


Visual Overview
Abstract
Antibiotic resistance is a major global health challenge and, worryingly, several key Gram negative pathogens can become resistant to most currently available antibiotics. Polymyxins have been revived as a last-line therapeutic option for the treatment of infections caused by multidrug-resistant Gram negative bacteria, in particular Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacterales. Polymyxins were first discovered in the late 1940s but were abandoned soon after their approval in the late 1950s as a result of toxicities (e.g., nephrotoxicity) and the availability of “safer” antibiotics approved at that time. Therefore, knowledge on polymyxins had been scarce until recently, when enormous efforts have been made by several research teams around the world to elucidate the chemical, microbiological, pharmacokinetic/pharmacodynamic, and toxicological properties of polymyxins. One of the major achievements is the development of the first scientifically based dosage regimens for colistin that are crucial to ensure its safe and effective use in patients. Although the guideline has not been developed for polymyxin B, a large clinical trial is currently being conducted to optimize its clinical use. Importantly, several novel, safer polymyxin-like lipopeptides are developed to overcome the nephrotoxicity, poor efficacy against pulmonary infections, and narrow therapeutic windows of the currently used polymyxin B and colistin. This review discusses the latest achievements on polymyxins and highlights the major challenges ahead in optimizing their clinical use and discovering new-generation polymyxins. To save lives from the deadly infections caused by Gram negative “superbugs,” every effort must be made to improve the clinical utility of the last-line polymyxins.
Significance Statement Antimicrobial resistance poses a significant threat to global health. The increasing prevalence of multidrug-resistant (MDR) bacterial infections has been highlighted by leading global health organizations and authorities. Polymyxins are a last-line defense against difficult-to-treat MDR Gram negative pathogens. Unfortunately, the pharmacological information on polymyxins was very limited until recently. This review provides a comprehensive overview on the major achievements and challenges in polymyxin pharmacology and clinical use and how the recent findings have been employed to improve clinical practice worldwide.
I. Introduction
The global mortality rate has rapidly declined over the last century after antibiotics were introduced into clinical practice (Armstrong et al., 1999). Unfortunately, resistance to the “magic bullet” antibiotics has become a major global health challenge since the 1990s. Furthermore, increasing development cost, regulatory stringency, scientific challenges, and financial hurdles have led to the disengagement of the pharmaceutical industry from antibiotic discovery and development (O’Neil, 2016). The Infectious Diseases Society of America, the Centers for Disease Control and Prevention, the World Health Organization, and many other major health authorities have highlighted the urgent unmet medical need due to the rapid emergence of resistance and diminishing antibiotic arsenal (Infectious Diseases Society of America, 2004; Centers for Disease Control and Prevention, 2013, 2019; World Health Organization, 2017). In the 2017 World Health Organization Priority Pathogen List, carbapenem-resistant Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacteriaceae are identified as the “Priority 1: Critical” pathogens, which desperately require new antibiotics (World Health Organization, 2017). Even though several new antibiotics have been approved by the US Food and Drug Administration (FDA) over the last few years, emergence of resistance has already been reported (Abdallah et al., 2015; de Man et al., 2018; Giddins et al., 2018; Karaiskos et al., 2019; Morrissey et al., 2020). Many clinicians worldwide have been forced to use polymyxins as the last-line therapy to treat life-threatening infections caused by the three aforementioned Gram negative “superbugs” because of their resistance to all currently available antibiotics (Wertheim et al., 2013; Andrei et al., 2019; Zhang et al., 2020). Polymyxins (i.e., polymyxin B and colistin) are an “old” class of lipopeptide antibiotics that were approved in the late 1950s (Li et al., 2006b). However, their use rapidly waned in the 1970s because of their potential nephrotoxicity and the availability of other antibiotics (e.g., aminoglycosides and fluoroquinolones). As neither polymyxin was ever evaluated with the contemporary drug development procedures, there was scarce pharmacological knowledge for polymyxins in the literature until recently. This review presents the latest achievements in the pharmacology of polymyxins, which have improved clinical practice globally. As an example, the first scientifically based dosing recommendations for intravenous colistin [as colistin methanesulfonate (CMS)] underpinned by modern pharmacokinetics/pharmacodynamics (PK/PD) research have been adopted by hospitals worldwide and contributed to the optimization of their use in patients (Nation et al., 2017; Tsuji et al., 2019). In addition, challenges in polymyxin pharmacology research and the recent discovery of new-generation polymyxin-like lipopeptides are also reviewed.
II. History and Chemistry of Polymyxins
A. History of Polymyxins
The naturally occurring lipopeptide polymyxins belong to a class of antibiotics isolated from the Gram positive spore-forming bacterium Paenibacillus polymyxa (also known as Bacillus polymyxa). In 1947, Benedict and Langlykke in the United States first reported the antibacterial activity of the crude mixture from P. polymyxa (Benedict and Langlykke, 1947). At the same time, Stansly, Shepherd, and White of the American Cyanamid Company described the isolation and purification of another antibiotic from the same bacterium, P. polymyxa, and named it as “polymyxin” (Stansly et al., 1947). In England, an antibiotic (aerosporin) from Bacillus aerosporus was isolated and identified by Brownlee and coworkers and was active against certain Gram negative bacteria (Ainsworth et al., 1947; Brownlee and Bushby, 1948). Further research disclosed that polymyxin and aerosporin were basic peptides and possessed very similar antimicrobial activities; therefore, they were classified as the same class of antibiotics (Shepherd et al., 1948; Brownlee et al., 1949a,b; Jones, 1949; White et al., 1949). As both antibiotics were isolated from P. polymyxa, this family of antibiotics was designated as polymyxin, and a nomenclature system was developed internationally (Brownlee, 1949; Stansly and Brownlee, 1949). Later on, the names polymyxin A and polymyxin D were determined for aerosporin and polymyxin, respectively, and other groups of polymyxins, such as polymyxin B, C, and E, were developed (Jones, 1948). Although another antibiotic, colistin, was isolated from Bacillus (Aerobacillus) colistinus in Japan and was initially described as a new class, eventually it was identified that both colistin and polymyxin E are the same compounds (Dautrevaux and Biserte, 1961; Wilkinson, 1963; Suzuki et al., 1965). Both colistin and polymyxin B showed very similar antimicrobial properties (Schwartz et al., 1959–1960; Wright and Welch, 1959–1960). In polymyxins (Table 1), the two major components of the polymyxin B, polymyxin B1 and B2, were first discovered in 1954 (Bell et al., 1949; Catch et al., 1949; Jones, 1949; Wilkinson, 1949; Hausmann and Craig, 1954). The chemical structures of different polymyxins, such as polymyxin B1, polymyxin B2, colistin A (polymyxin E1), colistin B (polymyxin E2), and polymyxin D1 and D2, were determined afterward (Hayashi et al., 1966) (Table 1). To date, polymyxin A to F, M, P, S, and T have been discovered from P. polymyxa strains (Wilkinson and Lowe, 1966; Parker et al., 1977; Shoji et al., 1977a,b; Niu et al., 2013) (Table 1).
Chemical structures of the naturally occurring polymyxins
Reprinted by permission from the Springer Nature: Springer, Adv Exp Med Biol, History, chemistry and antibacterial spectrum, Velkov T. et al. Copyright © 2019.
Soon after their discovery, reversible nephrotoxicity was reported for different polymyxins, and polymyxin B and colistin (polymyxin E) were the least nephrotoxic using in vivo models; however, all of them showed very similar antibacterial activity (Brownlee et al., 1952; Nord and Hoeprich, 1964; Storm et al., 1977). Consequently, both colistin and polymyxin B were further developed for clinical uses and evaluated for toxicity (Barnett et al., 1964; Nord and Hoeprich, 1964; Beveridge and Martin, 1967). For the first time, Stansly and Brownlee (1949) reported that the sulfomethyl derivatives of polymyxins are less toxic and irritant at the injection site compared with the parent antibiotic and possess similar in vivo antibacterial activity (Barnett et al., 1964; Beveridge and Martin, 1967). Since the 1950s, colistin has been commercially available for clinical use as a sulfomethylated derivative, CMS [8068-28-8 Chemical Abstracts Service (CAS) registry number], whereas colistin sulfate (1264-72-8) has been available for intravenous administration only in China. In the clinic, polymyxin B is available as polymyxin B sulfate (1405-20-5) for intravenous administration (Kwa et al., 2007). The commercial products of polymyxin B, CMS, and colistin available for therapeutic uses are in the form of mixtures of major components, such as polymyxin B1, polymyxin B2, colistin A, and colistin B (Kimura et al., 1981; Orwa et al., 2001b; Govaerts et al., 2002a,b). As a consequence, batch-to-batch variations in the abundance of individual components can be observed even in the same commercial products (Decolin et al., 1997; Orwa et al., 2001a,b; He et al., 2010). For example, according to the British Pharmacopeia, the proportions of colistin A and B have to be 50%–75% and 5%–20%, respectively, of the colistin product, and the proportion of polymyxin B1, B2, B3, and B1-I components should not be less than 80% of the total content of the polymyxin B product (British Pharmacopoeia Commission, 2018).
B. Chemistry of Polymyxin B and Colistin
To date, at least 10 individual polymyxin B lipopeptide components have been identified in the literature (Table 1) (Orwa et al., 2001b; Govaerts et al., 2002a; Shaheen et al., 2011). Polymyxin B1 to B6 possess branched and nonbranched N-terminal fatty acyl groups. Polymyxin B1-Ile is highly similar to polymyxin B1, with isoleucine (structural isomer of leucine) at position 7 instead of leucine. Polymyxin B1 and B2 are always the major lipopeptide components. Importantly, variations in proportions of the different lipopeptide components in commercially available polymyxin B products have been reported for different brands and batches of the same manufacturer (He et al., 2010).
For colistin, 11 individual components have been reported, and they have branched and nonbranched N-terminal fatty acyl groups (Table 1) (Orwa et al., 2001b; Govaerts et al., 2002b). Colistin A (polymyxin E1) and colistin B (polymyxin E2) are the two major components. In patients, colistin is administered as a sodium salt of an inactive prodrug, CMS, intravenously or by inhalation (Li et al., 2003a, 2004; Bergen et al., 2006; Garonzik et al., 2011; He et al., 2013). CMS is not stable and converts to its active form colistin in vitro and in vivo (Barnett et al., 1964; Beveridge and Martin, 1967). Because of the uncontrollable sulfomethylation of the Dab residues, the commercial products of CMS consist of a complex mixture of fully and partially sulfomethylated derivatives (He et al., 2013). Importantly, international regulatory compendiums recommend no specific limits on these sulfomethylated derivatives in CMS products (United States Pharmacopeial Convention, 2018, British Pharmacopoeia Commission, 2019, European Pharmacopoeia Commission, 2020).
Overall, the recent advancement of analytical tools for chemical identification and structure elucidation will expedite the development and discovery of new polymyxins with favorable pharmacological and pharmaceutical properties. New-generation polymyxins will strengthen our arsenals to combat antibiotic resistance due to problematic Gram negative pathogens.
III. Antibacterial Spectrum, Mechanisms of Activity, and Resistance
A. Antibacterial Spectrum
The two clinically available polymyxins, colistin and polymyxin B, demonstrate comparable spectra of antibacterial activity, mechanism of action, and resistance because of their similar structures (Kwa et al., 2007). Polymyxins are generally active against Gram negative bacilli and coccobacilli, including Acinetobacter spp. (Kuck, 1976; Catchpole et al., 1997; Gales et al., 2001, 2011, 2012; Tan and Ng, 2006b; Walkty et al., 2009; Queenan et al., 2012), P. aeruginosa (Catchpole et al., 1997; Gales et al., 2001, 2011, 2012; Schulin, 2002; Niks et al., 2004; Tan and Ng, 2006b; Cernohorska and Slavikova, 2009; Walkty et al., 2009; Bogiel et al., 2010; Jones et al., 2013; Zhanel et al., 2013), Haemophilus spp. (Kosakai and Oguri, 1976), Legionella spp. (Thornsberry et al., 1978), Bordetella pertussis (Li et al., 2005a), and Enterobacterales, such as Escherichia coli (Catchpole et al., 1997; Gales et al., 2006, 2011, 2012; Tan and Ng, 2006b; Walkty et al., 2009; Nakamura et al., 2014), Klebsiella spp. (Catchpole et al., 1997; Gales et al., 2006, 2011, 2012; Tan and Ng, 2006b; Walkty et al., 2009; Hawser, 2010; Sader et al., 2011; Nakamura et al., 2014), Salmonella spp. (Catchpole et al., 1997; Gales et al., 2006), Enterobacter spp. (Catchpole et al., 1997; Gales et al., 2006; Walkty et al., 2009), Citrobacter spp. (Catchpole et al., 1997; Gales et al., 2006), and Shigella spp. (Catchpole et al., 1997; Gales et al., 2006). Of these Gram negative bacteria, polymyxin resistance has been increasingly reported in Acinetobacter spp. (Ko et al., 2007; Falagas et al., 2008; Al-Sweih et al., 2011), P. aeruginosa (Landman et al., 2005; Falagas et al., 2008; Johansen et al., 2008), Klebsiella spp. (Antoniadou et al., 2007; Falagas et al., 2008; Suh et al., 2010; Mezzatesta et al., 2011), and E. coli (Liu et al., 2016b; Nang et al., 2019). Worryingly, A. baumannii (Li et al., 2006c; Lo-Ten-Foe et al., 2007; Tan et al., 2007; Hawley et al., 2008; Charretier et al., 2018; Srinivas et al., 2018), P. aeruginosa (Bergen et al., 2011a; Lin et al., 2019a), Klebsiella pneumoniae (Meletis et al., 2011; Jayol et al., 2015; Bardet et al., 2017; Cheong et al., 2019), and Enterobacter cloacae (Lo-Ten-Foe et al., 2007) have been shown to display heteroresistance to polymyxins, which is defined as the presence of a polymyxin-resistant subpopulation [minimum inhibitory concentration (MIC) > 2 mg/l] within a susceptible population (MIC ≤ 2 mg/l). Heteroresistance is of particular concern, as the minor resistant subpopulation would not be detected by the current susceptibility testing in clinical microbiology laboratories and can lead to suboptimal dosing in patients, which may select for the resistant population and result in treatment failure. Further investigations are warranted to examine the impact of heteroresistance on the efficacy of polymyxins in clinical settings, and it is crucial to optimize the current recommended dosage regimens to minimize the emergence of resistance.
Polymyxins have not been shown to be active against a number of Gram negative species, such as Providencia spp. (von Graevenitz and Nourbakhsh, 1972; Catchpole et al., 1997), Serratia spp. (Greenfield and Feingold, 1970; von Graevenitz and Nourbakhsh, 1972; Catchpole et al., 1997; Gales et al., 2006), Proteus spp. (von Graevenitz and Nourbakhsh, 1972; Gales et al., 2006), Vibrio spp. (Lesmana et al., 2002; Gales et al., 2006), Morganella morganii (Gales et al., 2006), Neisseria spp. (Doern and Morse, 1980; Gales et al., 2006), Brucella spp. (Gales et al., 2006), Helicobacter pylori (Glupczynski et al., 1988; García-Rodríguez et al., 1989; Gales et al., 2006), Edwardsiella tarda (Muyembe et al., 1973), Burkholderia cepacia (Gales et al., 2006, 2001), Pseudomonas pseudomallei (Dance et al., 1989), and Moraxella catarrhalis (Gales et al., 2006). Variable susceptibility has been reported against Stenotrophomonas maltophilia (Niks et al., 2004; Tan and Ng, 2006b) and Campylobacter spp. (Kiehlbauch et al., 1992; Aydin et al., 2001). Lastly, at clinically achievable concentrations, polymyxins are not active against Gram positive bacteria, anaerobes, fungi, or parasites (Schwartz et al., 1959–1960; Finland et al., 1976; Storm et al., 1977).
B. Mechanisms of Antibacterial Activity
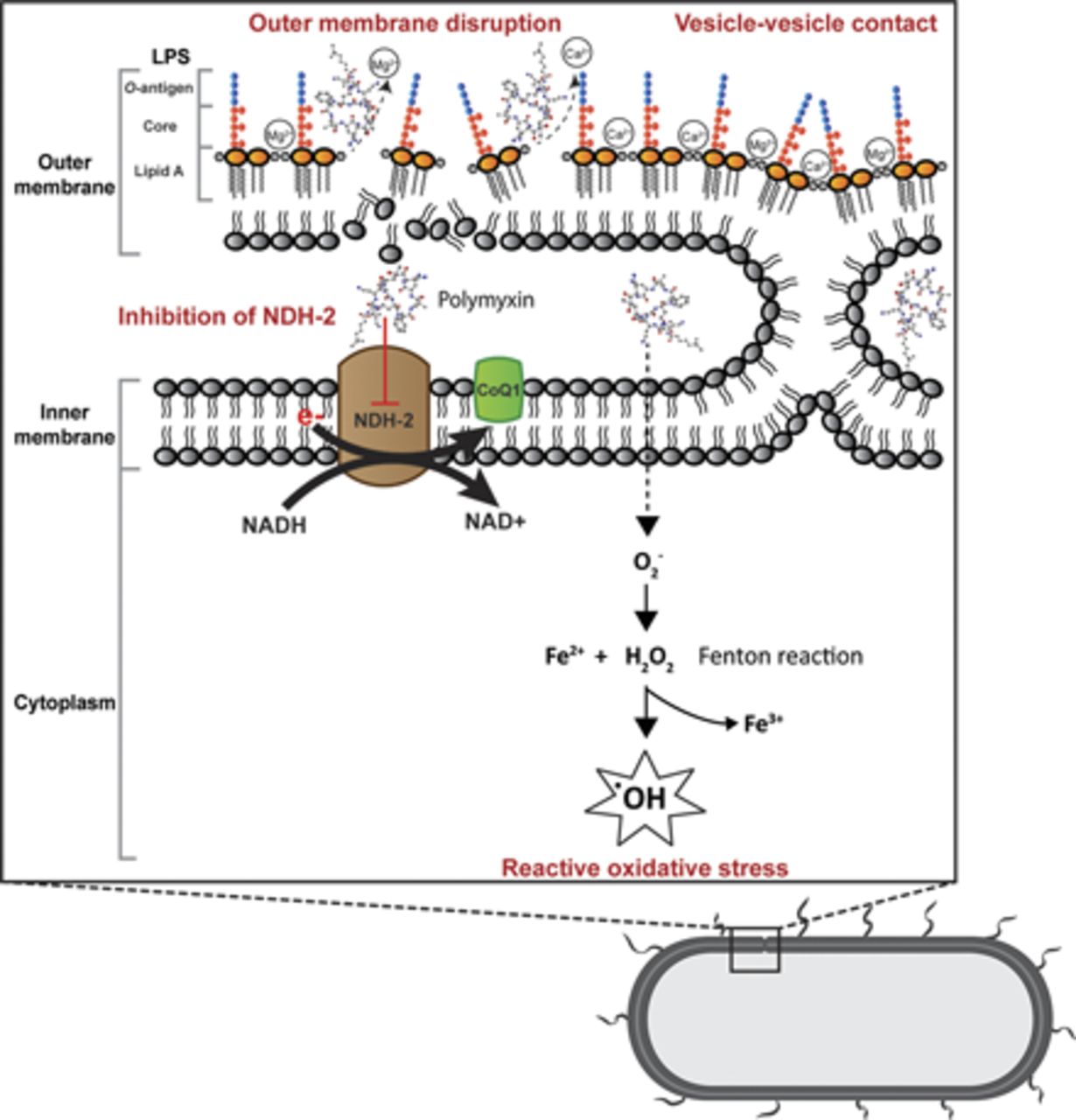
Gram negative bacteria consist of outer and inner membranes (Miura and Mizushima, 1968). The outer membrane is an important barrier against noxious components such as antimicrobial agents (Nikaido, 2003). It is an asymmetric bilayer with an inner leaflet of phospholipids and an outer leaflet that is composed of mainly lipopolysaccharide (LPS) (Zgurskaya et al., 2015). The LPS contains three key domains: O-antigen with an outer repeating polysaccharides unit, an inner core 2-keto-3-deoxyoctonoic acid (Kdo), and lipid A (β-1,6–linked d-glucosamine disaccharide that is phosphorylated at the 1 and 4′ positions with variable number of acyl chains) (Caroff and Karibian, 2003; Kim et al., 2016). The LPS molecules are bridged together by divalent cations (Mg2+ and Ca2+), which bind to the anionic lipid A phosphate groups via electrostatic interaction (Clifton et al., 2015). The primary target of polymyxins on Gram negative cells is the lipid A of the LPS in the outer membrane. Penetration of polymyxins across the outer membrane is highly dependent on the amphipathic property of polymyxins (Hancock, 1997; Clausell et al., 2007; Meredith et al., 2009). The initial binding of polymyxins to the outer membrane of Gram negative bacteria occurs via the initial electrostatic interaction between positively charged L-α,γ-Dab residues of polymyxins and the negatively charged lipid A phosphates. The aforementioned electrostatic interaction causes the displacement of divalent cations (Ca2+ and Mg2+) and destabilizes the outer leaflet of the outer membrane, thus allowing the hydrophobic N-terminal fatty acyl chain and position 6-7 motif (d‐Leu6‐l‐Leu7 for colistin or d‐Phe6‐l‐Leu7 for polymyxin B) to insert into the hydrophobic region of the outer membrane consisting of mainly fatty acyl chains of lipid A (Zhu et al., 2020). This hydrophobic interaction weakens the packing of adjacent fatty acyl chains and leads to expansion of the outer membrane. The insertion of polymyxins into the phospholipid bilayer disrupts the structural integrity of the membrane and promotes other polymyxin molecules to pass through the outer membrane (Velkov et al., 2010; Jiang et al., 2020b). It is postulated that this self-promoted uptake mechanism leads to an increased membrane permeability and leakage of cellular constituents, thus leading to cell death (Velkov et al., 2010; Yu et al., 2015) (Fig. 1). Both polar (e.g., Dab residues) and hydrophobic (N-terminal fatty acyl chain and position 6-7 motif) regions of polymyxins are essential for their interaction with LPS (Jiang et al., 2020b), and a structure-activity relationship model was developed based on the interaction with a single lipid A molecule (Velkov et al., 2010).

Mechanisms of antibacterial activity of polymyxins in Gram negative bacteria via disruption of the outer membrane, vesicle-vesicle contact, inhibition of respiratory enzyme NDH-2, and hydroxyl radical formation. CoQ1, coenzyme Q1.
Another proposed mode of action by polymyxins is via vesicle-vesicle contact, whereby polymyxins induce the contacts between outer and inner membranes and cause the exchange of phospholipids between both membranes (Fig. 1). This mechanism is believed to disrupt the phospholipid composition, which results in osmotic imbalance and cell lysis (Cajal et al., 1995, 1996; Clausell et al., 2007; Yu et al., 2015). The disruption of bacterial membrane is supported by transcriptomic and metabolomic analyses, which show the genes and metabolites associated with cell envelope biogenesis and membrane lipids, mainly fatty acids and glycerophospholipids, are significantly perturbated after polymyxin treatment (Henry et al., 2015; Maifiah et al., 2017; Han et al., 2019; Hussein et al., 2019; Lin et al., 2019c).
Accumulation of polymyxins has been observed in the inner membrane and cytoplasm in K. pneumoniae (Deris et al., 2014b). A secondary mechanism of polymyxin antibacterial activity involves the inhibition of NADH oxidases in bacterial respiratory chain in Bacillus subtilis and Mycobacterium smegmatis (Tochikubo et al., 1986; Deris et al., 2014a; Mogi et al., 2009). Notably, polymyxins have been demonstrated to inhibit the inner membrane type II NADH-quinone oxidoreductase (NDH-2) in E. coli, K. pneumoniae, and A. baumannii; and a slightly greater inhibition of NDH-2 by polymyxin B than colistin was reported (Deris et al., 2014a) (Fig. 1). The NDH-2 inhibitory effect was not observed with polymyxin B nonapeptide, colistin nonapeptide, or CMS; therefore, both hydrophobic N-terminal fatty acyl chain and positively charged moieties of polymyxins are important for the NDH-2 inhibitory activity of polymyxins (Deris et al., 2014a). Another secondary mode of action is the induction of endogenous production of reactive oxygen species (ROS) by polymyxins, thus leading to oxidative killing of the bacterial cell via hydroxyl radicals (Sampson et al., 2012; Yu et al., 2015) (Fig. 1). The killing of A. baumannii has been demonstrated via hydroxyl radical production, and inhibiting the formation of hydroxyl radicals attenuates polymyxin killing (Sampson et al., 2012). It is proposed that bactericidal antibiotics usually share a common killing activity via the disruption of metabolism, thus causing the formation of ROS, which eventually leads to bacterial cell death (Kohanski et al., 2007). However, bactericidal activity of polymyxins against P. aeruginosa has been reported to be an independent event of ROS production (Brochmann et al., 2014). The roles of ROS in bacterial killing by polymyxins are therefore inconclusive, and further studies are warranted.
In summary, increasing evidence has been reported on the potential secondary antibacterial mechanisms of polymyxins, including their effects on bacterial metabolism (Han et al., 2019). Elucidation of the exact antibacterial mechanism is crucial for the development of new-generation polymyxins with much-improved killing activity and higher therapeutic indices.
C. Mechanisms of Resistance
The outer membrane is the initial target of polymyxins, and most resistance mechanisms involve the alteration of the outer membrane to become less permeable to polymyxins. Although the resistance mechanisms vary in different bacterial species, the most common mechanism is LPS modification with the addition of positively charge moieties, such as phosphoethanolamine (pEtN) and 4-amino-l-arabinose (L-Ara4N) (Olaitan et al., 2014; Baron et al., 2016) (Fig. 2). These modifications result in reduced negative charges and impact the initial electrostatic interaction with the positively charged polymyxins (Jiang et al., 2020a).

Mechanisms of polymyxin resistance in Gram negative bacteria via lipid A modifications with L-Ara4N, pEtN, galactosamine, and palmitoylation; efflux pump systems, capsule shielding, loss of LPS, and polymyxin dependence.
Modification of the lipid A or Kdo with L-Ara4N has been observed in Gram negative Salmonella enterica (Vaara et al., 1981; Helander et al., 1994; Trent et al., 2001), E. coli (Nummila et al., 1995; Trent et al., 2001), K. pneumoniae (Kidd et al., 2017), P. aeruginosa (Moskowitz et al., 2004), Serratia marcescens (Lin et al., 2014), and B. cepacia (Shimomura et al., 2003). The biosynthesis and addition of L-Ara4N involve a number of enzymes: Ugd (PmrE), ArnB (PmrH), ArnC (PmrF), ArnA (PmrI), ArnD (PmrJ), ArnT (PmrK), ArnE (PmrL), and ArnF (PmrM) (Yan et al., 2007). The initial step for the synthesis of L-Ara4N takes place in the cytoplasm, where UDP-glucose is converted to UDP-glucuronic acid by UgpD, followed by oxidative decarboxylation by ArnA to UDP-4-keto-pyranose (Breazeale et al., 2002; Williams et al., 2005). The UDP-4-keto-pyranose is then converted by the transaminase ArnB to UDP-β-L-Ara4N, which subsequently undergoes formylation to form UDP-β-L-Ara4FN by ArnA (Breazeale et al., 2003; Williams et al., 2005). ArnC then transfers UDP-β-L-Ara4FN to undecaprenyl phosphate carrier located in the inner membrane, where it undergoes deformylation to undecaprenyl phosphate-α-L-Ara4N by ArnD (Breazeale et al., 2005). ArnE and ArnF function to flip the undecaprenyl phosphate-α-L-Ara4N to the outer surface of the inner membrane, where the L-Ara4N is finally transferred to the lipid A portion by the glycosyltransferase ArnT (Trent et al., 2001; Yan et al., 2007). The L-Ara4N moieties can be added to 1- and/or 4′-phosphate group; however, preference to 4′-phosphate has been demonstrated in S. enterica and E. coli (Zhou et al., 2001). The presence of 3′-acyloxyacyl–linked myristate group on the lipid A (conferred by LpxM) is crucial for the addition of L-Ara4N in S. enterica and E. coli (Tran et al., 2005). In contrast, lpxM deletion was reported to have no significant effect on lipid A modification in K. pneumoniae; however, increased susceptibility to polymyxins was observed, and it was postulated that the decreased abundance of acyl chains enhanced the penetration of polymyxins into the membrane (Clements et al., 2007). The same study also reported the occurrence of increased palmitoylation to compensate the loss of myristate (Clements et al., 2007). The outer membrane PagP is related to palmitoylation of lipid A, and its transcription can be stimulated in response to PhoPQ activation (Bishop et al., 2000). Palmitoylation of lipid A is proposed to increase the hydrophobicity of the outer membrane, which prevents the insertion of polymyxins (Fig. 2). The L-Ara4N modification has not been reported in A. baumannii; however, lipid A has been shown to be decorated with structurally similar galactosamine mediated by NaxD (Chin et al., 2015) (Fig. 2). Notably, homologs of UgpD and ArnBCADTEF are absent in A. baumannii.
The modifications of LPS with pEtN have been identified on several locations of LPS, which is dependent on the type of transferase. Addition of pEtN to lipid A has been observed in S. enterica (Zhou et al., 2001; Lee et al., 2004), E. coli (Kim et al., 2006; Liu et al., 2016b), K. pneumoniae (Jayol et al., 2014), and A. baumannii (Arroyo et al., 2011), mediated by the chromosomally encoded eptA (pmrC) and plasmid-mediated mcr (Liu et al., 2016b; AbuOun et al., 2017; Borowiak et al., 2017; Carattoli et al., 2017; Eichhorn et al., 2018; Kieffer et al., 2019; Wang et al., 2018, 2020; Yang et al., 2018); to Kdo in S. enterica (Gibbons et al., 2008) and E. coli (Reynolds et al., 2005) by chromosomally encoded eptB; and to heptose I in E. coli (Salazar et al., 2017) by chromosomally encoded eptC. Similar to the addition of L-Ara4N to lipid A, both 1- and 4′-phosphate groups can be decorated with pEtN moieties; however, in S. enterica, L-Ara4N is preferentially added to the 4′-phosphate and pEtN to the 1-phosphate of lipid A (Zhou et al., 2001). Interestingly, lack of L-Ara4N–modified lipid A has been demonstrated to result in greater decrease in resistance to polymyxins than the loss of pEtN modification in S. enterica, and vice versa in E. coli; this shows that different bacterial species could have different preference for the type of lipid A modifications (Lee et al., 2004; Herrera et al., 2010). Interestingly, modification of lipid A of Vibrio cholerae with glycine and diglycine, mediated by AlmEFG, has been demonstrated to be important for polymyxin resistance (Hankins et al., 2012).
Expression of the chromosomally encoded genes involved in lipid A modifications is commonly regulated by two-component regulatory systems (TCSs), and these can vary in different bacterial species (Fig. 3). In S. enterica, E. coli, and K. pneumoniae, exposure to polymyxins, low pH, and low Mg2+ induce PhoQ and lead to phosphorylation of PhoP and subsequently increase pmrD expression (Gunn et al., 2000; Cheng et al., 2010; Rubin et al., 2015). Inhibition of PmrA dephosphorylation by PmrD results in enhanced PmrA activity, thus leading to continuous transcription of the arn operon and eptA (Gunn et al., 2000; Kox et al., 2000; Cheng et al., 2010; Rubin et al., 2015; Jeannot et al., 2017). Unlike the indirect interaction between PhoPQ and PmrAB via PmrD, the phosphorylated PhoP activates PmrAB directly in P. aeruginosa, leading to the upregulation of arnBCADTEF-ugpD (Macfarlane et al., 1999; Jeannot et al., 2017). Additionally, in K. pneumoniae, the arn operon can be directly activated by PhoPQ (Cheng et al., 2010; Jeannot et al., 2017). Another molecular determinant that plays a key role in polymyxin resistance in K. pneumoniae is MgrB, which is a negative regulator of PhoPQ (Cannatelli et al., 2014). Functional inactivation of MgrB leads to PhoPQ upregulation and subsequently increases the transcription of the downstream genes involved in polymyxin resistance (Poirel et al., 2015). In addition to PhoPQ and PmrAB, CrrAB has been recently identified exclusively in K. pneumoniae as another TCS that participates in polymyxin resistance (Wright et al., 2015; McConville et al., 2020). Mutations in crrB lead to the upregulation of crrC, causing increased expression of arn operon and eptA via the PmrAB system (Cheng et al., 2016). The regulatory systems are far more complex in P. aeruginosa because of the presence of several other TCSs—namely, ParRS, CprRS, and ColRS. ParRS and CprRS induce the arn operon when exposed to antimicrobial peptides (Fernández et al., 2010, 2012). ColRS upregulates eptA but downregulates arnT in the presence of Zn2+; however, Zn2+-induced pEtN modification does not affect polymyxin resistance (Gutu et al., 2013; Nowicki et al., 2015). Thus far, only one TCS (PmrAB) has been identified in A. baumannii to control polymyxin resistance. The activation of PmrAB induces the expression of eptA and naxD, leading to lipid A modification with pEtN and galactosamine, respectively (Chin et al., 2015). The activation of PmrA also results in pmrR expression, which is responsible for the inhibition of lipid A phosphorylase LpxT and reduces the addition of phosphate group to lipid A. This process ultimately promotes resistance to cationic antimicrobial peptides such as polymyxins (Herrera et al., 2010). Additionally, in A. baumannii, polymyxin resistance has been shown to be mediated by increased expression of a homolog of pmrC via the inactivation of a global transcriptional regulator, histone-like nucleoid-structuring protein (Deveson Lucas et al., 2018).

Two-component systems regulating polymyxin resistance in E. coli, S. enterica, K. pneumoniae, A. baumannii, and P. aeruginosa.
A very interesting mechanism of polymyxin resistance is the loss of LPS in A. baumannii (Moffatt et al., 2010) (Fig. 2). The loss of LPS is a result of mutations in the lipid A biosynthesis genes lpxA, lpxC, and lpxD (Moffatt et al., 2010, 2011). LPS-deficient A. baumannii isolates lack the initial polymyxin binding target and demonstrate a high level of polymyxin resistance (MIC > 256 mg/l) (Moffatt et al., 2010). Fortunately, LPS-deficient outer membrane appears to be highly permeable and thus increases bacterial susceptibility to other antimicrobial agents, such as rifampicin, cefepime, teicoplanin, and azithromycin (Li et al., 2007; Moffatt et al., 2010). The ability of A. baumannii to survive in the absence of LPS is intriguing, and it has been suggested that this is attributed to the alteration in the production of lipoproteins, phospholipids, and surface polysaccharide poly-β-1,6-N-acetylglucosamine (Henry et al., 2012). Recently, an intriguing polymyxin resistance mechanism was reported in LPS-deficient A. baumannii due to the dependence on polymyxins (Zhu et al., 2020) (Fig. 2). On agar plates, polymyxin-dependent A. baumannii isolates can only be cultured in the presence of polymyxins or polymyxin-like peptides. Importantly, polymyxin-dependent isolates are capable of causing infections in neutropenic mice. This discovery highlights a significant concern in clinic, as polymyxin-dependent resistance phenotype is undetectable with the conventional clinical antibiotic susceptibility testing, which utilizes drug-free agar plates (Zhu et al., 2020).
Polymyxin resistance due to outer membrane proteins and efflux pumps has also been reported (Fig. 2). In K. pneumoniae, inactivation of the KpnGH efflux pump causes an increase in bacterial susceptibility toward several antibiotics, including polymyxin B (Srinivasan et al., 2014). The deletion of emrB in A. baumannii leads to an increased susceptibility to polymyxins, suggesting the role of Emr efflux pumps in polymyxin resistance (Tietgen et al., 2018). The MexAB-OprM efflux system is associated with unspecific adaptive polymyxin resistance in metabolically active subpopulations of P. aeruginosa in biofilm (Pamp et al., 2008). In addition, the efflux pump MtrC-MtrD-MtrE and outer membrane porin PorB are important for the intrinsic polymyxin resistance in Neisseria meningitidis (Tzeng et al., 2005). In Vibrio spp., the outer membrane protein OmpU contributes to polymyxin resistance by regulating sigmaE-associated periplasmic stress response (Mathur and Waldor, 2004; Mathur et al., 2007; Duperthuy et al., 2010). The RosA/RosB efflux pump/potassium antiporter system is identified as a polymyxin resistance contributor in Yersinia enterocolitica, and it is postulated that this system plays a role in the removal of polymyxins from cytoplasm and acidification of the cytoplasm, which leads to the inactivation of polymyxins (Bengoechea and Skurnik, 2000). In addition, the outer membrane remodeling plays a role in bacterial survival in response to polymyxin killing. Upregulation of the genes associated with the maintenance of lipid asymmetry (Mla) system in A. baumannii after colistin treatment suggests the importance of maintaining the asymmetry and integrity of the outer membrane (Malinverni and Silhavy, 2009; Henry et al., 2015).
Another mechanism of polymyxin resistance that is independent of the outer membrane is the involvement of capsule (Fig. 2). Certain capsulated strains of K. pneumoniae are more resistant to polymyxins by reducing the binding of polymyxin molecules to the bacterial outer membrane (Campos et al., 2004). Purified capsular polysaccharides from K. pneumoniae and P. aeruginosa have been demonstrated to bind directly to polymyxins, and the addition of the capsular polysaccharides enhanced polymyxin resistance of an acapsular K. pneumoniae (Llobet et al., 2008). Another mechanism of polymyxin resistance involves a putative serine protease colistinase that degrades colistin that is produced by polymyxin-producing P. polymyxa (Ito-Kagawa and Koyama, 1980). Although inconclusive, in view of the increased production of intracellular ROS in the presence of polymyxins, the colistinase might be necessary for the survival of P. polymyxa during polymyxin synthesis (Ito-Kagawa and Koyama, 1980). Additionally, the antioxidant superoxide dismutase A (SodA) has been reported to play a protective role against polymyxin-induced oxidative stress in P. polymyxa (Yu et al., 2017b). The contribution of Sod to polymyxin resistance has also been illustrated in A. baumannii, in which the knockout of sod2343 led to increased colistin susceptibility (Heindorf et al., 2014).
Overall, mechanisms of polymyxin resistance in Gram negative bacteria are multifaceted. Developing an in-depth understanding of different resistance mechanisms is the key toward minimizing the emergence of polymyxin resistance using combinations with other antibiotics or adjuvant compounds that maximize the killing and target the resistance pathway.
IV. Susceptibility Testing and Breakpoints
A. Susceptibility Testing
The optimal susceptibility testing method had not been developed for polymyxins until recently. It should be noted that colistin sulfate and polymyxin sulfate should be used for measurement of MICs (Clinical and Laboratory Standards Institute, 2015). As CMS is an inactive prodrug of colistin and is not stable in vitro (Bergen et al., 2006), it should not be used for measuring the MIC of colistin. Disk diffusion is among the classic antimicrobial susceptibility testing methods and is still being widely used (Sandle, 2016). However, the size of the inhibitory zone obtained using disk diffusion is often poorly correlated with MICs obtained using the reference broth microdilution method (Matsen et al., 1969). Additionally, many studies have also reported a high false susceptibility rate with disk diffusion and concluded its unreliability (Gales et al., 2001; Tan and Ng, 2006a; Lo-Ten-Foe et al., 2007; Moskowitz et al., 2010; Maalej et al., 2011). Another method for antibiotic susceptibility testing is the gradient diffusion with the use of strips impregnated with predefined gradient of antibiotic concentration (e.g., Etest, bioMérieux and MTS, Liofilchem). A number of studies compared the gradient diffusion method (mostly Etest) to other susceptibility testing methods for polymyxin B and colistin, and variable results have been reported (Arroyo et al., 2005; Lo-Ten-Foe et al., 2007; Tan and Ng, 2007; Behera et al., 2010; Moskowitz et al., 2010; Maalej et al., 2011; Hindler and Humphries, 2013; Landman et al., 2013; Matuschek et al., 2018) with high false susceptibility rates (Arroyo et al., 2005; Hindler and Humphries, 2013; Landman et al., 2013; Matuschek et al., 2018). Overall, disk diffusion and gradient diffusion methods are not recommended, as these methods are associated with relatively high false susceptibility results (Matuschek et al., 2018). Good concordance between agar dilution and broth microdilution was reported in three studies (Lo-Ten-Foe et al., 2007; Behera et al., 2010; Hindler and Humphries, 2013), but conflicting results have also been published (Hogardt et al., 2004; Moskowitz et al., 2010). Notably, a high agreement was demonstrated between broth microdilution and macrodilution methods (Haeili et al., 2019). To date, broth microdilution is the recommended reference method for determining the MICs of polymyxin B and colistin by the Clinical and Laboratory Standards Institute (CLSI) and the European Committee on Antimicrobial Susceptibility Testing (EUCAST) (Clinical and Laboratory Standards Institute, 2020, European Committee on Antimicrobial Susceptibility Testing, 2021).
Automated systems are available commercially for polymyxin MIC measurements in clinical microbiology laboratories. Underestimation of polymyxin resistance has been found with the Vitek 2 system (bioMérieux) and Phoenix automated system (BD Phoenix 100; BD Diagnostic) (Lo-Ten-Foe et al., 2007; Tan and Ng, 2007; Vourli et al., 2017; Jayol et al., 2018). The MicroScan system (Beckman Coulter Diagnostics) demonstrated a categorical agreement of 87% and 88% when compared with agar dilution and broth microdilution, respectively (Lee et al., 2013b; Chew et al., 2017). A high essential agreement with broth microdilution was seen with the use of Sensititre (96%; Thermo Fisher Scientific), MICRONAUT-S (96%; Merlin Diagnostika), and MICRONAUT MIC-Strip (99%; Merlin Diagnostika), whereas a slightly lower essential agreement was reported with SensiTest (88%; Liofilchem) (Matuschek et al., 2018). The same study also reported an essential agreement of 82% with UMIC Colistine kit (Biocentric), whereas a higher essential agreement of 94% was demonstrated in a separate study (Bardet et al., 2019).
Many experimental factors can affect the MIC values for polymyxins; hence, standardization is crucial when performing the susceptibility test for polymyxin B and colistin. The concentrations of Ca2+ and Mg2+ in cation-adjusted Mueller-Hinton broth (CAMHB) are accurately controlled, as these divalent cations influence the activity of polymyxins. The addition of Ca2+ and Mg2+ into broth media can reduce the in vitro activity of polymyxins (Newton, 1954; Davis et al., 1971; Chen and Feingold, 1972; D’Amato R et al., 1975). Very likely, the divalent cations interact with bacterial outer membrane, affecting the interaction between lipid A and polymyxins (Newton, 1954; Chen and Feingold, 1972). The bactericidal effect of polymyxin B against E. coli and P. aeruginosa was inhibited in the presence of 80 mg/l of Ca2+ or 24 mg/l of Mg2+ in Luria-Bertani broth (Chen and Feingold, 1972). Overall, the current standard protocol recommends adjusting CAMHB to final concentrations of 20–25 mg/l of Ca2+ and 10–12.5 mg/l of Mg2+ for the susceptibility testing of polymyxin B and colistin (Clinical and Laboratory Standards Institute, 2018). Another concern is the nonspecific binding of polymyxins to the surface of certain plasticware and glass due to its amphiphilic nature. An overall increase of 5.3-fold in colistin MICs [noncoated microtiter tray (0.54 ± 0.58 mg/l) and tissue culture–coated microtiter tray (2.84 ± 1.93 mg/l)] was observed when broth microdilution was conducted using negatively charged tissue culture microtiter plates coated with polystyrene due to nonspecific binding (Albur et al., 2014). It has also been demonstrated that polymyxin adsorption was higher at lower concentrations and substantial with microtiter plates made of polystyrene, glass, polypropylene, and low-protein-binding polypropylene (Karvanen et al., 2017). Polysorbate 80 (i.e., Tween 80) is a nonionic surfactant and is commonly used to prevent the binding of antimicrobial agents to plastic and other materials during susceptibility testing (Clinical and Laboratory Standards Institute, 2018). The addition of polysorbate 80 to a final concentration of 0.002% resulted in reduced polymyxin MICs; notably, a concentration-dependent binding effect was observed, and the magnitude of MIC reduction was variable in different bacterial strains (Sader et al., 2012; Hindler and Humphries, 2013). Notably, polysorbate 80 may have synergistic antibacterial activity with polymyxins (Brown and Winsley, 1968). The two postulated synergistic mechanisms are as follows: 1) the increased cell permeability by polysorbate 80 enhances the penetration of polymyxins in bacterial cells; 2) the destabilization of bacterial outer membrane by polymyxins facilitates the access of polysorbate 80 into the cell (Brown et al., 1979). However, both studies (Brown and Winsley, 1968; Brown et al., 1979) were conducted prior to the discovery of nonspecific binding of polymyxins to plastic and other materials; hence, the synergistic results reported could be merely an artefact of polymyxin binding to the surface of the experimental vessel used in these early studies (Brown and Winsley, 1968; Brown et al., 1979). Notably, the latest recommended broth microdilution method does not include polysorbate 80 (Clinical and Laboratory Standards Institute, 2020, European Committee on Antimicrobial Susceptibility Testing, 2021).
Overall, the current recommended susceptibility testing of polymyxins should be conducted using the broth microdilution method with CAMHB (final concentrations of 20–25 mg/l of Ca2+ and 10–12.5 mg/l of Mg2+).
B. Polymyxin Breakpoints
Determination of the susceptibility breakpoint for polymyxins has been a challenge. The CLSI (formerly known as National Committee on Clinical Laboratory Standards) published the first polymyxin breakpoints in 1976 based on disk diffusion, which was then abolished in 1980 because of concerns of polymyxin toxicity and the availability of other classes of safer antibiotics (National Committee for Clinical Laboratory Standards, 1976, 1980; Satlin et al., 2020). Revised polymyxin disk diffusion and MIC breakpoints were made available by the CLSI in 2003 (Satlin et al., 2020). Owing to the increasing threat from MDR Gram negative bacteria, colistin breakpoints were established for Enterobacteriaceae, Pseudomonas spp., and Acinetobacter spp. by the EUCAST in 2010 (European Committee on Antimicrobial Susceptibility Testing, 2010). In 2013, colistin breakpoints for P. aeruginosa and Acinetobacter spp. were revised by the CLSI; however, no breakpoint was made for Enterobacteriaceae (Clinical and Laboratory Standards Institute, 2013). Efforts have been made into the interpretation of MIC distribution statistically to separate bacteria into wild-type (susceptible) and non–wild-type (resistant) populations. This leads to the introduction of the epidemiologic cutoff value, referring to MIC values marking the upper end of the wild-type populations. Although PK/PD and clinical data were insufficient to generate a breakpoint for the polymyxins, an epidemiologic cutoff value of colistin (wild-type ≤ 2 mg/l and non–wild type ≥ 4 mg/l) is established by the CLSI for Enterobacteriaceae (E. coli, K. pneumoniae, Klebsiella aerogenes, E. cloacae, and Raoultella ornithinolytica) (Clinical and Laboratory Standards Institute, 2018). In 2020, the CLSI reviewed the polymyxin breakpoints and decided to include the breakpoints for Enterobacterales (previously as Enterobacteriaceae) (Clinical and Laboratory Standards Institute, 2020). However, in this latest CLSI guideline, the “susceptible” interpretive category has been removed, and ≤2 mg/l is set as an “intermediate” breakpoint due to limited clinical effectiveness (e.g., based upon 28-day mortality) of polymyxins, even against Gram negative bacteria with MIC ≤2 mg/l. This change has caused substantial confusions in clinical practice and is not in agreement with the latest polymyxin breakpoints provided by the EUCAST (Satlin et al., 2020). There are minor variations between the guidelines on the breakpoints from the CLSI and EUCAST (Table 2). The MIC breakpoints for colistin and polymyxin B are stated for Enterobacterales, P. aeruginosa, and Acinetobacter spp. by the CLSI and EUCAST (Clinical and Laboratory Standards Institute, 2020, European Committee on Antimicrobial Susceptibility Testing, 2021). The EUCAST has introduced the epidemiologic cutoff value of polymyxin B for Enterobacterales (2 mg/l), P. aeruginosa (4 mg/l), and Acinetobacter spp. (2 mg/l). Notably, polymyxin breakpoints based on the inhibition zone diameter are not established by either CLSI or EUCAST, as the disk diffusion method is not recommended for polymyxin susceptibility testing.
MIC breakpoints for colistin and polymyxin B established by CLSI (2020) and EUCAST (2021) guidelines
In November 2019, the US Committee on Antimicrobial Susceptibility Testing recommended an MIC of ≤2 mg/l as the susceptibility breakpoint for colistin and polymyxin B against P. aeruginosa, A. baumannii, and Enterobacterales (Pogue et al., 2020). Current clinical data are very limited on the efficacy (most with 28-day mortality as the primary outcome) of intravenous CMS and polymyxin B for the treatment of pulmonary infections caused by P. aeruginosa, A. baumannii, and Enterobacterales. Therefore, the US Committee on Antimicrobial Susceptibility Testing susceptibility breakpoint for colistin and polymyxin B is not applicable to lower respiratory tract infections (Pogue et al., 2020). Similarly, considering the very low urinary recovery after intravenous administration, no breakpoint is recommended for polymyxin B for lower urinary tract infections (UTIs) (Sandri et al., 2013a; Pogue et al., 2020). The susceptibility breakpoint for both colistin and polymyxin B after inhalation is not available either. Very limited PK information has shown that concentrations of formed colistin in the epithelial lining fluid (ELF) are within the range of 100–200 mg/l after inhalation of CMS in patients (Gkoufa et al., 2019). Therefore, it is very likely that the susceptibility breakpoint for both colistin and polymyxin B is higher than 2 mg/l for pulmonary infections after inhaled administration due to PK/PD considerations. Clearly, well designed clinical PK/PD studies are required to determine the accurate susceptibility breakpoint for the two polymyxins in different types of infections.
V. Pharmacokinetics and Pharmacodynamics
PK and PD data are essential for optimization of antibiotic use with maximal efficacy and minimal adverse effects. Significant advancements have been made over the last two decades toward the elucidation of PK/PD for polymyxins using in vitro and infected animal models (Li et al., 2006a; Bergen et al., 2012; Cheah et al., 2015; Kaye et al., 2016; Landersdorfer et al., 2018; Nation and Forrest, 2019).
A. Pharmacokinetics
With specific high-performance liquid chromatography and liquid chromatography–mass spectrometry assays, a number of studies investigated the PK of colistin, CMS, and polymyxin B in rats, mice, rabbits, pigs, baboons, and humans (Li et al., 2005a, 2006a,b; Marchand et al., 2019; Nation and Forrest, 2019). The PK of colistin and polymyxin B are comparable in animals, considering that there is only one amino acid difference (Sivanesan et al., 2017b). To date, it is not possible to compare their PK in patients, as an inactive prodrug CMS is used for colistin in the clinic. Colistin sulfate is only available for intravenous administration in China, and future clinical PK results will make the comparison of the two polymyxins possible. In rats, colistin A, colistin B, polymyxin B1, and polymyxin B2 display similar clearance, volume of distribution, elimination half-life, and very low urinary recovery (<1%) (Sivanesan et al., 2017b). Both positively charged Dab residues and the hydrophobic moieties (N-terminus and position 6/7) play key roles in the renal elimination of polymyxins, as polymyxin analogs with fewer Dab residues and polymyxin nonapeptide (i.e., lack of the N-terminus and Dab1) have a higher urinary recovery (Vaara et al., 2010a; Marchand et al., 2019; Vaara, 2019a). The difference in the length of N-terminal fatty acyl group and position 6 (d-Phe for polymyxin B and d-Leu for colistin) causes different plasma binding in rats (colistin A 56.6% ± 9.25%, colistin B 41.7% ± 12.4%, polymyxin B1 82.3% ± 4.30%, and polymyxin B2 68.4% ± 3.50%) (Sivanesan et al., 2017b).
CMS is a mixture of a large number of fully and partially methanesulfonated entities, plus possibly a very small portion of colistin (Li et al., 2019); therefore, determining the PK of CMS is challenging because of the potential conversion of CMS to colistin during the experiment (e.g., blood samples not placed on ice immediately after collection) and measurement of concentrations of CMS and formed colistin (e.g., suboptimal pretreatment procedure). As colistin is polycationic and CMS is polyanionic, their PK are significantly different (renal handling in particular) (Li et al., 2003b, 2004). After filtration by glomeruli, the negatively charged CMS is extensively secreted, whereas the positively charged colistin (and polymyxin B) undergoes extensive reabsorption by renal tubular cells (Li et al., 2004). In animals, CMS is cleared much faster than colistin, and approximately 60% of the CMS dose is eliminated in urine; the high concentration of formed colistin observed in urine is due to the ongoing conversion from CMS in the kidneys and bladder (Li et al., 2004; Marchand et al., 2019; Nation and Forrest, 2019). Limited information is available on the metabolism of polymyxins. A recent mass spectrometry imaging study identified 10 metabolites of polymyxin B1 in rat kidneys, and six of them were also detected in urine samples; similarly, three metabolites of colistin were discovered in urine and kidney homogenates (Nilsson et al., 2015). Metabolism of polymyxin B and colistin in kidneys is mainly via amide hydrolysis, demethylation, and oxidation (Nilsson et al., 2015).
Clinical PK data in healthy volunteers are not available for polymyxin B and only became available recently for CMS in ∼40 health subjects (Couet et al., 2011; Mizuyachi et al., 2011; Zhao et al., 2018). These studies reported higher plasma concentrations of CMS than formed colistin, with CMS achieving its maximum concentration at the end of the infusion, whereas the maximum concentration of formed colistin was achieved hours after the end of the infusion. The renal clearance of colistin was much lower than that of CMS because of the extensive renal tubular reabsorption (Nation and Forrest, 2019). Following normalization of CMS doses, a wide range of achievable plasma colistin concentrations (0.69–3.8 mg/l) were reported in humans across these three studies, and 40%–70% of the CMS dose was recovered in the urine as CMS and formed colistin (Couet et al., 2011; Mizuyachi et al., 2011; Zhao et al., 2018). Overall, the conversion of CMS to colistin in healthy subjects is low. There are at least two clinical PK studies on CMS and formed colistin in a small number of patients with cystic fibrosis using high-performance liquid chromatography assays, and both showed very similar disposition profiles (Li et al., 2003a; Yapa et al., 2014), as well as to those observed in healthy subjects (Couet et al., 2011; Mizuyachi et al., 2011; Zhao et al., 2018). The terminal half-lives of CMS and formed colistin are 2 to 3 hours and 3–8 hours, respectively, indicating that colistin is rate-limited by its own elimination, not the conversion from CMS (Couet et al., 2011; Mizuyachi et al., 2011; Zhao et al., 2018).
Over the last two decades, there have been a number of clinical PK studies for CMS and formed colistin in ∼250 critically ill patients (Li et al., 2005b; Markou et al., 2008, 2012; Plachouras et al., 2009; Karaiskos et al., 2015; Nation et al., 2017). After the first dose, a slow increase in the concentration of formed colistin was evident in most critically ill patients (Li et al., 2003a, 2005b; Markou et al., 2008, 2012; Plachouras et al., 2009; Yapa et al., 2014; Karaiskos et al., 2015; Nation et al., 2017; Nation and Forrest, 2019). Furthermore, flat plasma concentration versus time profiles of formed colistin were shown at steady state in critically ill patients without renal replacement therapy (Li et al., 2005b; Markou et al., 2008, 2012; Plachouras et al., 2009; Karaiskos et al., 2015; Nation et al., 2017) (Fig. 4). With the current recommended dosage regimens, the average steady-state plasma concentration (Css,avg) of formed colistin is approximately 2 to 3 mg/l in critically ill patients not on renal replacement therapy (Li et al., 2005b; Markou et al., 2008, 2012; Plachouras et al., 2009; Karaiskos et al., 2015; Nation et al., 2017). The mean unbound fraction of colistin in human plasma is 0.49 (Nation et al., 2017). In the largest clinical PK study in critically ill patients to date, significant interpatient variability (up to ∼10-fold) was revealed at a given creatinine clearance, and the apparent clearance of formed colistin was closely correlated to the renal function of the patient (Garonzik et al., 2011; Nation et al., 2017) (Fig. 5). With the current dosage regimens, it is very challenging to achieve Css,avg 2 mg/l of formed colistin in patients with good renal function, as most of the CMS dose is eliminated by the kidneys (Garonzik et al., 2011; Nation et al., 2017). For patients on renal replacement therapy (e.g., intermittent hemodialysis and continuous renal replacement), both CMS and formed colistin are able to be efficiently removed in the extracorporeal unit. As concentrations of CMS are much higher than that of formed colistin in plasma during most of the dosing interval, removal of CMS from blood by hemodialysis significantly decreases the proportion of CMS, which converts to colistin in vivo, thereby impacting the area under the plasma concentration curve (AUC) of formed colistin (Garonzik et al., 2011; Nation et al., 2017). There is very limited information on the PK of CMS and formed colistin in pediatric and burn patients, and prospective clinical studies are warranted to optimize CMS/colistin use in these types of patients (Nation and Forrest, 2019).

Steady-state plasma concentrations of colistimethate (A) and formed colistin (B) across a dosage interval in 215 critically ill patients. Patients were receiving colistimethate every 8, 12, or 24 hours. Each symbol represents the data from an individual. Permission obtained from Oxford University Press (Nation et al., 2017).
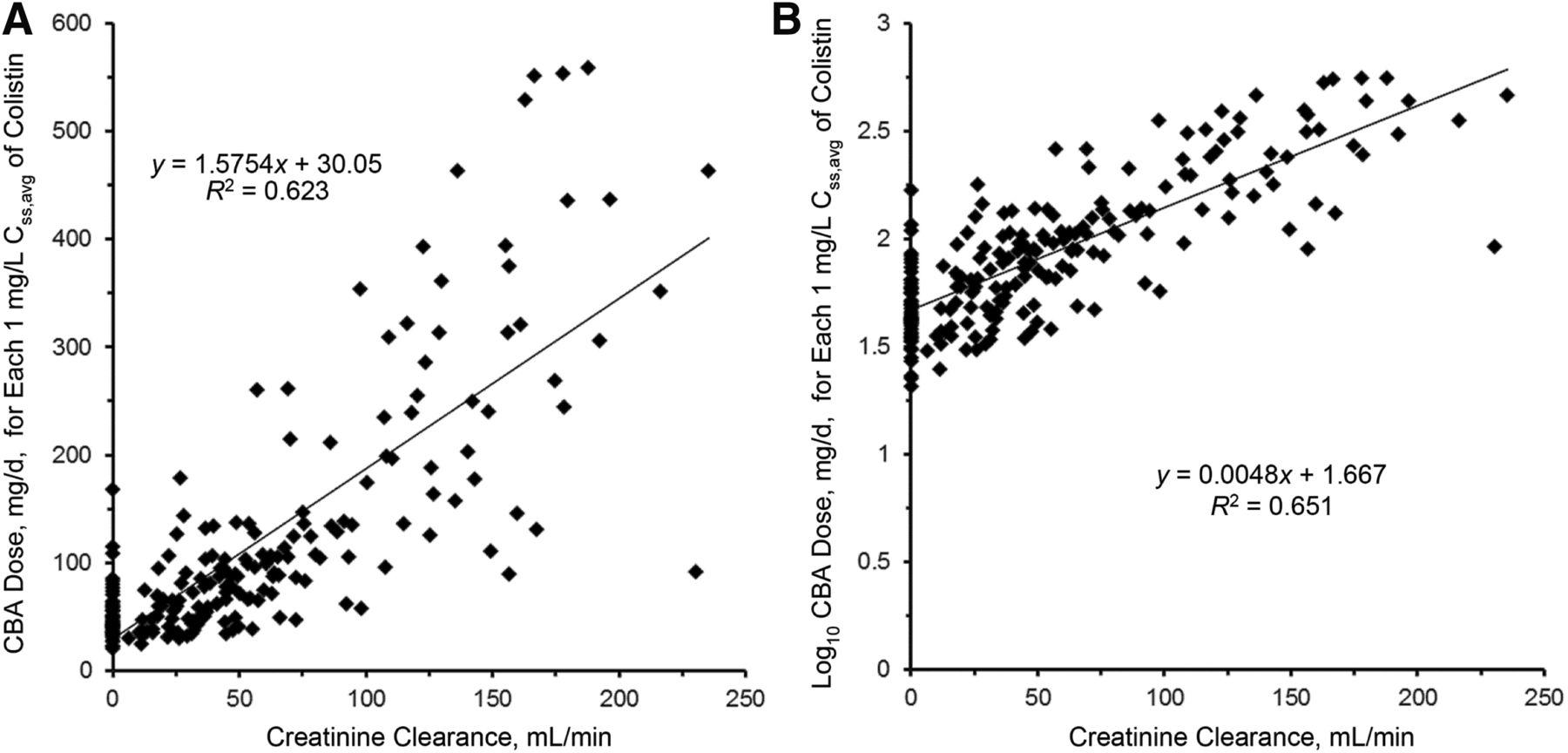
Linear (A) and log-linear (B) plots of the relationship between the daily dose of CBA needed for each 1 mg/l of the average steady-state plasma concentration of colistin (Css,avg) and creatinine clearance. The regression equation in (B) with the intercept adjusted from 1.667 to 1.825 is the renally based dosing algorithm. Permission obtained from Oxford University Press (Nation et al., 2017).
Since 2008 there have been at least seven clinical studies on the PK of polymyxin B in ∼150 patients after intravenous administration (Nation and Forrest, 2019). In a population PK study with 24 critically ill patients, the Css,avg of polymyxin B is ∼2.8 mg/l (range 0.68–4.88 mg/l) with dosage regimens of 0.45–3.38 mg/kg per day (Sandri et al., 2013a). Unlike the slow increase of formed colistin concentrations after intravenous CMS in critically ill patients (Nation et al., 2017) (Fig. 4), polymyxin B reached the peak concentration (∼2.4–14 mg/l) after the infusion (Sandri et al., 2013a) (Fig. 6). Compared with the PK of formed colistin after intravenous CMS, the interpatient variability of polymyxin B clearance (∼3-fold) was much smaller across a wide range of creatinine clearance (Nation and Forrest, 2019). The median unbound fraction of polymyxin B in human plasma is 0.42, and the half-life of polymyxin B in critically ill patients is approximately 12 to 13 hours (Nation and Forrest, 2019). Different from CMS, the urinary recovery of polymyxin B is less than 5%, which is consistent with the results in animals (Nation and Forrest, 2019). One of the most important differences between the PK of polymyxin B and CMS/colistin in critically ill patients is that creatinine clearance is not a covariate of the total body clearance of polymyxin B (Sandri et al., 2013a) (Fig. 7). Current clinical PK data indicate that daily doses of polymyxin B should not be adjusted based on the kidney function of patients due to PK/PD considerations. Therefore, caution is required when clinicians reduce the daily dose of polymyxin B in renally impaired patients based on the inaccurate and outdated product information. There is very limited information on the PK of polymyxin B in patients on renal replacement therapy. Data from two patients showed ∼5%–12% removal of the polymyxin B dose by continuous venovenous hemodialysis during the 12-hour dosing interval (Sandri et al., 2013b). In summary, polymyxin B and CMS have very different PK in patients, and their impacts on the clinical use will be discussed in the PK/PD and toxicities sections below.
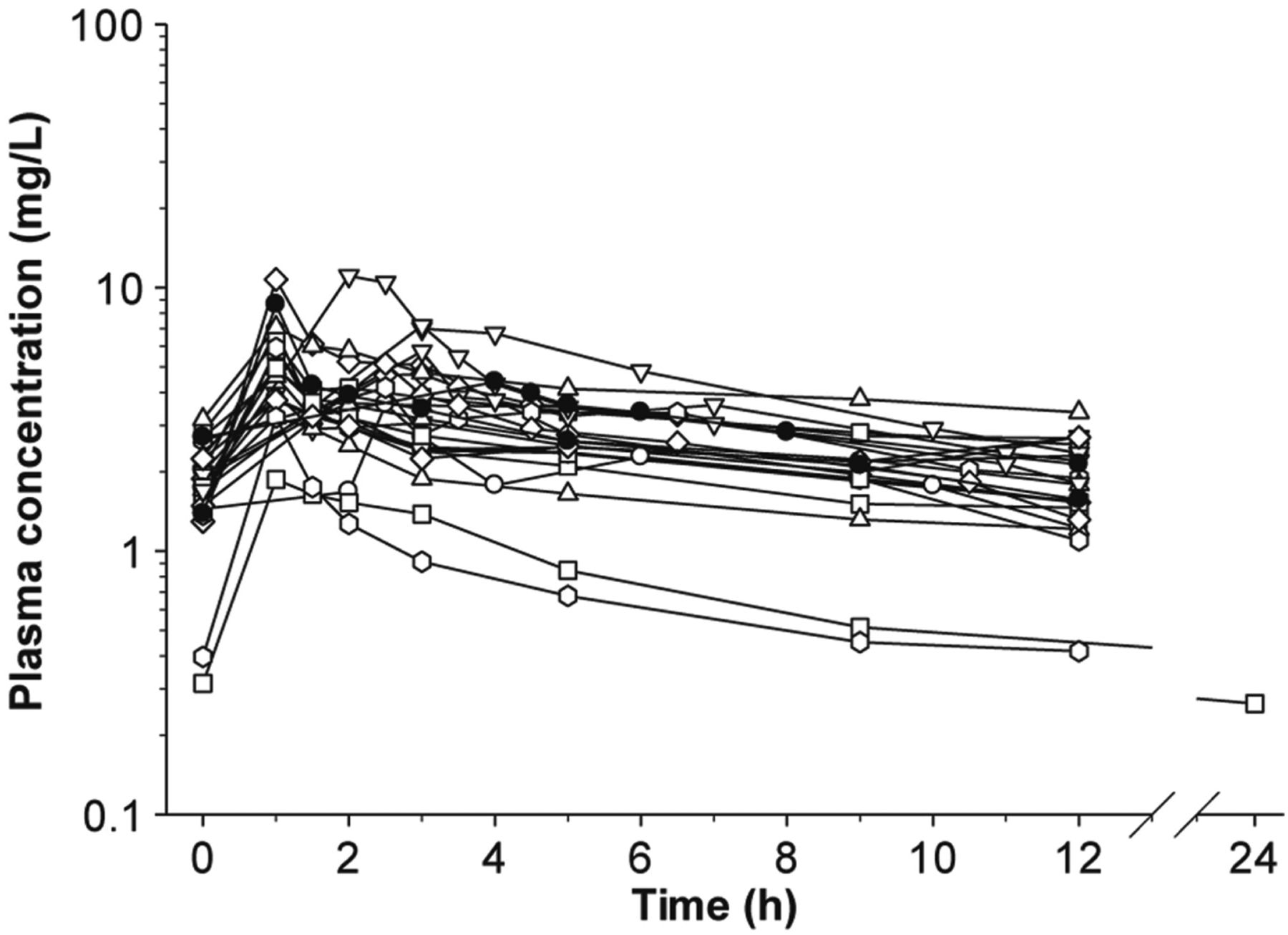
Plasma concentration-time profiles of polymyxin B in 24 critically ill patients. Permission obtained from Oxford University Press (Sandri et al., 2013a).

Individual polymyxin B clearance estimates vs. creatinine clearance in critically ill patients. Polymyxin B clearance was scaled by total body weight (liters per hour per kilogram). Open circles represent patients not on hemodialysis, the filled diamond represents the continuous venovenous hemodialysis patient who weighed 250 kg, and the filled triangle represents the lean continuous venovenous hemodialysis patient. Permission obtained from Oxford University Press (Sandri et al., 2013a).
B. Pharmacodynamics
Concentration-dependent killing against P. aeruginosa by polymyxins (most literature on colistin) has been demonstrated in static time-kill (Li et al., 2001; Tam et al., 2005; Bulitta et al., 2010; Bergen et al., 2011a), in vitro one-compartment dynamic model (IVM) (Bergen et al., 2011b), and hollow-fiber infection model (HFIM) (Tam et al., 2005). As P. aeruginosa is a notorious biofilm producer, it is important to note that concentration-dependent antibiofilm activity of colistin has also been demonstrated using biofilm models (Hengzhuang et al., 2011; Lora-Tamayo et al., 2014). However, eradication of biofilm formed by P. aeruginosa involves a higher colistin concentration and longer exposure as compared with planktonic cells, with mucoid biofilm being harder to eradicate than nonmucoid biofilm (Hengzhuang et al., 2011). Interestingly, in an in vitro model of THP-1 monocyte infection, colistin demonstrated concentration-dependent killing of P. aeruginosa both intracellularly and extracellularly; however, colistin exhibited poorer efficacy against intracellular P. aeruginosa (Buyck et al., 2013). The concentration-dependent killing of polymyxins has also been demonstrated against A. baumannii (Kroeger et al., 2007; Owen et al., 2007; Tan et al., 2007) and K. pneumoniae (Poudyal et al., 2008; Deris et al., 2012; Lin et al., 2019b). Variable postantibiotic effects (PAEs) have been demonstrated across different bacteria, including P. aeruginosa (Li et al., 2001; Bozkurt-Guzel and Gerceker, 2012), A. baumannii (Owen et al., 2007; Plachouras et al., 2007), and K. pneumoniae (Poudyal et al., 2008). It should be noted that at clinically achievable concentrations, the PAE of polymyxin B and colistin is not significant. In addition, the bacterial inoculum also affects the measurement of the killing kinetics and PAE of polymyxins. For example, the extent of killing by polymyxins has been significantly reduced at higher inocula (e.g., 108 cfu/ml) of P. aeruginosa and K. pneumoniae (Tam et al., 2005; Deris et al., 2012). The inoculum effect was also demonstrated in a mechanism-based mathematical model, with 6- and 23-fold slower killing rate at 107 cfu/ml and 109 cfu/ml of P. aeruginosa, respectively, as compared with 106 cfu/ml (Bulitta et al., 2010).
C. Pharmacokinetics/Pharmacodynamics
Mimicking the PK of formed colistin in patients with the recommended maximum daily dose of CMS, different dosing intervals of 8, 12, and 24 hours did not affect the magnitude of colistin antibacterial killing against P. aeruginosa in an IVM study (Bergen et al., 2008). This shows that the ratio of maximum concentration of unbound drug to MIC (fCmax/MIC) is not the most predictive PK/PD index for colistin efficacy (Bergen et al., 2008). An intensive IVM study involving a range of dosing regimens examined the relationship between the three PK/PD indices and efficacy in eradicating P. aeruginosa (Bergen et al., 2010). The results revealed the ratio of area under the plasma concentration curve of unbound drug to MIC (fAUC/MIC) (R2 = 93%) as the PK/PD index that best correlates with the killing activity of colistin, as compared with fCmax/MIC (R2 = 87%) and the duration that the unbound plasma concentration remains above the MIC (fT > MIC) (R2 = 79%). Using the gold-standard murine thigh and lung infection models, fAUC/MIC has also been demonstrated to be the best predictive PK/PD index for colistin and polymyxin B against P. aeruginosa (Hengzhuang et al., 2012; Cheah et al., 2015; Lin et al., 2017a,b), A. baumannii (Cheah et al., 2015; Lin et al., 2018), and K. pneumoniae (Landersdorfer et al., 2018).
The relationship between the magnitude of fAUC/MIC and AUC/MIC with bacterial killing has been evaluated for polymyxins (Table 3). Targeted values of fAUC/MIC for different magnitudes of antibacterial killing by colistin were determined in neutropenic mouse thigh and lung infection models using three strains of P. aeruginosa [American Type Culture Collection (ATCC) 27853, MIC 1 mg/l; PAO1, 1 mg/l; and a clinical MDR mucoid clinical isolate 19056, 0.5 mg/l) and three strains of A. baumannii (ATCC 19606, 1 mg/l, and two MDR clinical isolates: 248-01-C.248, 1 mg/l, and N-16870.213, 0.5 mg/l] (Cheah et al., 2015). Plasma binding of colistin in mouse plasma was determined within the concentration range of approximately 2–50 mg/l using two different methods, and the binding was 92.9% ± 3.3% (mean ± SD%) by ultracentrifugation and 90.4% ± 1.1% by equilibrium dialysis. In the mouse thigh infection model, an fAUC/MIC of 7.4–13.7 for P. aeruginosa and 7.4–17.6 for A. baumannii was required for 2-log10 kill (Cheah et al., 2015). In the mouse lung infection model, colistin showed much weaker efficacy due to poor disposition in the lungs with higher target values of fAUC/MIC (36.8–105) against two of the three P. aeruginosa strains and one strain of A. baumannii; colistin was not even bacteriostatic against the other two A. baumannii strains (Cheah et al., 2015).
Studies evaluating the fAUC/MIC targets of polymyxins with different magnitudes of bacterial killing
Similarly, over the range of approximately 0.9–37 mg/l, the binding of polymyxin B in mouse plasma, determined by ultracentrifugation, is 91.4% +/− 1.65% (Landersdorfer et al., 2018). The targeted values of fAUC/MIC for polymyxin B were determined in infected mice using three strains of K. pneumoniae (ATCC BAA-2146 and two clinical isolates, FADDI-KP042 and FADDIKP032; MIC = 0.5 mg/l for all). The fAUC/MIC targets for bacteriostasis and 1-log10 kill of polymyxin B against K. pneumoniae were 1.22–13.5 and 3.72–28.0, respectively, whereas 2-log10 kill was not observed with any strain (Landersdorfer et al., 2018). Polymyxin B and colistin show comparable efficacy against K. pneumoniae with equimolar doses in the mouse thigh infection model (Landersdorfer et al., 2018). In the mouse lung infection model, polymyxin B does not show any killing effect against any of the three K. pneumoniae strains examined, even at the highest subcutaneous dose tolerated by mice (i.e., 120 mg/kg per 24 hours) (Landersdorfer et al., 2018). Collectively, PK/PD results from recent mouse infection studies indicate that parenteral polymyxin B and colistin may not be efficacious against pulmonary infections due to poor PK in the lungs.
Importantly, PK/PD of pulmonary delivery of polymyxins has been recently reported and highlights the superiority of pulmonary delivery over parenteral administration in lung infection model with P. aeruginosa, A. baumannii, and K. pneumoniae (Lin et al., 2017a,b). Additionally, in comparison with colistin, higher fAUC/MIC values were obtained with polymyxin B, which was likely due to different binding affinities of colistin and polymyxin B to plasma and lung surfactant (Lin et al., 2017a,b). These preclinical PK/PD data are essential for the optimization of polymyxin dosing in patients.
Three small clinical studies conducted among 47 critically ill patients demonstrated that, because of the slow and low conversion of CMS to colistin in vivo, a loading dose is important for achieving reasonable concentrations of formed colistin as soon as possible after intravenous CMS (Plachouras et al., 2009; Mohamed et al., 2012; Karaiskos et al., 2015). To date, the largest National Institutes of Health–funded clinical PK study of intravenous CMS in critically ill patients (n = 215) revealed that creatinine clearance was an important covariate in the clearance of both CMS and formed colistin, and adequate plasma concentrations of formed colistin is difficult to achieve, in particular in patients with good renal function (Garonzik et al., 2011; Nation et al., 2017). As an example, for patients not on renal replacement therapy, Css,avg ≥2 mg/l was achieved in >80% of patients with creatinine clearance <80 ml/min, whereas Css,avg ≥2 mg/l was achieved in <40% in patients with creatinine clearance ≥80 ml/min, even with the maximum allowed daily dose [360 mg colistin base activity (CBA)]) (Garonzik et al., 2011; Nation et al., 2017). Considering the difficulty to achieve Css,avg ≥2 mg/l of formed colistin with CMS monotherapy, combination therapy is recommended for intravenous CMS, especially for a patient with a creatinine clearance of ≥80 ml/min and a bacterial pathogen with an MIC of >0.5 mg/l (Garonzik et al., 2011; Nation et al., 2017; Tsuji et al., 2019). Based on recent clinical PK and preclinical PK/PD findings in the literature, the first scientifically based dosing recommendation was proposed for intravenous CMS, and a dosing algorithm was designed for different categories of patients (based on renal function) and the determination of a loading dose based on body weight (Garonzik et al., 2011; Nation et al., 2017). To minimize potential describing errors due to the complex PK and different dose units of CMS around the world, a free clinician-friendly iPhone/iPad app has been developed based on the latest dosing recommendation to help clinicians calculate the dosage regimens of CMS in different types of patients (https://apps.apple.com/us/app/colistindose/id1336806844).
Even though high concentrations of polymyxin B (e.g., 3–14 mg/l) can be achieved after a short infusion of the first dose, Monte Carlo simulations indicated that lack of a loading dose resulted in substantially lower AUC0–24h of polymyxin B on day 1 than at steady state (Sandri et al., 2013a). In contrast to CMS, clinical studies demonstrated that the total body clearance of polymyxin B is not affected by renal function; hence, in terms of PK/PD, polymyxin B is a much better option for patients with good renal function, and its dosage regimens should not be adjusted according to the renal function (Zavascki et al., 2008; Kwa et al., 2011; Sandri et al., 2013a,b; Thamlikitkul et al., 2016; Manchandani et al., 2018; Miglis et al., 2018). The dosing guideline for polymyxin B is currently unavailable; however, a large National Institutes of Health–funded clinical PK, PD, and toxicodynamics (TD) study on polymyxin B is currently being undertaken for the optimization of its dosage regimens in critically ill patients (https://clinicaltrials.gov/ct2/show/NCT02682355). Considering the slow increase in the concentration of formed colistin in patients at the start of therapy (even with a loading dose), polymyxin B has significant PK/PD advantages for the treatment of bloodstream infections; whereas for UTIs, very high concentrations of formed colistin due to the high urinary recovery of CMS make it much more advantageous than polymyxin B. Pneumonia is often the most common infection in the majority of recent polymyxin clinical studies, and the reported poor clinical outcome with intravenous polymyxin B and CMS is very likely due to the suboptimal drug exposure at the infection site (e.g., lungs) and their binding to lung surfactant. As reviewed above, current mouse PK/PD data revealed the very limited efficacy of parenteral CMS and polymyxin B for the treatment of pneumonia and support the clinical results.
There are a number of challenges in conducting clinical PK/PD/TD studies on CMS and polymyxin B. First, most polymyxin-related clinical studies to date employed 28- or 30-day mortality as the primary endpoint. Because of major underlying diseases in critically ill patients, it has been difficult to obtain solid clinical PD data for intravenous polymyxin B and CMS. Microbiological outcome (e.g., bacterial eradication) may serve as a better endpoint for the clinical PK/PD evaluations of intravenous polymyxin B and CMS. Second, many patients were on combination therapy with other antibiotics and nephrotoxic drugs, which has complicated the evaluation of PK/PD and PK/TD of polymyxins. Clearly, future well-designed prospective clinical studies are warranted to optimize the clinical use of this last-line class of antibiotics.
VI. Labeling of Polymyxins
Two different labeling conventions have been used across the world for parenteral CMS products: international unit (IU) and CBA (Li et al., 2006a; Nation et al., 2014). It is important to note that both IU and CBA are formulated according to the antibacterial activity based on in vitro microbiological assays but not the absolute amount of CMS that is present in the product (Li et al., 2006a,b). For the conversion of these two different conventions, ∼80 mg of CMS is equivalent to ∼33.3 mg of CBA and corresponds to 1 MIU. Worryingly, the use of different conventions has been confusing, and an international harmonization has been urgently called upon. Several recommendations have been put in place to minimize confusions. For hospital guidelines and prescription orders, the CMS doses should be specified clearly in IU or CBA (Tsuji et al., 2019). It is recommended that an equivalence should be included for IU and CBA in clinical papers, and an absolute mass of CMS is essential in PK studies (Nation et al., 2014). Fortunately, only one labeling convention (i.e., unit) is made available for polymyxin B. One milligram of polymyxin B base is equivalent to 10,000 U and this corresponds to ∼1.2 mg of polymyxin B sulfate (Lightbown et al., 1973). Similar to CMS, absolute mass of polymyxin B base should be employed for PK studies. Overall, it is essential that clinicians and researchers are familiar with the different terms, labeling conventions and the conversion factors when dealing with polymyxins (Li et al., 2019).
VII. Clinical Utility
Colistin is commercially available in two forms: CMS (for intravenous and inhalation administration) and colistin sulfate (primarily for topical use) (Li et al., 2005a). Colistin sulfate is only available for intravenous administration in China (Li et al., 2019). It is important to note that unfortunately it can be difficult to ascertain the form of colistin used in some studies in the literature, as colistin has been used interchangeably with CMS. In contrast to colistin, polymyxin B is only available in the form of sulfate and can be administered via intravenous, inhalation, and topical routes (Goto and Al-Hasan, 2013; Avedissian et al., 2019; Li et al., 2019).
A. Pulmonary Infections
Lower respiratory infections caused by MDR Gram negative bacteria represent a major global health and economic burden. Polymyxins have been proven to be an important therapeutic option against these difficult-to-treat MDR P. aeruginosa, A. baumannii, and Enterobacterales; however, current clinical knowledge on intravenous administration of polymyxins for the treatment of pulmonary infections is limited, as the intravenous route is less preferable because of low drug exposure in the lungs based on current animal data (Antoniu and Cojocaru, 2012; Landersdorfer et al., 2018; Tsuji et al., 2019). Notwithstanding that, intravenous CMS at 2.5–5 mg/kg per day (with adjustment for patients with renal impairment) was shown to be effective in treating ventilator-associated pneumonia (VAP) caused by A. baumannii and P. aeruginosa that were susceptible to only colistin (Garnacho-Montero et al., 2003; Rios et al., 2007). Monotherapy of intravenous CMS at daily doses of 6–9 MIU (i.e., 480–720 mg) has been shown to be as effective as imipenem and ampicillin/sulbactam in treating VAP caused by MDR and pandrug-resistant A. baumannii and P. aeruginosa (Kallel et al., 2007; Betrosian et al., 2008; Zalts et al., 2016). Importantly, intravenous CMS at 2 MIU (i.e., 160 mg) three times daily was also effective in treating pulmonary exacerbations caused by P. aeruginosa in patients with cystic fibrosis (Conway et al., 1997; Ledson et al., 1998). A study involving 74 patients with nosocomial pneumonia who received intravenous polymyxin B at 1.5–3 mg/kg per day (with dose adjustment according to the renal function) reported a 47% favorable clinical response (Furtado et al., 2007). A high favorable response of 76% was also illustrated in another study with 29 cases of MDR Gram negative respiratory tract infections, in which 21 (72%) cases received intravenous polymyxin B (2.5–3 mg/kg per day with subsequent doses adjusted according to the renal function) with concomitant antibiotic therapy (Sobieszczyk et al., 2004). Both studies suggested that polymyxin B is a potential option for the treatment of MDR Gram negative respiratory infections in which therapeutic options are limited (Sobieszczyk et al., 2004; Furtado et al., 2007). On the contrary, a prospective cohort study was performed with 67 episodes of VAP and ventilator-associated tracheobronchitis treated with either polymyxin B (45 episodes; 67%) and other antimicrobials (22 episodes; 33%) (Rigatto et al., 2013). A higher crude 30-day mortality rate of 53% was observed in the polymyxin B group (median dose of 150 mg/day, ranges from 150 to 200 mg/day) as compared with the comparator group (ceftazidime, meropenem, cefepime, piperacillin-tazobactam, ciprofloxacin, levofloxacin, and imipenem) with 27% of mortality, suggesting that polymyxin B treatment could be inferior to other antibiotics (Rigatto et al., 2013). It is evident that current clinical data on the efficacy of intravenous polymyxin B or CMS against pulmonary infections are not consistent and that future large-scale prospective clinical studies are warranted.
MDR infections in the lower airways are often fatal and difficult to treat, as many parenteral or oral antibiotics cannot reach the bacterial site of infection in the deep lung. Animal and clinical PK/PD studies demonstrate the poor efficacy of parenteral polymyxins for the treatment of Gram negative lung infections (Garonzik et al., 2011), which is mainly due to the insufficient drug penetration from the blood circulation to the infection sites on airway surface with the current dosage regimens. Simply increasing the dose of parenteral polymyxin therapy is not a viable option because of the dose-limiting nephrotoxicity and neurotoxicity (Garonzik et al., 2011; Nation et al., 2017; Velkov et al., 2018a). Considering the PK/PD, inhalation of antibiotics such as polymyxins is very likely more efficacious for the treatment of bacterial lung infections. In clinical practice, inhaled antibiotic therapy is often a complementary treatment in addition to intravenous administration to reduce systemic toxicity (e.g., nephrotoxicity). For chronic lung infections, such as in patients with cystic fibrosis, inhaled therapy is recognized to be more effective than parental administration. A clinical PK study of nebulized CMS (4 MIU) in patients with cystic fibrosis demonstrated high concentrations of colistin in the sputum (Cmax 4–16 mg/l) for a prolonged period (>3 mg/l at 12 hours), with negligible systemic exposure (Cmax <0.5 mg/l in plasma) (Yapa et al., 2014).
Inhalation products of CMS [nebulization and dry powder inhalers (DPIs)] have been approved in several European countries and Australia, but no polymyxin products are approved in the United States for inhalation. Several clinical studies reported the efficacy of inhaled CMS therapy for lung infections in patients with bronchiectasis (Haworth et al., 2014), cystic fibrosis (Schuster et al., 2013), and pneumonia (Kwa et al., 2005). Nebulized CMS is increasingly used for MDR Gram negative lung infections in patients with cystic fibrosis as a complementary treatment to intravenous use and the use of nebulized CMS for lung infections in patients without cystic fibrosis is also common. Vardakas et al. (2018) systematically reviewed and conducted a meta-analysis on 12 clinical studies on nebulized CMS monotherapy for respiratory infections in adults with non–cystic fibrosis. Among these 12 studies (Kwa et al., 2005; Motaouakkil et al., 2006; Lin et al., 2010; Rattanaumpawan et al., 2010; Athanassa et al., 2012; Kuo et al., 2012; Lu et al., 2013; Chen et al., 2014; Choi et al., 2014; Maskin et al., 2015; Abdellatif et al., 2016; Hsieh et al., 2016), 10 included patients with pneumonia, and two included patients with ventilator-associated tracheobronchitis. MDR A. baumannii and P. aeruginosa were the major causative pathogens of the lung infections in these studies. It was revealed that nebulized CMS therapy is generally safe, and the clinical and microbiological efficacies in patients with non–cystic fibrosis are encouraging (Vardakas et al., 2018). It was recommended by the study that nebulized CMS therapy should be considered for the treatment of lung infections against polymyxin-susceptible MDR Gram negative bacteria (Vardakas et al., 2018). As compared with nebulization, DPI products become popular for inhaled therapy of high-dose antibiotics (Zhou et al., 2015). Nebulization often requires bulky devices, tedious preparation (frequent cleaning), oversight by professionals, and long administration time (Tiddens et al., 2014). Furthermore, traditional jet nebulization technologies generally have very low drug delivery efficiency (Reychler et al., 2004), with only 9%–15% of drug deposited in the lungs after CMS nebulization (Boisson et al., 2014). These issues can be overcome by DPIs, as generally they are cheaper, more portable, easier to use, less labor intensive, and have better patient compliance (Zhou et al., 2014). A phase 3 centrally randomized open-label study was conducted for a dry powder inhalation product, Colobreathe, in 380 patients with cystic fibrosis with chronic P. aeruginosa lung infections (Schuster et al., 2013). Patients in the Colobreathe group received CMS powder inhalation of 1,662,500 IU twice a day for 24 weeks, whereas the other cohort received 300 mg/5 ml tobramycin nebulized solution twice daily for three 28-day cycles with a 28-day off period after each tobramycin course; Colobreathe treatment has the same efficacy as tobramycin treatment in maintaining lung function (Schuster et al., 2013). The major benefit for such a DPI product is the convenience for use (90.7% for Colobreathe vs. 53.9% for tobramycin, P < 0.001) (Schuster et al., 2013). Although the total number of adverse events was similar in both groups, higher number of coughs, throat irritation, and treatment withdrawals due to adverse events were reported for the Colobreathe group (Schuster et al., 2013). Such respiratory adverse events have often been reported for inhaled polymyxins, likely because of the local toxicity to the airways. As suggested by the clinical studies to date, it appears that inhaled CMS has fewer adverse effects than inhaled polymyxin B and colistin sulfate (Velkov et al., 2015); however, there is a lack of well designed clinical study to substantiate such implications. It should be noted that CMS is an inactive prodrug and needs to be converted to active colistin in vivo (Bergen et al., 2006). The conversion of CMS to colistin after the inhalation is only 1.4% in the lung ELF (Boisson et al., 2014; Yapa et al., 2014). Although inhaled polymyxin therapies have been clinically used for several decades (e.g., in people with cystic fibrosis in Europe), it is worth noting that current inhaled polymyxin therapies have not been subjected to systematic PK/PD/TD optimization, and the dosage regimens are empirical (Velkov et al., 2015). Inhalation of injectable CMS and polymyxin B in the United States is of off-label use for the treatment of chronic MDR lung infections and has not been approved by the FDA (Moskowitz et al., 2008). Because of the current empirical dosing practice, excessive high doses of CMS have been used for nebulization (up to 6 MIU/day) and DPI (3.3 MIU/day) (Conole and Keating, 2014), which are comparable to the intravenous dosage regimens (Yapa et al., 2014). There is a lack of PK/PD targets for inhaled polymyxins even though the inhaled CMS products have been approved for many years in the United Kingdom and several European countries.
Recent PK/PD studies in the mouse lung infection model demonstrated that AUC/MIC in ELF is the most predictive PK/PD index for inhaled polymyxins (Lin et al., 2017a,b). However, because of the challenges in the inhalation dose translation from mice to humans and the lack of solid PK/PD data in patients, the AUC/MICELF target for various antibacterial magnitudes in patients is still unknown, and optimizing clinical dosage regimens for inhaled polymyxin B and CMS is urgently needed. Considering potentially different infection sites in the lungs, it may be prudent to use both intravenous and inhalation polymyxins for the treatment of pneumonia.
B. Bacteremia
The concern with high mortality rates associated with bacteremia has been escalated because of the rise of MDR Gram negative bacteria such as P. aeruginosa, A. baumannii, and K. pneumoniae (Anderson et al., 2006; Tumbarello et al., 2011; Nielsen, 2015; Zhou et al., 2019). A retrospective study investigated the impact of CMS dosage on the clinical and microbiological cure in 127 patients with bacteremia, 82 (65%) of whom received low-dosage regimens of CMS (≤4.4 mg CBA/kg per day), and 45 (35%) received high-dosage regimens (>4.4 mg CBA/kg per day) (Gibson et al., 2015). The high-dose CMS was significantly associated with greater 7-day clinical cure (40%) and microbiological cure (84%) and lower mortality rate (6.7%) as compared with the low-dose CMS (22%, 67%, and 22%, respectively) (Gibson et al., 2015). Similarly, the same group also reported the association between a higher CMS dose (2.9 mg CBA/kg per day (1.70–3.68)] with greater 7-day microbiological cure and lower 7-day mortality rate in a retrospective study with 76 patients with bacteremia at a large, academic, tertiary-care medical center in the United States between 2005 and 2010 (Vicari et al., 2013). Notably, CMS dose was not associated with the 28-day mortality rate in either study, whereas a higher CMS dose was associated with a higher rate of acute kidney injury (3.8 vs. 1.6 mg CBA/kg per day) (Vicari et al., 2013). Therefore, it is important to reconsider whether 28-day (or 30-day) mortality is an appropriate clinical endpoint for the evaluation of polymyxin efficacy in patients.
Another retrospective study from South Korea examined the effect of the desired target of colistin Css,avg on the efficacy and safety of intravenous CMS in 153 critically ill patients with pneumonia (112 patients; 73%) and bacteremia (41 patients; 27%) caused by P. aeruginosa and A. baumannii (Jung et al., 2019). A greater microbiological eradication in patients with bacteremia (28 patients; 58%) was observed as compared with those with pneumonia (20 patients; 42%). It should be noted that all 153 patients received intravenous CMS, and only 19 (12%) were given additional inhaled CMS therapy. Given that only intravenous CMS was administered in most of these pneumonia cases, this might lead to inadequate exposure of formed colistin in the lungs and lower bacterial eradication (Jung et al., 2019). Additionally, successful treatment of K. pneumoniae associated bacteremia was reported in another case study with intravenous CMS at 9 MIU per day in three divided doses (Karabinis et al., 2004). The impact of polymyxin B dosage regimens on in-hospital mortality was analyzed in a retrospective cohort study involving 276 patients receiving intravenous polymyxin B for ≥72 hours (Elias et al., 2010). Among 276 patients, 53 (19.2%) patients were presented with bacteremia caused by P. aeruginosa (32 patients; 60.4%) and A. baumannii (21 patients; 39.6%). Intravenous polymyxin B at dosage regimens of ≥200 mg per day resulted in lower mortality in patients with bacteremia (adjusted odds ratio 0.04; 95% confidence interval 0.006–0.28) but was associated with higher risk of severe renal impairment (Elias et al., 2010). Another retrospective study from the same group compared the efficacy of intravenous polymyxin B with other antibiotics for the treatment of P. aeruginosa bacteremia in 133 patients (Kvitko et al., 2011). In this cohort, 45 (34%) patients received polymyxin B, and 88 (66%) patients received other antibiotics. The mean average daily polymyxin B dose was 141 ± 54 mg in two divided doses, with 11 (24%) patients receiving a dose ≥200 mg per day. The inferiority of polymyxin B for the treatment of P. aeruginosa bacteremia was demonstrated, with a higher mortality associated with polymyxin B (30 of 45; 67%) in comparison with other antibiotics (25 of 88; 28%). Furthermore, a higher incidence of nephrotoxicity was revealed with the use of polymyxin B (Kvitko et al., 2011). A multicenter case study examined the risk factors and outcomes of extensively drug-resistant (XDR) A. baumannii bacteremia (Ng et al., 2014). For patients receiving polymyxin B, survivors at 30 days had been given a higher daily dose (median of 840,000 IU) of polymyxin B as compared with nonsurvivors (median of 700,000 IU) (Ng et al., 2014). Overall, higher concentrations of polymyxins have been associated with better clinical outcomes. However, increased concentrations have often resulted in renal impairment. These reports highlight the importance of using an appropriate dosage of polymyxins.
C. Urinary Tract Infections
UTI is one of the most common nosocomial infections with Enterobacterales, and carbapenems are often the first choice of therapeutic agents (Stamm and Norrby, 2001). Unfortunately, the increasing emergence of carbapenem-resistant Enterobacterales has limited the treatment options to polymyxins in many hospitals (Cassir et al., 2014; Dortet et al., 2014). The achievable concentrations of formed colistin and polymyxin B in urine are different in patients because of the different renal handling of CMS (mainly eliminated by the kidneys) from polymyxin B (extensively reabsorbed by the renal tubular cells) (see section V. Pharmacokinetics and Pharmacodynamics). A total number of 33 patients with symptomatic UTI (24 cystitis and 9 pyelonephritis) caused by P. aeruginosa were given intravenous CMS (5 ± 2.54 MIU per day) for at least 48 hours, 19 of whom (16 cystitis and 3 pyelonephritis) were treated with CMS monotherapy (Sorlí et al., 2019). A high clinical cure rate (17 patients; 90%) was achieved even though only five (30%) patients achieved the optimal plasma PK/PD target (fAUC/MIC ≥ 60), with one (6%) patient attaining the Css target in plasma (2.5 mg/l). Among patients receiving CMS monotherapy with clinical cure, five (30%) patients were presented with acute kidney injury. Based on the PK/PD of formed colistin in urine, lower CMS doses were suggested for the treatment of UTI with a reduced risk of nephrotoxicity (Sorlí et al., 2019). Another study also reported a high clinical cure of 83% with a low dose of intravenous CMS (1.9 ± 0.6 mg/kg; it is unclear whether the dose unit is mg CBA) for the treatment of UTI by MDR Gram negative bacteria; however, it was not stated whether CMS was used as monotherapy or in combination with other antibiotics (Zaidi et al., 2014).
In contrast to CMS, a much lower urinary recovery (<4%) was reported with intravenous polymyxin B in critically ill patients because of the low renal clearance (Zavascki et al., 2008; Sandri et al., 2013a). However, successful use of polymyxin B for the treatment of UTIs has been reported. Efficacious treatment of UTI with polymyxin B (1.5–2.5 mg/kg per day) was demonstrated among 92 solid organ transplant patients with 77.2% clinical cure and 100% microbiological cure (only 24 follow-up urine cultures from patients were obtained) (Mostardeiro et al., 2013). Notably, renal dysfunction was associated with the duration of polymyxin B treatment (Mostardeiro et al., 2013). Another retrospective study was performed with 87 cases of UTI caused by carbapenem-resistant K. pneumoniae, and 25 (29%) of these cases involved the use of polymyxin B (median of 2.25 mg/kg per day, range of 1.1–3.3 mg/kg per day) (Satlin et al., 2011). Polymyxin B demonstrated a superior microbiological clearance rate (64%) as compared with the untreated (36%) and tigecycline (43%) groups; however, it is inferior to aminoglycosides (88%) (Satlin et al., 2011). In another retrospective study, a clinical cure rate of 73% was observed among 23 patients treated with polymyxin B (1.5–2.5 mg/kg per day), including 10 patients with UTI caused by carbapenem-resistant K. pneumoniae (Dubrovskaya et al., 2013).
Bladder instillation is an alternative administration route for polymyxins to treat UTIs. A 7-day continuous intravesical administration of CMS in saline (3.5 mg/kg of CMS in 500 cm3) was effective in eradicating MDR A. baumannii while sparing the renal function in a patient (Volkow-Fernández et al., 2012). Efficacy of bladder instillation with CMS was also reported in three patients with UTI caused by MDR A. baumannii (100,000 IU in 50 ml via a single urinary catheter three times daily; 2 days for one patient and 7 days for two patients) (Giua et al., 2014). In another study with 17 patients with acute cystitis, an overall effectiveness rate (based on the degree of bacteriuria, pyuria, and pain on micturition) of 82% was observed with intravesical instillation of polymyxin B (Suzuki et al., 1989). Nonetheless, conflicting results were also presented in the literature showing that bladder irrigation with a combination of polymyxins and aminoglycosides (kanamycin-colistin/neomycin-polymyxin B) was ineffective (Pearman et al., 1988; Waites et al., 2006). With very limited pharmacological knowledge, intravesical instillation of polymyxins remains a potential alternative for the treatment of UTIs, especially in patients with severe renal impairment. Further investigations are warranted to optimize the administration method and dosing strategy of polymyxins for treating UTIs.
D. Central Nervous System Infections
Very limited therapeutic options are available for central nervous system (CNS) infections caused by MDR Gram negative bacteria, largely because of the blood-brain barrier. As intravenous polymyxins demonstrate poor CNS penetration, intrathecal and intraventricular routes have been employed to achieve high polymyxin concentrations in the CNS (Falagas et al., 2007; Jin et al., 2009, 2011). A case study examined intraventricular CMS for the treatment of ventriculitis caused by carbapenem-resistant A. baumannii in five patients with ventriculitis; two patients who received intraventricular CMS (5 mg twice daily) plus other intravenous antibiotics (sulbactam-tobramycin or tobramycin) survived, whereas the remaining three patients without intraventricular CMS died as a result of inadequate therapy (Fernandez-Viladrich et al., 1999). Similarly, intraventricular CMS at 125,000 IU (i.e., 10 mg) twice daily for 3 weeks successfully treated a patient with meningitis caused by MDR A. baumannii (Bukhary et al., 2005). Another successful case employed a lower daily dose of intrathecal CMS at 40,000 IU (i.e., ∼3.2 mg) for meningitis caused by A. baumannii (Benifla et al., 2004). Several other clinical studies showed that intrathecal and intraventricular CMS administration was effective against A. baumannii meningitis with daily doses of 3.2–20 mg (Vasen et al., 2000; Berlana et al., 2005; Al Shirawi et al., 2006) and P. aeruginosa ventriculitis, meningitis, and ventriculoperitoneal shunt infection with daily doses of 5–10 mg (Quinn et al., 2005; Yagmur and Esen, 2006; Baiocchi et al., 2010). The efficacy of intrathecal and intraventricular CMS with a daily dose of 125,000 IU was reported for the eradication of extended-spectrum β-lactamase (ESBL)-producing K. pneumoniae from the cerebrospinal fluid (Enfanto and Shin, 2007). A case series was conducted for the treatment of CNS infections caused by A. baumannii in 24 patients; 11 received intrathecal/intraventricular CMS [40,000–400,000 IU/day (3.2–32 mg/day)] as monotherapy, and 13 received coadministered systemic antibiotics (Khawcharoenporn et al., 2010). High clinical and microbiological cure rates were demonstrated in both groups of patients receiving intrathecal/intraventricular colistin monotherapy (91% and 91%, respectively) and combinations with systemic antibiotics (77% and 92%, respectively). Among the patients receiving intrathecal/intraventricular CMS, several patients experienced chemical ventriculitis (3 of 24; 13%) and treatment-associated seizures (1 of 24; 4%) (Khawcharoenporn et al., 2010). In another case series, six patients with meningitis and ventriculitis caused by XDR A. baumannii were successfully treated with intraventricular CMS with a loading dose of 500,000 IU (i.e., 40 mg) followed by 125,000–250,000 IU (i.e., 10–20 mg) every 24–48 hours in addition to intravenous CMS (Karaiskos et al., 2013). Notably, a patient (17%) developed chemical meningitis, and another one (17%) experienced chemical ventriculitis (Karaiskos et al., 2013). Several other clinical studies showed the efficacy of intrathecal/intraventricular CMS against difficult-to-treat CNS infections. A recurrent episode of meningitis was resolved only after the administration of the combination consisting of intraventricular amikacin, intraventricular CMS, and intravenous CMS for 6 weeks (Kasiakou et al., 2005b). The combination of intravenous and intrathecal/intraventricular CMS has been used by many clinicians as an effective and safe therapeutic option against CNS infections caused by MDR Gram negative bacteria, including in critically ill patients and children (Schina et al., 2006; Ho et al., 2007; Rodríguez Guardado et al., 2008; Dalgic et al., 2009; Ozdemir et al., 2010; Ziaka et al., 2013). There is very limited PK information on polymyxins after intrathecal/intraventricular administration in patients. In a study with nine patients, after administration of intraventricular CMS at 2.61–5.22 mg per day, cerebrospinal fluid concentrations of formed colistin were above 5.41 mg/l over 72 hours, which cannot be achieved with intravenous administration (Imberti et al., 2012). In addition, colistin did not accumulate in the CSF during this period, and intraventricular administration of CMS at doses of ≥5.22 mg per day seems appropriate in patients considering the PK/PD and safety of colistin. Given that the variable external cerebrospinal fluid efflux may affect the clearance of colistin, this study supports the recommended daily dose of 125,000 IU CMS (i.e., 10 mg), as suggested by the Infectious Diseases Society of America (Imberti et al., 2012).
In a systemic review (64 episodes), a high cure rate of 80% (51 of 64) was reported for treatment of Gram negative bacterial meningitis with intrathecal/intraventricular polymyxins, in which intrathecal polymyxin B was used in almost all episodes prior to 1974 (Falagas et al., 2007). Polymyxin B was intrathecally/intraventricularly administered with a daily dose of 50,000 IU (i.e., 5 mg) for most adults, and a CMS daily dose of ≥62,500 IU (i.e., 5 mg) was commonly used. A wide range of 5000–120,000 IU (in two divided doses) of polymyxins (CMS and polymyxin B) was reported for pediatric patients (Falagas et al., 2007). Additional case reports have also supported the use of intrathecal/intraventricular polymyxin B at a daily dose of 50,000 IU (i.e., 5 mg) in adults for the treatment of meningoencephalitis and ventriculitis caused by A. baumannii and P. aeruginosa (Macedo et al., 2011; Guo et al., 2018), whereas a lower dose of 40,000 IU (i.e., 4 mg) on alternate days was effective in treating neonatal meningoventriculitis caused by MDR A. baumannii (Piparsania et al., 2012). Unfortunately, there is still a lack of information on the cerebrospinal fluid PK of polymyxin B in patients.
Overall, the current literature on the treatment of meningitis and ventriculitis caused by MDR Gram negative bacteria supports a daily dosage of intrathecal/intraventricular CMS at 125,000 IU (i.e., 10 mg or 4.1 mg CBA) or polymyxin B at 50,000 IU (i.e., 5 mg) with concomitant intravenous administration of polymyxins. Additionally, CMS appears in favor over polymyxin B because of more clinical experience. Nonetheless, more clinical data on the safety and PK/PD/TD of intrathecal/intraventricular polymyxins are in need to optimize their dosage recommendations (Tsuji et al., 2019).
E. Selective Decontamination of Mucosal Surface
Selective digestive tract decontamination (SDD) and selective oropharyngeal decontamination (SOD) are preventive measures to reduce the incidence of nosocomial infections and ultimately decrease the mortality rate, especially among intensive care unit (ICU) patients. Topical forms of polymyxins, mostly colistin sulfate, have been commonly used as part of SDD and SOD regimens. The SDD and SOD approaches with polymyxins have been demonstrated to reduce mortality (Kerver et al., 1988; Rocha et al., 1992; Tetteroo et al., 1993; Luiten et al., 1995; Sánchez García et al., 1998; de Jonge et al., 2003; de Smet et al., 2009b; Melsen et al., 2012) and decrease the incidence of infections (Cockerill et al., 1992; Camus et al., 2005), bacteremia (Oostdijk et al., 2011), VAP (Pugin et al., 1991; Bergmans et al., 2001; Pneumatikos et al., 2002; Koeman et al., 2006), pneumonia (Stoutenbeek et al., 1987; Godard et al., 1990; Rodríguez-Roldán et al., 1990; Abele-Horn et al., 1997), and pancreatic infections (Luiten et al., 1995). However, SDD and SOD might also be associated with a high rate of post-ICU incidence of hospital-acquired infections (de Smet et al., 2009a). For pediatric patients, SDD has been reported effective in reducing Gram negative infections (Smith et al., 1993) and urinary tract and respiratory infections (Ruza et al., 1998); however, in severely burned pediatric patients, SDD was found ineffective in decreasing bacterial colonization and incidence of infection (Barret et al., 2001). The regimens that have been reported consist of topical amphotericin B, tobramycin, polymyxins, gentamicin, vancomycin, neomycin, nystatin, and netilmicin, usually in triple combinations (Daneman et al., 2013). Among these topical agents, colistin, tobramycin, and amphotericin B have been frequently used (Daneman et al., 2013). Although previous studies reported insignificant association between the use of SDD and SOD with increased antimicrobial resistance, concerns remain in regions where a high prevalence of MDR bacteria has been detected (Krueger et al., 2002; Heininger et al., 2006; de Smet et al., 2011; Ochoa-Ardila et al., 2011). Worryingly, the emergence of polymyxin-resistant Gram negative bacteria has been reported with the use of colistin for selective decontamination (Halaby et al., 2013; Lübbert et al., 2013). The administration of SDD to ICU patients has also been demonstrated to affect the gut microbiome by reducing the abundance of Enterobacterales, anaerobic Gram positive, butyrate-producing Clostridium clusters IV and XIVa bacteria, while elevating the abundance of Bacteroidetes and enterococci; however, the impact of altered microbiome on the physiologic condition of patients is currently unknown and requires further investigations (Benus et al., 2010; Buelow et al., 2017).
F. Ocular and Otologic Infections
The topical form of polymyxins has long been used for the treatment of ocular infections. An early study in 1969 reported successful treatment of Pseudomonas ocular infections using ophthalmic colistin sulfate (1.2 mg CBA/ml; average dose of one to two drops every one-half to 1 hour) with satisfactory response of 89% and minimal adverse effects (Lund, 1969). In 1991, a study was performed to compare the efficacy of 1% tetracycline ointment, combination of 1% tetracycline ointment and 0.1% dexamethasone eye drop, and Eubetal ointment (tetracycline 0.5%, dexamethasone 0.1%, chloramphenicol 1%, and colistin) for the treatment of chlamydial conjunctivitis (Maichuk, 1991). Similar efficacy was reported for both tetracycline-dexamethasone combination and Eubetal ointment, and they were more effective than tetracycline alone (Maichuk, 1991). In another study, the combination of polymyxin B (10,000 IU/g) and bacitracin (500 U/g) ointment enhanced bacterial eradication during the treatment of bacterial conjunctivitis (Gigliotti et al., 1984). Polymyxin B-trimethoprim has been demonstrated to be effective for conjunctivitis, with comparable efficacy to chloramphenicol (Behrens-Baumann et al., 1988), moxifloxacin (Williams et al., 2013), and polymyxin B-neomycin-gramicidin (Genée et al., 1982). Similar to ophthalmic formulations, polymyxins are often used in combination with other antibiotic(s) and corticosteroid for otologic infection, with the combination of polymyxins, neomycin, and hydrocortisone being the typical formulation. Polymyxin B-neomycin-hydrocortisone has been shown effective for acute otitis externa with comparable efficacy to ciprofloxacin, but it is inferior to ciprofloxacin-dexamethasone (Rahman et al., 2007; Drehobl et al., 2008).
G. Bone and Joint Infections
Because of the poor penetration of colistin into bones after parenteral administration, studies in the 1970s and 1980s reported the use of colistin by incorporation into orthopedic cement (Rosenthal et al., 1976; Murray, 1984). Successful treatment and prevention of deep-wound infection with the use of colistin-erythromycin–containing cement has been demonstrated (Waterman et al., 2012). Although the release of CMS from cement beads was adequate for the inhibition of bacterial growth in vitro, data on the appropriate concentrations for clinical use are lacking (Waterman et al., 2012). The current clinical experience with the use of intravenous polymyxins for bone- and joint-associated infections is limited. A case report of total knee arthroplasty infected by MDR P. aeruginosa was successfully treated with surgical removal of prosthesis, colistin-loaded cement, and intravenous CMS (Papagelopoulos et al., 2007). An initial CMS dose of 3 MIU (i.e., 240 mg) every 8 hours was given for 7 days, and the dose was adjusted to 2 MIU (i.e., 160 mg) every 6 hours for 5 weeks with the intention to prevent renal impairment. During the course of intravenous CMS, the creatinine level decreased to 2.2 mg/dl, which fortunately reverted to the baseline after discontinuation of the treatment (Papagelopoulos et al., 2007). Two cases of fixation device–related orthopedic infections by MDR A. baumannii were successfully treated with intravenous CMS [1 MIU (i.e., 80 mg) bolus followed by 6 MIU (i.e., 480 mg) in a 24-hour continuous infusion for 36 days or 2 MIU (i.e., 160 mg) every 8 hours for 22 days], and no colistin-associated toxicity was observed (Kasiakou et al., 2005a). In a retrospective study involving 19 patients, prolonged intravenous CMS at 50,000 IU/kg per day (i.e., 4 mg/kg per day) was reported to be effective against bone and joint infections caused by difficult-to-treat Gram negative bacteria (i.e., P. aeruginosa and Enterobacter spp.); however, a higher dose of 75,000 IU/kg per day (i.e., 6 mg/kg per day) might be required for implant-associated infections (Valour et al., 2013). Another retrospective study involving 34 patients reported success in treating difficult-to-treat osteoarticular infections with appropriate surgical treatment (implant removal) using the combination of CMS [2.8–6 MIU/day (i.e., 224–480 mg/day)] and β-lactam (Ribera et al., 2015). Overall, limited clinical data support the treatment of joint and bone infections with polymyxins; however, the optimal dosage regimens should be investigated with well designed prospective clinical PK/PD/TD studies.
In summary, preclinical and clinical PK/PD/TD of polymyxins, particularly at the infection sites, require extensive investigations that will provide the pharmacological evidence to develop scientifically based dosage regimens for different types of infections in patients.
VIII. Polymyxin Toxicity
Dose-dependent nephrotoxicity is the most reported adverse event of intravenous polymyxins, with 30%–60% occurrence in patients (Holmes, 1964; Ryan et al., 1969; Brown et al., 1970; Koch-Weser et al., 1970; Price and Graham, 1970; Devlieger et al., 1977; Evans et al., 1999; Falagas and Kasiakou, 2005, 2006; Li et al., 2005a; Pastewski et al., 2008; Elias et al., 2010; Garonzik et al., 2011; Kvitko et al., 2011; Dezoti Fonseca et al., 2012; Kubin et al., 2012; Mingeot-Leclercq et al., 2012). In critically ill patients, acute kidney injury can lead to high mortality. Intravenous polymyxins can also cause neurotoxicity and skin hyperpigmentation in patients, and inhalation can lead to toxicities in the respiratory tract (Grill and Maganti, 2011; Li et al., 2020b).
A. Nephrotoxicity
Acute kidney injury may occur with intravenous polymyxins in patients (e.g., within a week); fortunately, it can revert after the discontinuation of treatment (Pogue and Tam, 2019). Key clinical characteristics of polymyxin-associated nephrotoxicity include renal tubular damage, decreased glomerular filtration rate, and creatinine clearance, as well as increased serum urea and creatinine concentrations (Falagas and Kasiakou, 2005). As reviewed in section V. Pharmacokinetics and Pharmacodynamics, very extensive tubular reabsorption of polymyxin B and colistin occurs in the kidney (Zavascki et al., 2008; Abdelraouf et al., 2012; Manchandani et al., 2016; Sivanesan et al., 2017b), which plays a critical role in their nephrotoxicity. Molecular imaging [e.g., immunostaining, mass spectrometry imaging, fluorescence microscopy, and X-ray fluorescence microscopy (XFM)] revealed significant accumulation of polymyxins in renal tubules (Azad et al., 2015c; Manchandani et al., 2015; Nilsson et al., 2015; Yun et al., 2015a,b; Vattimo et al., 2016; Velkov et al., 2016b). Preferential accumulation of polymyxins in mouse renal cortex was observed by matrix-assisted laser desorption/ionizing mass spectroscopy imaging after subcutaneous administration of polymyxins (Azad et al., 2015b). Similarly, a study with polymyxin-specific monoclonal antibody–based immunostaining revealed substantial accumulation of polymyxins in renal cortex, particularly in the renal proximal tubular cells in mice treated with polymyxin B, and very low accumulation was observed in the cells from distal tubules (Yun et al., 2015a). No major accumulation was detected in the other organs, such as lungs, liver, or heart, in mice (Azad et al., 2015b).
A novel dual-module fluorescent probe, FADDI-96, was developed based on the polymyxin structure-activity relationship model (Velkov et al., 2010). This probe possesses a dansyl group in the N-terminus and an iodine fluorophore at position 6 d-Phe of polymyxin B and exerts comparable antibacterial activity and renal epithelial cell toxicity as polymyxin B. It should be noted that the commercially available fluorescent polymyxin probes BODIPY-polymyxin B and dansyl-polymyxin B are devoid of these pharmacological properties, as the modification of Dab residues with the fluorophores damages the structure-activity relationship of polymyxins (Velkov et al., 2010, 2013; Azad et al., 2014, 2015c). A correlative microscopic study employed synchrotron-based XFM, fluorescence microscopy, and scanning electron microscopy to reveal the remarkable dose- and time-dependent accumulation (∼2000- to 4760-fold) of polymyxins (as FADDI-96) in rat and human kidney proximal tubular NRK-52E and HK-2 cells, respectively (Azad et al., 2015c). The current literature indicates that the uptake of polymyxins in renal tubular cells is transporter-mediated (Yousef et al., 2012; Azad et al., 2015c; Sivanesan et al., 2017a).
Megalin, a key endocytic receptor in renal tubules, plays an important role in the accumulation of polymyxins in renal tubular cells (Suzuki et al., 2013; Eshbach and Weisz, 2017; Hori et al., 2017; Manchandani et al., 2017). Binding of cytochrome C, a megalin substrate, to megalin was competitively inhibited by colistin (Suzuki et al., 2013). A decreased accumulation of colistin in rat kidneys and increased urinary excretion in megalin-shed rats indicate the important role of megalin in tubular reabsorption of colistin (Suzuki et al., 2013). Coadministration of cytochrome C, fragment of albumin, or succinylated bovine gelatin polypeptides with colistin decreased the accumulation of colistin in the kidneys and reduced tubular damage marker N-acetyl-β-d-glucosaminidase in urine, indicating the attenuation of polymyxin-induced tubular damage by decreasing the megalin-mediated uptake (Suzuki et al., 2013; Hori et al., 2017; Sivanesan et al., 2017a). Members of the organic anion transporters (Jacquemin et al., 1994; Abe et al., 1998) and organic cation transporters (Busch et al., 1996; Okuda et al., 1996) have been discovered in kidney tubular cells and effectively mediate the transport of a diverse range of anions and cations. An ex vivo study using isolated perfused rat kidney demonstrated substantial tubular reabsorption of colistin from the perfusate (approximately 90%), suggesting the carrier-mediated renal tubular reabsorption (Ma et al., 2009); furthermore, this reabsorption was significantly inhibited by tetraethylammonium (substrate of organic cation transporter OCTN1), hydrogen ions, and glycine-glycine (substrate of peptide transporters) (Shen et al., 1999; Ma et al., 2009). In a recent study, human peptide transporter 2 (PEPT2) was demonstrated to mediate the uptake of polymyxins in HEK-293 cells (Lu et al., 2016). The human PEPT2-specific substrate [3H]glycylsarcosine was used, and the IC50 values of colistin and polymyxin B were 11.4 ± 3.1 µM and 18.3 ± 4.2 µM, respectively. Similar results were observed with [3H]polymyxin B1 and a fluorescent polymyxin probe MIPS-9541 when determining the PEPT2-mediated uptake of polymyxins in HEK-293 cells (Lu et al., 2016).
Information on the intracellular localization of polymyxins in renal tubular cells is limited. A confocal fluorescence imaging study with regioselectively labeled monodansylated polymyxin B probes (MIPS-9543 and MIPS-9544) revealed partial colocalization of polymyxins within the endoplasmic reticulum and mitochondria in rat NRK-52E cells (Yun et al., 2015b). High-resolution XFM imaging indicated preferential accumulation of polymyxins around the nuclear region compared with the cytoplasmic areas in rat NRK-52E and human HK-2 kidney proximal tubular cells (Azad et al., 2015c). Overall, deciphering the mechanisms of intracellular uptake and disposition of polymyxins in kidney tubular cells is critical for the optimization of their parenteral use, amelioration of nephrotoxicity, and the discovery of new-generation, safer polymyxins.
The present literature highlights the key role of DNA damage, metabolic and inflammatory perturbations, oxidative stress, cell cycle arrest, apoptosis, and autophagy in polymyxin-induced death of renal tubular cells in vitro and in vivo. A recent study showed the formation of micronuclei and multilobed nuclei, chromosome mis-segregation, and genome instability in HK-2 cells and mouse kidneys after polymyxin B treatment (Yun et al., 2018). Increased expression of kidney γH2AX (phosphorylated H2A histone family member X) suggested persistent DNA damage in kidneys by polymyxins. A transient cell cycle arrest followed by continuation with unrepaired DNA damage was speculated as a result of checkpoint perturbations by polymyxin B treatment (Yun et al., 2018). Intraperitoneal administration of colistin (16 mg/kg per day in two divided doses) caused the upregulation of CCNB1 and CDC2 genes in mouse kidneys, suggesting cell cycle arrest at the G2/M phase (Eadon et al., 2013). Furthermore, the translocation of cyclin B1 to the nucleus is consistent with the observed cell cycle arrest at the G2/M phase, and the upregulation of galectin-3 gene also highlighted the G1/S and G2/M as the points of arrest in the cell cycle (Eadon et al., 2013). A 3-day colistin treatment resulted in increased expression of proliferating cell nuclear antigen, which suggested the inhibition of DNA replication at S phase and kidney injury (Eadon et al., 2013). In addition, the activation of p53 and galectin-3 by colistin indicated the progression of the cell to the apoptotic pathways with unrepairable cellular damage (Sutton et al., 2013; Chen and Kuo, 2016).
Several studies revealed that polymyxins induced renal tubular apoptosis both in vitro and in vivo (Ozyilmaz et al., 2011; Yousef et al., 2011, 2012). Colistin treatment (a cumulative dose of 20.5 mg/kg over 5 days) caused an increase of terminal dUTP nick end labeling (TUNEL)-positive nuclei in rat kidneys and fragmentation of DNA, a biochemical signature of apoptosis (Yousef et al., 2012). Similar results were observed in rat and human proximal tubular cells treated with polymyxin B (Azad et al., 2013; Yun et al., 2018). Intravenous administration of colistin in mice (7.5 or 15 mg/kg per day in two doses for 7 days) caused a significant increase of cytochrome C, apoptosis-inducing factor, cleaved caspase-9, and cleaved caspase-3 with a simultaneous decrease of B-cell lymphoma (Bcl-2) (Dai et al., 2014). Furthermore, a significant increase in the expression of Fas, FasL, and Fas-associated death domain (FADD) and the cleavage of caspase-8 was observed. These results indicated the involvement of both death receptor and mitochondrial apoptotic pathways in polymyxin-induced apoptosis, and the increased expression of truncated Bax-inhibiting peptide (tBid) suggested the crosstalk between both cell death pathways. In addition, the endoplasmic reticulum pathway was also involved in colistin-induced apoptosis, as significant upregulation of glucose-regulated protein 78 kDa/Bax-inhibiting peptide (Grp78/Bip), cleaved activating transcription factor 6 (ATF6), DNA damage inducible gene 153/C/EBP-homologous protein (GADD153/CHOP), and caspase-12 was evident in colistin-treated mice (Dai et al., 2014).
In vitro cell culture studies using rat NRK-52E and human HK-2 kidney tubular cells demonstrated both concentration- and time-dependent apoptosis after the treatment with polymyxins (Azad et al., 2013, 2015c). NRK-52E cells showed a higher resistance to polymyxin B–induced cytotoxicity in comparison with HK-2 cells. In NRK-52 cells, polymyxin B treatment caused the concentration-dependent activation of caspase-3, -8, and -9; DNA damage; and translocation of membrane phosphatidylserine (Azad et al., 2013, 2015a). Furthermore, in NRK-52E cells, polymyxin B also caused concentration- and time-dependent mitochondrial damage, including mitochondrial morphology changes from filamentous to fragmented, loss of mitochondrial membrane potential, and generation of ROS (Azad et al., 2015a). A recent metabolomics study with NRK-52E cells treated with polymyxin B showed depletion of cellular metabolites taurine and hypotaurine, suggesting the oxidative stress induced by polymyxins (Azad et al., 2015b). Using transcriptomics, perturbations in both inflammatory and metabolic pathways were observed in polymyxin-induced damage in NRK-52E cells, as evidenced by the key differentially expressed genes Cxcl10, Cxcl1, Ccl2, Ccl20, Tnf, Tpi1, Pfkl, Ldh-A, and Gapdh (Azad et al., 2018).
There is limited information in the literature on the chemical biology of polymyxin-induced nephrotoxicity. The toxic effect of major components of polymyxin B (polymyxin B1 and polymyxin B2) and colistin (colistin A and colistin B) (Table 1) was assessed in vitro and in vivo (Roberts et al., 2015). The mild to moderate histologic damage observed in the mouse kidney treated with all four major components of polymyxins is comparable. However, polymyxin B1 and colistin A induced a >3-fold higher apoptotic effect on HK-2 cells than polymyxin B2 and colistin B. As there is only one methylene (-CH2-) group difference in the N-terminal fatty acyl group of polymyxin B1 and B2 and colistin A and B (Table 1), the hydrophobicity of this N-terminal fatty acyl group is critical in polymyxin-induced apoptosis. Furthermore, >2-fold differences in EC50 of polymyxin B and colistin indicate the importance of hydrophobicity at position 6 for polymyxin toxicity in renal tubular cells, considering that the only difference between polymyxin B and colistin is at position 6 (i.e., d-Phe vs. d-Leu) (Roberts et al., 2015; Azad et al., 2018). A recent molecular dynamic simulation study revealed the structure-interaction relationship of polymyxins with the membrane of human kidney proximal tubular cells at the atomic level and highlighted that the hydrophobicity at positions 6/7 and stereochemistry at position 3 regulated the interactions of polymyxins with the cell membrane (Jiang et al., 2020c).
Overall, recent studies on the mechanism of polymyxin-induced nephrotoxicity indicate a complex interplay of multiple biochemical processes in renal tubular cells (Fig. 8) (Dai et al., 2014), and further studies using systems pharmacology and chemical biology are warranted to decipher the kinetics of this interplay.

The proposed mechanisms of polymyxin-induced apoptotic cell death in renal tubular cells. AIF, apoptosis-induced factor; CAT, catalase; CDK2, cyclin-dependent kinase 2; cytC, cytochrome C; MAPK, mitogen-activated protein kinase; MDA, malondialdehyde; MR, endocytic receptor megalin; OCTN1, organic cation transporter 1; SOD, superoxide dismutase; tBid, truncated Bax-inhibiting peptide . Reproduced with permission (Dai et al., 2014). Copyright © American Society for Microbiology.
To alleviate polymyxin-associated nephrotoxicity in patients, the importance of optimization of the polymyxin dose, therapeutic drug monitoring, electrolyte balance, coadministration of nephrotoxic drugs, and early detection of kidney injury is paramount (Hartzell et al., 2009; Zavascki and Nation, 2017). Furthermore, several other approaches have been investigated to attenuate polymyxin-induced nephrotoxicity, including inhibiting the accumulation in renal tubular cells, coadministration of antioxidants to counteract polymyxin-induced oxidative stress, and structural modification of the polymyxins (Ma et al., 2009; Velkov et al., 2010; Ozyilmaz et al., 2011; Yousef et al., 2011, 2012; Ozkan et al., 2013; Azad et al., 2017a; Sivanesan et al., 2017a). A number of animal studies investigated the protective effect of antioxidants such as N-acetylcysteine, ascorbic acid, melatonin, grape seed proanthocyanidin extract, polyaspartic acid, and methionine on polymyxin-induced nephrotoxicity (Ozkan et al., 2013; Ozyilmaz et al., 2011; Yousef et al., 2011, 2012; Azad et al., 2017a,b). Methionine (100 or 400 mg/kg) attenuated polymyxin-induced kidney damage in mice (polymyxin B 35 mg/kg, twice daily over 3.5 days) and also significantly reduced mitochondrial oxidative stress in NRK-52E cells treated with polymyxin B (Azad et al., 2017a). Importantly, methionine did not alter the PK of polymyxin B in rats (Azad et al., 2017a). Similar protective activity by grape seed proanthocyanidin extract was observed in rats (Ozkan et al., 2013). Interestingly, coadministration of succinylated bovine gelatin polypeptides (Gelofusine) did not alter the pharmacokinetics of colistin in rats but decreased the accumulation of polymyxins in renal tissue and nephrotoxicity (Sivanesan et al., 2017a).
Information on the protection of polymyxin-associated nephrotoxicity in patients is scarce. A preliminary randomized controlled study with only 28 patients did not show the nephroprotective effect with ascorbic acid (2 g every 12 hours, intravenously) in patients treated with intravenous CMS (Sirijatuphat et al., 2015). No difference in the PK of formed colistin in plasma was observed in both groups. The low number of patients, administered doses of ascorbic acid, and the lack of resemblance of animal models to patients could account for the absence of protective effect from ascorbic acid against polymyxin-associated nephrotoxicity in patients (Sirijatuphat et al., 2015). In contrast, a prospective, observational cohort study revealed strong independent renal protection by intravenous ascorbic acid [3 (2–4) g every 12 hours] in critically ill patients treated with CMS (Dalfino et al., 2015). In the group without acute kidney injury, 76.9% (30 of 39) patients received concurrent ascorbic acid, whereas in the patients with acute kidney injury, the value was 41.9% (13 of 31); higher risk of acute kidney injury and greater probability of earlier onset of acute kidney injury were identified in the group without ascorbic acid as the adjuvant to CMS therapy (Dalfino et al., 2015). Several potential limitations were demonstrated in this study, including the small sample size, absence of randomization, and no characterization of patients between the groups (Dalfino et al., 2015). Nevertheless, well designed clinical studies are required to develop novel approaches to attenuate polymyxin-induced nephrotoxicity.
Detecting the early onset of acute kidney injury after intravenous administration of polymyxins is critical to alleviate polymyxin-induced nephrotoxicity in patients. Importantly, early biomarkers have been investigated in animal studies to predict polymyxin-induced kidney injury. Recently, a urinary proteomics study with colistin-treated mice identified multiple proteins, such as complements (C3 and CB-4), carbonic anhydrase 3, hexokinase, and kininogen-1, as the potential biomarkers for early detection of polymyxin-induced acute kidney injury (Sivanesan et al., 2017c). Furthermore, a correlation between polymyxin-induced kidney injury and urinary levels of kidney injury molecule-1 (KIM-1), neutrophil gelatinase-associated lipocalin, and α-glutathione S-transferase in rodents was demonstrated (Burt et al., 2014; Keirstead et al., 2014). Normalized urinary KIM-1 (ratio between urinary KIM-1 and creatinine) in patients appeared more sensitive for early prediction of acute kidney injury after polymyxin B treatments (25,000 U/kg loading dose and 12,500–15,000 U/kg per 12 hours) as compared with the level of serum creatinine (Babic et al., 2017). Comprehensive clinical validations are warranted to minimize polymyxin-induced nephrotoxicity in patients.
Overall, it is evident that polymyxin-induced nephrotoxicity involves substantial accumulation in renal tubular cells, oxidative stresses, induction of DNA damage, and apoptosis. Further systems and molecular investigations are required to elucidate the precise mechanisms of polymyxin-induced nephrotoxicity, which will not only facilitate the optimization of their clinical use but also expediate the discovery of next-generation polymyxins with better safety and efficacy.
B. Neurotoxicity
In old literature, neurotoxicity was also often associated with the use of polymyxins in patients (Koch-Weser et al., 1970). The incidence rate of neurotoxicity associated with polymyxins in patients is approximately 7% or less and substantially lower than that of nephrotoxicity (Bosso et al., 1991; Falagas and Kasiakou, 2006). The clinical features of polymyxin-induced neurotoxicity are often minor and include paresthesia, apnea, nausea and vomiting, neuropathy, myopathy, dizziness, psychosis, and seizures (Koch-Weser et al., 1970; Falagas and Kasiakou, 2006). One of the major risk factors associated with neurotoxicity is the extended exposure (duration and concentration) to polymyxins (Wolinsky and Hines, 1962; Falagas and Kasiakou, 2006). Other risk factors include the presence of medical conditions such as myasthenia gravis, renal impairment, and hypoxia; concomitant use of other medications (e.g., sedatives, anesthetics, narcotics, and muscle relaxants); and gender (Koch-Weser et al., 1970; Decker and Fincham, 1971). The onset of neurotoxicity usually occurs shortly after intravenous infusion (e.g., transient paresthesia and apnea) or within the first few days of polymyxin therapy (Koch-Weser et al., 1970; Decker and Fincham, 1971; Falagas and Kasiakou, 2006). Polymyxin-induced neurotoxicity is generally reversible with discontinuation of the therapy (Falagas and Kasiakou, 2006; Dalfino et al., 2012) or via a longer infusion duration. The exact mechanism of polymyxin-induced neurotoxicity is not clear, and the literature suggests that it could involve the inhibition of acetylcholine, acetylcholine release, prolonged depolarization, and calcium depletion (Falagas and Kasiakou, 2006).
Polymyxins can accumulate in neuron cells, generate mitochondrial ROS, and induce oxidative stress (Jin et al., 2009, 2011, 2012, 2013; Dai et al. 2016, 2018a,b, 2019; Wang et al., 2016). Similar to polymyxin-induced nephrotoxicity and pulmonary toxicity (Ahmed et al., 2017), polymyxin-induced neurotoxicity also involves autophagy. Both in vitro and in vivo results demonstrate the induction of substantial autophagy in neuron cells during polymyxin treatment as a protective mechanism against apoptosis (Dai et al., 2016, 2019). Polymyxin-induced autophagy is regulated by a complex signal network among the phosphatidylinositol-3-kinase/Akt, mitogen-activated protein kinase, nuclear factor-κB, p53, mammalian target of rapamycin, and nuclear factor erythroid 2-related factor 2/heme oxygenase-1 pathways (Dai et al., 2013, 2016, 2018a,b; Barcelos et al., 2016). Repurposing of clinically available drugs and food supplements such as minocycline, rapamycin, salidroside, and curcumin has shown to be protective against polymyxin-induced neurotoxicity in vitro and in animals (Dai et al., 2013, 2018a,b; Barcelos et al., 2016; Lu et al., 2017). Clinical evidence is required to develop novel combination therapies of polymyxins with neuroprotective agents to ameliorate the neurotoxicity (Dai et al., 2019). Additionally, several other approaches could potentially minimize polymyxin-induced neurotoxicity in patients, including early detection of toxicity and discontinuation of therapy, use of therapeutic drug monitoring, and employment of specific biomarkers (Jin et al., 2009, 2011; Dalfino et al., 2012; Justo and Bosso, 2015).
C. Skin Hyperpigmentation
Recent case reports highlight skin hyperpigmentation after treatments with intravenous polymyxin B in patients (Mattos et al., 2016, 2017; Zheng et al., 2018; Li et al., 2020b); however, no such reports have been published on CMS/colistin to date. Polymyxin B–induced skin hyperpigmentation has been reported in approximately 8%–15% of the subjects (Mattos et al., 2016, 2017; Zheng et al., 2018; Li et al., 2020b). The onset of skin hyperpigmentation can be noticed as early as 3 to 4 days after the initiation of polymyxin B treatment with the appearance of red rashes that gradually darken as the treatment progresses (Lahiry et al., 2017). For patients suffering from hyperpigmentation, skin color can be darkened to various shades of brown, particularly in the neck, face, and upper part of the body (Mattos et al., 2016); these symptoms have also been reported on the skin of lower body parts and limbs (Zheng et al., 2018). To date, polymyxin B–induced hyperpigmentation has been observed in patients of various age groups, ranging from neonates to adults (Gothwal et al., 2016; Zheng et al., 2018).
The mechanism of polymyxin B–induced skin pigmentation is not clear. Several possible mechanisms have been proposed, including 1) the production and accumulation of melanin within cutaneous cells, 2) accumulation of polymyxin B within the extracellular matrix, 3) synthesis of pigments such as lipofuscin, 4) deposition of iron and red blood cells from damaged dermal vessels, 5) extensive inflammation due to the release of histamine by polymyxin B, 6) increased proliferation of epidermal Langerhans cells and dermal dendritic cells, and 7) low expression of interleukin-6 after polymyxin B treatment (Miori et al., 1990; Voitenko et al., 1990; Fechner et al., 1993; Marone et al., 2003; Choi et al., 2012; Zavascki et al., 2015, 2016; Mattos et al., 2016, 2017). Decreasing the dose or discontinuation of the treatment allows gradual restoration of skin color to normal state (Zavascki et al., 2015; Zavascki et al., 2016). Clearly, position 6 d-Phe plays an important role in polymyxin B–induced skin pigmentation, as this is the only difference in the structure from colistin (i.e., d-Leu at position 6).
D. Pulmonary Toxicity
Inhalation of polymyxins has been widely used in people with cystic fibrosis for several decades and appears to be a safe and effective alternative to intravenous administration for the treatment of lung infections caused by MDR Gram negative pathogens (section V. Pharmacokinetics and Pharmacodynamics) (Min et al., 2018). However, adverse events can occur in approximately 10%–20% patients treated with inhaled polymyxins, and the common symptoms include cough, throat irritation, bronchospasm, bronchoconstriction, and abnormal taste (Marschke and Sarauw, 1971; Le Brun et al., 2002; Alothman et al., 2005; Falagas and Kasiakou, 2006; McCoy, 2007; Pereira et al., 2007; Wunsch et al., 2012; Velkov et al., 2015; Min et al., 2018; Biagi et al., 2019). A recent study reported the occurrence of pulmonary eosinophilia after inhalation of 75 mg CMS in an 83-year-old woman with a history of chronic bronchiectasis (Lépine et al., 2017). Hypersensitivity pneumonitis and eosinophilia were reported in a 69-year-old woman after treatment with a high dose of CMS [2 MIU (i.e., 160 mg) every 8 hours] (Leong et al., 2010). In another case study, a 29-year-old woman with cystic fibrosis and acute airway infection with P. aeruginosa developed acute respiratory distress syndrome after inhalation of 75 mg CMS twice daily (McCoy, 2007). In patients with cystic fibrosis (children and adults), inhaled CMS could induce chest tightness and a transient decrease in the respiratory function (Dodd et al., 1997; Cunningham et al., 2001; Alothman et al., 2005). Bronchoconstriction and decreased respiratory function are more commonly associated with inhalation of colistin sulfate than CMS (Westerman et al., 2004). Because of the potential conversion of CMS to colistin in vitro and sterility consideration, reconstituted CMS solution should be used within 24 hours to reduce any potential pulmonary toxicity, as recommended by the FDA and product information (Biagi et al., 2019). Prior to the use of inhaled polymyxins in patients, clinicians should consider the risk factors associated with pulmonary toxicity, such as the form of polymyxins (CMS or polymyxin B) administered, stability of the product, delivery techniques (nebulization or dry powder inhalation), and the presence of comorbidities.
The mechanism of polymyxin-induced pulmonary toxicity in patients is unknown. It is believed to be associated with local irritation of airways and histamine release (Marschke and Sarauw, 1971). In old literature, in vitro degranulation of mast cells and histamine release were reported in the presence of polymyxin B and colistin (Jasani et al., 1979). Recent studies evaluated the toxicity of polymyxins in human lung epithelial A549 cells and revealed the concentration- and time-dependent mitochondrial damage, such as the loss of mitochondrial membrane potential, mitochondrial fragmentation, generation of mitochondrial ROS, and activation of apoptotic pathways, by polymyxins at concentrations >2,000 mg/l (Ahmed et al., 2017). Colocalization of polymyxins with mitochondria supports its critical role in polymyxin-induced toxicities in lung epithelial cells (Azad et al., 2015c; Ahmed et al., 2019). Recent mouse studies showed that the ELF concentration after aerosolized polymyxins reached 607 mg/l and induced only minor lung inflammation (Lin et al., 2017a,c). In a recent PK study with 10 critically ill patients, the mean ELF concentrations of formed colistin were up to ∼300 mg/l after inhalation of 5 MIU (i.e., 400 mg) CMS, and no major adverse effect was observed (Gkoufa et al., 2019). In addition, a recent transcriptomics study revealed that polymyxin B significantly increased the expression of CD86 gene (encoding a costimulator of T- and B-lymphocyte function) and caused perturbations of metabolism, DNA repair and replication, and cell cycle in A549 cells (Azad et al., 2019).
Overall, the toxicity of polymyxins is a critical factor that needs to be considered for the optimization of their clinical use. Current clinical literature on polymyxin-induced toxicities has several limitations, including the lack of proper control groups, the presence of comorbidities, and coadministration of other toxic drugs. To maintain the clinical utility of this last-line class of antibiotics, several strategies have been proposed to minimize polymyxin-induced toxicities, such as therapeutic drug monitoring, personalized dosing with adaptive feedback control (Lakota et al., 2018), and avoidance of simultaneous administration of toxic medications.
IX. Polymyxin Combination Therapy
Considering potentially suboptimal PK/PD/TD, emergence of resistance, and narrow therapeutic windows, combination therapy is recommended for polymyxins even though there is a lack of strong clinical evidence (Tan et al., 2007; Bergen et al., 2008; Garonzik et al., 2011; Sandri et al., 2013a; Cheah et al., 2016; Nation et al., 2017; Tsuji et al., 2019). PK/PD principles have been largely overlooked in the current clinical literature on antibiotic combinations; in particular, drug exposure at the infection site is seldom considered. Another key factor that should be considered for evaluations of antibiotic combinations is whether synergy (e.g., >2-log10 killing than the more active monotherapy) or additive killing is acceptable given that decreased bacterial load can lead to lower potential of resistance development. Because of the different PK between CMS and colistin/polymyxin B and their poor disposition in the lungs after intravenous administration, PK/PD at the infection site should be considered when optimizing polymyxin combination therapies for pneumonia and other type of infections.
A. Combinations of Polymyxins and Carbapenems
Carbapenems have been widely used in combination therapy with polymyxins against MDR Gram negative bacteria. The combination of colistin or polymyxin B (concentrations at 2 × MIC) with doripenem (6 mg/l) resulted in synergistic killing against 4 polymyxin-susceptible KPC-3–producing K. pneumoniae (Lee and Burgess, 2013). An interesting study was performed to evaluate the efficacy of doripenem-colistin combination against KPC-producing K. pneumoniae based on the molecular resistance mechanisms (Clancy et al., 2013). An association was observed between higher doripenem MICs and reduced efficacy of colistin-doripenem with ompK36 (a gene encoding for the outer membraneporin OmpK36) mutations (Clancy et al., 2013). It was suggested that ompK36 genotyping could be used as an indicator for the efficacy of doripenem or colistin-doripenem treatment and facilitate the selection of therapeutic options (Clancy et al., 2013). Time-kill assays with clinically achievable concentrations of colistin (2 mg/l), doripenem (8 mg/l), and tigecycline (2 mg/l) also supported the use of colistin-doripenem combination against MDR and XDR A. baumannii, with greatest synergistic (53.6%) and bactericidal (75.4%) activities among those antibiotics tested in mono and double combinations (Park et al., 2016). The addition of colistin (0.12–16 mg/l) to doripenem (0.06–32 mg/l) demonstrated superior synergistic activity in comparison with the combination of doripenem with levofloxacin or amikacin against 25 A. baumannii isolates (Pankuch et al., 2010). The synergistic activity of colistin-doripenem combination was illustrated in an IVM study against MDR K. pneumoniae using clinically relevant concentrations of colistin (constant concentrations of 0.5 and 2 mg/l) and doripenem (Cmax of 2.5 and 25 mg/l over 8 hours) (Deris et al., 2012). Using the same combination regimens, increased bacterial killing activity was observed against colistin-heteroresistant and colistin-resistant MDR P. aeruginosa, whereas only the combinations consisting of colistin at 2 mg/l resulted in increased activity against colistin-resistant strains (Bergen et al., 2011b). Importantly, emergence of colistin-resistant subpopulations could be prevented with colistin-doripenem combination (Bergen et al., 2011b; Deris et al., 2012). The combination of colistin (constant concentrations of 2 or 5 mg/l) and doripenem (Cmax of 25 mg/l over 8 hours) has been shown to eliminate colistin-resistant subpopulations in addition to the synergistic killing activity using HFIM (Ly et al., 2015). The synergy of colistin (constant concentration of 3.5 mg/l) and doripenem (Cmax of 25 mg/l over 8 hours) combination was further extended to biofilm-associated infections using a dynamic biofilm model, and enhanced killing and minimized colistin emergence were observed against colistin-susceptible P. aeruginosa strains (Lora-Tamayo et al., 2014).
It is important to note that the combination of colistin (1.2, 2.5, or 4.2 mg/l) and meropenem (6.8 mg/l) was reported to exhibit limited efficacy against high-level carbapenem-resistant NDM-producing K. pneumoniae (meropenem MIC ≥16 mg/l) in vitro (Lagerbäck et al., 2016). In contrast, an IVM study demonstrated that the combination of meropenem (1 g in 3-hour infusion) and colistin (constant concentration of 1 mg/l) resulted in bactericidal activity against carbapenem-resistant A. baumannii (meropenem MIC of 128 mg/l), which was otherwise unachievable with monotherapies (Liu et al., 2016a). In another IVM study, enhanced bacterial killing was also reported with the combination of colistin (initial concentration of 0.5 mg/l; half-life of 4 hours) and meropenem (initial concentration of 40 mg/l; half-life of 1 hour) against P. aeruginosa and A. baumannii (Tängdén et al., 2017). The addition of imipenem (1–2 × MIC; 8–16 mg/l) to colistin (1–2 × MIC; 2–4 mg/l) was found to be synergistic and bactericidal against beta-lactamase–producing K. pneumoniae (Tang et al., 2015). However, the bactericidal effect was inoculum-dependent at the lower concentration of colistin-imipenem (Tang et al., 2015). Against 42 K. pneumoniae isolates, the combination of colistin sulfate (5 mg/l or 4 × MIC) and imipenem (10 mg/l or 4 × MIC) was synergistic against 50% of colistin-susceptible strains but antagonistic against 56% of colistin-resistant strains (Souli et al., 2009). This demonstrated that the colistin susceptibility should be considered and that colistin-imipenem combination may not be synergistic against colistin-resistant VIM-producing K. pneumoniae (Souli et al., 2009). Nonetheless, clinically relevant concentrations of colistin in combination with imipenem showed promising killing activity against MDR P. aeruginosa that were resistant to both colistin and imipenem (Bergen et al., 2011a). In addition to PK/PD considerations, the inconsistent in vitro results in the literature suggest that the effect of different mechanisms of carbapenem resistance on the synergy of polymyxin-carbapenem combinations may need further investigation.
Using a mouse sepsis model, the combination of colistin (1 mg/kg) and imipenem (20 mg/kg) resulted in a lower lethality rate (10%–15%) as compared with each monotherapy (colistin, 30%–40%; imipenem, 30%–80%) (Cirioni et al., 2007). In addition, the combination of intranasal CMS (10 mg/kg per day) and subcutaneous injection of imipenem (60 mg/kg per day) improved the survival rate in a mouse pneumonia model with MDR P. aeruginosa (Aoki et al., 2009). Some clinical data also support the use of polymyxin-carbapenem combination for the treatment of bacteremia caused by KPC-producing K. pneumoniae and carbapenem-resistant A. baumannii, with lower mortality rates observed for patients treated with polymyxin-carbapenem as compared with polymyxins alone (K. pneumoniae, 57.1% vs. 20%; A. baumannii, 47.5% vs. 25.8%) (Qureshi et al., 2012; Park et al., 2019).
A recent open-label, randomized, controlled trial evaluated colistin monotherapy versus colistin-meropenem combination therapy for the treatment of severe infections caused by carbapenem-resistant Gram negative bacteria (Paul et al., 2018). The results showed that, compared with colistin monotherapy, the combination therapy was not superior and did not improve clinical efficacy in severe A. baumannii infections (Paul et al., 2018). It should be noted that 99% and 98% of the pathogens in the combination group and colistin group, respectively, were meropenem-resistant, with MICs >8 mg/l (Paul et al., 2018). It would be valuable to examine the synergy of this combination against the pathogens even in vitro. In the same study, ∼50% of patients in each group had pneumonia for which intravenous polymyxins are not efficacious (section V. Pharmacokinetics and Pharmacodynamics); therefore, in this clinical study the colistin-meropenem combination is virtually equivalent to meropenem monotherapy against meropenem-resistant pathogens at the lung infection site (Paul et al., 2018). Overall, the conclusion can be specifically stated that colistin-meropenem combination therapy was not superior than colistin monotherapy for the treatment of pneumonia caused by meropenem-resistant A. baumannii (Paul et al., 2018). Nevertheless, well designed prospective clinical studies are urgently required to evaluate the clinical benefit of polymyxin combination therapy for the treatment of different types of infections.
B. Combinations of Polymyxins and Rifampicin
One of the most commonly studied antibiotics for polymyxin combination therapy is rifampicin. The in vitro synergistic effect of polymyxins and rifampicin has been reported against NDM-producing, KPC-producing K. pneumoniae, including polymyxin-resistant strains (Elemam et al., 2010; Tascini et al., 2013; Gaibani et al., 2014). In vitro time-kill kinetic studies showed that the combinations of colistin (1.2–4.2 mg/l) and rifampicin (1.7–2 mg/l) at clinically relevant concentrations were bacteriostatic or bactericidal against NDM-producing, KPC-producing K. pneumoniae (Gaibani et al., 2014; Lagerbäck et al., 2016). Against carbapenem-resistant Acinetobacter spp., polymyxin B-rifampicin combination exhibited bactericidal activity against 13 of 31 (polymyxin B 2 mg/l and rifampicin 2 mg/l) and two of three isolates (polymyxin B at concentration of 0.5 × MIC and rifampicin 2 mg/l) in two time-kill kinetic studies (Lim et al., 2009, 2011). In an IVM study, two MDR A. baumannii isolates were treated with three combination regimens of colistin (constant concentrations of 0.5, 2, and 5 mg/l) and rifampicin (Cmax of 5 mg/l) (Lee et al., 2013a). In comparison with each monotherapy, all combination regimens resulted in greater killing of A. baumannii at a low inoculum (∼106 cfu/ml), whereas only regimens containing colistin at 2 and 5 mg/l increased the killing activity against A. baumannii at a high inoculum (∼108 cfu/ml) (Lee et al., 2013a). Importantly, colistin-rifampicin combination was effective in suppressing the emergence of colistin resistance (Lee et al., 2013a). The in vivo efficacy of colistin (3 mg/kg intramuscularly) in addition to rifampicin (5 mg/kg intravenously) was demonstrated in a rat thigh infection caused by MDR A. baumannii (Pantopoulou et al., 2007). Colistin monotherapy and colistin-rifampicin resulted in a better median survival (4 days) in comparison with the untreated control (2 days) and rifampicin monotherapy (2.5 days) (Pantopoulou et al., 2007). Colistin-rifampicin combination was superior, with a lower 6-day mortality rate (70%) as compared with colistin monotherapy (100%) (Pantopoulou et al., 2007). In a mouse pneumonia model with MDR P. aeruginosa, the efficacy of CMS (10 mg/kg subcutaneously or 5 mg/kg per 12 hours intranasally) in combination with imipenem (30 mg/kg per 12 hours subcutaneously) or rifampicin (25 mg/kg per day orally) was examined (Aoki et al., 2009). Overall, an increased survival rate was observed for mice receiving intranasal CMS in combination with both imipenem (62.5%) and rifampicin (75%) (Aoki et al., 2009). It is important to note that a much greater survivability was observed after the combination therapy with intranasal CMS as compared with those with subcutaneous CMS because of higher drug exposure in the lungs via intranasal administration (Aoki et al., 2009). In addition, use of CMS (rather than colistin) and the lower subcutaneous dose (considering animal scaling) might also contribute to the lower survival rates with CMS alone and the combination via subcutaneous administration (Aoki et al., 2009). The efficacy of polymyxin-rifampicin combination is further extended against S. maltophilia, for which bactericidal effect was observed only with the combination of colistin (2 mg/l) and rifampicin (8 mg/l) (Betts et al., 2014b). The same study also demonstrated an increased survival of Galleria mellonella with colistin-rifampicin combination for the treatment of S. maltophilia infection in comparison with each monotherapy (Betts et al., 2014b). More importantly, clinical evidence on the efficacy of polymyxin-rifampicin combination has been reported against difficult-to-treat infections (meningitis, sepsis, pneumonia, and UTI) caused by P. aeruginosa [intravenous CMS 2 MIU/day (i.e., 160 mg/day) and intravenous rifampicin 600 mg/day; with additional aerosolized CMS at 1 MIU (i.e., 80 mg) twice daily for pneumonia], meningitis caused by A. baumannii (intrathecal CMS 10 mg and intravenous rifampicin 600 mg), and bacteremia and UTI caused by K. pneumoniae (intravenous CMS 100 mg/8 h and intravenous rifampicin 600 mg) (Tascini et al., 2004; Nastro et al., 2014). Large clinical studies are required to confirm the clinical benefit of polymyxin-rifampicin combination.
C. Combinations of Polymyxins and Tigecycline
Infections caused by carbapenem-resistant Gram negative bacteria (e.g., A. baumannii and Enterobacterales) often limit the therapeutic options to polymyxins and tigecycline. However, the use of these last-line antibiotics has been a concern due to the emergence of resistance, and studies have been undertaken to examine the combination of polymyxins and tigecycline. Against 15 carbapenem-resistant Enterobacterales (E. coli, K. pneumoniae, and Enterobacter spp.), a synergistic effect was demonstrated against 47% of the strains, and no antagonism was detected (Betts et al., 2014a). In contrast, a study reported that colistin-tigecycline combination did not enhance the bacterial killing against NDM-1–producing Enterobacterales; however, it should be noted that the colistin concentrations (0.1–0.29 mg/l) used in this study were extremely low (Albur et al., 2012). Colistin-tigecycline combination is promising in treating ESBL-producing K. pneumoniae infections, with a high rate of synergy (89%) at sub-MIC concentrations (0.25 × MIC) (Ku et al., 2017), and exhibits synergistic killing against colistin-susceptible (treated with colistin and tigecycline at 1, 2, and 4 × MIC) and colistin-resistant (treated with colistin 2 mg/l and tigecycline 1 mg/l) KPC-producing K. pneumoniae (Pournaras et al., 2011; Betts et al., 2014b). In another study, the combination of colistin (2 mg/l) and tigecycline (1 mg/l) was less effective than the combination of colistin (2 mg/l) and rifampicin (8 mg/l) against polymyxin-resistant S. maltophilia (MICs: 4–32 mg/l for colistin, 4–16 mg/l for rifampicin, and 0.5–8 mg/l for tigecycline) (Betts et al., 2014b). The combination of colistin-tigecycline also displayed synergistic killing against MDR A. baumannii (Moland et al., 2008; Sheng et al., 2011), as well as in an IVM study mimicking clinically relevant doses of colistin (5 mg/kg per day) and tigecycline (50 or 100 mg twice daily) (Cai et al., 2017). Another IVM study demonstrated the importance of tigecycline dosage regimen in determining the efficacy of polymyxin B-tigecycline combination against MDR A. baumannii, as synergy was achieved only when polymyxin B (1 mg/kg per 12 hours) was used in combination with tigecycline at a higher dose (200 mg/12 h) but not at 100 mg/12 h (Hagihara et al., 2014). In a mouse sepsis model, the combination of CMS (2.5 mg/kg per 12 hours) and tigecycline (10 mg/kg per 12 hours) did not result in better efficacy against carbapenem-resistant K. pneumoniae as compared with monotherapies, which is very likely due to the very low doses of both antibiotics considering animal scaling (Demiraslan et al., 2014). Similar PK/PD issues also occurred in a mouse pneumonia model caused by XDR A. baumannii, as the doses of colistin and tigecycline (CMS 1.25 mg/kg per 6 hours and tigecycline 10 mg/kg per 12 hours) were very low (Mutlu Yilmaz et al., 2012). In a small clinical study, treatment of bacteremia caused by KPC-producing K. pneumoniae with colistin and tigecycline as monotherapies resulted in mortality rates of 66.7% (4 of 7) and 40% (2 of 5), respectively, whereas no mortality (0 of 9) was observed with the colistin-tigecycline combination (Zarkotou et al., 2011). Similar results were also demonstrated in a small retrospective cohort study with mortality rates of 57.1% (4 of 7) and 80% (4 of 5) with polymyxin B (or CMS) and tigecycline as monotherapies, respectively, and only the one patient who received polymyxin-tigecycline survived (Qureshi et al., 2012). Despite the small sample size of these clinical studies, colistin-tigecycline combination therapy remains a promising therapeutic option, especially against KPC-producing K. pneumoniae bacteremia, and further clinical studies are warranted.
D. Other Polymyxin Combinations
Combinations of polymyxins with other antibiotics have also been conducted in preclinical and clinical studies. Synergy between colistin and vancomycin/teicoplanin has been demonstrated in vitro against MDR A. baumannii, including colistin-resistant strains (Gordon et al., 2010; Wareham et al., 2011; Percin et al., 2014); however, the limited clinical evidence did not support the use of this combination therapy (Leite et al., 2016). With the current recommended dosage regimens, colistin-vancomycin combination did not lower the mortality rate in patients (5 of 7; 71%) when compared with patients who received antibiotic therapy without vancomycin (8 of 14; 57%); however, definite conclusions could not be made because of the small sample size and study design (Leite et al., 2016). Furthermore, nephrotoxicity in patients is another major concern with this combination.
Colistin-fosfomycin is another promising combination that has shown in vitro synergy and bactericidal effect against P. aeruginosa (Di et al., 2015; Walsh et al., 2016); however, despite the increased killing activity, emergence of fosfomycin resistance was not eliminated (Walsh et al., 2016). An IVM study with clinically achievable concentrations of colistin and fosfomycin demonstrated the efficacy of the combination of colistin (Cmax/Cmin of 3.0/0.75 mg/l) and fosfomycin (Cmax/Cmin of 250/40 mg/l) against NDM-1–producing E. coli, K. pneumoniae, and Klebsiella oxytoca, with increased bacterial killing activity (Albur et al., 2015). Similar results were also demonstrated against KPC2-producing K. pneumoniae in an in vitro study for the combination of colistin (75,000 IU/kg per 12 hours) and fosfomycin (8 g/8 h) (Yu et al., 2017a). Furthermore, the combination of colistin (10 mg/kg) and fosfomycin (150 mg/kg) eradicated ESBL-producing E. coli biofilm in a guinea pig foreign-body infection model, suggesting the potential role of this combination for biofilm-related infections (Corvec et al., 2013). In a clinical study, 11 critically ill patients with carbapenem-resistant K. pneumoniae infection were treated with fosfomycin (2–4 g/6 h) in addition to CMS (six patients), gentamicin (three patients), or piperacillin/tazobactam (one patient) (Michalopoulos et al., 2010). Although a direct comparison between the effectiveness of these combinations could not be made, fosfomycin appeared as a safe adjuvant agent with no adverse events reported from these patients (Michalopoulos et al., 2010).
There are limited preclinical and clinical data on the combinations of colistin with other antibiotics such as doxycycline, daptomycin, ceftazidime, ciprofloxacin, aminoglycoside, and ampicillin/sulbactam. In an IVM study with XDR A. baumannii, daptomycin (10 mg/kg per day) and colistin [mimicking CMS 3 MIU/8 h (i.e., 240 mg/8 h)] combination therapy was found to be the most effective in comparison with colistin-imipenem (1 g/8 h) and imipenem-ertapenem (1 g/8 h and 1 g/day, respectively) (Córdoba et al., 2015). There is a lack of data supporting the in vitro synergistic activity of the combination of polymyxins and ampicillin/sulbactam; however, in a prospective, open-label, randomized study, the combination of colistin (as CMS) with a high dose of ampicillin/sulbactam showed favorable clinical response in 50 patients with VAP due to A. baumannii resistant to carbapenems but susceptible to colistin and ampicillin-sulbactam (Makris et al., 2018). In the same study, the combination of intravenous CMS [3 MIU/8 h (i.e., 240 mg/8 h)] and ampicillin/sulbactam ([6 (4/2)] g/6 h) demonstrated a favorable clinical response than those receiving CMS monotherapy (Makris et al., 2018). Combinations of colistin with ciprofloxacin or ceftazidime have been shown to exhibit good synergy against MDR P. aeruginosa using a checkerboard method (D'Souza et al., 2014). Additionally, the synergistic activity with the combination of colistin (Cmax 6 or 18 mg/l) and ceftazidime (constant Css 50 mg/l) was shown against MDR P. aeruginosa in an IVM study (Gunderson et al., 2003). Despite the positive results in the literature, a recent in vitro report suggested that the addition of colistin (2 mg/l) to ceftazidime-avibactam (concentrations at 0.25, 1, and 4 × MIC) was unable to potentiate the killing activity against most isolates of carbapenem-resistant Enterobacterales (Shields et al., 2018). Colistin combination with the next-generation aminoglycoside plazomicin showed synergy against 60% of 164 isolates of carbapenemase-producing Enterobacterales in a checkerboard study, which was superior over plazomicin-meropenem (20%) and plazomicin-fosfomycin (25%) (Rodríguez-Avial et al., 2015). Against KPC-producing K. pneumoniae, doxycycline-polymyxin B combination can also be a potential option with good synergistic activity (Elemam et al., 2010).
E. Triple Combination Treatment with Polymyxins
To date, no polymyxin double combinations can kill 100% of isolates of MDR Gram negative pathogens, and investigations on polymyxin triple combinations are emerging. In the clinic, triple combinations may be the only option for patients with bacterial infections that are resistant to all current antibiotics, including polymyxins. In a static time-kill study with polymyxin B, doripenem, and rifampicin at 0.25 × MIC, bactericidal activity was achieved with the triple combination against all carbapenem-resistant clinical isolates of P. aeruginosa (5 of 5) and E. coli (5 of 5), 80% of K. pneumoniae (4 of 5), and 60% of A. baumannii (3 of 5) (Urban et al., 2010). Among these isolates, double combinations were ineffective against 3 P. aeruginosa, 3 K. pneumoniae, 1 E. coli, and 1 A. baumannii (Urban et al., 2010). The potential of colistin triple combination with carbapenem and rifampicin was also demonstrated against VIM- and NDM-producing K. pneumoniae in a time-kill study (Tängdén et al., 2014). Although synergy was demonstrated by aztreonam (17 mg/l), fosfomycin (83 mg/l), or rifampicin (1.7 mg/l) in triple combinations with colistin (4 mg/l) and meropenem (6.8 mg/l), the colistin-meropenem-rifampicin combination was the most effective and the only one with synergistic and bactericidal effect against all four VIM- and NDM-producing K. pneumoniae (Tängdén et al., 2014). An IVM study stimulated clinically relevant concentrations of polymyxin B (Css 0.5, 1, or 2 mg/l), meropenem (1 or 2 g/8 h), and rifampicin (600 mg every 8 or 12 hours) in mono, double, or triple combination therapies and showed that only the triple combination was effective against two polymyxin-resistant KPC-producing K. pneumoniae (Diep et al., 2017). In an in vitro study, double combinations polymyxin B-rifampicin (0.25 mg/l and 0.5 mg/l, respectively) and polymyxin B-imipenem (0.25 mg/l and 8 mg/l, respectively) were bactericidal against seven out of eight MDR A. baumannii isolates, and only the triple combination of polymyxin B-imipenem-rifampicin (0.25 mg/l, 8 mg/l, and 0.5 mg/l, respectively) was bactericidal against all eight isolates (Yoon et al., 2004).
An in vitro study examined the triple combination of colistin (2 mg/l), doripenem (8 mg/l), and sulbactam (4 mg/l) against 18 A. baumannii isolates collected from nine patients with respiratory tract infections prior and after treatment with CMS and doripenem (Oleksiuk et al., 2014). An overall greater killing at 24 hours was observed with the triple combination than colistin-doripenem and colistin-sulbactam combinations, with the mean log10 killing of -5.74, -2.88, and -1.51, respectively (Oleksiuk et al., 2014). The effectiveness of polymyxin B, meropenem, and ampicillin/sulbactam triple combination against polymyxin-resistant, carbapenem-resistant A. baumannii was demonstrated in two HFIM studies (Lenhard et al., 2017a,b). With the triple combination of polymyxin B (1.43 mg/kg per 12 hours with a loading dose), meropenem (2 g/8 h), and ampicillin/sulbactam (8 or 4 g/8 h), a rapid eradication of A. baumannii was observed by 96 hours, which was otherwise unachievable with any mono and double combination therapies (Lenhard et al., 2017a,b). Importantly, the use of this triple combination is also supported by a small clinical study involving 17 patients with infections (mainly ventilator-associated pneumonia) caused by colistin-resistant A. baumannii (Qureshi et al., 2015). Among these patients, all seven patients who received CMS-carbapenem-ampicillin/sulbactam survived, whereas 6 out of the 10 patients who received other antibiotic regimens deceased within 30 days of infection (Qureshi et al., 2015). Another triple combination with clinical evidence is the combination of CMS (100 mg CBA/36 h), doripenem (250 mg/12 h), and tobramycin (40 mg/48 h) (Sun et al., 2010). A liver transplant recipient with bacteremia caused by MDR P. aeruginosa was successfully treated with this novel combination after treatment failure with conventional monotherapies of piperacillin/tazobactam, ciprofloxacin, and tobramycin, and even with a triple combination of piperacillin/tazobactam, tobramycin, and rifampicin (Sun et al., 2010). In a retrospective study conducted in three large Italian teaching hospitals, 125 patients with bacteremia caused by KPC-producing K. pneumoniae were treated with a range of antibiotics (Tumbarello et al., 2012). In a total of 52 (41.6%) nonsurvivors, 25 (48.1%), 23 (44.2%), and 4 (7.7%) patients were treated with mono, double, and triple combination therapies, respectively. Among the 125 patients (52 nonsurvivors and 73 survivors), 23 patients were treated with a triple combination; for the nonsurvivors, two were from the 16 (12.5%) patients who received CMS-meropenem-tigecycline, and two were from the seven (28.6%) patients who received gentamicin-meropenem-tigecycline (n = 6) or CMS-meropenem-gentamicin (n = 1). Overall, the multivariate analysis demonstrated that the triple combination of CMS-meropenem-tigecycline was associated with a reduced risk of mortality (Tumbarello et al., 2012).
It is evident that the clinical evidence on polymyxin combination therapy is limited, and the safety of polymyxin combinations has not been well evaluated, in particular the nephrotoxicity. Importantly, there is often a lack of PK/PD considerations, in particular at the infection site. A number of major challenges need to be addressed in clinical evaluations of antibiotic combination therapies (including polymyxins), such as ethics, appropriate endpoints, sample size, the optimal dosage regimen of each antibiotic, appropriate control groups, and PK/PD/TD modeling. Synergy occurs only when different antibiotics achieve optimal exposure at the infection site with the right timing.
F. Mechanistic Insights into Polymyxin Combination Therapy
It is generally believed that the synergy of polymyxin combinations is due to the disorganization of bacterial outer membrane by polymyxins, which facilitates the access of other antibiotics to their intracellular targets. In fact, at the transcriptomic and metabolomic levels the synergy mechanisms of polymyxin combinations are far more complicated. A pan-transcriptomic analysis revealed that polymyxin treatment caused rapid outer membrane remodeling, upregulation of efflux pumps, and downregulation of fatty acid biosynthesis in A. baumannii (Li et al., 2020a). Integrative modeling of correlative transcriptomics and metabolomics data using a strain-specific genome-scale metabolic model discovered the interplay of multiple key metabolic pathways in A. baumannii in response to colistin treatment: 1) downregulation of tricarboxylic acid cycle and biosynthesis of peptidoglycan and lipopolysaccharides; 2) upregulated fluxes through gluconeogenesis, pentose phosphate pathway, and biosynthesis of nucleotides and amino acids; and 3) altered fluxes over respiratory chain (Zhu et al., 2019). Using an IVM to mimic their PK in patients, colistin treatment (continuous infusion at 2 mg/l) within 1 hour caused >400 differentially regulated genes, in particular those for outer membrane biogenesis, phospholipid trafficking, and fatty acid metabolism in A. baumannii (Henry et al., 2015). Interestingly, no differentially regulated genes were shown after doripenem treatment (Cmax 25 mg/l; half-life 1.5 hours) at 15 minutes, but 45 genes were differentially expressed at 1 hour. The combination of colistin and doripenem led to >450 differentially expressed genes in A. baumannii at 1 hour, and >70% of the significant genes were also observed with colistin monotherapy (Henry et al., 2015). A metabolomics study demonstrated that the synergistic killing of A. baumannii by colistin and doripenem occurred via the inhibition of different key metabolic pathways in a time-dependent manner (Maifiah et al., 2017), which was consistent with the transcriptomics results (Henry et al., 2015). An early (15 minutes and 1 hour) disruption of outer membrane and cell wall was induced by colistin, and inhibition of cell wall synthesis by doripenem occurred subsequently (at 4 hours) (Maifiah et al., 2017). The colistin-doripenem combination significantly perturbed several key metabolic pathways in A. baumannii relative to either monotherapy, including the pentose phosphate pathway (induced initially by colistin at 15 minutes and 1 hour and by doripenem at 4 hours) and downregulation of cell wall biosynthesis (via d-sedoheptulose 7-phosphate) and nucleotide metabolism (via d-ribose 5-phosphate) (Maifiah et al., 2017). For colistin-aztreonam combination against A. baumannii, colistin caused an early (1 hour) disruption to the cell envelope, and aztreonam inhibited cell wall synthesis at later time points (4 and 24 hours) (Han et al., 2018). The level of two amino sugars, UDP-N-acetylglucosamine and UDP-N-acetylmuramate was significantly decreased by aztreonam alone and the combination at 4 and 24 hours (Han et al., 2018).
In P. aeruginosa, polymyxins disrupted cell envelope and induced significant perturbations in lipid and fatty acid metabolism pathways at early time points (15 minutes and 1 hour) (Hussein et al., 2019; Lin et al., 2019c). Amikacin monotherapy caused significant perturbations in amino acid metabolism in P. aeruginosa at 1 and 4 hours, and the synergistic killing by the polymyxin-amikacin combination involved the inhibition of cell envelope biogenesis and central carbohydrate metabolism, decreased levels of amino sugars, and a downregulated nucleotide pool (Hussein et al., 2019). The polymyxin B-enrofloxacin combination perturbed lipid metabolites in XDR P. aeruginosa at 1 hour, which was mainly driven by polymyxin B (Lin et al., 2019c). At 4 hours, the synergy was mainly driven by enrofloxacin through the inhibition of DNA replication; and importantly, polymyxin resistance was minimized by the combination via the inhibition of lipid A modification with L-Ara4N (Lin et al., 2019c). In K. pneumoniae, a transcriptomic study revealed that the synergy of the combination of polymyxin B (1 mg/l as continuous infusion) and chloramphenicol (Cmax 8 mg/l; half-life 4 hours) was persistent over 24 hours and mainly driven by chloramphenicol via perturbations of cell envelope synthesis and metabolism of carbohydrates, nucleotides, and amino acids (Abdul Rahim et al., 2020). Notably, chloramphenicol and its combination with polymyxin B significantly inhibited lipid A modification and decreased the resistance to polymyxin via downregulating the expression of the arn operon (Abdul Rahim et al., 2015, 2020). Overall, recent systems pharmacological studies highlight the significance of time-dependent synergistic effect of polymyxin combinations, and synergistic combinations may not be able to directly inhibit polymyxin resistance due to lipid A modifications. These results are crucial for the optimization of the dosage regimens of polymyxin combination therapy in patients.
In summary, the currently available preclinical and clinical data suggest a potential benefit with synergistic polymyxin combinations to treat MDR Gram negative pathogens. Ultimately, personalized combination therapy should be achieved based upon the condition of patient, type of infection, pathogen, rapid diagnosis, mechanisms of killing, resistance and toxicity, therapeutic drug monitoring, and PK/PD/TD using a systems approach.
X. Antiendotoxin Properties of Polymyxins
Endotoxins are the major component of Gram negative bacterial outer membrane that can cause fever, increased heart rate, inflammation, septic shock, and many other pathogenic effects (Peterson, 1996). Sepsis is a major global health problem, and Gram negative bacteremia and endotoxemia are often strong predictive of mortality in patients (Hurley et al., 2012). Thus, antiendotoxin therapy is an important therapeutic strategy in patients with Gram negative bacteremia. Owing to the ability of polymyxins to specifically bind to the lipid A (endotoxin) (Velkov et al., 2010), polymyxin B is a strong candidate as an antiendotoxin therapeutic agent (Morrison and Jacobs, 1976). The antiendotoxin activity of polymyxin B was illustrated in endotoxemia and septicemia animal models (Corrigan and Kiernat, 1979; Ingoldby, 1980; Flynn et al., 1987). Polymyxin B–immobilized fibers, in which polymyxin B is covalently bound and immobilized to polystyrene fiber, were introduced to selectively remove endotoxin in blood. Using an endotoxemia canine model, endotoxin was successfully removed from the blood by extracorporeal hemoperfusion with polymyxin B–immobilized fibers, and improved survivability was observed (Hanasawa et al., 1988). An increased survival rate was also reported in an E. coli sepsis canine model after extracorporeal hemoperfusion with polymyxin B–immobilized fibers (Hanasawa et al., 1989). Even though bactericidal activity was observed with polymyxin B–immobilized fibers against P. aeruginosa in vitro (Shoji et al., 1998), it is unclear whether the antibacterial killing was due to the release of polymyxin B from the immobilized fibers.
Toraymyxin, a blood purification cartridge containing polymyxin B–immobilized fibers, has been developed for the removal of endotoxin from blood in humans (Tani et al., 2019). The first clinical data with Toraymyxin were reported in 1994, showing a decreased level of circulating endotoxin and an improved hyperdynamic state in patients with septic multiple organ failure (n = 16) (Aoki et al., 1994). Since the employment of Toraymyxin for endotoxemia and septic shock treatment in Japan in 1994, the safety profile of Toraymyxin has been well established in more than 100,000 emergency and ICU cases in Japan (Shimizu et al., 2017). Since the Conformitè Europëenne (CE) mark approval in Europe in 1998, two randomized controlled trials were conducted in Europe in 2005 (n = 36) and 2009 (n = 64), and the results showed that Toraymyxin is safe, improves sepsis-associated cardiac and renal dysfunction, and reduces mortality caused by sepsis (Vincent et al., 2005; Cruz et al., 2009). A meta-analysis revealed 33%–80% reduction in plasma endotoxin level and significantly lower mortality risk with Toraymyxin with a pooled sample size of 1,425 patients from 28 studies; however, bias (e.g., preference of the publication of trials with positive results rather than negative and neutral results) and lack of blinding should be taken into consideration (Cruz et al., 2007). Notably, the randomized controlled trials published in 2015 (France; n = 243) and 2018 (North America; n = 450) reported conflicting results with no significant improvement in organ failure and survivability in favor of Toraymyxin over conventional treatments (Payen et al., 2015; Dellinger et al., 2018). Nevertheless, Toraymyxin remains a promising antiendotoxin therapy, especially in patients who are presented with severe septicemia; and further clinical evidence is required.
XI. Current Landscape of Polymyxin Drug Discovery
In addition to optimizing the clinical use of polymyxin B, colistin, and their combinations, the discovery of new-generation polymyxins is also crucial in combating infections caused by MDR Gram negative pathogens. There are a number of major challenges in the discovery of novel polymyxins, including the complex PK/PD, lack of understanding on the mechanisms of toxicities (e.g., acute toxicity, nephrotoxicity, and hyperpigmentation), and narrow chemical space due to the closely related relationships between the structure, activity, PK, and toxicity. Nevertheless, significant progress has been made over the last decade in the discovery of novel, safer polymyxins with several groups working actively in the field (Brown and Dawson, 2017; Vaara, 2019b; Velkov and Roberts, 2019). One of the leading groups at Monash University is the first to employ structure-activity-PK-toxicity relationship based mechanistic models to design novel polymyxins that target polymyxin resistance in Gram negative bacteria with reduced nephrotoxicity in comparison with polymyxin B and colistin (Velkov et al., 2014, 2018b; Velkov and Roberts, 2019). Their first-generation lipopeptide (FADDI-002) incorporated modifications of the core polymyxin scaffold (Table 1) with l-octylglycine at position 7 to increase the hydrophobic interactions with lipid A, thereby circumventing polymyxin resistance as a result of the modifications of the lipid A phosphates. Their novel polymyxins with lipidic groups at positions 6/7 and the N-terminus displayed significantly increased antimicrobial activity against polymyxin-resistant clinical isolates while also maintaining their activity against polymyxin-susceptible strains of P. aeruginosa, A. baumannii, and K. pneumoniae (Velkov et al., 2014). Notably, several lipopeptides showed, in addition to their activities against Gram negative superbugs, unexpected activity against the problematic Gram positive vancomycin-resistant Enterococcus faecium and methicillin- or vancomycin-resistant Staphylococcus aureus (Velkov et al., 2014; Zhao et al., 2016). In a neutropenic mouse lung infection model, FADDI-002 displayed improved in vivo efficacy against a polymyxin-resistant clinical isolate of P. aeruginosa as compared with colistin (Velkov et al., 2014). The kidneys of mice subcutaneously treated with lipopeptides FADDI-003 or FADDI-019 (accumulated dose 105 mg/kg) revealed no significant lesions in the cortex and medulla, and papilla regions essentially resembled the kidneys of mice treated with the saline control; no histologic grade was given (Velkov et al., 2014). One of their second-generation lipopeptides, QPX9003, is 4-fold more potent than colistin against MDR P. aeruginosa (n = 1000, including resistance to β-lactams/β-lactamase inhibitors) and carbapenem-resistant A. baumannii (n = 503) (Castanheira et al., 2019). Importantly, QPX9003 shows efficacy in mouse and rat pneumonia models that is superior to polymyxin B and is not inactivated by lung surfactant. QPX9003 is currently in development at Qpex Biopharma, Inc. (CA) with an investigational new drug submission in the near future (Sabet et al., 2019).
Northern Antibiotics (Helsinki, Finland) is developing several promising polymyxin analogs that possess only three positive charges (compared with the five carried by polymyxin B and colistin) (Vaara et al., 2008, 2010a,b, 2013; Ali et al., 2009; Vaara and Vaara, 2010, 2013; Vingsbo Lundberg et al., 2010; Vaara, 2013). The most promising lead compound reported was NAB739, which shared an identical cyclic core to that of polymyxin B and a modified linear segment consisting of octanoyl-[Thr1-d-Ser2]polymyxin B (Vaara et al., 2008). NAB739 displays good activity against E. coli (n = 66), with MIC90 values (1 to 2 mg/l) comparable to that of polymyxin B (Vaara et al., 2008; Vaara et al., 2013); and it can also sensitize A. baumannii strains to rifampicin, clarithromycin, and vancomycin by facilitating their entry into the cell (Vaara et al., 2008). In human renal proximal tubular HK-2 cells, NAB739 was 26-fold less toxic than polymyxin B and 7.5-fold less toxic than colistin (Vaara and Vaara, 2013).
Researchers at Hokuriku University reported some interesting des-fatty acyl polymyxin B analogs with potent antipseudomonal activity (MICs 0.5–1 mg/l) (Katsuma et al., 2009; Sato et al., 2011). Cubist Pharmaceuticals (MA) had a significant discovery program on novel N-terminal modified polymyxin analogs based on intellectual property developed by BioSource Pharmaceuticals (Leese, 2010). The Cubist polymyxin program centered around a novel semisynthetic methodology that involved enzymatically removing the N-terminal fatty acyl groups of polymyxin B or colistin mixtures to provide a single polymyxin core, of which the N-terminus was derivatized with individual novel haloaryl groups. The idea behind these modifications was to reduce nephrotoxicity by decreasing the hydrophobicity of the N-terminal fatty acyl group. Out of over 200 analogs, the Cubist lead compound was the 2-chlorophenylurea derivative CB-182,804 (Keith et al., 2010). In vivo studies in neutropenic mouse lung and thigh infection model showed that CB-182,804 had comparable or slightly improved in vivo efficacy to polymyxin B (Leese, 2010). In an in vitro cytotoxicity assay utilizing rat kidney proximal tubule cells, CB-182,804 displayed significantly reduced cytotoxicity (EC50 > 1000 µg/ml) compared with polymyxin B (EC50 318 µg/ml). Unfortunately, CB-182,804 failed in a phase 1 study as a result of severe nephrotoxicity (Blaskovich et al., 2018).
Pfizer (NY) also instigated a discovery program trying to alleviate polymyxin nephrotoxicity through modifications to the core and N-terminus of polymyxin B (Velkov et al., 2016a; Brown and Dawson, 2017; Velkov and Roberts, 2019). Through this work, it was demonstrated that lipopeptide 5a with the substitution of Dab3 with a diaminopropionic acid residue displayed a 2-fold improvement in MICs compared with polymyxin B against P. aeruginosa and A. baumannii strains, including several polymyxin-resistant strains. Furthermore, screening of lipopeptide 5a for in vitro nephrotoxicity utilizing human renal proximal epithelial cells showed a 2-fold decrease in cytotoxicity relative to polymyxin B. Based upon the nephrotoxicity results in rats and dogs, the overall therapeutic index of lipopeptide 5a is not better than that of polymyxin B (Brown and Dawson, 2017). Unfortunately, this polymyxin discovery program is discontinued by Pfizer (Velkov et al., 2016a).
Cantab Anti-Infectives (Hertfordshire, UK) has focused on the development of novel polymyxin nonapeptide derivatives to address the toxicity issues of the polymyxins (Saadi et al., 2013). Similar to the work of Sato and coworkers discussed earlier, Cantab compounds used the polymyxin nonapeptide core with the introduction of novel branched amine-containing acyl moieties with specific regio- and stereochemistry at the N-terminus. The lead lipopeptide from the Cantab discovery program was Cantab Ant-infectives (CA) CA1206, now SPR206, which is being developed by Spero Therapeutics (Brown et al., 2019; Vaara, 2019b). SPR206 contains a polymyxin B nonapeptide and a β-branched aminobutyrate N-terminus with an aryl substituent that displays low cytotoxicity, kidney exposure, and nephrotoxicity and an increased therapeutic window in mice compared with polymyxin B (Brown et al., 2019). A phase 1 clinical trial of SPR206 was conducted in 96 healthy volunteers (SPR206 or placebo) with seven multiple ascending dose cohorts (single intravenous doses from 10 to 400 mg) and 75–450 mg per day for 7 days plus 300 mg per day for 14 days across five multiple ascending dose cohorts. Preliminary data indicate that SPR206 was well tolerated in healthy subjects at doses up to 100 mg every 8 h for 14 days; and the PK was linear in the tested range, with mean plasma exposure of SPR206 consistent with preclinical models (https://investors.sperotherapeutics.com/news-releases/news-release-details/spero-reports-preliminary-findings-phase-1-clinical-trial-spr206).
MicuRx pharmaceuticals also has an active polymyxin discovery program that adopts a novel MRX-8 prodrug approach wherein the fatty acyl tail of the polymyxin core structure is linked to a cycle via an ester bond (Gordeev, 2018; Vaara, 2019b). This ester bond facilitates the breakdown of the parent compound in plasma into a des-fatty acyl less toxic nonapeptide form (Gordeev et al., 2016; Gordeev, 2018). Compared with polymyxin B, MRX-8 has higher MICs but better efficacy against lung infections caused by Gram negative pathogens in mice (Gordeev, 2018). Currently, an investigational new drug application of MRX-8 has been submitted to the FDA in the United States (http://micurxchina.com/project-progress).
In summary, in the wake of our increasing understanding of polymyxin PK/PD/TD, mode of action, and toxicity mechanisms, recent medicinal chemistry efforts have yielded several promising novel polymyxin lipopeptides with improved activity and/or toxicity compared with polymyxin B and colistin. Hopefully, new-generation, safer polymyxins will be approved for the treatment of Gram negative superbugs in patients in the near future.
XII. Conclusions
In stark contrast to most other antibiotic classes, the polymyxins did not follow the conventional antibiotic development pathway, with little preclinical development before being implemented for human use more than 60 years ago. Not surprisingly, the preclinical and clinical pharmacological knowledge base for polymyxins has largely been generated by academic research groups as opposed to pharmaceutical companies. Over the last two decades, polymyxins have virtually undergone a redevelopment phase, funded mainly by government agencies. The latest pharmacological research from academia led to the first scientifically based dosing guidelines for intravenous CMS. Clinicians are now in a much better position to optimize its use, and clinical practice continues to improve worldwide. Notwithstanding, there are still several major knowledge gaps that are crucial for the development of PK/PD/TD–informed dosing guidelines, in particular in the case of polymyxin B. This cannot be understated, as no novel classes of antibiotics will be available for Gram negative superbugs in the next few years. It is crucial to optimize the use of polymyxins in the clinic by maximizing their efficacy while minimizing the emergence of resistance and toxicity. Underpinned by the recent significant pharmacological achievements, several novel, safer polymyxin candidates are undergoing preclinical and clinical evaluations. It is without question that polymyxins will continue playing a major role in the battle against life-threatening infections caused by MDR Gram negative pathogens.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Nang, Azad, Velkov, Zhou, Li.
Footnotes
T.V., Q.T.Z., and J.L. are supported by the National Institutes of Health/National Institute of Allergy and Infectious Diseases under the award number [R01AI146160] and [R01AI132681]; and T.V. and J.L. under [R01AI132154].
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health. J.L. is an Australia National Health and Medical Research Council (NHMRC) Principal Research Fellow. The novel polymyxin candidates developed by J.L. and T.V.’s group were licensed to Qpex Biopharma. J.L. received seminar honoraria on the pharmacology of colistin and polymyxin B from Genentech, Inc.; DocMode; Healcare Pharmaceuticals; and a number of international conferences and research grants from Northern Antibiotics. No author has other actual or perceived conflicts of interest with the other contents of this article.
Abbreviations
- ATCC
- American Type Culture Collection
- AUC
- area under the plasma concentration curve
- CAMHB
- cation-adjusted Mueller-Hinton broth
- CAS
- Chemical Abstracts Service
- CBA
- colistin base activity
- cfu
- colony forming unit
- CLSI
- Clinical and Laboratory Standards Institute
- CMS
- colistin methanesulfonate
- CNS
- central nervous system
- Css
- steady-state concentration
- Css,avg
- average steady-state concentration
- Dab
- 2,4-diaminobutyric acid
- DPI
- dry powder inhaler
- ELF
- epithelial lining fluid
- ESBL
- extended-spectrum β-lactamase
- EUCAST
- European Committee on Antimicrobial Susceptibility Testing
- FADD
- Fas-associated death domain
- fAUC
- area under the curve of unbound drug
- fCmax
- maximum concentration of unbound drug
- FDA
- Food and Drug Administration
- HFIM
- hollow-fiber infection model
- ICU
- intensive care unit
- IU
- international unit
- IVM
- in vitro one-compartment dynamic model
- Kdo
- 2-keto-3-deoxyoctonoic acid
- KIM-1
- kidney injury molecule-1
- KPC
- Klebsiella pneumoniae carbapenemase
- L-Ara4N
- 4-amino-l-arabinose
- LPS
- lipopolysaccharide
- MDR
- multidrug-resistant
- MIC
- minimum inhibitory concentration
- MIU
- million international unit
- NDH-2
- type II NADH-quinone oxidoreductase
- NDM
- New Delhi metallo-β-lactamase
- PAE
- postantibiotic effect
- PD
- pharmacodynamics
- PEPT2
- peptide transporter 2
- pEtN
- phosphoethanolamine
- PK
- pharmacokinetics
- ROS
- reactive oxygen species
- SDD
- selective digestive tract decontamination
- SOD
- selective oropharyngeal decontamination
- TCS
- two-component regulatory system
- TD
- toxicodynamics
- UTI
- urinary tract infection
- VAP
- ventilator-associated pneumonia
- XDR
- extensively drug-resistant
- XFM
- X-ray fluorescence microscopy
- Copyright © 2021 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





{kind=link}
{kind=link}
{kind=link}
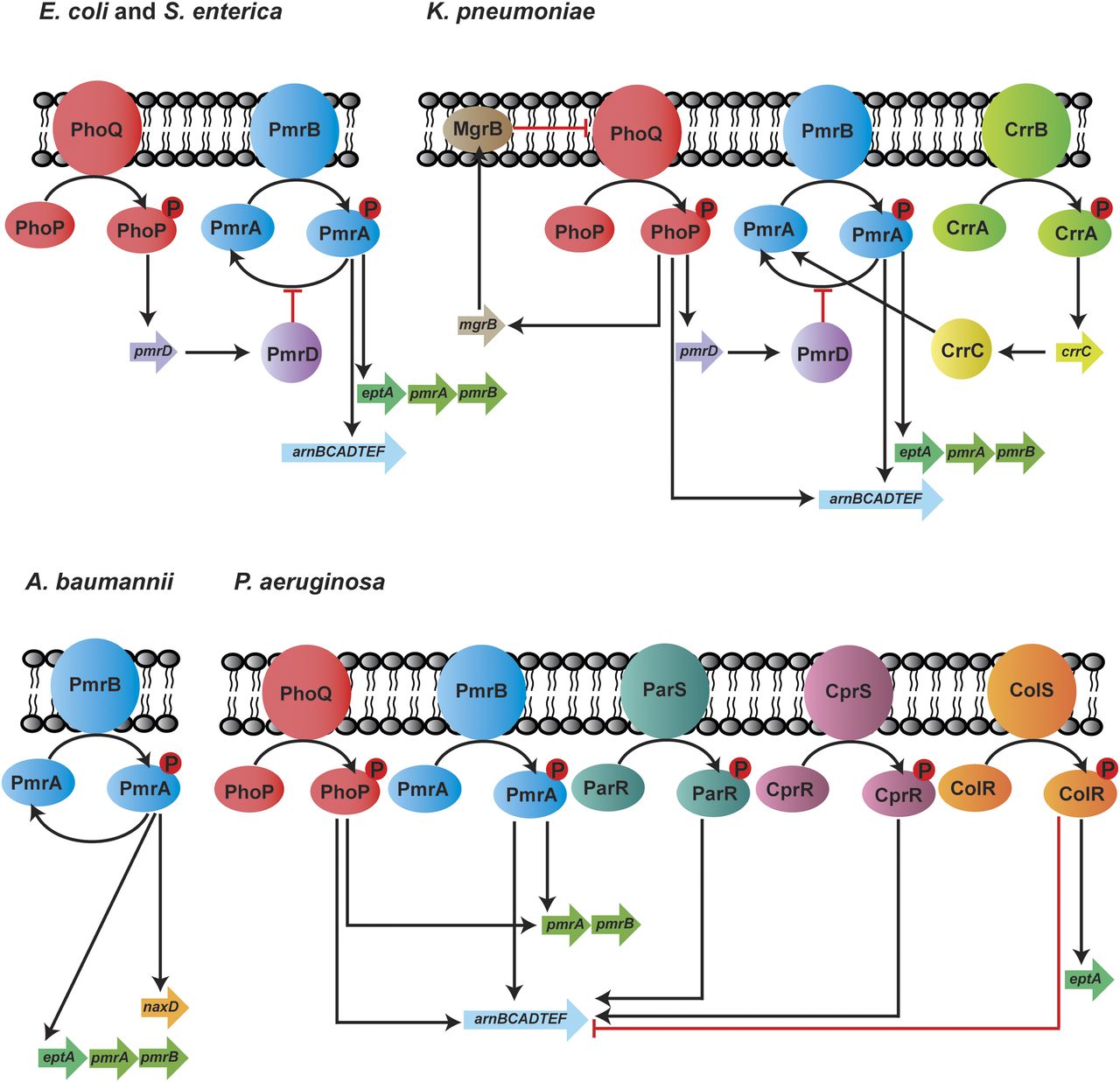{kind=link}
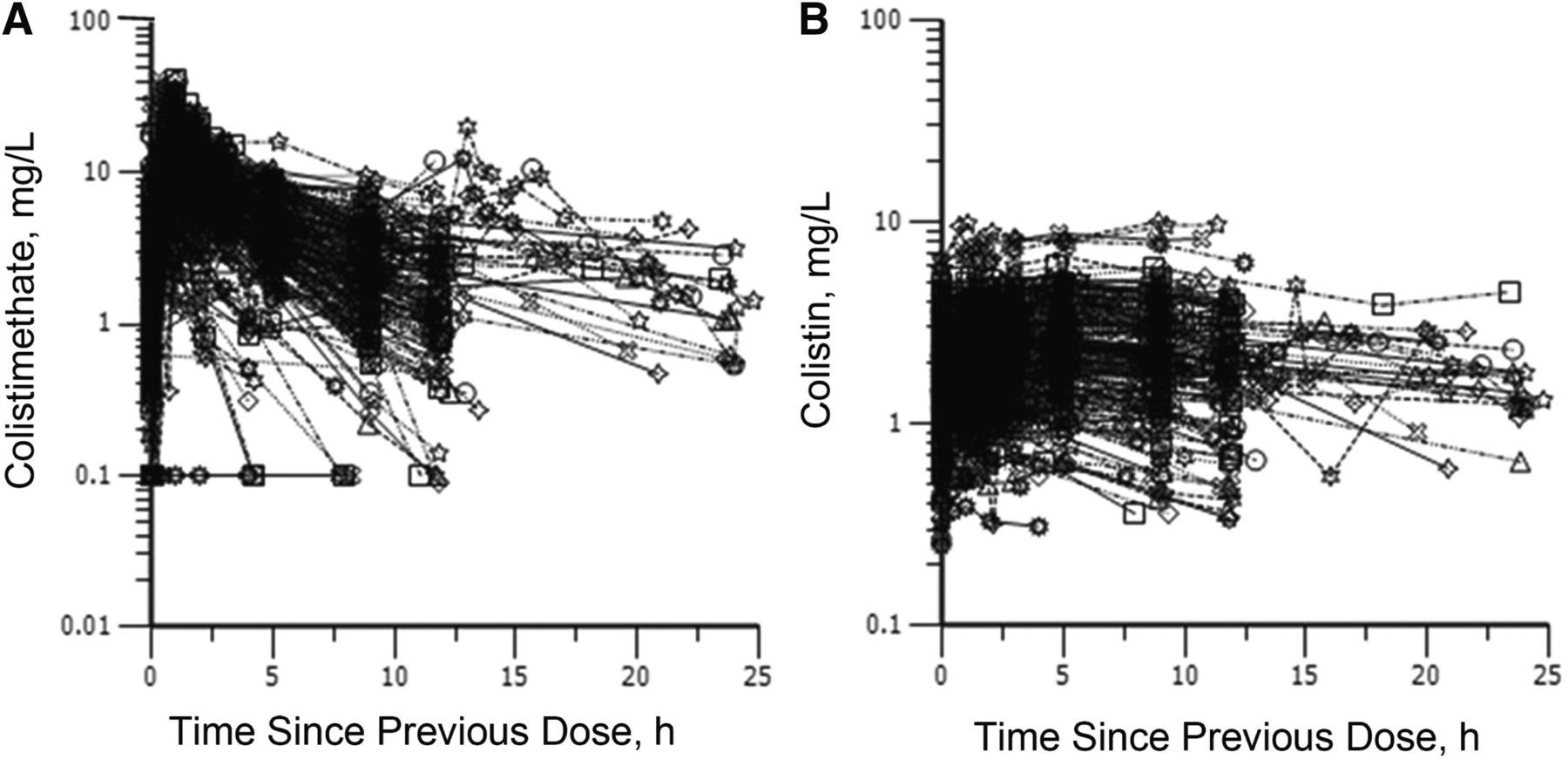{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Visual Overview
- Abstract
- I. Introduction
- II. History and Chemistry of Polymyxins
- III. Antibacterial Spectrum, Mechanisms of Activity, and Resistance
- IV. Susceptibility Testing and Breakpoints
- V. Pharmacokinetics and Pharmacodynamics
- VI. Labeling of Polymyxins
- VII. Clinical Utility
- VIII. Polymyxin Toxicity
- IX. Polymyxin Combination Therapy
- X. Antiendotoxin Properties of Polymyxins
- XI. Current Landscape of Polymyxin Drug Discovery
- XII. Conclusions
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters