


Visual Overview

Abstract
G protein–coupled receptors (GPCRs) are a large family comprising >800 signaling receptors that regulate numerous cellular and physiologic responses. GPCRs have been implicated in numerous diseases and represent the largest class of drug targets. Although advances in GPCR structure and pharmacology have improved drug discovery, the regulation of GPCR function by diverse post-translational modifications (PTMs) has received minimal attention. Over 200 PTMs are known to exist in mammalian cells, yet only a few have been reported for GPCRs. Early studies revealed phosphorylation as a major regulator of GPCR signaling, whereas later reports implicated a function for ubiquitination, glycosylation, and palmitoylation in GPCR biology. Although our knowledge of GPCR phosphorylation is extensive, our knowledge of the modifying enzymes, regulation, and function of other GPCR PTMs is limited. In this review we provide a comprehensive overview of GPCR post-translational modifications with a greater focus on new discoveries. We discuss the subcellular location and regulatory mechanisms that control post-translational modifications of GPCRs. The functional implications of newly discovered GPCR PTMs on receptor folding, biosynthesis, endocytic trafficking, dimerization, compartmentalized signaling, and biased signaling are also provided. Methods to detect and study GPCR PTMs as well as PTM crosstalk are further highlighted. Finally, we conclude with a discussion of the implications of GPCR PTMs in human disease and their importance for drug discovery.
Significance Statement Post-translational modification of G protein–coupled receptors (GPCRs) controls all aspects of receptor function; however, the detection and study of diverse types of GPCR modifications are limited. A thorough understanding of the role and mechanisms by which diverse post-translational modifications regulate GPCR signaling and trafficking is essential for understanding dysregulated mechanisms in disease and for improving and refining drug development for GPCRs.
I. Introduction
G protein–coupled receptors (GPCRs) are the largest family of cell surface signaling receptors expressed in mammalian cells and control vast physiologic responses. Agonist activation of GPCRs results in coupling to heterotrimeric G proteins at the plasma membrane and in signaling from endosomes. Signaling by GPCRs is tightly controlled by desensitization, internalization, and lysosomal sorting. Dysregulation of GPCR signaling is prevalent in disease and has been largely attributed to either a deficiency in signaling or an overabundance of signaling responses. These features, combined with the high druggability of GPCRs, have made this receptor class the largest target of drugs, currently representing over 34% of all Food and Drug Administration–approved therapeutics (Hauser et al., 2017; Sriram and Insel, 2018). Despite recent advances in GPCR structure and pharmacology, one aspect of GPCR regulation that has remained largely ignored is the contribution of post-translational modifications.
GPCRs are synthesized on ribosomes, large macromolecular structures that are responsible for translating mRNA into nascent polypeptides. All GPCR proteins are modified at least once during their lifetime, and this occurs either cotranslationally during biosynthesis or post-translationally after synthesis and delivery to the cell surface. Post-translational modifications (PTMs), including cotranslational modifications, enable proper GPCR folding and maturation in the biosynthetic pathway as well as regulation of receptor stability and degradation. PTMs occur at amino acid side chains of the GPCR present in the N-terminus, extracellular loops (ECLs), intracellular loops (ICLs), transmembrane domain, and the C terminus (Fig. 1) via enzymatic activity. PTM enzymes are currently known to represent 5% of the human proteome and perform over 200 different types of modifications mediated by kinases, ligases, transferases and is a reversible process. Numerous studies have documented GPCR phosphorylation, ubiquitination, glycosylation, and palmitoylation, whereas there are far fewer reports of GPCR tyrosine sulfation, methylation, small ubiquitin-like modifier (SUMO)-ylation, and nitrosylation (Fig. 1). Although the number of GPCRs shown to be modulated by PTMs has rapidly increased over the last two decades, our knowledge of the types of GPCR modifications as well as the regulation and function of PTMs is limited.

GPCR post-translational modifications. GPCRs are seven-transmembrane proteins subjected to multiple types of PTMs on ECLs, ICLs, and the C-terminal domain. Here we show the most common sites of GPCR PTMs. PTMs that occur on ECLs include the following: N-glycosylation at asparagine (N)-X-serine (S)/threonine (T) sites, where X is any amino acid other than proline; O-glycosylation at S or T residues; and tyrosine (Y) sulfation. Nitrosylation has been shown to occur at transmembrane cysteine (C) residues. PTMs on intracellular loops include the following: SUMOylation on K residues present in the motif ψ-K-X-(D/E), where ψ is aliphatic amino acid, X is any amino acid, aspartic acid is D, and glutamic acid is E; methylation at arginine (R) residues of R-G-G or R-X-R sites, where glycine is G and X is any amino acid; and palmitoylation at cysteine (C). GPCR C-terminal PTMs include phosphorylation on S or T, rarely on Y residues, and ubiquitination (Ub) on specific K residues.
A major function of GPCR post-translational modifications is to control the spatial and temporal dynamics of receptor signaling and appropriate physiologic responses. This is best characterized for GPCR phosphorylation. Similar to phosphorylation, GPCR modification with ubiquitination, glycosylation, and palmitoylation controls the dynamics of cellular signaling and is a reversible and finely regulated process. Although our knowledge of the role of phosphorylation in regulating GPCR biology is extensive, we have a limited understanding of the regulation and diverse functions by which other established PTMs control GPCR signaling. Here a comprehensive overview of post-translational modifications of GPCRs is presented with a focus on newer discoveries that control signaling, beginning with phosphorylation, followed by ubiquitination, glycosylation, and palmitoylation and other rare PTMs (Fig. 1). A discussion of the enzymology for each GPCR PTM, subcellular localization of modifying enzymes, the function of PTMs, methods to study PTMs, and GPCR PTM crosstalk is also provided. We conclude with a discussion of GPCR PTM implications in human disease and drug discovery.
II. GPCR Phosphorylation
Phosphorylation is a major regulator of GPCR cell signaling dynamics and is the best studied post-translational modification for this large receptor family expressed in mammalian cells. Phosphorylation occurs through the kinase-catalyzed transfer of γ-phosphate from ATP primarily to serine and threonine residues of GPCRs and rarely on tyrosine residues (Fig. 2A). GPCR phosphorylation is mediated primarily by GPCR kinases (GRKs), a widely studied family of kinases, as well as second messenger kinases such as protein kinase A (PKA) and protein kinase C. Phosphorylation of GPCRs is a reversible process and mediated by phosphatases through poorly understood mechanisms (Fig. 2A). GPCR phosphorylation and function have been comprehensively reviewed elsewhere (Pitcher et al., 1998; Reiter and Lefkowitz, 2006; Moore et al., 2007; Gurevich and Gurevich, 2019). Here, we briefly discuss GPCR phosphorylation with a focus on recent discoveries.

GPCR phosphorylation. (A) Agonist-activated GPCRs are phosphorylated at the cell surface primarily by GRKs, commonly at the C terminus on serine (S) or threonine (T) residues. Dephosphorylation of GPCRs is carried out by phosphatases. (B) Activation of PAR1 results in rapid phosphorylation as detected by immunoprecipitated (IP) [32]P-labeled PAR1 and autoradiography after 3 minutes of stimulation with peptide agonist 100 μM SFLLRN of PAR1 expressed in Rat1 fibroblasts. In PAR1-expressing cells stimulated with SFLLRN peptide for 3 minutes, followed by wash and chase for 27 minutes without agonist, PAR1 phosphorylation was no longer detectable, whereas continuous stimulation with SFLLRN for 30 minutes sustained phosphorylation. These findings suggest that PAR1 is subjected to phosphorylation and dephosphorylation. PAR1 protein from IPs detected by immunoblot with PAR1 antibody as shown in the bottom panel. ab, antibody; PM, plasma membrane; SFLLRN, Ser-Phe-Leu-Leu-Arg-Asn.
A. GPCR Phosphorylation, GRKs, and Phosphatases
A family of seven GRKs are primarily responsible for GPCR phosphorylation. GRKs are serine/threonine kinases that exhibit tissue-specific expression. GRK1 and 7 are expressed in the visual system, GRK4 is expressed in the testes, and GRK2 and 3 and GRK5 and 6 are ubiquitously expressed, although certain tissues and cell types exhibit preferential expression of specific GRK subtypes (e.g., GRK2 and GRK5 are highly expressed in the heart). GRKs are exquisitely regulated and activated upon binding to agonist-activated GPCRs enabling the phosphorylation of receptor-specific serine and threonine residues (Fig. 3) (Gurevich and Gurevich, 2019), unlike many kinases that recognize consensus sequences within targeted proteins. However, GRKs must be brought in close proximity to the GPCR substrate embedded in the plasma membrane to facilitate phosphorylation, and this is accomplished through different mechanisms. GRK1 and 7 are post-translationally modified by prenylation, and GRK4, 5, and 6 either are palmitoylated or contain an amphipathic helix that binds membrane phospholipids and thereby are constitutively associated with the plasma membrane. GRK2 and 3 are actively recruited to the GPCR after G protein activation and release of βγ-subunits, which bind the pleckstrin homology domain of GRK2 and 3. Besides GRKs, other kinases are known to specifically phosphorylate GPCRs on serine or threonine residues, such as the second messenger kinases PKA, protein kinase C, and casein kinase II, known to target conserved motifs for phosphorylation, that occur either basally or after agonist stimulation, but far less is known about unifying regulatory mechanisms, and phosphorylation appears to be receptor specific.

Model of GPCR regulation by phosphorylation. The schematic presents a classic view of GPCR regulation by phosphorylation in the cell. Agonist activation of a GPCR causes a conformational change that facilitates coupling to heterotrimeric G proteins (α, β, γ) and initiation of intracellular signaling cascades. Subsequently, GRKs are recruited and phosphorylate activated GPCRs at the C terminus, resulting in increased affinity and binding of β-arrestins (β-arr). β-arrestins compete with G protein binding to the same interhelical cavity localized within the cytoplasmic region of the GPCR. Once bound to the GPCR, β-arrestins prevent G protein coupling (desensitization) and facilitate association with clathrin and the endocytic machinery to promote internalization. Clathrin-coated pits bud inward and pinch off from the plasma membrane to form endocytic vesicles or endosomes. Once internalized, phosphorylation controls GPCRs’ capacity to nucleate the assembly of an endosomal β-arrestin signaling complex or, if dephosphorylated, GPCRs recycle from endosomes and return to the plasma membrane resulting in resensitization.
GPCRs are phosphorylated on multiple residues within the C-terminal tail and on the ICLs (Tobin et al., 2008). In some cases, GPCR phosphorylation is sequential as demonstrated for rhodopsin and other GPCRs. The chemokine receptor N-formyl peptide receptor C5a receptor is basally phosphorylated, which appears to prime the receptor for ligand-stimulated phosphorylation (Schreiber et al., 1994; Milcent et al., 1999). Similarly, basal phosphorylation of the bradykinin B2 receptor is required for subsequent agonist-induced phosphorylation (Blaukat et al., 2001). In contrast, several GPCRs display hierarchal agonist-induced phosphorylation, including the δ-opioid receptor (DOR) (Kouhen et al., 2000) and the muscarinic M3 receptor (Torrecilla et al., 2007).
Phosphorylation of GPCRs is reversible. As shown here, agonist-induced protease-activated receptor (PAR)-1 phosphorylation is rapid, occurs within minutes, and is reversed after agonist removal (Fig. 2B), whereas continuous exposure to agonist results in sustained phosphorylation for at least 30 minutes (Fig. 2B). Unlike agonist-induced GPCR phosphorylation, the regulatory mechanisms that control GPCR dephosphorylation remain poorly understood. This is due in part to the complexity of protein phosphatases (PPs) that exist as multisubunit enzymes and the variety of PPs expressed in mammalian cells, including PP1, PP2A, PP2B (calcineurin), PP3, PP4, and PP5. Protein phosphatase 2A was initially implicated in dephosphorylation of β2-adrenoreceptor after agonist stimulation (Pitcher et al., 1995; Krueger et al., 1997), whereas other activated GPCRs appear to be dephosphorylated by PP1α, β, and γ catalytic subunits as well as PP2A and PP2B phosphatases. Dephosphorylation of GPCRs can occur at the plasma membrane and/or on intracellular vesicles (Kliewer et al., 2017) and generally appears to regulate receptor recycling, resensitization, and cellular responsiveness (Fig. 3). Currently, however, there is limited understanding of the mechanisms of PP recruitment, regulation, and the impact on the spatial and temporal dynamics of GPCR signaling.
B. GPCR Phosphorylation, Desensitization, and Internalization
The major function of GPCR phosphorylation is to promote the recruitment of β-arrestins, which are multifunctional adaptor proteins that were first shown to mediate receptor uncoupling from G protein signaling or desensitization and subsequently demonstrated to promote receptor internalization (Fig. 3). The arrestin family includes two visual arrestins and two ubiquitously expressed β-arrestins, termed β-arrestin-1 and -2, that are highly conserved and share high sequence homology. After agonist activation, β-arrestins are recruited to activated phosphorylated GPCRs through a multistep process that results in β-arrestin conformational rearrangements that allow competition with G protein binding to the same interhelical cavity localized within the cytoplasmic region of the GPCR (Gurevich and Gurevich, 2006). In addition, β-arrestin engagement with activated phosphorylated GPCRs is also mediated by receptor-associated phosphates, indicating that two distinct features of the receptor control β-arrestin–GPCR engagement (Fig. 3). This creates high-affinity binding that easily competes with G proteins, which when bound to GTP readily dissociate from the receptor.
The diversity of β-arrestin function was established by the demonstration that β-arrestins not only control GPCR desensitization but also interact with the endocytic machinery to promote GPCR internalization (Fig. 3). During β-arrestin activation, the C-terminal bound to the polar core of β-arrestin is displaced by the binding of the GPCR phosphorylated C-tail (Peterson and Luttrell, 2017). The release of the β-arrestin C terminus then enables engagement with the clathrin endocytic machinery through binding to both clathrin and the clathrin adaptor protein complex-2 (AP-2) via recognition of consensus clathrin and AP-2 binding motifs present in the C terminus of β-arrestin (Goodman et al., 1996; Laporte et al., 1999). Ultimately, this results in activated GPCR recruitment to clathrin-coated pits and internalization from the plasma membrane (Fig. 3). Generally, internalization of activated GPCRs serves to terminate G protein signaling; however, new evidence suggests that at least for certain GPCRs, internalization permits a second phase of signaling mediated by either G proteins or β-arrestins on endocytic vesicles, as discussed below (Fig. 3) (Lobingier and von Zastrow, 2019; Weinberg et al., 2019).
C. Phosphorylation-Driven GPCR Signaling
Although the major function of agonist-induced GPCR phosphorylation historically has been ascribed to turning off signaling mediated by rapid desensitization via uncoupling from G proteins as well as internalization and downregulation, which removes the activated receptor from G protein effectors at the plasma membrane (Fig. 3) (DeWire et al., 2007), previous studies indicate that receptor phosphorylation can initiate a new type of signaling. The first example of GPCR phosphorylation initiation of signaling was described for β2-adrenoceptor C-tail phosphorylation by PKA, which results in uncoupling to Gs protein and increased coupling to Gi protein (Daaka et al., 1997; Zamah et al., 2002). In newer work, phosphorylation-driven GPCR signaling is mediated primarily by the engagement and conformation of the activated GPCR-bound β-arrestin protein (Fig. 3). A substantial literature supports the notion that β-arrestins can mediate signaling (Peterson and Luttrell, 2017; Gurevich and Gurevich, 2019); however, the precise mechanism that controls β-arrestin–dependent signaling remains controversial (O’Hayre et al., 2017; Luttrell et al., 2018). Regardless, β-arrestin–mediated signaling is diverse and differs for the receptor and cellular contexts. In some cases, activated GPCRs engage β-arrestins to promote Src-dependent signaling and function as scaffolds to promote activation of various mitogen-activated protein (MAP) kinase signaling cascades (Peterson and Luttrell, 2017; Gurevich and Gurevich, 2019). Interestingly, recent studies indicate that certain GPCRs can bind β-arrestin and G protein simultaneously via distinct interaction sites; this is particularly relevant for receptors that contain clusters of serine and threonine phosphorylation sites within the C-tail domain (Thomsen et al., 2016; Cahill et al., 2017). A subsequent study nicely demonstrates that an active chimeric β2-adrenoceptor fusion with the C-tail of the vasopressin V2 receptor is capable of activating both G protein and β-arrestin to facilitate sustained internalized G protein signaling from endosomes (Nguyen et al., 2019). Moreover, different phosphorylation patterns on the activated GPCR C-tail appear to generate distinct phosphorylation bar codes that can induce conformationally unique active states of arrestins that govern different cellular responses (Shukla et al., 2008; Butcher et al., 2011; Liggett, 2011; Mayer et al., 2019). These studies indicate that the spatial and temporal diversity of GPCR signaling is driven in part by β-arrestin–mediated signaling, which is primarily controlled by GPCR phosphorylation that regulates the recruitment and activation of β-arrestin functionality.
D. Detection and Study of GPCR Phosphorylation
The study of GPCR phosphorylation has been enabled by the ability to detect and control receptor phosphorylation using a variety of approaches. Computational methods to predict GPCR phosphorylation sites and cognate kinases such as NetPhos3.1, GPS (Group-Based Phosphorylation Scoring), Scansite, PHOSIDA (Phosphorylation Site Database), dbPAF (Database of Phospho-Sites in Animals and Fungi), and Musite are currently available (Blom et al., 1999, 2004; Obenauer et al., 2003; Xue et al., 2005; Shukla et al., 2008; Gnad et al., 2011; Ullah et al., 2016). One conventional and established methods for detection of GPCR phosphorylation is through metabolic labeling of cultured mammalian cells with [32P] orthophosphate (Fig. 2B). These assays require the use of radioactive inorganic phosphate and receptor-specific antibodies for immunoprecipitation, which is required for enrichment of phosphorylated receptors, provides a global assessment of GPCR phosphorylation, and has been demonstrated for numerous GPCRs, including PAR1 (Fig. 2B) (Trejo et al., 1996), and well documented in several classic reviews (Sibley et al., 1987; Benovic et al., 1990). A second approach to determine more detailed information about the pattern of phosphorylation of GPCR is two-dimensional peptide mapping as described by Prihandoko et al. (2015). This method also requires metabolic labeling with [32P] orthophosphate, immunoprecipitation, and proteolytic digestion and thin layer chromatography. However, the disadvantage of these techniques is that neither provides information about the actual sites of phosphorylation, which requires mass spectrometry.
Although significant advancements in mass spectrometry have enabled the detection of GPCR phosphorylation, it has been hampered by requiring large amounts of receptor, which is challenging for a receptor class that is typically expressed in cells at low levels. Nonetheless, mass spectrometry has been used to detect phosphorylation of several GPCRs, including the β2-adrenoceptor (Trester-Zedlitz et al., 2005) and muscarinic M3 receptor (Butcher et al., 2011). Indeed, a mass spectrometry–based quantitative proteomic approach was used to map β2-adrenoceptor phosphorylation sites induced by biased agonists and linked the distinct patterns phosphorylation sites to GRKs and β-arrestin function, establishing the “bar code” hypothesis (Nobles et al., 2011). The value of identifying specific sites of GPCR phosphorylation is that it permits the generation of GPCR phospho-specific antibodies, which will greatly increase the study of GPCR phosphorylation. In addition to phosphorylation, the study of GPCR dephosphorylation has been conducted using PP inhibitors such as okadaic acid and calyculin, which target both PP2A and PP1, siRNA-mediated depletion of specific phosphatases, and phospho-specific GPCR antibodies. Detailed methods for the determination of GPCR phosphorylation are described in Prihandoko et al. (2015).
III. GPCR Ubiquitination
Post-translational modification with ubiquitin is best known to target proteins for degradation via the 26S proteasome and the lysosome (Hershko and Ciechanover, 1998). Ubiquitin is a small 76 amino acid protein, ∼8 kDa protein, that is covalently linked to lysine residues of substrate proteins through the sequential action of three distinct enzymes, E1, E2, and E3 (Fig. 4A). Although most proteins are predicted to be modified with ubiquitin at least once during their lifetime, only a small subset of approximately 40 GPCRs have been reported to be ubiquitinated (Jean-Charles et al., 2016). In most of the studies, ubiquitin appears to function mainly as a signal that facilitates GPCR trafficking within the endosomal-lysosomal pathway or targeting to the proteasome (Petaja-Repo et al., 2000; Katzmann et al., 2001; Milojevic et al., 2006). However, new emerging studies indicate that for certain GPCRs, ubiquitin promotes direct interaction with signaling effectors (Grimsey et al., 2015). Thus, the function of ubiquitination may vary depending on the GPCR, cell type, and physiologic function, as discussed below.

Ubiquitin modifying enzymes, ubiquitin linkages and detection. (A) Ubiquitination of substrate proteins is carried out sequentially by a ubiquitin (Ub)–activating enzyme E1, ∼38 ubiquitin-conjugating enzyme E2s, and >600 ubiquitin ligase E3 enzymes. Ubiquitin is enzymatically cleaved by ∼100 deubiquitinases to release ubiquitin back to cytosolic pool. (B) GPCRs are modified with different ubiquitin conjugations, including monoubiquitin (single or multiple monoubiquitin) and K48- or K63-linked polyubiquitin, which regulate distinct functions. Nonlysine ubiquitination has also been reported to occur on GPCRs. (C) Ubiquitination of endogenous PAR1 ubiquitination in endothelial cells after 7-minute stimulation with 10 nM thrombin (α-Th) detected by immunoblotting of immunoprecipitated (IP) PAR1 using anti-pan ubiquitin P4D1 antibody that detects multiple Ubn species. N-terminal proteolytic cleavage of PAR1 by thrombin results in reduced protein size of total protein detected by immunoblotting with PAR1 antibody (ab), bottom panel.
A. Ubiquitination of GPCRs
Ubiquitination is mediated by three types of enzymes: ubiquitin-activating enzymes E1, ubiquitin-conjugating enzymes E2, and ubiquitin ligases E3 (Fig. 4A). Ubiquitin is initially covalently attached by its C-terminal glycine to a cysteine residue of the E1 enzyme and then shuttled to a cysteine residue of the ubiquitin-conjugating E2 enzyme. The E2 then binds to the E3 ligase, which directly interacts with the substrate protein and covalently attaches ubiquitin. E3 ubiquitin ligases are critical components of this system since they recognize substrates and thereby provide specificity to the ubiquitination reaction (Fig. 4A). The human genome encodes two E1 enzymes, at least 38 E2s, and >600 E3 ubiquitin ligases (Zheng and Shabek, 2017). The E3 ubiquitin ligases encoded in the human proteome are divided into three classes and include 1) really interesting new gene (RING)–type E3 ligases, which represents the largest family with >600 members; 2) homologous to E6AP C terminus (HECT)–type E3s, comprising 28 members; and 3) RING between RING–type E3s, with 14 members (Dove and Klevit, 2017; Reiter and Klevit, 2018). GPCRs are distinctly regulated by E3 ubiquitin ligases via diverse mechanisms.
1. E3 Ubiquitin Ligases and GPCRs
E3 ligases of the RING finger family are unique and simultaneously bind to both the charged E2 and the substrate, thereby facilitating the direct transfer of the ubiquitin moiety to the substrate. A notable member of RING family is Casitas B lineage lymphoma, which ubiquitinates protease-activated receptor-2 (Jacob et al., 2005). In contrast to RING-types, the HECT-type E3 ligases interact with the E2s, which facilitates the transfer ubiquitin to an active site cysteine residue within the E3 HECT catalytic domain. The ubiquitin is subsequently conjugated to lysine acceptor sites of the substrate protein. Of the HECT E3 ubiquitin ligases, the neural precursor cell expressed, developmentally downregulated (NEDD)-4 subfamily is best known to regulate GPCR trafficking. The NEDD4 family contains nine members, including NEDD4, NEDD4-2, atrophin-1–interacting protein-4 (AIP4), WW domain–containing E3 ubiquitin protein ligase (WWP) 1, WWP2, Sma- and Mad-related protein-specific E3 ubiquitin ligase proteins 1 and 2, and NEDD-like ubiquitin protein ligases 1 and 2 (Weber et al., 2019). All NEDD4 family members share a similar domain structure, including an amino-terminal C2 domain, three to four WW domains, and a carboxy-terminal catalytic HECT domain. The first authenticated examples of HECT-type E3 ligase–mediated ubiquitination of mammalian GPCRs include AIP4-mediated ubiquitination of the C-X-C chemokine receptor (CXCR) 4 (Marchese et al., 2003) and the β2-adrenoceptor (Shenoy et al., 2001, 2008). In both cases, agonist-induced GPCR ubiquitination mediates endolysosomal trafficking and receptor degradation. Finally, the RING between RING type of E3 ubiquitin ligases regulate diverse cellular processes (Dove and Klevit, 2017) with the most notable being Parkin, which expedites the clearance of damaged mitochondria via a process called mitophagy (Lazarou et al., 2012). In addition, Parkin mediates endoplasmic reticulum (ER)–associated protein degradation (ERAD) of the class A orphan G protein–coupled receptor 37 (GPR37) that regulates ER stress in Parkinson’s disease (Berger et al., 2017). GPR37 also functions as an ER chaperone for the Wnt co‐receptor lipoprotein receptor-related protein 6 in neuronal progenitor cells, emphasizing the importance of regulating GPR37 function at the ER (Imai et al., 2001; Berger et al., 2017).
2. GPCR Regulation of E3 Ligase Activity
Mammalian GPCRs are differentially modified with ubiquitin in space and time. This suggest that distinct mechanisms likely exist to control the diverse functions of E3 ligases in regulating GPCR ubiquitination. In most studies, regulation of HECT domain–containing NEDD4 E3 ubiquitin ligase activity appears to occur through recruitment of the E3 ligase to the GPCR substrate either directly through noncanonical WW domain–mediated interactions, as demonstrated for the CXCR4 and E3 ligase AIP4 (Bhandari et al., 2009), or indirectly through interactions with adaptor proteins, mainly β-arrestin recruitment of NEDD4 to β2-adrenoceptor (Shenoy et al., 2008) and metabotropic glutamate (mGlu)7 receptor (Lee et al., 2019). The less studied, mammalian α-arrestin domain-containing proteins (ARRDCs) may also function as adaptors to recruit E3 ligases to GPCRs (Alvarez, 2008). Several studies indicate that NEDD4 E3 ligases are exquisitely regulated through release of autoinhibition, which can occur through allosteric interactions (Rotin and Kumar, 2009) or via ligand-induced phosphorylation (Persaud et al., 2014) specifically at tyrosine residues (Chen et al., 2017b). A recent study demonstrated that endothelial GPCRs can also regulate NEDD4 E3 ligase activity by release of autoinhibition. In this work, thrombin-activated PAR1 stimulates c-Src–mediated phosphorylation of NEDD4-2 at tyrosine (Y)485 located within the autoinhibitory 2,3- linker peptide between WW domains 2 and 3, leading to its activation and ubiquitination of PAR1 (Fig. 4C) (Grimsey et al., 2018). This ultimately results in activated PAR1-stimulated p38 MAP kinase activation and regulation of endothelial inflammatory responses (Grimsey et al., 2018). The purinergic P2Y1 receptors also required c-Src and NEDD4 tyrosine phosphorylation for endothelial inflammatory signaling (Grimsey et al., 2018).
3. Ubiquitin Linkages and GPCRs
GPCR ubiquitination occurs on intracellular loops and on the C-terminal tail (Komander and Rape, 2012). GPCRs can be modified at one or multiple lysine residues with either monoubiquitin, as shown for the yeast α-factor receptor Sterile 2 (Ste2 or Ste2p), and CXCR4 or via polyubiquitin chains such as K63-linked ubiquitin for PAR1 (Fig. 4, B and C) (Grimsey et al., 2015), which can differentially affect receptor function. Ubiquitin chains are formed through ubiquitin linkages at several lysine sites within the ubiquitin molecule, including well characterized branched K48-, K63-, and linear N-terminal methionine–linked ubiquitin, offering numerous possibilities of ubiquitin polymer assembly (Fig. 4B) (Peng et al., 2003). In general, monoubiquitin and K63-linked ubiquitin predominate as sorting signals for GPCRs within the endocytic pathway (Terrell et al., 1998; Gulia et al., 2017). However, for certain GPCRs, K63 ubiquitin linkage has been recently implicated in regulating signaling from endosomes (Grimsey et al., 2015). Ubiquitin K48 and K11 linkages serve as a potent proteasomal degradation signals, whereas K29 and K63 linkages function to target substrate proteins for degradation via autophagy (Mukhopadhyay and Riezman, 2007). However, as an internalization signal, a single ubiquitin is exceptionally weak, and ubiquitin is likely operational only as polyubiquitin chains (Barriere et al., 2006). In the case of proteasomal degradation, proteins bearing chains of at least four ubiquitin molecules are the preferred substrates of the 26S proteasome (Chau et al., 1989). Recently, linkage of ubiquitin moieties to nonlysine nucleophilic residues such serine, cysteine, and threonine residues, as well as the free amino group of the N-terminus of proteins, has been demonstrated (Fig. 4B) (McDowell and Philpott, 2016). However, conjugates generated from ubiquitination on nonlysine residues are thermodynamically less stable than those generated on canonical lysine residues (McClellan et al., 2019). A recent study showed that a lysine-deficient dopamine D4 receptor was ubiquitinated on cytoplasmic serine and threonine residues that is important for regulating proteasomal degradation (Skieterska et al., 2015; Peeler et al., 2017). Whether nonlysine ubiquitination of GPCRs is common to other GPCRs remains to be determined.
B. Ubiquitin-Driven GPCR Trafficking
The control of GPCR cellular signaling dynamics is extensively regulated and ultimately governed by receptor expression and activity. Tight regulation of GPCR activity is achieved in part by ubiquitin-dependent receptor trafficking (Fig. 5). GPCRs are generally subjected to two modes of ubiquitination—constitutive or basal ubiquitination and ligand-induced ubiquitination—during their lifetime (Dores and Trejo, 2012).

Model of GPCR regulation by ubiquitination. Biosynthesis and folding of GPCRs is monitored with stringent quality-control machinery in the ER, which targets misfolded GPCRs for ubiquitination and degradation through the ERAD-proteosomal pathway that releases ubiquitin (Ub) back to the cytosol. Properly folded GPCRs are delivered to the plasma membrane, where GPCRs are targeted for ubiquitination either basally or after agonist stimulation. Ubiquitination of GPCRs by E3 ligases has been implicated in agonist-induced internalization or basal receptor retention at the plasma membrane. Once internalized, ubiquitinated GPCR has multiple fates, including 1) recycling back to the plasma membrane, initiated by the action of deubiquitinases; 2) targeting for lysosomal degradation; and 3) ubiquitin-driven endosomal signaling.
1. GPCR Biosynthesis and Cell Surface Expression
Constitutive or basal GPCR ubiquitination functions primarily to control receptor trafficking through the biosynthetic pathway. During biogenesis, newly synthesized GPCRs are folded in the ER with the assistance of chaperone proteins, undergo maturation in the Golgi, and then traffic to the plasma membrane (Fig. 5). However, misfolded or incompletely folded GPCRs are polyubiquitinated, retro-translocated from the ER to the cytosol via the ERAD quality control system, and then shuttled to the proteasome for proteolytic degradation (Fig. 5) (Petaja-Repo et al., 2001; Cook et al., 2003; Huang et al., 2006). Multiple GPCRs have been shown to be modified with K48 polyubiquitin chains during biogenesis and targeted to the 26S proteasome for degradation, often resulting from receptor mutations that can underlie the basis for disease, as reported for the visual GPCR rhodopsin and the vasopressin V2 receptor (Conn et al., 2007; Robben et al., 2009; Athanasiou et al., 2018). This has prompted the development of small molecules or pharmacoperones that bind to GPCRs and correct misfolding of mutant receptors (Nakamura et al., 2010) and has been well described for the gonadotropin-releasing hormone receptor (Bernier et al., 2004; Conn et al., 2007). Thus, at the very beginning of the GPCR life cycle, ubiquitination has an important role in regulating receptor biosynthesis.
In addition to biosynthesis, basal ubiquitination of GPCRs is important for regulating receptor expression at the plasma membrane. This has been illustrated for several GPCRs including PAR1, CXCR7, and chemokine (C-C motif) receptor (CCR) 7 (Moriyoshi et al., 2004; Wolfe et al., 2007; Canals et al., 2012). In the case of PAR1, ubiquitination occurs basally and negatively regulates constitutive internalization, thereby increasing cell surface expression (Wolfe et al., 2007). Constitutive internalization of PAR1 is mediated by the clathrin AP-2, where the μ2-adaptin subunit of AP-2 binds to the C-tail of PAR1 via interaction with a classic tyrosine-based motif (Y420KKL423) rather than β-arrestins (Paing et al., 2006; Wolfe et al., 2007). Ubiquitination of PAR1 occurs on C-tail lysine residues that has been confirmed by site-directed mutagenesis (Wolfe et al., 2007) and mass spectrometry high-throughput discovery-based analysis, PhosphoSitePlus (Hornbeck et al., 2015), that reside within the tyrosine-based motif and precludes binding of the μ2-adaptin subunit (Hornbeck et al., 2015). Moreover, a lysineless ubiquitin-deficient PAR1 mutant displayed enhanced internalization that was reversed by the fusion of a single ubiquitin moiety to the C-terminal tail, suggesting that ubiquitination is important for retaining PAR1 on the cell surface (Wolfe et al., 2007). Similar to PAR1, basal ubiquitination of the chemokine receptor CXCR7 occurs on a lysine residue located within the C-tail region and is deubiquitinated after agonist activation through a process that requires phosphorylation and β-arrestin recruitment (Canals et al., 2012). The immune cell expressed CCR7 chemokine receptor is also constitutively modified with K63-linked polyubiquitination and regulates basal trafficking of CCR7 (Schaeuble et al., 2012). A mutant CCR7 defective in ubiquitination alters the spatial distribution of the receptor and impairs immune cell migration (Schaeuble et al., 2012). Thus, basal ubiquitination of GPCRs is important for the appropriate spatial subcellular localization, which has a critical role in governing cellular behavior.
2. Endocytosis of GPCRs
GPCR trafficking through the endocytic pathway is a highly conserved process that includes internalization, recycling, and lysosomal sorting and is regulated by ubiquitination (Fig. 5). A function for ubiquitination in GPCR endocytosis was first described for the yeast Saccharomyces cerevisiae α-mating factor GPCR Ste2 (Rohrer et al., 1993). Subsequent studies identified Rsp5, an ortholog of Nedd4-like HECT domain E3 ubiquitin ligases, as the key regulator of ligand-induced Ste2p ubiquitination, internalization, and targeting to vacuoles, an organelle equivalent to the mammalian lysosome (Hicke and Riezman, 1996; Dunn and Hicke, 2001). Unlike yeast, most mammalian GPCRs are internalized through clathrin-coated pits via a β-arrestin–dependent pathway. β-arrestins act as endocytic adaptors by binding directly to the clathrin heavy chain and to the β-adaptin subunit of AP-2 (Goodman et al., 1996; Laporte et al., 1999; Gaidarov and Keen, 1999). Interestingly, yeast lack β-arrestins and rather express a family of ARTs (arrestin-related trafficking proteins) that mediate recruitment of E3 ligases to facilitate internalization of several membrane-spanning proteins, including Ste2 (Alvaro et al., 2014). However, mammalian GPCRs do not require ubiquitination for efficient endocytosis in most cases; in fact, the prevention of ubiquitination has minimal or no effect on endocytosis of a number of GPCRs, including the β2-adrenoceptor, DOR, and neurokinin 1 (NK1) receptor (Shenoy et al., 2001; Tanowitz and Von Zastrow, 2002; Hanyaloglu et al., 2005). However, not all GPCRs require β-arrestins for endocytosis, and this is best exemplified for PAR1 (Paing et al., 2002). Instead of β-arrestins, PAR1 requires the clathrin adaptors AP-2 and epsin-1 for efficient internalization (Chen et al., 2011). In this case, AP-2 recognizes activated PAR1 phosphorylation sites within the C-tail region rather than the tyrosine-based motif, whereas epsin-1 requires both the ubiquitin-binding motifs of epsin-1 and PAR1 ubiquitination to facilitate efficient endocytosis (Chen et al., 2011).
3. Lysosomal Sorting of GPCRs
The best characterized function of ubiquitination is to target activated receptors to lysosomes for degradation (Fig. 5). The β2-adrenoceptor was the first mammalian GPCR shown to exhibit ligand-dependent ubiquitination and degradation (Shenoy et al., 2001), and this was closely followed by a report of ligand-induced ubiquitination and degradation of the CXCR4 chemokine receptor (Marchese and Benovic, 2001). Initiation of ligand-induced ubiquitination of GPCRs occurs at the plasma membrane, generally requiring receptor phosphorylation and β-arrestin recruitment. Isoproterenol-stimulated β2-adrenoceptor is rapidly phosphorylated, which enhances β-arrestin–mediated recruitment of the NEDD4 E3 ubiquitin ligase (Shenoy et al., 2001, 2008). Mass spectrometry analysis of agonist-induced β2-adrenoceptor ubiquitination revealed the major sites for polyubiquitination reside in ICL3 at K263 and K270 and in the C-tail at K348, K372, and K375 also modified with polyubiquitination (Xiao and Shenoy, 2011), later identified to be K63-type ubiquitin linkages (El Ayadi et al., 2018). A β2-adrenoceptor variant in which all the phosphorylation sites are mutated showed impaired ubiquitination as well as significantly reduced β-arrestin interaction (DeWire et al., 2007). In contrast to the β2-adrenoceptor, CXCR4 displays ligand-induced monoubiquitination (Marchese and Benovic, 2001). The CXCR4 receptor contains two serine residues, S324 and S325, located within the C-tail degradation motif, which are rapidly phosphorylated by agonist activation (Busillo et al., 2010). Agonist-induced ubiquitination of CXCR4 mediated by the E3 ubiquitin ligase AIP4 targets CXCR4 for lysosomal degradation (Marchese et al., 2003). Similarly, thrombin activation of PAR1 results in rapid modification with K63-ubiquitin linkages mediated by the recruitment and activation of NEDD4-2 initiated at the plasma membrane (Fig. 4C) (Grimsey et al., 2015, 2018). Although ligand-induced ubiquitination of GPCRs ultimately controls lysosomal degradation, the time scales for GPCR degradation are vastly different. Activated PAR1 is sorted rapidly to lysosomes and degraded within minutes (Trejo et al., 1998; Trejo and Coughlin, 1999), whereas CXCR4 ubiquitination and degradation occurs much later, within 3–6 hours (Marchese and Benovic, 2001). In contrast, β2-adrenoceptor is rapidly ubiquitinated, but lysosomal degradation is protracted and occurs after prolonged 6–24 hours of isoproterenol stimulation (Shenoy et al., 2008). Ubiquitination of most classic GPCRs facilitates engagement with the endosomal sorting complex required for transport (ESCRT)-0, -I, -II, and -III machinery. The ubiquitin-binding ESCRT components function sequentially to sort GPCRs from endosomes to multivesicular bodies (MVBs) or lysosomes for degradation. The importance of ubiquitin and ESCRTs in receptor lysosomal degradation has been illustrated for several GPCRs. As discussed above, ubiquitination of CXCR4 facilitates lysosomal sorting and requires AIP4-mediated ubiquitination of the ESCRT-0 protein, hepatocyte growth factor–regulated tyrosine kinase substrate (HRS), and vacuolar protein sorting 4 (Vps4), an ATPases associated with diverse cellular activities (AAA)-ATPase (Marchese et al., 2003). Agonist-induced ubiquitination of PAR2 also requires HRS for lysosomal degradation (Hasdemir et al., 2007). Although ubiquitination plays an important role in GPCR lysosomal degradation, there are examples of receptors that can efficiently sort to lysosomes in a ubiquitin-independent manner, as exemplified by DOR. A ubiquitination-deficient DOR mutant is efficiently sorted to the limiting membrane of intralumenal vesicles (ILVs) of MVBs, where extensive proteolytic fragmentation of the receptor ectodomain occurs (Henry et al., 2011). Sorting of DOR to ILVs or MVBs requires HRS and Vps4 but not the ESCRT-I component, tumor susceptibility gene 101 (Hislop et al., 2004), indicating that ubiquitin-independent receptor sorting requires some but not all components of the ubiquitin-binding ESCRT machinery. Ubiquitination of μ-opioid receptor (MOR) mediates ESCRT-dependent degradation by controlling receptor distribution between the limiting endosome membrane and lumen but is not required for receptor delivery to the proteolytic compartments. Instead, this is dictated by the MOR C-terminal tail and is independent of receptor ubiquitination (Hislop et al., 2011). In contrast, the calcitonin-like receptor is not ubiquitinated after activation but nonetheless is sorted and degraded in the lysosomes via an ESCRT-0–dependent pathway, confirming that ubiquitination is not obligatory for GPCRs to enter the ESCRT pathway (Cottrell et al., 2007).
An alternative pathway for GPCR lysosomal sorting that bypasses the requirement for both receptor ubiquitination and ubiquitin-binding ESCRTs has been described for PAR1 and the purinergic P2Y1 receptor. Apoptosis-linked gene-2 (ALG-2)-interacting protein X (ALIX), an ESCRT-III–interacting protein, interacts directly with a highly conserved YPX3L motif present the second intracellular loop of PAR1 and the P2Y1 receptor via its central V domain to facilitate receptor lysosomal sorting. ALIX also directly binds to the ESCRT‐III complex, allowing receptors to bypass the ubiquitin‐binding ESCRTs and sort directly into ILVs of MVBs (Dores et al., 2012a, 2016). In recent work, ALIX activity was shown to be regulated by the ARRDC3, which facilitates ALIX ubiquitination and dimerization by the WWP2 HECT domain–containing E3 ubiquitin ligase (Gullapalli et al., 2006; Dores et al., 2012a,b, 2015, 2016). ALIX, ARRDC3, and WWP2 are essential for targeting activated PAR1 and P2Y1 to MVBs or lysosomes via an ECSRT-III charged MVB protein 4– and Vps4-dependent pathway (Dores et al., 2016). Besides PAR1 and P2Y1, six other mammalian GPCRs were found to possess conserved ALIX YPXnL binding motifs within their second ICL2, including the α1B-adrenoreceptor, angiotensin type 2 (AT2) receptor, galanin receptor, histamine H2 receptors, neuropeptide FF receptors, and neuropeptide S receptor (Dores et al., 2012a). Both ALIX and ARRDC3 exploit diverse pathways to capture receptors for endolysosomal sorting (Tian et al., 2016), suggesting that a vast number of other GPCRs may also be regulated by the ALIX-ARRDC3 pathway. Together, these studies illustrate that GPCRs have the capacity to use ubiquitin directly or indirectly to facilitate trafficking through the endolysosomal pathway.
C. Ubiquitin-Driven GPCR Signaling
Although the role of phosphorylation in regulating GPCR biology is extensive, as discussed above, there is a limited understanding of the diverse functions by which ubiquitination controls GPCR signaling. Here we discuss studies examining the role of ubiquitin in propagating GPCR signaling from the plasma membrane and endosomes and how ubiquitination of GPCRs may influence biased signaling.
1. Ubiquitin and Plasma Membrane GPCR Signaling
Ubiquitin-driven GPCR signaling was recently shown for mGlu7 receptor induction of extracellular signal–regulated protein kinase (ERK) 1/2 signaling in hippocampal neurons (Lee et al., 2019). In this scenario, agonist stimulation of mGlu7 receptor enables β-arrestin recruitment of NEDD4, which forms a complex at the plasma membrane and facilitates ubiquitination of the receptor. Both NEDD4 and β-arrestin are required for activated mGlu7 receptor-dependent ERK1/2 signaling, whereas induction of c-jun N-terminal kinase signaling occurs independently of NEDD4-mediated ubiquitination (Lee et al., 2019), suggesting that ubiquitin-driven mGlu7 receptor signaling is specific to ERK1/2. Similarly, activation of the chemokine receptor CXCR2 by interleukin-8 promotes ubiquitin-mediated proinflammatory signaling and proangiogenic responses in leukocytes and endothelial cells (Leclair et al., 2014). In this study, Leclair et al. mapped the site of CXCR2 ubiquitination to a single C-tail localized lysine K327 residue and showed that an ubiquitination-deficient CXCR2 mutant with a K327 to arginine (R) conversion failed to recruit β-arrestin-2 at the plasma membrane and blocked intracellular signaling including ERK1/2 phosphorylation (Leclair et al., 2014), suggesting that receptor ubiquitination is necessary for triggering signaling cascades. Agonist-induced ubiquitination of the parathyroid hormone receptor (PTHR) also requires phosphorylation and β-arrestin binding, but here ubiquitination appears to function selectively in promoting p38 MAP kinase activation and not cAMP accumulation (Zhang et al., 2018). The chemokine receptor CXCR4 has also been shown to mediate ubiquitin-dependent signaling; however, in this case the effect is indirect via modulation of signal transducing adapter molecule (STAM)-1, an ESCRT-0 component (Malik et al., 2012). In this study, agonist-induced CXCR4-mediated ERK1/2 signaling required the E3 ubiquitin ligase AIP4, which facilitates ubiquitination of STAM-1. Interestingly, CXCR4-promoted ERK1/2 activation is governed by a discrete subpopulation of STAM-1 and AIP4 localized to caveolae microdomains at the plasma membrane (Malik et al., 2012). This work expands the role of AIP4 and STAM-1 beyond regulation of CXCR4 lysosomal trafficking (Malik and Marchese, 2010) and provides a new link for ubiquitin-driven GPCR signaling between the trafficking machinery and signal propagation from the plasma membrane.
2. Ubiquitination and Endosomal GPCR Signaling
Several recent studies established that agonist-induced ubiquitination of endothelial GPCRs promotes p38 MAP kinase activation on endosomes via a noncanonical pathway (Fig. 5) (Grimsey et al., 2015, 2018, 2019). The activation and ubiquitination of PAR1 by NEDD4-2 initiates the recruitment of transforming growth factor-β–activated protein kinase 1–binding protein (TAB) 2 via an association with the TAB2 ubiquitin-binding domain, which specifically interacts with K63-linked ubiquitin (Kulathu et al., 2009). TAB2 is known to associate with TAB1 (Bouwmeester et al., 2004). TAB1 has also been shown to directly bind specifically to the p38α isoform, inducing a conformation change resulting in autophosphorylation and activation through a noncanonical pathway (Ge et al., 2002; DeNicola et al., 2013). Importantly, TAB1-dependent activation of p38α induced by PAR1 bypasses the requirement for upstream MAP2K of the canonical three-tiered kinase cascade. Moreover, although it is presumed that GPCRs activate p38 MAP kinase through the three-tiered kinase cascade, there is very limited supportive evidence, and rarely has the role of MAP kinase kinases been directly tested (Goldsmith and Dhanasekaran, 2007). The ubiquitin-driven PAR1 signaling pathway is specific to p38, as thrombin activation of ERK1/2 proceeds through the canonical three-tiered kinase cascade (Grimsey et al., 2015). Similar to PAR1, ubiquitination of the purinergic P2Y1 receptor also mediates p38 activation through a TAB1-TAB2–dependent pathway (Grimsey et al., 2015), indicating that this pathway is used by multiple GPCRs. Indeed, multiple endothelial GPCRs agonists including histamine (H1 or H2 receptor) and prostaglandin EP2 (EP4 prostanoid receptor) also activate p38 MAPK through a noncanonical TAB1-dependent pathway (Grimsey et al., 2019). This work was further advanced by showing that TAB1-dependent p38 activation was critical for PAR1-promoted endothelial barrier permeability in vitro and that p38 signaling was required for PAR1-induced vascular leakage in vivo (Grimsey et al., 2015). Dysfunction of the endothelial barrier is a hallmark of vascular inflammation and suggests that ubiquitin-driven p38 proinflammatory signaling is a common pathway used broadly by GPCRs at least in the context of the vascular endothelium.
3. Ubiquitination and Biased Signaling
Activation of the same GPCR by two or more distinct ligands can elicit different distinct responses, is referred to as biased agonism, and is an important emerging area for drug discovery. Several previous studies indicate that post-translational modification of GPCRs with ubiquitin is uniquely influenced by biased agonists and likely contributes to differential responses. A well studied example is μ-opioid receptor activation with morphine versus DAMGO that resulted in differential recruitment and utilization of β-arrestins and displayed remarkable differences in receptor ubiquitination. DAMGO stimulated robust ubiquitination of μ-opioid receptor, whereas morphine-induced receptor ubiquitination was negligible (Groer et al., 2011). As stated above, isoproterenol stimulates β2-adrenoceptor ubiquitination via a β-arrestin–NEDD4–mediated pathway (Shenoy et al., 2008); however, β2-adrenoceptor ubiquitination induced by the biased ligand carvedilol is mediated by membrane-associated RING-CH-type finger 2 (MARCH2), a RING-type E3 ligase, in the place of NEDD4 (Han et al., 2012). Clearly, biased ligands have the capacity to differentially regulate the ubiquitination machinery as well as GPCR ubiquitination and are important to consider for understanding the molecular basis of biased signaling and future drug discovery.
D. Deubiquitination of GPCRs
Ubiquitination is a reversible post-translational process, and deubiquitination is important for governing ubiquitin-dependent cellular responses such as endocytic trafficking and cell signaling. The accrual of ubiquitin on substrate proteins results from E1, E2, and E3 ubiquitin conjugating enzyme activities as well as by the activity of deubiquitinases or deubiquitinating enzymes (DUBs) (Fig. 4A). DUBs also control the biogenesis and steady state levels of ubiquitin within the cell. Ubiquitin is encoded by four genes that generate linear ubiquitin chains and released as single ubiquitin moieties by the action of DUBs (Grou et al., 2015). DUBs also recycle or reclaim ubiquitin from proteins targeted for degradation. The human genome encodes 99 deubiquitinases that are subdivided into seven families. Of the DUB subfamilies, six, including ubiquitin-specific proteases (USPs), are cysteine proteases, whereas one family comprises zinc-dependent metalloproteinases (Clague et al., 2019). DUBs function as proteolytic enzymes that cleave peptide or isopeptide bonds between linked ubiquitins or between the ubiquitin and substrate protein. DUBs are capable of discriminating between distinct ubiquitin chain linkages and chain length and can cleave from the end or within the ubiquitin chain. Thus, DUBs serve multiple functions by 1) removing ubiquitin from protein substrates, which can rescue proteins from degradation or modulate signaling; 2) editing ubiquitin chains, which can convert one type of ubiquitin chain linkage to another; and 3) recycling ubiquitin, which ensures that ubiquitin reenters the ubiquitin pool (Komander et al., 2009; Mevissen and Komander, 2017).
The regulation of DUB activity is important and occurs through controlling the abundance of DUBs expressed in a given cell, post-translational modification with ubiquitin and/or phosphorylation, and interaction with scaffolds or E3 ubiquitin ligases (Leznicki and Kulathu, 2017). An additional major determinant for controlling DUB function is subcellular localization, which permits access to specific substrates proteins. Some DUBs are highly restricted to organelles through transmembrane anchoring, such as localization of USP19 to the ER and USP30 to the mitochondrial outer membrane, whereas the vast majority of DUBs appear to be present in cytosol or nucleus and controlled at least in part by the presence of a nuclear export signal, as described for USP21 (Leznicki and Kulathu, 2017; Clague et al., 2019). Although ubiquitination of GPCRs is important for regulating receptor trafficking and cellular signaling, the role of DUBs is only beginning to emerge and is discussed below.
1. Constitutive GPCR Deubiquitiation
An emerging role for DUBs is in control of GPCR transport to the cell surface and thereby in preventing ubiquitin-proteasomal degradation (Fig. 5). The quality control machinery in the ER is stringent and prefers to err on the side of rapid degradation of proteins that, when given time, would fold into a properly functionally active protein. This has been shown for the GPCR adenosine A2A receptor. Deubiquitination of adenosine A2A receptor by USP4 relaxes quality control in the ER, enhances cell surface expression, and rescues the receptor from proteasomal degradation (Milojevic et al., 2006). DUBs show remarkably substrate specificity. USP4 binds to the adenosine A2A receptor C terminus and deubiquitinates the receptor but does not act on mGlu5 receptor, another GPCR that tends to accumulate intracellularly (Milojevic et al., 2006). Another role for DUBs in regulating GPCR cell surface expression occurs via regulation of receptor recycling as exemplified by Frizzled-4, a seven-transmembrane receptor for Wnt ligands (Mukai et al., 2010). Constitutive ubiquitination of Frizzled-4 promotes internalization and lysosomal degradation, whereas deubiquitination mediated by USP8 leads to recycling and increased surface expression, events that occur independently of stimulation with Wnt ligands (Mukai et al., 2010). Thus, the balance of ubiquitination and deubiquitination mediated by DUBs switch the receptor’s fate from lysosomal degradation to recycling and enhanced cellular resensitization.
2. Deubiquitination of Agonist-Activated GPCRs
The regulation of agonist-stimulated GPCR ubiquitination is ultimately important for regulating the biologic function of the receptor but has not been extensively studied. Shenoy et al. identified USP33 in a yeast-two hybrid screen with β-arrestin (Shenoy et al., 2009), suggesting that β-arrestins have a dual function to recruit not only the E3 ubiquitin ligases but also DUBs to regulate GPCR function. In a follow-up study, UPS33 as well as its homolog USP20 were shown to reverse agonist-activated β2-adrenoceptor ubiquitination and thereby switched the receptor fate from lysosomal degradation to recycling, resulting in enhanced cellular resensitization (Berthouze et al., 2009). In addition, phosphorylation of USP20 induced by β1-adrenoceptor is required for efficient lysosomal degradation of the receptor (Yu et al., 2019). The chemokine receptor CXCR4 has been shown to associate with USP14 in an agonist-dependent manner, which causes a decrease in receptor ubiquitination and lysosomal degradation (Mines et al., 2009). Interestingly, depletion of USP14 expression also results in loss of chemokine (C-X-C motif) ligand 12–CXCR4–induced cell migration but not of ERK1/2 signaling, suggesting that ubiquitin positively modulates certain aspects of CXCR4 signaling (Mines et al., 2009). Another study examined the effect of USP8 on CXCR4 ubiquitation and reported that loss of USP8 expression enhanced CXCR4 expression by preventing degradation without altering CXCR4 ubiquitination status and ERK1/2 signaling (Berlin et al., 2010). USP8 appears to modulate chemokine (C-X-C motif) ligand 12–CXCR4–induced ubiquitination of HRS, a component of ESCRT-0, which is mediated by AIP4 to control CXCR4 lysosomal trafficking (Berlin et al., 2010).
In contrast to most GPCRs, metabotropic GABAB receptor is insensitive to agonist-induced internalization but undergoes constitutive ubiquitination at the cell surface followed by internalization and lysosomal degradation. Overexpression of USP14 decreased GABAB1 receptor ubiquitination, which appears to occur at a postendocytic site, and consequently regulates lysosomal degradation independently of USP14 catalytic activity (Lahaie et al., 2016), suggesting that GPCR deubiquitination occurs at multiple subcellular locations. Indeed, the subcellular localization of DUBs is an important spatial-temporal regulatory mechanism for ubiquitinated proteins, especially for signaling receptors, but remains poorly understood for GPCRs (Coyne and Wing, 2016). Although PAR2 is proteolytically activated like PAR1, ubiquitination of activated PAR2 is mediated by a Casitas B lineage lymphoma, a RING-type E3 ligase, rather than NEDD4 HECT E3 ligase, as has been demonstrated for PAR1 and numerous other GPCRs (Grimsey et al., 2015; Jean-Charles et al., 2016). In addition, agonist-induced ubiquitination of PAR2 is required for lysosomal degradation, unlike PAR1 (Hasdemir et al., 2009). To understand how ubiquitination regulates PAR2 trafficking and signaling, which occurs from the plasma membrane via G proteins and on endosomes via β-arrestins, one study focused on the function of two endosomal DUBs, associated molecule with the Src homology 3 domain of STAM (AMSH) and ubiquitin-specific protease Y (UBPY), also known as USP8. This study showed that deubiquitination of activated PAR2 is mediated by both AMSH and UBPY and occurs in the endocytic pathway. Moreover, perturbation of either AMSH or UBPY function results in accumulation of ubiquitinated PAR2 in endosomes and slowed lysosomal degradation but failed to alter activated PAR2–β-arrestin association or β-arrestin–dependent signaling (Hasdemir et al., 2009). Given the preponderance of ubiquitinated receptors, there is no doubt that DUBs will have important roles in regulating receptor function by modulating the spatial and temporal dynamics of receptor signaling. However, most studies to date have failed to use comprehensive approaches to identify and study the physiologically relevant DUBs that control cellular responses.
E. Detection and Study of GPCR Ubiquitination
Unlike phosphorylation, the study and interrogation of GPCR ubiquitination is more difficult. Several ubiquitin prediction tools have been recently developed, including UbPred and ESA-UbiSite (Radivojac et al., 2010; Wang et al., 2017); however, comparative analysis of the software concluded that no universal algorithm exists for predicting ubiquitination consensus sites across all species (Chen et al., 2015). Current, widely used strategies for the detection of GPCR ubiquitination include target protein immunoprecipitation followed by immunoblotting and mass spectrometry (Shenoy et al., 2001; Caballero and Marchese, 2011; Dores et al., 2015; Grimsey et al., 2015, 2018; Lee et al., 2019). However, studying endogenous GPCR ubiquitination is challenging due to the dynamic nature of ubiquitination, lack of consensus sites, low abundance of ubiquitinated proteins, rapid degradation, and the large size of ubiquitin compared with other PTMs, which increases the difficulty of detection by mass spectrometry (Mann and Jensen, 2003; Jadhav and Wooten, 2009; Helbig et al., 2010; Danielsen et al., 2011). Often, epitope-tagged GPCRs coupled with site-directed mutagenesis of targeted lysine residues are employed to define the sites and function of ubiquitination (Wolfe et al., 2007; Xiao and Shenoy, 2011). Due to the difficulty of detecting GPCR ubiquitination by immunoblotting, many studies employ ectopic expression of epitope-tagged ubiquitins (Caballero and Marchese, 2011; Giordano et al., 2011), and rarely is endogenous ubiquitination detected, as shown for PAR1 (Fig. 4C). A more rigorous approach to identify the precise sites of GPCR ubiquitination has been determined by mass spectrometry and was shown recently for the β2-adrenoreceptor and PTHR (Xiao and Shenoy, 2011; Zhang et al., 2018). Ubiquitination of multiple GPCRs, including PAR1, have also been detected by curation of high-throughput proteomic mass spectrometry and low-throughput data sources and published on PhosphoSitePlus (Hornbeck et al., 2015). To identify type of ubiquitin linkages, several modified ubiquitin expression constructs with specific lysine mutants are available to detect lysine-specific ubiquitin linkages (Raasi and Pickart, 2005; Avagliano Trezza et al., 2017; Rinaldi et al., 2019). A major advantage of using K48 and K63 linkage-specific antibodies is the ability to identify ubiquitination status of endogenous proteins under physiologic conditions (Grimsey et al., 2015). GPCR ubiquitination can also be probed using fluorescence and bioluminescence techniques that allow the monitoring of ubiquitination dynamics. Bioluminescence resonance energy transfer–based techniques have also been used to detect ubiquitination of GPCRs in intact cells and in real time (Perroy et al., 2004; Nagi and Shenoy, 2019). Currently, biochemical approaches remain a feasible approach for the detection and study of GPCR ubiquitination. Despite the large number of ubiquitinated GPCRs identified, far less is known about the cognate E3 ligases, and rarely have DUBs for specific GPCR been determined. If such information is known, small interfering RNA (siRNA) depletion of specific E3 ligases or DUBs can be performed to assess function. This should be followed by a rescue approach with an siRNA-resistant E3 ligase wild-type and mutant form as recently demonstrated by Grimsey et al. (2018). Clearly, there is a need to develop better methods to monitor GPCR ubiquitination dynamics and to improve of mass spectrometry–assisted ubiquitinome profiling as well as to develop a repertoire of probes for superresolution cellular imaging (van Wijk et al., 2019).
IV. GPCR Glycosylation
Most if not all mammalian GPCRs are post-translationally modified with glycosylation at their extracellular N-terminus or on ECLs. A nascent GPCR undergoes constitutive glycosylation modifications as it traffics from the ER-Golgi to the plasma membrane (Fig. 6A). The role of glycosylation in regulating GPCR biology is expansive and has been attributed to receptor folding, trafficking, ligand binding, signaling, and dimerization (Fig. 7). The ubiquitous roles of glycosylation are due in part to the diversity of glycosylation linkages that differ for a given receptor as well as receptor types and can vary in different cellular contexts. Here we discuss the current knowledge of glycosylation for GPCRs.
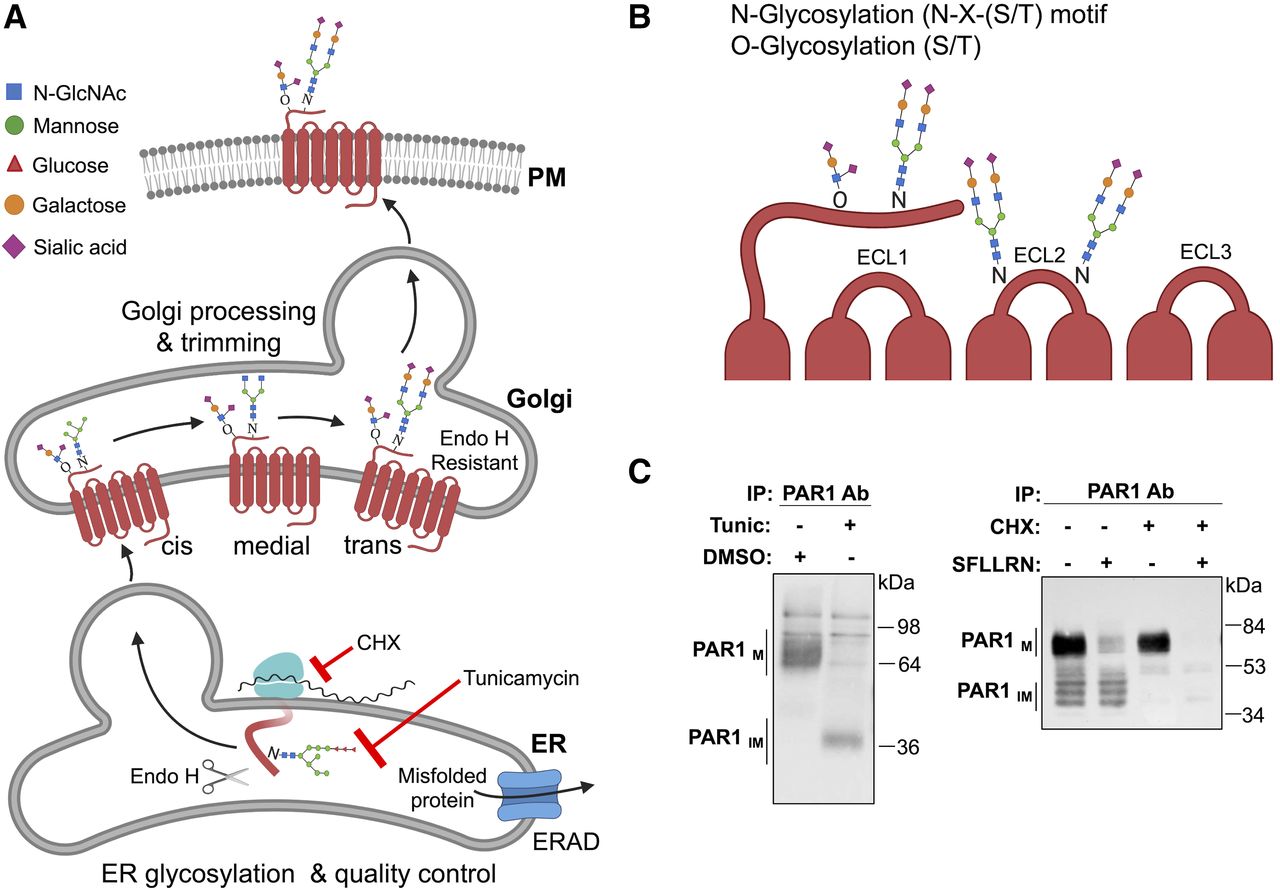
GPCR maturation by glycosylation modifying enzymes and detection of glycosylation. (A) Glycans are covalently linked to GPCRs cotranslationally in the ER, mediate proper maturation, and facilitate expression at the plasma membrane (PM). N-glycans consist of N-acetylglucosamine (GlcNAc) attached to Asn (N) residues at the consensus Asn-X-Ser/Thr site, whereas O-glycosylation occurs at serine or threonine residues. Glycans are extensively trimmed in the Golgi and heterogeneous in nature. Misfolded GPCRs in the ER are cleared through endoplasmic-reticulum-associated protein degradation (ERAD)-proteasomal pathway. (B) Glycosylation of GPCRs occurs preferentially at the N-terminus and ECL2. (C) PAR1 expressed in HeLa cells treated with or without tunicamycin (Tunic), a global inhibitor of glycosylation, left panel. Mature PAR1 (PAR1M) migrates as multiple high-mobility bands, whereas treatment with tunicamycin results in a marked size shift of PAR1 to the predicted molecular weight, representative of unmodified or immature receptor (PAR1IM). PAR1 expressed in Rat1 fibroblasts treated with or without cycloheximide (CHX), a global inhibitor of protein synthesis, right panel. Mature PAR1M migrates predominantly as a high molecular weight species, with several lower migrating bands of partially modified or immature PAR1IM. Incubation with 100 μM SFLLRN agonist peptide for 2 hours results in mature PAR1M degradation but not PAR1IM. In non–SFLLRN-stimulated cells treated with CHX, immature PAR1IM is no longer detectable compared with mature PAR1M, which remains sensitive to SFLLRN-induced degradation. ab, antibody; endoH, endoglycosidase H; IP, immunoprecipitation; SFLLRN, Ser-Phe-Leu-Leu-Arg-Asn.
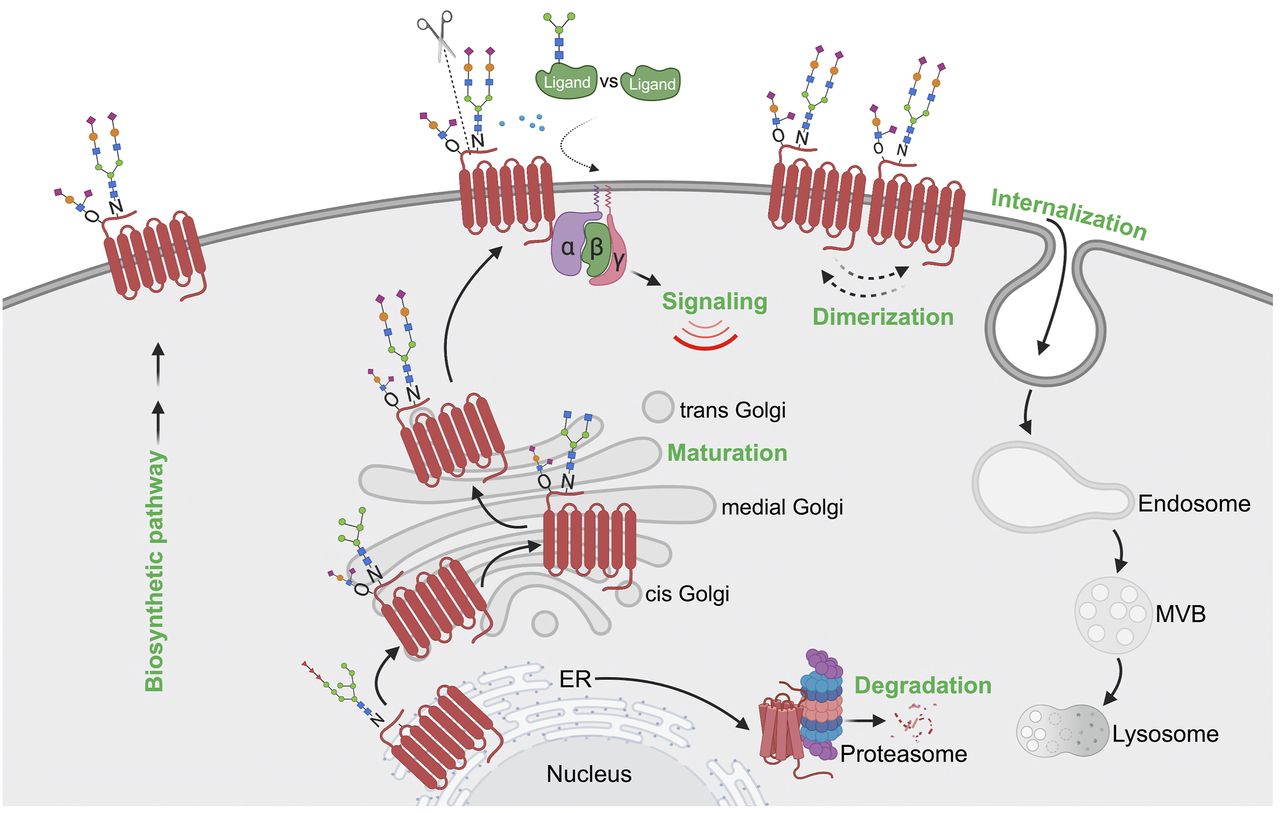
Model of GPCR regulation by glycosylation. GPCRs are extensively modified with N-glycosylation and O-glycosylation during biosynthesis and transport through the ER-Golgi en route to the plasma membrane. Glycosylation-deficient or misfolded GPCRs undergo ER-associated proteasomal degradation pathway. Glycosylation controls GPCR folding and maturation in the biosynthetic pathway, transport to the cell surface, signaling, ligand affinity, N-terminal cleavage, receptor dimerization, and internalization. Importantly, glycosylation controls GPCR biased signaling through direct modulation of the GPCR or in some cases the GPCR ligand. Glycosylation also regulates metalloprotease-mediated N-terminal cleavage of GPCR to influence biased signaling and modulates ligand-binding affinity by providing a larger and potentially more flexible binding surface of the GPCR. Dimerization of certain GPCRs is also positively modulated by glycosylation.
A. GPCRs and N-Linked and O-Linked Glycosylation
Glycosylation is mediated by a complex multistep process involving hundreds of modifying enzymes and results in different types of glycoconjugates covalently linked to lipids or proteins. Glycoconjugates are heterogeneous in nature and differ in their glycan sequences, connections, and length of carbohydrate structures. N-linked glycosylation is abundant and well described to occur on most GPCRs, and new developments have led to the recent identification of O-linked glycosylation sites in numerous GPCRs (Goth et al., 2020).
1. N-Glycosylation of GPCRs
N-glycosylation is initiated in the ER and occurs cotranslationally by the actions of an oligosaccharide transferase (Fig. 6A). The N-linked glycan structure then undergoes extensive trimming during protein transport from the Golgi to the plasma membrane resulting in significant heterogeneity of the glycan structure. N-glycosylation is one of the most common types of glycosylation, where a complex glycan structure is linked to the amide nitrogen on the side chain of an asparagine (N) residue at the consensus sequence N-X-serine (S)/threonine (T), where X is any amino acid other than proline. N-linked glycan structures are often capped with negatively charged sialic acids. The vast majority of mammalian GPCRs contain at least one N-linked glycosylation consensus sites (N-X-S/T) present in the extracellular N-terminal domain (Wheatley and Hawtin, 1999; Wheatley et al., 2012). GPCRs also often contain N-linked glycosylation N-X-S/T sites in ECL2 (Fig. 6B). PAR1 contains five N-linked glycosylation sites—three in the N-terminus and two in ECL2—and is subjected to extensive glycosylation (Fig. 6C) (Soto and Trejo, 2010). Treatment of cells with tunicamycin, an inhibitor of glycosylation, causes a marked mobility shift of PAR1 to ∼39 kDa, its predicted size based on amino acid sequence (Fig. 6C). Interestingly, continuous agonist stimulation caused degradation of mature PAR1 (Fig. 6C), whereas inhibition of protein synthesis with cycloheximide resulted in loss of nascent forms of PAR1 but not the mature form (Fig. 6C). In-depth analysis of GPCRs using computational methods revealed that the consensus N-X-S/T sequence is present on ECL2 (66%), ECL1 (14%), and ECL3 (20%) (Lanctot et al., 2005). Overall, N-glycosylation of GPCR is abundant and occurs at the extracellular N-terminal domain as well as on ECL2 in most receptors and is capable of performing various GPCR functions.
2. O-Linked Glycosylation of GPCRs.
O-glycosylation is initiated by the transfer a N-acetylgalactosamine (GalNAc) to the hydroxyl group of serine or threonine residues, rarely on tyrosine, and occurs in the Golgi after protein folding (Stanley et al., 2009) (Figs. 6A and 7). This reaction is catalyzed by 20 different GalNAc transferases, which can produce eight different core structures (Mulloy et al., 2015). Elongation occurs by the addition of monosaccharides to yield higher-order linear and branched glycan structures, which are capped with negatively charged sialic acids. Unlike N-linked glycosylation, O-glycosylation occurs at serine or threonine residues, usually in stretches rich in hydroxy amino acids, but there is no consensus sequence. Currently, over 60 GPCRs have detected O-linked glycosylation sites within the N-terminal domain, whereas more than 350 GPCRs have predicted O-glycosylation sites based on the use of the NetOGlyc 4.0 model for prediction of O-glycosylation (Steentoft et al., 2013). The quantity and quality of glycosylation depends both on the GPCR itself and on the cell type expressing the protein. However, validation of these predicted sites on the GPCRs and the regulatory functions of O-glycosylation is only beginning to emerge.
B. N-Linked Glycosylation and GPCR Trafficking
1. GPCR Biosynthesis and Cell Surface Expression
The functional effects of N-glycosylation on GPCRs generally control biosynthesis and cell surface expression (Fig. 7). However, for certain receptors, there seem to be no detectable deficits in receptor function if N-glycosylation is blocked, as has been described for the muscarinic M2 receptor, H2 histamine receptor, dopamine D1 receptor, class A orphan GPR61, α1-adrenoreceptor, vasopressin V2 receptor, PTHR, and others (van Koppen and Nathanson, 1990; Fukushima et al., 1995; Kozielewicz et al., 2017), whereas for many other GPCRs, disrupting N-glycosylation or expression of aglycosylated mutant perturbs receptor surface expression, as shown for the β2-adrenoceptor, angiotensin type 1 (AT1) receptor, dopamine D5 receptor, smoothened (SMO), PAR2, GPR176, and others (Karpa et al., 1999; Lanctôt et al., 1999; Compton et al., 2002; Michineau et al., 2004; Marada et al., 2015; Wang et al., 2020). Notably, mutation of all angiotensin AT1 receptor N-glycosylation sites resulted in loss of plasma membrane expression, where the nonglycosylated receptors accumulated in the ER (Deslauriers et al., 1999). However, preservation of AT1 receptor Asn176 in the second extracellular loop enabled surface expression similar to wild-type receptors. A glycosylation-defective gonadotropin-releasing hormone receptor also displayed decreased expression (Davidson et al., 1995). Other studies showed that the final processing for N-glycans for DOR occurs in the trans-Golgi network, whereas O-linked glycosylation is mediated in the trans-Golgi cisternae (Petaja-Repo et al., 2000). Two N-glycosylation sites in the N-terminus of DOR were subsequently shown to enhance transport through the ER but resulted in loss of receptor surface expression due to increased internalization and lysosomal degradation (Markkanen and Petäjä-Repo, 2008). Similarly, the β2-adrenoceptor N-terminus harbors two N-glycosylation sites that have been implicated in receptor trafficking to cell surface but not in ligand binding or G protein coupling (Rands et al., 1990; He et al., 2002). A more recent report indicates that a mutant purinergic P2Y2 receptor deficient in glycosylation undergoes ER-associated proteasomal degradation pathway, possibly due to retention in ER lipid rafts and failure of traffic to the Golgi (Nakagawa et al., 2017). A similar observation was made for α1D-adrenoreceptor, where a glycosylation-deficient mutant displays impaired plasma membrane expression likely because of degradation via ERAD (Janezic et al., 2020). Collectively, substantial evidence suggests that N-glycosylation of GPCRs during maturation in the biosynthetic pathway is essential to achieve optimal cell surface expression.
2. GPCR Plasma Membrane Compartmentalization and Internalization
Once GPCRs reach the cell surface, they can partition into plasma membrane subdomains, including clathrin-coated pits and caveolar microdomains (Guo et al., 2015), which appears to be governed in part by N-glycosylation. The sphingosine 1-phosphate (S1P1) receptor is modified at an N-terminal site with N-glycosylation, and a mutant receptor lacking glycosylation fails to efficiently partition in caveolin-enriched microdomains (Kohno et al., 2002), suggesting that N-glycans may function in plasma membrane compartmentalization. Another study examining the role of N-glycosylation in trafficking of the dopamine D2 and D3 receptor showed that glycosylation regulates not only receptor transit through the biosynthetic pathway but also receptor uptake within microdomains at the plasma membrane (Min et al., 2015). Specifically, glycosylation on the N-terminus was shown to mediate internalization of dopamine D2 receptor through caveolae, whereas glycosylation of dopamine D3 receptor mediates internalization via clathrin-coated pits, which is regulated through direct interactions with caveolin-1 and clathrin (Min et al., 2015). These new findings support a role for N-glycans in mediating GPCR internalization via clathrin-mediated and caveolae-dependent pathways, the major internalization routes of GPCRs (Guo et al., 2015).
In addition to the dopamine D2 and D3 receptor, N-glycan functions have been linked to the chemokine receptor CCR7 internalization (Hauser et al., 2016). Using N-glycosylation prediction software NetNGlyc 4.0, two potential N‐glycosylation sites were identified: one in the N-terminus and one within ECL3 of human CCR7. Surprisingly, only the CCR7 receptor variant with mutation of the N-terminal site had a significantly reduced endocytic rate, whereas the ECL3 mutant variant behaved similar to the wild type (Hauser et al., 2016). S1P1 receptor containing a mutation in an N-terminal N-glycosylation site also exhibited impaired endocytosis (Kohno et al., 2002). Thus, N-glycosylation of S1P1 receptor is required not only for association with caveolae but also for agonist-induced internalization (Kohno et al., 2002). In contrast, a glycosylation-deficient NK1 receptor displayed enhanced internalization after agonist-stimulated compared with wild-type glycosylated NK1 receptor, suggesting that glycosylation may function to retain NK1 receptor at the cell surface (Tansky et al., 2007). In many cases, N-glycosylation has multiple purposes in controlling biosynthesis, export to the cell surface, and internalization through clathrin-coated pits. This has been exemplified for PAR1, which is extensively modified by N-linked glycosylation on both the N-terminus and ECL2 (Fig. 6C). Although N-terminal glycosylation was shown to function in export to the cell surface, glycosylation of PAR1 at ECL2 caused a modest impact on agonist-induced internalization, whereas constitutive internalization remained intact (Soto and Trejo, 2010).
C. N-Linked Glycosylation and GPCR Signaling
Given that N-glycosylation occurs on the extracellular regions of GPCRs, it is not surprising that glycosylation can influence GPCR signaling at multiple levels, including ligand binding, G protein coupling, and biased signaling.
1. N-Glycosylation, Ligand Binding, and GPCR Signaling
In early studies of mammalian GPCRs, perturbation of N-glycosylation resulted in either nonfunctional receptors or receptors with severe functional deficits in ligand binding, as was demonstrated for the thyrotropin receptor, parathyroid hormone, shekel somatostatin receptor type 3, and rhodopsin (Russo et al., 1991; Kaushal et al., 1994; Nehring et al., 2000). In the case of PAR1, loss of N-glycosylation at ECL2 caused a marked increase in signaling compared with wild-type receptor (Soto and Trejo, 2010), suggesting that the lack of glycosylation in this region allows the ligand to bind the receptor in a manner that induces an active receptor conformation that is more efficient in coupling to G protein signaling. Glycosylation may hinder stabilization of the active conformation of the loop or may orientate the loop to prevent ligand access to binding pocket. Similarly, glycosylation of the human chemokine receptor CCR7 at the N-terminus and ECL3 reduces responsiveness, where the lack of glycosylation enhances chemokine signaling (Hauser et al., 2016). In contrast, N‐glycosylation of CXCR4 at the N-terminus is necessary for high‐affinity binding of the chemokine ligand (Wang et al., 2004). Constitutive signaling by GPR30, an emerging player in breast cancer and cardiometabolic regulation, is regulated by N-glycosylation. A recent study demonstrated that one of the three N-glycosylation sites in the N-terminus is important for receptor-stimulated ERK1/2 activity (Gonzalez de Valdivia et al., 2019), suggesting that a single site is critical for receptor structure and activity. In addition to diffusible ligands, recent work suggests that GPCRs can act as mechanosensors activated by mechanical stimulus that appears to be governed by N-glycans (Langenhan et al., 2016). In this case, activation of the β2-adrenoceptor expressed in endothelial cells occurs during infection with the bacteria meningococcus, where the filamentous structures appear to trigger receptor signaling by exerting direct mechanical traction forces via the exposed N-glycans (N6 and N15) present in the N-terminus of β2-adrenoceptor (Virion et al., 2019). This is the first example of a glycan-dependent mode of allosteric mechanical activation of a GPCR.
2. N-Linked Glycosylation and GPCR Biased Signaling
GPCRs are dynamic molecules that assume different conformational states. Consequently, different ligands can stabilize unique active conformations of the same GPCR and facilitate activation of distinct signaling effectors such as G proteins or β-arrestins (Walker et al., 2003; Kenakin and Christopoulos, 2013). This process is termed biased agonism or functional selectivity. New work suggests that N-glycosylation can control GPCR biased signaling. The first report to show a role for N-glycosylation in controlling biased signaling was demonstrated for PAR1. In this study, PAR1 N-glycosylation at ECL2 was shown to direct differential coupling of PAR1 to Gq versus G12/13 signaling (Soto et al., 2015). A fully glycosylated activated PAR1 wild type displayed greater efficacy at coupling to G12/13-dependent Ras homolog family member A signaling than the glycosylation-deficient mutant. In contrast, activation of PAR1 mutant lacking glycosylation at ECL2 exhibited a greater capacity to elicit Gq signaling compared with G12/13 signaling. Both PAR1 wild type and glycosylation-deficient mutant were equally effective at coupling to Gi signaling and β-arrestin recruitment. These findings suggest that N-glycosylation at ECL2 contributes to the stabilization of an active PAR1 state that preferentially couples to G12/13 versus Gq and defines a previously unappreciated function for N-glycosylation of GPCRs in regulating G protein signaling bias (Soto et al., 2015). Similarly, N-glycosylation of SMO, a GPCR that contains seven predicted glycosylation sites, functions as the signal transducer of the Hedgehog pathway and can bias SMO signaling. SMO signals via a noncanonical pathway mediated by Gαi as well as through a canonical route mediated by Gli transcriptional factors. In this study, an SMO mutant rendered glycosylation deficient via mutation of four N-glycosylation sites failed to induce a noncanonical signal through Gαi, whereas it retained normal receptor trafficking, ligand binding, and canonical Gli signaling (Marada et al., 2015). These studies demonstrate that modification of PAR1 and SMO with N-glycosylation can regulate biased signaling.
D. N-Linked Glycosylation and GPCR Dimerization
Substantial evidence supports the notion that GPCRs self-associate or associate with other GPCRs, resulting in dimeric complex formation that modulates receptor function. A plethora of biochemical and pharmacological evidence supports the idea that class A GPCRs exist as homodimers or heterodimers when expressed exogenously or endogenously in native tissues (Milligan, 2009). In addition, several high-resolution crystal structures of class A GPCR homodimeric complexes have been solved, including CXCR4 (Wu et al., 2010), DOR (Granier et al., 2012), and κ-opioid receptor (Wu et al., 2012) receptors. Although heterodimerization of class C GPCRs such as GABAB receptor and taste receptors is obligate and essential for expression and function, more recent studies indicate that class A GPCRs also form functionally significant heterodimers (Milligan, 2009). However, in many cases, the role of class A GPCR dimerization remains elusive. Post-translational modifications are likely to influence GPCR heterodimerization, which can alter specific receptor active conformations, resulting in unique signaling responses, and is important to understand for drug development. Here, we briefly discuss studies showing N-glycosylation control of GPCR dimerization (Fig. 7).
The first reported study showed that N-glycosylation of the β1-adrenoreceptor at the N-terminus reduces dimerization compared with wild-type receptors (He et al., 2002), suggesting that glycosylation positively modulates homodimerization. Similarly, mutating two N-glycosylation sites in the N-terminus of the β2-adrenoceptor decreased receptor dimerization as well as receptor function (Li et al., 2017). N-glycosylation has also been reported to negatively regulate receptor heterodimerization between the β1-adrenoreceptor and α2A-adrenoreceptor (Xu et al., 2003). Moreover, a study showed that introduction of an N‐glycan at the GABAB2 obligate dimer interface prevents the association of the two subunits and abolishes all activities of GABAB2, including agonist activation of G protein signaling (Rondard et al., 2008). These studies suggest that the capacity of GPCRs to form dimers is regulated by their state of N-glycosylation.
E. O-Linked Glycosylation and GPCR Function
In addition to N-glycosylation, new studies predict that over 350 GPCRs undergo O-linked glycosylation. O-glycosylation occurs in a stepwise fashion where the process is initiated by the attachment of α-linked GalNAc residues primarily to the hydroxyl group of serine, threonine, and tyrosine residues. O-glycans are usually capped with terminal negatively charged sialic acids and vary in structure and size. O-glycosylation is initiated in the Golgi after protein folding, where newly synthesized proteins or recycled membrane proteins serve as substrates (Figs. 6A and 7). Despite the vast number of GPCRs predicted to be modified by O-linked glycosylation, in most cases, experimental validation is lacking, and the role of O-linked glycosylation of GPCRs in trafficking, ligand binding, and signaling is largely unexplored. In this section, we discuss recent advances and challenges in understanding functions of O-glycosylation in GPCR biology.
1. O-Glycosylation and GPCR Cell Surface Expression
The study of O-glycosylation is challenging; nonetheless, mass spectrometry has been used to detect O-glycans on rhodopsin (Nakagawa et al., 2001) and human opsins (Nakagawa et al., 2001), but in neither case was the function determined. In more recent work, using an improved prediction algorithm for O-glycosylation (NetOGlyc 4.0), five potential O-glycosylated sites were predicted to reside in the N-terminal domain of human DOR, with three sites—S6, S25, and S29—experimentally validated and shown to regulate DOR transport to the cell surface (Lackman et al., 2018). This study further identified the enzyme GalNAc-transferase 2, one of 20 GalNAc transferase isoforms expressed in mammalian cells, as the specific regulator of DOR O-glycosylation using a human embryonic kidney (HEK293) cell knockout system (Lackman et al., 2018). However, it should be noted that GPCRs are often modified with both N- and O-glycosylation simultaneously, which may perform different and overlapping functions (Sadeghi and Birnbaumer, 1999; Park et al., 2017; Goth et al., 2018; Salom et al., 2019).
2. O-Glycosylation and GPCR Ligand Binding
Similar to N-glycosylation, modification of the N-terminus of GPCRs with O-glycosylation influences ligand binding. O-linked glycosylation of the DP2 and EP2 prostanoid receptors is important for maintaining high-affinity ligand-binding activity (Morii and Watanabe, 1992), as was similarly demonstrated for N-linked glycosylation of the same receptors. The chemokine receptor system controls fundamental biologic processes such as inflammation and cell migration; however, there are far more ligands then receptors, and various ligands bind to multiple receptors; thus, understanding critical features of ligands and receptors that dictate ligand-binding specificity is important. One such determinant that contributes to the specificity of chemokine binding is the post-translational modification of the CCR5 chemokine receptor by the addition of O-linked glycans and tyrosine sulfates (Bannert et al., 2001). These modifications provide not only a larger and potentially more flexible binding surface but also supply an array of negative charges that allow electrostatic interactions with the generally positive receptor-binding interface of the chemokines (Bannert et al., 2001). Although there is no consensus sequence for O-glycosylation, a high prevalence of serine, threonine, and tyrosine residues in the N-terminal domain of chemokine receptors suggests that O-glycosylation may function broadly to modulate chemokine receptor function.
3. O-Glycosylation, GPCR N-Terminal Cleavage, and Signaling
In recent work, O-glycosylation has been implicated in N-terminal cleavage of certain GPCRs (Fig. 7) (Goth et al., 2017, 2018; Park et al., 2017). Interestingly, almost all GPCRs reported to undergo N-terminal cleavage possess identified or predicted O-glycosylation modifications in close proximity to the reported cleavage sites (Goth et al., 2018). Two studies examined the role and function of O-glycosylation on β1-adrenoreceptor N-terminal cleavage and signaling (Goth et al., 2017; Park et al., 2017). Using in vitro O-glycosylation assays, synthetic peptides representing the β1-adrenoreceptor N-terminus and recombinantly expressed GalNAc-transferase 2 identified O-glycosylation at several serine residues. Moreover, loss of β1-adrenoreceptor O-glycosylation in cells, using GalNAc-transferase 2–deficient cells, resulted in a decrease in isoproterenol-induced Gs-mediated cAMP formation (Goth et al., 2017), suggesting that O-glycosylation is important for signaling. In contrast, other work showed that different serine residues of the β1-adrenoreceptor N-terminus are O-glycosylated in GalNAc-transferase 2–expressing CHO cells (Park et al., 2017). This study also demonstrated that β1-adrenoreceptor N-terminal cleavage controlled by O-glycosylation functions alters the balance of β1-adrenoreceptor signaling between the Gs/cAMP and ERK signaling, with a preference for cAMP signaling (Park et al., 2017). N-terminal proteolysis of PAR2 by neutrophil elastase is also inhibited by the presence of O-glycosylation (King et al., 2017), indicating control of proteolytic activation of certain GPCRs. Finally, modulation of CCR7 on immune cells with multiple sialic acids attached to both the N- and O-linked glycans is important for maintaining immune cell responsiveness and immune cell trafficking (Kiermaier et al., 2016) (Figs. 6 and 7). Thus, similar to N-glycosylation, O-glycosylation has important functions in regulating GPCR biology.
F. Detection and Study of GPCR Glycosylation
Recent advancements in the field of bioinformatics, mass spectrometry, molecular biology, and genetic engineering have led to an enormous expansion in the identification of glycosylated GPCRs. NetNGlyc 4.0 and NetOGlyc 4.0 softwares are most widely used to predict N-linked and O-linked glycosylation of GPCRs, respectively (Steentoft et al., 2013; Hauser et al., 2016; Lackman et al., 2018). Standard molecular biology and biochemical techniques to study N-glycosylation of GPCRs include mutagenesis of known or predicted glycosylation sites as well as enzymes that cleave glycans from protein substrates such as peptide:N-glycosidase F (PNGaseF), endoglycosidase H (EndoH), neuraminidase, and O-glycosidase treatment to discriminate between terminally and core-glycosylated N-glycans. Another commonly used reagent is tunicamycin, an antibiotic derived from Streptomyces lysosuperificus, that functions as a global inhibitor of N-linked glycosylation (Fig. 6C) (Rands et al., 1990; Sadeghi and Birnbaumer, 1999; Roy et al., 2010). Typically, GPCR post-translational modification with glycan moieties changes the electrophoretic mobility of the protein, which can be readily observed by immunoblotting with GPCR-specific antibodies. As demonstrated for PAR1, treatment with tunicamycin results in the loss of multiple high-mobility bands and the appearance of a major aglycosylated receptor that migrates near the predicted molecular weight (Fig. 6C). Typically, GPCR post-translational modification with glycan moieties changes the electrophoretic mobility of the protein, which can be analyzed by gel electrophoresis and immunoblotting with GPCR-specific antibodies.
The study of GPCR O-glycosylation is more challenging because O-glycans are complex and heterogenous and there are no known consensus sequence sites. Second, unlike peptide:N-glycosidase F (PNGaseF) for N-glycans, there is not a single universal glycosidase that is able to specifically remove O-linked glycan structures (Yang et al., 2018; Salom et al., 2019). The structures of glycans released after digestion may be determined by a combination of liquid or gas chromatography, mass spectrometry, and nuclear magnetic resonance spectroscopy (Mulloy et al., 2015). Moreover, glycan heterogeneity and flexibility can prevent formation of ordered GPCR crystals (Milić and Veprintsev, 2015). To avoid effects of heterogeneous glycosylation in overexpression system, some researchers have used cell lines lacking N-acetylglucosaminyltransferase I activity. These cells express proteins with homogeneous N-linked glycosylation, which proved beneficial for the expression of rhodopsin for applications such as crystallization (Reeves et al., 2002; Standfuss et al., 2011).
In the last decade, X-ray crystallography has provided a significant contribution to our understanding of GPCR structure and pharmacology. However, in most if not all cases, GPCR structures lacked modification with glycans because of the complexity and heterogeneity of glycan structures that interfere with GPCR crystallization (Milić and Veprintsev, 2015). Thus, future work on the development of more advanced techniques is important to study native structures with their full repertoire of post-translational modifications, including N-glycosylation.
V. GPCR Palmitoylation
Palmitoylation of GPCRs occurs via the covalent attachment of a 16-carbon fatty acid palmitate to one or more cysteine residues, generally in the C-terminal tail (Fig. 8, A and C), specifically termed S-palmitoylation. More than 70% of mammalian GPCRs contain at least one cysteine residue located 10–14 amino acids carboxyl to the seventh transmembrane helical domain. The rhodopsin and β2-adrenoceptor were the first GPCRs shown to be palmitoylated, and early work indicated that palmitoylation of rhodopsin results in the formation of a fourth cytoplasmic loop (O’Brien and Zatz, 1984; O’Dowd et al., 1989; Palczewski et al., 2000). The preponderance of predicted palmitoylation of GPCRs suggests that palmitoylation serves broad functions in receptor regulation. Here, we discuss the mechanism of GPCR palmitoylation and the role of palmitoylation in regulating spatial and temporal aspects of receptor signaling (Qanbar and Bouvier, 2003).

Palmitoylation modifying enzymes and detection of GPCR palmitoylation. (A) Palmitoyl-CoA, a derivative of palmitic acid, is a substrate of DHHC PATs, which catalyzes substrate palmitoylation through a two-step process, where a cysteine intermediate within a DHHC domain is autopalmitoylated; the palmitoyl moiety is then transferred to cysteine residues of the target protein. Palmitoylation is reversible. APTs remove the palmitoylation moiety from substrate proteins. (B) HeLa cells expressing PAR1 wild type (WT) and mutant, in which cysteine (C)387 and C388 were converted to alanine (A), were metabolically labeled with [3H]-palmitate and either left untreated or treated with 100 μM SFLLRN peptide agonist for various times. Cells were lysed, PAR1 was immunoprecipitated (IP) and subjected to autoradiography to visualize [3H]palmitate-labeled PAR1 or immunoblot (IB) to detect total PAR1 protein with PAR1 antibody (ab). (C) A DHHC PATs are located at the ER, Golgi, and plasma membrane. The juxtaposition of the PAT to the target GPCR facilitates palmitoylation. GPCR palmitoylation on C-terminal tail cysteines embeds a region in the membrane creating a fourth intracellular loop and, in some cases, facilitates GPCR localization to lipid rafts. Depalmitoylation of substrate proteins including GPCRs is mediated by APT, which itself may be subjected to palmitoylation to facilitate membrane localization. SFLLRN, Ser-Phe-Leu-Leu-Arg-Asn.
A. Regulation of GPCR Palmitoylation
GPCR palmitoylation is a reversible process. Although early studies of rhodopsin suggested that spontaneous transfer of palmitate can occur in vitro, newer studies clearly indicate that palmitoylation occurs via an enzymatic catalytic process (O’Brien and Zatz, 1984; Korycka et al., 2012). Moreover, GPCRs are basally modified with palmitoylation, and in some cases GPCR palmitoylation is induced by agonist stimulation.
1. Enzymology of GPCR Palmitoylation
A family of palmitoyl acyl transferases (PATs) catalyze the attachment of a palmitoyl group to cytosolic cysteine (C) residues and include at least 23 enzymes (Fig. 8, A and C). PATs contain a conserved D-H-H-C (Asp-His-His-Cys) cysteine-rich domain, designated as DHHC 1–23, that mediates the transfer of palmitoyl to substrate proteins (Fukata et al., 2006; Korycka et al., 2012; De and Sadhukhan, 2018). DHHC proteins are localized in the ER and Golgi, and some are targeted to the plasma membrane (Ohno et al., 2006; Petäjä-Repo et al., 2006; Korycka et al., 2012). Several GPCRs have been shown to be palmitoylated by various DHHCs in different subcellular compartments. GPCR palmitoylation in the ER-Golgi has been shown for DOR, β2-adrenoceptor, PAR2, CCR5, thyrotropin receptor, and vasopressin V1A receptor, whereas other GPCRs appear to be palmitoylated at the plasma membrane, including DOR and S1P1 receptor (Petäjä-Repo et al., 2006; Adams et al., 2011; Adachi et al., 2016; Badawy et al., 2017). The substrate specificity of DHHCs is due in part to subcellular localization (Roth et al., 2006; Linder and Deschenes, 2007; Ohno et al., 2012). Palmitoylation is a reversible process, and depalmitoylation of substrate proteins is catalyzed by the action of acyl-protein thioesterase (APT) 1, APT2, and APT1-like and palmitoyl-protein thioesterase-1 and -2 (Fig. 8, A and C) (Linder and Deschenes, 2003). Currently, only APT1 and APT2 have been reported to mediate depalmitoylation of β2-adrenoceptor and melanocortin MC1 receptor, respectively (Adachi et al., 2016; Chen et al., 2019).
2. Basal and Agonist-Induced GPCR Palmitoylation
The majority of newly synthesized GPCRs are subjected to palmitoylation basally during biosynthesis. This appears to be the case for PAR1, where the detection of palmitoylation occurs basally and is not further modified after agonist stimulation (Fig. 8B). However, a few GPCRs appear to be palmitoylated after agonist stimulation. This has been demonstrated for the β2-adrenoceptor, which is basally palmitoylated at C341, whereas agonist-induced palmitoylation and depalmitoylation occurs predominantly on C265 (O’Dowd et al., 1989; Adachi et al., 2016). Several other GPCRs undergo agonist-induced palmitoylation and depalmitoylation, including but not limited to the vasopressin V1A receptor, dopamine D1 receptor, α1B-adrenoreceptor, 5-hydroxytryptamine (HT)4 receptor, S1P1 receptor, α2A-adrenoreceptor, and muscarinic M2 receptor (Ponimaskin et al., 2001; Badawy et al., 2017; Naumenko and Ponimaskin, 2018).
B. Palmitoylation and GPCR Trafficking
An important function of GPCR palmitoylation is the efficient transport of receptors through the biosynthetic pathway and delivery to the cell surface, compartmentalization in plasma membrane microdomains, dimerization, and trafficking through the endocytic pathway.
1. GPCR Surface Expression
A role for palmitoylation in regulating GPCR surface expression has been demonstrated for several GPCRs (Fig. 9). Mutation of three C-tail cysteine residues of the chemokine CCR5 receptor resulted in retention largely in the ER and Golgi complex (Blanpain et al., 2001; Percherancier et al., 2001). The authors further showed that the nonpalmitoylated CCR5 mutant displays impaired diffusion properties within the ER. Similarly, defects in palmitoylation caused a marked loss of endogenous PAR2 expression at the cell surface (Adams et al., 2011) as well as diminished surface expression of the thyrotropin receptor, vasopressin V2 receptor, adenosine A1 receptor, histamine H2 receptor, and the dopamine D1 and D2 receptors (Schülein et al., 1996; Sadeghi et al., 1997; Tanaka et al., 1998; Fukushima et al., 2001; Ebersole et al., 2015). The mechanism by which defects in palmitoylation diminish GPCR surface expression is attributed mainly to receptor misfolding and proteasomal degradation. This has been shown for deficiencies in palmitoylation of CCR5 and adenosine A1 receptor, which enhance degradation (Gao et al., 1999; Percherancier et al., 2001). Follicle-stimulating hormone receptor contains three cytosolic cysteine residues; however, mutation of a single C269 was sufficient to impair cell surface expression, likely due to misfolding and degradation (Uribe et al., 2008). These findings are consistent with an important role for palmitoylation in facilitating the proper folding and maturation of GPCRs.
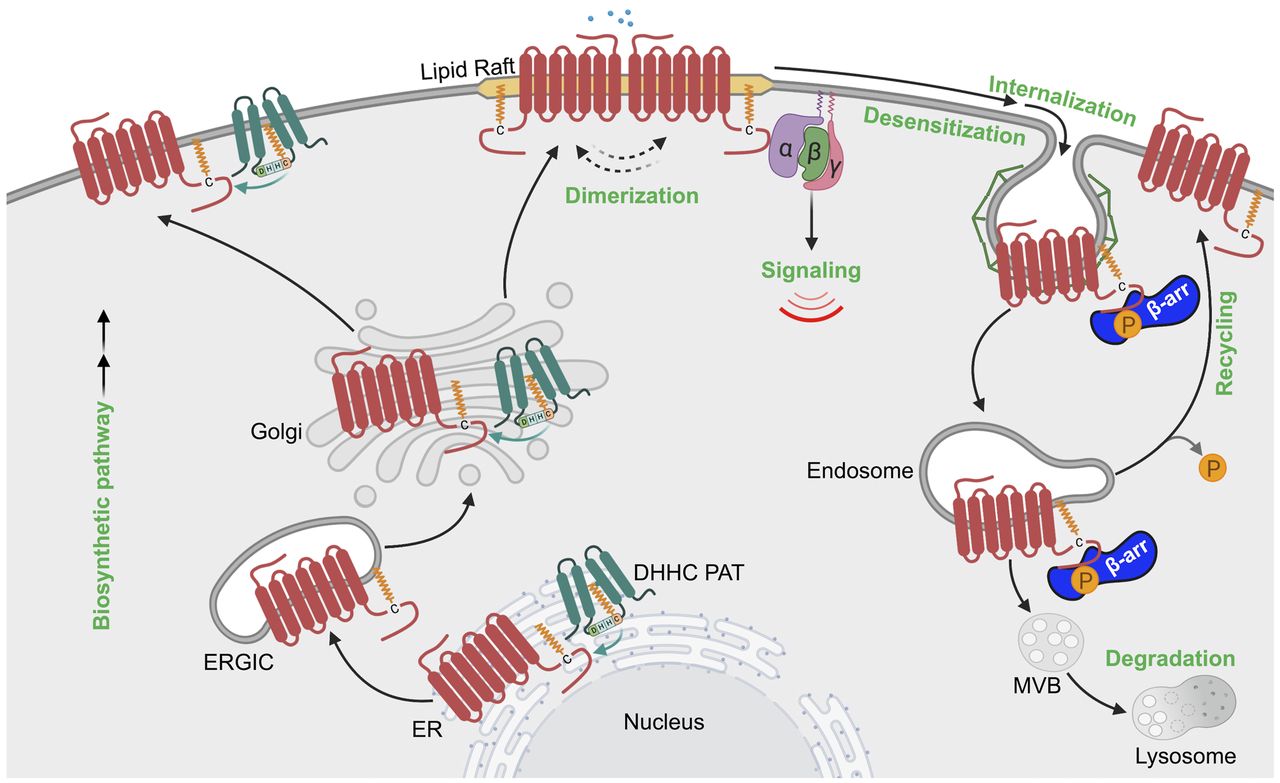
Model of GPCR regulation by palmitoylation. GPCRs are palmitoylated during biosynthesis and can occur at the endoplasmic reticulum (ER), endoplasmic-reticulum–Golgi intermediate compartment (ERGIC), Golgi and the plasma membrane, where DHHC PATs are known to be localized. GPCR palmitoylation regulates partitioning into membrane microdomains enriched in cholesterol such as lipid rafts and caveolae. GPCR palmitoylation has also been implicated in receptor dimerization as well as G protein coupling. Palmitoylation of GPCRs can further influence β-arrestin (β-arr) recruitment and receptor internalization. GPCR palmitoylation is also important for regulating receptor recycling and thereby prevents lysosomal degradation.
2. GPCR Dimerization and Lipid Rafts
GPCRs partition into lipid raft plasma membrane microdomains enriched in cholesterol and is regulated by palmitoylation (Figs. 8C and 9) (Barnett-Norris et al., 2005; Villar et al., 2016). The serotonin 5-HT1A receptor defective in palmitoylation showed decreased association with lipid rafts (Papoucheva et al., 2004; Renner et al., 2007). Similarly, dopamine D1 receptor (Tiu et al., 2020) and cannabinoid receptor type 1 (Oddi et al., 2012, 2018) mutants deficient in palmitoylation exhibited impaired lipid raft association. Interestingly, the crystal structure of the human β2-adrenoceptor revealed a receptor dimer complex, where lipid-mediated contacts via palmitic acid and cholesterol are the major interactions (Cherezov et al., 2007). In addition, palmitoylation of several other GPCRs has been shown to promote lipid raft association and dimerization, including the MOR (Zheng et al., 2012), rhodopsin (Seno and Hayashi, 2017), and the serotonin 5-HT1A receptor (Kobe et al., 2008). These results indicate that for certain GPCRs, palmitoylation facilitates receptor compartmentalization in lipid rafts and dimerization.
3. GPCR Internalization, Recycling, and Lysosomal Degradation
In addition to GPCR plasma membrane localization, palmitoylation has been shown to regulate GPCR internalization, recycling, and lysosomal degradation (Fig. 9). Several studies have documented a role for palmitoylation in GPCR internalization. Defects in cannabinoid receptor type 1 palmitoylation inhibited agonist-induced internalization and coassociation with caveolin 1 (Oddi et al., 2017). Similarly, defects in palmitoylation of the prostanoid thromboxane A2 receptor, PAR2, and thyrotropin receptor perturbed agonist-induced β-arrestin recruitment and receptor internalization (Tanaka et al., 1998; Reid and Kinsella, 2007; Adams et al., 2011). In contrast, a vasopressin V1A receptor mutant deficient in palmitoylation displayed an increased rate of agonist-induced internalization without affecting intracellular signaling (Hawtin et al., 2001). Similarly, a palmitoylation-deficient dopamine D1 receptor mutant exhibited an enhanced rate of internalization compared with wild type and showed preferentially internalization via a clathrin-dependent pathway over caveolae (Kong et al., 2011).
Once internalized, GPCRs are either recycled back to the cell surface or targeted to lysosomes for degradation. Although palmitoylation of PAR2 is required for efficient internalization and lysosomal degradation (Adams et al., 2011), palmitoylation has an opposing function for PAR1, as a palmitoylation-deficient PAR1 mutant exhibited an enhanced rate of internalization and lysosomal degradation (Canto and Trejo, 2013). The defects in trafficking caused by alteration of PAR1 palmitoylation are due to inappropriate utilization of C-tail tyrosine-based sorting motifs for endocytic adaptor proteins (Canto and Trejo, 2013). In the absence of palmitoylation, PAR1 sorting motifs appear to be more accessible to the clathrin adaptor binding proteins AP-2 and adaptor protein complex 3, which accelerate the rate of internalization from the plasma membrane as well as enhanced sorting from endosomes to lysosomes and degradation (Canto and Trejo, 2013). Similarly, a CCR5 palmitoylation mutant exhibits rapid lysosomal degradation and a reduced half-life (Percherancier et al., 2001).
C. Palmitoylation and GPCR Signaling
In addition to GPCR trafficking, palmitoylation is important for regulating activated GPCR coupling to G protein signaling. In several cases, deficiencies in GPCR palmitoylation fail to affect ligand binding but impact G protein coupling or alter the specificity of coupling to certain G protein subtypes. Studies of a β2-adrenoceptor C341 mutant showed defects in coupling to Gs and impaired cAMP production (O’Dowd et al., 1989). In other studies, defects in GPCR palmitoylation were shown to compromise G protein coupling of the agonist-activated serotonin 5-HT1A receptor, human somatostatin receptor type 5, human endothelin ETA receptor, α2A-adrenoreceptor, dopamine D1 receptor, human adenosine A1 receptor, and the human thyrotropin receptor (Hukovic et al., 1998; Doi et al., 1999). This is not surprising since GPCR localization in lipid rafts is known to facilitate the assembly of signaling ensembles (Barnett-Norris et al., 2005; Villar et al., 2016). Indeed, methyl-β-cyclodextrin, a cholesterol-chelating agent that disrupts lipid rafts, reduced the localization of serotonin 5-HT1A receptor to lipid rafts and G protein coupling (Papoucheva et al., 2004; Renner et al., 2007). Thus, palmitoylation-driven lipid raft localization of certain GPCRs is important for regulating signaling. However, some studies suggest that conformational changes induced by modulating lipid interaction of preexisting dimers may alter G protein coupling preferences. Although the β2-adrenoceptor couples to both Gs and Gi proteins, depletion of cholesterol resulted in preferential coupling to Gs proteins (Xiang et al., 2002). Moreover, β2-adrenoceptor coupled to Gs protein was shown to occur with receptor monomers (Whorton et al., 2007), indicating that dimers are not a prerequisite for Gs coupling. In mice, treatment with palmostatin B, a cell-permeable inhibitor of APT1, increased MC1 receptor palmitoylation and enhanced the MC1 receptor–stimulated cAMP production, which provided protection against progression of melanoma (Chen et al., 2017a). Thus, the impacts of palmitoylation are partly due to defects in compartmentalization, receptor conformation, and receptor capacity to couple to G protein activation.
D. Detection and Study of GPCR Palmitoylation
Although there are no consensus sites for palmitoylation, several palmitoylation prediction tools are currently available, such as CSS-Palm, GPS-Lipid, PalmPred, and SwissPalm (Ren et al., 2008; Kumari et al., 2014; Blanc et al., 2015; Xie et al., 2016). Although palmitoylation of GPCRs has been reported, the detection of GPCR palmitoylation is challenging and includes the use of metabolic labeling with [3H] palmitate and more recently with click chemistry. The traditional method to study GPCR palmitoylation uses [3H] palmitate metabolic labeling followed by autoradiography and is reliable but has limited sensitivity (Fig. 8B) (O’Dowd et al., 1989; Ponimaskin et al., 2002; Petäjä-Repo et al., 2006). In recent years, bio-orthogonal labeling or click chemistry has been employed to study palmitoylation of GPCRs (Ebersole et al., 2014). In this method, a cell-permeable chemical probe that mimics palmitic acid is covalently attached to proteins by PATs. The GPCR modified with the chemical probe is then detected using bioorthogonal azide-labeled fluorescent chromophore or biotin azide via click chemistry (Hannoush and Sun, 2010; De and Sadhukhan, 2018). If using biotin, this method allows streptavidin-mediated pulldown of the modified GPCR, which can be subjected to mass spectrometry analysis, whereas a fluorescent chromophore allows in-gel fluorescence visualization of palmitoylation (Hannoush and Sun, 2010; Broncel et al., 2015; De and Sadhukhan, 2018). Acyl-biotin exchange coupled with mass spectrometry is an approach used to identify and characterize protein palmitoylation on a proteomewide scale (Drisdel and Green, 2004; Collins et al., 2017; Gorinski et al., 2019). Acyl–polyethylene glycol (PEG) exchange (APE) shift assay is a modification of the acyl-biotin exchange method, where acyl-PEG exchange utilizes cysteine chemistry to exchange S-palmitoylation sites with different PEG mass tags of defined size, which can be observed by immunoblotting and can determine the number of S-palmitoylation sites (Percher et al., 2016).
To study the function of GPCR palmitoylation, multiple approaches have been taken, including site-directed mutagenesis of key cysteine residues as shown for PAR1, where mutation of two C-tail C387 and C388 residues to alanine resulted in a loss of [3H] palmitate incorporation (Fig. 8B) (Canto and Trejo, 2013), as well as knockdown-rescue of palmitoyl transferase enzymes (Zuckerman et al., 2011). Another common approach includes the use of 2-bromopalmitate, a general inhibitor of palmitoylation, that is converted to 2-bromopalmitoyl-CoA and is known to inhibit palmitoyltransferase activity of all the DHHC enzymes (Adams et al., 2011; Davda et al., 2013). Due to the lack of available more feasible methods, the extent, function, and dynamic nature of GPCR palmitoylation remain poorly understood.
VI. Other GPCR Post-Translational Modifications
In addition to PTMs discussed above, a few reports indicate that GPCRs are targets of other types of PTMs such as SUMOylation, S-nitrosylation, tyrosine sulfation, and methylation (Fig. 1). SUMOylation, which is mediated by the covalent conjugation of a 11 kDa SUMO protein to lysine residues present in the consensus motif [ψ-K-X-(D/E), where ψ is aliphatic amino acid and X is any amino acid; Geiss-Friedlander and Melchior, 2007]. SUMOylation occurs via an enzymatic cascade mediated by a dedicated set of SUMO E1, E2, and E3 enzymes targeting intracellular domains GPCRs (Flotho and Melchior, 2013); this is analogous to and reminiscent of ubiquitin-catalyzed reactions. SUMOylation was believed to be a predominantly nuclear process, but recent advancements found connection with integral membrane proteins, such as GPCRs (cannabinoid receptor type 1, SMO, mGlu7 receptor, serotonin 5-HT1A receptor) (Luo et al., 2013; Choi et al., 2016; Zhang et al., 2017; Xu et al., 2019). A recent notable example of GPCR SUMOylation was described for the muscarinic M1 receptor, which is SUMOlyated on K327 within the intracellular loop (IL3) and increases ligand-binding affinity to the muscarinic M1 receptor, resulting in enhanced signaling efficiency and receptor endocytosis (Xu et al., 2019). In other work, agonist treatment was shown to increase expression of SUMOylated serotonin 5-HT1A receptors in specific regions of the rat brain and was postulated to regulate receptor endocytosis (Li and Muma, 2013). GPCRs are also subjected to S-nitrosylation, most likely in the transmembrane domain, which is mediated by the covalent attachment of a nitric oxide moiety to specific cysteine thiol groups of the receptor. This has been shown for the α1-adrenoreceptor using an in vitro biotin switch assay; however, the actual sites of S-nitrosylation among the possible 14 cysteine residues were not determined (Jaffrey et al., 2001; Nozik-Grayck et al., 2006). The function of α1-adrenoreceptor S-nitrosylation has been attributed to decreases in the vasoconstrictor response in response to agonist stimulation (Nozik-Grayck et al., 2006). S‐nitrosylation of the angiotensin AT2 receptor decreases the binding affinity of angiotensin and occurs on C289 located in the seventh transmembrane domain of the receptor (Leclerc et al., 2006).
In contrast to SUMOlyation and S-nitrosylation, tyrosine sulfation has been described for a select group of chemokine GPCRs and occurs in the Golgi through attachment of a negatively charged sulfate group to an exposed tyrosine residue, yielding tyrosyl O-sulfate. Tyrosine sulfation has been experimentally confirmed for several human chemokine GPCRs, including CCR2, CCR3, CCR5, and CCR8, as well as CXCR3, CXCR4, and CX3CR1 receptors (Colvin et al., 2006; Liu et al., 2008; Zhu et al., 2011; Ludeman and Stone, 2014) and shown to enhance the affinity, potency, and specificity of chemokine ligands (Ludeman and Stone, 2014; Stone et al., 2017). An additional new PTM for GPCRs is provided by studies suggesting that arginine methylation of GPCRs is prevalent and contributes to the regulation of GPCR function. A bioinformatics analysis has identified 300 human GPCRs with greater than 583 predicted methylation motifs (RGG or RXR, where arginine is R, glycine is G, and X is any amino acid), localized within ICL3 (Likhite et al., 2015). A study of the human dopamine D2 receptor revealed methylation modification at ICL3 mediated by an arginine methyltransferase 5, which resulted in attenuation of dopamine D2-mediated inhibition of cAMP signaling in cultured human cells in vitro and in vivo in C. elegans (Likhite et al., 2015; Bowitch et al., 2018). The ICL3 of the dopamine D2 receptor facilitates coupling to G proteins and signaling effectors, consistent with the idea that methylation may perturb receptor–G protein coupling. Despite these findings, the role of other types of PTMs, including SUMOylation, S-nitrosylation, tyrosine sulfation, and methylation, remains relatively unexplored for most members of the vast GPCR superfamily and is important to consider to thoroughly understand mechanisms of GPCR regulation and for drug discovery.
VII. GPCR PTM Crosstalk
Clearly, post-translational modifications of GPCRs are essential for function. GPCRs are also subjected to multiple diverse types of post-translational modifications at a given time, which is critical for expanding their function. Although over 200 types of PTMs have been identified (Olsen and Mann, 2013), the best characterized PTMs for GPCRs include phosphorylation, ubiquitination, glycosylation, and palmitoylation. Moreover, different PTMs can either positively or negatively influence each other. Here, we briefly discuss examples of GPCR post-translational modification crosstalk.
A. GPCR Phosphorylation and Ubiquitination Crosstalk
The crosstalk between ligand-induced GPCR phosphorylation and ubiquitination has been well described. The yeast Ste2 GPCR is hyperphosphorylated and ubiquitinated after α-factor stimulation (Hicke et al., 1998). Phosphorylation occurs on the Ste2 C-terminal tail and positively regulates ubiquitination at neighboring lysine residues via recruitment of the E3 ligase Rsp5, which is required for both constitutive and ligand-induced receptor internalization. The precise mechanism by which Ste2 phosphorylation regulates ubiquitination is unclear, but the ability of WW domains of Rsp5p might serve to recognize phosphoserine (Lu et al., 1999). Certain mammalian GPCRs also require phosphorylation for ubiquitination. The β2-adrenoceptor requires phosphorylation, which occurs mainly within the C-terminal tail region, for agonist-induced ubiquitination. In this case, β2-adrenoceptor phosphorylation is important for β-arrestin association, which facilitates recruitment of the E3 ligase NEDD4-1 (Shenoy et al., 2001; DeWire et al., 2007; Han et al., 2013). Similarly, activated CXCR4 phosphorylation occurs at two critical serine residues within the C-tail region and is required for the recruitment of the E3 ligase AIP4. AIP4 binds to activated and phosphorylated CXCR4 and ubiquitinates adjacent lysine residues (Marchese and Benovic, 2001; Bhandari et al., 2009). Other studies have shown that the PTHR and PAR1 require phosphorylation for ubiquitination (Chen et al., 2011; Zhang et al., 2018); however, the mechanistic details are lacking. Thus, the vast majority of ligand-activated GPCR ubiquitination is likely to require phosphorylation for either direct recruitment of the E3 ligase or an adaptor protein that mediates E3 ligase recruitment. However, the role of phosphorylation is expansive and likely to regulate GPCR ubiquitination at multiple levels, including the activity of the adaptor proteins as well as the ubiquitination machinery itself (Song and Luo, 2019). In the case of GPCRs, this has been demonstrated recently for PAR1-induced NEDD4-2 tyrosine phosphorylation and activation as described above (Grimsey et al., 2018).
B. GPCR Phosphorylation and Palmitoylation Crosstalk
Another prominent studied GPCR post-translational modification crosstalk occurs between phosphorylation and palmitoylation. Palmitoylation has a critical role in generating a GPCR fourth ICL through membrane insertion and is thus likely to impact receptor structure and major domains that serve as sites for phosphorylation. Indeed, deficiencies in palmitoylation have been shown to impair phosphorylation of multiple GPCRs. A palmitoylation-deficient vasopressin V1A receptor exhibited lower phosphorylation both basally and after agonist activation (Hawtin et al., 2001). Similar observations were made for the chemokine receptor CCR5 (Kraft et al., 2001). In contrast, a palmitoylation-deficient mutant of the β2-adrenoceptor is hyperphosphorylated and constitutively desensitized (Moffett et al., 1993). In other work, β2-adrenoceptor palmitoylation of C341 was shown to control PKA-dependent C-tail phosphorylation and receptor responsiveness (Moffett et al., 1996). Such observations were also reported for serotonin 5-HT4 receptor where a palmitoylation-deficient mutant exhibited enhanced receptor phosphorylation under basal conditions and after agonist stimulation (Ponimaskin et al., 2002). Moreover, in vitro studies confirmed that for certain GPCRs the lack of palmitoylation renders the receptor more susceptible to phosphorylation. In vitro studies of depalmitoylated β2-adrenoceptor and rhodopsin were found to be robustly phosphorylated by PKA (Moffett et al., 1996) and rhodopsin kinase (Karnik et al., 1993), respectively. Overall, these studies indicate that crosstalk exists between GPCR palmitoylation and phosphorylation.
VIII. Conclusions
Although modulation of the GPCR phosphorylation state is likely to occur in multiple disease settings, phosphorylation of β2-adrenoceptor in heart disease is well described. In this case, chronic stimulation of β1-adrenoceptor and β2-adrenoceptor, which are critical modulators of contractile function, in heart failure results in GRK2-mediated phosphorylation, desensitization, and ultimately degradation of the vast majority of receptors (Sato et al., 2015).
In addition to phosphorylation, several well described GPCRs modified with ubiquitination have been implicated in disease, including the β2-adrenoceptor, CXCR4, and PAR1. Carvedilol is a well described nonselective β blocker used for the treatment of heart failure and shown to induce ubiquitination of β2-adrenoceptor via a unique mechanism mediated by the RING-type membrane-associated RING-CH-type finger (MARCH) E3 ubiquitin ligase (Han et al., 2012), but the precise contribution of ubiquitination to regulation of β2-adrenoceptor in cardiac myocytes and heart failure remains poorly understood. PAR1 is an important drug target for treatment of thrombotic cardiovascular events and has also been implicated in metastatic cancer (Hamilton and Trejo, 2017; Arakaki et al., 2018a,b). Ubiquitination of PAR1 is clearly important for regulating the temporal and spatial dynamics of signaling in various cells types (Wolfe et al., 2007; Grimsey et al., 2015, 2018, 2019). In fact, metastatic breast carcinoma displays aberrant PAR1 trafficking, resulting in increased surface expression, persistent signaling, and tumor cell invasion (Booden et al., 2004; Arora et al., 2008; Arakaki et al., 2018b). However, the contribution of ubiquitination to dysregulated PAR1 function in metastatic cancer has not been explored and represents an opportunity for drug development. CXCR4 is also overexpressed in various types of cancer, including breast carcinoma, and contributes to breast cancer progression. Interestingly, the oncogene human epidermal growth factor receptor 2 enhances CXCR4 expression in breast cancer by increasing translation and inhibiting ubiquitination and degradation of CXCR4 (Li et al., 2004; Luker and Luker, 2006), whereas epidermal growth factor receptor increases transcription of CXCR4 and diminishes AIP4 and β-arrestin activity and reduces CXCR4 degradation (Rahimi et al., 2010). Thus, CXCR4 expression in breast cancer is controlled by ubiquitination at multiple levels.
Similar to phosphorylation and ubiquitination, the vast majority of GPCRs contain potential sites for palmitoylation within their C-terminal tail domain, suggesting it may represent a general feature of this receptor family. As discussed above, palmitoylation is important for GPCR structural conformation, trafficking, plasma membrane localization, and signaling. The link between palmitoylation and disease is best described in the brain, which express multiple types of GPCRs, including adrenergic, serotonin, dopamine, opioid, muscarinic, vasopressin, adenosine, melatonin, and cannabinoid GPCRs that function in various cellular processes such as signal transduction and synaptic plasticity (Naumenko and Ponimaskin, 2018). Importantly, studies have documented alterations in palmitoylation in the brain associated with various pathologic disorders such as Alzheimer’s, Huntington’s disease, schizophrenia, and mental retardation (Sanders and Hayden, 2015; Cho and Park, 2016). However, the precise link to specific defects in GPCR palmitoylation remains to be determined. Glycosylation has been linked primarily to GPCR maturation protein folding and controls transport of GPCRs via the biosynthetic pathway to the cell surface. Interestingly, the vast majority of GPCR mutations linked to disease have been associated with defects in maturation and folding. This is best documented for rhodopsin, where naturally occurring mutations in N-glycosylation consensus sequences of rhodopsin have been linked to retinitis pigmentosa (Sullivan et al., 1993; van den Born et al., 1994), indicating that modulation of GPCR glycosylation status is directly linked to disease progression. In recent work, Trypanosoma cruzi infection was shown to decrease β1-adrenergic receptor sialylation and N-terminal cleavage that resulted in enhanced signaling and adverse effects on cardiac remodeling (Freire-de-Lima et al., 2015; Park et al., 2017). Given the expansive roles of GPCRs in physiology and disease, coupled with the critical function of post-translational modifications in regulating GPCR function, more work is needed to address how modulation of specific GPCRs by PTMs alters the spatial and temporal dynamics of signaling leading to disease progression.
Clearly, post-translational modifications offer novel and diverse mechanisms for regulation of GPCR biology and opportunities for new or refined drug development. Although there has been major progress in understanding the role and function of ubiquitination, glycosylation, and palmitoylation in GPCR function, we have very limited knowledge about other PTMs. A major barrier to generally studying GPCR PTMs is the lack of ability to predict PTMs and technology for detecting PTM dynamics in response to agonist stimulation. One promising approach is the development of mass spectrometry–based quantitative proteomics that can rapidly and accurately detect the dynamics of PTMs and has shown significant advancements, particularly for clinical applications (Pagel et al., 2015). Although the characterization of PTMs is challenging, particularly for GPCRs, a thorough understanding of the molecular mechanisms by which key regulators and mediators of PTMs regulate GPCR signaling is essential for understanding dysregulated mechanisms in disease and for identifying new targets for drug development. GPCRs are important drug targets, with 108 GPCRs representing the target of over 475 of Food and Drug Administration–approved drugs (Hauser et al., 2018). Given the large number of GPCRs >800 and vast functions, GPCRs continue to present an enormous opportunity for drug development. Similar to natural genetic variations within GPCRs that alter or cause adverse drug responses, post-translational modification of GPCRs vary between individuals as well as in different tissues and cell types due to genetic variation and/or epigenetic factors and are likely to influence drug responses. Despite the fact that PTMs of GPCRs has an essential role in receptor folding, conformation, stability, activity, and ultimately function, the role and function of PTMs in GPCR drug response have been largely ignored and require greater attention.
Acknowledgments
We thank the Trejo laboratory and especially Isabel Canto, Antonio Soto, and Neil Grimsey for their contribution to work related to PAR1 palmitoylation, glycosylation, and ubiquitination. Illustrations in the figures were created with BioRender.com.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Patwardhan, Cheng, Trejo.
Footnotes
This work is supported by the National Institutes of Health National Institute of General Medical Sciences [Grants R35 GM GM127121, T32 GM007752] and the University of California, Tobacco-Related Disease Research Program Postdoctoral Award [T31FT1574].
Abbreviations
- AIP4
- atrophin-1–interacting protein-4
- ALIX
- apoptosis-linked gene 2–interacting protein X
- AMSH
- associated molecule with the Src homology 3 domain of STAM
- AP-2
- adaptor protein complex-2
- APT
- acyl protein thioesterase
- ARRDC
- α-arrestin domain-containing protein
- AT1
- angiotensin type 1
- AT2
- angiotensin type 2
- CCR
- chemokine (C-C motif) receptor
- CXCR
- C-X-C chemokine receptor
- DOR
- δ-opioid receptor
- DUB
- deubiquitinase or deubiquitinating enzyme
- ECL
- extracellular loop
- ER
- endoplasmic reticulum
- ERAD
- endoplasmic reticulum–associated protein degradation
- ERK
- extracellular signal–regulated kinase
- ESCRT
- endosomal sorting complex required for transport
- GalNAc
- N-acetylgalactosamine
- GPCR or GPR
- G protein–coupled receptor
- GRK
- G protein–coupled receptor kinase
- HECT
- homologous to E6AP C terminus
- HRS
- hepatocyte growth factor–regulated tyrosine kinase substrate
- HT
- hydroxytryptamine
- ICL
- intracellular loop
- ILV
- intralumenal vesicle
- MAP
- mitogen-activated protein
- MC1
- melanocortin 1
- mGlu
- metabotropic glutamate
- MOR
- μ-opioid receptor
- MVB
- multivesicular body
- NEDD
- neural precursor cell expressed, developmentally downregulated
- NK1
- neurokinin 1
- PAR
- protease-activated receptor
- PAT
- palmitoyl acyl transferase
- PEG
- polyethylene glycol
- PKA
- protein kinase A
- PP
- protein phosphatase
- PTHR
- parathyroid hormone receptor
- PTM
- post-translational modification
- RING
- really interesting new gene
- S1P1
- sphingosine 1-phosphate
- siRNA
- small interfering RNA
- SMO
- smoothened
- STAM
- signal transducing adapter molecule
- Ste2 or Ste2p
- Sterile 2
- SUMO
- small ubiquitin-related modifier
- TAB
- transforming growth factor β–activated protein kinase 1–binding protein
- UBPY
- ubiquitin-specific protease Y
- USP
- ubiquitin-specific protease
- Vps4
- vacuolar protein sorting 4
- WWP
- WW domain–containing E3 ubiquitin protein ligase
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵





{kind=link}
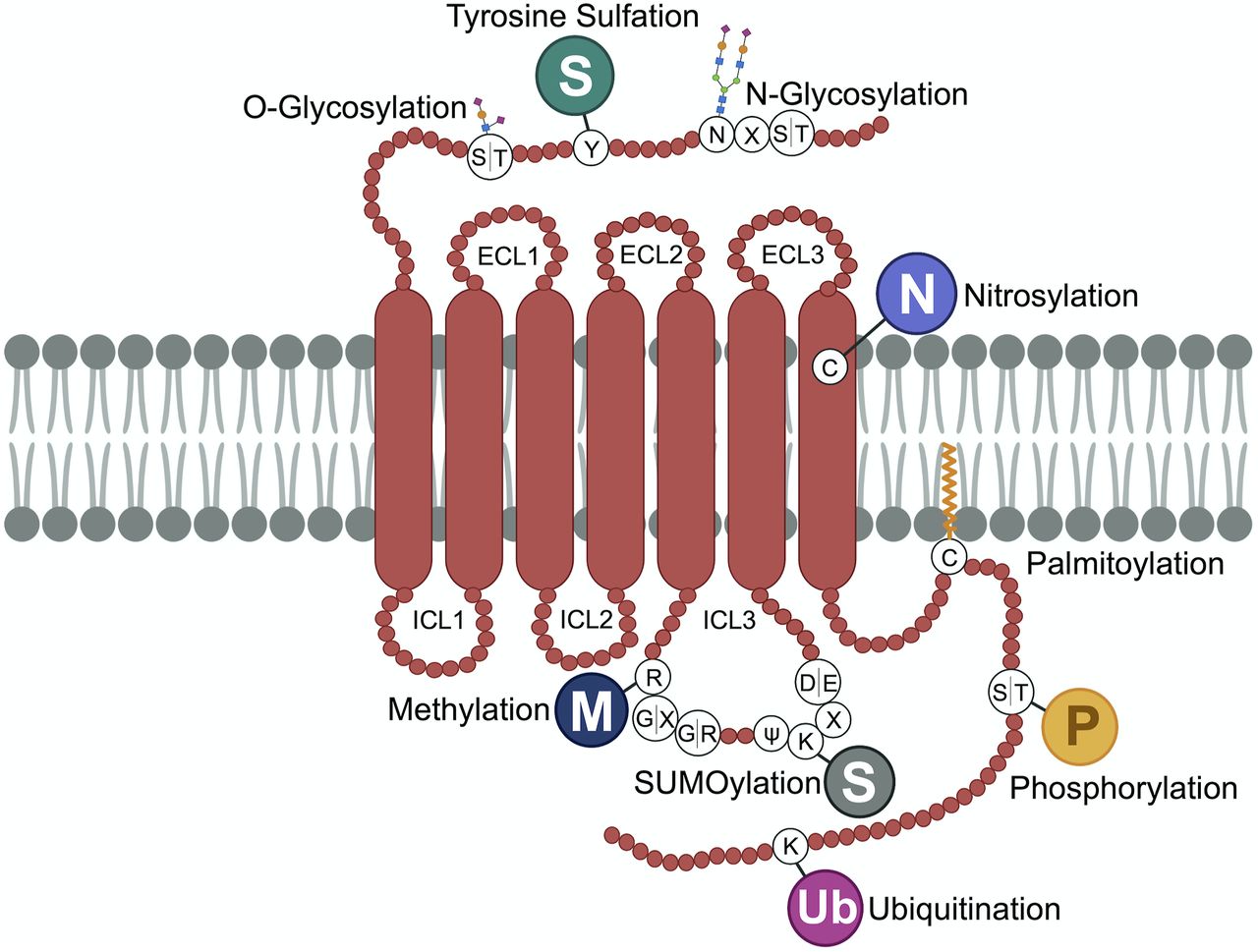{kind=link}
{kind=link}
{kind=link}
{kind=link}
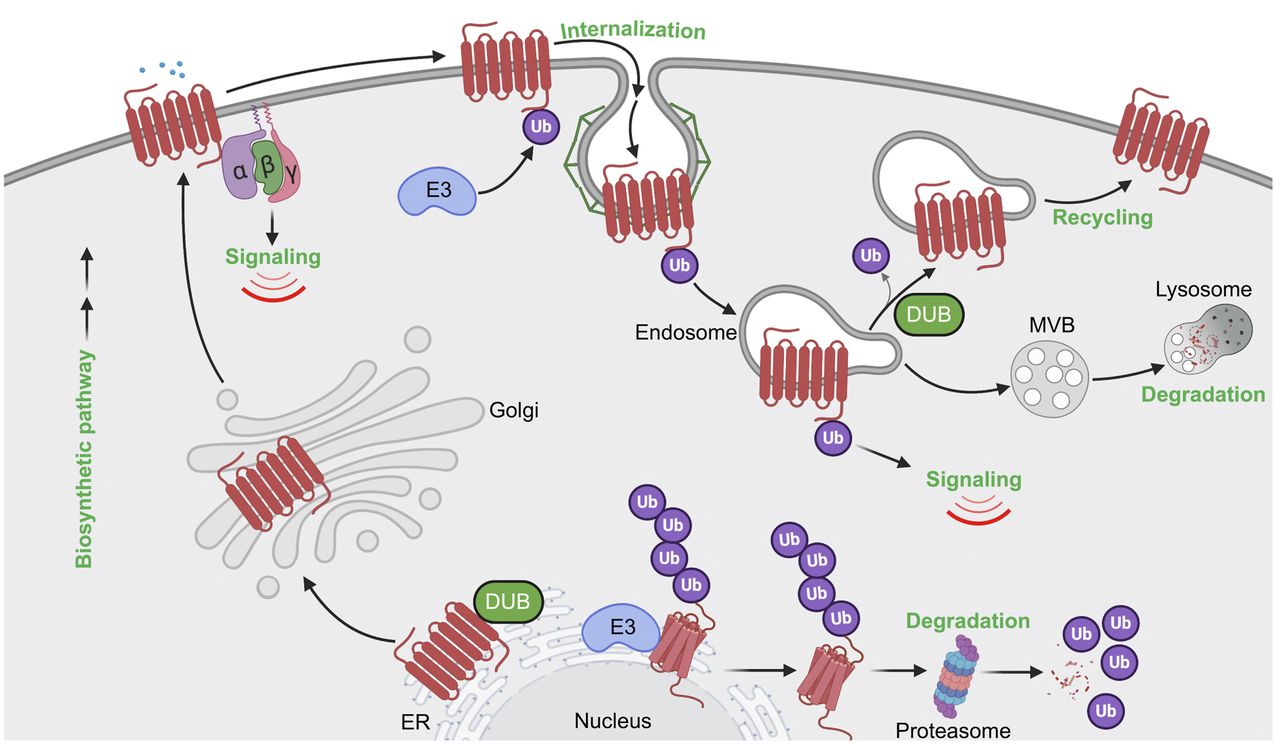{kind=link}
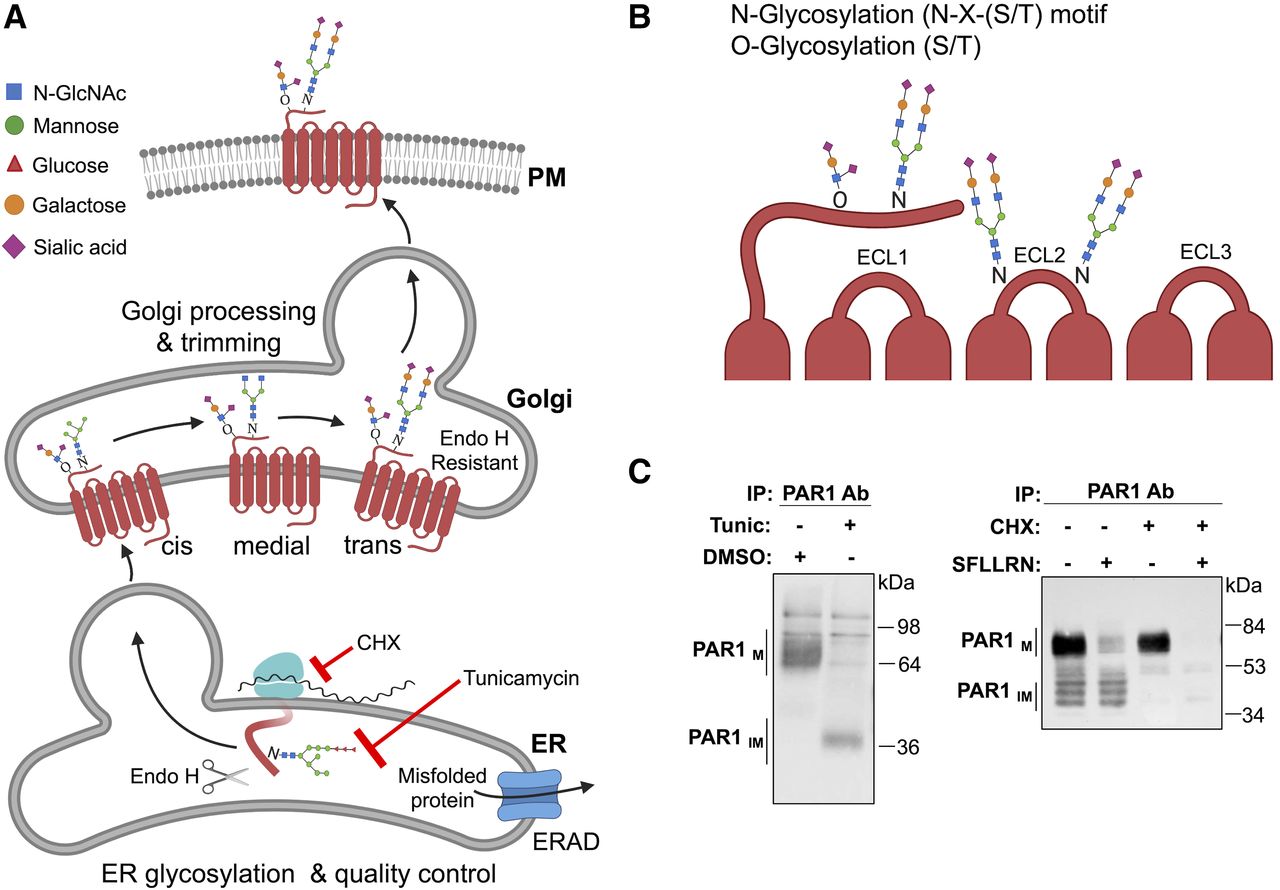{kind=link}
{kind=link}
{kind=link}
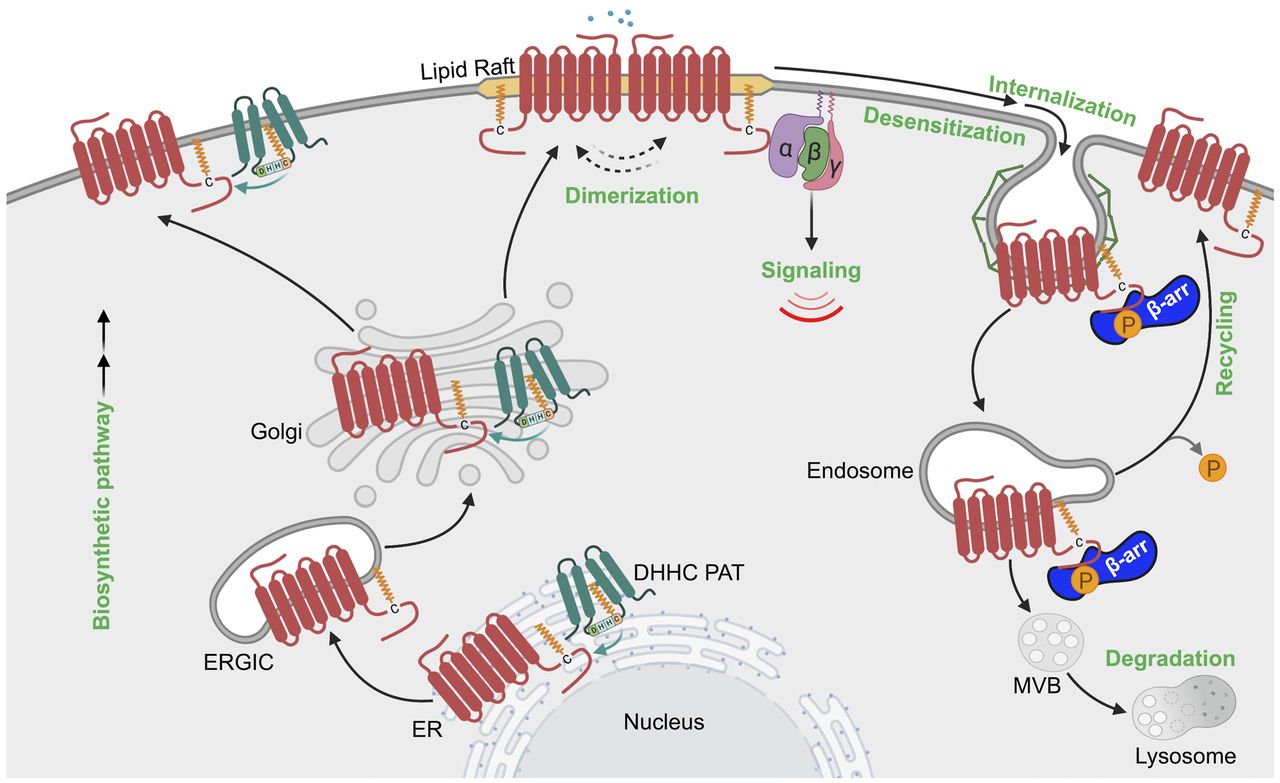{kind=link}