulman and Harper, 2009). Structurally, E1 enzymes adopt a monomeric (UBA1, UBA6, and UBA7), heterodimeric (NAE and SAE), or homodimeric (UBA4, UBA5, and ATG7) architecture (Fig. 2).

E1 enzymes and their structural domains. There are eight E1 enzymes known so far, including canonical (UBA1, UBA6, UBA7, SAE, and NAE) and noncanonical (UBA4, UBA5, and ATG7) E1 enzymes. Of these, UBA1, UBA6, and UBA7 are monomeric; SAE and NAE are heterodimeric; and UBA4, UBA5, and ATG7 are homodimeric. The AAD, catalytic cysteine half-domains (FCCD and SCCD), and UFD are conserved among canonical E1s. Noncanonical E1s do not possess discernible CCD, and the catalytic cysteine is located on the AD. They also comprise noncanonical E1-specific domains and sequences such as the RHD of UBA4. CC, catalytic cysteine; FCCD, first catalytic cysteine half-domain; SCCD, second catalytic cysteine half-domain; UFCBD, UFC1-binding sequence.
To carry out their multistep catalytic function, eukaryotic canonical E1 enzymes possess multiple domains, including the adenylation domain (AD), the catalytic cysteine domain (CCD), and the ubiquitin-fold domain (UFD), with several variations that exist in the noncanonical E1 enzymes (Huang et al., 2004b).
In canonical E1s, the AD is pseudosymmetric, with one active adenylation domain (AAD) involved in identifying and adenylating the C terminus of cognate UBLs and another inactive adenylation domain (IAD) that may provide structural stability (Schulman and Harper, 2009; Lv et al., 2017b). Similarly, the CCD is divided into the first catalytic cysteine half-domain and the second catalytic cysteine half-domain and is involved in thioester bond formation with UBLs (Lv et al., 2017b). The UFD is involved in the interaction with cognate E2s for UBL transfer via transthiolation. Of these, the AD is the most conserved domain and is homologous to adenylation domains in the ancestral prokaryotic enzymes molybdopterin biosynthetic enzyme B (MoeB) and thiamine biosynthesis protein F (ThiF), involved in molybdopterin and thiamine biosynthesis, respectively, suggesting that adenylation of UBLs is the most conserved functionality of all E1s (Huang et al., 2004b). Other domains might have evolved to accommodate the more complex catalytic activity carried out by eukaryotic E1s as opposed to their prokaryotic homologs (Cappadocia and Lima, 2018).
On the other hand, noncanonical E1s are homodimeric with symmetric ADs as well as other noncanonical E1-specific domains. In addition, their catalytic cysteines are situated close to the adenylation pocket without having dedicated CCDs as opposed to canonical E1s (Cappadocia and Lima, 2018). UBA4 possesses a distinctive rhodanese homology domain (RHD) at its C terminus, which is involved in the sulfur transfer to URM1, the cognate UBL of UBA4 (Termathe and Leidel, 2018). Each of these E1s activates specific UBL(s) and transfers them to one or more of its cognate E2s, establishing distinct UBL→E1→E2→E3→substrate cascades to influence a broad range of cellular functions. Of these, the UBA1-initiated cascade is the most branched, with tens of E2s, hundreds of E3, and thousands of substrates (Clague et al., 2015) (Table 1).
E1 enzymes, their cognate E2 enzymes, UBLs, and UBL proteases (Groettrup et al., 2008; Schulman and Harper, 2009)
B. Biochemical and Structural Mechanisms of E1-Catalyzed Ubiquitin-Like Protein Activation
UBL activation is a multistep ATP-dependent enzymatic process whereby the UBL moiety is attached to the catalytic cysteine of E1 enzyme via a thioester bond for subsequent transfer to cognate E2s (Schulman and Harper, 2009). With their multiple functional domains, particularly the AD, E1s are catalytically competent to perform such activation as opposed to the simpler E2 enzymes (Stewart et al., 2016). The catalytic steps of UBL activation have been well characterized with canonical E1s, particularly the archetypal UBA1 enzyme (Bohnsack and Haas, 2003). There exist, however, some variations in the UBL activation mechanisms by noncanonical E1s (Schulman and Harper, 2009; Cappadocia and Lima, 2018). In general, the catalytic cascade of UBL activation and transfer by canonical E1s includes four major steps: 1) first adenylation, 2) thioester formation, 3) second adenylation, and 4) UBL transfer to E2 by transthiolation.
UBL activation starts with the C-terminal adenylation of a free UBL molecule in the presence of ATP and Mg2+, followed by binding of the adenylated UBL to the AAD of E1, forming an E1-bound UBL∼adenylate (UBL∼AMP) intermediate and releasing PPi. This is followed by a nucleophilic interdomain reaction whereby the catalytic cysteine of the CCD attacks the adenylated UBL, forming a UBL-bound E1 via a covalent thioester bond (E1∼UBL), coupled with the release of free AMP. Subsequently, E1 catalyzes another round of adenylation of a second UBL molecule, releasing PPi and forming a double-loaded E1 with two UBL molecules at two sites: one bound to the CCD via a covalent thioester bond and another bound to the AAD via a noncovalent bond (Haas and Rose, 1982). From energetic and conformational perspectives, this form is likely more competent for subsequent transfer of UBL (Schulman and Harper, 2009; Cappadocia and Lima, 2018). The double-loaded E1 then interacts with the cognate E2 enzyme through the UFD to transfer the thiol-bound UBL molecule to the corresponding thiol in the CCD of E2 via a transthiolation reaction, leaving an E1-bound UBL∼AMP intermediate that is used in other rounds of UBL activation (Schulman and Harper, 2009) (Fig. 3).

Cascade of canonical E1-catalyzed UBL activation and substrate-assisted inhibition by adenosine sulfamates. (A) Canonical E1-catalyzed UBL activation is a multistep catalytic process that involves UBL adenylation in an ATP-dependent manner forming an E1-AMP∼UBL intermediate and releasing PPi. The UBL at the adenylation domain is then attacked by the sulfhydryl (-SH) group to form an E1-S∼UBL thioester intermediate associated with the release of AMP. The adenylation step is then repeated with another ATP molecule to form E1 doubly loaded with two UBL molecules at two distinct sites. The UBL at the catalytic cysteine is then attacked by the -SH group of the cognate E2 enzyme to transfer the UBL in a transthiolation reaction. ADSs inhibit E1 enzymes by attacking the E1-S∼UBL intermediate (highlighted in a green box) and forming an ADS-UBL covalent adduct, which binds to the nucleotide-binding site of E1, preventing its utilization in subsequent reactions. R and R′ correspond to the side chains in different ADSs. (B) The four catalytic steps of UBL activation and transfer to cognate E2s (first UBL adenylation, E1∼UBL thioester formation, second UBL adenylation, and E1-E2 transthiolation) are illustrated.
The catalytic activity of E1s is enabled by several conformational changes that facilitate different steps of UBL activation. Of particular importance is the thioester bond formation facilitated by the rotation of the CCD to come in close proximity to the UBL∼AMP and attack the UBL moiety (Olsen et al., 2010). The rotation is associated with other conformational changes that lead to remodeling of the AAD of E1 and switching from an open to a closed conformation (Streich and Lima, 2014). These conformational changes are transient, as the E1 restores the open conformation after forming the E1∼UBL thioester. These structural insights were partly derived by using E1 inhibitors that serve as UBL∼AMP mimetics, such as SUMO-AMSN and SUMO-AVSN (Olsen et al., 2010). Although all steps of UBL activation are bidirectional, the progression of these reactions is maintained in one direction by several biochemical and structural factors, including the abundance of ATP, active site remodeling to promote thioester bond formation and PPi release, and double loading with a second UBL molecule to regain the open conformation of the AAD and drive subsequent UBL transfer to cognate E2s (Cappadocia and Lima, 2018).
C. Druggability of E1 Enzymes
E1 enzymes catalyze the multistep UBL activation exploiting their multidomain structure, with at least two active sites that are readily amenable to therapeutic targeting (Fig. 4). The first active site is the nucleotide-binding pocket in the AAD to which the UBL∼AMP binds. Historically, small-molecule ATP mimetics targeting mutant tyrosine kinases have been among the first classes of molecularly targeted agents to be developed for cancer therapy (Capdeville et al., 2002; Cohen, 2002). Therefore, E1 enzymes with such ATP-binding pockets can serve as typical druggable targets for modulating different UBL conjugation pathways. Moreover, the unique catalytic mechanism involving the interaction of UBL∼AMP rather than ATP to the nucleotide-binding pocket sets a clear distinction between E1s and other ATP-dependent enzymes and offers an opportunity to develop selective inhibitors with little impact on the kinome or other ATP-dependent targets (Hubbard and Till, 2000). The challenge lies, however, in defining selectivity among the eight E1 enzymes that use conserved or closely related mechanisms of UBL activation. In this respect, the involvement of UBLs in the interaction with the nucleotide-binding pocket can be perceived as another source of selectivity even among related E1s owing to the structural variations of different UBLs. Therefore, structural information on these UBLs can be exploited to develop inhibitors that selectively target individual E1s.

Inhibitors of E1 enzymes and their sites of action. A diagram of the E1 enzyme showing different E1 inhibitors and the structural domains or active sites they are reported to target.
For example, semisynthetic UBL∼AMP analogs have been used as selective chemical probes to inhibit and interrogate the structural biology of E1s (Wilkinson et al., 1990; Olsen et al., 2010). With more structural and biochemical information revealed on different E1s, small-molecule E1 inhibitors with more favorable drug-like properties have been developed. Standing out among these are the adenosine sulfamate E1 inhibitors that form a UBL∼AMP–like intermediate in situ after permeation into the cells overcoming the drug delivery issue experienced with semisynthetic analogs (Ciavarri and Langston, 2017).
The second active site is the catalytic cysteine in the CCD to which the UBL binds via a thioester bond. This catalytic residue with its redox-sensitive thiol group offers another opportunity for developing thiol-reactive electrophilic inhibitors that covalently modify the active site (Visscher et al., 2016). Such cysteine-directed covalent agents are expected to exert irreversible and potent inhibition, which is desirable in several contexts, including cancer therapy. Although many of these covalent inhibitors may have off-target effects because of their promiscuous reactivity, a number of Food and Drug Administration–approved agents, including the kinase inhibitors rociletinib and osimertinib, target cysteine residues with a high level of selectivity (Visscher et al., 2016). For E1 enzymes, several drugs that target the CCD have been reported, most prominently the nitropyrazone-based UBA1 inhibitors PYR-41 and PYZD-4409 (Yang et al., 2007; Xu et al., 2010). Interestingly, COH000 is a recently discovered SAE inhibitor that targets a cysteine residue in the AAD without affecting the catalytic cysteine (Lv et al., 2018b; Li et al., 2019).
The third, and yet more challenging, site to target in E1 enzymes is the UFD through which they interact with their cognate E2s for subsequent UBL transfer. The difficulty of targeting E1-E2 interaction, like other protein-protein interactions, lies in the large surface area involved and the lack of pockets to which small-molecule inhibitors can bind (Jin et al., 2014). Added to these difficulties, many inhibitors targeting protein-protein interactions are peptide-based with less favorable drug-like properties (Vlieghe et al., 2010). An example of these inhibitors is UBC12N26, a 26–amino acid peptide that inhibits the interaction between NAE and its cognate E2 enzyme, UBC12. UBC12N26 corresponds to the N terminus of UBC12 and has been used to gain structural insights into NAE-UBC12 interaction (Huang et al., 2004a).
With all these druggable sites on E1s, there is ample opportunity to target these enzymes with structurally, mechanistically, and pharmacologically diverse drugs, including irreversible inhibitors that are well-suited for use in cancer therapy.
D. Rationale for Targeting E1 Enzymes in Cancer
As a genetic disease, cancer is initiated and maintained by tumor-specific oncogenic alterations of cellular proteins, a state known as oncogene addiction (Weinstein and Joe, 2006). However, the cellular stresses created as a result of malignant transformation and growth, such as proteotoxic, replicative, oxidative and metabolic stresses, need to be supported by other nonmutated, broadly acting cellular machineries that are essential for both normal and cancer cells (Luo et al., 2009). With these stresses, cancer cells become much more dependent on these machineries compared with normal cells, a state known as nononcogene addiction, which results in higher vulnerability to therapeutic agents that target these machineries. Although molecularly targeted agents directed mostly against tumor-specific oncogenes are relatively safer compared with cytotoxic agents, the emergence of resistance is a common pitfall that compromises the efficacy of such agents (Dobbelstein and Moll, 2014). Therefore, it has become increasingly important to explore targeting nononcogene addictions of cancer with drugs that are potentially more effective than molecularly targeted agents and less toxic compared with cytotoxic therapy (Luo et al., 2009; Dobbelstein and Moll, 2014).
Proteasome inhibitors constitute a classic example of these drugs (Dobbelstein and Moll, 2014). They target the proteasome, which is an essential cellular machinery with broad cellular functions in both normal and cancer cells, including a key role in protecting against proteotoxic stress, particularly in myelomas that are engaged in immunoglobulin production (Hoeller and Dikic, 2009). Despite this essentiality and the expected toxicity of proteasome inhibition, bortezomib—the prototype of this class—is effective in malignancies that are highly dependent on the UPS, such as multiple myeloma (MM) and mantle cell lymphoma, with a therapeutic window that allows for a clinically acceptable safety profile in selected patient cohorts (Manasanch and Orlowski, 2017).
Similarly, several E1 enzymes are essential for supporting cellular stresses of cancer cells, including proteotoxic and DNA damage stress (Schulman and Harper, 2009). Of all E1s, UBA1 is the most essential enzyme whose loss-of-function is anticipated to be most deleterious to the survival and growth of cancer cells (Clague et al., 2015; Groen and Gillingwater, 2015). As assessed by the analysis of publicly available datasets of cancer cell line dependencies, only UBA1 and UBA2 (encoding the active subunit of SAE) genes are considered to be essential upon knockdown in large pancancer RNA interference screens (Tsherniak et al., 2017). However, the analysis of CRISPR/Cas9 knockout screens extends essentiality to UBA3 (encoding the active subunit of NAE), UBA4 and UBA5, as well as SAE1 and NAE1, encoding the nonactive subunits of SAE and NAE, respectively (Fig. 5; Supplemental Figs. 1 and 2).

Cancer dependence of E1 genes. Essentiality of E1 enzymes was assessed by pancancer genome-wide loss-of-function screens conducted in 710 (RNAi) and 582 (CRISPR) cancer cell lines. Donut charts reveal the percentage of cell lines dependent on the indicated genes encoding E1 or E1 subunits. A cell line is regarded as “dependent” when it has a probability of dependence greater than 0.5. These data were obtained by the analysis of publicly available datasets on the depmap portal at https://depmap.org/portal. RNAi, RNA interference.
As highlighted above, E1 enzymes lie at the apex of the UBL conjugation cascade, and their targeting is expected to disrupt a variable range of biologic processes regulated by different UBLs (Schulman and Harper, 2009). For instance, UBA1 activates ubiquitylation required for both degradative and nondegradative cellular functions (Ulrich and Walden, 2010). Compared with proteasome inhibitors, UBA1 inhibitors are anticipated to induce broader, and thus more efficacious, biologic effects, as they disrupt proteasomal degradation as well as ubiquitin-regulated signaling pathways, such as DNA repair and nuclear factor-kappa B (NF-κB) signaling (Bedford et al., 2011). Such higher efficacy may be needed in malignancies in which proteasome inhibitors suffer from intrinsic or acquired resistance (Manasanch and Orlowski, 2017). Targeting SAE is similarly expected to induce broad, yet less profound, effects as UBA1 inhibitors because of the indispensable role of SUMOylation in many signaling pathways (Gareau and Lima, 2010; Seeler and Dejean, 2017). Since neddylation is required to regulate a subset of ubiquitin E3 ligases, targeting NAE induces a narrower spectrum of ubiquitylation-dependent biologic effects and is perhaps associated with higher tolerability compared with UBA1 and SAE inhibitors. This is consistent with the development of an NAE inhibitor, pevonedistat, as the first E1-targeted drug to enter clinical trials (Soucy et al., 2009a).
Given the important biologic functions of UBL conjugation pathways initiated by E1 enzymes and the perceived therapeutic window resulting from the higher dependence of cancer cells on these pathways, E1 enzymes can thus serve as attractive therapeutic targets in cancer. These specific biologic roles and their relevance to cancer biology are detailed below and in sections IV. Canonical E1 Enzymes and V. Noncanonical E1 Enzymes.
E. Biologic Functions of E1 Enzymes and Their Implications in Cancer Therapy
Cancer is supported by a number of biologic hallmark capabilities and cellular stress phenotypes that enable tumor development and progression (Luo et al., 2009; Hanahan and Weinberg, 2011). As E1 enzymes initiate more than a dozen post-translational modifications that influence a wide array of cellular substrates, and with the functional diversity and breadth of such modifications, it is not surprising that E1s play a paramount role in almost every aspect of cancer cell biology (Welchman et al., 2005) (Fig. 6). In this section, we briefly summarize the biologic functions of E1-initiated post-translational modifications and their impact on key signaling pathways and cellular processes of relevance to cancer.

Biologic functions of E1 enzymes and their implications in cancer therapy. E1 enzymes comprise eight members: UBA1, NAE, SAE, UBA6, UBA7, UBA4, UBA5, and ATG7. They initiate more than a dozen post-translational modifications, including ubiquitylation, neddylation, SUMOylation, FATylation, ISGylation, URMylation, UFMylation, and ATGylation. These modifications influence the biologic hallmark capabilities and cellular stress phenotypes (e.g., oxidative and proteotoxic stress) of cancer by modulating the activity, expression, stability, and/or localization of a wide range of signaling molecules. Therefore, targeting E1 enzymes may elicit useful antitumor effects in a context-dependent manner.
1. Proliferative Signaling and Growth Suppressors
Over the course of tumor development and progression, tumor cells accumulate genetic aberrations and other alterations that promote their proliferation and allow them to evade growth suppressors (Hanahan and Weinberg, 2011). The E1-initiated post-translational modifications are implicated in such rewiring of cancer cell signaling via proteolytic and nonproteolytic mechanisms (Pérez-Benavente et al., 2020). In this respect, the activity of several growth factor receptors (e.g., EGFR, Met, platelet-derived growth factor receptor, and prolactin receptor) and their downstream kinases is normally regulated by ubiquitylation and subsequent proteasomal degradation. Several oncogenic alterations involve the stabilization of these proteins by downregulating their degradation, resulting in constitutive mitogenic signaling (Huangfu and Fuchs, 2010). In these contexts, UBA1 and UBA6, which initiate ubiquitylation, and NAE, which initiates the neddylation of a subset of ubiquitin E3 ligases enhancing their function, are particularly important. Therapeutic inhibition of these E1 enzymes in cancers driven mainly by such alterations may cause further activation of mitogenic signaling, resulting in unfavorable outcomes. Conversely, the antitumor activity of several tumor-suppressive proteins is reduced by alterations that enhance their ubiquitin-dependent proteasomal degradation (Huangfu and Fuchs, 2010). In such contexts, therapeutic inhibition of the ubiquitin-activating E1s or NAE is anticipated to stabilize these tumor suppressors and slow tumor progression. For instance, the well known tumor suppressors p53 and retinoblastoma-associated protein (Rb) are susceptible to proteasomal degradation after interaction with the ubiquitin E3 ligase double minute 2 protein (MDM2), which facilitates their ubiquitylation (Chène, 2003; Uchida et al., 2005). SUMOylation has also been reported to regulate the activity of tumor suppressors [e.g., p53 and phosphatase and tensin homolog (PTEN)] via nonproteolytic mechanisms, including the alteration of cellular localization (Stindt et al., 2011; Bassi et al., 2013).
2. Cell Cycle Progression
Ubiquitylation, neddylation, and SUMOylation play highly coordinated roles in orchestrating DNA replication, chromosome segregation, and mitosis in a spatiotemporal manner and mainly through nonproteolytic mechanisms (Eifler and Vertegaal, 2015; Gilberto and Peter, 2017). These modifications span a broad spectrum of cell cycle regulators, including, among others, cyclin-dependent kinases, cyclins, topoisomerases, and E3 ligases (Nakayama and Nakayama, 2006; Teixeira and Reed, 2013; Cuijpers and Vertegaal, 2018). As cancer cells proliferate at much higher rates compared with normal cells, they are highly dependent on these UBL conjugation machineries including E1 enzymes to support their proliferation. Therefore, inhibition of UBA1, NAE, and SAE is anticipated to cause cell cycle defects, replicative stress, and mitotic stress, and if sustained and excessive, these will ultimately lead to cell death (Hoeller et al., 2006; Luo et al., 2009). Specific cell cycle defects ensuing from the inhibition of different E1s are detailed in Sections IV. Canonical E1 Enzymes, V. Noncanonical E1 Enzymes, VI. Dual and Multi-E1 Inhibitors, and VII. Adenosine Sulfamate E1 Inhibitors.
3. DNA Damage Response
After exposure to genotoxic stress, ubiquitylation and SUMOylation are particularly important for coordinating the DNA damage response that comprises lesion sensing, DNA repair, and/or damage tolerance (Bergink and Jentsch, 2009). They are actively implicated in the different DNA repair pathways, including base-excision repair, nucleotide-excision repair, double-strand break repair, Fanconi anemia pathway, and translesion synthesis (Ulrich and Walden, 2010; Schwertman et al., 2016). These modifications alter protein stability and/or localization and guide repair proteins to the site of the lesion (Ulrich, 2014). Examples of DNA damage response proteins that are amenable to such modifications are proliferating cell nuclear antigen (PCNA), Fanconi anemia group D2 protein (FANCD2), MDC1, xeroderma pigmentosum group C-complementing protein (XPC), thymine-DNA glycosylase, and H2A/B (Huang and D’Andrea, 2006; Jackson and Durocher, 2013; Dantuma and van Attikum, 2016). Inhibition of UBA1 and SAE is associated with the disruption of the DNA damage response and induction of DNA damage stress (Luo et al., 2009; Hyer et al., 2018).
4. Apoptosis and Autophagy
The signaling pathways that govern different forms of cell death are also amenable to regulation by E1-initiated post-translational modifications, particularly ubiquitylation. In this respect, the levels of pro- and antiapoptotic proteins are controlled by ubiquitin-dependent proteasomal degradation. Caspases are also regulated by nondegradative ubiquitylation (Broemer and Meier, 2009; Vucic et al., 2011). Tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) receptors DR4 and DR5, which are involved in the extrinsic apoptosis pathway, may be endocytosed and/or downregulated in response to their ubiquitylation (Huangfu and Fuchs, 2010). Therefore, the stabilization of proapoptotic proteins [e.g., Bcl-2-associated X protein (Bax), Bcl-2 homologous antagonist killer (Bak), Bcl-2-interacting mediator of cell death (Bim), BH3-interacting domain death agonist (Bid), p53 up-regulated modulator of apoptosis (Puma), Bcl-2-modifying factor (Bmf), Bcl-2-associated death promoter (Bad), Noxa, caspases, and DR4/5] upon UBA1 or NAE inhibition may be associated with useful anticancer effects in certain contexts (Chen et al., 2016a; Hyer et al., 2018). Post-translational modifications initiated by UBA1 and ATG7 are known to play a central role in autophagy, including the oncogenic and tumor-suppressive forms (Xiong, 2015; Gómez-Díaz and Ikeda, 2019); therefore, therapeutic targeting of these E1s may elicit autophagy-related anticancer effects in a context-dependent manner.
5. Inflammation, Immune Responses, and Tumor Microenvironment
Ubiquitylation plays an integral role in regulating inflammatory and immune signaling pathways, including those implicated in cancer pathogenesis (Bhoj and Chen, 2009; Hu and Sun, 2016; Fujita et al., 2019). Of these pathways, the NF-κB signaling pathway is particularly important. NF-κB is a transcription factor that plays multifunctional roles in regulating survival, inflammation, and immune responses (Skaug et al., 2009). The transcriptional activity of NF-κB is kept in check by the inhibitor of NF-κBα (IκBα), which sequesters NF-κB in an inactive state in the cytosol (Hoeller et al., 2006). This inhibitory activity is released after ubiquitin-dependent proteasomal degradation of IκBα, resulting in the nuclear translocation and transcriptional activation of NF-κB (DiDonato et al., 2012). Therefore, UBA1 inhibition is anticipated to stabilize IκBα and inhibit NF-κB, leading to useful anticancer effects, particularly in malignancies with aberrant NF-κB activity such as acute myeloid leukemia (AML) (Birkenkamp et al., 2004; Bosman et al., 2014). Of note, the NF-κB signaling pathway is also regulated by nondegradative ubiquitylation and SUMOylation (Hoeller et al., 2006; Skaug et al., 2009). There is an emerging role of SUMOylation in modulating the innate and adaptive immune responses with potential implications in cancer therapy (see section IV. C. Small Ubiquitin-Like Modifier–Activating Enzyme). In addition to immune cells, other components of the tumor microenvironment, including cancer-associated fibroblasts and endothelial cells, are susceptible to regulation by UBL modifications including neddylation (Zhou et al., 2019a).
6. Angiogenesis and Metastasis
E1-initiated post-translational modifications have been reported to regulate angiogenesis via direct and indirect mechanisms (Rahimi, 2012; Rabellino et al., 2020). In this respect, vascular endothelial growth factor receptor signaling is known to be regulated by ubiquitylation and SUMOylation (Simons et al., 2016; Zhou et al., 2018). In the context of cancer, ubiquitylation may indirectly promote tumor angiogenesis by enhancing p53 degradation and thus stabilizing hypoxia-inducible factor (HIF)-1 (Ravi et al., 2000). Similarly, ubiquitylation has been reported to regulate metastasis by multiple molecular mechanisms, such as the induction of Von Hippel–Lindau (VHL) degradation and thus HIF-1 stabilization (Jung et al., 2006; Gallo et al., 2017; Chen et al., 2019). Genetic and pharmacologic inhibition of ubiquitylation and neddylation have been reported to suppress tumor angiogenesis and metastasis (Tan et al., 2014; Yao et al., 2014; Jin et al., 2018).
7. Cellular Stress Phenotypes
Cancer cells are characterized by several stress phenotypes, including proteotoxic, oxidative, metabolic, replicative, mitotic, and DNA damage stress (Luo et al., 2009). Such stresses are supported by normal cellular machineries, including the UBL conjugation systems, but to a greater extent in tumor versus normal cells, creating a myriad of therapeutic opportunities through stress overload and stress sensitization (Luo et al., 2009; Dobbelstein and Moll, 2014). Therapeutic inhibition of UBA1 and NAE is known to induce unfolded protein response (UPR) and proteotoxic stress, particularly in hematologic malignancies (Soucy et al., 2009a; Hyer et al., 2018). Ubiquitylation and SUMOylation have been reported to respond to and modulate cellular production of reactive oxygen species (ROS) (Bossis and Melchior, 2006; Swords et al., 2010; Shang and Taylor, 2011; Graves et al., 2020; Ma et al., 2020). Thus, inhibition of UBA1 and NAE has been associated with the induction of oxidative stress (Nawrocki et al., 2013; Zhuang et al., 2019). Similar regulatory roles have been identified with cellular metabolism (Agbor and Taylor, 2008; Flick and Kaiser, 2012; Lavie et al., 2018; Thapa et al., 2020), replication, mitosis and DNA damage, as highlighted above. Dependent on the context, therapeutic targeting of E1s is anticipated to exacerbate one or more of these stresses, with potentially detrimental effects on cancer cell survival.
F. Targeting E1 Versus E3 Enzymes
E1 enzymes initiate the UBL conjugation cascade and thus lie upstream of the E3 enzymes that are responsible for substrate specificity (Schulman and Harper, 2009). Although targeting certain E3 enzymes is anticipated to elicit selective antitumor effects as a result of modulating one or a few substrates (Bedford et al., 2011; Huang and Dixit, 2016), targeting E1 enzymes offers several advantages over E3 enzymes. From a drug discovery perspective, E1 enzymes possess well defined pockets that are readily druggable—as opposed to most E3 enzymes, which have multiple subunits and lack active pockets—and many E3s are not functionally annotated (Huang and Dixit, 2016). In addition, E3-oriented drug discovery endeavors have been mainly focused on targeting protein-protein interactions, which is a more challenging approach compared with targeting catalytic/active sites; therefore, the field witnessed slower progress until recently, as novel targeted protein degradation strategies that exploit ligands of E3 ligases to form heterobifunctional degraders have been developed (Cohen and Tcherpakov, 2010; Schapira et al., 2019; Barghout, 2020).
Although several ubiquitin E3 ligases are deregulated and thus implicated in oncogenic transformation and tumor progression, their selective therapeutic targeting may not be highly effective because of the functional redundancy among ubiquitin E3 ligases and the activation of compensatory signaling pathways (Nakayama and Nakayama, 2006; Senft et al., 2018; Wu et al., 2020). In contrast, targeting UBA1 that initiates the ubiquitylation cascade and regulates many ubiquitin-dependent signaling pathways may be more effective and less likely to suffer from the development of adaptive resistance, and this approach is useful in several contexts, particularly in malignancies highly dependent on ubiquitylation and where a sufficient therapeutic window can be attained (Dobbelstein and Moll, 2014). This has been the case with proteasome inhibitors that, despite the lack of selectivity and perceived toxicity, proved to be more therapeutically useful compared with investigational E3 ligase inhibitors in several malignancies (Nakayama and Nakayama, 2006).
III. Tools and Assays for Exploring E1 Biology and Drug Discovery
E1 enzymes possess multiple domains that enable them to perform their multistep catalytic activity as UBL-activating enzymes (Schulman and Harper, 2009; Cappadocia and Lima, 2018). This catalytic activity comprises adenylation, thioester formation, and transthiolation and, in concert with downstream E2 and E3 enzymes, leads to the conjugation of specific UBL modifiers to selected substrates. Numerous assays and tools have been developed to interrogate the biology of E1 enzymes at different catalytic steps and to assist in the discovery and characterization of drugs and chemical probes that modulate the activity of these enzymes. In this section, we briefly highlight a number of these tools and their utility in E1 biology and the discovery of E1 inhibitors.
A. ATP:PPi and ATP:AMP Exchange Assays
E1 enzymes resemble aminoacyl–transfer RNA (tRNA) synthetases and other adenylate-forming enzymes in their catalytic mechanisms (Dusha and Denes, 1977; Wilson and Aldrich, 2010). In this respect, they activate UBLs by catalyzing a three-step biochemical mechanism that involves the utilization of ATP and release of PPi in the first and second adenylation steps and release of AMP in the thioester formation step (Fig. 3B).
In ATP:PPi exchange assays, the catalytic reaction is assembled under cell-free conditions using a recombinant E1 enzyme, recombinant UBL, ATP, and radiolabeled PPi. The incorporation of radiolabeled PPi into ATP and formation of radiolabeled ATP, representing the reverse adenylation reaction, are quantitatively monitored using an appropriate detection method (Haas and Rose, 1982; Bruzzese et al., 2009). This assay can be used to assess the UBL specificity of E1 enzymes and to quantify their adenylate-forming activity by calculating several kinetic parameters, such as rate and equilibrium constants for the ternary E1-AMP∼UBL complex formation and the turnover number for the enzyme—the number of E1-catalyzed reactions per unit time (Alontaga et al., 2012). On the other hand, ATP:AMP exchange assays are used to obtain information on both the adenylation and thioester formation steps of E1-catalyzed UBL activation (Alontaga et al., 2012). In these assays, the catalytic reaction is also assembled under cell-free conditions using recombinant E1 enzyme, recombinant UBL, ATP, PPi, and radiolabeled AMP (Ciechanover et al., 1981; Wang et al., 2009). The generation of radiolabeled ATP from radiolabeled AMP is then monitored and quantified after the separation of both species by thin-layer chromatography. ATP:PPi and ATP:AMP exchange assays have been widely used to characterize the catalytic activity of different E1 enzymes (Ciechanover et al., 1981, 1982; Haas and Rose, 1982; Bohnsack and Haas, 2003; Bruzzese et al., 2009; Wang et al., 2009). They have also been used in the context of biochemical characterization of novel E1 inhibitors to gain mechanistic insights into their mode of action and mechanisms of resistance (Brownell et al., 2010; Chen et al., 2011; Milhollen et al., 2012; Ungermannova et al., 2012a; Xu et al., 2014; Hyer et al., 2018).
B. E1∼ Ubiquitin-Like Protein Thioester Formation and Transthiolation Assays
E1∼UBL thioester formation assays determine the formation of E1∼UBL thioesters under cell-free or cell-based conditions using detection methods such as immunoblotting with antibodies against either the E1 or UBL. In cell-free assays, the thioester formation reaction is assembled using recombinant E1, recombinant UBL, and ATP (Wang et al., 2007; Olsen et al., 2010). Since the thioester bond is sensitive to reducing agents such as dithiothreitol, the detection of E1∼UBL thioesters requires nonreducing conditions (Alontaga et al., 2012). On the other hand, transthiolation assays determine the transfer of UBL from a certain E1 to its cognate E2 enzyme to form an E2∼UBL thioester. In addition to the reaction components used in the E1∼UBL thioester formation assays, a recombinant E2 is also included to assess transthiolation. In addition, these assays are conducted in two steps and can provide quantitative information on both UBL activation and transfer (Alontaga et al., 2012). Recently, high-throughput luminescence-based assays have been developed to evaluate thioester formation with UBA1 and its E2, radiation sensitivity protein 6 (RAD6), in a quantitative manner (Fenteany et al., 2020). E1∼UBL thioester formation and transthiolation assays have been used to identify and characterize the activity of different E1 inhibitors in various platforms including large-scale drug discovery screens (Yang et al., 2007; Brownell et al., 2010; Hyer et al., 2018; Lv et al., 2018b).
C. Substrate Ubiquitin-Like Protein Conjugation Assays
Substrate UBL conjugation assays determine the conjugation of specific UBLs to their target substrates and are conducted under cell-free or cell-based conditions. For example, candidate proteins such as histones H2A/H2B and PCNA are known to be common targets for monoubiquitylation, cullin-1 for neddylation, and Ran GTPase-activating protein 1 (RanGap1) for SUMOylation (Gareau and Lima, 2010; Enchev et al., 2015; Uckelmann and Sixma, 2017; Fenteany et al., 2020). In addition, the global changes in UBL conjugation to cellular proteins can be assessed by immunoblotting using antibodies against the UBL modifier or by large-scale proteomic approaches (Visconte et al., 2016). In cell-free assays, the whole UBL conjugation system including a recombinant E3 ligase needs to be assembled. However, this is not an absolute requirement, as for instance, RanGAP1 can be SUMOylated in the absence of an E3 ligase (Alontaga et al., 2012). Substrate UBL conjugation assays are not specific for E1 enzymes, as they evaluate the combined activity of E1, E2, and E3 enzymes. In addition to their use in drug discovery screens for E1 inhibitors, these assays are routinely used in evaluating the downstream biologic effects of potential E1 inhibitors on UBL conjugation to cellular substrates (Hyer et al., 2018; Barghout et al., 2019; Li et al., 2019).
D. Detection of Ubiquitin-Like Protein–Drug Adduct Formation
Adenosine sulfamates and related E1 inhibitors act by forming a covalent adduct with their cognate UBLs, and these adducts serve as the inhibitory species (Ciavarri and Langston, 2017). To detect the formation of these adducts, specific anti–UBL-drug antibodies have been generated and used in immunoblot-based assays to evaluate the pharmacodynamic profile of these drugs (Soucy et al., 2009a; Hyer et al., 2018). Similarly, a radiolabeled form of the adenosine aminosulfonamide TAS4464 has been used to detect the formation of these adducts by immunoblotting and autoradiography (Yoshimura et al., 2019). However, it remains important to adapt these assays to provide quantitative information on the rate of adduct formation, which in turn depends on E1 activity.
In the following sections, we discuss in more detail the different E1s, their roles in cancer, the inhibitors developed so far to target these enzymes, and how these assays have been used to discover and characterize these inhibitors. In our discussion, we will emphasize mechanism-based E1 inhibitors, particularly those advanced to clinical trials, and the common principles shared among these agents. We will start with the NAE for which the prototypical clinical E1 inhibitor has been developed.
IV. Canonical E1 Enzymes
A. Neural Precursor Cell–Expressed Developmentally Downregulated Protein 8–Activating Enzyme
Neddylation is the process of conjugating NEDD8 to cellular substrates (Enchev et al., 2015). NEDD8 is 59% identical to ubiquitin—the highest level of similarity observed among all UBLs, albeit with structural differences that are sufficient to mediate distinct functions (Watson et al., 2011b; Enchev et al., 2015). Neddylation cascade is initiated by NAE that catalyzes NEDD8 activation. NAE is a heterodimeric enzyme composed of a regulatory subunit, NAE1 (amyloid β precursor protein-binding protein 1), and a catalytic subunit, NAE2 (UBA3), with similar domain structure as other canonical E1s (Fig. 2) (Schulman and Harper, 2009). Although the AAD is located on NAE2, the CCD half-domains are located on both subunits (Schulman and Harper, 2009; Cappadocia and Lima, 2018). Interestingly, the ubiquitin-specific UBA1 enzyme can activate NEDD8 under stress conditions, such as heat shock and oxidative stress (Leidecker et al., 2012). Two neddylation E2 enzymes have been reported in metazoans, UBC12 and UBE2F, which function in concert with E3 ligases including ubiquitin ligases (Enchev et al., 2015). Neddylation targets many substrates, of which cullins are the best characterized and the most established class of neddylated proteins (Enchev et al., 2015). Cullins serve as scaffold proteins upon which the largest class of RING ubiquitin E3 ligases, known as cullin-RING ubiquitin ligases (CRLs), are assembled (Watson et al., 2011b). Cullin neddylation plays a pivotal role in the ubiquitylation of a subset of cellular proteins by inducing conformational changes that increase the activity of CRLs and enhance ubiquitin conjugation (Lydeard et al., 2013; Baek et al., 2020). Other noncullin substrates include signaling molecules such as p53, p73, E2F1, epidermal growth factor receptor (EGFR), tumor growth factor β receptor, NF-κB essential modulator (NEMO), Von Hippel–Lindau (VHL) tumor suppressor, and HIFs, which play diverse roles in normal and cancer cell biology (Watson et al., 2011b; Enchev et al., 2015). Therefore, neddylation regulates numerous pathways, including proteasomal degradation, cell cycle progression, receptor tyrosine kinase signaling, apoptosis, DNA damage response, inflammatory/immune responses, oxidative stress, hypoxia, and nucleolar stress signaling (Soucy et al., 2009b; Mathewson et al., 2013, 2016; Brown et al., 2015; Enchev et al., 2015; Balachandran et al., 2016).
1. Role of Neural Precursor Cell–Expressed Developmentally Downregulated Protein 8–Activating Enzyme in Cancer
As neddylation plays a salient role in numerous signaling pathways, dysregulation of NAE and/or downstream components of neddylation cascade is anticipated to contribute to the development and progression of several malignancies (Soucy et al., 2010; Watson et al., 2011b). In most cases, expression data from published studies or publicly available datasets indicate a negative correlation between the expression of either NAE1 or NAE2 or both and clinical outcomes. For instance, NAE1/2 are upregulated in hepatocellular carcinoma and are associated with an aggressive phenotype and poor overall and relapse-free survival in patients (Barbier-Torres et al., 2015; Uhlen et al., 2017; Yu et al., 2018a). Similar data has been reported in lung cancer (Li et al., 2014b), glioblastoma (Hua et al., 2015), squamous cell carcinoma (Li et al., 2014b), intrahepatic cholangiocarcinoma (Gao et al., 2014), MM (Huang et al., 2015), esophageal squamous cell carcinoma (Chen et al., 2016a), uveal melanoma (Jin et al., 2018), pancreatic cancer (Misra et al., 2017; Li et al., 2018), clear cell renal cell carcinoma (Tong et al., 2017), and chronic myeloid leukemia (Liu et al., 2018). In contrast, patients with thyroid cancer show a favorable prognosis with high NAE1 expression (Uhlen et al., 2017). It appears, however, that no significant mutations are known to implicate NAE1/2 in cancer pathogenesis (Tate et al., 2019).
2. Neural Precursor Cell–Expressed Developmentally Downregulated Protein 8–Activating Enzyme Inhibitors
NAE is one of the first human E1 enzymes to have its crystal structure elucidated, and this provided valuable structural insights and a common framework for mechanisms of activation and transfer of NEDD8 as well as other UBLs (Walden et al., 2003a,b; Huang et al., 2004a, 2007). This structural information contributed, in part, to guiding drug discovery endeavors for E1 inhibitors, and it is not surprising that the first reported mechanism-based E1 inhibitor is targeted against NAE (Soucy et al., 2009a; Brownell et al., 2010). The hyperactive neddylation pathway or high reliance on one or more of its components in cancer is a therapeutic avenue that has garnered attention in recent years, particularly after the development of selective NAE inhibitors (Ying et al., 2018). Compounds targeting NAE comprise the adenosine sulfamate pevonedistat, the related adenosine aminosulfonamide TAS4464 — both are selective clinical candidates — and several experimental inhibitors including NEDD8 adenylate analogs and NEDD8-mimicking peptides (APN, pND20, and pND22), natural and semisynthetic NAE inhibitors (6,6′′-biapigenin, flavokawain A, and piperacillin), as well as other inhibitors with diverse chemical structures [M22, LZ3, ZM223, and rhodium (III)-based complexes]. So far, pevonedistat is the most extensively studied E1 inhibitor, with a large number of preclinical and clinical studies in numerous malignancies. Apart from pevonedistat and TAS4464, most known NAE inhibitors have been discovered by in silico approaches and possess less favorable potency and selectivity profiles with activity in the micromolar range. Nonetheless, they can potentially serve as useful chemotypes to boost drug discovery endeavors and develop novel NAE inhibitors (Fig. 7; Table 2).
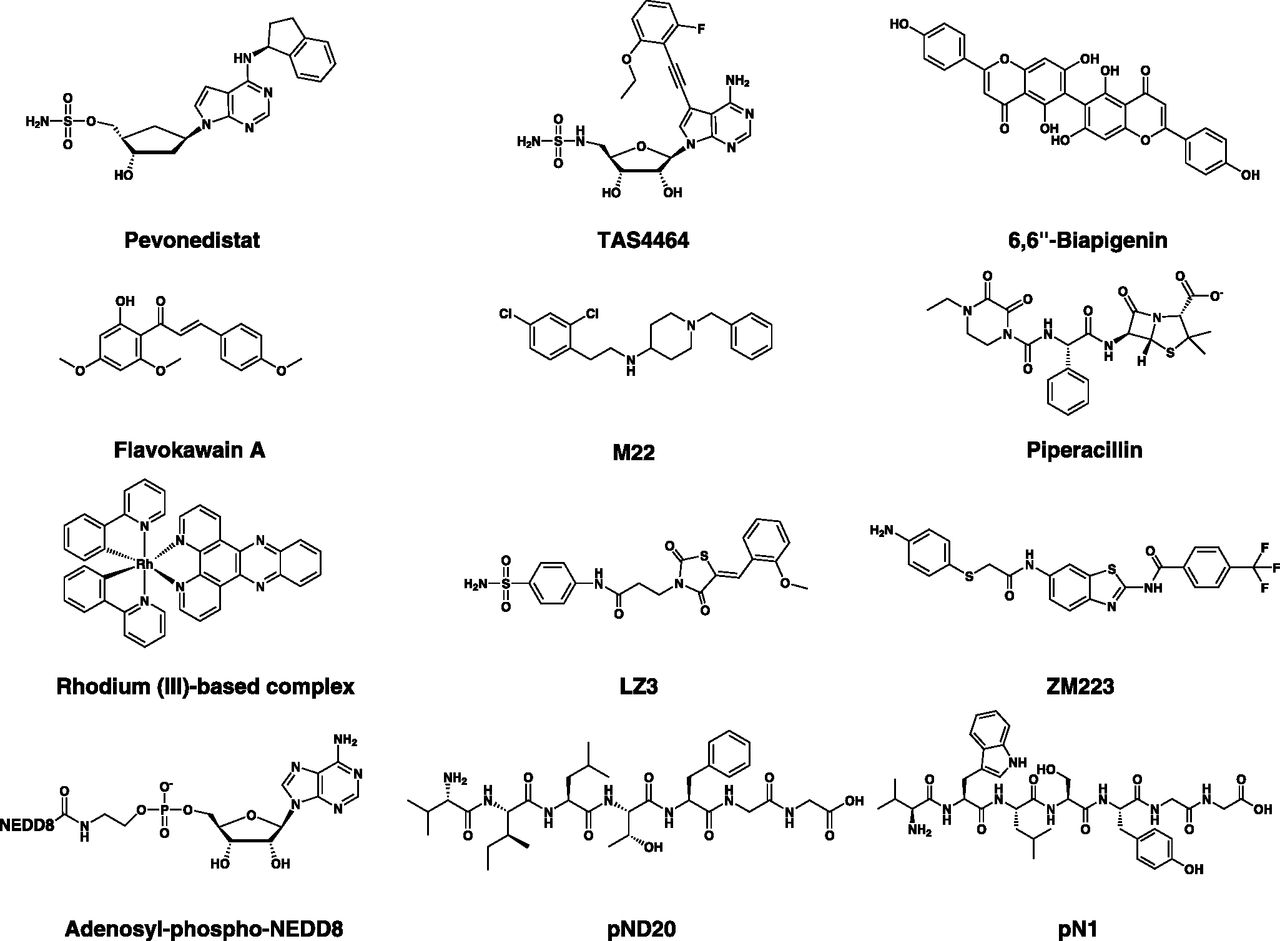
Chemical structures of NAE inhibitors. NAE inhibitors belong to diverse classes, including adenosine sulfamates and related analogs (e.g., pevonedistat and TAS4464), natural compounds (e.g., 6,6′′-Biapigenin), semisynthetic compounds (piperacillin), NEDD8 adenylate analogs (e.g., adenosyl-phospho-NEDD8), NEDD8-mimicking peptides (e.g., pND20), and other inhibitors.
NAE inhibitors, their chemical structures, and pharmacologic properties
a. Pevonedistat
Pevonedistat (MLN4924/TAK-924) is the prototypical adenosine sulfamate E1 inhibitor (Soucy et al., 2009a). A high-throughput screen (HTS) identified N6-benzyl adenosine as an NAE inhibitor, and further optimization by iterative medicinal chemistry led to the development of pevonedistat, a first-in-class mechanism-based NAE inhibitor (Soucy et al., 2009a; Ciavarri and Langston, 2017). Mechanism-based inhibition is a form of enzyme inhibition whereby an analog of the enzyme substrate is processed through its normal catalytic mechanism, producing a species that inhibits the enzyme (Copp, 2003). Pevonedistat is structurally related to AMP and thereby acts as an AMP mimetic (Brownell et al., 2010). It exerts its NAE inhibitory activity by a form of mechanism-based inhibition termed “substrate-assisted inhibition,” whereby it forms a covalent adduct with NEDD8 in a reaction catalyzed by NAE itself. This covalent adduct tightly binds to the nucleotide-binding site and inhibits NAE in an ATP-competitive manner (Brownell et al., 2010). Substrate-assisted inhibition serves as a common mechanism of action among E1 adenosine sulfamate inhibitors (Ciavarri and Langston, 2017).
Although compounds of this structural class may have inhibitory effects on adenylate-forming enzymes such as tRNA synthetases (Brown et al., 1999; Forrest et al., 2000; Lux et al., 2019), or potentially other ATP-dependent enzymes such as kinases, the action of pevonedistat is highly selective for NAE even compared with other E1 enzymes (Soucy et al., 2009a). In this context, it selectively inhibits neddylation with little or no effect on the conjugation of related UBLs with IC50 values in the nanomolar range (Table 2).
Given the role of neddylation in regulating the activity of CRLs, pevonedistat induces a potent inhibition of these enzymes with a subsequent reduction in the turnover of a subset of cellular proteins (Soucy et al., 2009a,b). Prominent among these proteins is the DNA replication licensing factor CDT1, which is essential for prereplication complex assembly and plays a key role in DNA replication and mitosis (Milhollen et al., 2011). Stabilization of CDT1 in the S phase of the cell cycle is primarily responsible for the re-replication phenotype and mitotic defects observed after pevonedistat treatment (Soucy et al., 2009a; Lin et al., 2010; Milhollen et al., 2011). DNA re-replication is characterized by ≥4 N DNA content as a result of unscheduled repeated rounds of DNA synthesis and leads to replication stress, DNA damage, and apoptosis (Petropoulos et al., 2019). Consistent with these cell cycle–specific effects, actively proliferating cells are more susceptible to pevonedistat cytotoxicity (Soucy et al., 2009a). TAK-243, the related UBA1 inhibitor, does not induce re-replication, possibly because of the stabilization of both CDT1 and its endogenous inhibitor geminin (Hyer et al., 2018).
With the narrower spectrum of proteins influenced by disrupting neddylation, it is conceivable that NAE inhibitors will exhibit a wider therapeutic window compared with proteasome and UBA1 inhibitors (Buac et al., 2013). Pevonedistat is well tolerated in vivo with broad antitumor activity in solid and hematologic malignancies (Table 3).
Preclinical and clinical studies of pevonedistat
Apart from the induction of re-replication and DNA damage (Swords et al., 2010; Zhang et al., 2015b; Guo et al., 2017; Tong et al., 2017; Zheng et al., 2017b), a plethora of diverse mechanisms have been reported to mediate the antitumor effects of pevonedistat. These include the inhibition of the NF-κB pathway (Milhollen et al., 2010; Swords et al., 2010; Godbersen et al., 2014; Li et al., 2014a; Khalife et al., 2015); stabilization and upregulation of proapoptotic proteins such as Noxa (Dengler et al., 2014; Knorr et al., 2015; Misra et al., 2017; Wang et al., 2017); generation of ROS (Swords et al., 2010), stabilization of cell cycle regulators such as p21, p27, and WEE1 (Yang et al., 2012; Blank et al., 2013; Mackintosh et al., 2013; Huang et al., 2015; Liu et al., 2018; Wang et al., 2016); inhibition of the mammalian/mechanistic target of rapamycin (mTOR) pathway (Gu et al., 2014; Li et al., 2014a); induction of endoplasmic reticulum (ER) stress (Kuo et al., 2015; Leclerc et al., 2016); induction of immunomodulatory effects (Best et al., 2020); and activation of death receptor signaling (Chen et al., 2016a; Paiva et al., 2017). In addition to apoptosis, pevonedistat induces senescence in several malignancies (Jia et al., 2011; Wang et al., 2015; Benamar et al., 2016). Moreover, it exerts antiangiogenic and antimetastatic effects in preclinical models of pancreatic cancer (Yao et al., 2014) and uveal melanoma (Jin et al., 2018). While inhibiting migration in human urothelial (Kuo et al., 2015), clear cell renal (Tong et al., 2017) and gastric cancer cells (Lan et al., 2016), pevonedistat has been reported to display promigratory effects in glioblastoma and prostate cancer cells through induction of caveolin-1 phosphorylation (Park et al., 2018), suggesting control of migration by neddylation is context-dependent.
With the multiple effects exerted by pevonedistat, it is not surprising that additive or synergistic antitumor effects are observed with mechanistically diverse antineoplastic agents including conventional cytotoxic drugs (Kee et al., 2012; Jazaeri et al., 2013; Nawrocki et al., 2013; Garcia et al., 2014; Li et al., 2014b; Ho et al., 2015; Czuczman et al., 2016; Xu et al., 2018b), radiotherapy (Wei et al., 2012; Wang et al., 2016; Xie et al., 2017; Vanderdys et al., 2018), differentiation therapy (Tan et al., 2011), and targeted therapies such as Bcl-2 inhibitors (Knorr et al., 2015), epigenetic modulators (Visconte et al., 2016; Zhou et al., 2016), monoclonal antibodies (Czuczman et al., 2016), immunomodulatory drugs (Liu et al., 2019; Zhou et al., 2019b), poly (ADP-ribose) polymerase inhibitors (Guo et al., 2017), BRAF inhibitors (Benamar et al., 2016), and other investigational therapies (Chen et al., 2015; Sumi et al., 2016; Cooper et al., 2017; Ishikawa et al., 2017). In certain malignancies, the cytotoxicity of pevonedistat is ameliorated by the induction of cytoprotective autophagy (Luo et al., 2012), an effect that can be overcome by combination with autophagy inhibitors leading to synergistic antitumor effects (Zhao et al., 2012; Chen et al., 2015).
Since its discovery in 2009, pevonedistat has been and is currently evaluated in different phases of over 30 clinical trials alone and in combination with other drugs (Table 4). Approximately half of these are in AML and/or myelodysplastic syndromes (MDS).
Clinical trials of pevonedistat
Phase 1 clinical trials in AML/MDS has shown pevonedistat is generally well tolerated, with pyrexia as the most common adverse event and some dose-limiting toxicities, such as hepatotoxicity and sepsis syndromes with multiorgan failure, that are mostly observed at doses beyond 50 mg/m2 (Swords et al., 2015, 2017). In AML, the rates of complete and partial responses ranged from 10% to 17%, with an overall response rate of 13% (Swords et al., 2015, 2017). When evaluated in combination with azacitidine, the overall response rate increased up to 83%, with a complete response/partial response rate of 80% in patients with TP53 mutations (Swords et al., 2018). These encouraging results with azacitidine prompted several phase 2 clinical trials, and a phase 3 clinical trial, PANTHER, is currently ongoing to evaluate pevonedistat-azacitidine combination versus azacitidine alone as a frontline therapy for specific forms of MDS and AML (clinicaltrials.gov identifier: NCT03268954). In other hematologic (relapsed/refractory MM and lymphoma) and advanced solid malignancies, pevonedistat has also shown similar tolerability with potential therapeutic benefit in these populations (Bhatia et al., 2016; Sarantopoulos et al., 2016; Shah et al., 2016; Lockhart et al., 2019).
b. TAS4464
TAS4464 is an adenosine aminosulfonamide that has structural features similar to adenosine sulfamates but differs in having an aminosulfonamide group in lieu of the sulfamate moiety (Yoshimura et al., 2019). Still, it needs to form a covalent adduct with NEDD8 through this functional group, and this adduct in turn serves as the potent inhibitory species of NAE (Yoshimura et al., 2019). TAS4464 retains the ribose sugar moiety found in AMP, as is the case with several dual and pan E1 inhibitors (see below). Despite this similarity, it exhibits a highly selective activity toward NAE. Accordingly, it induces similar biologic effects on neddylation and downstream signaling, yet with up to 64-fold higher potency and more durable effects compared with pevonedistat. TAS4464 displays a broad-spectrum activity on cancer cell lines of different origin, particularly those of hematologic malignancies. It also displays a superior activity on primary patient samples as well as patient-derived xenografts, including treatment-resistant samples. TAS4464 has been advanced to a phase 1/2 clinical trial in MM and non-Hodgkin lymphoma (clinicaltrials.gov identifier: NCT02978235).
c. Neural Precursor Cell–Expressed Developmentally Downregulated Protein 8 Adenylate Analogs and Neural Precursor Cell–Expressed Developmentally Downregulated Protein 8–Mimicking Peptides
Adenosyl-phospho-NEDD8 (APN) has been synthesized as a chemical probe to assist in elucidating the mechanism of NAE inhibition by pevonedistat (Brownell et al., 2010). Similar to the ubiquitin adenylate analog APU (see below), APN acts as a nonhydrolyzable mimetic of NEDD8∼AMP. It also resembles pevonedistat-NEDD8 adduct in tightly binding to NAE and inhibiting its activity in an ATP-competitive manner; however, NAE exhibits a slower recovery from pevonedistat-NEDD8 adduct, which is consistent with tighter binding compared with APN (Brownell et al., 2010).
As the C-terminal sequence of NEDD8 plays a fundamental role in its recognition by NAE, phage display technology has been exploited to screen mutant versions of this sequence for variants with different kinetics and enzyme reactivities (Zhao et al., 2013b). In this respect, pND20 and pND22 have been identified as NEDD8-mimicking heptameric peptides that can still be recognized and activated by NAE, yet with a higher affinity to the enzyme compared with the wild-type NEDD8. Despite successful activation by NAE, these mutant variants cannot be subsequently used in the neddylation cascade, thereby serving as mechanism-based inhibitors that block NEDD8 activation and conjugation. NEDD8 is the closest UBL to ubiquitin in terms of sequence homology, and UBA1 can activate NEDD8 under certain conditions (Leidecker et al., 2012). Therefore, NEDD8-mimicking peptides have also been derived from mutant C-terminal sequences of ubiquitin, resulting in similar inhibitory properties against NAE (Zhao et al., 2013a).
d. Natural and Semisynthetic Neural Precursor Cell–Expressed Developmentally Downregulated Protein 8–Activating Enzyme Inhibitors
6,6′′-Biapigenin is a semisynthetic flavonoid derivative whose NAE inhibitory activity was identified through virtual screening of a 20,000-compound library using the crystal structure of the quaternary NAE1-UBA3-NEDD8-ATP complex (Leung et al., 2011). Based on molecular modeling studies, 6,6′′-biapigenin is predicted to bind to NAE at a binding site distinct from pevonedistat, resulting in reversible inhibition. Another flavonoid with anti-NAE activity is flavokawain A (Li et al., 2015). Specifically, it belongs to the chalcone subclass of flavonoids and is isolated from the kava extract (Zi and Simoneau, 2005). Flavokawain A inhibits NAE after binding to the enzyme at the nucleotide-binding pocket, resulting in the deneddylation of cullins and inducing proteasome-dependent degradation of S-phase kinase-associated protein 2 (Skp2). As a result, it shows antitumor activity against Rb-deficient cells that are dependent on Skp2 for survival, as well as antitumor and antimetastatic effects in vivo (Li et al., 2015).
Piperacillin, a Food and Drug Administration–approved semisynthetic β-lactam antibiotic, is also reported to competitively inhibit NAE at the nucleotide-binding pocket, suppressing neddylation of downstream targets and inducing accumulation of p27kip1 in cells (Zhong et al., 2014). Another virtual screen of a 90,000-compound library identified a dipeptide-conjugated alkaloid derivative with anti-NAE properties in cell-free and cellular enzymatic assays (Zhong et al., 2012a).
e. Other Natural and Semisynthetic Neural Precursor Cell–Expressed Developmentally Downregulated Protein 8–Activating Enzyme Inhibitors
M22 is a piperidin-4-amine–based NAE inhibitor identified through a structure-based virtual screening of 50,000 compounds (Lu et al., 2016). Molecular modeling studies predict M22 binds to the nucleotide-binding site in a similar conformation to that of ATP. As assessed by biochemical analyses, M22 reversibly inhibits NAE in an ATP-competitive manner, resulting in the stabilization of CRL substrates. These effects are associated with antiproliferative activity in cancer cells of different origin both in vitro and in vivo, as well as synergistic effects with bortezomib. Using zebrafish as an acute toxicity model, M22 is tolerated even at high micromolar concentrations (Lu et al., 2016).
To identify covalent NAE inhibitors, Zhang et al. (2014) leveraged structural information on NAE and pharmacologic data available on pevonedistat’s mode of action. Specifically, they applied a ligand- and structure-based pharmacophore modeling approach combined with covalent docking and screened a focused library of free sulfamoyl-containing compounds for such irreversible inhibitors. These endeavors led to the identification of LZ3 as the top hit. LZ3 is a sulfonamide derivative that covalently binds to NAE, inhibiting its activity in cell-free and cell-based assays. It is unclear, however, whether it forms a covalent adduct with NEDD8 to exhibit such irreversible inhibition. On the other hand, target-based virtual screening approaches have been performed using structural data of the noncovalent interaction between pevonedistat and NAE to identify reversible NAE inhibitors. This led to the identification of a series of reversible nonsulfamate-based inhibitors, including ZM223, a benzothiazole with activity in the low micromolar range (Ma et al., 2017).
A rhodium (III)-based complex is reported to possess a similar inhibitory activity against NAE in cell-free assays and Caco-2 cells, with a low micromolar potency partly attributed to its octahedral coordination geometry (Zhong et al., 2012b).
B. Ubiquitin-Like Modifier–Activating Enzyme 1
UBA1 is the major ubiquitin-activating enzyme that catalyzes the first step in ubiquitin conjugation cascade, and it is involved in the ubiquitylation of most cellular proteins (Hyer et al., 2018). It is an evolutionarily conserved protein and plays an indispensable role in regulating numerous cellular functions (Groen and Gillingwater, 2015). Structurally, UBA1 is a canonical monomeric E1 enzyme with several domains, including the AAD, IAD, UFD, and CCD (Lee and Schindelin, 2008; Schulman and Harper, 2009). Among the eight E1 enzymes identified so far, only UBA1 and UBA6 can activate ubiquitin, but the functions of UBA6-mediated ubiquitylation are not well characterized compared with those mediated by UBA1 (Chiu et al., 2007; Jin et al., 2007; Pelzer et al., 2007; Groettrup et al., 2008). In addition, several differences exist between UBA1 and UBA6 (Table 5).
Properties of ubiquitin-activating enzymes: UBA1 and UBA6 (McGrath et al., 1991; Groettrup et al., 2008; Kulkarni and Smith, 2008; Clague et al., 2015; Hyer et al., 2018)
Ubiquitin itself can be modified on one or more of its seven lysine residues, N terminus, and/or 11 phosphorylation sites by ubiquitin or other post-translational modifications, creating highly diverse ubiquitin signals with multiple topologies (Swatek and Komander, 2016). These distinct forms of ubiquitylation can be simply divided into mono- and polyubiquitylation, and this latter comprises K48- and K63-linked polyubiquitin chains as the most common forms of ubiquitylation (Swatek and Komander, 2016). The diverse ubiquitin-based modifications constitute a “ubiquitin code” that is written by the concerted action of ubiquitin E1-E2-E3 enzymes, erased by a set of DUBs, and read by cellular proteins that specifically recognize this code and translate it into a certain biologic response (Swatek and Komander, 2016). Ubiquitylation is important for protein homeostasis through proteasomal degradation and autophagy and is implicated as a post-translational modification in regulating and fine-tuning a myriad of nondegradative cellular processes, such as cell cycle progression, DNA replication and repair, protein translation, cell death, endocytosis, chromatin architecture and epigenetic regulation, cellular differentiation, inflammation, and immune signaling (Bhoj and Chen, 2009; Ulrich and Walden, 2010; Swatek and Komander, 2016; Dikic, 2017; Varshavsky, 2017; Rape, 2018).
Given its role in almost every aspect of cell biology, ubiquitylation is tightly regulated by multiple mechanisms, including other post-translational modifications and DUBs to specifically conjugate target proteins in a spatiotemporal manner (Grabbe et al., 2011). The ubiquitin system is “pyramidal” in design, as only two E1 enzymes transfer ubiquitin to over 30 E2s, which in turn interact with hundreds of E3 ligases to conjugate the respective protein substrate (Nalepa et al., 2006). The specificity of ubiquitin conjugation is mainly controlled downstream at the level of ubiquitin E3 ligases. Therefore, pharmacologic manipulation of the upstream enzyme UBA1 is anticipated to indiscriminately influence numerous biologic processes.
1. Role of Ubiquitin-Like Modifier–Activating Enzyme 1 in Cancer
Although many mutations and copy number variations (CNVs) have been reported to affect UBA1, there is no known evidence that these genetic aberrations are functionally important in the pathogenesis or progression of cancer (Tate et al., 2019; UniProt Consortium, 2019). Physiologically, UBA1 is among the most abundant proteins in the cell; however, variations in its expression levels may occur in several cancers (Clague et al., 2015). The analysis of patient survival data has demonstrated that UBA1 expression may correlate with the prognosis of cancer. Although it is associated with poor prognosis in liver cancer, high expression of UBA1 is associated with a favorable prognosis in prostate cancer (Uhlen et al., 2017). On the other hand, AML cell lines and primary samples exhibited equal levels of UBA1 protein compared with normal hematopoietic cells while having a higher abundance of ubiquitylated proteins (Xu et al., 2010). This increased ubiquitylation is likely attributable to the more active utilization of UBA1 in AML versus normal cells, constituting a potential therapeutic vulnerability in AML and other malignancies (Yang et al., 2007; Hyer et al., 2018). Ubiquitin conjugation is a multienzymatic process, and cancer-associated dysregulation of ubiquitylation may occur at the level of downstream E2 or E3 enzymes with no changes in the ubiquitin E1 enzymes. The role of these other components of the ubiquitin system in cancer has been extensively reviewed (Nakayama and Nakayama, 2006; Fulda et al., 2012; Alpi et al., 2016; Senft et al., 2018).
2. Ubiquitin-Like Modifier–Activating Enzyme 1 Inhibitors
The crystal structure of human UBA1 has not been elucidated until recently (Lv et al., 2018a), and most of the structural information on UBA1 and ubiquitin activation was derived from published structures of yeast, humanized yeast, mouse Uba1, and related bacterial enzymes (Lake et al., 2001; Szczepanowski et al., 2005; Lee and Schindelin, 2008; Olsen and Lima, 2013; Lv et al., 2017a,b; Hyer et al., 2018). Nonetheless, drug discovery endeavors started earlier using several approaches, including structure-based rational design, relying on the close similarity between human UBA1 and related orthologs (Hyer et al., 2018). These efforts have led to the identification of several inhibitors that act by diverse mechanisms (Table 6). The inhibitors reported thus far can be structurally classified into ubiquitin adenylate analogs, natural compounds, disulfides, nitro-based compounds, and adenosine sulfamates (Fig. 8). Of these, the adenosine sulfamate TAK-243 (MLN7243) is the only drug that has been advanced to phase 1 clinical trials.
UBA1 inhibitors and their pharmacologic properties

Chemical structures of UBA1 inhibitors. Classes of UBA1 inhibitors include adenosine sulfamates (e.g., TAK-243), nitro-based compounds (e.g., PYR-41, PYZD-4409, and JS-K), natural compounds (e.g., panepophenanthrin, himeic acid A, hyrtioreticulins A/B, and largazole), and ubiquitin adenylate analogs (e.g., adenosyl-phospho-ubiquitinol, Ub-AMSN, and Ub-AVSN), and disulfides (e.g., NSC624206).
a. TAK-243
TAK-243 (MLN7243) is a first-in-class, mechanism-based UBA1 inhibitor (Hyer et al., 2018). It was identified through a screen of more than 700 chemical candidates using an in vitro transthiolation assay that assesses ubiquitin transfer to the ubiquitin-specific E2 enzyme UBCH10 (also known as UBE2C). It is a second-in-class adenosine sulfamate E1 inhibitor developed after the NAE inhibitor pevonedistat, the prototype of this structural class (Soucy et al., 2009a). TAK-243 has been reported to be partially selective for UBA1, as it displays activity against UBA6 and NAE but severalfold higher IC50, as assessed by transthiolation assays (Ciavarri and Langston, 2017; Hyer et al., 2018). However, it displays minimal or no activity against kinases and carbonic anhydrases. It forms a covalent adduct with ubiquitin (through a covalent linkage between the sulfamate nitrogen of TAK-243 and glycine 76 of ubiquitin), which tightly binds to the nucleotide-binding site and inhibits UBA1 by substrate-assisted inhibition (Misra et al., 2017; Hyer et al., 2018).
In cells of different origins, TAK-243 reduces the abundance of mono- and polyubiquitylated proteins and induces the accumulation of ubiquitin-regulated short-lived proteins, including p53, c-Jun, c-Myc, MCL1, and X-linked inhibitor of apoptosis protein (XIAP) (Hyer et al., 2018; Barghout et al., 2019; Best et al., 2019a; Zhuang et al., 2019). These changes are associated with a multitude of ubiquitylation-dependent biologic effects that lead to cell death, including cell cycle arrest, proteotoxic stress, and DNA damage stress (Hyer et al., 2018). In this respect, TAK-243 induces cell cycle arrest at the G2/M phase or both G1 and G2/M phases at high concentrations. Notably, the cell cycle phenotype observed after TAK-243 treatment resembles that observed after proteasome inhibitors and differs from NAE inhibitors, as TAK-243 exhibits no induction of re-replication phenotype (Hyer et al., 2018) or a slight induction of this phenotype at high concentrations (Best et al., 2019a). Given the role of UPS in the degradation of misfolded and unfolded proteins in the ER, TAK-243 induces the accumulation of such proteins, leading to sustained ER stress associated with morphologic ER changes, a terminal UPR, and apoptosis (Wang and Kaufman, 2014; Hyer et al., 2018; Best et al., 2019a; Zhuang et al., 2019). Moreover, ubiquitylation plays a pivotal role in several DNA repair pathways, such as translesion synthesis, Fanconi anemia pathway, double-strand break repair, and nucleotide-excision repair (Milhollen et al., 2015); thus, TAK-243 impairs DNA repair and induces DNA damage stress under irradiated and unirradiated conditions (Hyer et al., 2018; Barghout et al., 2019). Of note, genomic and proteomic analysis of TAK-243 effects demonstrates a pleiotropic impact on diverse signaling pathways, including proliferative signaling, inflammatory responses, apoptosis, hypoxia, oxidative stress, and oxidative phosphorylation (Zhuang et al., 2019).
As expected with these pleiotropic effects, TAK-243 demonstrates a broad antitumor activity in several malignancies, including solid tumors (Hyer et al., 2018), AML (Hyer et al., 2018; Barghout et al., 2019), myeloma (Hyer et al., 2018; Zhuang et al., 2019), B-cell lymphoma (Best et al., 2019a), cutaneous squamous cell carcinoma (McHugh et al., 2018), chronic lymphocytic leukemia (Best et al., 2019b), mantle cell lymphoma (Best et al., 2019b), and small-cell lung cancer (Majeed et al., 2019). Most tested cell lines of these malignancies are sensitive to TAK-243, with low nanomolar IC50 values in vitro and robust responses in vivo, and only a few of them display intrinsic resistance (Hyer et al., 2018; Majeed et al., 2019; Zhuang et al., 2019). In contrast, data with normal human fibroblasts and in vivo studies show a lesser effect on normal versus cancer cells, suggesting TAK-243 possesses a therapeutic window in several malignancies (Hyer et al., 2018; Barghout et al., 2019). Although seemingly narrow, this therapeutic window may lead to beneficial therapeutic outcomes in selected malignancies with dose optimization and careful patient selection, as is the case with proteasome inhibitors (Ruschak et al., 2011; Hyer et al., 2018). Based on preclinical data including efficacy and toxicology studies, TAK-243 has been advanced to phase 1 clinical trials in advanced solid tumors and AML (Table 7).
Clinical trials of TAS4464, TAK-243, and TAK-981
TAK-243 also exhibits additive and/or synergistic effects with radiotherapy and DNA damaging agents in preclinical models of breast cancer and lung cancer (Milhollen et al., 2015; Hyer et al., 2018; Majeed et al., 2019), as well as histone deacetylase (HDAC) inhibitors in MM (Zhuang et al., 2019). Because of its different mechanism of action and biologic effects that are distinct from proteasome inhibitors, TAK-243 can overcome specific forms of resistance to these drugs (McHugh et al., 2018; Zhuang et al., 2019). Interestingly, B-cell lymphoma cells with Myc overexpression are more sensitive to TAK-243, suggesting higher dependence on ubiquitylation in such cells, resembling the effects observed with SUMOylation inhibitors (Kessler et al., 2012; Best et al., 2019a).
Although many aspects of TAK-234 action in several malignancies have been studied, the determinants of sensitivity and resistance remain largely unknown (Barghout and Schimmer, 2018). With the drug being tested in clinical trials, it is important to identify patient cohorts that are likely to respond and to maximize the efficacy and minimize unnecessary toxicity from the drug. Such endeavors require a deeper understanding of the drug action at the cellular and molecular levels to identify transporters, metabolic factors, signaling molecules, and/or (epi)genetic mechanisms that may influence response. In this context, unbiased large-scale genetic and chemical screens can serve as a rapid and adaptable tool to achieve this goal and provide, with appropriate validation, useful insights into TAK-243 response (Barghout et al., 2018). Of note, correlating TAK-243 sensitivity with UBA1 expression levels in cell lines has been attempted; however, the data are conflicting, and the number of cell lines tested is not large enough for conclusive findings (Hyer et al., 2018; McHugh et al., 2018). Equally important are the development and optimization of biomarkers to monitor drug response in these clinical trials.
b. Ubiquitin Adenylate Analogs
Adenosyl-phospho- ubiquitinol (APU) was synthesized in 1990 and is reportedly the first experimental inhibitor of UBA1 (Wilkinson et al., 1990). It is a stable analog of the ubiquitin∼AMP intermediate, formed during the adenylation step of ubiquitin activation, with a methylene group replacing the carbonyl of the C-terminal glycine residue (Brownell et al., 2010). Biochemically, it inhibits ubiquitin activation in a competitive manner with ATP and a noncompetitive manner with ubiquitin and tightly binds to the nucleotide-binding site of the free rather than ubiquitin-bound form. APU also inhibits protein degradation in cell-free assays; however, its structural features are not suggestive of appreciable cell permeability (Brownell et al., 2010). As such, the biochemical properties of this inhibitor, particularly affinity and ATP competitiveness, are not sufficiently favorable to produce a physiologically relevant UBA1 inhibition (Brownell et al., 2010). However, APU or related analogs could potentially serve as chemical probes to interrogate ubiquitin biology. In this respect, ubiquitin (Ub)-AMSN and Ub-AVSN have been developed as semisynthetic mechanism-based UBA1 inhibitors by chemical ligation of truncated ubiquitin to a synthetic cysteylglycylglycyl tripeptide with a C-terminal 5′-sulfonyladenosine and an N-terminal cysteine (Lu et al., 2010). Although they are structurally related, Ub-AMSN and Ub-AVSN possess distinct features that allow them to inhibit different steps of ubiquitin activation. Specifically, Ub-AMSN inhibits the adenylation step because of its nonhydrolyzable sulfamide, which mimics the phosphate group in the ubiquitin∼AMP intermediate. On the other hand, Ub-AVSN inhibits the ubiquitin thioester formation step because of its electrophilic vinyl sulfonamide, which traps the nucleophilic cysteine of UBA1. In cell-free assays, Ub-AMSN and Ub-AVSN selectively inhibit Uba1∼ubiquitin thioester formation in a concentration-dependent manner. In addition, Ub-AVSN forms a covalent adduct with Uba1 that is resistant to hydrolysis by dithiothreitol as opposed to the native Uba1∼ubiquitin thioester. Importantly, these inhibitors have been exploited to trap ubiquitin activation intermediates to gain insights into the biochemical and structural basis of ubiquitin activation (Olsen et al., 2010).
c. Natural Compounds
This class comprises panepophenanthrin, himeic acid A, hyrtioreticulins A/B, and largazole. The UBA1 inhibitory activities of these compounds were not extensively characterized or verified in other studies. In addition, all of them except largazole were not tested in cellular contexts. Panepophenanthrin is a bioactive compound derived from the fermented broth of the mushroom strain Panus rudis Fr. IFO 8994. It is reported to possess UBA1 inhibitory activity in a cell-free thioester formation assay (Sekizawa et al., 2002). Himeic acid A is another natural compound isolated from the marine fungus Aspergillus sp. In analogy to panepophenanthrin, it reportedly inhibits cell-free thioester formation at a high micromolar concentration; however, further characterization of its UBA1 inhibitory activity has not been pursued in independent studies (Tsukamoto et al., 2005). Hyrtioreticulins A and B are indole alkaloids isolated from the marine sponge Hyrtios reticulatus. When tested in vitro, they showed a more potent UBA1 inhibitory activity compared with panepophenanthrin and himeic acid A; however, they displayed no activity against HeLa cells (Yamanokuchi et al., 2012). Based on these reports, himeic acid A is predicted to inhibit UBA1 at the ubiquitin adenylation step, whereas hyrtioreticulins A and B inhibit UBA1 at the thioester formation step (Yamanokuchi et al., 2012).
Largazole is a macrocyclic compound isolated from the Floridian marine cyanobacterium Symploca sp. It demonstrates a potent and preferential antiproliferative activity against breast, osteosarcoma, colon, and neuroblastoma cancer cell lines, with relatively less activity against nontransformed cells (Taori et al., 2008). Although the mechanism of this antiproliferative action has been attributed to HDAC inhibition in several studies (Liu et al., 2010; Ghosh et al., 2012; Hong and Luesch, 2012; Law et al., 2013; Poli et al., 2017), a study has reported that largazole and select analogs also possess UBA1-inhibitory activity (Ungermannova et al., 2012b). Specifically, largazole was identified as a top hit through a cell-based screen for compounds that stabilize GFP-labeled p27 in a lung epithelial cell line with potent activity in the nanomolar range. Further investigation using cell-free ubiquitylation assays demonstrated that largazole and ketone/ester analogs inhibit UBA1 at the adenylation step, resulting in decreased ubiquitin charging of ubiquitin-conjugating enzyme E2 R1 (CDC34) and reduced ubiquitylation of p27 with no effect on SUMOylation. Nonetheless, the higher micromolar IC50 required for in vitro UBA1 inhibition as opposed to the nanomolar cellular activity suggests that largazole-induced cytotoxicity is mediated through HDAC inhibition and subsequent cell cycle arrest rather than UBA1 inhibition (Ungermannova et al., 2012b; Wu et al., 2013).
d. Nitropyrazones and Nitropiperazines
Nitro-based UBA1 inhibitors include pyrazones (PYR-41 and PYZD-4409) and piperazines (JS-K). They share a nitro group that is susceptible to displacement by nucleophilic attack and covalent interaction with the catalytic cysteine of the CCD, resulting in irreversible inhibition (Yang et al., 2007; Xu et al., 2010).
PYR-41, the prototype of this class, was reported in 2007 as the first cell-permeable inhibitor of UBA1. It was discovered through a secondary screen of E3 ligase inhibitors for hits with reactivity to UBA1’s catalytic cysteine (Yang et al., 2007). In vitro, PYR-41 demonstrates activity against the E1 enzyme in addition to some activity against homologous to the E6-AP carboxyl terminus (HECT) E3s. Biochemically, it inhibits thioester formation with free UBA1 without impact on ubiquitin transfer to E2s from ubiquitin-charged UBA1. It also inhibits degradative and nondegradative forms of ubiquitylation and cytokine-induced activation of NF-κB. p53 is an important transcription factor and tumor suppressor protein whose stability is prone to regulation by degradative ubiquitylation (Vucic et al., 2011). PYR-41 induces the stabilization of p53 with a subsequent increase in transcriptional activity and preferential cytotoxicity to transformed cells with wild-type p53. Cellular effects of PYR-41 include reductions in ubiquitin charging of UBA1 and the abundance of ubiquitylated proteins. Interestingly, increases in ubiquitylated proteins with high molecular mass (>250 kDa) as well as total SUMOylation are observed after PYR-41 treatment with no effect on neddylation (Yang et al., 2007; Kapuria et al., 2011). This paradoxical accumulation of ubiquitylated proteins is ascribed to the protein cross-linking activity of PYR-41 resulting in covalent interaction with several DUBs (e.g., USP5), kinases [breakpoint cluster region protein-Abelson tyrosine-protein kinase (BCR-ABL) and Janus kinase 2 (JAK2)], and possibly other proteins, forming adducts with high molecular weight. Therefore, it appears that PYR-41 is a promiscuous inhibitor with multiple targets, and its antineoplastic effects are likely due to the combined inhibition of UBA1 as well as other off-target effects, particularly after prolonged treatment (Kapuria et al., 2011).
PYZD-4409 is a structural analog of PYR-41 that was discovered through a focused screen of a pyrazolidine pharmacophore-based chemical library (Xu et al., 2010). In addition to cancer cell lines, it has been tested in primary cancer cells and a mouse model of murine leukemia. It is cytotoxic to cell lines of hematologic and solid malignancies, with MM cell lines being particularly sensitive. It also displays preferential cytotoxicity to AML versus normal hematopoietic primary cells (Xu et al., 2010). Mechanistically, it stabilizes the short-lived proteins p53 and cyclin D3 and induces ER stress resulting in cell death. Nonetheless, the structural similarities to PYR-41 suggest that PYZD-4409 may also induce cytotoxicity through off-target effects besides UBA1 inhibition.
JS-K is a nitric oxide (NO•)-releasing prodrug that is metabolically activated by glutathione S-transferases in the presence of glutathione. Therefore, it is preferentially cytotoxic to glutathione S-transferase–overexpressing malignancies, such as MM (Kiziltepe et al., 2007). In this context, it is cytotoxic to MM cell lines and primary cells, with a lesser effect on normal hematopoietic cells. It has also been tested in a mouse xenograft model of MM without significant toxicity. Mechanistically, JS-K induces apoptosis and activates DNA damage response and c-Jun N-terminal kinase (JNK) pathways. The apoptotic effects of JS-K in preclinical models of MM have been attributed to NO• generation without implicating UBA1 (Kiziltepe et al., 2007). A later study, however, showed that JS-K induces S-nitrosylation of UBA1 in immortalized human cells, resulting in UBA1 inhibition along with other effects similar to those reported with nitropyrazones in transformed cells (Kitagaki et al., 2009). The effects of JS-K on ubiquitylation and p53 levels have been further corroborated in prostate cancer cells (Tan et al., 2017). Therefore, it appears that the nitro group of JS-K serves as a source of NO• that causes inhibition of multiple targets and is likely a leaving group at which covalent adduct formation with UBA1 occurs in a similar manner to nitropyrazones.
e. Disulfides
As with largazole, the disulfide compound NSC624206 was discovered through a cell-based screen of a National Cancer Institute library for compounds that prevent degradation of p27 (Ungermannova et al., 2012a). As the inhibitory activity is lost with an analog lacking the disulfide group, this group is thought to be essential for activity (Ungermannova et al., 2012a). In a similar manner to nitropyrazones, NSC624206 inhibits UBA1∼ubiquitin thioester formation, with little or no effect on the adenylation step. It is speculated that the disulfide group of NSC624206 may undergo a nucleophilic attack by the -SH group of the catalytic cysteine in a thiol-disulfide exchange reaction, forming a covalent adduct coupled with UBA1 inhibition. NSC624206 is a cell-permeable inhibitor, as it stabilizes p27 in lung epithelial and liver cancer cells (Ungermannova et al., 2012a).
C. Small Ubiquitin-Like Modifier–Activating Enzyme
SUMOylation is a post-translational modification that regulates several cellular processes, including nuclear transport, gene expression, chromosome segregation, quality control and proteasomal degradation of proteins, DNA damage response, and cell cycle progression (Gareau and Lima, 2010; Eifler and Vertegaal, 2015). SAE is the E1 enzyme that catalyzes SUMO activation, which is followed by SUMO conjugation to a variety of cellular proteins (Schulman and Harper, 2009). Structurally, SAE is a canonical heterodimeric enzyme composed of two subunits, SAE1 and SAE2 (UBA2), with several domains analogous to UBA1 and NAE (Fig. 2). Of the four SUMO isoforms known so far, SUMO1–3 are activated by SAE; however, it is not known whether SUMO4 is conjugated to cellular proteins (Gareau and Lima, 2010). Compared with the ubiquitylation cascade that involves tens of E2 enzymes, hundreds of E3 ligases, and over 100 DUBs, the SUMOylation cascade is much less complex, as it involves a single E2 enzyme, UBC9, as well as few E3 ligases and SUMO-specific proteases (SENPs) (Gareau and Lima, 2010). Among the substrates that are regulated by SUMOylation are RanGAP1, IκBα, PCNA, p53, c-Jun, c-Myc, and promyelocytic leukemia (PML) nuclear bodies (Seeler and Dejean, 2003; Wilkinson and Henley, 2010). Of these, RanGAP1 SUMOylation is commonly used in SUMOylation assays to identify and characterize SUMOylation modulators (Sarge and Park-Sarge, 2009).
1. Role of Small Ubiquitin-Like Modifier–Activating Enzyme in Cancer
Given its critical role in many cellular processes, SUMOylation is tightly coordinated to maintain normal cellular functions (Seeler and Dejean, 2017). In cancer, however, SUMOylation may be dysregulated, and mostly the activation of the SUMOylation pathway is associated with an adverse outcome. The expression, activity, and stability of SAE1/2 are among the key regulators of SUMOylation, and either SAE1, SAE2, or both may be altered in several malignancies. For example, primary mantle cell lymphoma samples and cell lines have been reported to display higher SAE1/2 expression as well as a higher abundance of global SUMOylated proteins compared with normal counterpart cells (Hanel et al., 2019). In addition, gastric and small-cell lung cancer show a high expression of SAE2, which promotes tumor progression and correlates with a poor prognosis (Shao et al., 2014; Liu et al., 2015). SAE1 is upregulated in glioblastoma tumors compared with normal brain samples (Bellail et al., 2014) and is a part of a 75-gene signature associated with lymph node metastasis in lung adenocarcinomas (Inamura et al., 2007). SAE expression is also reported to be important for Myc-driven tumor progression in solid and hematologic malignancies. For example, elevated SAE1/2 expression has been reported in Myc-overexpressing lymphoma, and as assessed by genetic and pharmacologic approaches, SAE is functionally important for Myc-driven tumorigenesis (Hoellein et al., 2014). Consistent with these findings, low SAE1/2 expression in Myc-overexpressing breast cancers has been associated with favorable survival outcomes in patients (Kessler et al., 2012). In these contexts, targeting SAE is expected to be synthetically lethal with hyperactive Myc, demonstrating SUMOylation is crucial to tolerate aberrant Myc signaling and maintain tumor progression, possibly through alleviating mitotic stress. Similarly, higher dependence on SUMOylation enzymes, including SAE, has been observed in breast cancer cells with Notch1 activation (Licciardello et al., 2015). Moreover, high SAE1 expression is associated with unfavorable prognosis in liver, renal, and thyroid cancers, with similar prognostic outcomes with SAE2 in liver and renal cancers (Uhlen et al., 2017). In preclinical models of colorectal cancer, SAE has been found to play a critical role in cancer stem cell self-renewal and maintenance (Du et al., 2016). The roles of other components of the SUMO pathway in cancer have been reviewed elsewhere (Eifler and Vertegaal, 2015; Seeler and Dejean, 2017). These data provide compelling evidence that SUMOylation may be hyperactive in several cancers compared with normal cells to support cellular stresses associated with the malignant phenotype. This form of nononcogene addiction can be therapeutically targeted at different levels of the SUMOylation machinery including SAE that initiates the cascade (Luo et al., 2009).
2. Role of SUMOylation in Differentiation Therapy of Acute Promyelocytic Leukemia
Acute promyelocytic leukemia (APL) is a subtype of AML characterized by a chromosomal translocation [t(15;17)(q22;q12)] involving the PML gene and the retinoic acid receptor α (RARA) gene, producing the fusion oncoprotein PML/RARA (de Thé et al., 2017). Biologically, PML/RARA contributes to APL pathogenesis by the deregulation of RARα-mediated transcriptional control and disruption of PML nuclear bodies (NBs), which are spherical, stress-sensitive nuclear structures involved in regulating many cellular functions and are mainly organized by PML protein (Bernardi and Pandolfi, 2007; de Thé et al., 2017). These effects lead to resistance to apoptosis, differentiation block, and enhancement of self-renewal of myeloid progenitors (de Thé and Chen, 2010). This oncoprotein is the main oncogenic driver in several types of APL, providing a unique opportunity for targeted therapy aimed at inducing differentiation. Currently, differentiation therapy comprising ATO plus ATRA is a frontline therapeutic option that has transformed the outlook of APL in many patients, with cure rates up to 90% (Lallemand-Breitenbach et al., 2012; Lo-Coco et al., 2013; Sanz et al., 2019). Mechanistically, ATO and ATRA bind to PML/RARA to induce its degradation, resulting in the reformation of PML NBs, p53 activation, loss of self-renewal, and differentiation (Jeanne et al., 2010; Zhang et al., 2010; Ablain et al., 2014; de Thé et al., 2017). ATO-induced degradation of PML/RARA is dependent on both SUMOylation and ubiquitylation. Specifically, ATO targets the PML moiety of the fusion oncoprotein, inducing oxidative and conformational changes that enhance recruitment and tight binding of UBC9, leading to hyperSUMOylation of PML. This form of PML actively recruits the SUMO-dependent ubiquitin E3 ligase RING finger protein 4 (RNF4) followed by K48-linked polyubiquitylation and proteasomal degradation (Lallemand-Breitenbach et al., 2008; Tatham et al., 2008). Of note, genetic and chemical inhibition of SAE in colon cancer cells has been reported to reduce SUMOylation and cause disruption of PML NB organization and dissociation of the PML-interacting protein death-associated protein 6 (DAXX), leading to antitumor effects (He et al., 2015, 2017). Therefore, it appears that the induction of either hyper- or hypoSUMOylation can be leveraged in different contexts to obtain beneficial therapeutic outcomes.
3. Antitumor Immunomodulatory Roles of SUMOylation
Interferons (IFNs) are small secreted proteins that play a crucial role in innate and adaptive immunity. They include three classes: type I (IFN-α, IFN-β, IFN-ε, IFN-κ, IFN-ω), type II (IFN-γ), and type III IFNs (IFN-λ, interleukin-28 [IL-28], and interleukin-29 [IL-29]), which initiate three distinct signaling cascades (Schneider et al., 2014). SUMOylation has been reported to regulate type I IFN signaling (Decque et al., 2016; Crowl and Stetson, 2018). Type I IFN signaling is initiated by binding of IFNs to a heterodimeric cell surface receptor complex composed of IFN-α receptor (IFNAR) 1 and IFNAR2, leading to the activation of Janus kinase-signal transducer and activator of transcription protein (JAK-STAT) signaling and formation of a heterotrimeric complex termed IFN-stimulated gene factor 3 (ISGF3), which is composed of STAT1, STAT2, and IFN-regulatory factor 9 (IRF9) (Schneider et al., 2014). ISGF3 undergoes nuclear translocation, in which it activates the expression of ISGs by modulating IFN-stimulated response elements. Recently, SUMOylation has been reported to negatively regulate the innate inflammatory response by silencing IFN-β expression and attenuating the priming of inflammatory cytokine production by type I IFNs (Decque et al., 2016). Accordingly, inhibiting SUMOylation by UBC9 knockdown enhanced the innate immune responses. Loss of SUMOylation after SAE or UBC9 knockout was also found to induce a type I IFN response, however, through a noncanonical mechanism independent of known IFN-regulating pathways (Crowl and Stetson, 2018). Among the inducers of type I IFN signaling is the cGAS–stimulator of interferon genes protein (cGAS-STING) pathway, which acts as a sensor of aberrant cytosolic DNA and activates the expression of type I IFNs (Schneider et al., 2014; Chen et al., 2016b). SUMOylation has been reported to suppress the activity of cGAS, and this suppression is reversed by the SUMO-specific protease SENP7, which catalyzes deSUMOylation of cGAS, triggering type I IFN-dependent innate immune responses (Cui et al., 2017). The importance of SUMOylation is not confined to regulating innate immunity, as it also plays a key role in adaptive immune responses. In this context, SENP3 has been found to maintain the stability and function of regulatory T cells through deSUMOylation of BTB and CNC homolog 2 (BACH2), a regulatory T-cell–specific transcription factor, facilitating its nuclear retention and function (Yu et al., 2018b). Therefore, it appears that SUMOylation restrains a subset of innate and adaptive immune responses; thus, targeting the SUMOylation machinery including SAE can serve, in specific contexts, as a potential therapeutic strategy to obtain useful antitumor and antimicrobial immune effects.
4. Small Ubiquitin-Like Modifier–Activating Enzyme Inhibitors
The structural basis of SUMO activation by human SAE and interaction with UBC9 has been elucidated since 2005, and most SAE inhibitors reported later are natural compounds whose SAE-inhibiting activity was discovered without regard to the crystal structure of SAE (Lois and Lima, 2005; Wang et al., 2007; Olsen et al., 2010). These endeavors started with Gam1, an adenoviral protein that interacts with SAE and enhances SAE-UBC9 proteasomal degradation (Boggio et al., 2004). This was followed by the discovery of a series of small-molecule SAE inhibitors with diverse structures until the first mechanism-based selective SAE inhibitor, ML-792, was developed more than a decade later (He et al., 2017). SAE inhibitors reported so far can be classified into SUMO adenylate analogs and SUMO-mimicking peptides, natural compounds, urea-based derivatives, oxabicycloheptadienes, and adenosine sulfamates (Fig. 9; Table 8). A first-in-class SAE activator, N106, was reported in 2015 as a potential modulator of heart failure (Kho et al., 2015).
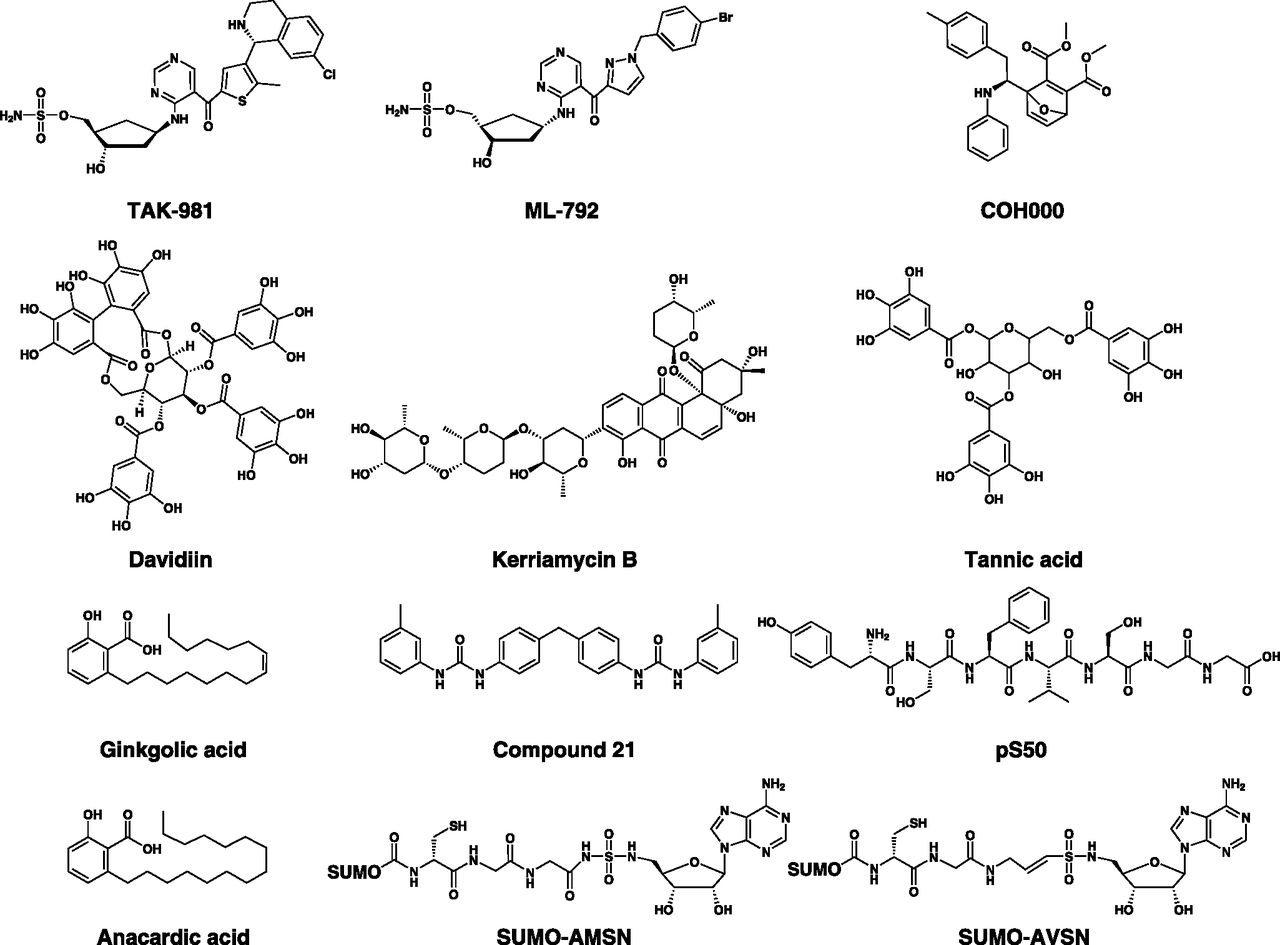
Chemical structures of SAE inhibitors. SAE inhibitors can be classified into adenosine sulfamates (e.g., TAK-981 and ML-792), oxabicycloheptadienes (e.g., COH000), natural compounds (e.g., ginkgolic acid, anacardic acid, davidiin, tannic acid, and kerriamycin B), SUMO adenylate analogs and SUMO-mimicking peptides (e.g., SUMO-AMSN, SUMO-AVSN, and pS50), and urea-based inhibitors (e.g., compound 21).
SAE inhibitors and their pharmacologic properties
a. ML-792 and TAK-981
ML-792 and TAK-981 are adenosine sulfamate SAE inhibitors, and they share similar profiles but differ in terms of their in vivo properties (Huszar, 2019). In pursuit of developing SAE inhibitors that are analogous to pevonedistat, ML-792 was discovered through a drug development program that used a pyrazole-carbonylpyrimidine–based scaffold (He et al., 2017). In analogy with other adenosine sulfamates, ML-792 acts by substrate-assisted inhibition and forms a covalent adduct with SUMO. In cell-free and cell-based assays, ML-792 demonstrates a potent and selective SAE-inhibitory activity, with little or no activity against related E1s or other ATP-dependent enzymes. ML-792 is also cytotoxic to colorectal, breast, and melanoma cancer cell lines, with Myc-overexpressing cell lines being particularly sensitive. Despite the importance of the SUMOylation pathway in regulating gene expression, ML-792 only induces modest transcriptional and proteomic changes, with no impact on splicing in different cell lines. As opposed to TAK-243, ML-792 has little or no effect on DNA repair, suggesting that DNA damage plays no role in the observed cytotoxicity (He et al., 2017; Hyer et al., 2018). Instead, ML-792 induces mitotic disruption (multinucleation, cell enlargement, and endoreduplication), senescence, and chromosome segregation defects (decrease in the anaphase/telophase of mitosis and formation of DNA bridges). Cytotoxicity and cell cycle changes are evident only in actively cycling cells and can be rescued by the overexpression of ML-792–resistant UBA2 S95N M97T mutant, suggesting that mitotic disruption is functionally important for ML-792 cytotoxicity (He et al., 2017).
Despite the excellent profile of ML-792 ex vivo, it shows poor stability in vivo, making it suitable only as a chemical probe for interrogating SUMOylation biology (He et al., 2017; Huszar, 2019). Specifically, high doses are required to maintain in vivo responses, as ML-792-SUMO adducts are rapidly released from SAE and degraded after in vivo administration (Huszar, 2019). Conversely, TAK-981—developed thereafter to overcome this limitation—forms SUMO adducts that are stable and causes sustained inhibition of SUMOylation after administration of small doses of the drug (Huszar, 2019). TAK-981 selectively inhibits SAE∼SUMO and UBC9∼SUMO thioester formation as well as global SUMOylation, with IC50 values in the low nanomolar range, and without significant effects on neddylation or ubiquitylation (Berger et al., 2019; Hanel et al., 2019; Huszar, 2019). In analogy with ML-792, TAK-981 treatment in mantle cell lymphoma cells induces cell cycle arrest and mitotic disruption, as evidenced by the accumulation of polyploid and aneuploid cells (Hanel et al., 2019).
Interestingly, prolonged effects of TAK-981 in vivo have allowed the observation of several immune responses that are associated with the antitumor effects of the drug, particularly against tumor models with intrinsic resistance to TAK-981 in vitro (Huszar, 2019). The molecular basis of these responses partly derives from the role of SUMOylation in regulating type I IFN signaling (Decque et al., 2016; Crowl and Stetson, 2018). This is consistent with the reduced antitumor effects of TAK-981 with prior administration of anti-IFN-α/β receptor 1 antibody (Berger et al., 2019). The effects of TAK-981 mediated by type I IFN signaling are pleiotropic and implicate several effectors of innate and adaptive arms of immunity (Berger et al., 2019; Khattar et al., 2019; Lam et al., 2019; Nakamura et al., 2019). Direct microinjection of TAK-981 into tumors is also associated with both local and abscopal immune responses, which are consistent with those observed after systemic dosing (Hatton et al., 2019). In addition, TAK-981 exerts synergistic antitumor effects when administered in combination with other immunotherapies, such as rituximab, an anti-CD20 monoclonal antibody, and immune checkpoint inhibitors (Huszar, 2019; Nakamura et al., 2019). Based on these preclinical data, TAK-981 is currently evaluated in phase 1 clinical trials for patients with metastatic solid tumors and lymphomas (Table 7).
b. COH000
Most SAE inhibitors reported so far target either the nucleotide-binding site or the catalytic cysteine of the enzyme. Through an HTS of a library of ∼300,000 small molecules for compounds that inhibit RanGAP1 SUMOylation, Li et al. (2019) discovered COH000, an inhibitor that targets a novel, highly conserved allosteric site in the SAE. COH000 is an oxabicycloheptadiene-based compound that selectively and potently inhibits SUMOylation in cell-free and cell-based assays with no effects on ubiquitylation. In this respect, COH000 inhibits SAE∼SUMO thioester formation in a noncompetitive manner with neither ATP nor SUMO, and this is associated with a very slow off-rate, suggesting irreversible inhibition via covalent binding to the SAE (Li et al., 2019). Mass spectrometry data demonstrate covalent adduct formation after the binding of COH000 to a hidden cysteine residue (Cys30), without modifying the other SAE’s non–disulfide-bonded cysteines, including the catalytic cysteine (Cys173). Chemically, a Michael addition reaction is involved in the covalent bond formation between the Cys30 and an electrophilic center within the 7-oxabicyclohept-2-ene group of COH000. However, the selectivity observed with COH000 implies that noncovalent interactions precede covalent adduct formation.
Cys30 is located in the AAD at a site proximal to the active adenylation site; however, as per published SAE crystal structures, it is among the inaccessible cysteines, suggesting that COH000 binding is associated with a set of conformational changes that expose this residue (Li et al., 2019). This has been confirmed by a detailed structural analysis of SAE in complex with COH000, in which the drug was shown to target a novel cryptic allosteric site located specifically between the AAD and IAD (Lv et al., 2018b). Although the associated conformational changes involving AAD, IAD, and CCD are an extension of those observed during the normal catalytic mechanism of SAE, they are differently orchestrated in a way that leads ultimately to locking the enzyme in an inactive conformation. Interestingly, COH000 induces ubiquitylation and degradation of SAE2, suggesting that this inactive conformation is susceptible to proteasomal degradation.
The anti-SAE activity of COH000 is associated with reduced proliferation, apoptosis, increased miR-34b expression, and decreased c-Myc expression in lymphoma and colorectal cancer cell lines and primary cells as well as reduced tumor growth in an esterase-deficient mouse xenograft model of colorectal cancer (Li et al., 2019). Since COH000 contains two ester bonds, it is susceptible to hydrolysis by plasma esterases; however, it is unclear whether cleavage of these bonds will compromise the efficacy of the drug in vivo.
In addition to the unique mechanism of action and potent biologic activities, COH000 has enabled a chemically assisted discovery of a novel cryptic pocket that was not previously reported and provided new insights into the structural biology of SAE and possibly other E1 enzymes (Li et al., 2019).
c. Small Ubiquitin-Like Modifier Adenylate Analogs and Small Ubiquitin-Like Modifier–Mimicking Peptides
SUMO-AMSN and SUMO-AVSN are semisynthetic mechanism-based SAE inhibitors that are structurally and biochemically analogous to Ub-AMSN and Ub-AVSN (Lu et al., 2010). They were similarly exploited as chemical probes to interrogate the structural basis and biochemistry of SUMO activation in cell-free assays (Lu et al., 2010; Olsen et al., 2010).
Based on the SUMO C-terminal sequences that can be activated by SAE, as profiled by phage display technology, Zhao et al. (2014) developed pS50 and pS90, two other mechanism-based SAE inhibitors. pS50 and pS90 are heptameric SUMO-mimicking peptides that can be activated by SAE, conjugated to UBC9, and transferred to protein substrates at a higher rate compared with the wild-type SUMO, but with little or no functionality. Therefore, these peptides competitively inhibit SUMOylation at every step of the cascade, starting with the SAE-catalyzed activation step (Zhao et al., 2014).
d. Natural Small Ubiquitin-Like Modifier–Activating Enzyme Inhibitors
This class of SAE inhibitors includes ginkgolic acid, anacardic acid, davidiin, tannic acid, and kerriamycin B, which have diverse chemical structures and molecular mechanisms of SAE inhibition. A cell-based screen of herbal extracts and nutraceuticals for compounds that inhibit SUMOylation identified ginkgolic acid, anacardic acid, and davidiin as SAE inhibitors (Fukuda et al., 2009a; Takemoto et al., 2014). Ginkgolic acid is an alkylphenol derivative and one of the active ingredients of ginkgo (Gingko biloba) leaf extract, and anacardic acid is a structurally related analog found in cashew nut (Anacardium occidentale) shells and immature ginkgo seeds (Fukuda et al., 2009a; Takemoto et al., 2014). Both compounds reduce cell-free protein-specific and cell-based global SUMOylation in a concentration- and time-dependent manner, with little or no effect on ubiquitin conjugates. Structure-activity relationship (SAR) and molecular docking studies suggest that the carboxylic acid and long aliphatic side chains are essential for activity and that these compounds potentially target the AAD of the enzyme (Hamdoun and Efferth, 2017). Although known to interact with multiple targets, inducing numerous molecular activities, it appears that the effects of ginkgolic and anacardic acid on the SAE are achievable at low concentrations (Fukuda et al., 2009a). A subset of these other targets may themselves be subject to regulation by SUMOylation. For instance, the NF-κB pathway, regulated partly by SUMOylation of NEMO and IκBα (Seeler and Dejean, 2017), is modulated by anacardic acid, resulting in antitumor effects in cancer cell lines of solid and hematologic malignancies (Sung et al., 2008). Inhibition of NEMO SUMOylation by ginkgolic acid in cell-free assays has also been reported (Hamdoun and Efferth, 2017). Therefore, the inhibition of SAE and subsequent effects on global SUMOylation may mediate the pleiotropic effects of these two compounds on other targets.
Ginkgolic and anacardic acids have also been reported to display antitumor effects in cancers addicted to oncogenes that promote a higher reliance on SUMOylation. In this respect, anacardic acid selectively induces growth arrest, apoptosis, and aneuploidy in B-cell lymphoma cell lines with Myc activation (Hoellein et al., 2014). Similarly, ginkgolic acid reduces proliferation and induces apoptosis in engineered and patient-derived breast cancer cell lines with Notch1 activation, without cytotoxicity in isogenic cell lines with normal Notch1 activity (Licciardello et al., 2015). Consistent with these findings, ginkgolic acid reduces migration in wound-healing assays of MCF7 and MDA-MB 231 breast cancer cell lines without inducing cytotoxicity (Hamdoun and Efferth, 2017).
Inhibition of SUMOylation by anacardic acid has been reported to overcome the resistance of AML to conventional therapies. In this context, treatment with daunorubicin, cytarabine, or etoposide induced ROS-dependent deSUMOylation that was associated with transcriptional alterations and apoptosis in chemosensitive AML cells (Bossis et al., 2014). Intrinsically chemoresistant AML cells, however, failed to undergo deSUMOylation after chemotherapy. Treatment with anacardic acid in vitro and in vivo induced deSUMOylation and circumvented AML chemoresistance. Most AML subtypes, except for APL, are not responsive to differentiation therapy with ATRA. Baik et al. (2018) reported that this was partly due to the SUMOylation that repressed ATRA-induced expression of genes involved in myeloid differentiation, proliferation, and apoptosis. Suppression of SUMOylation by anacardic acid and other SUMOylation inhibitors sensitized non-APL AML cells to ATRA, suggesting a potential utility of differentiation therapy in other subtypes of AML in addition to APL (Baik et al., 2018). ATRA is mostly used in APL in combination with ATO that acts partly by inducing hyperSUMOylation and subsequent degradation of PML/RARA. Therefore, SUMOylation inhibitors may be beneficial when combined with ATRA therapy in non-APL AML, although in theory they may cause adverse outcomes when combined with ATO therapy in APL.
Davidiin is an ellagitannin found in the botanical extract of Davidia involucrata and Polygonum capitatum (Wang et al., 2014). Compared with other known naturally occurring SUMOylation inhibitors, it displays a more potent inhibition of SUMOylation in vitro, with an IC50 in the nanomolar range and antiproliferative activity in several cancer lines in the micromolar range. The antitumor activity of davidiin in vivo is reported to be associated with the downregulation of enhancer of zeste homolog 2 (EZH2), a histone-lysine N-methyltransferase that itself is a target for SUMOylation (Riising et al., 2008; Wang et al., 2014).
Suzawa et al. (2015) performed a cellular, gene expression–based, phenotypic screen of a 1600-compound library for drugs that inhibit human liver receptor homolog-1 (hLRH-1) SUMOylation using two genes whose expression undergoes a robust induction upon hLRH-1 deSUMOylation. They identified tannic acid, a polyphenolic gallotannin related to davidiin that is extracted from several botanical sources, as an SAE inhibitor. Compared with ginkgolic acid, tannic acid exhibits much less cytotoxicity in liver cancer cells; however, its effects on the SAE appear to be specific and not due to promiscuous binding or modulation of ROS levels, known with similar polyphenols.
An earlier cell-based screen of samples from microbial cultured broth identified kerriamycin B as a potential inhibitor of SUMOylation (Fukuda et al., 2009b). Kerriamycin B is a polycyclic antibiotic produced by certain strains of actinomycetes and is reported to reduce substrate-specific and global SUMOylation by targeting the SAE and inhibiting the formation of E1∼SUMO thioesters.
e. Urea-Based Small Ubiquitin-Like Modifier–Activating Enzyme Inhibitors
The discovery of this class of SAE inhibitors was informed by the crystal structure of the SAE as opposed to natural compounds reported earlier (Kumar et al., 2013b). A structure-based virtual screening of a 78,000-compound library for molecules that docked to the nucleotide-binding site of the SAE identified a series of hits with IC50 values comparable to ginkgolic acid, including compound 21. However, the structural features of these compounds were suggestive of unfavorable solubility properties. Computational interrogation of chemical databases and drug libraries identified structurally related compounds with a better solubility profile and comparable potency (Kumar et al., 2013a, 2016). Notwithstanding the potential of this class, further chemical optimization is still needed to generate more potent compounds with enhanced drug-like properties.
D. Ubiquitin-Like Modifier–Activating Enzyme 6
UBA6 is a canonical monomeric E1 enzyme with a domain structure similar to UBA1 (Fig. 2). It is 40% identical to UBA1, and compared with other E1 enzymes, it possesses the highest level of similarity to UBA1 at the nucleotide-binding C-terminal domain (Groettrup et al., 2008). The discovery that UBA6 can also activate ubiquitin has challenged the conventional wisdom that UBA1 is the only ubiquitin-activating enzyme, leading to a paradigm shift in the field and serving as the first example in which two distinct E1s can activate the same UBL (Chiu et al., 2007; Jin et al., 2007; Pelzer et al., 2007; Groettrup et al., 2008). Despite having similar ubiquitin affinities and ubiquitin activation kinetics, there exist several differences between the two enzymes, including the spectrum of E2 and E3 enzymes to which they transfer activated ubiquitin (Table 5) (Jin et al., 2007; Schelpe et al., 2016). Although the substrate pools for UBA1 and UBA6 are partially overlapping, many substrates are specifically targeted by only one of the two enzymes, suggesting that they are required for nonredundant biologic functions (Liu et al., 2017c). This notion is consolidated by the lethal knockout phenotypes observed with both enzymes (Groettrup et al., 2008).
As is the case with SAE and ATG7, which can activate more than one UBL, UBA6 can activate both ubiquitin and FAT10 (Chiu et al., 2007). FAT10 is an 18-kDa protein composed of two ubiquitin-like domains, and conjugation of FAT10 to protein substrates (known as FATylation) plays poorly understood roles in immune responses, apoptosis, NF-κB activation, and mitosis (Gong et al., 2010; Li et al., 2011; Merbl et al., 2013; Schmidtke et al., 2014; Theng et al., 2014). Interestingly, a subset of FATylated proteins can be identified and degraded by the proteasome; therefore, the role of UBA6 in proteasomal degradation is mediated by both ubiquitylation and FATylation (Schmidtke et al., 2014). There also exist several differences between ubiquitin and FAT10 as signals for proteasomal degradation. As opposed to ubiquitylation, monoFATylation is sufficient to induce effective proteasomal degradation (Schmidtke et al., 2014). Despite having similar mechanisms of UBA6-mediated activation, there is conflicting evidence regarding the preferential affinity of UBA6 toward FAT10 and ubiquitin (Chiu et al., 2007; Gavin et al., 2012). Nevertheless, the expression of FAT10 is known to be induced in response to proinflammatory cytokines such as IFN-γ and TNF-α, which may shift the competition in favor of FAT10 in certain contexts (Raasi et al., 1999; Groettrup et al., 2008). Although Uba6-knockout mice are embryonically lethal, FAT10-knockout mice are viable, suggesting UBA6-mediated ubiquitylation rather than FATylation is responsible for the lethal phenotype (Canaan et al., 2006, 2014).
1. Role of Ubiquitin-Like Modifier–Activating Enzyme 6 in Cancer
UBA6 is affected by somatic mutations and CNVs, but their role in cancer is uncertain (Tate et al., 2019). Analysis of patient survival data has shown that high UBA6 expression is associated with unfavorable prognosis in a subset of patients with renal cancer (Uhlen et al., 2017). Apart from these, the data on the role of UBA6 in cancer is limited. Nevertheless, other components of the UBA6-initiated cascade have been implicated in the pathogenesis of cancer. For instance, FAT10 has demonstrated protumorigenic functions in hepatic and gastrointestinal malignancies (Lee et al., 2003; Yuan et al., 2014). Similarly, the UBA6-specific E2 enzyme UBA6-specific E2 conjugating enzyme 1 (USE1) is overexpressed in a subset of patients with lung cancer and promotes tumor growth, migration, and invasiveness (Kim et al., 2017). In contrast, UBA6 is inversely related to invasiveness in a subset of breast cancer in which it suppresses mammary carcinogenesis of breast cancer cells by inhibiting epithelial-mesenchymal transition (Liu et al., 2017b). Therefore, UBA6 appears to display context-dependent oncogenic and tumor-suppressive functions in different cancers.
2. Ubiquitin-Like Modifier–Activating Enzyme 6 Inhibitors
So far, there are no reports of the crystal structure of UBA6. However, biochemical studies show the unique ability of the active site of UBA6 to engage both ubiquitin and the larger diubiquitin-like FAT10 (Gavin et al., 2012). A pan-E1 adenosine sulfamate E1 inhibitor, compound 1, has been reported to inhibit several E1s, including UBA6 (Brownell et al., 2010). In a similar manner to other adenosine sulfamates, compound 1 forms an adduct with both ubiquitin and FAT10 in a reaction catalyzed by UBA6 (Gavin et al., 2012). This adduct serves as the active inhibitory species for the enzyme (Brownell et al., 2010). However, the inhibitory effects of compound 1 on UBA6-mediated FATylation are much weaker compared with its effects on UBA6-mediated ubiquitylation. This weaker potency has been attributed to the slow rate at which the FAT10–compound 1 adduct is formed and the low affinity with which the adduct binds to the enzyme (Gavin et al., 2012). A selective adenosine sulfamate UBA6 inhibitor has been developed, but there are no biochemical or biologic data available for this inhibitor (Fig. 10) (Amidon et al., 2017).

Chemical structures of UBA6, UBA5, and pan/dual E1 inhibitors. (A) Chemical structure of a selective adenosine sulfamate UBA6 inhibitor with the chemical name ((1S,2S,4R)-4-(4-(1H-indol-2-yl)-7H-pyrrolo[2,3-day]pyrimidin-7-yl)-2-hydroxycyclopentyl)methylsulfamate (here referred to as UBA6i) is shown. Moreover, the reported UBA5 inhibitors include the adenosine sulfamate compound 8.5 and the chloroacetamide DKM 2-93. (B) Multi-E1 inhibitors include adenosine sulfamates (e.g., adenosine 5′-sulfamate, compound 1, ABP1, and ABPA3), which are pan-E1 inhibitors, as well as nonnucleoside compounds, e.g., LP0040, that serves as a dual UBA1/NAE inhibitor.
E. Ubiquitin-Like Modifier–Activating Enzyme 7
UBA7 is a canonical monomeric E1 enzyme with a domain structure that resembles both UBA1 and UBA6 (Fig. 2). With a 46% sequence identity, UBA7 possesses the highest overall similarity to UBA1 among all E1 enzymes (Table 1). The function of UBA7 is to activate ISG15, initiating its conjugation to protein substrates, a process termed ISGylation (Krug et al., 2005). ISG15 is a 15-kDa UBL composed of two ubiquitin-like domains and is 29.7% identical to ubiquitin (Narasimhan et al., 2005; Hermann and Bogunovic, 2017). Historically, it was the second member of type 1 UBLs to be identified after ubiquitin (Loeb and Haas, 1992; Schwartz and Hochstrasser, 2003). As opposed to ubiquitin, the sequence of ISG15 is much less conserved among different species (Zhao et al., 2013c). ISG15 is initially produced as a 17-kDa precursor polypeptide that undergoes post-translational processing into the 15-kDa functional form (Blomstrom et al., 1986; Knight et al., 1988). In addition to its function as a post-translational modifier of other proteins, ISG15 can serve as a free intracellular molecule and a secreted cytokine (Knight and Cordova, 1991; Sadler and Williams, 2008; Jeon et al., 2010).
As the name suggests, the expression of ISG15 and ISGylation is induced in response to type I IFN signaling, specifically IFN-α and IFN-β (Hermann and Bogunovic, 2017). In addition, they are induced by other stimuli, including infection, radiation, ischemia, aging, and activation of the NF-κB and p53 pathways (Villarroya-Beltri et al., 2017). Similarly, the expression of UBA7 is inducible by IFNs, supporting the enhanced ISGylation upon exposure to IFN-α/β (Fan et al., 2015). ISGylated proteins are deconjugated by USP18, an ISG15-specific protease, and a negative regulator of type I IFN signaling by a mechanism that is independent of its protease activity (Malakhov et al., 2002; Ketscher et al., 2015). USP18 is also induced in response to viral infections, IFN signaling, and DNA damage (Malakhov et al., 2002). ISGylation has been implicated in numerous cellular functions, including immune responses against viral and bacterial infections, DNA repair by translesion synthesis, regulation of cytoskeletal dynamics, exosome secretion, protein translation, autophagy, proteasomal degradation, and the hypoxic response (Villarroya-Beltri et al., 2017). In a subset of these processes, ISGylation is associated with context-dependent opposite functions (Hermann and Bogunovic, 2017).
There is substantial evidence of cross talk between the ISG15 and ubiquitin conjugation systems. In this respect, the two ubiquitin E2 enzymes, UBCH6 and UBCH8, have been reported to serve as E2 enzymes that transfer ISG15 from UBA7 to cellular substrates (Kim et al., 2004; Zhao et al., 2004; Takeuchi et al., 2005). A model has also been proposed wherein proteasomal degradation influences the substrate pool available for ISGylation (Liu et al., 2003). In addition, increased expression of ISG15 in tumor cells has been found to antagonize ubiquitylation and subsequent proteasomal degradation of a subset of cellular proteins (Desai et al., 2006). Interestingly, ISGylation has been reported to act as a signal for proteasomal degradation of misfolded dominant-negative forms of p53 independent of ubiquitylation-regulated degradation (Huang et al., 2014).
1. Role of Ubiquitin-Like Modifier–Activating Enzyme 7 in Cancer
Reduced expression of UBA7 has been reported in lung cancer cell lines compared with normal cells, suggesting that it may possess tumor-suppressive functions in a subset of lung cancers (McLaughlin et al., 2000; Yin et al., 2009). This is corroborated by the observation that frequent genomic lesions affect the chromosomal region 3p21.3, where the UBA7 gene is located (Hesson et al., 2007). Mechanistic studies of this tumor-suppressive activity have implicated cyclin D1 as a target that is repressed after ISGylation by UBA7, resulting in antitumor effects (Pitha-Rowe et al., 2004b; Feng et al., 2008). Similar tumor-suppressive activity has been reported in APL, in which UBA7 acts as a retinoid-inducible target that causes PML-RARA degradation and apoptosis (Kitareewan et al., 2002; Pitha-Rowe et al., 2004a). In addition, UBA7 and other components of the ISGylation system exert tumor-suppressive functions in cervical, hepatic, and renal cell cancer cells (Jeon et al., 2012; Yeh et al., 2013; Zhou et al., 2017). In contrast, the upregulation of the ISGylation system has been associated with oncogenic functions in models of breast, pancreatic, and prostate cancer (Kiessling et al., 2009; Burks et al., 2014; Sainz et al., 2014). These contrasting reports support the context-dependent oncogenic and tumor-suppressive functions of UBA7 and ISG15 in cancer (Desai, 2015; Zuo et al., 2016). Consistent with these observations, the analysis of patient survival data has correlated UBA7 expression to unfavorable outcomes in renal cancer and favorable outcomes in urothelial and breast cancer (Uhlen et al., 2017). Of note, the ISG15-specific deconjugase USP18 has also been shown to exert opposite functions in several malignancies (Mustachio et al., 2018).
2. Ubiquitin-Like Modifier–Activating Enzyme 7 Inhibitors
The crystal structure of UBA7 has not been elucidated thus far. However, a docking model of ISG15 onto the published structure of the NAE-NEDD8 complex allowed the prediction of several specific interactions, including residues critical for the binding between the C-terminal domain of ISG15 and the AAD of UBA7 (Narasimhan et al., 2005). With the exception of the pan-E1 inhibitor compound 1, there are no inhibitors reported to target UBA7 so far. As assessed by ATP-PPi exchange assays, compound 1 inhibits UBA7 with a potency that is comparable to UBA1 and SAE but less than NAE and UBA6 (Chen et al., 2011). Even after the development of clinical UBA7 inhibitors/activators, it is reasonable to expect that the use of such agents will be determined by the context-dependent roles of UBA7 and other ISGylation components in different cancers. A model has been proposed in which secreted ISG15 stimulates antitumor immune responses; thus, UBA1-initiated ISGylation may reduce the pool of secreted ISG15 and constitute a mechanism by which tumor cells evade immune surveillance (Desai, 2015). In these contexts, UBA7 inhibitors are anticipated to trigger useful antitumor immune responses.
V. Noncanonical E1 Enzymes
A. Autophagy-Related Protein 7
ATG7 is a noncanonical E1 enzyme that plays a key role in the regulation of autophagy, a process by which cellular proteins and organelles are targeted to the lysosome to be degraded by the lysosomal enzymes (Schulman and Harper, 2009; Choi et al., 2013; Xiong, 2015; Levine and Kroemer, 2019). The UPS and the autophagy-lysosome system are the two major cellular machineries involved in protein degradation and are highly coordinated by ubiquitylation and other UBL modifications (Klaips et al., 2018). ATG7 is essential for the autophagic cell death program, a role that is regulated by signaling molecules involved in apoptosis, such as caspases 8 and 9 (Yu et al., 2004; Han et al., 2014). Moreover, ATG7 is implicated in several autophagy-independent functions, including cellular proliferation, cytokine and exosome secretion, and regulation of immune responses (Galluzzi and Green, 2019). Of note, ATG7 has been reported to bind p53 to regulate cell cycle progression and survival under conditions of metabolic stress, a function that appears to be independent of its E1 enzymatic activity (Lee et al., 2012).
In autophagy, ATG7 lies at the center of two UBL conjugation systems, where it activates two classes of UBLs, ATG12 and ATG8 family proteins, initiating two pathways that are important for the formation of autophagosomes, which are vesicular structures that sequester and transport target proteins to the lysosomes (Nakatogawa, 2013) (Fig. 11A). In humans, ATG8 family proteins (simply referred to as ATG8) comprise two subfamilies: microtubule-associated protein 1 light chain 3 proteins and γ-aminobutyric acid receptor–associated proteins (Abdollahzadeh et al., 2017). In the ATG12 conjugation pathway, ATG7 activates ATG12 and transfers it to the E2-like enzyme ATG10, which in turn conjugates ATG12 to ATG5. The ATG12-ATG5 conjugate forms a complex with ATG16, and the complex serves as an E3-like enzyme that plays a key role in ATG8 conjugation. On the other hand, the ATG8 conjugation pathway requires prior processing of the precursor form of ATG8 into the functional form through proteolytic cleavage mediated by the protease ATG4. After activation by ATG7, ATG4 is transferred to the E2-like enzyme ATG3, which in turn conjugates this UBL to phosphatidylethanolamine (PE) in a reaction facilitated by the E3-like enzyme complex ATG12-ATG5-ATG16, which is formed by the ATG12 conjugation pathway (Geng and Klionsky, 2008; Nakatogawa, 2013). The resultant ATG8-PE conjugate plays an important role in autophagosome formation and serves as a receptor for the selective recognition of cargos during autophagy (Suzuki et al., 2017). In addition to its role in the proteolytic cleavage of pro-ATG8, ATG4 can also serve as a deconjugase for the ATG8-PE conjugate (Maruyama and Noda, 2018). Of note, the ATG12 conjugation pathway has been implicated in the regulation of mitochondrial homeostasis and cell death, a function that appears to be independent of its role in autophagy (Radoshevich et al., 2010).
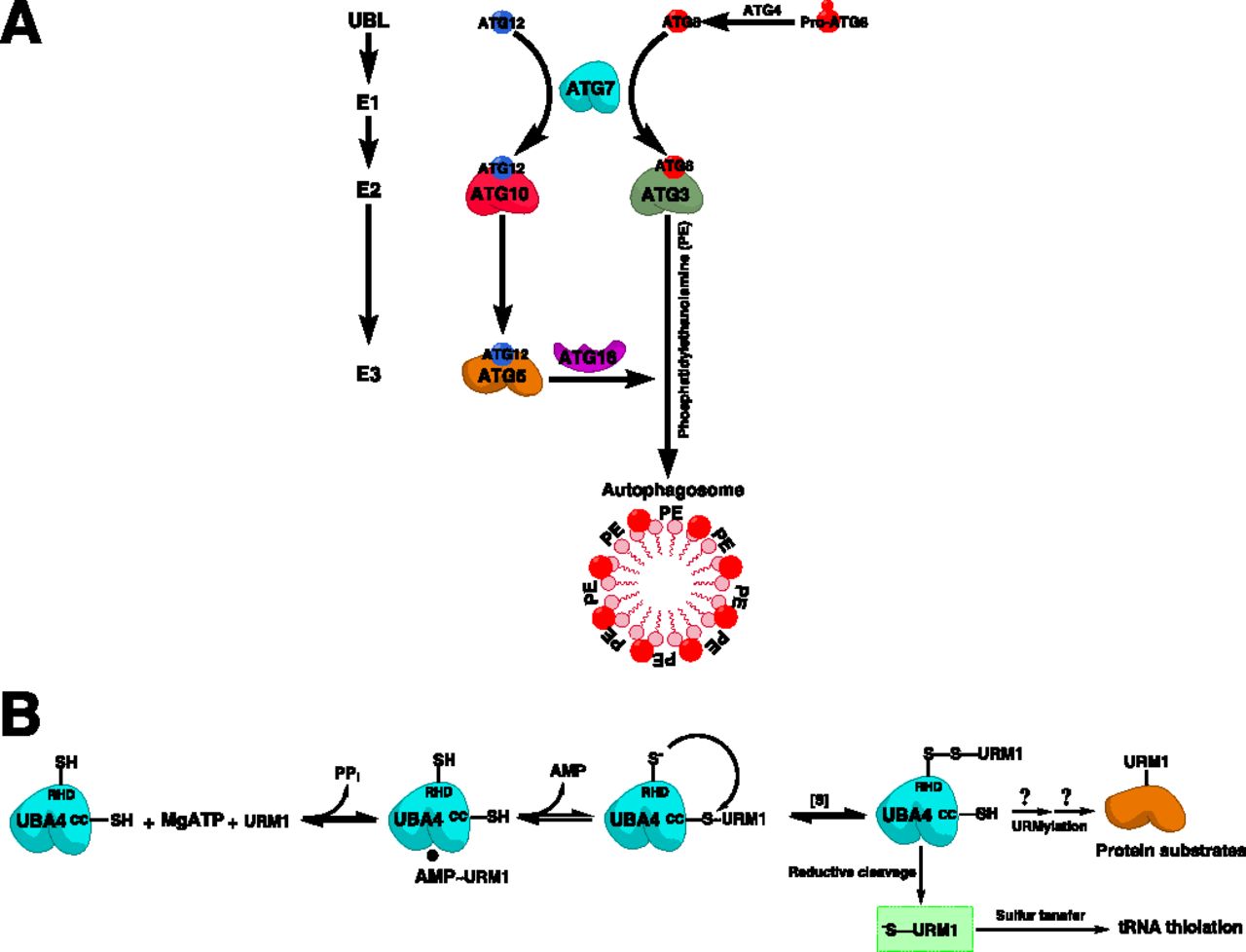
Noncanonical UBL activation by ATG7 and UBA4. (A) ATG7 activates two UBLs, ATG12 and ATG8, which lie at the apex of two cascades that exhibit significant cross talk. After activation, ATG12 is transferred to the E2-like enzyme ATG10, which in turn transfers ATG12 to ATG5. ATG12-ATG5 conjugate then associates with ATG16, forming an E3-like complex, ATG12-ATG5-ATG16, which is catalytically important in the ATG8 conjugation arm. In this other arm, ATG8 is first proteolytically activated by the protease ATG4 and is then activated and transferred by ATG7 to the E2-like enzyme ATG3. ATG8 is then conjugated to PE in a reaction facilitated by the ATG12-ATG5-ATG16 complex. ATG8-PE conjugate is functionally important for the formation of autophagosomes and the identification of cargos during autophagy. The general scheme of UBL activation (UBL→E1→E2→E3) is shown in parallel to the corresponding enzymes in the cascade. (B) UBA4 activates URM1 by a noncanonical mechanism that enables its dual function as a post-translational modifier and a sulfur carrier. After adenylation of URM1 and subsequent thioester formation at the catalytic cysteine (CC), URM1 is transferred by an intramolecular reaction to the other catalytic cysteine located on the RHD of UBA4 to form UBA4∼URM1 acyl persulfide intermediate. The formation of the persulfide bond is enabled by the presence of a sulfur donor [S], and the activated URM1 thiocarboxylate species (highlighted in a green box) is released by a reductive cleavage step for subsequent use in sulfur transfer reactions, such as tRNA thiolation. The E2 and E3 enzymes involved in URMylation are unknown.
1. Role of Autophagy-Related Protein 7 in Cancer
Similar to other genes encoding E1 enzymes, ATG7 is affected by a number of mutations and CNVs; however, they do not appear to play a significant role in cancer development and/or progression (Grossman et al., 2016; Tate et al., 2019). As assessed by the analysis of survival data in a subset of cancer patients, high expression of ATG7 is associated with favorable prognosis in renal and colon cancer and unfavorable prognosis in liver cancer (Luo et al., 2016; Uhlen et al., 2017). ATG7 has been implicated in both oncogenic and tumor-suppressive functions in different malignancies. This derives partly from the role of ATG7 in autophagy, which has dual functions in tumor development and progression in a context-dependent manner (Wen and Klionsky, 2019). In this respect, ATG7 has been reported to promote the tumorigenesis of several malignancies, including prostate cancer (Santanam et al., 2016), chronic myeloid leukemia (Karvela et al., 2016), skin cancer (Xie et al., 2015; Qiang et al., 2017), and bladder cancer (Zhu et al., 2019b), and this protumorigenic role occurs mostly in cooperation with other genetic aberrations, such as phosphatase and tensin homolog (PTEN) loss, Adenomatous polyposis coli gene (APC) inactivation, BCR-ABL fusion, as well as BRAF and Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations (Guo et al., 2013b). On the other hand, ATG7 has been associated with contextual oncogenic and tumor-suppressive functions in liver cancer (Takamura et al., 2011; Gong et al., 2012; Luo et al., 2016), colon cancer (Zhao et al., 2010; Lévy et al., 2015), and lung cancer (Zhao et al., 2010; Guo and White, 2013; Guo et al., 2013a; Strohecker et al., 2013; Strohecker and White, 2014; Han et al., 2018; Cao et al., 2020). Given the reported role of ATG7 in the maintenance of hematopoietic stem and progenitor cells, genetic inactivation of ATG7 has been found to cause dysregulated myeloid proliferation, suggesting a tumor-suppressive role in the early stages of MDS and AML development (Mortensen et al., 2011a,b; Watson et al., 2011a; Gomez-Puerto et al., 2016). In contrast, ATG7 is required to support tumor progression and resistance to chemotherapy in established AML neoplasms (Piya et al., 2016). Therapeutic targeting of ATG7 has been reported to sensitize several malignancies to cytotoxic chemotherapy and targeted agents, including tyrosine kinase inhibitors, BRAF inhibitors, and monoclonal antibodies (Li and Fan, 2010; Han et al., 2011; Xie et al., 2015; Karvela et al., 2016; Piya et al., 2016).
2. Autophagy-Related Protein 7 Inhibitors
The crystal structure of human ATG7 has not been solved to date; however, significant structural and biochemical information has been obtained from Atg7 of other organisms, particularly yeast (Hong et al., 2011; Noda et al., 2011; Taherbhoy et al., 2011; Kaiser et al., 2012; Yamaguchi et al., 2012, 2018). Based on these studies, ATG7 is a homodimeric enzyme with each subunit composed of two domains that are connected by a short flexible linker: 1) N-terminal domain (NTD) and 2) C-terminal domain (CTD). As a homodimer, ATG7 possesses two nucleotide-binding sites and two catalytic cysteine residues (Noda et al., 2009). Moreover, it assumes a bird-like three-dimensional structure in which the CTD dimer occupies a central position corresponding to the body, and the two NTDs connected to the CTD dimer correspond to the wings (Kaiser et al., 2013). The NTD is structurally unique and does not show significant sequence homology with other E1 enzymes. The CTD is composed of two subdomains: a homodimeric AD that is conserved among all E1s and an extreme CTD (ECTD) that is specific to ATG7 (Noda et al., 2011). The ECTD subdomain comprises a C-terminal tail that identifies ATG7-specific UBLs and subsequently transfers them to the AD subdomain that harbors the adenylation active site and the catalytic cysteine that are important for the UBL-activating function of the enzyme. The NTD is involved in the interaction of ATG7 with E2 enzymes for UBL transfer (Noda et al., 2011; Taherbhoy et al., 2011). There are several unique aspects of the noncanonical mechanisms of ATG7 catalytic activity, including the ability to activate UBLs without undergoing significant active site remodeling (Hong et al., 2011). Given the unique architecture and catalytic mechanism of ATG7, it is not surprising that the pan-E1 inhibitor compound 1 does not display significant activity against this enzyme (Brownell et al., 2010). Apart from genetic ablation approaches, no selective chemical inhibitors have been reported to target ATG7 thus far. It is noteworthy that the utility of ATG7 inhibitors in cancer therapy depends on the specific malignancy and the context (e.g., tumor stage) in which they are used. For example, in contexts in which autophagy is perceived as tumor-suppressive, mainly through induction of autophagic cell death, inhibiting ATG7 is anticipated to cause adverse therapeutic outcomes and vice versa. Moreover, the cytoprotective effects of ATG7-mediated autophagy in normal cells should be considered to minimize the untoward effects of ATG7 inhibitors.
B. Ubiquitin-Like Modifier–Activating Enzyme 4
UBA4 [also known as molybdenum cofactor synthesis 3 (MOCS3)] is a noncanonical E1 enzyme that activates URM1, a UBL that acts as a post-translational modifier conjugated to other proteins (by a process termed URMylation) and a sulfur carrier in the thiolation of cytosolic tRNAs (Schlieker et al., 2008; Leidel et al., 2009; Wang et al., 2011). These two functions of URM1 are highly conserved among eukaryotes (Jüdes et al., 2015). Evolutionarily, URM1 is the most ancient UBL and is closely related—in the structure and mechanism of activation—to the prokaryotic sulfur carrier proteins molybdopterin synthase sulfur carrier subunit (MoaD) and thiamine biosynthesis protein S (ThiS), which play a role in molybdopterin and thiamine biosynthesis, respectively (Furukawa et al., 2000; Xu et al., 2006; Pedrioli et al., 2008; Petroski et al., 2011). Therefore, URM1 is regarded as the evolutionary link between prokaryotic sulfur carriers and UBL eukaryotic modifiers (Iyer et al., 2006; Schlieker et al., 2008; Schmitz et al., 2008). The repertoire of human substrates that are modified by URMylation include proteins implicated in the ubiquitylation pathway (e.g., USP15 and 47), nuclear transport (e.g., RanGAP1), and oxidative stress [e.g., carbonyl reductase (NADPH) 1]; however, the biologic roles of URM1 modifications of these substrates are not well understood (Van der Veen et al., 2011). Nonetheless, few biologic functions have been linked to protein URMylation and tRNA thiolation. For example, genetic ablation of URM1 has been reported to induce cytokinesis defects, suggesting a role of URMylation in cell cycle progression (Schlieker et al., 2008). Moreover, tRNA thiolation plays a regulatory role in translation, metabolic homeostasis, and control of cellular growth (Laxman et al., 2013). Of note, URMylation and tRNA thiolation are both stimulated in response to oxidative stress, suggesting a role for these URM1 modifications in sensing and/or protecting against oxidative damage (Leidel et al., 2009; Van der Veen et al., 2011).
1. Role of Ubiquitin-Like Modifier–Activating Enzyme 4 in Cancer
The expression of UBA4 has been associated with unfavorable prognosis in liver and endometrial cancers and a favorable prognosis in renal cancer (Uhlen et al., 2017). Apart from these data, there are no published reports that directly implicate UBA4 in cancer development and/or progression. However, it has been reported that several enzymes involved in the tRNA modification pathway are upregulated in breast cancer and are important in supporting tumor metastasis, suggesting UBA4 can potentially serve as a therapeutic target in this context (Delaunay et al., 2016; Termathe and Leidel, 2018).
2. Ubiquitin-Like Modifier–Activating Enzyme 4 Inhibitors
As is the case with other noncanonical E1 enzymes, UBA4 is a homodimeric enzyme composed of two symmetric subunits (Schulman and Harper, 2009). Each of these subunits comprises E1-like domains at its N terminus that harbor the active adenylation and catalytic cysteine sites (Schulman and Harper, 2009; Termathe and Leidel, 2018). Different from all other E1 enzymes, UBA4 possesses a characteristic RHD at its C terminus that also harbors a catalytic cysteine and plays an important catalytic role in URM1 activation (http://www.rcsb.org/structure/3I2V; Schulman and Harper, 2009). These structural features enable a noncanonical mechanism of UBL activation that is unique to UBA4 and allow for the dual function of URM1 as a UBL and a sulfur carrier (Wang et al., 2011). As with other UBLs, URM1 undergoes initial adenylation and thioester formation steps to form a UBA4∼URM1 thioester intermediate in which URM1 is linked to the catalytic cysteine located on the E1-like domain of UBA4. However, an intramolecular reaction follows whereby URM1 is transferred to the catalytic cysteine located on the RHD, forming a UBA4∼URM1 acyl persulfide intermediate. This reaction requires the presence of a sulfur donor, such as cysteine, to form the persulfide linkage (Termathe and Leidel, 2018). The final step involves reductive cleavage and release of URM1 thiocarboxylate that serves as the activated species for subsequent sulfur transfer reactions (Fig. 11B). To date, there are no known E2 or E3 enzymes that catalyze URMylation (Pedrioli et al., 2008). UBA4 similarly activates molybdenum cofactor synthesis protein 2A (MOCS2A), a polypeptide structurally related to UBLs, and forms a MOCS2A thiocarboxylate that serves as a sulfur donor in molybdenum cofactor biosynthesis (Chowdhury et al., 2012). No inhibitors have been reported to target UBA4 thus far; however, the possession of two catalytic cysteines and the unique catalytic mechanism of this enzyme may create valuable opportunities to develop inhibitors that selectively target UBA4 at the expense of related E1 enzymes.
C. Ubiquitin-Like Modifier–Activating Enzyme 5
UBA5 (also known as ubiquitin-activating enzyme E1-domain containing 1) is a noncanonical E1 enzyme that catalyzes the activation of UFM1, a metazoan-specific UBL that is conjugated to cellular substrates—by a process termed UFMylation—to modulate their functions (Komatsu et al., 2004; Daniel and Liebau, 2014; Yoo et al., 2015; Wei and Xu, 2016; Gerakis et al., 2019). After activation of UFM1 by UBA5, UFMylation of target substrates is mediated by UFM1-conjugating enzyme 1 (UFC1) and UFM1-specific ligase 1 (UFL1), which serve as UFM1-specific E2 and E3 enzymes, respectively (Komatsu et al., 2004; Dou et al., 2005; Mizushima et al., 2007; Tatsumi et al., 2010). UFMylation is subject to regulation by UFSP1 and 2, UFM1-specific proteases that activate the precursor form of UFM1 and deconjugate UFMylated substrates by proteolytic cleavage (Ha et al., 2008, 2011; Kim et al., 2018). In humans, it appears that UFSP2 is the only functional UFM1-specific protease (Yoo et al., 2014). UBA5 has also been reported to activate SUMO2 in vitro and in vivo (Zheng et al., 2008). The repertoire of cellular substrates that is modified by UFMylation comprises few known targets, including activating signal cointegrator 1/thyroid receptor-interacting protein 4 (ASC1/TRIP4), which acts as a transcriptional coactivator of nuclear receptors (e.g., estrogen receptor α); ribophorin 1 (RPN1), which serves as an ER quality control factor; and 60S ribosomal protein L26 (RPL26), which is a ribosomal protein and is reportedly the main target of UFMylation in the cell (Lee et al., 1999; Yoo et al., 2014, 2015; Walczak et al., 2019; Liang et al., 2020). Nonetheless, UFMylation is functionally important in regulating several cellular functions, including DNA damage response (Qin et al., 2019; Wang et al., 2019), transcriptional regulation (Yoo et al., 2014), cell cycle control (Yu et al., 2020), autophagy (DeJesus et al., 2016), and ribosomal functions (Simsek et al., 2017; Walczak et al., 2019). Of particular importance are the roles played by UFMylation in regulating hematopoiesis and ER-associated proteostasis (Gerakis et al., 2019). In this respect, UBA5 and other components of the UFMylation cascade have been reported to be essential for hematopoietic stem cell survival, plasma cell development, and erythroid differentiation, as evidenced by the development of anemic phenotype in mice upon genetic ablation of these components (Tatsumi et al., 2011; Cai et al., 2015, 2016; Zhang et al., 2015a; Zhu et al., 2019a). Moreover, UFMylation plays a key regulatory role in ER homeostasis; thus, the disruption of this pathway is associated with the induction of ER stress (DeJesus et al., 2016; Liu et al., 2017a; Walczak et al., 2019; Wang et al., 2020). UFMylation of ER-resident proteins has been implicated in the autophagy of ER (also known as ER-phagy) and regulation of the UPR (Liang et al., 2020). Of note, a subset of UFMylation system components are subject to transcriptional changes in response to ER stress, establishing a regulatory feedback loop that coordinates UFMylation and ER homeostasis (Zhang et al., 2012).
1. Role of Ubiquitin-Like Modifier–Activating Enzyme 5 in Cancer
The analysis of publicly available datasets has shown that UBA5 is affected by genetic aberrations, particularly amplifications, in several malignancies, including lymphoma, lung, ovarian, and esophageal cancers (Wei and Xu, 2016). As assessed by chemical and genetic approaches, inhibition of UBA5 has revealed that pancreatic and breast cancer cells are dependent on UFMylation for survival and growth in vitro and in vivo (Yoo et al., 2014; Roberts et al., 2017). Besides, a genome-wide CRISPR/Cas9 knockout screen in patient-derived glioblastoma stem cells identified UBA5, among other UFMylation pathway components, as a regulator of glioblastoma stem cell survival (MacLeod et al., 2019). However, in preclinical models of lung cancer, inhibition of UFMylation pathway components including UBA5 has been found to induce a prosurvival UPR that promotes resistance to EGFR inhibition by tyrosine kinase inhibitors (Terai et al., 2018). These reports suggest that UBA5 can serve as a potential therapeutic target in glioblastoma, pancreatic, and breast cancer; however, in the context of targeted therapy in lung cancer, UBA5 inhibition may result in adverse therapeutic outcomes by fostering adaptive resistance signaling.
2. Ubiquitin-Like Modifier–Activating Enzyme 5 Inhibitors
As a noncanonical E1 enzyme, UBA5 is predicted to possess a homodimeric organization with two symmetric subunits (Schulman and Harper, 2009; Cappadocia and Lima, 2018). However, compared with other E1s, each of these subunits displays minimalistic structural features that are shared between the more complex canonical E1s and the simpler, ancestral E1-like enzymes. Specifically, UBA5 comprises an AD that harbors both the nucleotide-binding site as well as the catalytic cysteine residue (Cys250), with no dedicated domain for this catalytic cysteine. It also comprises a small UFD-like region at the C terminus—termed UFC1-binding sequence—that facilitates the interaction with UFC1 and subsequent transfer of activated UFM1 (Bacik et al., 2010; Oweis et al., 2016). UBA5 also possesses characteristic structural motifs, including a UFM1-interacting sequence (UIS) involved in UFM1 interaction, and an N-terminal extension that exists in one of the two known UBA5 isoforms and enhances UFM1 activation (Padala et al., 2017; Soudah et al., 2019). In addition to UFM1, it appears that UBA5 can also interact with γ-aminobutyric acid receptor–associated proteins, and this interaction is required to facilitate the recruitment of UBA5 to the ER membrane (Habisov et al., 2016; Huber et al., 2020).
As opposed to canonical E1s that activate their cognate UBLs via a three-step mechanism, UBA5 activates UFM1 in two steps—adenylation and thioester bond formation—with no second adenylation step, forming a binary rather than a ternary complex from which UFM1 is transferred to UFC1. Besides, UBA5 displays a slower catalytic activity compared with canonical E1s (Gavin et al., 2014). Of note, UBA5 activates UFM1 by a trans-binding mechanism whereby UFM1 interacts with the UIS of one subunit and undergoes adenylation by the AD of the other subunit of the UBA5 homodimer, suggesting that homodimerization is essential for UFM1 activation (Oweis et al., 2016; Mashahreh et al., 2018).
As with other E1 enzymes, UBA5 can be targeted either at the nucleotide-binding site or the catalytic cysteine, taking into consideration the UBA5-specific structural features to maximize the selectivity of UBA5 inhibitors. To date, only two experimental UBA5 inhibitors have been reported: compound 8.5 and DKM 2−93 (Fig. 10; Table 9).
UBA5 inhibitors and their pharmacologic properties
Compound 8.5 is an adenosine-based organometallic UBA5 inhibitor that was identified by a focused screen of compounds that incorporate adenosine and zinc(II)cyclen coordination complex. As assessed by structural and biochemical analyses, compound 8.5 is predicted to target the nucleotide-binding pocket, and with respect to ATP, it serves as a noncompetitive inhibitor that displays selectivity for UBA5 over other E1 enzymes and kinases. Nonetheless, high concentrations are required to induce cytotoxicity in UBA5-overexpressing cancer cell lines, suggesting that further optimization is required to enhance the potency of this inhibitor (da Silva et al., 2016).
DKM 2−93 is a UBA5 inhibitor identified through a screen for cysteine-reactive covalent compounds that inhibit pancreatic cancer cell proliferation and subsequent target identification by activity-based protein profiling. DKM 2−93 covalently inhibits UBA5 by targeting the catalytic cysteine, preventing UFM1 charging of UBA5, and resulting in antitumor effects in pancreatic and glioblastoma cancer cells in vitro and in vivo (Roberts et al., 2017; MacLeod et al., 2019). With the high concentrations required to induce cytotoxicity, it is anticipated that DKM 2−93 may have off-target effects; however, it can potentially serve as a useful chemotype for covalent UBA5 inhibitors with higher potency and selectivity (Roberts et al., 2017).
Adenosine 5′-sulfamate (ADS), a pan-E1 inhibitor, has been reported to inhibit UBA5 by targeting the UBA5∼UFM1 thioester intermediate and forming an ADS-UFM1 covalent adduct that inhibits UBA5 at the nucleotide-binding site (Table 10). ADS has also displayed cell-based activity in colon cancer cells, as it inhibited UBA5∼UFM1 thioester formation with a subsequent reduction in the abundance of UFMylated proteins (Gavin et al., 2014).
Nonselective E1 inhibitors and their pharmacologic properties
VI. Dual and Multi-E1 Inhibitors
E1 inhibitors discussed so far have variable selectivity profiles toward E1 enzymes or other targets of the proteome. Of these, most adenosine sulfamates, particularly those in clinical trials, TAS4464 and COH000 exhibit a high level of selectivity toward a specific E1 enzyme, with little or no activity against other E1 members. On the other hand, other E1 inhibitors, particularly those with relatively high IC50 values, are expected to demonstrate multiple off-target effects besides their E1 activities. In between, there exists a third subclass whose members possess potent activity toward two or more E1 enzymes. These include the non-nucleoside derivative LP0040 and the adenosine sulfamates compound 1, activity-based probe (ABP) 1, and activity-based probe A3 (ABPA3) (Fig. 10; Table 10). In addition, several investigational adenosine sulfamates with such activity—but not as well characterized—have been identified during the chemical optimization stages of pevonedistat and TAK-243 development, such as compound 15 and compound 46 (Ciavarri and Langston, 2017).
A. Multi-E1 Adenosine Sulfamate Inhibitors
Members of this class retain several structural features of AMP, including the ribose sugar and the adenine base. Compound 1 is a pan-E1 inhibitor that has been exploited to elucidate important mechanistic aspects of substrate-assisted inhibition (Brownell et al., 2010). While having variable potencies against E1 enzymes in ATP-PPi exchange assays, with the highest potency observed with NAE, compound 1 is equipotent against UBA1, NAE, and SAE in E1-E2 transthiolation assays (Brownell et al., 2010; Chen et al., 2011). Of note, compound 1 displays no appreciable activity against ATG7 in ATP-PPi exchange assays. Replacing the indane moiety of compound 1 with a propargyl moiety generates another nonselective E1 inhibitor, ABP1, with a free alkyne group that has been leveraged for covalent conjugation to fluorescent or biotin tags using click chemistry (An and Statsyuk, 2013). This advantage has enabled the use of ABP1 as a probe to quantify the activity of E1 enzymes, as the adducts are formed by the enzymes themselves and thus the rate of adduct formation is dependent on E1 activity (see below). In addition, it has been used to provide further insights into the biochemistry of UBL conjugation and evaluate the potency and selectivity of investigational E1 inhibitors (An and Statsyuk, 2013). Experience with ABP1 has been exploited to develop a related analog, ABPA3, which acts as a dual UBA1/NAE inhibitor. ABPA3 is also structurally related to compound 1, with phenylacetylene replacing the indane moiety (An and Statsyuk, 2015). ABPA3 displays anti-UBA1/NAE activity in lung cancer cells, which results in the induction of UPR and prevents cytoprotective aggresome formation leading to cell death. Of note, ABPA3 paradoxically causes increases in SUMOylation and UFMylation, which are likely compensatory responses activated in response to UPR induction (An and Statsyuk, 2015).
B. LP0040
LP0040 is a non-nucleoside dual UBA1/NAE inhibitor developed by rational drug design and molecular modeling based on the structure of the NAE inhibitor M22 (Lu et al., 2018). Replacing the benzyl group of M22 with biphenyl-substituted 2H-chromen-2-one in LP0040 led to an improved antiproliferative activity associated with cell cycle arrest and apoptosis in cancer cell lines of different origin with a low micromolar IC50. LP0040 has also displayed synergistic cytotoxicity with bortezomib (Lu et al., 2018).
VII. Adenosine Sulfamate E1 Inhibitors
This class comprises pevonedistat, TAK-243, ML-792, TAK-981, compound 1, ABPA3, and ABP1, which act as mechanism-based inhibitors of E1 enzymes. They are structurally related to AMP, which tightly binds to E1 enzymes during the catalytic cascade of UBL activation (Soucy et al., 2009a). The structures of adenosine sulfamates share three major chemical features: 1) a ribose or cyclopentane moiety, 2) a sulfamate group analogous to the phosphate group in AMP, and 3) a nucleobase extension analogous to the adenine base in AMP (Misra et al., 2017) (Fig. 12).
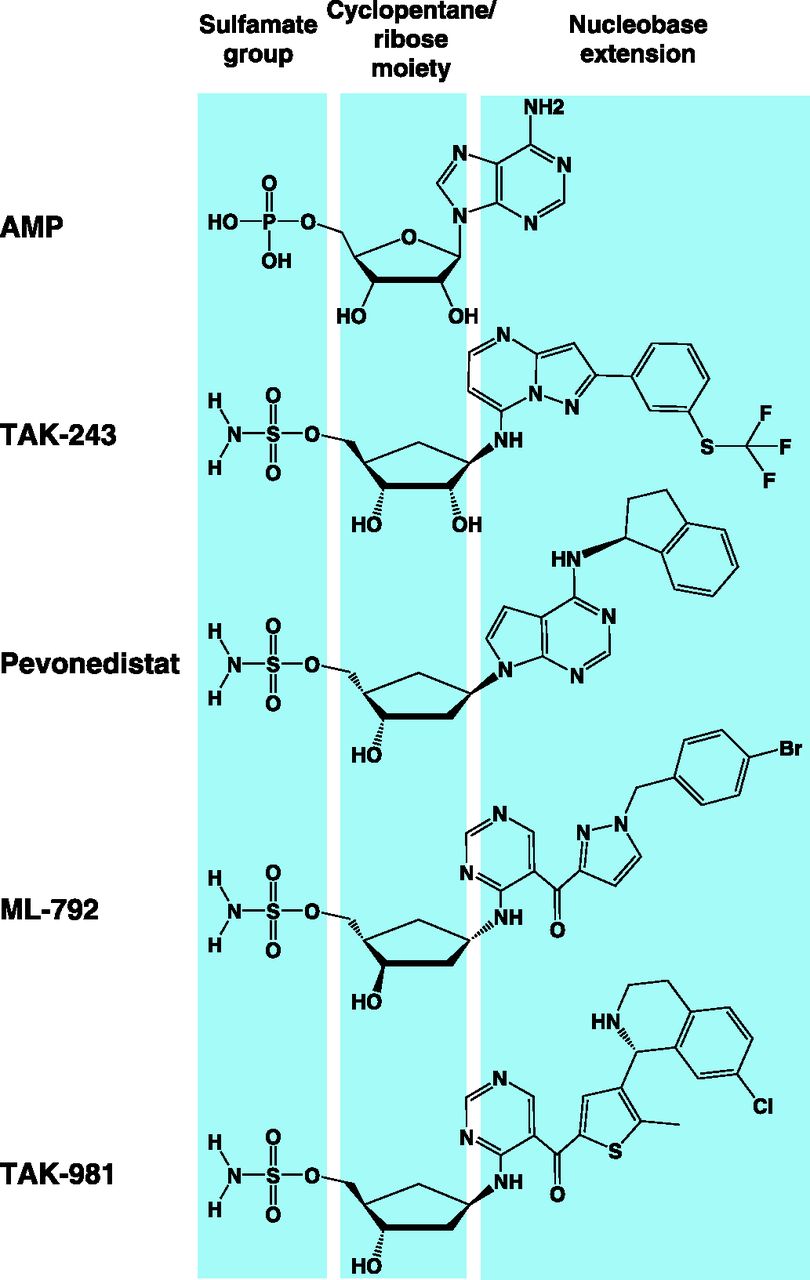
Structural features of adenosine sulfamates. The chemical structures of TAK-243, pevonedistat, ML-792, and TAK-981 are shown in comparison with AMP. Adenosine sulfamates share common chemical features—mainly 1) sulfamate group, 2) cyclopentane/ribose moiety, and 3) nucleobase extension.
A. Discovery and Structure-Activity Relationship
The discovery and SAR of adenosine sulfamates have been reviewed in detail; here, we briefly highlight the main features of these discovery programs (Ciavarri and Langston, 2017). An HTS identified N6-substituted adenosine derivatives as initial chemotypes with weak NAE inhibitory activity. Further chemical optimization was required to enhance potency, cellular activity, selectivity, and stability (Ciavarri and Langston, 2017) (Fig. 13). Although phosphate derivatives demonstrated sub-micromolar potency, they did not permeate into cells and thus lacked cellular activity (Ciavarri and Langston, 2017). Replacement with the neutral sulfamate group retained potency and yielded a series of cell-active E1 inhibitors. The sulfamate functional group is the closest bioisostere to phosphate and does not undergo promiscuous thioester formation in cells as a result of its low intrinsic reactivity (An and Statsyuk, 2015). Compared with sulfamate, sulfonamide and sulfamide groups are less favorable in many aspects, including cellular activity, potency, and selectivity; therefore, the sulfamate group remained as a common chemical feature of adenosine sulfamates (Ciavarri and Langston, 2017). In this series, certain compounds were prone to an intramolecular cyclization reaction, resulting in the loss of activity. In addition, several compounds lacked selectivity for individual E1 enzymes. A combination of chemical modifications in the ribose sugar, purine base, and stereochemical orientation of the sulfamate group solved these stability and selectivity issues and retained or even improved potency and cellular activity, leading to pevonedistat as the prototype of this class (Soucy et al., 2009a; Ciavarri and Langston, 2017).
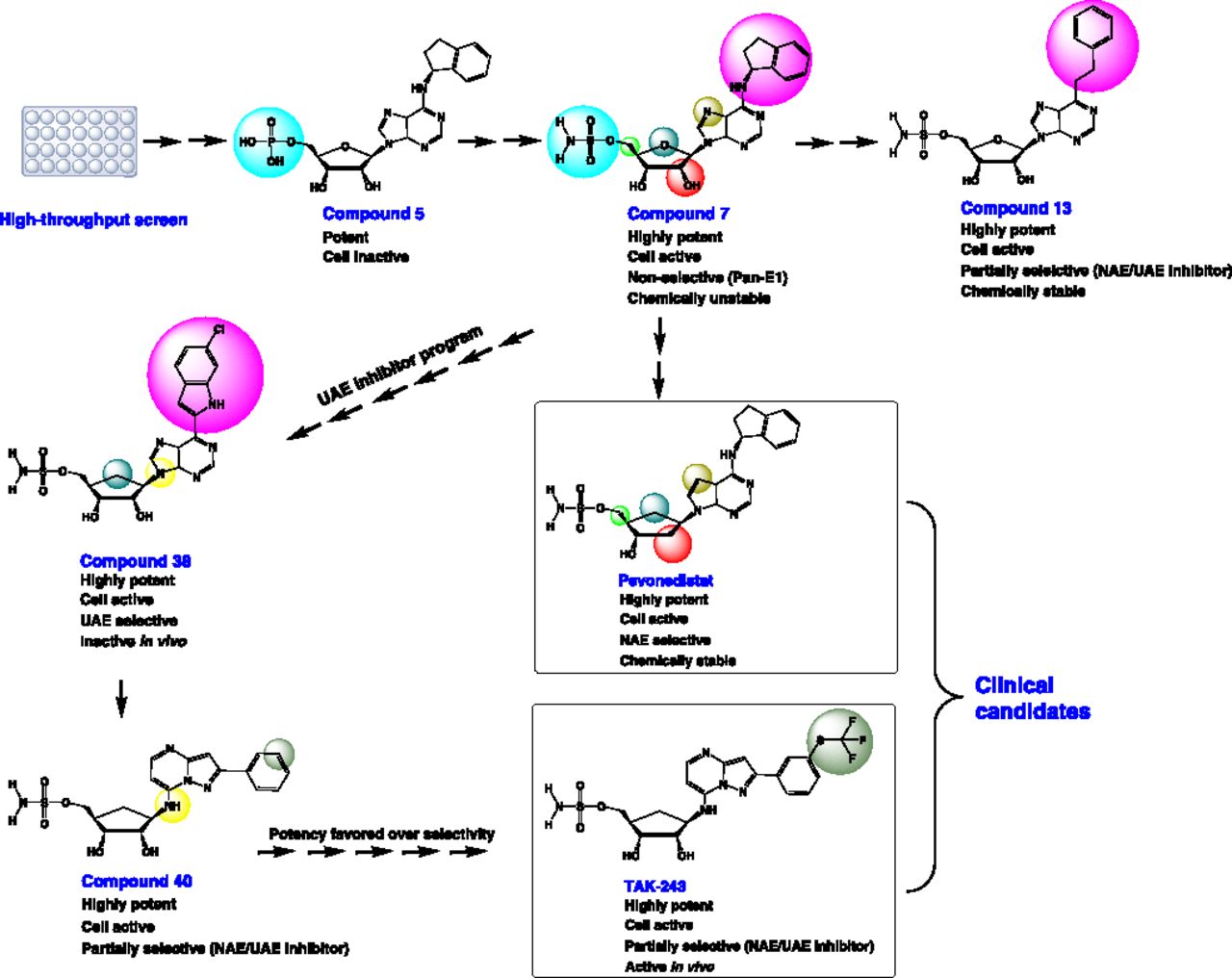
Discovery of adenosine sulfamates. A diagram showing the main features of the drug discovery program of pevonedistat and TAK-243. Compounds, their chemical structures, and pharmacologic properties are shown. Chemical moieties that are changed between intermediate compounds are shaded in similar colors. UAE, ubiquitin-activating enzyme.
The success of the NAE inhibitor program inspired the initiation of another program to develop UBA1 inhibitors by exploring the adenosine sulfamate series and building on the previous SAR and mechanistic studies done with pevonedistat (Brownell et al., 2010). Although chemical modifications of the ribose sugar and aromatic side chain led to improved selectivity to UBA1, the pharmacodynamic profile in vivo was not optimal. The replacement of a purine base with a pyrazolo-pyrimidine scaffold and the placement of the N9-nitrogen in an exo position improved the in vivo profile and significantly enhanced selectivity over the SAE; however, the selectivity over the NAE was only partial (Ciavarri and Langston, 2017). Further modifications enhanced selectivity to UBA1, however, at the expense of potency, which was significantly compromised. As NAE regulates the activity of only a subset of ubiquitin E3 ligases downstream of UBA1, it is anticipated that a dual UBA1/NAE inhibitor will demonstrate the same biologic activity and phenotype as a selective UBA1 inhibitor (Ciavarri and Langston, 2017b). Therefore, chemical optimization was pursued with priority given to potency over selectivity, leading ultimately to the selection of TAK-243 as a clinical candidate.
B. Substrate-Assisted Inhibition
The development of pevonedistat involved an extensive characterization of its mode of action using various approaches including crystallographic studies to elucidate the structural basis of NAE inhibition by this drug (Brownell et al., 2010). These studies showcased a continuous electron density between the C terminus of NEDD8 and the sulfamate group of pevonedistat when bound to NAE, suggesting that a pevonedistat-NEDD8 adduct is formed and possibly it is the species that inhibits NAE (Brownell et al., 2010). Given that the UBL activation mechanism is highly conserved among canonical E1 enzymes, this finding has stimulated further studies that revealed this mode of inhibition—termed substrate-assisted inhibition—is a general mechanism shared by other adenosine sulfamates, including those targeting UBA1 and SAE (Chen et al., 2011; He et al., 2017; Hyer et al., 2018).
Substrate-assisted inhibition is unique in that the UBL substrate of E1 enzymes is involved in the enzyme inhibition through a chemical reaction with adenosine sulfamates to form a covalent adduct that itself serves as the active inhibitory species (Brownell et al., 2010). This mode of inhibition may be pharmacologically beneficial, as it confers selectivity among E1 enzymes because most E1s activate only one or closely related UBLs. Besides, it reduces the likelihood of interaction with other ATP-dependent enzymes, including kinases, which have different catalytic mechanisms and thus are likely less susceptible to these UBL-based inhibitory species (Brownell et al., 2010).
To form these adducts, adenosine sulfamates act as AMP mimetics and exploit the multistep catalytic mechanism of UBL activation by E1 enzymes (detailed in section II. B. Biochemical and Structural Mechanisms of E1-Catalyzed Ubiquitin-Like Protein Activation). They specifically target the E1∼UBL thioester intermediate, a binary complex in which E1 is loaded with a single UBL molecule at the catalytic cysteine, and bind to the nucleotide-biding site in the AAD. This is followed by a nucleophilic attack of the thioester bond by the sulfamate amino group of adenosine sulfamates to form a covalent adduct with UBL (Fig. 3). Consistent with a mechanism-based inhibitory mode, the adduct formation is catalyzed by the E1 enzyme itself and requires the presence of magnesium, ATP, and an active catalytic cysteine, mimicking the conditions of AMP∼UBL formation and implicating most sites of interaction with the E1 enzyme (Brownell et al., 2010; Chen et al., 2011). Although E1∼UBL intermediate formation is the rate-limiting step in UBL activation under physiologic conditions, biochemical studies suggest adduct formation is the rate-limiting step in E1 inhibition by adenosine sulfamates (Chen et al., 2011). As opposed to the binary complex, the ternary complex loaded with two ubiquitin molecules is not susceptible to inhibition by adenosine sulfamates, as the nucleotide-binding site, to which these drugs need to bind first, is occupied by AMP∼UBL. After formation, adenosine sulfamate adducts are stable and tightly bind—with a picomolar affinity—to E1 enzymes in an ATP-competitive manner. By competing for E1 binding, high ATP concentrations are thus anticipated to slow the binding rate of adenosine sulfamates with a resultant increase in their IC50 values (Chen et al., 2011).
Of note, the inhibitory activity of stable AMP∼UBL mimetics against E1 enzymes has been known several years before the development of adenosine sulfamates as with APU, and later with UBL-AMSN/AVSN which are preformed semisynthetic UBL phospho- and sulfamoyl-adenosine derivatives (Wilkinson et al., 1990; Olsen et al., 2010). Adenosine sulfamates, however, differ in that they form adducts in situ after binding to their cognate E1 enzymes, allowing for a preferential activity against cancer cells that display higher activity of these enzymes to support their cellular stresses (Luo et al., 2009; Dobbelstein and Moll, 2014). It is also noteworthy that—in cell-free assays—preformed adenosine sulfamate adducts can inhibit E1 enzymes with much higher potency compared with unbound adenosine sulfamates (Chen et al., 2011). This may be ascribed to their different inhibition kinetics, as the adducts rapidly bind to and inhibit unloaded E1 enzymes, halting all subsequent steps, whereas unbound adenosine sulfamates bind at a slower rate to the E1∼UBL thioester intermediate with no effect on the first step of UBL activation (Chen et al., 2011).
C. Selectivity and Potency of Adenosine Sulfamate E1 Inhibitors
Biochemical studies suggest that the potency of adenosine sulfamates is a function of the rate of adduct formation and the affinity of this adduct to E1 enzymes, and these biochemical parameters in turn depend on their chemical structure (Chen et al., 2011). Although adenosine sulfamates share general chemical features, they still have several variations that dictate—among other properties— potency and selectivity to E1 enzymes. These include chemically diverse nucleobase extensions, different stereochemical orientation of the sulfamate group, as well as varying numbers and stereochemical orientations of the hydroxyl groups on the cyclopentane/ribose moiety (Misra et al., 2017). For instance, TAK-243 possesses a characteristic thio(trifluoromethyl) group in its nucleobase extension, which is absent in other adenosine sulfamates (Fig. 11). This group serves as a hook that engages TAK-243 in several tight contacts with several amino acid residues within a hydrophobic pocket in the UBA1 enzyme, contributing to the drug selectivity compared with related analogs (Misra et al., 2017). On the other hand, the NAE has an extended hydrophobic pocket, and the presence of a hydrophobic indane cycle in the structure of pevonedistat partly contributes to its selectivity through hydrophobic interactions with the extended pocket of the enzyme (Misra et al., 2017). As they exhibit a wider selectivity profile, dual- or pan-specific E1 inhibitors such as ABPA3 and compound 1 retain the adenine base and the ribose sugar with its two hydroxyl groups that are present in the natural substrate, AMP, thereby increasing their capacity to interact with multiple E1 enzymes with a comparable affinity (Misra et al., 2017). Although the interactions with AMP are generally conserved among different E1 enzymes, there still exist some variations that are reflected in their interactions with panspecific E1 inhibitors with different nucleobase extensions, leading to narrower/wider selectivity profile. For example, ABP1 possesses a small propargyl group linked to the exocyclic amino group of the adenine as opposed to the larger phenylacetylene in the related adenosine sulfamate ABPA3. With its small size, this propargyl can fit into the SAE pocket leading to an inhibitory activity against SAE, which is lacking with ABPA3 (Misra et al., 2017).
D. Resistance to Adenosine Sulfamate E1 Inhibitors
The emergence of acquired anticancer drug resistance is a common problem, particularly with molecularly targeted agents, including drugs targeting the ATP-dependent tyrosine kinases (Holohan et al., 2013; Dobbelstein and Moll, 2014). Mechanisms of such resistance include the activation of adaptive/secondary signaling pathways that maintain survival by compensating for the inhibitory effects on the targeted pathway, overexpression of multidrug resistance proteins that reduce the intracellular concentrations of the drug by extracellular efflux, and the acquisition of on-target mutations that interfere with binding to or preclude inhibition of the intracellular target (Ellis and Hicklin, 2009). Although the first mechanism is less likely to develop with adenosine sulfamates, especially those targeting UBA1—given their pleiotropic effects, the other two mechanisms are still potential contributors to resistance because of the on-target selectivity and structural similarity of adenosine sulfamates to ATP (Dobbelstein and Moll, 2014).
Preclinical resistance studies on adenosine sulfamates involved the development of laboratory-evolved resistance models under the selective pressure of exposure to the drug in tissue culture and in vivo. Such models have led to the identification of several on-target mutations in the AADs of UBA1 and NAE that preclude the binding of TAK-243 and pevonedistat, respectively (Milhollen et al., 2012; Toth et al., 2012; Xu et al., 2014; Barghout et al., 2019).
Milhollen et al. (2012) and Toth et al. (2012) independently identified A171T missense mutation as a mechanism of resistance to pevonedistat. Milhollen et al. also identified other mutations, such as A171D, C324Y, G201V, E204K, and N209K. These mutations map to the AAD either within the nucleotide-binding pocket (A171T and A171D) or within/close to the NEDD8-binding cleft (C324Y, G201V, E204K, and N209K), reducing the binding of pevonedistat or NEDD8 to NAE, respectively (Milhollen et al., 2012). Consistent with the heterozygosity of these mutations, pevonedistat-NEDD8 adduct can still be formed by the wild-type copy of the enzyme; however, the mutant form displays a dominant effect, resulting in a resistant phenotype.
Of these mutation sites, A171 is particularly important, as it constitutes a hotspot where two-thirds of the reported mutations are located. A171 serves as a gatekeeper residue that plays a key role in controlling ATP and pevonedistat access to the nucleotide-binding site (Toth et al., 2012). In this respect, the alanine residue with its small side chain contributes to shaping an edge of the nucleotide-binding pocket without engaging in direct contact with ATP (Toth et al., 2012). Structural and computational studies suggest the replacement of alanine with threonine (A171T) or aspartic acid (A171D), which possess larger side chains, leads to conformational changes and steric clashes with the aminoindane group of pevonedistat, particularly with the A171D-mutant NAE, which is completely insensitive to pevonedistat because of the bulkier aspartic acid residue (Milhollen et al., 2012; Toth et al., 2012; Verma et al., 2014).
These mutations alter two fundamental biochemical properties required for potent NAE inhibition (Brownell et al., 2010; Chen et al., 2011). Specifically, they slow the rate of pevonedistat-NEDD8 adduct formation and reduce the adduct affinity to the NAE, resulting in the loss of tight binding and faster recovery of enzyme activity with 2- to >2000-fold reduction in pevonedistat potency. Compound 1, which exhibits tighter binding properties to several E1 enzymes, including the NAE, could circumvent pevonedistat-resistant NAE mutations, suggesting that second-generation NAE inhibitors with higher affinities may offer a potential opportunity to overcome acquired pevonedistat resistance. Using a similar approach, Xu et al. (2014) identified two other mutations, I310N and Y352H, that decrease the affinity of NAE to NEDD8 and confer resistance of leukemia cell lines to pevonedistat.
The A171 residue of NAE is highly conserved among E1 enzymes; thus, mutation of the corresponding alanine residues in other E1s is expected to alter the response to adenosine sulfamates selective for these enzymes. In this respect, A580T mutation generated in recombinant UBA1 conferred resistance to TAK-243 in cell-free thioester formation assays (Misra et al., 2017). Consistent with these findings, Barghout et al. (2019) identified A580S mutation in a laboratory-evolved TAK-243–resistant leukemia cell line. They also identified another mutation in a close residue, Y583C. This is a TAK-243–specific mutation that was not reported with pevonedistat as TAK-243 possesses a thio(trifluoromethyl) hook through which it extends in the nucleotide-binding pocket further than pevonedistat and undergoes a favorable interaction with Y583 (Misra et al., 2017; Barghout et al., 2019). Although not highly conserved among E1 enzymes, the 583 position is mostly occupied with amino acid residues that possess bulky side chains. Therefore, replacement with cysteine is anticipated to perturb the hydrophobic core and eliminate hydrogen bonds with neighboring residues, leading to changes in the binding pocket that preclude the efficient binding of TAK-243 to UBA1 (Barghout et al., 2019).
Drug resistance is known to be multifactorial, and the resistance phenotype observed after clonal selection with adenosine sulfamates can result from a combination of several mechanisms. In this context, moderate upregulation of ATP-binding cassette transporter proteins is observed in pevonedistat-resistant cells, suggesting that they may contribute to drug resistance, particularly at higher expression levels (Milhollen et al., 2012). Recently, pevonedistat has been reported to serve as a substrate for breast cancer resistance protein/ATP-binding cassette sub-family G member 2 (BCRP/ABCG2), and breast cancer resistance protein upregulation confers resistance to pevonedistat in ovarian, colorectal, and non–small-cell lung cancer cells (Kathawala et al., 2020; Wei et al., 2020).
Although A580T-mutant UBA1 exhibits a 6-fold resistance to TAK-243 in cell-free assays, TAK-243–resistant cell lines with A580S and Y583C mutations exhibit 28- to 33-fold resistance, suggesting that other resistance mechanisms may coexist in these cells (Misra et al., 2017; Barghout et al., 2019). Apart from these mechanisms, genetic knockout of the transcriptional regulator BEN domain-containing protein 3 is also reported to confer resistance to TAK-243 in a leukemia cell line; however, it remains unknown how it alters the response to this drug (Barghout et al., 2018).
E. Pharmacodynamic Activity of Adenosine Sulfamate E1 Inhibitors
Adenosine sulfamates targeting UBA1, NAE, and SAE are currently investigated in different phases of clinical trials in various malignancies (Tables 4 and 7). To maximize the benefit from such trials, pharmacodynamic biomarkers need to be assessed either as evidence of mechanism or to inform clinical decision-making in the trial (Schilsky et al., 2012). These biomarkers are usually pathway-specific, and in the case of adenosine sulfamates, adduct formation, E1∼UBL thioesters, and/or reduction in the levels of UBL conjugates could serve as potential biomarkers (Bhatia et al., 2016; Sarantopoulos et al., 2016). For preclinical research, qualitative or semiquantitative approaches can be used to assess the pharmacodynamic effects of these drugs as a proof of principle (Soucy et al., 2009a; Hyer et al., 2018). However, for clinical trials, it is important to develop reliable pharmacodynamic assays that can assess and monitor the response to these investigational agents, preferably in a quantitative manner.
The pharmacodynamic evaluation of adenosine sulfamate activity has been performed at different levels pre- and post-E1 inhibition. For example, the cellular thermal shift assay—a biophysical assay based on the thermostabilization of proteins after binding specific ligands, has been used to assess the binding of TAK-243 to UBA1 in cells and tumors after drug injection into mice (Martinez Molina et al., 2013; Barghout et al., 2019). As the formation of adenosine sulfamate adducts is a prerequisite for potent inhibition of E1 enzymes, antibodies raised against adducts with ubiquitin (e.g., MIL90 antibody), NEDD8 and SUMO have been used to assess drug activity in vitro and in vivo (Yan et al., 2013; He et al., 2017; Hyer et al., 2018; Berger et al., 2019). In addition, levels of E1∼UBL thioesters, global protein conjugates, and conjugates of specific proteins such as ubiquitylated H2A, neddylated Cul1, and SUMOylated RanGAP1 have been used (Yan et al., 2013; He et al., 2017; Hyer et al., 2018; Barghout et al., 2019).
In such preclinical studies, semiquantitative assays such as immunoblotting and immunohistochemistry have been used as a proof of principle for E1 engagement and pathway inhibition. To achieve a more quantitative evaluation of these parameters, other platforms have been developed, such as the AlphaScreen format, a nonradioactive amplified luminescent proximity homogenous assay that provides a faster and more robust assessment as well as higher throughput (Yan et al., 2013). Moreover, a mass spectrometry–based profiling of SDS-PAGE fractionated lysates of biologic samples has been used to provide absolute quantification of the NAE and NEDD8-pevonedisatat adducts in these samples (Yang et al., 2013). In addition to gaining quantitative insights into the dynamics of UBL conjugation, the molar ratio of adduct to E1 enzyme can be calculated to provide an indirect pharmacodynamic measure of pathway inhibition (Yang et al., 2013).
Since the adduct is formed by the E1 enzyme itself, the rate of adduct accumulation can provide valuable information on the activity of the enzyme, which in turn can potentially serve as a predictive biomarker of response to these drugs (Brownell et al., 2010). In this context, the availability of specific antibodies against these adducts can be exploited to develop flow cytometry–based quantitative assays that can—with appropriate analytical and clinical validation—be employed to quantitatively measure the rate of adduct formation after administration of a single dose of the drug. This may help with decision-making as to whether patients should continue therapy with these agents, particularly in hematologic malignancies for which blood samples can be easily obtained. It remains challenging, however, to determine the cutoffs of enzyme activity that will be used to make such decisions.
F. Cell Cycle Effects of Adenosine Sulfamate E1 Inhibitors
Post-translational modifications, particularly with ubiquitin, SUMO, and NEDD8, play an important role in regulating cell cycle progression (Teixeira and Reed, 2013; Eifler and Vertegaal, 2015). Therefore, the disruption of these post-translational modifications is anticipated to result in the deregulation of the cell cycle. However, the ultimate phenotype varies among different adenosine sulfamates. Although the inhibition of neddylation with pevonedistat causes cell cycle arrest and accumulation of cells in the S phase, selective inhibition of ubiquitylation with a selective UBA1 inhibitor causes cell cycle arrest in the G2/M phase. Interestingly, dual inhibition of ubiquitylation and neddylation with TAK-243 also results in G2 arrest, suggesting that the effect on ubiquitylation is more dominant (Ciavarri and Langston, 2017). Although TAK-243 resembles pevonedistat in inducing the accumulation of CDT1, it also stabilizes its endogenous inhibitor, geminin, offsetting CDT1 effects (Hyer et al., 2018). As a result, pevonedistat induces CDT1-mediated re-replication, which is not observed with TAK-243. ML-792, on the other hand, causes mitotic disruption with a decrease in the number of cells in the anaphase/telophase subphases of mitosis, without affecting the number of those in the prometaphase and metaphase. These mitotic defects are associated with the induction of endoreduplication and the formation of DNA bridges (He et al., 2017).
G. Pharmacokinetics of Adenosine Sulfamate E1 Inhibitors
Among adenosine sulfamates, pevonedistat is the most extensively studied agent in clinical settings; thus, substantial human pharmacokinetic (PK) data are available on this drug (Table 11). After intravenous infusion, pevonedistat shows biphasic PK with a rapid distribution from the plasma to peripheral tissues, followed by a slower elimination from the body. In addition, it displays dose-dependent increases in plasma concentrations and remains detectable at quantifiable concentrations up to 72 hours after infusion; however, it shows little or no accumulation in the plasma after repeated intermittent or daily infusion (Bhatia et al., 2016; Sarantopoulos et al., 2016; Swords et al., 2015, 2017, 2018; Shah et al., 2016). The terminal elimination half-life time (t1/2) of pevonedistat is approximately 7.5–10 hours after intravenous infusion. These PK parameters are not affected when pevonedistat is administered in combination with azacytidine, docetaxel, or gemcitabine, suggesting no significant interactions with these drugs. Conversely, higher plasma concentrations of pevonedistat have been observed after combination with carboplatin plus paclitaxel (Swords et al., 2018; Lockhart et al., 2019).
Pharmacokinetic parameters of pevonedistat after 1 h intravenous infusion
The PK of the second-in-class adenosine sulfamate, TAK-243, has also been evaluated in immunodeficient mice bearing subcutaneous diffuse, large B-cell lymphoma tumors after intravenous administration of the drug at doses of 12.5–25 mg/kg. As opposed to the short t1/2 (0.2–0.4 hour) displayed by TAK-243 in the plasma, the tumor t1/2 is much longer (16.9–20.6 hour), reflecting the high volume of distribution and the higher exposure to the drug in tumors versus plasma (Hyer et al., 2018). The human PK of TAK-243 has also been reported among secondary outcome measures in a dose escalation study of the drug in advanced solid tumors (clinicaltrials.gov identifier: NCT02045095). Based on this study, the plasma t1/2 of TAK-243 is 7–9.4 hours after intravenous infusion of the drug at doses of 4–18 mg (Table 12). With respect to the SAE inhibitor TAK-981, there is no available PK data on this drug thus far.
Pharmacokinetic parameters of TAK-243 after 10 min intravenous infusiona
H. Insights into Clinical Trials of E1 Inhibitors
Of the four E1 inhibitors that have entered clinical trials so far (pevonedistat, TAK-243, TAK-981, and TAS4464), pevonedistat is the only drug for which clinical trial data has been published. It appears that pevonedistat shows more promising results when used in combinatorial versus single-agent regimens. In this respect, pevonedistat showed superior clinical benefits in combination with azacytidine in patients with AML and carboplatin plus paclitaxel in patients with advanced solid tumors (Swords et al., 2018; Lockhart et al., 2019). Many of these combinations are supported with preclinical studies showing synergistic antitumor effects with cytotoxic drugs as well as azacytidine (Smith et al., 2011; Jazaeri et al., 2013; Li et al., 2014b; Visconte et al., 2016). This suggests similar results could be obtained with other drug combinations that have shown preclinical evidence of synergism, such as belinostat, olaparib, vemurafenib, pomalidomide, and other conventional anticancer agents (Benamar et al., 2016; Leclerc et al., 2016; Zhou et al., 2016; Guo et al., 2017; Liu et al., 2019).
Better results have been observed with longer treatment durations of pevonedistat, and this could be achieved by dose adjustments and combination with low-toxicity drugs or drugs known to display toxicity profiles distinct from those of pevonedistat, as is the case with azacytidine (Swords et al., 2018). As E1 inhibitors are anticipated to have less favorable toxicity profile compared with other anticancer agents that selectively target aberrant oncogenic molecules, it is important to develop predictive biomarkers that identify patient cohorts most likely to benefit from these drugs and spare potential nonresponders from unnecessary toxicities. Clinical trials of pevonedistat in advanced solid tumors have shown that the expression of excision repair cross-complementation group 1 could potentially serve this role, as high expression levels positively correlated with clinical benefits and continuation of treatment when pevonedistat was combined with paclitaxel plus carboplatin. The need for such biomarkers is even more urgent with TAK-243 and TAK-981, which are expected to have more adverse effects compared with pevonedistat as a result of their broader pharmacologic effects (Lockhart et al., 2019). Potential biomarkers for E1 inhibitors are highlighted in section VII. E. Pharmacodynamic Activity of Adenosine Sulfamate E1 Inhibitors.
VIII. Concluding Remarks
Despite the anticipated toxicity of proteasome inhibitors, they have proved clinically useful in several malignancies, particularly MM. These clinical successes have fueled subsequent research endeavors to explore other components of the UPS including E1 enzymes that initiate the cascade and possess readily druggable active sites, two key attributes that position E1s as attractive therapeutic targets. Although extensively evaluated in preclinical settings and clinical trials, the first-in-class NAE inhibitor pevonedistat has not been approved for clinical use yet. However, it has reached an advanced stage of clinical development as a combinatorial therapy with azacitidine in certain types of AML. The clinical approval of pevonedistat will encourage further exploration of other E1 inhibitors, particularly those that target SAE and UBA1. It must be noted, however, that the adverse drug reactions expected with these inhibitors—given the broad and essential biologic roles of ubiquitylation and SUMOylation—may present several challenges that dictate identifying the patient cohorts who are most likely to benefit from these therapies. Equally encouraging is the unprecedented appreciation of immunotherapy as a promising cancer treatment and the emerging immunomodulatory functions of SUMOylation, ubiquitylation, neddylation, and ISGylation. Identifying these potential immunotherapeutic effects is actively pursued with TAK-981 and pevonedistat, and further investigation may be warranted with other E1 inhibitors. Although possible with UBA1, for which a selective, clinical-grade inhibitor is available, it may be challenging to interrogate these effects with other E1s with no known inhibitors available thus far. Therefore, the discovery of selective inhibitors of UBA6, UBA7, and noncanonical E1s is needed to interrogate many unexplored areas of E1 biology and therapeutic potential in cancer.
Authorship Contributions
Performed data analysis: Barghout.
Wrote or contributed to the writing of the manuscript: Barghout, Schimmer.
Footnotes
This work was supported by the Leukemia & Lymphoma Society of Canada, the Canadian Institutes of Health Research (CIHR), the Princess Margaret Cancer Centre Foundation, and the Ontario Ministry of Health and Long-Term Care. A.D.S. holds the Ronald N. Buick Chair in Oncology Research. S.H.B. was supported by the Ontario Trillium Scholarship, Department of Medical Biophysics Fellowship, Graduate Studies Endowment Fund (GSEF) Scholarship, and Queen Elizabeth II Scholarship from the Faculty of Medicine, University of Toronto, Canada.
A.D.S. has received honorariums or consulting fees from Novartis, Jazz, Otsuka, and Takeda Pharmaceuticals and research support from Medivir AB and Takeda Pharmaceuticals. A.D.S. owns stock in Abbvie Pharmaceuticals and is named on a patent application for the use of DNT cells for the treatment of leukemia.
↵
 This article has supplemental material available at pharmrev.aspetjournals.org.
This article has supplemental material available at pharmrev.aspetjournals.org.

Abbreviations
- AAD
- active adenylation domain
- ABP
- activity-based probe
- ABPA3
- activity-based probe A3
- AD
- adenylation domain
- ADS
- adenosine 5′-sulfamate
- AML
- acute myeloid leukemia
- AMSN
- 5′-(sulfonylaminodeoxy)adenosine
- APL
- acute promyelocytic leukemia
- APN
- adenosyl-phospho-NEDD8
- APU
- adenosyl-phospho-ubiquitinol
- ATG
- autophagy-related protein
- ATO
- arsenic trioxide
- ATRA
- all-trans retinoic acid
- CCD
- catalytic cysteine domain
- CDT1
- chromatin licensing and DNA replication factor 1
- cGAS
- cGMP-AMP synthase
- CNV
- copy number variation
- CRL
- cullin-RING ubiquitin ligase
- CTD
- C-terminal domain
- DUB
- deubiquitylase
- ECTD
- extreme C-terminal domain
- EGFR
- epidermal growth factor receptor
- ER
- endoplasmic reticulum
- FAT10
- human leukocyte antigen F-associated transcript 10
- HDAC
- histone deacetylase
- HIF
- hypoxia-inducible factor
- hLRH-1
- human liver receptor homolog-1
- HTS
- high-throughput screen
- IAD
- inactive adenylation domain
- IκBα
- inhibitor of NF-κBα
- IFN
- interferon
- IFNAR
- IFN-α receptor
- ISG
- interferon-stimulated gene
- MDS
- myelodysplastic syndrome
- MM
- multiple myeloma
- MOCS3
- molybdenum cofactor synthesis protein 3
- NAE
- NEDD8-activating enzyme
- NB
- nuclear body
- NEDD8
- neural precursor cell-expressed developmentally downregulated protein 8
- NEMO
- NF-κB essential modulator
- NF-κB
- nuclear factor κB
- NTD
- N-terminal domain
- PCNA
- proliferating cell nuclear antigen
- PE
- phosphatidylethanolamine
- PK
- pharmacokinetic
- PML
- promyelocytic leukemia
- RanGAP1
- Ran GTPase-activating protein 1
- RARA
- retinoic acid receptor α
- RHD
- rhodanese homology domain
- RING
- really interesting new gene
- ROS
- reactive oxygen species
- SAE
- SUMO-activating enzyme
- SAR
- structure-activity relationship
- SENP
- SUMO-specific protease
- SUMO
- small ubiquitin-like modifier
- t1/2
- terminal elimination half-life time
- TRAIL
- tumor necrosis factor–related apoptosis-inducing ligand
- UBA
- ubiquitin-like modifier–activating enzyme
- UBL
- ubiquitin-like protein
- UFC1
- UFM1-conjugating enzyme 1
- UFD
- ubiquitin-fold domain
- UFM1
- ubiquitin-fold modifier 1
- UFSP
- UFM1-specific protease
- UIS
- UFM1-interacting sequence
- ULP
- UBL-specific protease
- UPR
- unfolded protein response
- UPS
- ubiquitin-proteasome system
- URM1
- ubiquitin-related modifier 1
- USP
- ubiquitin-specific protease
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
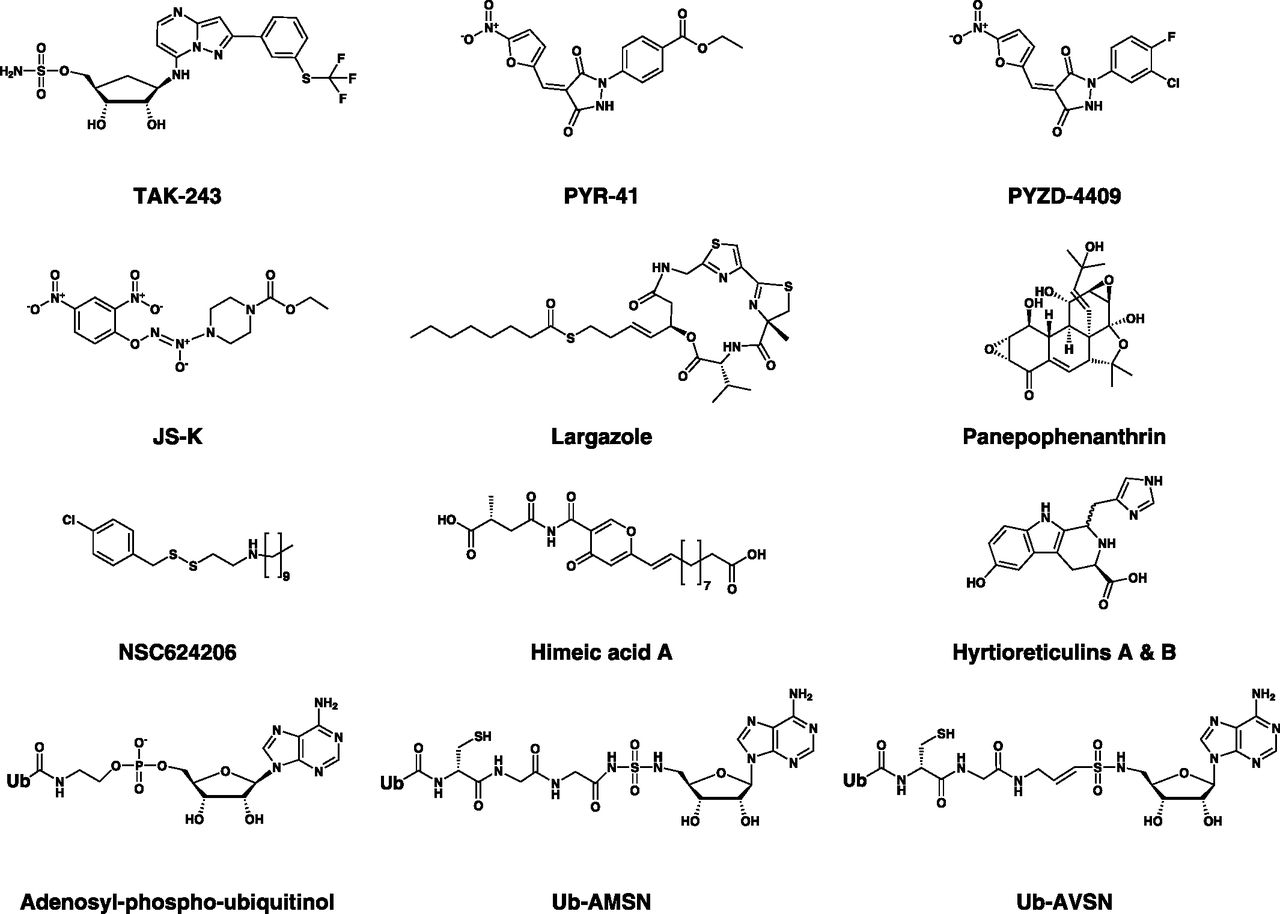{kind=link}
{kind=link}
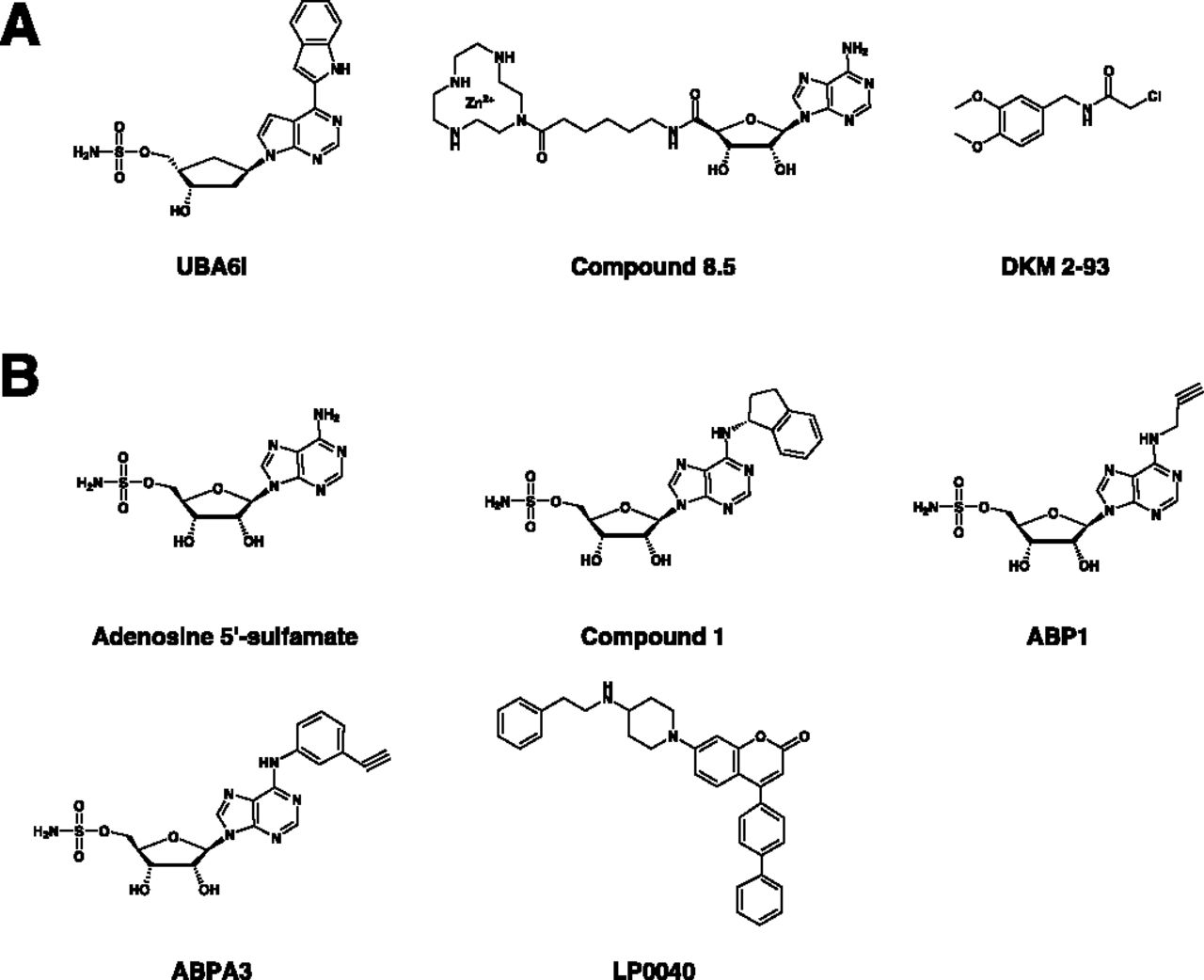{kind=link}
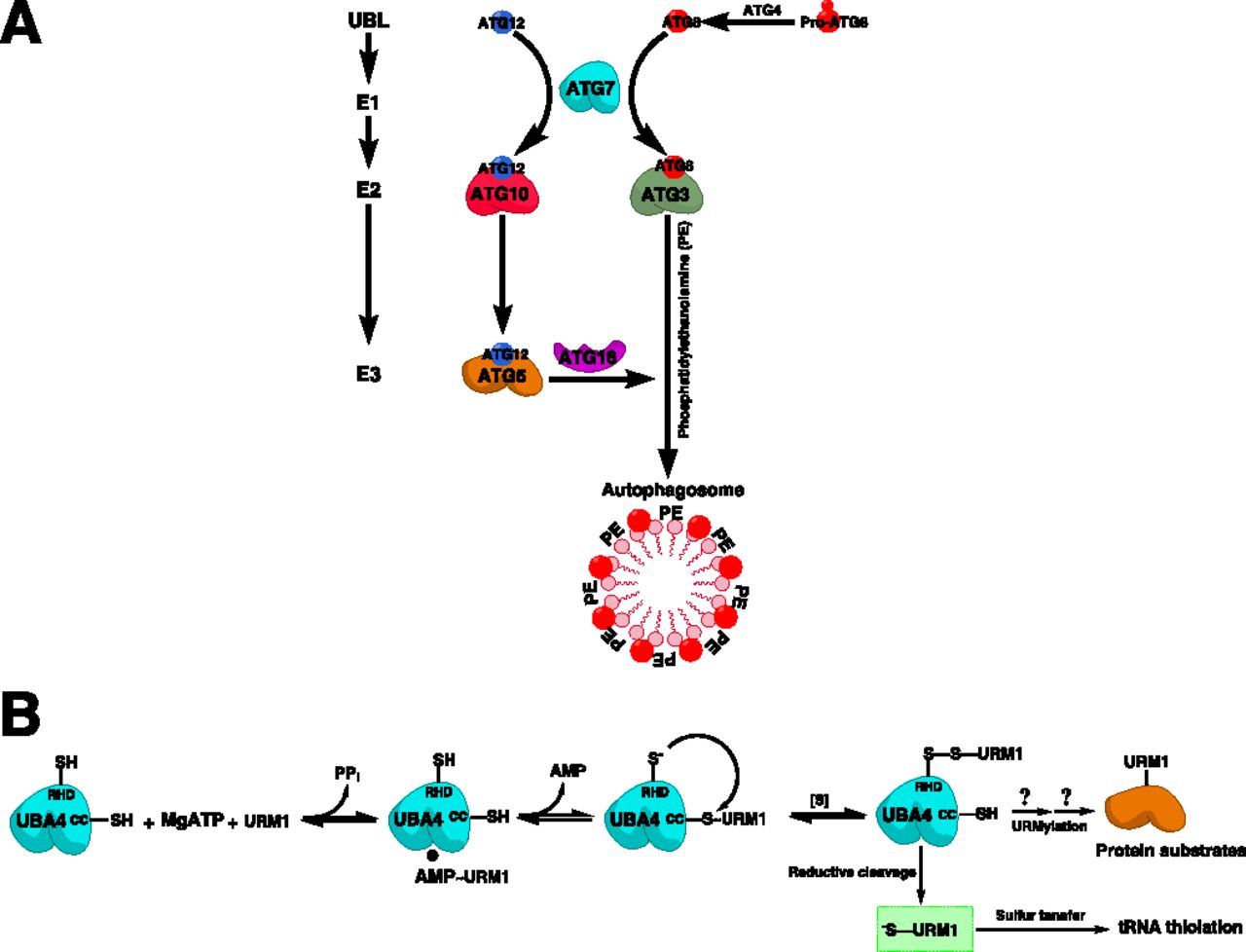{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Visual Overview
- Abstract
- I. Ubiquitin-Like Protein Conjugation System
- II. E1 Enzymes
- III. Tools and Assays for Exploring E1 Biology and Drug Discovery
- IV. Canonical E1 Enzymes
- V. Noncanonical E1 Enzymes
- VI. Dual and Multi-E1 Inhibitors
- VII. Adenosine Sulfamate E1 Inhibitors
- VIII. Concluding Remarks
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters
- PDF + SI