


Visual Overview

Abstract
The field of cAMP signaling is witnessing exciting developments with the recognition that cAMP is compartmentalized and that spatial regulation of cAMP is critical for faithful signal coding. This realization has changed our understanding of cAMP signaling from a model in which cAMP connects a receptor at the plasma membrane to an intracellular effector in a linear pathway to a model in which cAMP signals propagate within a complex network of alternative branches and the specific functional outcome strictly depends on local regulation of cAMP levels and on selective activation of a limited number of branches within the network. In this review, we cover some of the early studies and summarize more recent evidence supporting the model of compartmentalized cAMP signaling, and we discuss how this knowledge is starting to provide original mechanistic insight into cell physiology and a novel framework for the identification of disease mechanisms that potentially opens new avenues for therapeutic interventions.
Significance Statement cAMP mediates the intracellular response to multiple hormones and neurotransmitters. Signal fidelity and accurate coordination of a plethora of different cellular functions is achieved via organization of multiprotein signalosomes and cAMP compartmentalization in subcellular nanodomains. Defining the organization and regulation of subcellular cAMP nanocompartments is necessary if we want to understand the complex functional ramifications of pharmacological treatments that target G protein–coupled receptors and for generating a blueprint that can be used to develop precision medicine interventions.
I. Introduction
Over 60 years ago, the discovery by Earl Sutherland that cAMP is responsible for hormone-induced glycogen breakdown in the liver led to the formulation of the fundamental concept of the intracellular second messenger: a small molecule that relays into the cell the information carried by a hormone, a neurotransmitter, or another extracellular cue and triggers the cascade of biochemical reactions required to achieve the appropriate cellular response to that specific stimulus (Sutherland and Rall, 1958). In follow-up studies, cAMP was found to be synthetized at the cell membrane by adenylyl cyclases (ACs) after activation of seven transmembrane receptors that couple with GTP-binding proteins [G protein–coupled receptors (GPCRs)] (Levitzki, 1988; Mons et al., 1998; Selbie and Hill, 1998). G proteins are trimeric αβγ complexes, in which the α subunit can either stimulate AC enzymatic activity (Gαs) or inhibit it (Gαi), resulting in either an increase or reduction of intracellular cAMP levels (Rodbell et al., 1971; Bloom et al., 1978; Ross et al., 1978). Although Gs and Go were thought for a long time to be the only heterotrimeric G proteins, several additional variants have since been identified (Hepler and Gilman, 1992; Ross and Wilkie, 2000; Wettschureck and Offermanns, 2005). Mammalian cells express an assortment of ACs from a set of nine membrane-integral isoforms (AC1–9) participating in the Gs protein-coupled receptor (GsPCR) signaling (Dessauer et al., 2017) and one soluble AC (AC10) that is not directly linked to the GsPCR signaling but responds to bicarbonate (Wiggins et al., 2018). cAMP signals are translated into action by several effector proteins, including protein kinase A (PKA) (Walsh et al., 1968; Krebs and Beavo, 1979), the exchange protein directly activated by cAMP (EPAC) (de Rooij et al., 1998), cyclic nucleotide–gated ion channels (Fesenko et al., 1985), and the more recently identified Popeye domain containing proteins (Brand, 2005). Phosphodiesterases (PDEs) are a superfamily of enzymes that comprises eight different families with cAMP-degrading activity (PDE1, 2, 3, 4, 7, 8, 10, 11). These enzymes are transcribed from multiple genes that in turn can generate several isoforms (Beavo et al., 1971; Cheung, 1971; Marchmont and Houslay, 1980; Conti et al., 1984; Lugnier, 2006; Francis et al., 2011a). Phosphatases counteract PKA-mediated phosphorylation, terminating the cAMP signal (Khandelwal et al., 1976; Ingebritsen and Cohen, 1983). Evidence generated over the years shows that this pathway triggers a plethora of cellular effects, at times with opposing functional outcomes, including cell proliferation and differentiation (Stork and Schmitt, 2002); gene expression (Yamamoto et al., 1988); apoptosis (Weissinger et al., 1997; Suen et al., 2008); several metabolic processes, such as insulin secretion (Brisson et al., 1972), glycogen synthesis (Rous, 1970), and lipogenesis (Geelen and Vaartjes, 1977); and many others.
When Mayer and colleagues observed that in cardiac myocytes catecholamines and prostaglandins raise cellular cAMP to similar levels but have distinct functional effects (Hayes et al., 1980; Beavo and Brunton, 2002), it became clear that the idea that the cellular response to hormones depends on a linear transduction of the message from the receptor to the intracellular effector via cAMP was too simplistic. Those early studies showed that hearts challenged with the β-adrenergic receptor (AR) agonist isoproterenol contract more forcefully (positive inotropy), whereas prostaglandin has no effect on cardiac contractility, even when both stimuli produce similar amounts of cAMP and comparable levels of PKA activation. Clearly, a “linear cascade” model linking hormone binding to its receptor with generation of cAMP, activation of PKA, and substrate phosphorylation could not explain those observations. Although the pleiotropic role of cAMP in the regulation of cellular function became increasingly apparent (Kuo and Greengard, 1969), the question of how cAMP mediates hormone-specific effects remained unanswered for several decades. The last 20 years, however, have seen exciting developments demonstrating that the components of the cAMP signaling pathway are compartmentalized within the cell and that spatial confinement and local regulation are critical for hormonal specificity (Houslay and Milligan, 1997; Steinberg and Brunton, 2001; Beavo and Brunton, 2002; Zaccolo and Pozzan, 2002; Nikolaev et al., 2006; Scott and Pawson, 2009; Houslay, 2010). Here, we review some of the most recent literature to illustrate our current understanding of cAMP subcellular compartments, with a focus on those studies that link local regulation of cAMP with function.
II. Imaging cAMP Nanodomains
The development of genetically encoded molecular tools for monitoring cAMP in real time provided a key methodological breakthrough, as it allowed direct imaging of compartmentalized cAMP signaling in living cells (Adams et al., 1991; Zaccolo et al., 2000; Nikolaev et al., 2004; Ponsioen et al., 2004). This technique exploits the phenomenon of fluorescence resonance energy transfer (FRET) and is based on optical imaging of intact cells expressing genetically encoded probes that provide a readout of cAMP dynamics as they happen in the complex intracellular environment. The biosensors typically include a cAMP binding domain from PKA (Zaccolo et al., 2000; Zaccolo and Pozzan, 2002) or EPAC (DiPilato et al., 2004; Nikolaev et al., 2004; Ponsioen et al., 2004) sandwiched between two spectral variants of the GFP. On cAMP binding to the cAMP binding domain, a conformational change occurs, and the distance between the two fluorophores is modified, affecting the efficiency of energy transfer. The change in FRET can be detected with a conventional optical microscope, making the methodology easy to apply (Stangherlin et al., 2014). The subcellular resolution afforded by these sensors and the possibility to quantitatively monitor the second messenger in real time provided information on cAMP dynamics with unprecedented level of detail (Koschinski and Zaccolo, 2015). With these sensors, it was possible to demonstrate that the cAMP response to activation of GPCRs is not homogeneous within the cell, but the concentration of cAMP is higher within confined subcellular sites (Zaccolo and Pozzan, 2002). The heterogeneous change in cAMP results in only a limited subset of effector proteins being activated (Di Benedetto et al., 2008). Other studies showed that different GPCRs couple with different ACs (Bundey and Insel, 2006) and generate distinct local pools of cAMP (Houslay and Milligan, 1997). The spatial confinement of these pools is disrupted on inhibition of PDEs, indicating the key role of these enzymes in cAMP compartmentalization (Jurevicius and Fischmeister, 1996; Terrin et al., 2006; Iancu et al., 2007; Shcherbakova et al., 2007; Chen et al., 2008, 2009; Leroy et al., 2008; Feinstein et al., 2012; Mika et al., 2012; Stangherlin and Zaccolo, 2012; Cooper and Tabbasum, 2014; Guellich et al., 2014; Froese and Nikolaev, 2015).
In a recent advancement of the FRET-based imaging technique, the cAMP sensors have been genetically modified to target them to different subcellular compartments and monitor cAMP signaling events at specific sites (DiPilato et al., 2004; Allen and Zhang, 2006; Liu et al., 2011; Lefkimmiatis et al., 2013; Sprenger et al., 2015; Agarwal et al., 2017; Pendin et al., 2017; Surdo et al., 2017). This methodological refinement further enhanced the spatial resolution of the approach, providing a more detailed map of the organization of the cAMP signaling network. The new targeted tools have contributed additional strong evidence for compartmentalized cAMP signaling, have uncovered novel complexity and interconnectivity in the mechanisms that regulate cAMP subcellular domains, and are starting to reveal the functional relevance of individual cAMP pools. One aspect that has emerged from studies using targeted reporters is that the radius of individual cAMP domains can be as small as a few tens of nanometers. In cardiac myocytes, for examples, these sensors revealed that the plasmalemma, sarcoplasmic reticulum, and the myofilaments, structures that are at most 300 nm apart, experience distinct amplitude and kinetics of the cAMP signal in response to catecholamine stimulation (Surdo et al., 2017). Another study in neurons showed that application of the guidance molecule ephrin-A5 generates a cAMP signal in the sub–plasma membrane space adjacent to lipid rafts but not in the space proximal to nonraft regions of the membrane (Averaimo et al., 2016), indicating that the size of the functional cAMP pools generated must be in the range of 20–200 nm, which is the estimated size of lipid rafts. These are important observations, as they imply that individual functionally relevant cAMP pools can be below the resolution limit of an optical microscope and that cAMP compartments can go undetected unless analyzed using a targeted reporter.
III. Contribution to Compartmentalization of Individual Pathway Components
A. Gs Protein Coupled Receptors
An individual cell detects the message from a huge number of extracellular cues via a multitude of specific GsPCRs at the plasma membrane, and the unique informational content of each extracellular stimulus is retained even though all these signals converge toward raising intracellular cAMP. Clear evidence shows that the different localization of GsPCRs at the plasma membrane contributes to triggering a stimulus-specific cellular response (Averaimo et al., 2016; Halls, 2019). The localization of GsPCRs within different plasma membrane domains (e.g., rafts versus nonraft domains) can physically couple the receptor with diverse components of the signaling cascade, e.g., specific G proteins and AC isoforms (Insel et al., 2005; Pontier et al., 2008; Magalhaes et al., 2012; Agarwal et al., 2018). For example, in the neuroblastoma cell line PC12, in which AC6 and AC7 are the two predominant cyclase isoforms, the cAMP response to activation of the pituitary adenylate cyclase–activating polypeptide type I receptor by its selective endogenous neuropeptide agonist pituitary adenylate cyclase–activating polypeptide 38 was unaffected by knockdown of AC7 but abolished by knockdown of AC6, indicating that coupling of pituitary adenylate cyclase–activating polypeptide type I receptor and AC6 is an exclusive requirement for pituitary adenylate cyclase–activating polypeptide signaling (Emery et al., 2015). In adult mouse cardiac myocytes, caveolin-3 was found to direct the localization of AC5 predominantly to T-tubular invaginations of the plasmalemma, whereas AC6 was found to reside outside the T-tubules, where it is responsible for β1-AR–mediated enhancement of the L-type Ca2+ current (Timofeyev et al., 2013). Another study found that the cAMP signal generated by activation of β2-AR is mainly confined to T-tubules, where its localization requires caveolin-3 (Wright et al., 2014), whereas β1-AR–induced cAMP signals are more diffuse across the entire cell (Nikolaev et al., 2010). The arrangement of receptors at the plasma membrane and their preferential coupling with ACs is not static, and this can significantly contribute to fine-tuning the cellular response to a given stimulus. That activation can affect the localization of GPCRs at specific plasma membrane domains has long been recognized, as studies have shown, for example, that treatment with agonist leads to a marked decline in the abundance of β2-AR (but not β1-AR) in caveolae (Rybin et al., 2000). In addition, GPCRs can switch their coupling to ACs in a regulated manner. For example, in uterine smooth muscle cells, α2-AR stimulation is inhibitory in early pregnancy via predominant Gαi-mediated inhibition of ACs but becomes stimulatory after midterm pregnancy via selective Gβγ activation of AC2 (Zhou et al., 2000, 2007). Evidence also suggests that the spatial arrangement of GPCRs can be disrupted in disease. For example, in chronic heart failure, β2-ARs redistribute from the T-tubules to nontubular portions of the plasmalemma, leading to loss of compartmentalization of the β2-AR–dependent cAMP signal (Nikolaev et al., 2010). Selective stimulation of β1-AR generates a more global cAMP signal and activates proapoptotic pathways and hypertrophy (Engelhardt, 2007), whereas selective stimulation of β2-AR results in a more confined cAMP signal, is antiapoptotic, and has protective effects on the myocardium (Yano et al., 2008). It is possible that the redistribution of β2-AR observed in heart failure may contribute to the failing phenotype as a consequence of disrupted cAMP compartmentation, although direct experimental evidence for a causative role is still missing.
Evidence supports confinement of GsPCRs to membrane hot spots defined by actin fibers, microtubules, and clathrin-coated pits where both receptors and G proteins are concentrated and where G protein coupling and signaling preferentially occur (Magalhaes et al., 2012). Activation of the relaxin family peptide receptor 1 (RXFP1) is an example of how differential coupling of GsPCR with distinct signaling pathway components can result in subcellular context-specific responses. Studies have shown that picomolar relaxin can generate cAMP by binding to a preformed complex involving the relaxin receptor RXFP1 and Gαs/AC2. A slower second wave of cAMP is then generated upon binding of nanomolar relaxin to RXFP1; activation of Gα13 leads to cAMP synthesis via a pathway involving Gβγ-phosphoinositide 3-kinase/protein kinase C (PKC) and AC5 (Nguyen and Dessauer, 2005; Halls et al., 2006, 2007; Dschietzig et al., 2011). However, evidence supports distinct functional outcomes downstream of these different RXFP1-activated pathways, with the Gαs-coupled pathway promoting cAMP response element–controlled gene transcription and the Gα13 pathway regulating nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB)-mediated gene transcription (Halls et al., 2007). Interestingly, the expression level of different Gα13 subunits can vary significantly—for example, in disease conditions (Dschietzig et al., 2011)—providing further mechanisms whereby the functional outcome of receptor activation and cAMP signaling depends on the contingent cellular state.
A recent study connects differential expression levels of Gα subunits and sub–plasma membrane location of GPCR as factors that contribute to the response output downstream of a certain receptor. The data show that the level of expression of different Gα subunits affects efficacy and potency of β-AR ligands, a feature that the authors attribute to Gα subunit–directed partitioning of the receptors in different sub–plasma membrane domains (Onfroy et al., 2017). The hypothesis is that the impact of Gα subunits on receptor membrane distribution could account for receptor-biased pharmacology such that a specific ligand could promote different conformations of the GPCR/G protein complexes, with different outputs depending on their compartmentalization at the cell surface. How biased signaling intersects with compartmentalized cAMP signaling is an important question that remains largely unexplored (Shen et al., 2019).
In addition to differential expression and localization at the plasma membrane, the ability of at least some Gαs-coupled receptors to trigger cAMP production after internalization provides additional mechanisms for diversification of the cAMP message at a temporal and spatial level (Irannejad et al., 2013; Gidon et al., 2014). The conventional paradigm is that, upon activation, receptors are internalized as part of the GPCR desensitization process, which involves receptor phosphorylation and recruitment of β-arrestin followed by GPCR endocytosis (Freedman and Lefkowitz, 1996). Receptor internalization and its degradation in the lysosome provides a mechanism that protects the cell from excessive stimulation, and receptor recycling back to the plasma membrane induces resensitization and protects cells from prolonged hormone resistance (Zhang and Eggert, 2013). However, studies combining imaging of real-time cAMP signaling and GPCR trafficking events in the intact cell have challenged the dogma that internalized GPCRs are inactive by showing that sustained cAMP signaling can occur from GsPCR embedded in internalized vesicles (Freedman and Lefkowitz, 1996; Ferrandon et al., 2009; Calebiro et al., 2010; Lan et al., 2012; Irannejad et al., 2013; Stoddart et al., 2015; Namkung et al., 2016; Pavlos and Friedman, 2017), where they couple with ACs that are internalized with the receptor (Lazar et al., 2020).
The first direct evidence that internalized ligand/GPCR complexes can stimulate synthesis of cAMP in mammalian cells came from two independent studies showing that thyroid stimulating hormone receptor (TSHR) and parathyroid hormone receptor, respectively, are able to elicit a “second wave” of Gs protein activation after ligand-induced internalization (Calebiro et al., 2009; Ferrandon et al., 2009). Conformation-specific single-domain antibodies that can directly probe the activated state of β2-AR were used to demonstrate that the adrenergic agonist isoproterenol promotes an immediate activation of β2-AR and Gs protein at the plasma membrane and, within several minutes after agonist application, a delayed activation at the membrane of early endosomes (Irannejad et al., 2013). The temporal shift of these two waves of cAMP signals provides opportunities for encoding complex biologic information that the cell can translate into specific cellular effects (Grundmann and Kostenis, 2017). Evidence indicates that signaling by internalized GPCR is functionally relevant and is responsible, for example, for meiosis resumption in the oocyte within the follicle (Lyga et al., 2016) or regulation of transcription of certain cAMP-dependent genes (Tsvetanova and von Zastrow, 2014; Godbole et al., 2017). One study used a bacteria-derived adenylyl cyclase that allows cAMP production to be acutely induced by light and that was selectively targeted either to the plasma membrane or to the membrane of endosomes. cAMP generated on light stimulation at the endosomes, but not at the plasma membrane, triggered expression of the phosphoenolpyruvate carboxykinase 1 gene, a cAMP-regulated enzyme, confirming that information can be encoded in the spatial pattern of cAMP signals and supporting the notion that cAMP synthesis at the endosomes can effectively uncouple transcriptional control from cAMP generated at the plasma membrane (Tsvetanova and von Zastrow, 2014). Different Gs-coupled receptors can adopt distinct modalities of signaling after internalization. For example, whereas internalized parathyrod hormone receptors or β2-adrenergic receptors appear to signal from the early endosome compartment (Ferrandon et al., 2009; Irannejad et al., 2013) and their retrograde trafficking to the trans Golgi coincides with signal termination (Feinstein et al., 2011), signaling from internalized TSHR occurs only at the Golgi/trans Golgi network (TGN) and not at early endosome membranes (Godbole et al., 2017). For the latter modality, the model is that thyroid stimulating hormone (TSH)/TSHR complexes traffic to the TGN and there activate Gs proteins to induce cAMP synthesis. This leads to activation of a local PKA subset and regulation of gene transcription. Importantly, inhibition of TSHR internalization shows no effect on the overall amplitude of the cAMP response but prevents cAMP/PKA signaling at the Golgi as well as phosphorylation of the cAMP response element binding protein (CREB) and gene transcription. In line with this model, subcellular anchoring of PKA to A-kinase anchoring proteins (AKAPs) appears to be required for TSH-dependent regulation of gene transcription (Calebiro et al., 2006), although the detailed architecture of this Golgi/TGN signalosome and its regulation remain largely to be defined. Thus, receptor-specific signaling after internalization can potentially occur at distinct intracellular sites, providing additional opportunities to achieve response specificity via compartmentalization of cAMP.
A number of studies have also documented cAMP signaling from intracellular GPCRs that are located at intracellular membranes independently from receptor internalization (Kumar et al., 2008; Suofu et al., 2017; Dahl et al., 2018; Nash et al., 2019). For example, studies show that a pre-existing β1-AR receptor pool that is not delivered from the cell surface resides at the Golgi, where it can be activated by endogenous ligands that enter the cell by facilitated transport via the organic cation transporter 3 (Irannejad et al., 2017). Activation of this internal pool of receptors induces Epac-phospholipase Cε (PLCε)–dependent phosphatidylinositol-4-phosphate (PI4P) hydrolysis in a process that has been linked to catecholamine-dependent cardiomyocyte hypertrophy (Nash et al., 2019).
B. Adenylyl Cyclases
Two classes of mammalian ACs, the nine transmembrane (AC1 to AC9) and the soluble (AC10) enzyme, generate cAMP from ATP either in response to GsPCR activation or bicarbonate, respectively, or via cross talk with other signaling pathways (Hindson, 2003). All transmembrane ACs share the same general architecture, yet they vary considerably outside their catalytic and transmembrane domains and are subject to complex regulation by a number of different modulators, including G proteins, PKA, protein kinase C, and calcium (Sunahara et al., 1996; Dessauer et al., 2017). The specific regulation and subcellular localization of different transmembrane AC isoforms provide an ideal mechanism for compartmentalization of cAMP signaling (Cooper and Tabbasum, 2014; Halls and Cooper, 2017; Johnstone et al., 2018). For example, AC1, AC3, and AC8 are Ca2+ activated, whereas AC5 and AC6 are inhibited by Ca2+ (Wachten et al., 2010; Halls and Cooper, 2017). Ca2+ regulation, however, can be complex, as AC3, for example, is activated by Ca2+/calmodulin and inhibited by the Ca2+/calmodulin protein kinase CaMKII (Wei et al., 1998). Although both AC5 and AC6 are Ca2+-inhibited, disruption of AC5 increases longevity and protects against aging-induced cardiomyopathy (Yan et al., 2007), whereas deletion of AC6 has opposite effects, leading to increased mortality upon β-AR stimulation–induced cardiomyopathy (Tang et al., 2013). These opposite effects have been ascribed to the different localization of these two enzymes at the plasma membrane. In arterial smooth muscle cells, AC6 and, to some extent, AC3 have been associated with vasodilatory pathways via regulation of K+ channels (Ostrom et al., 2002; Nelson et al., 2011), whereas AC5 was shown to mediate vasoconstriction in response to high glucose. Super-resolution analysis of arterial myocytes revealed a nanometer proximity between subpopulations of AC5 and the L-type Ca2+ subunit Cav1.2. Glucose-induced AC5 activity was found to mediate the activation of an anchored PKA pool, phosphorylation of Cav1.2, potentiation of L-type Ca2+ channel activity, increased [Ca2+]i, and vasoconstriction, a chain of events that the authors suggest underpins increased myogenic tone during diabetic hyperglycemia (Syed et al., 2019).
In other studies, targeted FRET-based sensors were used to detect cAMP in different subcellular domains in GH3B6 cells, a pituitary cell line that secretes prolactin in response to thyrotropin-releasing hormone (TRH) and vasoactive intestinal peptide (VIP). These two hypothalamic hormones act via different signaling pathways: TRH activates a Gq-coupled receptor and signals via inositol 1,4,5-trisphosphate (IP3)-dependent Ca2+ release from the endoplasmic reticulum (ER), whereas VIP activates a Gs-coupled receptor that signals via cAMP. The authors could measure an increase in cAMP at the plasma membrane in response to VIP when using a sensor targeted via a myristylation and palmitoylation motif, but no cAMP signal was detected when using a sensor targeted to the plasma membrane via fusion to AC8, suggesting a coupling of the VIP receptor with ACs other than AC8. In contrast, when the same cells were stimulated with TRH, a dramatic increase in cAMP was registered by an AC8-targeted sensor, whereas the cAMP response at regions of the plasma membrane away from AC8 was significantly smaller (Wachten et al., 2010). The authors found that the different local cAMP response was due to Ca2+-dependent activation of AC8 and to Ca2+-dependent inhibition of AC5 and AC6 (as well as to concomitant stimulation of cytosolic PDE1 by Ca2+) (Wachten et al., 2010). These findings illustrate how different stimuli can generate locally disparate cAMP responses via selective engagement of individual transmembrane ACs, which respond according to their contingent regulatory status.
The structural basis for the ability of different transmembrane ACs to participate in distinct signaling pathways appears to be their unique coupling with signaling elements at the plasma membrane (Fig. 1). A number of transmembrane ACs have been found to be part of multiprotein complexes organized by AKAP (see below) (Bauman et al., 2006; Dessauer, 2009; Li et al., 2012) and including GPCRs/G proteins (Dupré et al., 2007), PDEs, and phosphatases (Crossthwaite et al., 2006) (see below). These complexes provide a local platform for a compartmentalized source of cAMP at the plasma membrane, where ACs can undergo feedback inhibition via PKA (e.g., AC5, 6, and 8) or feed-forward regulation via PKC (e.g., AC2) (Bauman et al., 2006; Willoughby et al., 2012; Efendiev et al., 2013; Shen and Cooper, 2013). In addition, interaction of an AC to an AKAP complex can lead to greater local increases in cAMP. For example, anchoring of an AC to AKAP5 was shown to sensitize the PKA phosphorylation of the anchored transient receptor potential cation channel subfamily V member 1 (TRPV1) channel to the effects of cAMP by shifting the forskolin dose-response curve by ∼100-fold (Efendiev et al., 2013). Similarly, coexpression of AC9 and the AKAP Yotiao was found to sensitize phosphorylation of the α subunit [potassium voltage-gated channel subfamily Q member 1 (KCNQ1)] of the slowly activating delayed rectifier potassium channel I(Ks) to lower concentrations of β-AR stimulation compared with cells expressing only AC9 or Yotiao (Li et al., 2012).

Plasma membrane and primary cilium cAMP signalosomes. A selection of the signalosomes that localize at the plasma membrane and primary cilium are illustrated. PKA-dependent phosphorylation is indicated in yellow. The cellular function that each signalosome controls is indicated by the gray arrows. Ub indicates ubiquitination. The red gradient indicates cAMP. The white halo surrounding the phosphodiesterases indicates reduced local concentration of cAMP. For details of the different protein components, see the main text. ICa, calcium current; ICl, cloride current; INa, sodium current; Hh, hedgehog protein; NHERF1, Na+/H+ exchanger regulatory factor 1; P, phosphate group; PCM1, pericentriolar material 1; SAP-97, synapse-associated protein 97.
In contrast to AC1–9, AC10 is not regulated by G proteins and is activated by bicarbonate (Chen et al., 2000; Bitterman et al., 2013) and ATP (Zippin et al., 2013) and is stimulated by Mn2+ and Ca2+ (Braun, 1975; Braun and Dods, 1975). Although originally identified as a soluble enzyme, soluble AC has been subsequently reported to be present at specific subcellular sites, including mitochondria, nucleus, centrioles, mitotic spindles, and midbodies (Zippin et al., 2003; Valsecchi et al., 2014; Wiggins et al., 2018). At these sites, AC10 is thought to provide a local source of cAMP that is triggered in response to changes in cellular metabolic state. A number of cellular functions have been associated to AC10-generated cAMP, including sperm capacitation and fertility (Esposito et al., 2004), ATP production (Acin-Perez et al., 2009; Di Benedetto et al., 2013), regulation of lysosomal acidification (Rahman et al., 2016), apoptosis (Ladilov and Appukuttan, 2014), and the downstream effects from the trafficking corticotropin-releasing hormone receptor (Inda et al., 2016).
C. Protein Kinase A
PKA is the most extensively studied effector of cAMP (Walsh et al., 1968; Taskén and Aandahl, 2004). In its inactive form, PKA is a heterotetramer composed of two regulatory (R) and two catalytic (C) subunits (Walsh et al., 1968; Corbin and Keely, 1977; Sowadski et al., 1985). Four types of R subunits (RIα, RIβ, RIIα, and RIIβ) and three C subunits (Cα, Cβ, and Cγ) have been described (Diskar et al., 2010). In the absence of cAMP, a dimer of R subunits binds to and blocks the activity of two C subunits. Binding of cAMP to the R subunits causes a conformational change that removes their inhibitory action on C (Taylor et al., 2005, 2013), allowing phosphorylation of protein targets preferentially at a [R/K][R/K/X]X[pS/pT] consensus motif (Taylor et al., 1990). The most significant difference between RI and RII isoforms is the autophosphorylation potential of the inhibitor site in RII subunits at S114, which affects the interaction between RII and C subunits and which, when phosphorylated, results in a reduced activation threshold for PKA (Taylor et al., 1990). PKA tetramers containing RI-type subunits are activated at lower concentrations of cAMP compared with those including RII, providing a mechanism for concentration-dependent selective activation of different PKA subsets. (Dostmann et al., 1990; Moll et al., 2008). Genetic studies demonstrate that PKA subunits are nonredundant (Brandon et al., 1995; Cummings et al., 1996; Hensch et al., 1998), and the different sensitivity to cAMP can contribute to signal compartmentalization, as it provides a means for differential activation of PKA isoforms based on the local cAMP concentration. This is achieved via anchoring of PKA to the multiscaffolding proteins AKAPs, which tether PKA at defined subcellular locations in proximity to specific PKA substrates (see below).
Other mechanisms involving PKA can further contribute to compartmentalize the action of this enzyme. For example, the E3 ubiquitin ligase really interesting new gene-H2 (RING-H2) protein praja2, a component of the ubiquitin proteasome system (UPS) (Yu et al., 2002), has been reported to form a stable complex with, and to be phosphorylated by, PKA. cAMP elevation followed by PKA activation leads to praja2 phosphorylation and activation of UPS-mediated degradation of R subunits. By reducing the R-to-C ratio, this positive-feedback mechanism can promote sustained PKA substrate phosphorylation in response to cAMP (Lignitto et al., 2011). Praja2 targets PKA to distinct cell compartments, including plasma membrane, cellular organelles, and perinuclear membrane, thus potentially providing a means for local regulation of PKA activity. The Praja2-dependent effect on the amplitude and duration of PKA signaling was shown to impact the phosphorylation level of the transcription factor CREB and nuclear gene transcription, significantly contributing to synaptic plasticity and long-term memory (Lignitto et al., 2011; Rinaldi et al., 2015). An additional mechanism involving ubiquitination of PKA C subunits has been reported (Eccles et al., 2016). ARHGAP36, a member of the Rho GTPase-activating protein family, was shown to directly bind PKA C via a pseudosubstrate motif and to potently inhibit PKA activity. The interaction of ARHGAP36 with PKA C quickly promotes their ubiquitination and targets them for endolysosomal degradation, leading to effective suppression of PKA signaling. ARHGAP36-mediated endolysosomal degradation of C subunits can therefore provide a mechanism for timely inactivation and selective degradation of a restricted pool of C subunits and, as described for Paja2-dependent ubiquitination of R subunits, may contribute to the compartmentalization of cAMP/PKA signaling (Eccles et al., 2016).
D. A-Kinase Anchoring Protein
The AKAPs play a fundamental role in signal compartmentalization, as they are responsible for tethering PKA holoenzyme to specific subcellular locations in physical proximity to PKA targets, increasing the specificity and efficiency of signaling (Lees-Miller and Anderson, 1989; Scott et al., 1990; Hulme et al., 2003; Lygren et al., 2007; Skroblin et al., 2010). All AKAPs identified so far (>30) share a short characteristic motif that serves to anchor PKA R subunits. The interaction between AKAPs and PKA occurs via binding of the dimerization-docking domain, located at the N terminus of the PKA R subunit, to an amphipathic α-helix, generally encompassing 14–18 residues, that is conserved among the AKAP family members. All four R subunit isoforms contain the conserved dimerization-docking domain (Banky et al., 1998; Gold et al., 2006). AKAPs include additional interaction sites that are responsible for their subcellular targeting and mediate interaction with other components of the cAMP pathway, including GPCRs (Malbon et al., 2004; Malbon, 2007), ACs (Piggott et al., 2008; Dessauer, 2009), phosphatases (Coghlan et al., 1995; Dodge-Kafka et al., 2010), PDEs (Dodge et al., 2001; Scott et al., 2013), as well as other kinases, including PKC, mitogen-activated protein kinase kinase (MEK)1/2, extracellular signal-regulated kinases (ERK)1/2, polycystic kidney disease (PKD), and others (Klauck et al., 1996; Tasken et al., 2001; Scott et al., 2013). The diverse subcellular targeting domains present in AKAPs recruit kinase activities to specific subcellular compartments, such as the plasma membrane, nucleus, mitochondria, endo/sarcoplasmic reticulum, the primary cilium, and other organelles (Langeberg and Scott, 2015; Torres-Quesada et al., 2017) (see below; illustrated in Figs. 1–3). Given the ability of each AKAP to bind and compartmentalize a different assortment of kinases, their substrates, and a variety of regulatory enzymes, a large diversity of PKA-focused signaling complexes (signalosomes) can be assembled at different subcellular locations where specific cAMP signaling is implemented locally (Greenwald and Saucerman, 2011).
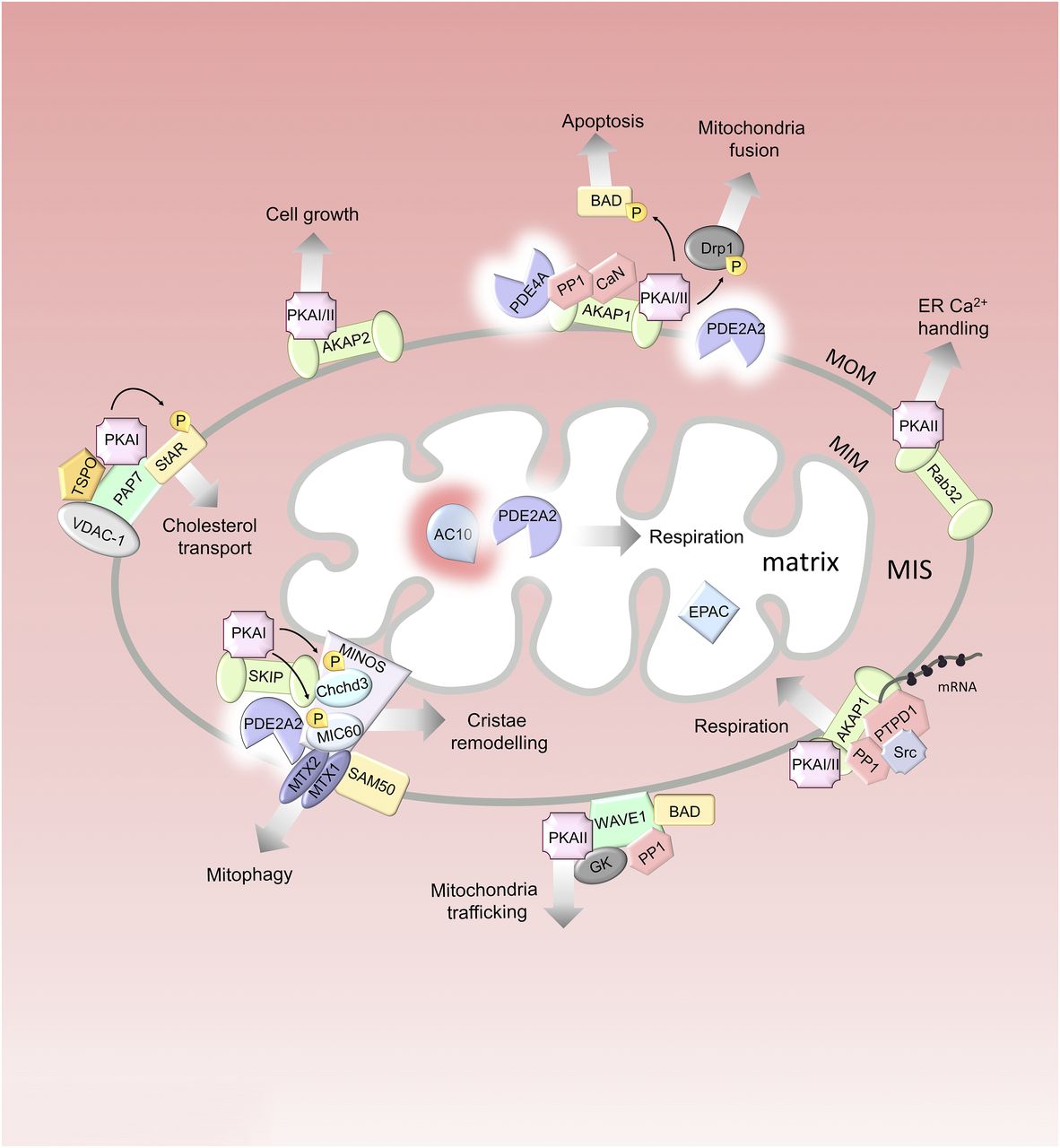
Mitochondrial cAMP signalosomes. Multiple cAMP signalosomes localize to the mitochondria. Red gradients illustrate cAMP. The MOM is permeable to cytosolic cAMP, whereas the MIM is not. In the matrix, AC10 generates the second messenger locally. For the individual protein components of the signalosomes, see the main text. GK, glucokinase; P, phosphate group; PTPD1, protein tyrosine phosphatase D1; SAM50, sorting and assembly machinery component; StAR, steroidogenesis acute regulatory protein; TSPO, translocator protein; VDAC-1, voltage-dependent anion channel 1.

cAMP signalosomes at the endoplasmic reticulum, nucleus, and centrosome. For clarity, multiprotein complexes illustrated here may not include all components that have been described in the literature. For identification of individual protein components, please refer to the main text. The red gradient illustrating cAMP highlights the fact that the nuclear membrane is permeable to cAMP, whereas the ER membrane is not. Lighter red halos surrounding PDEs indicate low local cAMP. P, phosphate group; SEC24, secretion 24.
RII subunits show higher affinity for AKAPs than RI subunits (Banky et al., 2003; Kritzer et al., 2012). This seems at odds with the idea of compartmentalized signaling. An untargeted, predominantly cytosolic and freely moving type I kinase that is activated at a lower cAMP concentration than required to activate the AKAP-bound type II PKA, (Taylor et al., 2012) would appear to nullify the advantage of having pools of cAMP and effector PKAII enzymes localised at at the same site. Multiple studies, however, have identified several type I selective AKAPs with high affinity for RI subunits—including sphingosine kinase interacting protein (SKIP) (Kovanich et al., 2010), small membrane AKAP (smAKAPs) (Means et al., 2011; Burgers et al., 2012, 2016), and G protein-coupled receptor 161 (Gpr161) (Bachmann et al., 2016). In addition, a number of so-called dual-specific AKAPs (e.g., AKAP1/2, ezrin, merlin, and AKAP11) can also interact with RI dimers, although typically with lower affinity than with RII dimers (Herberg et al., 2000; Pawson and Scott, 2010; Skroblin et al., 2010; Torres-Quesada et al., 2017). Experiments using fluorescence recovery after photobleaching showed that in cardiomyocytes, RI, similarly to RII, is largely anchored to AKAPs (Di Benedetto et al., 2008). In the same study, cAMP FRET-based sensors selectively targeted to RI- or RII-binding AKAPs were used to demonstrate that PKA type I and type II define distinct intracellular signaling compartments in these cells. Activation of β-AR generates a cAMP response that is selectively confined to AKAP-anchored PKA type II. This cAMP pool leads to phospholamban (PLN) and troponin I phosphorylation and is sensitive to rolipram (a PDE4 selective inhibitor). Conversely, prostaglandin 1 and glucagon stimulation of cardiac myocytes generates a cAMP signal that is selectively confined to AKAP-anchored PKA type I. This pool of cAMP is selectively sensitive to the PDE2A inhibitor erythro-9-Amino-β-hexyl-α-methyl-9H-purine-9-ethanol (EHNA) and does not affect PLN or troponin I phosphorylation (Di Benedetto et al., 2008).
The study of the functional role of PKA-AKAP interactions has been facilitated by the development of several disruptor peptides and small molecules (Carr et al., 1992; Christian et al., 2011). For example, the peptide super A Kinase anchoring protein-in silico (SuperAKAP-IS) is 10,000-fold more selective for RII over RI (Gold et al., 2006), whereas the peptide RI anchoring disruptor shows 1000-fold higher affinity for RI over RII (Gold et al., 2006). Disruptor peptides that displace PKA specifically from individual AKAPs have been more difficult to generate. Some success has been achieved using a screening approach based on high-resolution structural information. In this study, several engineered RII subunit fragments that show some preference in disrupting interaction of RII with specific AKAPs were isolated. These peptides, named RSelect, were shown to have some selectivity for targeting the cognate AKAP in living cells, although their affinity of binding remains lower than endogenous full-length RII subunits, limiting for the moment their applicability (Gold et al., 2013). One recent example that successfully used PKA-AKAP–disrupting peptides to study the multifaceted role that AKAPs play in the regulation of cAMP/PKA signaling has been reported by Gorshkov et al. (2017), who observed cAMP gradients in developing hippocampal neurons, with higher cAMP levels in more distal regions of the axon compared with other regions of the cell; disruption of PKA-AKAP interactions using competing peptides enhanced the cAMP gradient and promoted axon elongation. The data suggest that, during development, AKAPs localized in the elongating axons organize a negative-feedback loop whereby AKAP-bound PKA activates a local PDE, resulting in reduced local cAMP levels (Gorshkov et al., 2017). Thus, depending on the different signaling elements that they assemble locally, AKAPs can play a complex role in the regulation of the cAMP/PKA pathway, in some cases facilitating and in others quenching signaling.
The localization of some AKAPs has been shown to be dynamic and to respond to changes in cellular conditions, resulting in translocation of the cAMP signalosome across the cell (Fields et al., 2016). One example is AKAP79/150, which targets both PKA and protein phosphatase 2B (PP2B)/calcineurin (CaN) to the plasma membrane to regulate the phosphorylation status of glutamate receptor 1. Activation of glutamate receptors results in the redistribution of AKAP79/150 and its anchored PKA-RII, but not of PP2B/CaN, from postsynaptic membranes to the cytoplasm in hippocampal slices (Smith et al., 2006).
Evidence is also emerging that alteration of AKAP-PKA interactions is linked to disease (Suryavanshi et al., 2018). Analysis of large datasets has uncovered genetic variation in AKAPs that correlates with susceptibility to a variety of conditions, including neurologic disorders, hypertension, cardiac arrhythmias, and certain cancers (Kammerer et al., 2003; Chen et al., 2007; Logue et al., 2014; Reggi and Diviani, 2017; Kjällquist et al., 2018). A systematic investigation of the properties of polymorphic variants in AKAPs that elicit changes of single amino acids in the helical region that binds to regulatory subunits type II of PKA has shown that a number of these amino acid substitutions disrupt PKA anchoring. In one example, a valine-to-methionine variant in the PKA-anchoring helix of AKAP18 was sufficient to reduce RII-binding affinity by over 8-fold, with functional consequences on the cAMP-dependent potentiation of L-type Ca2+ currents (Smith et al., 2018).
Although a large body of evidence supports a key role of AKAPs in directing context-specific cellular outputs of cAMP/PKA signals, one long-standing question is how, on PKA activation, dissociation of the C subunits from the R subunits dimer does not result in uncontrolled phosphorylation of PKA targets that are distal to the PKA-AKAP complex, thus nullifying the advantage of locally anchoring the kinase. A few recent studies have provided some possible answers. One hypothesis is that, for PKA to phosphorylate its substrates, complete dissociation of C from R subunits is not necessary. This idea was originally suggested after the observation that the rotational mobility of labeled C subunits complexed with dimeric R subunits was unchanged on addition of cAMP (Yang et al., 1995). More recent work by Smith et al. has provided fresh evidence in support of this hypothesis (Smith et al., 2013, 2017). Using pull-down experiments and native mass spectrometry of PKA holoenzyme in complex with AKAPs, the authors observed that, in the presence of cAMP, the AKAP-bound PKA holoenzyme remains largely intact (Smith et al., 2017). Using single-particle electron microscopy analysis of the AKAP-PKA complex, they observed that intrinsically disordered domains within the RII subunits allow a significant level of conformational plasticity within the AKAP-PKA complex (Smith et al., 2013). This molecular plasticity of the R subunit would provide a radius of motion to the associated C subunits of 15–25 nm, constraining phosphorylation to targets that are within the immediate vicinity of the AKAP-anchored PKA (Smith et al., 2013). An alternative hypothesis to explain how activation of PKA results in preferential phosphorylation of local targets is that C subunits that dissociate upon cAMP binding are rapidly recaptured by R subunits that are present at high density in the same neighborhood. A study revealed that the R subunits can be expressed in large molar excess of C subunits: up to 17-fold in the cells analyzed in that investigation (Walker-Gray et al., 2017). Clustering of AKAP complexes has also been reported—for example, at the plasma membrane (Mo et al., 2017)–which would effectively increase the local concentration of PKA R subunits. With increased local buffering capacity provided by free R subunits, the range over which the released C subunit can phosphorylate substrates would depend on its rate of diffusion and the rate of recapture.
In addition to AC species, AKAPs can also bind PDEs, thus providing the signaling nodes with machinery that allows them to both form and shape cAMP microenvironments (Sherpa et al., 2019).
E. Exchange Protein Directly Activated by cAMP
EPAC1 and EPAC2 proteins function as guanine nucleotide exchange factors for both Rap1 and Rap2 (de Rooij et al., 1998, 2000). Rap, a small Ras-like GTPase, belongs to the Ras superfamily of small G proteins, which cycle between an inactive GDP-bound state and an active GTP-bound state. Rap, whose activation by cAMP occurs independently of PKA, is activated by several extracellular stimuli and is involved in a large variety of cellular processes, including cell proliferation and adhesion, actin cytoskeleton dynamics, cell polarity, exocytosis, membrane protein recycling, and cell differentiation (Bos, 2006). Several studies support the involvement of EPAC in cell migration and proliferation in multiple types of cancer, making this protein a potential therapeutic target for cancer treatments (Wang et al., 2017; Kumar et al., 2018).
As for PKA, EPAC proteins are highly compartmentalized by different anchoring mechanisms. In response to cAMP, EPAC1 translocates and anchors to the plasma membrane, where it tethers to phosphatidic acid or to phosphorylated cytoskeleton proteins to induce its downstream effectors (Ponsioen et al., 2009; Gloerich and Bos, 2010; Gloerich et al., 2010; Consonni et al., 2012). EPAC2 is targeted to the plasma membrane after binding to activated Ras proteins independently of its interaction with cAMP (Li et al., 2006). In thyroid cells, EPAC1 and PKA simultaneously bind to radixin, an actin filament binding protein that belongs to the ezrin/radixin/moesin family, and form a functional cAMP-sensing sub–plasma membrane domain in which the two effectors act synergistically to mediate efficient proliferation in response to the mitogenic action of thyroid stimulating hormone (Hochbaum et al., 2011). Epac1 and Epac2 show distinct activation patterns in response to cAMP-raising stimuli, indicating that, similarly to PKA, differently localized EPAC isoforms are under the control of distinct pools of cAMP. For example, in cardiac myocytes, EPAC1 has been shown to localize to the plasmalemma, sarcoplasm, and nuclear/perinuclear region (Cazorla et al., 2009; Pereira et al., 2015). Stimulation of β1-AR, but not β2-AR, induces the recruitment of a β-arrestin-EPAC1 signaling complex at the plasma membrane, whereby it activates a pro-hypertrophic signaling cascade involving the small GTPases Rap2, Ras, and CaMKII, triggering histone deacetylase (HDAC) type 4 nuclear export and initiation of a pro-hypertrophic gene program (Berthouze-Duquesnes et al., 2013). By contrast, EPAC2 has been found to localize at the T-tubules (Pereira et al., 2015), and its activation by cAMP in response to β1-AR stimulation leads to CaMKII-dependent diastolic sarcoplasmic reticulum (SR) calcium release and arrhythmias via a pathway that involves EPAC-phosphoinositide 3-kinases (PI3K)-protein kinase B (Akt)-nitric oxide synthase (NOS1) (Pereira et al., 2013, 2017).
F. Phosphodiesterases
The different PDE isoforms have distinct tissue distribution, and each individual cell typically expresses several PDE isoforms. The diversity of binding affinities and regulatory and targeting features of the various PDE isoforms provides a sophisticated system for unique regulation of amplitude and duration of cAMP signals at different sites (Houslay and Milligan, 1997; Houslay, 2001, 2010; Houslay and Adams, 2003; Houslay et al., 2005; Lugnier, 2006; Zaccolo et al., 2006; Conti and Beavo, 2007; Francis et al., 2011a). The large number of PDE isoforms arises from a combination of multiple genes, alternative splicing sites, and multiple transcription starting points. Each member shares a similar overall structure but is functionally unique. The catalytic domain located at the C-terminal region of each PDE enzyme is highly conserved (Conti and Beavo, 2007). The N terminus of these proteins is quite diverse and confers unique regulatory properties and targeting sequences that result in tethering of PDEs to specific subcellular locations (Bender and Beavo, 2006; Zaccolo, 2009; Kokkonen and Kass, 2017). The N-terminal region can carry post-translational modification sites. For example, phosphorylation of PDE3, PDE4, and PDE10 modulates their enzymatic activity and the ability of these enzymes to form complexes with other proteins. In addition, some PDEs can be regulated by cGMP (e.g., PDE2 and PDE3) (Zaccolo and Movsesian, 2007; Kokkonen and Kass, 2017). The UPS is also involved in the regulation of PDE degradation (e.g., PDE4D), which leads to upregulation of cAMP levels and to activation of PKA (Zhu et al., 2010). For long PDE4D isoforms, direct binding of the protein phosphatase calcineurin has been shown to inhibit proteasomal degradation of the PDE. Such degradation may occur in specific cellular compartments, including the nucleus, suggesting a complex regulatory mechanism in which both phosphatases and kinases are involved in the control of cAMP signaling (Zhu et al., 2010; Rinaldi et al., 2015). A detailed discussion of the regulatory mechanisms that control the activity of PDEs is beyond the scope of this article, but excellent reviews are available elsewhere (Conti and Beavo, 2007; Houslay, 2010; Cheepala et al., 2013; Baillie et al., 2019; Chao et al., 2019).
A large body of experimental evidence (Jurevicius and Fischmeister, 1996; Rich et al., 2001; Zaccolo and Pozzan, 2002; Mongillo et al., 2004; Rochais et al., 2004; Houslay et al., 2005, 2007; Lynch et al., 2006; Baillie, 2009; Houslay, 2010; Perera and Nikolaev, 2013; Lefkimmiatis and Zaccolo, 2014; Kokkonen and Kass, 2017) as well as computational studies (Oliveira et al., 2010; Saucerman et al., 2014; Chay et al., 2016; Yang et al., 2016; Lohse et al., 2017) indicate that PDEs play an important role in compartmentalization of cAMP signaling. However, how PDEs can accomplish this is still debated (Feinstein et al., 2012; Saucerman et al., 2014; Lohse et al., 2017). The heart of the problem is in the apparent contradiction between the high diffusivity of cAMP and the somewhat sluggish enzymatic activity of the PDEs (Koschinski and Zaccolo, 2017). The diffusion coefficient of cAMP has been experimentally calculated to be around 40 μm2/s, only approximately one order of magnitude lower than in water (Nikolaev et al., 2004; Richards et al., 2016). Based on the fast intracellular diffusion of cAMP, one would predict fast equilibration of cAMP levels across the cell, in apparent conflict with the notion of cAMP compartmentalization (Terrin et al., 2006; Lefkimmiatis and Zaccolo, 2014; Kokkonen and Kass, 2017). Based on the reported KM and Vmax values for PDEs, it is difficult to envisage how PDEs may be able to maintain the concentration of cAMP below the activation threshold of PKA even at basal cAMP levels, measured to be around 1 μM in a number of cell types (Beavo et al., 1974; Börner et al., 2011; Koschinski and Zaccolo, 2017), let alone contribute to compartmentalization of high concentrations of fast diffusing cAMP generated on hormonal stimulation. Inside a living cell, a number of factors may however come into play that slow down cAMP diffusion, including cAMP buffering (Chen et al., 1999; Lefkimmiatis et al., 2009), cytosol viscosity (Feinstein et al., 2012), and physical barriers (Richards et al., 2016). These factors may be particularly relevant within the restricted volume of a nanodomain, in which the stoichiometry of the molecules involved may be close to equimolar and enzymatic reactions do not follow Michaelis-Menten kinetics. In the confined volume of a nanodomain, local PDEs may be able to effectively reduce the concentration of free cAMP, keeping it below the level achieved a few tens of nanometers farther away (Musheshe et al., 2018; Chao et al., 2019).
Early evidence of a role for PDEs in compartmentalization of cAMP was provided by Hohl and Li, who showed that although approximately 45% of the cAMP generated upon isoproterenol stimulation of cardiac myocytes is found in the cellular particulate fraction, combining isoproterenol stimulation with inhibition of PDEs results in a decline to less than 20% of the proportion of total cAMP residing in the particulate fraction, in spite of a 3.4-fold total increase in cAMP. The authors concluded that this was a result of compartmentalized cAMP in the particulate fractions due to high PDE activity in the cytosol (Hohl and Li, 1991; Méry et al., 1991). cAMP-sensitive FRET-based sensors confirmed the central role of PDEs in shaping the intracellular cAMP nanodomains (Zaccolo and Pozzan, 2002; Nikolaev et al., 2004; Leroy et al., 2008; Surdo et al., 2017). PDE family–selective inhibitors are available and have contributed significantly to our understanding of the role of different PDEs in the compartmentalization of cAMP (Houslay et al., 2005; Lugnier, 2006; Francis et al., 2011b; Page and Spina, 2012; Kokkonen and Kass, 2017). For example, in rat cardiac myocytes, PDE3 and PDE4 enzymes show a very distinct localization, and inhibition of PDE4, but not inhibition of PDE3, results in significant increase in cAMP levels generated by β-agonists (Mongillo et al., 2004; Bedioune et al., 2017; Conti, 2017). Currently available pharmacological inhibitors do not discriminate between different isoforms within the same PDE family. To dissect the role of individual PDE isoforms in compartmentalization, an approach that has been used successfully relies on using isoform point mutants that are catalytically inactive. When overexpressed in the cell, the mutant PDE has a dominant negative effect by displacing the endogenous active enzyme from its anchor sites, effectively resulting in local ablation of cAMP hydrolytic activity (Lynch et al., 2007). Studies using this approach have shown that different isoforms of the same PDE family have a nonredundant and specific role in shaping local cAMP gradients, and their displacement can effectively disrupt local cAMP nanodomains (Baillie et al., 2003; Lynch et al., 2005; McCahill et al., 2005; Zoccarato et al., 2015). For example, expression of the catalytically inactive mutant PDE4D5-D556A in cardiac myocytes was used to demonstrate that β-AR activation recruits a β-arrestin–sequestered subpopulation of the PDE4D5 isoform that specifically regulates the phosphorylation of the receptor by PKA and induces a switch of the receptor from Gs to Gi coupling (Baillie et al., 2003). In another study, overexpression in neonatal rat cardiac myocytes of PDE2A2, PDE3A2, and PDE4D3 catalytically inactive mutants showed that displacement of endogenous PDE4D3 and PDE3A2 results in cardiac myocyte hypertrophy, whereas displacement of PDE2A2 counteracts hypertrophic growth induced by chronic β-AR stimulation (Zoccarato et al., 2015).
G. Phosphatases
The phosphorylation state of the PKA targets in a subcellular domain depends on the dynamic balance between kinase activity and phosphatase activity available at that specific site (Virshup and Shenolikar, 2009), and phosphoprotein phosphatases (PPPs) are the last checkpoint control for termination of the cAMP/PKA signaling cascade (Heijman et al., 2013). A key defining feature of all PPPs is that they are multimeric enzymes in which the catalytic subunit is associated with numerous regulatory subunits. These regulators are key determinants of PPP subcellular localization, substrate specificity, and enzymatic activity, conferring high complexity to the phosphatase system and the ability to exert a highly diversified local action (Virshup and Shenolikar, 2009). Dephosphorylation of PKA targets is accomplished by the three most abundant serine/threonine phosphatases–namely, protein phosphatase 1 (PP1), PP2A, and CaN (or PP2B) (Virshup and Shenolikar, 2009). In cardiac myocytes, for example, PP1, PP2A, and CaN account for >90% of the total phosphatase activity (Heijman et al., 2013).
PP1 is ubiquitously expressed. It includes three types of catalytic subunits that are encoded in different genes (PP1α, PP1γ, and PP1δ, also known as PP1β) (Herzig and Neumann, 2000). Each PP1α and PP1γ isoform has two splice variants (PP1α1 and PP1α2; PP1γ1 and PP1γ2), and different isoforms localize to distinct cellular regions. For example, in the heart, the myofibrillar fraction contains mainly PP1α (Herzig and Neumann, 2000), whereas PP1β predominantly localizes to the longitudinal SR, and PP1γ is enriched in the junctional SR (Aoyama et al., 2011). PP1 catalytic subunits can dimerize with more than 200 different regulatory subunits (Heroes et al., 2013). Notably, PP1 has been found to be associated with AKAPs, in which it can locally modulate the phosphorylation status of PKA targets within the same AKAP complex. For example, in cardiac myocytes, PP1 interacts with the AKAP Yotiao and regulates the PKA-mediated phosphorylation of the KCNQ1 subunit of the slowly activating potassium channel I(Ks) (Marx et al., 2002). PP1 interaction with AKAP11 (also called AKAP220) has been suggested to play a role in regulating the activity of glycogen synthase kinase 3b (Schillace et al., 2001), whereas anchoring of PP1 to AKAP1 has been proposed to be involved in efficient nuclear envelope assembly after mitosis (Steen et al., 2000). PP1 also interacts with AKAP7 (also called AKAP18), although indirectly. At this AKAP complex, PP1 binds Inhibitor-1 to promote its phosphorylation by PKA, and the net effect of this later phosphorylation event is to promote local inhibition of PP1c (Singh et al., 2011).
PP2A can form a dimer containing a catalytic (PP2A-C) and a structural scaffold (PP2A-A) subunit or a trimer consisting of scaffold, catalytic, and regulatory subunits. The scaffold and catalytic subunits consist of two isoforms each (PP2A-Aα and PP2A-Aβ; PP2A-Cα and PP2A-Cβ), whereas the regulatory PP2A subunits are organized into four families (PP2A-B, PP2A-B′, PP2A-B′′, and PP2A-B′′′), most of which contain multiple isoforms with different splice variants (Shi, 2009; DeGrande et al., 2013; Heijman et al., 2013). Cells can assemble more than 200 distinct protein complexes containing different combinations of PP2A-A, PP2A-B, PP2A-C, and other proteins (Virshup and Shenolikar, 2009). Although regulatory subunit α and β are mainly cytosolic, the γ subunit is enriched in the cytoskeletal fraction, conferring different subcellular localizations to the PP2A holoenzyme (Strack et al., 1998). In addition, PP2A localization can be mediated by interaction with AKAPs. For example, in striated muscle, PP2A is part of a complex organized by muscle-specific A-kinase anchoring protein (AKAP6 or mAKAP) and also includes multiple other signaling components (Dodge-Kafka et al., 2010).
CaN is a Ca2+-dependent phosphatase consisting of one catalytic (CNA) and one regulatory (CNB) subunit. CNA is expressed by three genes (CNAα, CNAβ, and CNAγ), whereas CNB is expressed by two genes (CNBα and CNBβ) (Shi, 2009). Regulation of CaN activity toward specific substrates is accomplished primarily through protein-protein interactions, which notably include AKAP5 (AKAP79/150) (Dell'Acqua et al., 2002) and AKAP6 (mAKAP) (Li et al., 2010). For example, in the hippocampus, AKAP5-bound CaN modulates the phosphorylation state of Ser845 of the glutamate A1 (GluA1) subunit of the amino-3-hydroxy-5-methylisoxazole-4-propionate (AMPA) receptor and modulates synaptic plasticity (Woolfrey and Dell'Acqua, 2015). In the heart, the localization of CaN at the AKAP6 complex appears to be involved in efficient dephosphorylation of the nuclear factor of activated T-cells (NFAT), leading to increased gene transcription (Li et al., 2010).
A study comparing the spatiotemporal dynamics of PKA target phosphorylation in the bulk cytosol and at the mitochondrial outer membrane (MOM) using FRET reporters for cAMP and PKA activity showed that the bulk cytosol contains significantly higher phosphatase activity than the MOM (Burdyga et al., 2018). Based on this observation, the authors suggest that the excess of phosphatases in the cytosol may play a role in the compartmentalization of cAMP/PKA signaling by efficient suppression of accidental phosphorylation of PKA targets that may occur outside the designated signaling domain.
IV. cAMP Subcellular Nanodomains
Below, we describe cAMP signalosomes for which there is experimental evidence, grouping them based on their association with different cellular organelles or structures. The aim here is not to provide an exhaustive account of all multiprotein complexes reported in the literature, and relevant reviews have been published elsewhere (Malbon et al., 2004; Taskén and Aandahl, 2004; Skroblin et al., 2010; Langeberg and Scott, 2015; Torres-Quesada et al., 2017). With the following examples, we intend to highlight the fact that compartmentalization of cAMP occurs on a nanometer scale (macromolecular complex dimension) rather than within the micrometer range (organelle or subcellular structure dimension). Individual organelles within the same cell host multiple cAMP nanodomains, each potentially undergoing independent regulation and controlling different functions.
A. Plasma Membrane
The plasma membrane has been shown to host multiple cAMP signaling domains, which are often organized by large multiscaffolding protein complexes. The specific localization at the plasma membrane of individual Gs-coupled receptors affects the subcellular compartment that experiences the rise in cAMP and determines, at least in part, the functional response to receptor activation. This is supported, for example, by experiments in which membrane rafts are disrupted via cholesterol depletion (Calaghan et al., 2008; DiPilato and Zhang, 2009; Wright et al., 2018). How the arrangement of the receptors at the plasma membrane can affect compartmentalized cAMP signaling is not completely understood, but a contributing factor appears to be the coupling of a given receptor with a specific AC.
1. A-Kinase Anchoring Protein 9
The preferential coupling of receptor and AC at the plasma membrane can be mediated by isoform-specific interaction of the cyclase with an AKAP (Baldwin and Dessauer, 2018). For example, AKAP9 interacts with several ACs (AC1, AC2, AC3, and AC9), contributing to their regulation. Although binding of AKAP9 to AC2 does not alter the basal cyclase activity, this interaction significantly decreases AC2 activity in the presence of cAMP-raising agents (Piggott et al., 2008). The smallest splicing variant of AKAP9, called Yotiao, organizes a macromolecular complex containing AC9, PKA, PP1, PDE4D3, the NR1 subunit of the N-methyl-D-aspartate (NMDA) receptor, and the PKA-regulated α subunit (KCNQ1) of the slowly activating delayed rectifier potassium channel I(Ks) (Malbon et al., 2004; Li et al., 2012) (Fig. 1). In the heart, recruitment of PKA to the Yotiao-I(Ks) complex facilitates phosphorylation of KCNQ1, whereas PP1 anchored to the same complex opposes the actions of PKA for tight temporal control (Lin et al., 1998; Marx et al., 2002; Kurokawa et al., 2004). Within the same signalosome, phosphodiesterase PDE4D3 locally degrades cAMP to both temporally and spatially regulate channel function (Terrenoire et al., 2009). AKAP9 is required for sympathetic regulation of I(Ks) currents, which shape the duration of action potentials (Chen and Kass, 2006), and inherited mutations in KCNQ1 and/or AKAP9 that disrupt their interaction are associated with long QT syndrome, a disease characterized by cardiac arrhythmias and sudden death (Chen et al., 2007). It should be noted that not all the proteins that can interact with AKAP9 will do so simultaneously. In particular, it is unlikely that NMDA and KCNQ1 are in the same complex; more likely, AKAP9 organizes a complex with NR1 in neurons and KCNQ1 in heart.
2. A-Kinase Anchoring Protein 5
In neurons, Yotiao binds directly to the NR1 subunit, but a second signalosome is organized by AKAP5 (or AKAP79/150) that interacts indirectly with NMDA receptors via postsynaptic density protein 95 (PSD-95) (Colledge et al., 2000). Both scaffolds, in addition to PKA, also anchor a phosphoprotein phosphatase—PP1 and CaN, respectively—that opposes the action of PKA on NMDA receptor (NMDAR) function. This allows a complex, tightly balanced, local regulation of NMDAR function by the interplay of PKA and these phosphatases. A study showed that, under basal conditions, constitutively active PP1 keeps NMDARs in a state of dephosphorylation and low activity. Upon activation by cAMP, PKA phosphorylates PP1, decreasing its activity. This, combined with direct phosphorylation of the channel, leads to a shift in the balance of NMDARs to a higher phosphorylation state and thus a higher activity state (Westphal et al., 1999).
AKAP5 can function as a scaffold for multiple signalosomes. In addition to the NMDA receptors, this AKAP anchors β-adrenergic receptors, AC5/6, PKA, PKC, the phosphatase CaN, and a variety of channels, including the calcium channel voltage-dependent (Cav1.2), the AMPA-type glutamate receptors, M-type K+ channels, and the capsaicin receptor. Given its multivalent nature, the AKAP5 signalosome coordinates different enzyme combinations to modulate the activity of anchored channels depending on the specific context (Dessauer, 2009). This cluster of proteins enables a negative-feedback loop that temporally regulates cAMP production in the heart and in the brain (Fuller et al., 2014; Murphy et al., 2014) (Fig. 1). In cardiac myocytes, PKA stimulates Cav1.2 currents while also negatively regulating the action of adrenergic receptors and AC5/6. In neurons, AKAP5 forms a complex with synaptic glutamate receptor ion channels through the intermediary scaffolding proteins postsynaptic density protein 95 (PSD-95) and synapse-associated protein 97 (SAP-97). At this molecular hub, AKAP5 anchors PKA and CaN, whose opposing actions regulate AMPA current through glutamate receptor 1 (Scott and Pawson, 2009; Welch et al., 2010).
3. Ezrin
In some circumstances, AKAPs can recruit both PKA and EPAC at the plasma membrane and control their coordinated activity. One example is the regulation of the cystic fibrosis transmembrane conductance regulator (CFTR) channel. A signalosome including the CFTR, the AKAP ezrin (or AKAP78), the Na+/H+ exchanger regulatory factor, PKA, and EPAC1 has been described at the plasma membrane (Sun et al., 2000). Both cAMP effectors regulate CFTR activity, although via distinct molecular mechanisms: PKA phosphorylates CFTR and promotes opening of the channel, whereas EPAC1 stabilizes CFTR at the plasma membrane (Moyer et al., 2000; Lobo et al., 2016). In human airway epithelial cells, cAMP-mediated regulation of CFTR activity requires the integrity of the actin cytoskeleton and compartmentalized cAMP and PKA activity in the sub–plasma membrane domain. CFTR expression is required to maintain an organized cortical cytoskeleton (Favia et al., 2010), and in turn the cortical cytoskeleton is essential for maintaining compartmentalized cAMP and PKA activity for optimal CFTR regulation (Monterisi et al., 2012). The subcortical cytoskeleton, with its densely packed structure, may contribute to limiting the diffusion of cAMP from the plasma membrane to the bulk cytosol, thus promoting activation of membrane-anchored subsets of PKA and EPAC (Rich et al., 2000; Monterisi et al., 2012). In patients with cystic fibrosis, mutations in the CFTR result in a misfolded protein and its defective trafficking to the cell surface, affecting chloride efflux. Pharmacological correctors that rescue the channel expression at the membrane and its activity were shown to also restore the subcortical actin organization as well as the subplasma membrane compartmentalization of cAMP and the local activity of PKA (Abbattiscianni et al., 2016).
4. A-Kinase Anchoring Protein 12
AKAP12 (or gravin) is targeted to lipid rafts, where it appears to interact with the β2-AR (Fan et al., 2001). In prostate cancer cells, AKAP12 was shown to anchor PKA, PKC, calmodulin, cyclins, and Src-tyrosine kinase and to mediate suppression of the metastatic effects of the oncogenic kinase (Su et al., 2013). It has been suggested that in the presence of oxidative stress the lipids in the membrane are oxidized and change their biophysical properties, resulting in relocalization of AKAP12; as a consequence, Src-tyrosine kinase is released from lipid rafts, and the metastatic potential of the cell is increased (Su et al., 2013). In the forebrain, AKAP12 recruits a signaling complex containing PKA, PKC, calmodulin, and PDE4D to the β2-adrenergic receptor that regulates long-lasting forms of hippocampal synaptic plasticity and long-term memory storage (Havekes et al., 2012).
5. A-Kinase Anchoring Protein 7
AKAP7 (or AKAP18) comprises a family of four isoforms: AKAP18α, -β, -γ, and -δ, (Trotter et al., 1999; Henn et al., 2005). The α- and β-isoforms of AKAP15/18 localize at the plasma membrane in complex with L-type Ca2+ channels (Gray et al., 1997; Fraser et al., 1998), whereas the a high-molecular-weight isoform AKAP15/18δ localizes at the sarcoplasmic reticulum (see below). A lipid modification of three residues in the N terminus of the protein mediates interaction with the inner leaflet of the plasma membrane, whereas a leucine zipper–like motif at its C terminus interacts with the cytoplasmic domain of the L-type Ca2+ channel (Hulme et al., 2002). Experimental evidence demonstrates that, in this arrangement, the PKA subset anchored to AKAP15/18 is optimally positioned to phosphorylate the Ca2+ channel in the heart (Gray et al., 1998) (Fig. 1) and the epithelial Na+ channel in the brain (Johnson et al., 2012). However, the knockout of AKAP7 had no effect on L-type calcium channel currents in cardiac myocytes, and some question whether the α splice variant is expressed in heart. More recent work (Yu et al., 2018) suggests that the AKAP Cypher/Z-band alternatively spliced PDZ-motif (Zasp) is important for PKA regulation of Cav1.2 in the heart. Electrophysiological measurements demonstrated that a mutant of AKAP15/18α that lacks the sites of lipid modification reduced the rate of cAMP-dependent stimulation of L-type Ca2+ currents (Fraser et al., 1998), and disruption of the AKAP15/18α-channel interaction with a leucine zipper peptide mimetic had similar effects (Hulme et al., 2002). Compared with AKAP18α, AKAP18β carries an insert of 23 amino acid residues in its N terminus, which leads to apical plasma membrane localization of the AKAP-PKA complex; however, its function remains unknown.
B. Primary Cilium
The primary cilium, a nonmotile, microtubule-based protrusion of the plasma membrane present in most cells in the body, functions as a highly specialized sensory structure with a primary role in cell fate specification and cell proliferation (Ingham et al., 2011). In normal proliferating cells, it is transiently observed in G1 phase and is reabsorbed when the cell enters the cell cycle (Plotnikova et al., 2008). The primary cilium uses cAMP to transduce multiple signals, and given the spatially confined nature of this structure, it is not surprising to find that it hosts compartmentalized cAMP/PKA signaling modules (Moore et al., 2016; Mukherjee et al., 2016; Jiang et al., 2019; Sherpa et al., 2019).
1. G Protein Coupled Receptor 161
Multiple components of the cAMP signaling cascade localize to the cilium, including GPCRs, ACs, PKA, and PDEs. Recently, the orphan GPCR Gpr161 has been reported to act as an AKAP that selectively recruits PKA type to the cilium (Fig. 1). The complex has been shown to be involved in cAMP/PKA-dependent antagonism of Hedgehog-initiated signaling, a pathway that is essential for normal embryonic development (Chen et al., 2011; Bachmann et al., 2016). Gpr161 is the first example of a GPCR that directly recruits PKA holoenzymes to its C-terminal tail, and the receptor itself is a PKA substrate containing a PKA phosphorylation site at the C terminus. A phosphomimetic mutant of Gpr161 was found to significantly reduce the ciliary localization of the Gpr161/PKA complex, suggesting that PKA phosphorylation of Gpr161 affects PKA compartmentalization (Mukhopadhyay et al., 2013; Bachmann et al., 2016; Torres-Quesada et al., 2017).
2. A-Kinase Anchoring Protein 5
A protein complex comprising the protein polycystin-2 (PC-2), a transient receptor potential (TRP)-type, Ca2+-permeable nonselective cation channel; AC5/6; PKA; PDE4C; and the A-kinase–anchoring protein AKAP5 has been described within the primary cilium of renal epithelial cells (Choi et al., 2011) (Fig. 1). PKA directly phosphorylates PC-2, which increases the channel mean open time and regulates PC-2–mediated cation transport. Mutations in PC-2 result in PKD, a genetic disorder characterized by the formation of cysts within the kidney tubules, altered molecular architecture of the cilium, and abnormally elevated cAMP production. Interestingly, treatment with PDE4 activator compounds was shown to lower intracellular cAMP levels and inhibit cyst formation in both animal and human cell models of the disease (Omar et al., 2019), suggesting that targeting cAMP signaling within this domain may be a valuable therapeutic strategy.
3. Pericentriolar Matrix Protein 1
A distinct multimeric complex nucleated by the pericentriolar matrix protein 1 is localized at the base of the cilium. This signalosome includes, in addition to PKA, the kinase NEK10 and the E3 ubiquitin ligase carboxy-terminus of Hsc70 interacting protein (CHIP). Within this multiprotein complex, PKA phosphorylation primes NEK10 for CHIP-mediated ubiquitination and proteolysis, resulting in cilium reabsorption (Fig. 1), a control mechanism for cilium dynamics that might be affected in specific human cancers and genetic disorders (Porpora et al., 2018).
C. Mitochondria
Mitochondria have been increasingly recognized as organelles with tightly regulated cAMP signaling. Indeed, every mitochondrial compartment, including the MOM, the mitochondrial intermembrane space (MIS), and the mitochondrial matrix, harbors one (or more) spatially and functionally distinct cAMP signalosome. cAMP signaling at the mitochondria contributes to the extreme adaptability of these organelles in response to changes in metabolic demands (Carlucci et al., 2008; Reggi and Diviani, 2017) via direct modulation of a variety of functions, including mitochondria biogenesis and morphology, gene expression, metabolism, steroidogenesis, and survival (Zhang and Xu, 2016). Several components of the cAMP signaling pathway, including source of cAMP, effectors, terminators, and targets, have been identified in mitochondria (Monterisi and Zaccolo, 2017; Rinaldi et al., 2018).
PKA R and C subunits were isolated from purified mitochondria over 30 years ago (Schwoch et al., 1990; Papa, 1999). Mass spectrometry analysis of the mitochondrial phosphoproteome identified more than 100 potential PKA targets in the mitochondrial phosphopeptide pool (Grimsrud et al., 2012). Most of these phosphorylation sites are yet to be validated, and it is therefore possible that the mitochondria may harbor additional cAMP signalosomes that remain to be characterized (Monterisi and Zaccolo, 2017). At least five different AKAPs have been found to associate with the MOM: AKAP1 (also known as AKAP121, AKAP149, and spermatid (S)-AKAP84, depending on the species) (Ma and Taylor, 2002; Affaitati et al., 2003), AKAP2 (Wang et al., 2001), Rab32 (Alto et al., 2002), peripheral-type benzodiazepine receptor–associated protein 7 (PAP7) (Li et al., 2001), and Wiskott-Aldrich syndrome protein verprolin homologous (WAVE) (Danial et al., 2003) (Fig. 2). The presence of several AKAPs at the MOM implies that this structure hosts several cAMP signaling domains with different roles in the regulation of mitochondrial function.
1. A-Kinase Anchoring Protein 1
AKAP1 has two N-terminal splice variants, with the shorter form being targeted to the MOM through a functional tubulin-binding motif, which is essential for mitochondrial localization. A longer splice variant has been described that includes a hydrophobic segment upstream to the mitochondrial targeting domain and generates a protein that targets to the ER and may be potentially involved in coordinating Ca2+ homeostasis and cell respiration (Carlucci et al., 2008), although the existence of this longer variant is controversial (Merrill and Strack, 2014). A feature of AKAP1 is its ability to bind both type I and type II PKA holoenzymes (Huang et al., 1999; Ma and Taylor, 2002; Merrill et al., 2011; Torres-Quesada et al., 2017). Other components of the cAMP cascade that have been reported to interact with AKAP1 are CaN (Abrenica et al., 2009), PP1 (Bridges et al., 2006; Rogne et al., 2009), and PDE4A (Asirvatham et al., 2004) (Fig. 2). AKAP1 appears to be involved in multiple signalosomes at the MOM. Whether these are independent multiprotein complexes all simultaneously present on the mitochondria or whether AKAP1 can interact with different components in a dynamic manner remains to be established.
AKAP1 contains at its C terminus a heterogeneous nuclear ribonucleoprotein K-homology (KH) domain, which interacts with nuclear-encoded mRNAs (Chen et al., 1997). Tethering mRNA and ribosomes to the MOM promotes efficient translation and coimport of mitochondrial proteins into the organelle, with important implications for global protein synthesis and mitochondrial physiology (Ranganathan et al., 2002; Dyson et al., 2008; Unal et al., 2008; Gabrovsek et al., 2020). This AKAP1 complex includes, in addition to PKA and mRNA, the protein tyrosine kinases Src and the phosphatases PP1 and protein tyrosine phosphatase D1 (PTPD1) (Fig. 2). This transduction unit supports mitochondrial homeostasis, metabolism, respiration, and survival (Cardone et al., 2004; Livigni et al., 2006).
AKAP1 overexpression stimulates PKA-dependent phosphorylation of the proapoptotic protein BCL2-associated agonist of cell death (BAD), inhibits release of cytochrome c from mitochondria, and protects cells from apoptosis (Harada et al., 1999; Affaitati et al., 2003) (Fig. 2). In addition to BAD, PKA can phosphorylate Bcl-2–associated X protein (BAX) and bcl-2–interacting mediator of cell death (BIM), two other proteins involved in regulation of apoptosis. PKA phosphorylation of these targets, however, has opposite effects. Although PKA phosphorylation of BAD inhibits its apoptotic activity, PKA-mediated phosphorylation of BAX promotes its translocation to mitochondria and triggers cytochrome c release and apoptosis (Srivastava et al., 1998; Kumar et al., 2009). Similarly, phosphorylation of BIM increases its stability and protects it from proteasome-dependent degradation, promoting apoptosis (Moujalled et al., 2011). This is an example of how cAMP can regulate opposing effects even when the targets involved are in relatively close proximity and supports the existence of functionally independent cAMP/PKA signalosomes operating at the mitochondria. Although AKAP1-tethered PKA has been linked to regulation of BAD, whether and how PKA activity is selectively targeted to BAX and BIM remains to be elucidated.
Overexpression of AKAP1 also promotes recruitment of PKA to the MOM and phosphorylation at serine 637 of dynamin-1–like protein (Drp1), a GTPase involved in regulating mitochondrial fusion-fission balance, resulting in inhibition of mitochondrial fission and cell survival (Fig. 2). Studies in cardiac myocytes have shown that downregulation or displacement of AKAP1 results in mitochondrial dysfunction and cell death, confirming the prosurvival actions of AKAP1-tethered PKA (Livigni et al., 2006; Perrino et al., 2010). A similar role has been described in neurons, in which the PKA/AKAP1 complex is involved in maintenance of mitochondrial integrity and protection from cell injury. In invasive breast cancer cells, disruption of this complex affects cell motility and mitochondrial movement toward the leading edge. Analysis of primary and metastatic samples revealed an inverse correlation between AKAP1 expression and mesenchymal markers, and depletion of this signalosome from the MOM was suggested to contribute to the metastatic growth of breast cancer (Aggarwal et al., 2019).
AKAP1 presence at the mitochondria is dynamic and is modulated by the UPS. Such regulation impacts mitochondrial dynamics and reduces mitochondrial oxidative stress in conditions of low oxygen levels, promoting mitochondrial adaptation to metabolic and environmental challenges. After a hypoxic insult, the Really Interesting New Gene (RING) domain E3-ubiquitin ligase seven in absentia homolog 2 (Siah2) ubiquitinates AKAP1, resulting in its proteolysis. Degradation of AKAP1 by the UPS rapidly attenuates the oxidative metabolism, promoting adaptation of cells to accidental hypoxic conditions (Carlucci et al., 2008). Similarly, proteolysis of AKAP1 by the hypoxia-induced Siah2 pathway removes the inhibitory constraint of PKA on the Drp1/fission machinery and leads to mitochondria fragmentation and cell death (Kim et al., 2011).
2. A-Kinase Anchoring Protein 2
AKAP2 also localizes to the MOM and anchors both type I and type II PKA (Wang et al., 2001) (Fig. 2). This signalosome may be cell type–dependent, as AKAP2 was not found in the mitochondria at the neuromuscular junction (Perkins et al., 2001). AKAP2 can also interact with components of the cytoskeleton via its C terminus (Sarma et al., 2015), and its overexpression promotes cell growth and is required for calcitonin-mediated migration of cancer cells (Thakkar et al., 2016; Li et al., 2017).
3.Ras-Related Protein 32
Ras-related protein 32 (Rab32), a member of the Ras superfamily of small-molecular-weight G proteins, was initially described as an AKAP for PKA type II involved in mitochondrial dynamics (Alto et al., 2002) (Fig. 2). More recent studies have shown that Rab32 localizes to mitochondria-associated membranes, a specialized subdomain at the interface between mitochondria and ER that acts as an intracellular signaling hub (Bui et al., 2010; van Vliet et al., 2014). At the mitochondria-associated membrane, Rab32 is involved in the modulation of ER Ca2+ handling; it regulates the levels of the chaperon protein calnexin and is involved in the regulation of the phosphorylation status of both BAD and Drp1 via PKA, affecting mitochondrial dynamics and cell death (Bui et al., 2010).
4. Peripheral-Type Benzodiazepine Receptor–Associated Protein
PAP7 is a mitochondrial AKAP that selectively binds and targets PKA type I to the MOM (Li et al., 2001; Liu et al., 2003). PAP7 is present in all mammalian tissues and cells, with predominant expression in steroidogenic tissues, such as testis and adrenal gland, and organizes a complex that includes translocator protein, the voltage-dependent anion channel 1 (VDAC-1), and type I PKA (Fig. 2). PAP7 myristylation is required for binding to MOM. At this site, PAP7 promotes PKA-directed phosphorylation and catalytic activation of steroidogenesis acute regulatory protein, leading to cholesterol transport into the organelle. Interference with PAP7-mediated cAMP signaling profoundly affects hormone-induced cholesterol transport into mitochondria and downregulates steroidogenesis in endocrine cells. In adrenal tumor cells, PAP7 participates in PKA-dependent mitogenic signaling, supporting tumorigenesis and hormone-independent hypercortisolism (Miller, 2013).
5. Wiskott-Aldrich Syndrome Protein Verprolin Homologous-1
WAVE1 controls actin polymerization and cytoskeletal remodeling (Kim et al., 2006). In neurons, WAVE1 is dephosphorylated and activated upon NMDA stimulation and translocates from the cytoskeleton to the mitochondria, where it induces organelle fission and trafficking along filopodia, promoting filopodia outgrowth and spine morphogenesis, a prerequisite for synaptic activity and plasticity. The mechanisms that regulate localization of WAVE1 to mitochondria seem to involve interaction with mitochondrial BAD family members. These nucleate multivalent complexes on mitochondria that include glucokinase, WAVE1, and other PKA-regulated substrates/effectors (Fig. 2). The complexes assembled by BAD on mitochondria act as metabolic sensors for nutrient availability and stress stimuli, optimally integrating glycolytic machinery and respiration with the organelle network and apoptotic pathway (Danial et al., 2003; Kim et al., 2006; Cheng et al., 2007).
6. Sphingosine Kinase Interacting Protein
The protein SKIP has been identified as an AKAP localized in the MIS. SKIP was shown to specifically interact with PKA RI subunits with high affinity (Kovanich et al., 2010; Means et al., 2011). As the MOM is permeable to cAMP (Acin-Perez et al., 2009), MOM and MIS experience the same level of cAMP. The presence of a SKIP-PKA type I complex in the MIS, however, provides a means for these two compartments to potentially act as distinct cAMP domains whereby the MIS-targeted PKA type I responds to levels of cAMP that are not sufficient to activate the MOM-tethered pools of PKA type II (Lefkimmiatis and Zaccolo, 2014). SKIP interacts with several proteins, including coiled-coil-helix-coiled-coil-helix domain containing (ChChd) 3, ChChd6, sorting and assembly machinery 50 (SAM50), metaxin 1 (MTX1), and MTX2. ChChd3 and ChChd6 are components of the mitochondrial inner membrane organizing system (MINOS), a multiprotein complex that is involved in the regulation of mitochondrial membrane architecture and formation of cristae junctions and contact sites between the outer and inner membranes (Pfanner et al., 2014). PKA phosphorylates ChChd3, and SKIP is required for this phosphorylation to take place (Schauble et al., 2007; Darshi et al., 2011; Means et al., 2011). PKA also phosphorylates another component of MINOS, the protein mitofilin [MICOS complex subunit 60 (MIC60)] (Akabane et al., 2016). A recent study showed that PKA-mediated phosphorylation of MIC60 and ChChd3 reduces phosphatase and tensin homolog (PTEN)-induced putative kinase 1 levels at the MOM and, consequently, negatively regulates Parkin recruitment and mitochondrial clearance by mitophagy (Akabane et al., 2016). These findings suggest that cAMP in the MIS may regulate mitophagy via the MINOS complex, whereas the cAMP signalosomes at the MOM are mainly involved in the regulation of mitochondria fission-fusion dynamics, effected by Drp1, and apoptosis.
7. Mitochondrial Matrix
The mitochondrial matrix appears to define a cAMP compartment that is isolated from the rest of the cell. Cytosolic cAMP cannot reach the mitochondrial matrix, as unlike the MOM, the mitochondrial inner membrane (MIM) is not permeable to cytosolic cAMP (Acin-Perez et al., 2011; Di Benedetto et al., 2013), and studies suggest that the second messenger is generated within mitochondria by the carbon dioxide/bicarbonate-activated and Ca2+-regulated AC10 in response to metabolically generated carbon dioxide (Fig. 2) (Acin-Perez et al., 2009). Therefore, MOM and MIS are physically separated from the mitochondrial matrix, which forms an independent cAMP domain distinct from the cytosol. Evidence supports a mechanism whereby a Ca2+ increase in the mitochondrial matrix stimulates a local cAMP increase that promotes mitochondrial ATP synthesis (Di Benedetto et al., 2013; Lefkimmiatis et al., 2013; Wang et al., 2016). These findings suggest that the mitochondrial matrix may host a cAMP signaling pathway that responds to metabolic changes and is not directly dependent on hormonal stimulation. The fact that PKA subunits lack any obvious mitochondrial targeting sequence challenges the idea of their presence in the matrix (Valsecchi et al., 2014). Indeed, presence of PKA as the effector of cAMP signaling in the matrix remains controversial, and experiments using FRET-based sensors targeted to the matrix failed to detect any PKA enzymatic activity in this compartment (Lefkimmiatis et al., 2013). Other studies also questioned the presence of a PKA pathway in the matrix, as no significant effect of Ca2+ stimulation on oxidative phosphorylation was observed when applying membrane-permeable cAMP analogs or PKA inhibitors (Covian et al., 2014).
8. Mitochondrial Exchange Protein Directly Activated by cAMP
EPAC1 was originally reported to localize to the mitochondria in a cell cycle–dependent manner, although its precise submitochondrial localization and function were unclear (Qiao et al., 2002). Treatment of isolated mitochondria with EPAC-specific activators showed no effect on oxygen consumption and oxidative phosphorylation activity. A recent study demonstrates that during hypoxia/reoxygenation, EPAC1 is activated by AC10-generated cAMP to increase Ca2+ uptake and reactive oxigen species (ROS) production, thereby promoting permeability transition pore (mitochondrial permeability transition pore) opening, cytochrome c release, and caspase-9 and -3 activation (Fazal et al., 2017). Another report, however, provides evidence that both isoform 1 and 2 of EPAC are present in the mitochondrial matrix in rat cardiac myocytes and that cAMP generated by AC10 activates EPAC1 and prevents Ca2+-induced opening of the mitochondrial permeability transition pore (Wang et al., 2016). Reports also suggest that during ischemia or acidosis, cytosolic AC10 translocates to the mitochondria and activates a local pool of PKA that phosphorylates and enhances the activity of the proapoptotic BAX protein (Kumar et al., 2009; Appukuttan et al., 2012).
9. Mitochondrial Phosphodiesterases
Mitochondria are also provided with their own supply of PDEs. The first report documenting localization of PDEs at this organelle dates back to 1982, when Cercek and Houslay, using cell fractionation experiments and PDE activity assays, found two distinct PDEs associate with the MOM and the MIM, respectively (Cercek and Houslay, 1982). More recent studies have confirmed that both PDE2A2 and PDE8 localize to mitochondria (Acin-Perez et al., 2011; Shimizu-Albergine et al., 2012). Although little is known on the specific role of PDE8 at the mitochondria, several recent studies have focused on mitochondrial PDE2A2. A study reports the presence of PDE2A2 in the matrix of mitochondria isolated from liver and brain lysates (Acin-Perez et al., 2011), in keeping with the idea of a cAMP pool confined to the mitochondrial matrix. These investigations showed that targeting of PDE2A2 to the matrix is mediated by its N terminus and that selective inhibition of PDE2A enhances oxidative phosphorylation and ATP synthesis (Acin-Perez et al., 2011). In contrast with these findings, another study based on protein fractionation, electron microscopy, and super-resolution imaging showed localization of PDE2A2 to the MOM and MIS rather than the mitochondrial matrix (Monterisi et al., 2017). No detectable PDE2A activity in the mitochondrial matrix was found using targeted FRET reporters, in agreement with other investigations in which nonspecific PDE inhibition had no effect on cAMP levels in the matrix (Di Benedetto et al., 2013; Lefkimmiatis et al., 2013). PDE2A2 at the MOM was shown to regulate local cAMP levels and PKA-dependent phosphorylation of Drp1, affecting mitochondria morphology and apoptotic cell death, effects that were recapitulated in AC10−/− cells, confirming a role of PDE2A2 at the mitochondria outside the matrix (Monterisi et al., 2017). A PDE2A2 interactome analysis also revealed interaction of this PDE isoform with the MINOS components MIC60 and ChChd3, consistent with its localization in the MIS. At this site, PDE2A2 was shown to regulate local levels of cAMP and control Parkin recruitment to the mitochondria and mitophagy (Lobo, 2020).
D. Golgi Apparatus
Several components of the cAMP signaling pathway have been found to localize at the Golgi, and several studies support compartmentalized cAMP/PKA signaling at this organelle (Godbole et al., 2017; Nash et al., 2018). For example, Yang et al. showed that in cardiac myocytes, where the Golgi encircles or is adjacent to the nucleus, activation of β1-AR stimulates Ca2+ release from this organelle. Release of Ca2+ from the Golgi seems to require activation of both EPAC and PKA and results in enhanced trafficking of proteins to the cell surface. The perinuclear release of Ca2+ appears to be distinct from the release of Ca2+ from the SR, an event that is also under the control of β1-AR/cAMP and that regulates myocyte contraction. The amount of agonist required to induce release of Ca2+ from the Golgi is larger than the amount that promotes Ca2+ release from the SR. However, such difference is obliterated on inhibition of PDE3 or PDE4 (Yang et al., 2015), two enzymes that have also been shown to be associated with the Golgi fraction (Pooley et al., 1997; Geoffroy et al., 2001), but not by inhibition of PDE2 (Yang et al., 2015), which instead affects the amplitude of the Ca2+ transient in response to catecholamines (Mongillo et al., 2006). These findings are consistent with compartmentalization of cAMP signaling in the microenvironment surrounding the Golgi apparatus attained by local degradation of cAMP by PDE3 and PDE4 (Yang et al., 2015). Yang et al. did not investigate whether an AKAP-dependent signalosome is involved in the regulation of Ca2+ release from the Golgi. Interestingly, AKAP9 [AKAP350/AKAP450/centrosome and golgi localized protein kinase N-associated protein (CG-NAP)/Yotiao] has been found to localize to the Golgi apparatus in cerebellar granule cells (Shanks et al., 2002), where it brings PKA-RIIβ in close proximity to the IP3 receptors (Collado-Hilly and Coquil, 2009). Several studies, predominantly in cell lines, have shown that Ca2+ efflux from the Golgi is mediated by IP3 receptors (Pinton et al., 1998), and PKA is known to phosphorylate and activate IP3 receptors and enhance Ca2+ release from the ER (Betzenhauser and Yule, 2010). However, in cardiac myocytes, Ca2+ release from the Golgi appears to be mediated by ryanodine receptors rather than IP3 receptors (Yang et al., 2015).
E. Centrosome
The centrosome is the major microtubule-nucleating organelle in animal cells and is usually composed of a pair of centrioles surrounded by a protein matrix (Doxsey et al., 1994). In mitosis, the centrosome ensures fidelity of chromosome segregation through the formation of a bipolar spindle, whereas in differentiating cells, the centrosome acts as a platform for the formation of the primary cilium (Joukov and De Nicolo, 2019). Centrosomes contain all the components of the cAMP signaling pathway, including PKA (Nigg et al., 1985), at least two AKAPs (Schmidt et al., 1999; Takahashi et al., 1999; Witczak et al., 1999; Diviani et al., 2000), PDEs, and targets of PKA regulation, such as dynein, MAP2, and centrin, which are necessary for spindle formation, chromosome movement, and cytokinesis (Diviani and Scott, 2001).
1. A-Kinase Anchoring Protein 9
AKAP-450, one of the splice variants of AKAP9, is one of the anchoring proteins found at the centrosome and has been shown to be involved in microtubule nucleation (Schmidt et al., 1999; Takahashi et al., 1999; Witczak et al., 1999) and to affect cell migration but not cell polarization (Rivero et al., 2009). At the centrosome, AKAP9 organizes a multiprotein complex involving PKA type II, PDE4D3, PP1, and PP2A (Takahashi et al., 1999; McCahill et al., 2005; Taskén et al., 2001) (Fig. 3) and is required for centrosome integrity, centriole duplication, and cell cycle progression (Keryer et al., 2003). AKAP9 contains two distinct RII-binding domains, which could potentially recruit two PKA holoenzymes per scaffolding unit. Anchoring of PKA type II to AKAP9 was shown to lower the activation threshold of the enzyme as a consequence of increased RII autophosphorylation at S114. The AKAP9-anchored PDE4D3 provides local modulation of cAMP levels at the centrosome in a cell cycle–dependent manner (Terrin et al., 2012). Displacement of PDE4D3 from AKAP9 was shown to lead to accumulation of cells in prophase, indicating that cell cycle progression relies on unique regulation of centrosomal cAMP/PKA signals at the AKAP9 signalosome (Terrin et al., 2012).
2. Pericentrin
Another centrosomal protein that anchors PKA is pericentrin, although this interaction does not involve the canonical amphipathic helix found in other AKAPs (Diviani et al., 2000). Pericentrin is a highly conserved component of the centrosomal matrix and provides a structural scaffold for proper centrosomal architecture and microtubule nucleation during mitosis and meiosis (Dictenberg et al., 1998). In addition to PKA, pericentrin anchors γ-tubulin and dynein, the latter being a target for PKA-dependent phosphorylation (Inaba et al., 1998). Impairment of dynein function results in spindle fragmentation and disorganization (Purohit et al., 1999), defects that are also observed on disruption of AKAP/PKA interactions, and it has been speculated that PKA anchored at the centrosome through association with pericentrin might be involved in the regulation of dynein function (Diviani and Scott, 2001).
3. Centrosomal Exchange Protein Directly Activated by cAMP
Overexpression studies have shown dynamic localization of EPAC at the centrosome. Although in interphase EPAC is predominantly localized to mitochondria and the perinuclear region, in metaphase it dissociates from the nuclear membrane and localizes to the mitotic spindle and centrosomes. When cell division has completed, EPAC reassociates with the nuclear envelope and concentrates around the contractile ring. In breast cancer cells, inhibition of EPAC1 was shown to disrupt the association of AKAP9 with the microtubule cytoskeleton and to delocalize AKAP9 from the centrosome, with effects on cell migration and cell death (Kumar et al., 2017). Immunolocalization studies have identified AC10 at the spindle poles and centrioles, where it can potentially act as a source of cAMP to direct centriole separation and mitotic spindle formation (Zippin et al., 2003).
F. Endo/Sarcoplasmic Reticulum
1. A-Kinase Anchoring Protein 7
A well studied example of compartmentalized cAMP signaling occurs at the SR of cardiac myocytes. A critical event that occurs in the flight-or-fight response upon adrenergic stimulation is cAMP/PKA-dependent enhancement of the amplitude of the Ca2+ transient during systole. One mechanism that contributes to this is more efficient Ca2+ reuptake in the SR during diastole via PKA-dependent modulation of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA2) activity. The pump is part of a signalosome organized by AKAP18δ that also includes PKA type II, PDE3A1, PDE4D, and the SERCA2-associated protein PLN (Fig. 3) (Kerfant et al., 2007; Lygren et al., 2007; Ahmad et al., 2015). In unstimulated myocytes, SERCA2 is inhibited by unphosphorylated PLN. Upon activation of β-ARs, PKA phosphorylates PLN and removes its inhibitory effect on SERCA2, promoting Ca2+ refilling of the SR and relaxation (Henn et al., 2004; Rigatti et al., 2015; Götz et al., 2016). Disruption of the PKA-AKAP18δ interaction using competing peptides or via AKAP18δ knockdown was shown to blunt phosphorylation of PLN and to abolish the effect of catecholamines on SR Ca2+ reuptake (Lygren et al., 2007), demonstrating that clustering of PKA next to PLN via anchoring to AKAP18δ is necessary for PLN phosphorylation and that other PKA subsets, either free in the cytosol or anchored to other AKAPs, cannot carry out this function. The cAMP pool that controls SERCA2 activity is modulated by PDE3A1 and PDE4D. Both enzymes have been shown to interact with SERCA2 and to modulate basal cAMP levels in SR microdomains containing SERCA2A-PLN, thus regulating basal cardiac contractility (Schwoerer et al., 2008; Beca et al., 2011, 2013). The hydrolytic activity of both PDE3A1 and PDE4D can be enhanced by PKA-dependent phosphorylation (Grant et al., 1988; Fertig and Baillie, 2018), providing a local negative-feedback loop. Furthermore, phosphorylation of PDE3A1 at a PKA site in its unique N-terminal region promotes incorporation of this PDE into the SERCA2/AKAP18δ complex by modulating phosphorylation-dependent protein-protein interactions between PDE3A1 and SERCA2 and between PDE3A1 and AKAP18δ (Ahmad et al., 2015).
PP1β is the most significant PP1 isoform involved in regulating SR Ca2+ levels (Aoyama et al., 2011). Phosphatase inhibitor-1 (I-1) regulates phosphatase activity and has also been described to be part of the signaling complex organized by AKAP18δ at the SR. PKA-mediated phosphorylation of I-1 induces potent inhibition of PP1, providing an additional level of local regulation of SERCA2 activity (Livigni et al., 2006; Nicolaou et al., 2009). As mentioned above, AKAP18δ is the longest isoform of the AKAP18 family (Henn et al., 2004), which consists of four splice variants with various involvement in intracellular Ca2+ regulation. A knockout mouse model for AKAP18 did not show altered regulation of Ca2+ handling in cardiomyocytes (Jones et al., 2012), questioning the unique involvement of AKAP18 in these processes. Potential confounding effects deriving from expression in the knock out model of a truncated AKAP18 protein lacking the PKA-binding domain have been suggested as a possible explanation for these contrasting findings (Smith et al., 2018).
2. A-Kinase Anchoring Protein 13
A signalosome organized by AKAP13 [also known as AKAP–lymphocyte blast crisis (Lbc)] has recently been described to operate at ER exit sites, the specialized region at the ER membrane where export of newly synthesized proteins is facilitated via coat protein complex II–coated vesicles. At these sites, AKAP13 is involved in a complex signaling cascade that includes Gα12, AC7, PDE3B, PKA, and other kinases. cAMP generated locally by AC7 activates the AKAP13 anchored PKA pool, and the ensuing substrate phosphorylation contributes to activate cargo export (Subramanian et al., 2019). The role of this signalosome is thought to be to ensure quick response to fluctuations in protein synthesis that might occur under different conditions and that, if not handled properly, may lead to aberrant cargo processing and sorting and potentially to disease.
G. Nucleus
Multiple components of the cAMP signaling cascade have been shown to localize to the nucleus and/or nuclear membrane, including GPCRs, G proteins, ACs, AKAPs, and PKA (Vaniotis et al., 2011).
1. A-Kinase Anchoring Protein 6
AKAP6 (or mAKAP) is expressed in excitable cells as two splice variants: mAKAPα is found in neurons, and mAKAPβ is found in striated muscle cells. Earlier studies described a macromolecular complex involving mAKAP and localized at the SR (Pare et al., 2005); however, subsequent studies have firmly established a localization of mAKAPβ at the outer nuclear membrane via interaction with the protein Nesprin-1α (Marx et al., 1998; Yang et al., 1998; Kapiloff et al., 1999). This signalosome is complex, and multiple elements have been found in different studies to associate with mAKAPβ. Whether all components are simultaneously present within this signalosome remains to be determined. In addition to PKA, there is evidence of direct interaction of mAKAPβ with RyR2 (Marx et al., 2001), PP2A (Dodge-Kafka et al., 2010) and CaN (PP2B) (Li et al., 2010), AC5 (Kapiloff et al., 2009), PLCε (Zhang et al., 2011), the nuclear HDAC5 (Dodge-Kafka et al., 2018), myocyte enhancer factor-2 (MEF2) (Vargas et al., 2012), 3-phosphoinositide-dependent kinase-1 (Michel et al., 2005), the p90 ribosomal S6 kinase 3, (Passariello et al., 2015), and PDE4D3 (Dodge et al., 2001) (Fig. 3). It should be noted that although some groups have claimed that PDE4D3 interacts natively with mAKAP, in these studies, the identification of PDE4D3 relied solely on comigration of recombinant PDE4D3 with an immunoreactive species in cardiac cell extracts, as detected using “pan-PDE4” antisera able to detect all PDE4 isoforms. However, such studies were unable to take into account the subsequent identification, cloning, and characterization of several other PDE4D isoforms that were found to have a similar molecular weight to PDE4D3 and, indeed, were found to comigrate with PDE4D3 on SDS-PAGE—namely, PDE4D8, PDE4D9, and PDE4D11 (for a review, see Bolger, 2006). Thus, the PDE4D species in cardiac tissue that interacts with AKAPs could be any of PDE4D3, PDE4D8, PDE4D9, and PDE4D11. Furthermore, given the rarity of PDE4D3 transcripts, it is highly unlikely that the PDE4D interacting in heart tissues with mAKAP is PDE4D3. The concomitant assembly by mAKAP of cAMP-sensing and terminating enzymes is the basis for a negative-feedback loop regulation demonstrated in cardiac myocytes. PKA-mediated phosphorylation of Ser13 in PDE4D3 enhances its binding to mAKAP (Carlisle Michel et al., 2004), whereas phosphorylation of Ser54 enhances the cAMP hydrolytic activity of the catalytic domain of PDE4D3 (Sette and Conti, 1996) (but see comments above). Interestingly, mAKAP is expressed at very low levels in myocytes, such that loss of mAKAP does not even affect the localization of perinuclear PKA (Passariello et al., 2015). However, replacement of endogenous mAKAP with a mAKAP mutant that cannot bind PKA is sufficient to inhibit the induction of myocyte hypertrophy (Pare et al., 2005). AC5 activity is inhibited by PKA feedback phosphorylation when the cyclase is associated with the mAKAP complex. Disruption of mAKAP-AC5 interaction in neonatal cardiac myocytes results in increased cAMP and hypertrophy in the absence of agonist stimulation (Kapiloff et al., 2009). Lastly, through Rap1, EPAC1 can inhibit extracellular signal-regulated kinase 5 (ERK5) activity, thus preventing PDE4D3 inhibition by MAPK signaling, resulting in higher PDE4D3 activity due to concomitant PKA phosphorylation. As a result, EPAC1 provides a third negative-feedback loop that can attenuate cAMP levels in the proximity of mAKAP complexes (Dodge-Kafka et al., 2005; Passariello et al., 2015). Interestingly, mAKAP proteins containing nonsynonymous polymorphisms display different binding affinities for PKA and PDE4D3 (Rababa’h et al., 2013), suggesting potential individual differences on how cAMP signaling is handled at mAKAP by PDE4D3. Binding of PP2A to the C terminus of mAKAP provides an additional layer of regulation. PP2A dephosphorylates PDE4D3 at Ser54, inhibiting the PDE in the absence of upstream stimuli (Dodge-Kafka et al., 2010; Passariello et al., 2015) (Fig. 3).
As perhaps expected, given its perinuclear localization, the signalosome organized by mAKAPβ is involved in the modulation of transcriptional regulation (Dodge-Kafka et al., 2019). In cardiac myocytes, the mAKAPβ signalosome has been proposed to nucleate the effect of distinct pools of cAMP, which either activate or block transcription of pro-hypertrophic genes (Zhang et al., 2013; Zhang and Eggert, 2013; Kritzer et al., 2014). mAKAPβ-bound PKA can phosphorylate a small pool of perinuclear RyRs promoting Ca2+ released from perinuclear stores and local activation of CaN, which in turn induces nuclear factor of activated T-cells (NFAT) and MEF2-dependent gene expression associated with hypertrophy (Li et al., 2010). A distinct pro-hypertrophic pathway involves EPAC-PLCε–dependent PI4P hydrolysis at the Golgi, which results in diacylglycerol-dependent activation of mAKAPβ-bound PKCε and PKD, phosphorylation, and nuclear export of class IIa HDACs and induction of hypertrophy (Zhang et al., 2013). On the other hand, cAMP, via PKA activation, can mediate inhibition of PLCε-dependent PI4P hydrolysis, counteracting hypertrophy (Nash et al., 2018). mAKAPβ thus appears to act as a platform where the complex action of multiple pathways is locally coordinated to control the development of cardiac hypertrophy, although the full details of how cAMP/PKA/EPAC pathways operate in this context remains to be fully elucidated (Diviani et al., 2016).
2. A-Kinase Anchoring Protein 8
AKAP8 (or AKAP95) has been described to anchor PKA in the nuclear matrix in a cell cycle–dependent manner (Coghlan et al., 1994; Eide et al., 1998) (Fig. 3). Several different functions have been attributed to AKAP8, including DNA replication via interaction with minichromosome maintenance 2 protein, a function that appears to be independent of PKA (Eide et al., 2003), chromatin condensation through association with the RII subunit of PKA (Collas et al., 1999; Bomar et al., 2002), recruitment of HDACs to mitotic chromosomes (Li et al., 2006), and regulation of histone methylation and gene expression (Jiang et al., 2013; Hu et al., 2016). Additionally, AKAP8 has been shown to bind PDE4A (Asirvatham et al., 2004) and to be involved in the PKA-mediated phosphorylation of CREB (Lu et al., 2017). Tyrosine phosphorylation of AKAP8 promotes its dissociation from chromatin and the nuclear matrix, although the impact on local signaling remains unclear (Kubota et al., 2015).
Activation of the transcription factor CREB via PKA-mediated phosphorylation is one of the long-recognized functional effects of cAMP signals (Yamamoto et al., 1988). The conventional view is that upon cAMP binding to PKA holoenzyme, C subunits dissociate and translocate to the nucleus via diffusion (Harootunian et al., 1993). Once in the nucleus, PKA-mediated phosphorylation of CREB promotes its interaction with CREB binding protein, followed by enhanced transcription of target genes. Binding of protein kinase inhibitor to the C subunits induces exit of the complex from the nucleus (Dalton and Dewey, 2006), followed by dephosphorylation of CREB mainly by PP2A (Wadzinski et al., 1993). Recent studies, however, suggest an alternative mechanism. Sample et al. reported the activation of a resident pool of PKA holoenzyme in the nuclei of HEK-293 cells by AC10. These studies suggest a model whereby the nuclear AKAP8 assembles PDE4D3 or PDE4D5 (Clister et al., 2019) and PKA to form a signaling complex in the nucleus that controls local cAMP levels and nuclear PKA activity. The nuclear envelope is permeable to cAMP produced at the plasma membrane (Terrin et al., 2006). When cAMP generated by ACs at the plasma membrane reaches the nucleus, PDE4D3/5 keeps cAMP concentration below the activation threshold of the local PKA holoenzyme; when cAMP is produced locally by AC10 and above that threshold, activation of the local PKA occurs, resulting in faster activation kinetics compared with slow kinetics associated with translocation of C subunit to the nucleus (Sample et al., 2012). Other studies support the presence of AC10 in the nucleus (Dodge-Kafka et al., 2005; Passariello et al., 2015). However, Yang et al. failed to reproduce these observations in primary rat neonatal cardiac myocytes. The authors propose that the different kinetics of nuclear PKA are explained by diffusion and differences in the nuclear expression of protein kinase inhibitor rather than being dependent on a nuclear pool of PKA holoenzyme (Yang et al., 2014).
V. Novel Insight into Cell Physiology from Nanodomain cAMP Signaling
The recent methodological advances allow analysis of cAMP dynamics and PKA activity with unprecedented spatial and temporal resolution. The data that emerge from studies using targeted versions of the cAMP probes suggest that cAMP signaling is complex. Compartmentalization of cAMP is more extreme than initially thought, as evidence suggests that distinct handling of cAMP is confined to domains that can have submicrometer dimensions. Whatever the source of cAMP (whether an AC at the plasma membrane, an AC localized at internal membranes, or the soluble AC), its activation generates a gradient of second messenger that propagates away from the site of synthesis and that dissipates as distance increases. The size of this gradient is variable and depends on the AC involved and the amplitude and duration of the stimulus. In some cases, the gradient may dissipate rapidly, and a significant amount of cAMP is achieved only at sites proximal to the site of synthesis. If the trigger persists and the activity of the AC is sufficiently high to counteract cAMP degradation by PDEs, the level of second messenger can progressively increase distally from the site of synthesis, and over time, cAMP equilibrates across the entire cell. However, superimposed to this overall cytosolic gradient, a more variegated pattern of multiple and diverse cAMP pools develops in which the level of the second messenger in each nanocompartment is dictated by the local activity of PDEs and other contributing factors, including local buffering, high viscosity, and physical obstacles. The amount of cAMP within these multiple subcellular nanodomains can be either lower or higher than the level of the second messenger in the bulk cytosol, depending on which modulators prevail. It is the concentration of cAMP within each of these nanodomains, then, that dictates whether the local cAMP effectors are activated and whether a certain function is triggered. Bulk cytosolic cAMP may be functionally irrelevant (Bers et al., 2019), or its unique spatiotemporal dynamics may also encode information that serves particular roles.
What is clear is that cAMP signal nanoheterogeneity is essential for regulation of function, and as research uncovers new details, novel facets of cell physiology emerge. A recent investigation in retinal ganglion cells analyzed the role of cAMP in the regulation of growth cone response to ephrine-A, a cue that during development triggers refinement of retinal axons that initially overshoot in the most caudal part of the superior colliculus. cAMP plays an important role in neuronal development, as it mediates the response to a variety of axon guidance molecules, including Netrin-1, Semaphorin-3A, and ephrin-A5. The appropriate development of neuronal connectivity relies on axon guidance molecules that promote axonal growth and orient the axon toward appropriate synaptic partners. Initially, axonal branches develop exuberant projections that must be pruned to establish a correct projection map. Using a combination of real-time imaging of cAMP and molecular tools to either generate or buffer cAMP within restricted domains, the study found that it is specifically a decrease in cAMP occurring at the plasma membrane proximally to lipid rafts that is responsible for the ephrine-A–induced axon retraction in vitro and for pruning of retinal ganglion cells arbors in the superior colliculus in vivo. Manipulation of cAMP at nonraft plasma membrane domains, in contrast, showed no effect on pruning. Interestingly, selective manipulation of cAMP at lipid rafts had no effect on axon outgrowth and early development of the retinal pathway, both process that are also dependent on cAMP, confirming in vivo that distinct pools of cAMP generated at the plasma membrane exhibit clear functional specificity (Averaimo et al., 2016).
Recent studies using targeted reporter also demonstrate that not only does activation of different GPCRs result in different cAMP signals in distinct subcellular compartments, but activation of a single receptor can generate multiple cAMP signals with different amplitude and kinetics at different subcellular locations. In ventricular myocytes, cAMP mediates the response to sympathetic activation of β-AR. Strength of contraction and efficiency of relaxation are regulated via PKA-dependent phosphorylation of the L-type Ca2+ channel at the plasmalemma and PLN at the sarcoplasmic reticulum, which rapidly enhance the amplitude of the Ca2+ transients and contraction, and phosphorylation of troponin I at the myofilaments, which reduces the affinity of the troponin complex for Ca2+, promoting relaxation. A recent study using targeted cAMP FRET-based reporters demonstrated that β-AR stimulation results in distinct signals at these sites and that the cAMP increase is delayed and attenuated at the myofilaments compared with the plasmalemma and sarcoplasmic reticulum (Surdo et al., 2017). The compartmentalized nature of the cAMP signal generated on β-AR activation appears to be required to achieve maximal effect of sympathetic stimulation, as demonstrated by the fact that upon application of PDE inhibitors, a maneuver that generates overall a larger increase in cAMP but abolishes local differences, a weaker contraction was detected. Interestingly, in experimental models of cardiac disease, such as cardiac hypertrophy and heart failure, which are characterized by downregulation of the adrenergic pathway, the cAMP signal triggered by β-AR stimulation was nearly abolished at the myofilament but was well preserved at plasmalemma and sarcoplasmic reticulum. These findings have important implications for cardiac pathophysiology and potentially for the treatment of cardiac disease (Bers et al., 2019). Firstly, the data suggest that maximal stimulated contractility is achieved at the cost of an intrinsic predisposition of the myofilament to Ca2+ sensitization, which derives from attenuated local cAMP signaling. The propensity of the myofilament to experience lower levels of cAMP may become dangerous in conditions of reduced adrenergic drive (as experienced in pathologic conditions) when no effective cAMP signal may be generated at the troponin complex, resulting in persistent Ca2+ binding to the myofilament and diastolic dysfunction. Secondly, the findings may explain the well established long-term adverse effects of PDE inhibitors in the treatment of heart failure (Packer et al., 1991). The data show that PDE inhibition equalizes cAMP levels in the different compartments and reduces efficiency of cardiac myocyte contraction: in a failing heart, this results in further reduction of cardiac reserve and may contribute to pathogenesis. These unexpected findings also provide a new perspective on how to approach treatment of heart failure, as they suggest that therapeutic interventions aimed at normalizing Ca2+ sensitivity may prove beneficial, particularly in early stages of the pathologic process.
VI. Open Questions and Future Directions
Although the recent findings represent important progress in understanding novel aspects of cAMP signaling, significant gaps remain to be bridged, and important questions remain to be answered. For example, additional factors that contribute to compartmentalizing cAMP (e.g., buffering, high viscosity, or physical obstacles) have been postulated, but what exactly is their nature and how they impact different subcellular sites remain largely to be defined. How activation of a given GPCR orchestrates the activity of downstream modulators to determine what part of the signaling network is activated to achieve the required pattern of cAMP nanosignals remains puzzling. Answers to these questions will emerge as we learn more about the organization, regulation, and function of individual signaling compartments. Defining how many cAMP domains there are in a cell, what their size is, what their architecture and molecular composition are, and how they are affected in pathologic conditions is now feasible. High-resolution imaging approaches combined with biochemical studies (Schleicher and Zaccolo, 2020) applied to different cell types will allow us to work out the unique topography and function of cell type–specific cAMP nanodomains. As probes that can monitor cAMP signaling with single-cell resolution in moving animals are being developed (Ma et al., 2018), the physiologic relevance of subcellular signaling will be established at the organ and organism level. Integration of this information will certainly benefit from development of mathematical models that incorporate local handling of cAMP at the nanometer scale.
Defining the details of local cAMP signaling is important. Multiple drugs currently in clinical use target this signaling pathway, and understanding the subcellular ramifications of these pharmacological interventions would facilitate dissection of the mechanisms leading to side effects and help devise mitigation strategies. Although assessment of global cellular cAMP responses is still the prevalent approach used in screening programs for new drugs that target GPCRs, consideration of subcellular local effects at early stages would help reduce the high attrition rates. Finally, the compartmentalized nature of the cAMP signaling pathway offers the unique opportunity to develop molecules that target individual cAMP domains rather than global intracellular cAMP (Baillie et al., 2019), and a detailed understanding of this complex signaling systems and its nanodomain organization will provide a framework for the development of precision medicine strategies.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Zaccolo, Zerio, Lobo.
Footnotes
This study was supported by the British Heart Foundation (BHF) [Grant PG/10/75/28537], [Grant PG/15/5/31110], [Grant RG/12/3/29423], and [Grant RG/17/6/32944]; the BHF Centre of Research Excellence, Oxford [Grant RE/18/3/34214] to M.Z.; and the Medical Research Council [Grant MR/K501256/1] to M.J.L.
No author has an actual or perceived conflict of interest with the contents of this article.
Abbreviations
- AC
- adenylyl cyclase
- AKAP
- A-kinase anchoring protein
- AMPA
- amino-3-hydroxy-5-methylisoxazole-4-propionate
- AR
- adrenergic receptor
- ARHGAP36
- Rho GTPase activating protein 36
- BAD
- BCL2-associated agonist of cell death
- BAX
- Bcl-2-associated X protein
- BIM
- bcl-2-interacting mediator of cell death
- CAMKII
- calcium/calmodulin-dependent protein kinase II
- CaN
- calcineurin
- Cav1.2
- voltage-dependent, L type, alpha 1C subunit
- CFTR
- cystic fibrosis transmembrane conductance regulator
- ChChd
- coiled-coil-helix-coiled-coil-helix domain containing
- CREB
- cAMP response element binding protein
- Drp1
- dynamin-1–like protein
- ER
- endoplasmic reticulum
- EPAC
- exchange protein directly activated by cAMP
- FRET
- fluorescence resonance energy transfer
- GPCR
- G protein–coupled receptor
- GsPCR
- Gs protein-coupled receptor
- HDAC
- histone deacetylase
- IP3
- inositol 1,4,5-trisphosphate
- KCNQ1
- potassium voltage-gated channel subfamily Q member 1
- mAKAP
- muscle-specific A-kinase anchoring protein
- MEF2
- myocyte enhancer factor-2
- MINOS
- mitochondrial inner membrane organizing system
- MIS
- mitochondrial intermembrane space
- MOM
- mitochondrial outer membrane
- MTX
- metaxin
- NMDA
- N-methyl-D-aspartate
- NMDAR
- NMDA receptor
- PAP7
- peripheral-type benzodiazepine receptor–associated protein 7
- PC-2
- polycystin-2
- PDE
- phosphodiesterase
- PI4P
- phosphatidylinositol-4-phosphate
- PKA
- protein kinase A
- PKC
- protein kinase C
- PKD
- polycystic kidney disease
- PLCε
- phospholipase Cε
- PLN
- phospholamban
- PP1
- protein phosphatase 1
- PP2B
- protein phosphatase 2B
- PPP
- phosphoprotein phosphatase
- Praja2
- E3 ubiquitin-protein ligase
- RyR
- ryanodine receptor
- RXFP1
- relaxin family peptide receptor 1
- SERCA2
- sarco/endoplasmic reticulum Ca2+-ATPase 2
- SKIP
- sphingosine kinase interacting protein
- SR
- sarcoplasmic reticulum
- TGN
- trans Golgi network
- TRH
- thyrotropin-releasing hormone
- TSH
- thyroid stimulating hormone
- TSHR
- thyroid stimulating hormone receptor
- UPS
- ubiquitin proteasome system
- VIP
- vasoactive intestinal peptide
- WAVE
- Wiskott-Aldrich syndrome protein verprolin homologous
- Copyright © 2020 by The Author(s)
This is an open access article distributed under the CC BY Attribution 4.0 International license.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





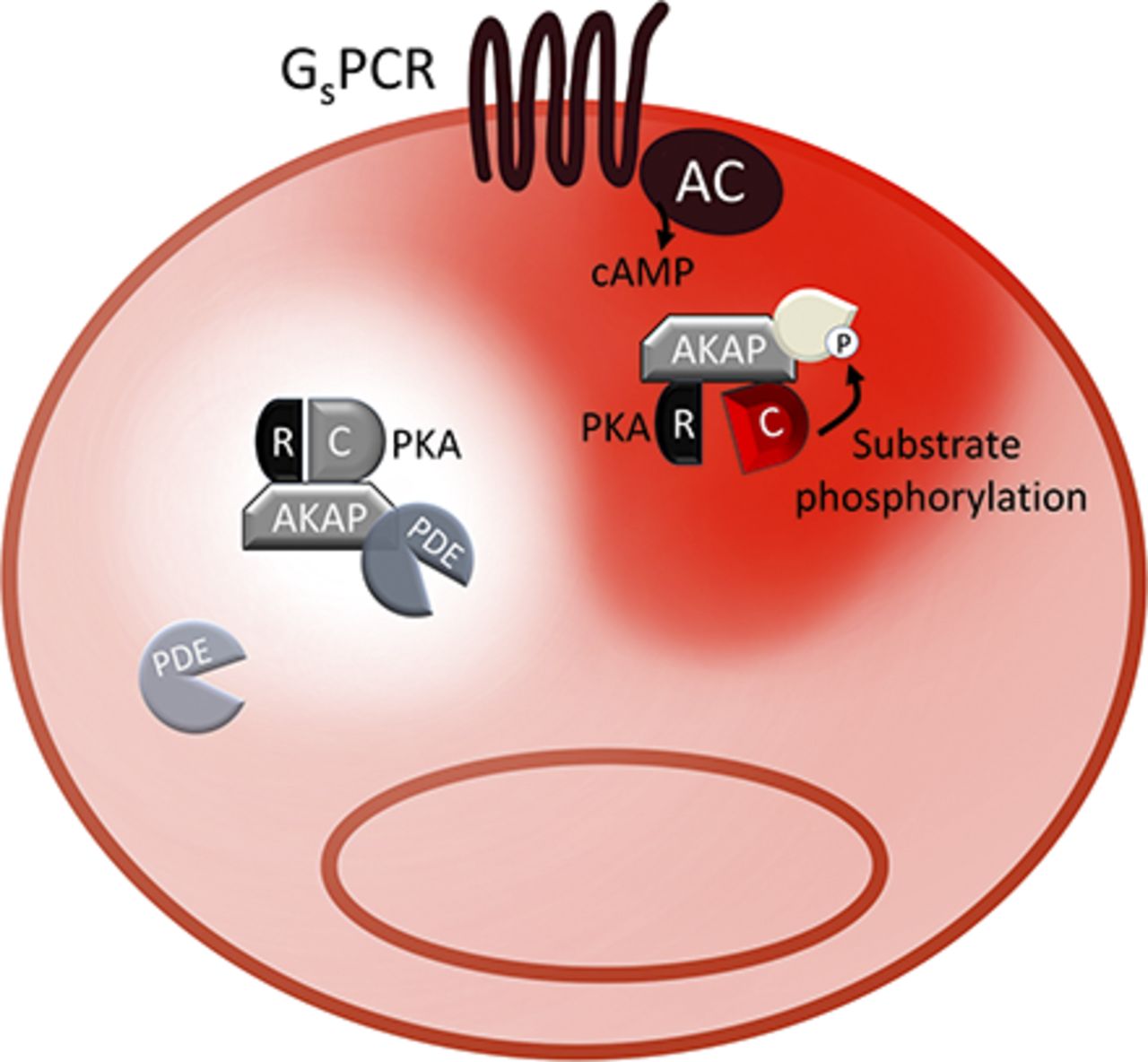{kind=link}
{kind=link}
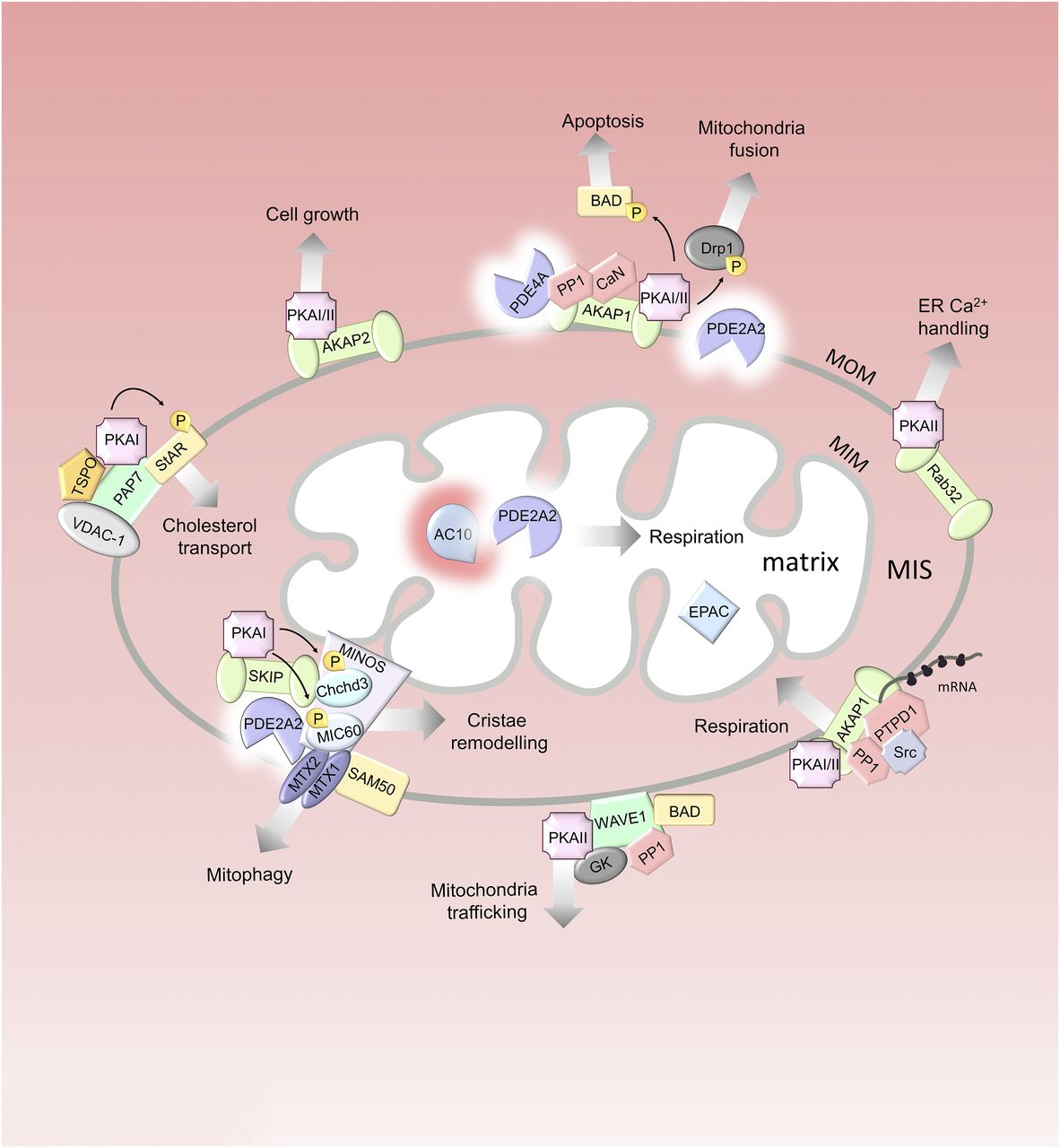{kind=link}
{kind=link}
Jump to section
- Article
- Visual Overview
- Abstract
- I. Introduction
- II. Imaging cAMP Nanodomains
- III. Contribution to Compartmentalization of Individual Pathway Components
- IV. cAMP Subcellular Nanodomains
- V. Novel Insight into Cell Physiology from Nanodomain cAMP Signaling
- VI. Open Questions and Future Directions
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters