


Abstract
Hydroxynorketamines (HNKs) are formed in vivo after (R,S)-ketamine (ketamine) administration. The 12 HNK stereoisomers are distinguished by the position of cyclohexyl ring hydroxylation (at the 4, 5, or 6 position) and their unique stereochemistry at two stereocenters. Although HNKs were initially classified as inactive metabolites because of their lack of anesthetic effects, more recent studies have begun to reveal their biologic activities. In particular, (2R,6R)- and (2S6)-HNK exert antidepressant-relevant behavioral and physiologic effects in preclinical models, which led to a rapid increase in studies seeking to clarify the mechanisms by which HNKs exert their pharmacological effects. To date, the majority of HNK research has focused on the actions of (2R,6R)-HNK because of its robust behavioral actions in tests of antidepressant effectiveness and its limited adverse effects. This review describes HNK pharmacokinetics and pharmacodynamics, as well as the putative cellular, molecular, and synaptic mechanisms thought to underlie their behavioral effects, both following their metabolism from ketamine and after direct administration in preclinical studies. Converging preclinical evidence indicates that HNKs modulate glutamatergic neurotransmission and downstream signaling pathways in several brain regions, including the hippocampus and prefrontal cortex. Effects on other neurotransmitter systems, as well as possible effects on neurotrophic and inflammatory processes, and energy metabolism, are also discussed. Additionally, the behavioral effects of HNKs and possible therapeutic applications are described, including the treatment of unipolar and bipolar depression, post-traumatic stress disorder, chronic pain, neuroinflammation, and other anti-inflammatory and analgesic uses.
Significance Statement Preclinical studies indicate that hydroxynorketamines (HNKs) exert antidepressant-relevant behavioral actions and may also have analgesic, anti-inflammatory, and other physiological effects that are relevant for the treatment of a variety of human diseases. This review details the pharmacokinetics and pharmacodynamics of the HNKs, as well as their behavioral actions, putative mechanisms of action, and potential therapeutic applications.
I. Introduction
Hydroxynorketamines (HNKs) are metabolites of (R,S)-ketamine [ketamine; a racemic mixture of (R)- and (S)-ketamine] formed in vivo after its administration to humans and other mammalian species (Leung and Baillie, 1986; Moaddel et al., 2010, 2016; Zarate et al., 2012; Zhao et al., 2012; Zanos et al., 2016; Farmer et al., 2020). Twelve individual HNK stereoisomers (Fig. 1; characterized by the position of cyclohexyl ring hydroxylation and their unique stereochemistry at two stereocenters) have been identified in the plasma of humans (Moaddel et al., 2010; Zarate et al., 2012; Zhao et al., 2012; Farmer et al., 2020) and in the plasma and brains of rodents (Leung and Baillie, 1986; Moaddel et al., 2016; Zanos et al., 2016) after administration of racemic ketamine. Although the parent compound ketamine is a potent N-methyl-d-aspartate receptor (NMDAR) antagonist and anesthetic (Lodge et al., 1982; Anis et al., 1983; MacDonald et al., 1987), HNKs were initially classified as “inactive” ketamine metabolites because of their lack of anesthetic activity (Leung and Baillie, 1986) mediated by NMDAR inhibition. However, more recent preclinical studies have established various pharmacological and biologic effects of HNKs.

Metabolic formation of hydroxynorketamines from ketamine. (R,S)-ketamine (KET) is N-demethylated to form (R,S)-norketamine (norKET), which is then further metabolized to form the HNKs and dehydronorketamine (DHNK). Via this pathway, (R,S)-norKET is hydroxylated to form the HNKs as shown [see Portmann et al. (2010); Desta et al. (2012)]. Ketamine additionally undergoes direct hydroxylation to form the 6-hydroxyketamines (HKs), which are then N-demethylated to form the (2,6)-HNKs (Portmann et al., 2010; Desta et al., 2012).
Expanding upon its use as an anesthetic, a growing number of studies have reported that ketamine can be used for the treatment of pain, inflammation, and psychiatric conditions, including unipolar and bipolar depression, post-traumatic stress disorder, and obsessive-compulsive disorder [reviewed in Zanos et al. (2018)]. Of particular interest, human clinical trials have provided evidence that ketamine mitigates symptoms of depression more rapidly than many existing antidepressants (acting within hours rather than weeks) [e.g., Berman et al. (2000); Zarate et al. (2006); DiazGranados et al. (2010); Price et al. (2014)]. These findings led to investigations of the mechanisms underlying the antidepressant effects of ketamine. At least partly because of the lack of robust antidepressant-like actions of other NMDAR antagonists, it was hypothesized that ketamine exerts antidepressant actions via an NMDAR inhibition-independent mechanism and that the antidepressant effectiveness of ketamine could be mediated at least in part by the actions of HNKs. Further, it is possible that individual HNKs could exert such effects themselves. This hypothesis has led to several studies seeking to examine the pharmacological properties and potential clinical indications of HNKs. An initial study identified the antidepressant potential of HNKs and revealed that the (2,6)-HNKs have a critical role in mediating at least the long-lasting antidepressant-relevant behavioral effects of ketamine. Additionally, this preclinical study also identified that, when administered directly, (2R,6R)-HNK—and, to a lesser extent, (2S,6S)-HNK—exerts antidepressant-related effects similar to those of ketamine (Zanos et al., 2016). Importantly, in rodents, (2R,6R)-HNK lacks the dissociative side effects and abuse potential that limit the antidepressant application of ketamine (Krystal et al., 1994; Sassano-Higgins et al., 2016; Zanos et al., 2016; Highland et al., 2019; Zanos and Gould, 2018). It appears that (2R,6R)-HNK lacks many of the adverse effects of ketamine since (2R,6R)-HNK does not sufficiently block the NMDAR at pharmacologically relevant concentrations (Suzuki et al., 2017; Zanos et al., 2018; Lumsden et al., 2019; Abbott and Popescu, 2020), a property that led to its early classification as an inactive metabolite (Leung and Baillie, 1986; Zanos et al., 2018).
In addition to exerting rapid antidepressant-relevant effects (Nelson and Trainor, 2007; Zanos et al., 2016, 2019b; Chou et al., 2018; Highland et al., 2019; Pham et al., 2018; Fukumoto et al., 2019; Lumsden et al., 2019; Ko et al., 2020; Rahman et al., 2020; Yokoyama et al., 2020), (2R,6R)- and (2S,6S)-HNK may also have prophylactic stress-protective actions (Chen et al., 2020) and analgesic properties (Kroin et al., 2019), as well as effects on aggression and social behaviors (Ye et al., 2019; Chou, 2020). Although the full mechanisms underlying the preclinically observed behavioral effects of HNKs are still being elucidated, a number of potential sites of action have been identified.
Based upon the presumption that NMDAR inhibition is the primary mechanism of ketamine’s antidepressant actions, it was initially hypothesized that the antidepressant effects of HNKs were also due to NMDAR inhibition. Current evidence suggests that (2R,6R)-HNK does not inhibit NMDAR activity at brain concentrations associated with its preclinical antidepressant-like behavioral effects, but the relevance of NMDAR function to HNKs’ biologic effects remains a subject of debate (Moaddel et al., 2013; Zanos et al., 2016, 2017; Morris et al., 2017; Suzuki et al., 2017; Kavalali and Monteggia, 2018; Lumsden et al., 2019; Abbott and Popescu, 2020).
Numerous studies have reported that (2R,6R)-HNK increases α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR)-dependent synaptic transmission, in part by increasing glutamate release (Pham et al., 2018; Riggs et al., 2019) and AMPAR expression (Zanos et al., 2016; Ho et al., 2018; Shaffer et al., 2019). There is also evidence indicating that the effects of (2R,6R)-HNK converge with metabotropic glutamate (mGlu) receptor signaling (Wray et al., 2019; Zanos et al., 2019b) and activate mammalian target of rapamycin (mTOR) and brain-derived neurotrophic factor (BDNF) signaling (Paul et al., 2014; Zanos et al., 2016; Fred et al., 2019; Fukumoto et al., 2019; Lumsden et al., 2019; Aguilar-Valles et al., 2020; Anderzhanova et al., 2020). There are additional reports demonstrating that (2R,6R)-HNK increases the release of other neurotransmitters, including serotonin and norepinephrine (Ago et al., 2019), and promotes structural plasticity via dendritic outgrowth (Cavalleri et al., 2018; Collo et al., 2018). Recently, (2R,6R)-HNK was also shown to affect other processes, such as inflammatory responses (Ho et al., 2019; Xiong et al., 2019; Rahman et al., 2020) and mitochondrial function (Faccio et al., 2018; Rahman et al., 2020).
Although most studies have focused on the antidepressant potential of HNKs, a variety of possible therapeutic applications exist. These include, but are not limited to, indications for which the parent compound ketamine has shown promise, such as anxiety and mood disorders, post-traumatic stress disorder, obsessive-compulsive disorder, neurologic and peripheral inflammation, and chronic pain or other analgesic applications (Zanos et al., 2018). In this review, we discuss the pharmacokinetic profiles of HNKs after their metabolism from ketamine in humans and other mammals, as well as in rodents treated with HNKs directly. We also review the various biologic effects of HNKs, including the observed synaptic and nonsynaptic pharmacodynamic actions, as well as behavioral effects.
II. Pharmacokinetics
A. Ketamine Metabolism to Hydroxynorketamines
Ketamine undergoes rapid and extensive metabolism, primarily catalyzed by the cytochrome P450 liver enzymes (Adams et al., 1981; Kharasch and Labroo, 1992; Desta et al., 2012). Several metabolites have been identified, including norketamine, dehydronorketamine, and HNKs (Fig. 1) (Adams et al., 1981; Woolf and Adams, 1987; Kharasch and Labroo, 1992; Desta et al., 2012; Farmer et al., 2020). Twelve individual HNK stereoisomers have been detected after ketamine administration to both humans and other animals (Fig. 1) (Leung and Baillie, 1986; Moaddel et al., 2010, 2015, 2016; Zarate et al., 2012; Zhao et al., 2012; Can et al., 2016; Zanos et al., 2016, 2018, 2019b; Fassauer et al., 2017; Pham et al., 2018; Yamaguchi et al., 2018; Farmer et al., 2020).
Ketamine and the HNKs diverge structurally through the demethylation of nitrogen and the addition of a hydroxyl group to the cyclohexanone ring. The relative molecular mass of ketamine is similar to HNK (237.7 vs. 239.1 Da, respectively). In addition, from a physicochemical standpoint, the calculated pKa values of ketamine’s and HNK’s protonated amine groups are similar (∼6.99 vs. ∼6.75, respectively; calculated in PerkinElmer Chemdraw 19). Differences exist, however, in their polar surface area and partitioning coefficient values. The total polar surface area for ketamine is lower than for (2R,6R)-HNK (29.1 vs. 63.3 Å2, respectively; calculated in PerkinElmer Chemdraw 19), whereas ketamine’s calculated partitioning coefficient is somewhat higher than (2R,6R)-HNK’s partitioning coefficient (calculated log P of 2.93 vs. 1.58, respectively; PerkinElmer Chemdraw 19). These differences are driven by the presence of the hydroxyl group, which creates an alternate molecular dipole and another hydrogen bond donor/acceptor. For the HNKs, these physicochemical differences, and the defined stereochemical positioning of substituents on the cyclohexyl ring, influence the overall molecular properties (solubility, lipophilicity, protein binding, etc.) and the potential pharmacological interactions that may influence the pharmacokinetic and pharmacodynamic properties of these agents.
Microsomal studies have reported that ketamine initially undergoes stereoselective N-demethylation, primarily catalyzed by CYP2A6, CYP2B6, and CYP3A4, to form (R,S)-norketamine, which is further metabolized to form dehydronorketamine and HNKs (Fig. 1) (Woolf and Adams, 1987; Kharasch and Labroo, 1992; Portmann et al., 2010; Desta et al., 2012; Dinis-Oliveira, 2017). The cyclohexyl ring of (R,S)-norketamine can be hydroxylated at the 4, 5, or 6 position, resulting in the (2,4)-, (2,5)-, and (2,6)-HNKs, respectively (Woolf and Adams, 1987; Portmann et al., 2010; Desta et al., 2012). Through a minor metabolic pathway, ketamine additionally undergoes direct hydroxylation to form the 6-hydroxyketamines (HKs), primarily catalyzed by CYP2A6 and CYP3A5 (Portmann et al., 2010; Desta et al., 2012), which are then N-demethylated to form the (2,6)-HNKs (Fig. 1) (Woolf and Adams, 1987; Portmann et al., 2010; Desta et al., 2012). Via this pathway, the (2R,6R;2S,6S)-HK metabolite is readily demethylated to form (2R,6R;2S,6S)-HNK (Desta et al., 2012). The predominant cytochrome P450 isoforms involved in the production of the HNKs, via either of the aforementioned pathways, are CYP2A6, CYP2B6, and CYP3A5 (Portmann et al., 2010; Desta et al., 2012). Although both Desta et al. (2012) and Portmann et al. (2010) made tentative assignments for the HNK diastereomers, as standards were not available, both studies concluded that CYP2A6 and CYP2B6 are the predominant cytochrome P450 isoforms involved in the production of the HNKs, via either pathway (Portmann et al., 2010; Desta et al., 2012). Desta et al. (2012) additionally identified CYP3A5 as a predominant isoform involved in the production of HNKs. The analytical method from Desta et al. (2012) was recently updated to allow for conclusive peak assignment (Farmer et al., 2020). Both (2R,6R;2S,6S)-HNK and (2R,5R;2S,5S)-HNK were previously correctly assigned; however, (2R,6S;2S,6R)-HNK was incorrectly assigned and was in fact coeluting with (2R,4R;2S,6S)-HNK (Farmer et al., 2020), indicating that (2R,6S;2S,6R)-HNK is also metabolized from norketamine, consistent with an earlier report from Portmann et al. (2010).
After ketamine administration, HNK metabolites have been detected in humans (Moaddel et al., 2010; Zarate et al., 2012; Zhao et al., 2012; Fassauer et al., 2017; Hasan et al., 2017; Grunebaum et al., 2019; Farmer et al., 2020; Kurzweil et al., 2020) and other animal species, including mice (Zanos et al., 2016, 2018, 2019a; Pham et al., 2018; Yamaguchi et al., 2018), rats (Leung and Baillie, 1986; Moaddel et al., 2015, 2016; Tůma et al., 2020), dogs (Sandbaumhüter et al., 2016, 2017b; Theurillat et al., 2016), and horses/ponies (Lankveld et al., 2006; Schmitz et al., 2009; Sandbaumhüter et al., 2017b). Rapid metabolic conversion of ketamine to HNKs appears to be conserved across species, despite differences in experimental design. HNKs have been detected at the earliest time points studied to date—that is, within 2.5–20 minutes of intraperitoneal or intravenous ketamine dosing in rodents, dogs, and horses (Leung and Baillie, 1986; Moaddel et al., 2015; Zanos et al., 2016, 2019a; Pham et al., 2018; Yamaguchi et al., 2018; Tůma et al., 2020) and immediately upon completion of an intravenous ketamine infusion in humans (Zarate et al., 2012; Zhao et al., 2012; Kurzweil et al., 2020).
In humans, the (2,6)-HNKs are the most abundant HNK stereoisomers in plasma after intravenous ketamine administration (Moaddel et al., 2010; Zarate et al., 2012; Farmer et al., 2020). In particular, (2R,6R;2S,6S)-HNK accounted for approximately 80% of the total plasma concentrations of HNKs after ketamine (0.5 mg/kg, 40-minute i.v. infusion) administration (Farmer et al., 2020). In studies comparing human plasma levels of the (2R,6R)- and (2S,6S)-HNK enantiomers, concentrations of the (2S,6S)-HNK enantiomer were higher than (2R,6R)-HNK at their peak and at 24 hours post–ketamine infusion (Hasan et al., 2017; Grunebaum et al., 2019). However, assessment of changes in metabolite concentration as a function of time [i.e., area under the curve (AUC)] revealed that total plasma concentrations of (2R,6R)-HNK were higher compared with (2S,6S)-HNK, consistent with a longer half-life observed for (2R,6R)-HNK (Hasan et al., 2017).
Similar to findings from human studies, the (2,6)-HNKs are the most abundant HNKs detected in the plasma of dogs and horses after intravenous ketamine injection (Sandbaumhüter et al., 2016, 2017b) and in the plasma and brains of mice and rats after intraperitoneal or intravenous ketamine administration (Moaddel et al., 2016; Zanos et al., 2016). In mice and rats, the (2R,6R)- and (2S,6S)-HNK stereoisomers are more abundant than (2R,6S)- and (2S,6R)-HNK after racemic ketamine dosing (Moaddel et al., 2016; Zanos et al., 2016). Additionally, total plasma concentrations of (2S,6S)-HNK after (S)-ketamine administration exceed that of (2R,6R)-HNK achieved after (R)-ketamine dosing (Moaddel et al., 2015; Zanos et al., 2016, 2018). Studies in dogs and horses have quantitated the (2R,6R)- and (2S,6S)-HNK stereoisomers separately, revealing that the predominant HNK in circulation after racemic ketamine dosing is (2R,6R)-HNK in dogs (Sandbaumhüter et al., 2016, 2017b; Theurillat et al., 2016) and (2S,6S)-HNK in horses (Sandbaumhüter et al., 2017b).
B. Hydroxynorketamine Pharmacokinetics
1. After Ketamine Administration
In preclinical studies, HNKs have been detected in the plasma at the earliest time points tested, as early as 2.5–10 minutes after an intraperitoneal ketamine injection (10 mg/kg) to mice (Zanos et al., 2016, 2019a; Yamaguchi et al., 2018; Lumsden et al., 2019), within 2–10 minutes of an intravenous bolus of ketamine (20–40 mg/kg) to rats (Leung and Baillie, 1986; Moaddel et al., 2015, 2016; Tůma et al., 2020), 2–9 minutes after the start of an intravenous ketamine infusion (1 mg/kg infused over 10 minutes or 1.5/mg/kg per hour infused over 5.3 hours) in horses (Lankveld et al., 2006), and 20 minutes after intravenous ketamine injection (bolus, 2–4 mg/kg) in dogs (Sandbaumhüter et al., 2016). After an intraperitoneal ketamine injection, circulating HNK concentrations reach their maximum within 5–10 minutes in mice (Zanos et al., 2016, 2019a; Yamaguchi et al., 2018). After intravenous ketamine injection, circulating HNK levels peak within 10–36 minutes in rats (40 mg/kg) (Leung and Baillie, 1986; Moaddel et al. 2016) and within 49–68 minutes in dogs (4 mg/kg) (Sandbaumhüter et al., 2016). In mice, (2R,6R)-HNK was also detected in exhaled breath after ketamine dosing (30 mg/kg, i.p.), and peak levels were measured 25–30 minutes after ketamine treatment (Martinez-Lozano Sinues et al., 2017).
In humans receiving an infusion of ketamine (0.5 mg/kg over 40 minutes), the (2R,5R;2S,5S)- and (2R,6R;2S,6S)-HNK stereoisomers were measurable in plasma at the earliest sampling time point (at the end of the infusion period, at 40 minutes) (Zarate et al., 2012; Zhao et al., 2012; Farmer et al., 2020). After the same dose of ketamine (0.5 mg/kg over 40 minutes), peak plasma concentrations were observed at 80 minutes (1.33 hours) postinfusion for (2R,5R;2S,5S)-HNK and at 230 minutes (3.83 hours) postinfusion for (2R,6R;2S,6S)-HNK (Zhao et al., 2012; Farmer et al., 2020). We note that Farmer et al. (2020) provided an updated analytical method from the two earlier studies (Zarate et al., 2012; Zhao et al., 2012), and the results of the updated method are described above. In humans that received an escalating intravenous dose of racemic ketamine (0.25 mg/kg per hour from 0 to 60 minutes, 0.57 mg/kg per hour from 60 to 120 minutes, and 1.14 mg/kg per hour from 120 to 180 minutes) or (S)-ketamine (0.14 mg/kg per gram from 0 to 60 minutes, 0.28 mg/kg per hour from 60 to 120 minutes, and 0.57 mg/kg per hour from 120 to 180 minutes), peak plasma HNK levels (total HNK levels from all HNK stereoisomers) were observed approximately 4 hours [4.03 ± 0.93 hours after racemic and 4.18 ± 0.98 hours after (S)-ketamine; mean ± S.D.] after the end of the infusion period (Kamp et al., 2020). After an intravenous infusion of (S)-ketamine (0.22 mg/kg infused over 40 minutes after an initial bolus injection of 0.11 mg/kg), (2S,6S)-HNK was also detected at the earliest time point studied (at the end of the infusion period) (Kurzweil et al., 2020).
HNKs readily cross the blood-brain barrier and can be detected in the brains of rodents as early as 5–10 minutes after intraperitoneal ketamine dosing in mice and 2–10 minutes after intravenous ketamine dosing in rats (at the earliest time points tested) (Leung and Baillie, 1986; Zanos et al., 2016, 2019a; Yamaguchi et al., 2018). Of note, local metabolism of ketamine to HNKs does not appear to occur in the brain (Moaddel et al., 2015; Zanos et al., 2018); thus, brain levels reflect penetrance from the periphery. Maximum brain concentrations of HNKs are achieved within 10–15 minutes of ketamine dosing in mice (intraperitoneal) and rats (intravenous) (Leung and Baillie, 1986; Moaddel et al., 2015; Zanos et al., 2016, 2019a). The ratio of brain-to-plasma AUCs for both (2R,6R)- and (2S,6S)-HNK is approximately 1:1 (reported between 0.9 and 1.3) in rodents (Moaddel et al., 2015; Zanos et al., 2016; Yamaguchi et al., 2018). After a dose of ketamine that is commonly reported to induce behavioral antidepressant-relevant actions in mice (10 mg/kg, i.p.), peak brain concentrations of (2R,6R;2S,6S)-HNK were reported to be 1.54–2.46 µmol/kg (Table 1) (Zanos et al., 2016, 2019a).
CNS concentrations of (2R,6R)-HNK relevant to antidepressant actions
(2R,6R)-HNK has also been measured in the cerebrospinal fluid (CSF) of mice as early as 5 minutes after (R)-ketamine (intraperitoneal) injection, with peak CSF levels observed at 15 minutes postinjection (Yamaguchi et al., 2018). In the same study, the ratio of total CSF-to-plasma concentrations was reported to be between 0.5 and 0.6 (Yamaguchi et al., 2018). Although brain or CSF levels have not been studied directly in humans, one study used pharmacokinetic modeling to estimate the maximum concentration of unbound (2R,6R;2S,6S)-HNK in the human brain after intravenous administration of a typical antidepressant dose of ketamine (0.5 mg/kg per 40 minutes). This study estimated peak (2R,6R;2S,6S)-HNK levels in the human brain to be to be ≤37.8 ± 14.3 nM (mean ± S.D.; Table 1), which corresponds to previously measured peak plasma levels of 157 ± 59.2 nM (mean ± S.D.; Table 1) (Shaffer et al., 2019).
HNKs undergo glucuronide conjugation, at least partly catalyzed by the UDP- glucuronosyltransferase isoform UGT2B4 (Moaddel et al., 2010), and are eliminated both in their unconjugated and conjugated forms in urine and bile (Chang and Glazko, 1974; Lankveld et al., 2006; Turfus et al., 2009; Moaddel et al., 2010; Dinis-Oliveira, 2017; Sandbaumhüter and Thormann, 2018). In mice, HNKs are detected up to 2–4 hours in the plasma and brain after intraperitoneal ketamine administration (Zanos et al., 2016, 2019a). One study reported that, in rats, (2R,6R;2S,6S)-HNK could be detected (≥5.0 ng/ml) in the plasma up to 48 hours after a single intraperitoneal dose of ketamine, whereas other HNK stereoisomers were below levels of quantitation within 4 hours of ketamine dosing (Moaddel et al., 2016). After a single intravenous ketamine dose, HNKs were detected in the plasma of dogs for at least 7.5 hours (Sandbaumhüter et al., 2016; Theurillat et al., 2016) and in the plasma of horses up to 2 hours after ketamine dosing (Lankveld et al., 2006).
The half-life of (2R,6R;2S,6S)-HNK elimination from the plasma during the terminal phase after a single intraperitoneal ketamine dose was determined to be 13.2 ± 3.7 hours (mean ± S.D.) in rats (Moaddel et al., 2016). After intraperitoneal (R)-ketamine injection, the half-life of (2R,6R)-HNK was 0.56 hours in the plasma and 0.32 hours in the brain of mice (Zanos et al., 2019a). In horses receiving an intravenous infusion of ketamine, the plasma half-life of HNK was approximately 1 hour (median 56.95 minutes), and the clearance was determined to be 2.0–6.8 ml/min per kilogram (median 3.1 ml/min per kilogram) (Lankveld et al., 2006). In humans, circulating (2R,5R;2S,5S)-HNK, could be detected up to 230 minutes (3.83 hours) after a single ketamine infusion (0.5 mg/kg, 40-minute infusion) (Zarate et al., 2012), whereas (2R,6R;2S,6S)-HNK was measurable in plasma (Zarate et al., 2012; Zhao et al., 2012; Grunebaum et al., 2019) and urine (Fassauer et al., 2017) for 1–3 days postinfusion. After intravenous ketamine infusion in humans, the plasma half-life of (2R,6R;2S,6S)-HNK was determined to be approximately 14 hours (14.25 ± 3.95 hours, mean ± S.D.) (Farmer et al., 2020), although (2R,6R;2S,6S)-HNK half-lives were previously reported to show considerable interindividual variability between human subjects (Zhao et al., 2012).
Thus, the HNKs, and in particular (2R,6R;2S,6S)-HNK, are rapidly produced after the administration of ketamine in all species assessed (Leung and Baillie, 1986; Lankveld et al., 2006; Zarate et al., 2012; Zhao et al., 2012; Moaddel et al., 2015, 2016; Sandbaumhüter et al., 2016; Zanos et al., 2016, 2019a; Yamaguchi et al., 2018; Lumsden et al., 2019; Farmer et al., 2020; Tůma et al., 2020). This metabolism occurs mostly, if not entirely, in the liver (Adams et al., 1981; Kharasch and Labroo, 1992; Desta et al., 2012), as there is no documentation of HNK production locally in the brain or other organs. In rodent studies, HNKs readily penetrate the brain (Leung and Baillie, 1986; Zanos et al., 2016, 2019a; Yamaguchi et al., 2018). The elimination of HNKs is variable between species, with reported plasma half-lives ranging from <1 hour in mice (Zanos et al., 2019a) to approximately 14 hours in humans (Farmer et al., 2020).
2. Direct Administration of Hydroxynorketamines
The pharmacokinetic profiles of (2R,6R)-HNK and (2S,6S)-HNK have been studied after their direct administration to mice (Zanos et al., 2016, 2018; Highland et al., 2019; Pham et al., 2018; Yamaguchi et al., 2018; Lumsden et al., 2019), rats (Leung and Baillie, 1986; Moaddel et al., 2015; Highland et al., 2019; Lilius et al., 2018), and dogs (Highland et al., 2019). To date, human pharmacokinetic data for direct HNK administration are not available.
(2R,6R)-HNK and (2S,6S)-HNK are rapidly absorbed after their intraperitoneal or oral administration. After a single intraperitoneal injection to mice, (2R,6R)-HNK and (2S,6S)-HNK have been detected at peak levels in the plasma at the earliest time points sampled (2.5–10 minutes post-treatment) (Zanos et al., 2016; Highland et al., 2019; Yamaguchi et al., 2018; Lumsden et al., 2019). After oral administration, (2R,6R)-HNK and (2S,6S)-HNK have also been detected in the plasma at the earliest time points studied, i.e., 10 minutes postadministration in mice and rats (Moaddel et al., 2015; Highland et al., 2019). Similarly, in dogs, (2R,6R)-HNK was measurable in the plasma at the earliest time point studied, 10 minutes after oral dosing (Highland et al., 2019). After oral administration, peak plasma concentrations of (2R,6R)-HNK occurred within 5–10 minutes for mice and rats (maximum levels observed occurred at the earliest time points studied) and within 30 minutes for dogs (concentrations at 30 minutes surpassed those observed at 10 minutes in dogs) (Highland et al., 2019). Peak plasma concentrations after oral (2S,6S)-HNK occurred at approximately 25 minutes after dosing in rats (Moaddel et al., 2015). The oral bioavailability of (2S,6S)-HNK was reported to be 46.3% in rats (Moaddel et al., 2015), and that of (2R,6R)-HNK was 46.52% in mice, 42% in rats, and 58% in dogs (Highland et al., 2019).
After their intraperitoneal or intravenous administration, (2R,6R)- and (2S,6S)-HNK rapidly cross the blood-brain barrier and have been detected in the brains of rodents as early as 2.5–15 minutes after dosing (at the earliest time points studied) (Leung and Baillie, 1986; Moaddel et al., 2015; Zanos et al., 2016; Highland et al., 2019; Lilius et al., 2018; Yamaguchi et al., 2018; Lumsden et al., 2019), with levels rapidly decreasing thereafter. Although peak brain levels and total concentrations are higher for (2S,6S)-HNK than (2R,6R)-HNK after equivalent dosing in mice (Zanos et al., 2016), the ratio of brain-to-plasma AUCs for both enantiomers is approximately 1:1 in mice and rats (Leung and Baillie, 1986; Moaddel et al., 2015; Highland et al., 2019; Yamaguchi et al., 2018).
(2R,6R)-HNK has also been detected in mouse CSF 5 minutes after systemic (10 mg/kg, i.p.) injection (at the earliest time point studied), and the ratio of CSF-to-plasma AUC was determined to be approximately 1:2 (reported between 1:1.4 and 1:2) (Yamaguchi et al., 2018). After a single intraperitoneal injection in mice, concentrations of (2R,6R)-HNK have additionally been monitored in the extracellular hippocampal compartment, where peak levels were observed 10 minutes after dosing (Lumsden et al., 2019). After a dose of (2R,6R)-HNK frequently reported to exert behavioral effects in mice (10 mg/kg, i.p.), the peak concentrations were reported to be between 10.66 and 18.70 µmol/kg in the brain tissue (Zanos et al., 2016; Pham et al., 2018; Yamaguchi et al., 2018; Lumsden et al., 2019), 7.57 ± 2.13 µM (mean ± S.E.M.) in the extracellular hippocampal space (Lumsden et al., 2019), and 18.40 µM in the CSF (Table 1) (Yamaguchi et al., 2018).
(2R,6R)- and (2S,6S)-HNK remain detectable in the plasma and brain up to 2–4 hours after a single dose in mice (Zanos et al., 2016; Highland et al., 2019), 8–24 hours in rats (Moaddel et al., 2015; Highland et al., 2019), and 12–24 hours in dogs (Highland et al., 2019). The plasma half-life of (2R,6R)-HNK was determined to be 0.2–0.8 hours in mice, 6.9–8.0 hours in rats, and 1.5–1.6 hours in dogs (Highland et al., 2019). The plasma half-life of the (2S,6S)-HNK enantiomer was 8.0 hours in rats (Moaddel et al., 2015). In the brain, the half-life of (2R,6R)-HNK was determined to be 0.4–0.7 hours in mice and 5.7–8.8 hours in rats (Highland et al., 2019). After a single intravenous dose, the clearance of (2R,6R)-HNK was determined to be 51–74 in mice, 27 in rats, and 21 ml/min per kilogram in dogs (Highland et al., 2019), whereas that of (2S,6S)-HNK was approximately 32 ml/min per kilogram in rats (Moaddel et al., 2015).
C. Factors Altering Hydroxynorketamine Pharmacokinetics
Several factors impact the metabolism of ketamine to form HNKs, including biologic sex (Zanos et al., 2016; Highland et al., 2019), circadian phase (Martinez-Lozano Sinues et al., 2017), and prior treatment with other compounds (Sandbaumhüter et al., 2016, 2017a; Lilius et al., 2018; Yamaguchi et al., 2018). For instance, peak and total plasma concentrations of (2R,6R;2S,6S)-HNK after ketamine administration (Zanos et al., 2016) and of (2R,6R)-HNK after direct dosing (Highland et al., 2019) are higher in female than male mice. The circadian phase has also been reported to alter ketamine metabolism to HNKs in mice, with approximately 2-fold higher concentrations of (2R,6R)-HNK being measured in exhaled breath of mice receiving a ketamine injection (30 mg/kg, i.p.) in the evening compared with those injected with the same dose in the morning (Martinez-Lozano Sinues et al., 2017). This effect was determined to be dependent on liver circadian rhythms, as liver-specific deletion of the core clock gene Bmal1 prevented this effect (Martinez-Lozano Sinues et al., 2017). However, this finding has not been confirmed with direct assessment of plasma or other tissues.
Prior treatment with or coadministration of various compounds has also been reported to alter ketamine metabolism and total plasma levels of HNK (Sandbaumhüter et al., 2016, 2017a; Lilius et al., 2018; Yamaguchi et al., 2018). Namely, peak plasma levels of (2R,6R)- and (2S,6S)-HNK were achieved sooner, and total plasma levels were lower in dogs sedated with the α2-adrenoceptor agonist medetomidine prior to ketamine administration compared with those sedated with sevoflurane, despite similar maximum concentrations between the two groups (Sandbaumhüter et al., 2016). This finding may suggest that both metabolism of ketamine to (2R,6R;2S,6S)-HNK and elimination of (2R,6R;2S,6S)-HNK were faster with medetomidine treatment (Sandbaumhüter et al., 2016). However, it is also possible that sevoflurane-induced vasodilation (Ebert et al., 1995; Ebert, 1996) increases tissue distribution of ketamine and HNK, thereby contributing to the earlier peak concentrations observed. An in vitro study also suggested that α2-adrenoceptor agonists can alter ketamine metabolism to HNKs, demonstrating that the formation of (2R,6R;2S,6S)-HNK from ketamine is attenuated in liver microsomes incubated with medetomidine (Sandbaumhüter et al., 2017a). However, the same study found that three other α2-adrenoceptor agonists, detomidine, xylazine, and romifidine, had the opposite effect, resulting in higher levels of (2R,6R;2S,6S)-HNK (Sandbaumhüter et al., 2017a). The interactions with medetomidine are likely cytochrome P450–mediated, as CYP3A plays an important role in the metabolism of both medetomidine and ketamine (Duhamel et al., 2010; Desta et al., 2012; Sandbaumhüter et al., 2015). Additionally, one study demonstrated that administration of ketamine (10 mg/kg, s.c.) resulted in higher serum and brain levels of (2R,6R;2S,6S)-HNK in morphine-treated rats (6 days of continuous morphine administration, 9.6 mg/d via osmotic minipumps) compared with vehicle-treated rats (Lilius et al., 2018). Finally, as expected, prior treatment with the cytochrome P450 inhibitors ticlopidine and 1-aminobenzotriazole led to a robust attenuation of (2R,6R)-HNK formation after (R)-ketamine administration (10 mg/kg, i.p.) in mice (Yamaguchi et al., 2018). A chemical modification of the structure of ketamine has also been used to alter its metabolism to HNKs; namely, di-deuterium substitution at the C6 position of the hydroxyl ring of ketamine or (R)-ketamine [designated as d2-ketamine and (R)-d2-ketamine, respectively] results in a selective and robust attenuation of the metabolism of ketamine to the (2,6)-HNKs or (2R,6R)-HNK, respectively (Zanos et al., 2016, 2019a; Zhang et al., 2018b). Of note, one study examined whether CYP2B6 genotype variants altered the N-demethylation of ketamine and subsequent formation of HNKs but reported that no statistically significant differences were observed for HNK plasma concentrations, AUCs, elimination half-lives, or clearance between CYP2B6 genotypes (Rao et al., 2016).
III. Pharmacodynamics
A. Synaptic Effects
1. Glutamatergic Actions
a. N-methyl-d-aspartate receptor function
NMDARs are ionotropic glutamate receptors involved in fast excitatory synaptic transmission [reviewed in Vyklicky et al. (2014); Hansen et al. (2018); Scheefhals and MacGillavry (2018)]. A higher Ca2+ permeability of NMDARs compared with other ionotropic glutamatergic receptors makes the NMDAR an essential transducer of intracellular Ca2+, which acts as a second messenger to modulate the efficacy of synaptic transmission [reviewed in Vyklicky et al. (2014); Hansen et al. (2018)]. Ketamine is a well established NMDAR antagonist that acts as an open channel blocker at hyperpolarized membrane potentials [e.g., Lodge et al. (1982); Anis et al. (1983); MacDonald et al. (1987)]. NMDAR inhibition underlies the anesthetic effects of ketamine and is also linked to its dissociative properties and abuse potential [see Zanos et al. (2018)]. The lack of NMDAR inhibition at concentrations generated by antidepressant-relevant doses of (2R,6R)-HNK initially suggested that HNKs may exert their antidepressant-relevant effects through NMDAR inhibition–independent pathways (Zanos et al., 2016; Morris et al., 2017). However, the question as to whether NMDAR inhibition plays a role in the effects of HNKs continues to be debated (Morris et al., 2017; Suzuki et al., 2017; Zanos et al., 2017; Kavalali and Monteggia, 2018; Lumsden et al., 2019; Abbott and Popescu, 2020).
Binding affinity studies demonstrated that some HNKs are capable of binding to the same NMDAR binding site as ketamine, albeit at higher concentrations than ketamine (Ki 0.25–1.06 µM). Specifically, (2S,6S)-HNK exhibits a greater NMDAR binding affinity (Ki between 7.34 and 21.19 µM) than any other HNK stereoisomer [Ki of (2R,6R)-, (2R,6S)-, (2S,6R)-, (2R,5R)-, (2S,5S)-, (2R,5S)-, (2S,5R)-, (2R,4R)-, (2S,4S)-, (2R,4S)-, and (2S,4R)-HNK are all >100 µM] (Moaddel et al., 2013; Morris et al., 2017). Consistent with this, Zanos et al. (2016) demonstrated that neither (2R,6R)- nor (2S,6S)-HNK displaced NMDAR binding of radiolabeled MK-801 at the concentration of 10 µM. Thus, although (2S,6S)-HNK does displace MK-801 with lower affinity than ketamine, other HNK stereoisomers have substantially lower capacity to bind NMDARs than either (2S,6S)-HNK or ketamine.
It is possible that HNKs may bind NMDARs at a site distinct from that of MK-801. Lumsden et al. (2019) demonstrated that, when compared with (2R,6R)-HNK, (2S,6S)-HNK has a 12- to 22-fold greater potency to block glutamate-evoked NMDAR-mediated currents in Xenopus laevis oocytes, regardless of subunit composition (Table 2). Similarly, Abbott and Popescu (2020) demonstrated that (2R,6R)-HNK inhibits glutamate-evoked whole-cell currents in HEK-293 cells ectopically expressing GluN2A- and GluN2B-containing NMDARs with approximately 100-fold lower potency than ketamine (Table 2). The same study reported that, although inhibition of NMDARs by high concentrations of (2R,6R)-HNK is voltage-dependent (similar to inhibition by ketamine), (2R,6R)-HNK has slower association and dissociation constants compared with ketamine (Abbott and Popescu, 2020). Further, this study highlighted that (2R,6R)-HNK–induced inhibition of NMDARs is reduced in the presence of extracellular Mg2+ (Abbott and Popescu, 2020), suggesting that (2R,6R)-HNK may interact with both the open and closed states of NMDARs, as has previously been reported for ketamine (Orser et al., 1997).
NMDAR ligand binding and functional inhibition
Ketamine Ki and IC50 values are only reported from studies that compared the effects of ketamine to HNKs under similar experimental conditions.
In mouse hippocampal slices, (2R,6R)- and (2S,6S)-HNK inhibited NMDAR-mediated field excitatory potentials (fEPSPs) with IC50 values of 211.9 and 47.2 μM, respectively, both higher than that of ketamine (IC50 = 4.5 µM) (Lumsden et al., 2019). Similarly, in rat hippocampal slices, (2R,6R)-HNK reduced the mean amplitude of NMDAR-mediated miniature excitatory postsynaptic currents (mEPSCs) with an IC50 of 63.7 μM, an apparent potency approximately 10-fold lower than that of ketamine (IC50 = 6.4 μM; see Table 2) (Lumsden et al., 2019). Consistent with these findings, Suzuki et al. (2017) reported that at 50 µM, (2R,6R)-HNK reduced the amplitude of NMDAR-mEPSCs in cultured hippocampal neurons, whereas no effect was observed at a concentration of 10 µM. Likewise, the potency of (2R,6R)-HNK to attenuate NMDA-induced lethality in mice (ED50 = 227.8 mg/kg), a historical measure of in vivo NMDAR inhibition, is approximately 12- and 35-fold lower than the observed potencies of (2S,6S)-HNK (ED50 = 18.6 mg/kg) or ketamine (ED50 = 6.4 mg/kg) (Lumsden et al., 2019). We note there are discrepancies among the published IC50 values, which may be explained by the different preparations used, distinct cellular location and receptor subtypes assessed, and the use of varying extracellular Mg2+ concentrations. Altogether, these data show that the observed potency of (2S,6S)-HNK and other HNKs to displace ligand binding from, and inhibit the activity of, NMDARs is lower than that of ketamine (Table 2). Of note, the lack of robust NMDAR inhibition by (2R,6R)-HNK may underlie its lower apparent adverse effect burden and abuse potential in preclinical studies, in contrast to ketamine (described in section IV.A.4. Characterization of Adverse Behavioral Effects).
A study revealed that a 2-hour preincubation of mouse hippocampal slices with ketamine, (2S,6S)-HNK, or (2R,6R)-HNK attenuated long-term potentiation at Schaffer collateral synapses with an IC50 of 1.6, 1.0, and 7.8 μM, respectively (Kang et al., 2020). It was concluded that the NMDAR is the site of action for these compounds, as synaptic long-term potentiation (LTP) requires NMDAR-mediated synaptic transmission to be intact. However, considering the above-described effects of (2R,6R)-HNK on NMDAR function in these concentration ranges (Table 2), an alternative explanation is that LTP was occluded after functional and structural enhancements that occurred during incubation (see next sections). It is also unclear why (2S,6S)-HNK exerted a more potent effect than ketamine while being a weaker inhibitor of NMDAR function (Table 2). Although the authors showed that fEPSPs did not differ among the groups at baseline, a more detailed examination of the input-output relationship with regard to AMPAR- and NMDAR-mediated currents would be needed to rule this possibility out.
b. Presynaptic glutamatergic mechanisms
A growing body of evidence suggests that, largely via facilitation of synaptic glutamate release, (2R,6R)-HNK causes a rapid increase of glutamatergic neurotransmission mediated by AMPARs (see Table 3) (Chou et al., 2018; Pham et al., 2018; Riggs et al., 2019), which is followed by a delayed enhancement in AMPAR expression (Zanos et al., 2016; Ho et al., 2018; Shaffer et al., 2019). AMPARs, similar to NMDARs, are ionotropic glutamate receptors that participate in fast synaptic transmission [reviewed in Chater and Goda (2014); Scheefhals and MacGillavry (2018)]. AMPARs are an essential regulator of synaptic plasticity, with increases in their expression and/or function leading to a long-lasting increase in synaptic transmission efficacy [reviewed in Derkach et al. (2007); Chater and Goda (2014)].
Effects on synaptic glutamatergic transmission
Contained in this table are synaptic effects other than direct effects on NMDAR function, which are shown in Table 2. Studies and experiments that did not identify an effect are not summarized in this table; please refer to the text for these details.
Systemic administration of (2R,6R)-HNK (10 mg/kg, i.p.) leads to an increase (measured 24 hours post-treatment) in extracellular glutamate concentrations in the prefrontal cortex of mice (Pham et al., 2018). These effects are independent of changes in glutamate reuptake and are mimicked by local intracortical perfusion of (2R,6R)-HNK (bilateral infusion, 1 nmol per side) (Pham et al., 2018). Similarly, systemically administered (2R,6R)-HNK (30 mg/kg, i.p.) induces an increase (observed 24 hours post-treatment) in the frequency and amplitude of hypocretin- and serotonin-evoked spontaneous excitatory postsynaptic currents recorded from pyramidal neurons in the medial prefrontal cortex (Fukumoto et al., 2019).
In the hippocampus, (2R,6R)-HNK induces a concentration-dependent potentiation of glutamatergic synaptic transmission (EC50 = 3.3 µM), which is correlated with reductions in paired-pulse facilitation (IC50 = 3.8 µM) and occluded by conditions of high release probability (Riggs et al., 2019). Additionally, (2R,6R)-HNK increases the frequency, but not amplitude, of mEPSCs recorded from CA1 pyramidal neurons (Riggs et al., 2019). Altogether, these results support the conclusion that (2R,6R)-HNK increases the probability of glutamate release in the hippocampus, which does not appear to require NMDAR activation (Riggs et al., 2019) (effects on NMDAR function are discussed in section III.A.1.a. N-methyl-d-aspartate receptor function). Interestingly, the maintenance of hippocampal Schaffer collateral long-term potentiation is impaired in vivo in stress-susceptible Wistar-Kyoto rats (Aleksandrova et al., 2019), which is restored within 3.5 hours of systemic (2R,6R)-HNK (5 mg/kg, i.p.) administration (Aleksandrova et al., 2020).
In contrast to the findings above, one study reported that a low dose of (2R,6R)-HNK (0.075 mg/kg, i.p.) led to an attenuation of both AMPAR- and NMDAR-mediated hippocampal pyramidal burst firing one week post-treatment, whereas (2S,6S)-HNK (0.075 mg/kg, i.p.) attenuated only NMDAR-mediated bursts (Chen et al., 2020). It is unclear whether differences in doses or experimental preparation contributes to these seemingly contradictory findings (i.e., attenuation of AMPAR-mediated transmission at lower doses versus enhancement of AMPAR-mediated transmission at higher doses/concentrations) in the hippocampus. Additionally, Hare et al. (2020) reported that (2R,6R)-HNK (30 mg/kg, i.p.) did not enhance optically recorded bulk Ca2+ signals in pyramidal neurons of the ventromedial prefrontal cortex in vivo. However, the data collectively suggest that HNKs exert AMPAR-dependent effects on glutamatergic synaptic transmission (Zanos et al., 2016; Yao et al., 2018; Riggs et al., 2019; Ye et al., 2019; Chen et al., 2020). Importantly, these effects are not mediated by direct actions on AMPARs, as neither (2R,6R)- nor (2S,6S)-HNK (10 µM) directly bind to or activate AMPARs (Riggs et al., 2019; Shaffer et al., 2019).
In addition to the prefrontal cortex and hippocampus, (2R,6R)-HNK exerts effects on glutamatergic synaptic activity in midbrain structures, including the ventrolateral periaqueductal gray. Systemic administration of (2R,6R)-HNK (10 mg/kg, i.p.) restores the expression of Ca2+-permeable AMPARs, the magnitude of inward AMPA-evoked currents, the probability of glutamate release, and the frequency and amplitude of mEPSCs in excitatory ventrolateral periaqueductal gray projection neurons after chronic stress in rats (Chou et al., 2018; Ye et al., 2019). Additionally, these effects are mimicked by bath application of (2R,6R)-HNK (10 µM) in ventrolateral periaqueductal gray slices prepared from chronically stressed rats (Chou et al., 2018). These findings are consistent with a postsynaptic component in addition to presynaptic actions described in this section, which could be brain region–dependent. Of note, these actions in the ventrolateral periaqueductal gray are thought to underlie the proaggressive and antidepressant-relevant behavioral effects observed in (2R,6R)-HNK–treated mice (Chou et al., 2018; Ye et al., 2019), as discussed in section IV.A.4. Characterization of Adverse Behavioral Effects.
The effects of (2R,6R)-HNK on AMPAR-mediated synaptic activity have also been studied in the nucleus accumbens and ventral tegmental area (Yao et al., 2018). (2R,6R)-HNK (10 mg/kg, i.p.) administration in mice leads to a reduction in frequency and amplitude of mEPSCs in dopaminergic neurons in the ventral tegmental area, along with a decrease in AMPAR, relative to NMDAR, transmission 24 hours after treatment (Yao et al., 2018). We note that these effects are generally opposite of what has been observed in the hippocampus (Zanos et al., 2016; Riggs et al., 2019). This is in line with the finding that increased plasticity in the hippocampus is associated with antidepressant-like actions, whereas decreased plasticity is associated with similar effects in the ventral tegmental area to nucleus accumbens pathway (Berton and Nestler, 2006; Yu and Chen, 2011). Consistent with this, in the nucleus accumbens, (2R,6R)-HNK (10 mg/kg, i.p.) attenuated the magnitude and prevalence (i.e., proportion of slices where observed) of LTP, but without altering paired-pulse ratios or input-output curves (Yao et al., 2018). These data suggest that (2R,6R)-HNK can modulate the efficacy with which plasticity can be induced while leaving basic properties of synaptic transmission intact. Altogether, the literature supports that (2R,6R)-HNK exerts both rapid and sustained synaptic effects in various regions of the mesocorticolimbic system. These synaptic effects of (2R,6R)-HNK may underlie changes in network-wide excitation, as systemically administered (2R,6R)-HNK (10 mg/kg, i.p.) leads to an increase in electrocorticographic high-frequency γ oscillations (30–80 Hz) in mice (Zanos et al., 2016, 2019b), a marker of cortical activation. Similarly, in humans that received an infusion of ketamine for the treatment of depression, peak plasma (2R,6R;2S,6S)-HNK levels were associated with increased resting-state whole-brain magnetoencephalography γ power 6–9 hours postinfusion (Farmer et al., 2020). However, in contrast to this, one study failed to detect an increase in cortical γ power after administration of a racemic mixture of (2R,6R;2S,6S)-HNK (20 mg/kg, i.p.) to mice (Kohtala et al., 2019). Future studies are required to fully understand the effects of (2R,6R)- versus (2S,6S)-HNK on γ power and the relevance of this measure to therapeutic outcomes.
There is evidence to suggest that cAMP-dependent mechanisms (Wray et al., 2019), possibly via group II metabotropic glutamate (mGlu2) receptor signaling (Zanos et al., 2019b), are implicated in the (2R,6R)-HNK–induced synaptic potentiation of excitatory synapses. mGlu2 receptors are presynaptic autoreceptors that are negatively coupled to adenylyl cyclase and suppress cAMP-dependent glutamate release under normal conditions [reviewed in Schoepp (2001)]. It has been demonstrated that (2R,6R)-HNK has a convergent mechanism of action with mGlu2 receptors; in particular, the (2R,6R)-HNK–induced increase in cortical γ oscillations is blocked by mGluR2/3 activation, whereas inhibiting mGluR2/3 activity enhances the effects of a subeffective antidepressant-relevant (2R,6R)-HNK dose (1 mg/kg, i.p.) (Zanos et al., 2019b). (2R,6R)-HNK (10 mg/kg, i.p.) also prevents mGlu2/3 receptor agonist–induced hyperthermia in mice, a physiologic assay of mGlu2/3 receptor activity (Zanos et al., 2019b). Despite this evidence, we note that no specific binding of (2R,6R)-HNK to the mGlu2 receptor or direct effects of (2R,6R)-HNK on mGlu2 receptor have been identified. Thus, the convergent effects may be upstream or downstream of the actual receptor. Furthermore, the behavioral effects of (2R,6R)-HNK in preclinical tests thought to predict antidepressant efficacy can be bidirectionally altered by mGlu2 receptor activity modulation (Zanos et al., 2019b), as described in section IV.A.1. Antidepressant-Relevant Behaviors. As (2R,6R)-HNK (10 µM; 15-minute exposure) has been shown to induce redistribution of the G protein GαЅ to nonlipid raft regions, resulting in a robust increase in cAMP accumulation in C6 cells (Wray et al., 2019), it is possible that (2R,6R)-HNK acts through an mGlu2 receptor– and cAMP-dependent pathway to modulate glutamate release and enhance network activity.
c. Targets downstream of glutamate receptor activation
(2R,6R)- and (2S,6S)-HNK have been shown to modulate the activity of targets downstream of glutamate receptors, including BDNF (Zanos et al., 2016; Fukumoto et al., 2019; Lumsden et al., 2019; Anderzhanova et al., 2020) and mTOR (Paul et al., 2014; Fukumoto et al., 2019; Lumsden et al., 2019). These effects are summarized in Table 4. For instance, hippocampal BDNF levels are increased in mice 30 minutes (Lumsden et al., 2019) and 24 hours after (2R,6R)-HNK treatment (10 mg/kg, i.p.) (Zanos et al., 2016). (2S,6S)-HNK (10 mg/kg, i.p.) has also been demonstrated to increase extracellular BDNF levels in the prefrontal cortex of mice 30–90 minutes after treatment (Anderzhanova et al., 2020). Additionally, (2R,6R)-HNK (10 and 50 nM) induced a rapid (after 1-hour exposure) AMPAR-dependent increase in BDNF release in rat primary cerebral cortical cultures (Fukumoto et al., 2019). (2R,6R)-HNK (10–100 µM) may also promote maintenance of tropomyosin receptor kinase B (TrkB) at the cell surface by inhibiting interactions between TrkB and the AP-2 adaptor protein complex (involved in vesicular endocytosis), thereby enhancing BDNF signaling (Fred et al., 2019). As discussed in section IV.A.1. Antidepressant-Relevant Behaviors, BDNF/TrkB signaling is thought to have an essential role in mediating the antidepressant-relevant behavioral effects of (2R,6R)-HNK in mice (Fukumoto et al., 2019). This conclusion is supported by the finding that ketamine and (2R,6R)-HNK, but not (2S,6S)-HNK, binds directly to the TrkB receptor, leading to increases in BDNF signaling (Casarotto et al., 2021). It was also observed that a single administration of (2R,6R)-HNK (10 mg/kg i.p.) induced plasticity in the visual cortex of adult mice as measured by ocular dominance following monocular deprivation, which was prevented in mice harboring a genetic mutation of TrkB (Casarotto et al., 2021).
Biochemical effects implicating glutamatergic neurotransmission
Studies and experiments that did not identify an effect are not summarized in this table; please refer to the text for these details.
The mTOR pathway, a downstream target of BDNF/TrkB, has also been shown to be increased by HNKs (Paul et al., 2014; Singh et al., 2016; Fukumoto et al., 2019; Lumsden et al., 2019). In particular, increased mTOR phosphorylation was observed in vivo in the cerebral cortex 20–60 minutes after intravenous treatment of rats with (2S,6S)-HNK treatment (20 mg/kg) and in vitro in PC12 cells exposed for 1 hour to (2S,6S)-HNK (0.01–1 nM) (Paul et al., 2014). Similarly, mTOR phosphorylation was increased in the hippocampus of mice 30 minutes after an intraperitoneal injection of (2R,6R)-HNK (10 mg/kg) (Lumsden et al., 2019) and in the prefrontal cortex of mice 30 minutes after an intraperitoneal injection of (2R,6R)-HNK (30 mg/kg) (Fukumoto et al., 2019).
The effects of HNKs on downstream synaptic signaling proteins have also been studied. In particular, 20–60 minutes after administration of (2S,6S)-HNK (20 mg/kg, i.v.) to rats, levels of the phosphorylated forms of eukaryotic initiation factor 4E binding protein (p4E-BP1) and p70S6 kinase in the prefrontal cortex were significantly higher than those in the same brain region of control rats (Paul et al., 2014). In addition, a 1-hour exposure of PC12 cells to (2S,6S)-HNK concentrations ranging from 0.1 to 1 nM significantly increased 1) p4E-BP1 expression and 2) phosphorylation of extracellular signal–related kinases (pERK) and protein kinase B (Paul et al., 2014). Similarly, pERK was increased in rat primary cerebral cortical cultures after a 1-hour exposure to (2R,6R)-HNK (1–50 nM), an effect that was dependent on TrkB and AMPAR activity (Fukumoto et al., 2019). Evidence indicates that synthesis through eukaryotic elongation factor 2 (eEF2) dephosphorylation has been linked to ketamine’s antidepressant action (Kavalali and Monteggia, 2020). (2R,6R)-HNK induced a decrease in eEF2 phosphorylation in the hippocampus of mice 1 hour and 24 hours after injection (Zanos et al., 2016) and after 30 minutes of exposure to mouse cultured primary hippocampal neurons (Suzuki et al., 2017).
Notably, there are some discrepancies in the reported (2R,6R)-HNK–induced changes in mTOR and BDNF pathway activation. Although Fukumoto et al. (2019) observed increases in mTOR phosphorylation in the mouse prefrontal cortex at 30 minutes post-treatment (30 mg/kg, i.p.), they found no changes at 60 minutes post-treatment. Likewise, no changes in mTOR expression or phosphorylation were detected in the hippocampus or prefrontal cortex 1 or 24 hours after treatment of mice with (2R,6R)-HNK (10 mg/kg, i.p.) (Zanos et al., 2016). Furthermore, one study reported no changes in TrkB-glycogen synthase kinase 3β (GSK-3β) signaling in mice 20 minutes after treatment with a 1:1 mixture of (2S,6S)-HNK and (2R,6R)-HNK (20 mg/kg, i.p.) (Kohtala et al., 2019). Finally, it has been reported that 3 days after treatment of mice with (2R,6R)-HNK (10 mg/kg, i.p.), BDNF mRNA expression in the dentate gyrus and CA3 field of the hippocampus was comparable to that measured in control mice (Herzog et al., 2020). Likewise, BDNF expression in the prefrontal cortex of mice 1 or 24 hours after treatment with (2R,6R)-HNK (10 mg/kg, i.p.) was comparable to that measured in control mice (Zanos et al., 2016). However, differences in brain regions, rodent species and/or strain, time course of treatment and testing, or assessment methods may explain these discrepancies.
In addition to the rapid AMPAR-dependent effects mediated by HNK-induced glutamate release, several studies have provided evidence supporting the conclusion that the sustained antidepressant-relevant effects of HNKs may be a result of an increase in the synaptic expression of AMPARs and a resulting sustained increase in synaptic strength (Table 5) (Zanos et al., 2016; Ho et al., 2018; Shaffer et al., 2019). Expression of AMPAR GluA1 and GluA2 subunits is increased in the hippocampal synaptic fractions 24 hours, but not 1 hour, after systemic administration of (2R,6R)-HNK (10 mg/kg, i.p.) to mice (Zanos et al., 2016). A time- and concentration-dependent increase in AMPAR surface expression has also been observed after either a 90-minute (1–10 µM) or 180-minute (0.1–10 µM) exposure (but not a 30-minute exposure) of rat primary hippocampal cultures to (2R,6R)-HNK (Shaffer et al., 2019). However, in the same study, no changes in AMPAR surface expression were observed after a 180-minute exposure of the primary cultures to (2S,6S)-HNK (0.1–10 µM) (Shaffer et al., 2019).
Changes in AMPAR expression
Studies and experiments that did not identify an effect are not summarized in this table; please refer to the text for these details.
In U251-MG human glioblastoma cells, 24-hour exposures to (2R,6R)-HNK or (2S,6S)-HNK increased expression of GluA1 (200–400 nM), GluA2 (400 nM), and GluA4 (400 nM) mRNA but had no significant effect on GluA3 mRNA expression (Ho et al., 2018). In primary cultures of human astrocytes, (2S,6S)-HNK (400 nM), but not (2R,6R)-HNK (400 nM), increased GluA1 and GluA2 expression, and both HNKs increased expression of GluA4 (Ho et al., 2018). Further, application of estradiol in conjunction with (2R,6R)- and (2S,6S)-HNK increased the expression of AMPAR subunits in primary cultures of human astrocytes and in cultures of U251-MG cells, an effect that was attributed to the HNKs acting as estrogen receptor α (ERα) ligands (Ho et al., 2018). In particular, this study demonstrated that 1) the expression of ERα mRNA in U251-MG cells was increased by (2R,6R)- and (2S,6S)-HNK, 2) HNK-induced AMPAR subunit changes were prevented by ERα knockdown, and 3) (2R,6R)- and (2S,6S)-HNK were capable of binding to ERα (Ho et al., 2018).
As discussed in section IV. Behavioral Effects, AMPAR activity appears to be required for (2R,6R)-HNK to exert its antidepressant-relevant behavioral outcomes (Zanos et al., 2016) and effects on aggressive behavior (Ye et al., 2019; Chou, 2020). Likewise, HNK-induced actions on several antidepressant-relevant signaling pathways are AMPAR-dependent.
2. Other Neurotransmitters
In addition to increasing the activity of the glutamatergic system, HNKs have been shown to affect several other neurotransmitter systems (Can et al., 2016; Pham et al., 2018; Ago et al., 2019). For instance, (2R,6R)-HNK, but not (2S,6S)-HNK, increased serotonin (10–20 mg/kg, i.p.) (Pham et al., 2018; Ago et al., 2019) and norepinephrine (20 mg/kg, i.p.) (Ago et al., 2019) levels in the mouse medial prefrontal cortex. Notably, the finding that, in vitro, neither (2R,6R)-HNK nor (2S,6S)-HNK affected functional activity on serotonin and norepinephrine transporters at concentrations up to 10 μΜ (Can et al., 2016) suggests that (2R,6R)-HNK–induced increase in serotonin and norepinephrine levels are not accounted for by reduced activity of the transporters.
In vivo, neither (2R,6R)- nor (2S,6S)-HNK treatment (20 mg/kg, i.p.) altered extracellular dopamine levels in the prefrontal cortex of mice (Ago et al., 2019). In addition, in vitro, (2R,6R)-, (2S,6S)-, (2S,6R)-, or (2R,6S)-HNK (up to 10 µM) had no significant effect on ligand binding to or functional activity of D1-5 dopamine receptors or monoamine transporters (Can et al., 2016). Consistent with this, (2R,6R)-HNK (10 mg/kg, i.p.) did not alter the intrinsic properties of mouse ventral tegmental area dopaminergic neurons or the basic properties of synaptic transmission in the nucleus accumbens, which also receives substantial input from the ventral tegmental area (Yao et al., 2018). These data suggest that (2R,6R)-HNK likely exerts indirect effects on monoaminergic transmission through dynamic regulation of glutamate signaling. This finding may be relevant to the lack of abuse potential of (2R,6R)-HNK compared with ketamine observed in preclinical tests (see section IV.A.4. Characterization of Adverse Behavioral Effects).
The effects of (2R,6R)-HNK and (2S,6S)-HNK on multiple subtypes of neuronal nicotinic receptors have also been assessed. Up to 100 μM, neither (2R,6R)-HNK nor (2S,6S)-HNK induced activation of α3β4 nicotinic receptors ectopically expressed in the KXa3b4R2 cell line (Moaddel et al., 2013). At high micromolar concentrations, both (2R,6R)-HNK and (2S,6S)-HNK inhibited (S)-nicotine–induced whole-cell currents in this cell line. Their IC50 values were ∼350 μM, indicating that these compounds had no effect on α3β4 nicotinic acetylcholine receptor activity at antidepressant-relevant concentrations (see Table 1). However, in the KXα7R1 cell line expressing α7 nicotinic receptors, (2R,6R)-HNK and (2S,6S)-HNK at both 100 and 1000 nM inhibited acetylcholine-evoked currents (Moaddel et al., 2013). Application of the whole-cell mode of the patch-clamp technique to interneurons in the stratum radiatum in the CA1 field of the hippocampus [as described in Alkondon et al. (1999)] revealed that (2R,6R)-HNK (10 µM) is also devoid of agonistic activity on native α7 nicotinic receptors. Specifically, U-tube application of (2R,6R)-HNK (10 µM, 5-second pulses) evoked no whole-cell current in CA1 stratum radiatum interneurons that at −60 mV had responded to 5-second pulses of acetylcholine (1 mM) or choline (10 mM) with rapidly decaying currents that were inhibited by the α7 nicotinic receptor–selective antagonist methyllycaconitine (10 nM). In addition, 20-minute superfusion of hippocampal slices with artificial cerebrospinal fluid containing 10 µM (2R,6R)-HNK had no significant effect on the amplitude of choline-evoked (10 mM) whole-cell currents recorded from CA1 stratum radiatum interneurons voltage clamped at −60 mV (M. Alkondon, unpublished data).
3. Effects on Morphology and Structural Plasticity
Changes in the efficacy of synaptic transmission can correspond to structural modifications to synaptic connections, including changes in the size of the cell body; number and/or length of dendrites (i.e., dendritic arborization/retraction); and/or number, size, and shape of dendritic spines. It has been demonstrated that (2R,6R)-HNK can promote such structural and morphologic changes (see Table 6) (Cavalleri et al., 2018; Collo et al., 2018).
Effects of (2R,6R)-HNK on cellular morphology
Subeffective or ineffective concentrations or doses are not included in this summary table. Please refer to the text for full details.
Using a translational in vitro approach, Cavalleri et al. (2018) investigated the time-dependent effects of (2R,6R)-HNK on neuronal morphology, given that it remains in circulation at sub-micromolar concentrations for several hours after ketamine administration in humans (Zhao et al., 2012). The authors demonstrated that (2R,6R)-HNK (0.5 µM; 1- to 6-hour exposure) leads to dendritic outgrowth in primary cultures of mouse mesencephalic dopamine neurons and in cultures of induced pluripotent stem cell (iPSC)-derived dopamine neurons from healthy donors. A 1-hour exposure to (2R,6R)-HNK increased the number and length of dendrites, as well as the size of dopaminergic neuronal cell bodies, effects which were more robust after a 6-hour incubation period (Cavalleri et al., 2018). This process was later shown to be AMPAR- and mTOR-dependent (Collo et al., 2018). These data support that (2R,6R)-HNK can induce structural plasticity of mouse- and human-derived dopaminergic neurons, with a time course and concentration dependence relevant to those observed after ketamine administration in patients with depression.
Additional studies have investigated whether HNKs induce structural changes in other brain regions and in cell cultures. In particular, although Michaëlsson et al. (2019) found a ketamine-induced (10 mg/kg, i.p.) increase in hippocampal cell proliferation in the dentate gyrus in vivo in rats, neither ketamine nor a racemic mixture of (2R,6R;2S,6S)-HNK (1–100 µM) altered cell proliferation in neurosphere cultures. However, the effects of racemic mixtures of HNKs, as well as of the individual HNK stereoisomers, on in vivo cell proliferation were not investigated. Another study evaluated the spine density of medial prefrontal cortical pyramidal neurons in mice 24 hours after systemic treatment with (2R,6R)-HNK (30 mg/kg, i.p.) but did not observe any HNK-induced changes in proximal or distal apical dendrites (Fukumoto et al., 2019). Collectively, the literature suggests that (2R,6R)-HNK initiates intracellular signaling, potentiates synaptic transmission, promotes BDNF release, activates downstream signaling pathways, and induces selective changes in neuronal structures that can support sustained increases in synaptic transmission.
B. Nonsynaptic Effects
1. Effects on Inflammatory Processes
Ketamine exhibits anti-inflammatory properties, reducing the levels of both proinflammatory cytokines, such as interleukin-6 and tumor necrosis factor-α, and nitric oxide [reviewed in Zanos et al. (2018)]. Consequently, several investigators have sought to understand whether HNKs also exert anti-inflammatory effects.
Recent work by Ho et al. (2019) delved into the potential anti-inflammatory effects of ketamine and HNKs. Using cultures of a human microglial cell line (HMC3 cells), they conducted transcriptome analysis after a 24-hour exposure to (2S,6S)-HNK and (2R,6R)-HNK (400 nM). The authors reported a significant increase in indicators of type I interferon pathway activity induced by both HNKs. Exposure of HMC3 cell cultures to (2S,6S)-HNK and (2R,6R)-HNK also increased expression and nuclear translocation of signal transducer and activation of transcription 3 (Ho et al. (2019)), a transcription factor important for interferon pathway regulation and gene expression. Notably, signal transducer and activation of transcription 3 binds to eEF2 in the cytoplasm prior to translocating to the nucleus, which may explain, at least in part, the reduction in eEF2 phosphorylation and subsequent increase in BDNF, as well as changes in other synaptic proteins observed after treatment of mice with (2R,6R)-HNK (10 mg/kg, i.p.) (Zanos et al., 2016; Suzuki et al., 2017).
In contrast to ketamine, (2R,6R)-HNK (10 mg/kg, i.p.) had no significant effect on several systemic markers of inflammation in socially defeated mice (Xiong et al., 2019). Specifically, no changes were observed in the plasma levels of the bone inflammatory markers osteoprotegerin, receptor activator of nuclear factor κB ligand, and osteopontin (Xiong et al., 2019). However, a recent proteomics study demonstrated that, in mice, (2R,6R)-HNK (10 mg/kg, i.p.) decreased hippocampal expression of peptidyl prolyl cis-trans isomerase A, a mediator of inflammation and immunosuppression (Rahman et al., 2020). Potential anti-inflammatory effects of HNKs require further study, including assessment of potential effects on cytokines or other anti-inflammatory measures.
2. Effects on Mitochondrial Function and Energy Metabolism
There is some evidence indicating that HNKs can alter mitochondrial function and energy metabolism, which may contribute to their antidepressant actions; namely, using a metabolomics approach, Faccio et al. (2018) demonstrated that a 36-hour exposure of PC12 cells to (2R,6R)-HNK (5 nM) and, to a lesser extent, (2S,6S)-HNK (0.5 nM), (R)-, and (S)-ketamine (1 µM) altered the mitochondrial metabolome. Specifically, at the concentrations tested, (2R,6R)-HNK robustly increased markers of glycolysis, including glycerate, fructose-6-phosphate, and glucose-6-phosphate. (2S,6S)-HNK also elicited a significant increase in glycerate and fructose-6-phosphate but had little effect on glucose-6 phosphate (Faccio et al., 2018). Compared with (2S,6S)-HNK, (2R,6R)-HNK enhanced activity of the pyrimidine salvage pathway, indicated by decreases in uridine and cytidine, concurrent with elevated uracil (Faccio et al., 2018). Further, incubation with (2R,6R)-HNK, as compared with (2S,6S)-HNK, induced a more robust decrease in fatty acids, including linoleic acid and elaidic acid, suggesting a shift in fatty acid and lipid metabolism toward the β-oxidation pathway (Faccio et al., 2018). (2R,6R)-HNK, but not (2S,6S)-HNK, robustly increased both glycine and serine while decreasing threonine production. Both (2R,6R)-HNK and (2S,6S)-HNK impacted purine metabolism, as evidenced by a reduction in inosine and guanosine signals and increase adenine and guanine, suggesting an overall increase in the generation of GTP and ATP (Faccio et al., 2018).
A more recent proteomic study examined the effect of (2R,6R)-HNK (10 mg/kg, i.p.) administration to mice on hippocampal protein expression after exposure to a forced swim session (Rahman et al., 2020). Consistent with the prior report, indicators of increased glycolysis—namely, pyruvate kinase isoenzymes M1/M2, pyruvate dehydrogenase E1, and triosephosphate isomerase—were observed in (2R,6R)-HNK–treated mice. The same study reported that (2R,6R)-HNK also increased the expression of several other proteins involved in energy metabolism, including creatine kinases, ATP synthase α, and mitochondrial cytochrome b-c1 complex subunit 6, but decreased expression of fructose-bisphosphate aldolase A, isocitrate dehydrogenase subunit α, and malate dehydrogenase (Rahman et al., 2020). Of note, both of the described studies tested only a single concentration or dose of each of the (2,6)-HNKs; thus, concentration-dependent differences on the outcomes studied may exist and require further investigation.
IV. Behavioral Effects
A. Preclinical Behavioral Studies
1. Antidepressant-Relevant Behaviors
To date, HNKs have primarily received attention for their therapeutic potential as novel antidepressant compounds. This is largely due to a growing number of preclinical studies demonstrating that (2R,6R)- and/or (2S,6S)-HNK induce behavioral effects in preclinical tests that predict antidepressant effectiveness (see Table 7 for a list of tests and outcomes; results are summarized in Table 8) (Nelson and Trainor, 2007; Zanos et al., 2016, 2019b; Chou et al., 2018; Highland et al., 2019; Pham et al., 2018; Fukumoto et al., 2019; Lumsden et al., 2019; Aguilar-Valles et al., 2020; Chen et al., 2020; Elmer et al., 2020; Rahman et al., 2020; Yokoyama et al., 2020).
Example behavioral tests predictive of antidepressant effectiveness
Preclinical behavioral effects
Subeffective doses and studies that did not identify an effect are not summarized in this table; please refer to the text (section IV.A.1.b. Direct antidepressant-relevant effects of hydroxynorketamines) for these details.
a. Potential role of hydroxynorketamines in mediating ketamine’s antidepressant-like effects
One of the first indications of the antidepressant-relevant behavioral effects of HNKs was the finding that metabolism of ketamine to form the (2,6)-HNKs is critical for its sustained actions in mouse models (Zanos et al., 2016). In particular, it was demonstrated that deuterium substitution at the C6 position of ketamine, a chemical modification that selectively and robustly hinders ketamine metabolism to the (2,6)-HNKs (described in section II.C. Factors Altering Hydroxynorketamine Pharmacokinetics), prevents the sustained actions of ketamine in the learned helplessness and forced swim tests assessed 24 hours post-treatment in mice (Zanos et al., 2016). It was later demonstrated that the analogous deuterium substitution to (R)-ketamine [(R)-d2-ketamine that specifically prevents metabolism to (2R,6R)-HNK; see section II.C. Factors Altering Hydroxynorketamine Pharmacokinetics] resulted in a pronounced shift in the dose-response curve (Zanos et al., 2019a). Namely, a 2-fold higher dose of (R)-d2-ketamine (10 mg/kg, i.p.) was necessary to reduce immobility time in the forced swim test and reverse learned helplessness escape deficits compared with unmodified (R)-ketamine (5 mg/kg, i.p.) in mice (Zanos et al., 2019a). Although one study reported that (R)-d2-ketamine did not attenuate the ability of the drug to induce antidepressant-relevant effects at 10 mg/kg (i.p.) in a chronic social defeat stress paradigm (Zhang et al., 2018b), the study design using a single dose does not allow comparison of the dose-response relationships between the two compounds [(R)- versus (R)-d2-ketamine], which may also be altered by differences in study design (i.e., strain, behavioral testing methods, etc.).
In addition to studies with d2-ketamine, it was also demonstrated that ketamine more potently induced decreases in immobility time in the forced swim test in female mice (effective doses 3–10 mg/kg, i.p.) compared with males (effective dose 10 mg/kg, i.p.), concomitant with approximately 3-fold higher levels of (2R,6R;2S,6S)-HNK detected in the plasma and brain of females (Zanos et al., 2016). This finding may further support a critical role of the (2,6)-HNKs in the antidepressant-relevant behavioral effects of ketamine, although one study reported that there were neither sex-dependent differences in the behavioral effects of (R)-ketamine in the forced swim test nor in the plasma (2R,6R)-HNK levels after (R)-ketamine dosing (Chang et al., 2018). However, inconsistent with the findings above, one study demonstrated that pretreatment of mice with a cytochrome P450 inhibitor cocktail (ticlopidine and 1-aminobenzotriazole) prior to (R)-ketamine administration [such pretreatment robustly decreases (2R,6R)-HNK levels; see section II.C. Factors Altering Hydroxynorketamine Pharmacokinetics] enhanced, instead of suppressing, the antidepressant-relevant behavioral effects of (R)-ketamine (3 mg/kg, i.p.) (Yamaguchi et al., 2018). Of note, cytochrome P450 inhibitor pretreatment also resulted in a robust increase in the peak plasma concentrations of (R)-ketamine (Yamaguchi et al., 2018), which have been demonstrated to exert HNK-independent behavioral effects (Zanos et al., 2019a) and, therefore, may explain the enhancement of antidepressant-relevant effects.
b. Direct antidepressant-relevant effects of hydroxynorketamines
Independent of the role of HNKs in mediating ketamine’s antidepressant effectiveness, a growing number of studies have demonstrated that direct administration of (2R,6R)-HNK and, to a lesser extent, (2S,6S)-HNK induces both rapid (observed within hours) and long-lasting (persisting days to weeks after treatment) behavioral effects in preclinical rodent studies used to predict antidepressant effectiveness (Nelson and Trainor, 2007; Zanos et al., 2016, 2019b; Chou et al., 2018; Highland et al., 2019; Pham et al., 2018; Fukumoto et al., 2019; Lumsden et al., 2019; Chen et al., 2020; Elmer et al., 2020; Ko et al., 2020; Rahman et al., 2020; Yokoyama et al., 2020); namely, a single systemic (intraperitoneal) injection of (2R,6R)-HNK reduces immobility time in the mouse forced swim test, an effect which has been observed at the following post-treatment times: 1 hour (5–125 mg/kg, i.p.) (Zanos et al., 2016, 2019b; Highland et al., 2019; Lumsden et al., 2019; Aguilar-Valles et al., 2020; Rahman et al., 2020), 24 hours (5–125 mg/kg, i.p.) (Zanos et al., 2016, 2019b; Highland et al., 2019; Fukumoto et al., 2019; Lumsden et al., 2019; Chen et al., 2020), 3 days (10 mg/kg, i.p.) (Zanos et al., 2016), 5 days (30 mg/kg, i.p.) (Fukumoto et al., 2019), and 2 weeks (0.025 mg/kg, i.p., in females vs. 0.075 mg/kg, i.p., in males) (Chen et al., 2020). In addition, (2R,6R)-HNK decreased escape failures in the learned helplessness test 24 hours post-treatment (5–125 mg/kg, i.p.) (Zanos et al., 2016; Highland et al., 2019) and reduced latency to eat in the novelty-suppressed feeding test 30 minutes (10 mg/kg, i.p.) (Lumsden et al., 2019) and 1 hour post-treatment (10 mg/kg, i.p.) (Zanos et al., 2016; Aguilar-Valles et al., 2020) in mice. Moreover, Elmer et al. (2020) demonstrated that, in mice, a single injection of (2R,6R)-HNK (20 mg/kg, i.p.) reversed helpless behavior (behavioral despair; footshock escape failures) after exposure to live predator stress (snake) in adolescence. Administration of (2R,6R)-HNK (10–20 mg/kg, i.p.) also exerts anti-anhedonic effects in chronically stressed mice, as evidenced by the reversal of chronic corticosterone–induced sucrose and female urine sniffing preference deficits (10 mg/kg, i.p.) (Zanos et al., 2016), as well as the reversal of chronic social defeat stress–induced sucrose preference (10 mg/kg, i.p.) and social interaction (20 mg/kg, i.p.) deficits (Zanos et al., 2016, 2019b). Likewise, in rats, (2R,6R)-HNK (10 mg/kg, i.p.) reduced forced swim test immobility time 24 hours after treatment (Chou, 2020) and also rescued chronic footshock stress–induced sucrose preference deficits and behavioral despair (forced swim test immobility), effects which lasted at least 21 days post-treatment (Chou et al., 2018). Importantly, oral administration of (2R,6R)-HNK reduced immobility time in the forced swim (15–150 mg/kg, administered orally) and learned helplessness (50–150 mg/kg, administered orally) paradigms in mice (Highland et al., 2019), which suggests that oral (2R,6R)-HNK may have clinical utility. These behavioral effects of (2R,6R)-HNK are unlikely to be mediated by changes on the hypothalamic pituitary adrenal axis activity, since an acute administration of 10 mg/kg (2R,6R)-HNK to rats neither changed plasma or cerebral cortex corticosterone levels nor altered the expression of the Sgk1 glucocorticoid responsive gene expression 1 hour after dosing (Wegman-Points et al., 2020).
Of note, there are some inconsistencies in the reported effects of (2R,6R)-HNK in tests predictive of antidepressant effectiveness. In particular, Hashimoto and colleagues have not detected effects of (2R,6R)-HNK in several preclinical models, including chronic social defeat stress (10 mg/kg, i.p.) (Yang et al., 2017a; Xiong et al., 2019), chronic corticosterone (10 mg/kg, i.p.) (Yokoyama et al., 2020), lipopolysaccharide-induced inflammation (10 mg/kg, i.p.) (Yamaguchi et al., 2018), and the learned helplessness paradigm (10 and 50 mg/kg, i.p.) (Shirayama and Hashimoto, 2018) in mice. Additionally, Herzog et al. (2020) did not detect any behavioral changes in (2R,6R)-HNK–treated (10 mg/kg, i.p.) mice compared with vehicle-treated controls in the forced swim test 24 hours post-treatment, although the ketamine positive control also failed to exert significant effects in the same study. Finally, Aleksandrova et al. (2020) failed to detect any changes in the forced swim test 30 minutes or 24 hours after (2R,6R)-HNK (5 mg/kg, i.p.) treatment in stress-susceptible Wistar-Kyoto rats, despite restoring novel object location recognition and Schaffer collateral LTP in vivo. However, the lack of an analysis of the dose-response relationship (i.e., testing only a single dose) in most of these studies precludes a more informed conclusion on the functional significance of these results, which contrast those from numerous other laboratories [described above; also reviewed in Hillhouse et al. (2019)].
The (2S,6S)-HNK stereoisomer has also been shown to exert antidepressant-relevant behavioral actions in mice (Zanos et al., 2016; Yokoyama et al., 2020), albeit typically with lower potency compared with (2R,6R)-HNK. In particular, (2S,6S)-HNK reduced immobility in the forced swim test 1 and 24 hours post-treatment (25 mg/kg, i.p) and escape deficits in the learned helplessness paradigm 24 hours post-treatment (75 mg/kg, i.p.) in mice (Zanos et al., 2016). Additionally, (2S,6S)-HNK (20 mg/kg, i.p.) reversed chronic corticosterone–induced behavioral despair in mice (assessed via forced swim test immobility time) 30 minutes post-treatment (Yokoyama et al., 2020). The minimally effective doses reported for the effects of (2S,6S)-HNK in mice are typically higher compared with (2R,6R)-HNK (see Table 8) (Nelson and Trainor, 2007; Zanos et al., 2016, 2019b; Pham et al., 2018; Fukumoto et al., 2019; Lumsden et al., 2019; Chou, 2020; Rahman et al., 2020), suggesting greater antidepressant potency of the (2R,6R)-HNK stereoisomer. Consistent with this, it has also been reported that (2R,6R)-, but not (2S,6S)-HNK, reduces escape failures in the learned helplessness test in both rats (10 mg/kg, i.p.) (Chou et al., 2018) and mice (10 mg/kg, i.p.) (Zanos et al., 2016), reduces immobility in the mouse forced swim test (10 mg/kg, i.p.) (Zanos et al., 2016; Chou, 2020), and increases female urine sniffing preference in mice (10 mg/kg, i.p.) (Chou, 2020) when tested at equivalent doses.
The greater antidepressant-relevant potency of (2R,6R)-HNK compared with (2S,6S)-HNK is consistent with preclinical data demonstrating that (R)-ketamine [the parent compound of (2R,6R)-HNK] is more potent than (S)-ketamine [the parent compound of (2S,6S)-HNK] in behavioral tests predictive of antidepressant efficacy (Zhang et al., 2014; Yang et al., 2015, 2017b, 2018; Zanos et al., 2016; Fukumoto et al., 2017). Nonetheless, at least one study has reported that (2S,6S)-, but not (2R,6R)-HNK, reversed chronic corticosterone–induced immobility in the forced swim test (30 minutes post-treatment) and anhedonia in the female mouse encounter test (24 hours post-treatment) when tested at equivalent doses (20 mg/kg, i.p.) (Yokoyama et al., 2020). It is unclear whether this discrepancy may be explained by differences in experimental methods. Another exception is a study conducted by Chen et al. (2020) in which low doses of (2S,6S)-HNK (0.025–30 mg/kg, i.p.) administered 2 weeks prior to testing attenuated learned fear in male, but not female mice, suggesting that sex and time elapsed between treatment and testing may affect dose-response relationships.
Consistent with a site of action localized in the central nervous system, direct administration of (2R,6R)-HNK into the brain mimicked the effects of systemic treatment in several rodent behavioral tests. For instance, intracerebroventricular administration of (2R,6R)-HNK (3–10 nmol per side) reversed escape deficits in the learned helplessness test conducted 24 hours (3–10 nmol per side) and 5 days (10 nmol per side) post-treatment (Zanos et al., 2019a). Of note, a U-shaped curve was demonstrated for intracerebroventricular (2R,6R)-HNK administration, where doses ≤1 nmol per side or ≥30 nmol per side did not reverse inescapable shock-induced escape deficits (Zanos et al., 2019a) or behavioral despair and sucrose preference deficits after chronic social defeat stress (Zhang et al., 2018a). Direct infusion into the medial prefrontal cortex (0.036–1 nmol per side) also reduced immobility measured 24 hours later in the mouse forced swim test (0.036–1 nmol per side) (Pham et al., 2018; Fukumoto et al., 2019). In addition, after chronic footshock stress, administration of (2R,6R)-HNK into the ventrolateral periaqueductal gray reduced immobility in the forced swim test and reversed sucrose preference deficits in rats, effects that were observed between 1 hour (3.62–36.2 pmol per side, forced swim test; 36.2 pmol per side, sucrose preference) and 21 days (3.62 pmol per side) post-treatment (Chou et al., 2018). Administration of (2R,6R)-HNK (1 pg) into the ventrolateral periaqueductal gray also reversed sucrose preference deficits and decreased immobility time in the tail suspension test, both assessed 1 hour post-treatment, in a mouse model of inflammatory bowel disease that was shown to induce behavioral despair and anhedonia (Ko et al., 2020).
To date, several putative mechanisms of action have been linked to some of the described antidepressant-like behavioral effects of (2R,6R)-HNK, including increased AMPAR activity, downstream activation of the BDNF/TrkB and mTOR pathways, and reduced mGlu2/3 receptor activity (mechanisms discussed in detail in section III. Pharmacodynamics). In particular, it has been demonstrated that AMPAR activity, both at the time of (2R,6R)-HNK administration and at the time of behavioral testing, is necessary for the antidepressant-like behavioral effects of (2R,6R)-HNK in the mouse forced swim test (Zanos et al., 2016). Specifically, pretreatment with the AMPAR antagonist 2,3-dioxo-6-nitro-7-sulfamoyl-benzo[f]quinoxaline either 10 minutes prior to (2R,6R)-HNK (10 mg/kg, i.p.) treatment (i.e., AMPAR blockade at the time of treatment) or 23.5 hours after treatment and 30 minutes prior to testing (i.e., AMPAR blockade at the time of testing) prevented the (2R,6R)-HNK–induced reduction in forced swim test immobility time measured 24 hours after treatment (Zanos et al., 2016).
In addition to AMPAR activity, there is also evidence to support a role of the BDNF/TrkB pathway in the effects of (2R,6R)-HNK. For instance, mice carrying the Met/Met BDNF allele, in which activity-dependent release of BDNF is abolished, do not exhibit (2R,6R)-HNK–induced (30 mg/kg, i.p.) decreases in forced swim test immobility time or novelty-suppressed feeding test latency to eat, measured 1 and 3 days post-treatment, respectively (Fukumoto et al., 2019). Additionally, infusion of a neutralizing BDNF antibody into the medial prefrontal cortex (Fukumoto et al., 2019) or ventrolateral periaqueductal gray (Chou, 2020) blocked the behavioral effects of (2R,6R)-HNK (10–30 mg/kg, i.p.) in the forced swim (Fukumoto et al., 2019; Chou, 2020) and novelty-suppressed feeding tests (Fukumoto et al., 2019). Infusion of the TrkB receptor antagonist K252a into the ventrolateral periaqueductal gray also blocked the effects of (2R,6R)-HNK (10 mg/kg, i.p.) in the forced swim test (Fukumoto et al., 2019), as did infusion of the mTOR pathway inhibitor rapamycin into the medial prefrontal cortex (Fukumoto et al., 2019) or ventrolateral periaqueductal gray (Chou, 2020). The involvement of mTOR signaling is further implicated by the finding that eukaryotic initiation factor 4E-binding proteins 1 and 2, a target of mammalian target of rapamycin complex 1 (mTORC1) kinase, are critical for the behavioral antidepressant actions of (2R,6R)-HNK as measured in both the forced swim and novelty-suppressed feeding tests (Aguilar-Valles et al., 2020). Of note, mice lacking eukaryotic initiation factor 4E-binding protein 1 or 2 in either excitatory (glutamatergic) or inhibitory (GABAergic) neurons lacked antidepressant responses to (2R,6R)-HNK as measured with the forced swim test.
In addition to activation of the BDNF/TrkB and mTOR pathways, it has recently been demonstrated that behavioral effects of (2R,6R)-HNK are convergent with mGlu2 receptor signaling (Zanos et al., 2019b). In particular, the behavioral effects of (2R,6R)-HNK (10 mg/kg, i.p.), including reduction of immobility time in the forced swim test, decrease of escape failures in the learned helplessness paradigm, and reversal of sucrose preference deficits in the chronic social defeat mouse model, were prevented by pretreatment with an mGlu2/3 receptor agonist. Furthermore, (2R,6R)-HNK’s behavioral effects were absent in mice lacking the Grm2, but not Grm3, gene (Zanos et al., 2019b). In addition, combined subeffective doses of an mGlu2/3 receptor antagonist and (2R,6R)-HNK (1 mg/kg, i.p.) exerted synergistic effects in the forced swim test (Zanos et al., 2019b).
Another factor that has been reported to modulate the antidepressant-relevant effects of (2R,6R)-HNK is ovarian-derived hormones (Chen et al., 2020). In particular, the long-term effects of (2R,6R)-HNK in the forced swim test observed 3 weeks after dosing were shown to be dependent on ovarian-derived hormones in female mice, as they were absent in ovariectomized mice and the effects were reinstated after estradiol or estradiol/progesterone replacement (Chen et al., 2020).
2. Analgesic Effects
There is some preclinical evidence of possible analgesic effects of (2R,6R)- and (2S,6S)-HNK in rodents (Table 8) (Lilius et al., 2018; Kroin et al., 2019). Although these findings await further replication, they may suggest that HNKs could have analgesic applications, including in the treatment of chronic or neuropathic pain. In particular, Lilius et al. (2018) reported that acute administration of racemic (2R,6R;2S,6S)-HNK (10–30 mg/kg, s.c.) did not induce antinociceptive effects as assessed 30–90 minutes post-treatment in the tail-flick, hot-plate, or paw pressure tests in rats. In the same study, neither acute (10–30 mg/kg, s.c.) nor repeated (20 mg/kg per day, for 6 days, s.c.) treatment with (2R,6R;2S,6S)-HNK altered the development of tolerance to the antinociceptive actions of morphine (after a 6-day morphine treatment). However, in contrast to these findings, acute administration of (2R,6R)-HNK (10 mg/kg, i.p.) elevated von Frey thresholds in a mouse model of neuropathic pain (spared nerve injury model) 24 hours post-treatment (Kroin et al., 2019). In addition, daily injections of (2R,6R)-HNK (10 mg/kg per day for 3 days, i.p.) reduced mechanical allodynia in the neuropathic pain model, the complex regional pain syndrome type 1 (CRPS1) model, and a postoperative pain model (left plantar hind paw incision) during the 3-day treatment period (Kroin et al., 2019). These effects extended to 4 and 5 days post-treatment in the CRPS1 and postoperative pain models, respectively (Kroin et al., 2019). Notably, naloxone administration did not prevent (2R,6R)-HNK’s effects in the postoperative pain model (Kroin et al., 2019), indicating that this metabolite likely does not act through opioid receptors to exert analgesic properties.
3. Effects on Aggression and Social Behaviors
There are some reports indicating that (2R,6R)- and (2S,6S)-HNK alter aggression and social behaviors in rats (Table 8) (Ye et al., 2019; Chou, 2020). In particular, it has been demonstrated that a single treatment of male and female rats with (2R,6R)-HNK (10 mg/kg, i.p., or 1–10 pg, intraventrolateral periaqueductal gray), but not (2S,6S)-HNK (3–10 mg/kg, i.p.), enhances aggressive behaviors (i.e., threats and bites in the resident-intruder test) assessed 1 hour (Ye et al., 2019) and 24 hours (Ye et al., 2019; Chou, 2020) post-treatment, and this effect lasts for up to 28 days after treatment (Ye et al., 2019). In addition, treatment of male rats with (2R,6R)-HNK (10 mg/kg, i.p.) enhances social dominance assessed 24 hours after treatment (Ye et al., 2019). The (2R,6R)-HNK–induced enhancement of aggression was determined to be dependent on BDNF signaling and AMPAR activity in the ventrolateral periaqueductal gray, as an infusion of an AMPAR antagonist into this region prevented aggressive behaviors (Ye et al., 2019; Chou, 2020) and infusion of a neutralizing BDNF antibody, the TrkB receptor antagonist K252a, or sustained application of BDNF ribonucleic acid interference (RNAi) in the same region reversed the proaggressive effects of the (2R,6R)-HNK (Chou, 2020).
Although “aggressive” behaviors could be characterized as an adverse effect of (2R,6R)-HNK, it could also be interpreted as a positive outcome in terms of being an adaptive response of rodents in the presence of an intruder to cope with competition [see Nelson and Trainor (2007)]. However, the effects of HNKs on social behaviors require further clarification. As discussed in section IV.A.1. Antidepressant-Relevant Behaviors, (2R,6R)-HNK restores social behaviors in chronically stressed mice, reversing chronic social defeat stress–induced social interaction deficits (Zanos et al., 2016, 2019b), but at least one study found that (2R,6R)-HNK (10 mg/kg, i.p.) did not alter social preference in nonstressed mice (Herzog et al., 2020).
4. Characterization of Adverse Behavioral Effects
A number of preclinical studies have examined adverse behavioral effects after treatment with (2R,6R)- and (2S,6S)-HNK, with the former demonstrating an innocuous adverse effect profile in rodents (Zanos et al., 2016; Highland et al., 2019; Lilius et al., 2018). Of note, one of the earliest studies evaluating in vivo HNK dosing demonstrated that the (2,6)-HNKs did not exert anesthetic actions (loss of righting reflex) after an intravenous bolus injection (40 mg/kg) in rats, in contrast to equivalent dosing of ketamine (Leung and Baillie, 1986). More recently, it was demonstrated that (2R,6R)-HNK administration did not alter locomotor activity in mice (assessed in an open-field arena) up to 125 mg/kg when administered intraperitoneally (Zanos et al., 2016) and 450 mg/kg when administered orally (Highland et al., 2019). In rats, no changes in locomotor activity were observed after an intraperitoneal injection of 5 mg/kg (2R,6R)-HNK (Aleksandrova et al., 2020). However, in mice, (2S,6S)-HNK induced hyperlocomotion at doses >25 mg/kg (i.p.) (Zanos et al., 2016), an effect likely attributed to its potency to block NMDARs [discussed in section III.A.1.a. N-methyl-d-aspartate receptor function; also see Morris et al. (2017)]. In addition, in mice, (2R,6R)-HNK administration did not alter sensorimotor gating, as assessed via disruption of prepulse inhibition, up to 375 mg/kg, i.p. (Zanos et al., 2016). Neither racemic (2R,6R;2S,6S)-HNK (10–30 mg/kg, s.c.) (Lilius et al., 2018) nor (2R,6R)-HNK (up to 125 mg/kg, i.p.) (Zanos et al., 2016) induced motor incoordination in the rotarod test (30–60 minutes post-injection and 5–60 minutes post-injection, respectively) in mice. In contrast, (2S,6S)-HNK (125 mg/kg, i.p.) induced motor incoordination during the first 20 minutes postinjection (Zanos et al., 2016). After oral treatment of mice with 450 mg/kg of (2R,6R)-HNK (a dose at least 9× higher than is required to induce antidepressant-relevant behavioral effects after oral dosing), no behavioral changes indicative of discomfort, sickness, or stereotypies were observed (Highland et al., 2019). Administration of (2R,6R)-HNK (10 and 50 mg/kg, i.p.) to mice trained to discriminate (R,S)-ketamine (10 mg/kg, i.p.) did not produce ketamine-like discrimination responses (Zanos et al., 2016). In addition, mice did not self-administer (2R,6R)-HNK (up to 3.2 mg/kg/infusion, i.v.) (Zanos et al., 2016). (2R,6R)-HNK (10 mg/kg, i.p.) did not alter memory performance in the spatial object and novel object recognition tasks in mice (Herzog et al., 2020). Notably, it was reported that (2R,6R)-HNK (5 mg/kg, i.p.) reverses long-term memory deficits in the novel object recognition task in stress-susceptible Wistar-Kyoto rats (Aleksandrova et al., 2020). Overall, it has been demonstrated that (2R,6R)-HNK lacks overt adverse effects, including the NMDAR inhibition–mediated anesthetic actions, dissociative properties, or abuse liability, characteristic of ketamine. Consistent with the higher potency of (2S,6S)-HNK to bind to and inhibit NMDAR function (Table 2), some adverse effects have been noted for (2S,6S)-HNK, although this compound has not been as well characterized as (2R,6R)-HNK. Thus, adverse actions of (2S,6S)-HNK, as well as potential actions of other HNKs, require further investigation.
B. Associations between Hydroxynorketamine Levels and Human Clinical Outcomes
Although there are currently no human clinical studies that have directly assessed the behavioral profile of HNK administration, retrospective studies have evaluated the associations between the antidepressant response and plasma levels of ketamine and its metabolites in patients that received ketamine as a treatment of depression. The first clinical study conducted by Zarate et al. (2012) examined 67 patients suffering from either unipolar (n = 45) or bipolar (n = 22) treatment-resistant depression, who were administered a 40-minute infusion of (R,S)-ketamine (0.5 mg/kg, i.v.). Although mean plasma levels of (2R,6R;2S,6S)-HNK at 120 and 230 minutes post infusion were higher for responders compared with nonresponders in patients with bipolar depression, this effect did not reach statistical significance (Zarate et al., 2012). There was no difference between (2R,6R;2S,6S)-HNK plasma levels between responders and nonresponders within the patients with unipolar major depressive disorder (Zarate et al., 2012). Higher levels of (2R,5R;2S,5S)-HNK were associated with nonresponse in the group with bipolar depression, and levels of (2R,5R;2S,5S)-HNK were inversely associated with psychotic and dissociative symptoms (Zarate et al., 2012).
More recently, correlations between plasma levels of ketamine and its metabolites and the clinical antidepressant effectiveness of ketamine administration (0.5 mg/kg, i.v., 40-minute infusion) were assessed in 53 patients suffering from major depression with clinically significant suicidal ideation (Grunebaum et al., 2019). In this study, higher (2R,6R)-HNK, but not (2S,6S)-HNK, levels measured in plasma obtained at the end of the infusion period correlated with less improvement in depression and suicidal ideation at 24 hours post–ketamine infusion (Grunebaum et al., 2019). Plasma levels of (2R,6R)-HNK were also inversely associated with longer-term symptom improvements, with higher (2R,6R)-HNK associated with less reduction in depression scores at 1, 2, 4, and 5 weeks postketamine and in suicidal ideation at 1–4 and 6 weeks post-treatment (Grunebaum et al., 2019). Likewise, levels of (2S,6S)-HNK were inversely associated with improvements in depression scores at 1 and 3 weeks post-treatment and with suicidal ideation at 3 to 4 weeks post-treatment (Grunebaum et al., 2019). Of note, neither (2R,6R)-HNK nor (2S,6S)-HNK plasma levels correlated with the acute dissociative or anxiogenic effects induced by ketamine administration (Grunebaum et al., 2019).
Lastly, Farmer et al. (2020) examined the relationship between plasma metabolite levels and the antidepressant effects of (R,S)-ketamine (0.5 mg/kg, i.v., 40-minute infusion) in 34 treatment-resistant patients with unipolar major depressive disorder. Higher plasma levels of (2R,6R;2S,6S)-HNK were associated with poorer antidepressant responses, especially at 3 and 7 days post–ketamine infusion (Farmer et al., 2020), contradicting preclinical findings. Conversely, peak (2R,6R;2S,6S)-HNK levels were associated with increased resting-state whole-brain magnetoencephalography γ power 6–9 hours post–ketamine infusion (Farmer et al., 2020), consistent with preclinical findings of increased γ power in mice after (2R,6R)-HNK administration (Zanos et al., 2016, 2019b) and suggesting that (2R,6R;2S,6S)-HNK enhances γ electroencephalographic oscillations that may be directly involved in ketamine’s rapid antidepressant response (Farmer et al., 2020). The dichotomy in the associations of HNK with antidepressant response may result from several factors, including possible lack of correlation between plasma and brain (or specific brain region) levels of HNKs, especially in light of the small number of subjects participating in the studies (Abdallah, 2020). In addition, peak concentrations for the HNK metabolites may actually occur before the end of the 40-minute infusion of (R,S)-ketamine, prior to the earliest time points tested in the clinical studies. Alternatively, it is possible that ketamine treatment results in both HNK-dependent and HNK-independent effects, which may confound the interpretation of correlations between plasma HNK and clinical outcomes after ketamine infusion. The possibility that direct administration of (2R,6R)-HNK, or possibly other HNKs, has antidepressant effects in patients suffering from treatment-resistant depression awaits testing in clinical trials.
V. Summary and Conclusions
Although originally deemed to be inactive ketamine metabolites because of their lack of anesthetic effects (Leung and Baillie, 1986), HNKs have more recently gained attention for their antidepressant potential (Nelson and Trainor, 2007; Zanos et al., 2016, 2019b; Chou et al., 2018; Highland et al., 2019; Pham et al., 2018; Fukumoto et al., 2019; Lumsden et al., 2019; Rahman et al., 2020; Yokoyama et al., 2020), leading to a growing interest in understanding their biologic activities (Abdallah, 2017; Aleksandrova et al., 2017; Gould et al., 2019). Of the twelve individual HNK stereoisomers that have been identified after ketamine administration (Fig. 1) (Moaddel et al., 2010; Zarate et al., 2012; Zhao et al., 2012; Farmer et al., 2020), studies thus far have focused on (2R,6R)-HNK and, to a lesser extent, (2S,6S)-HNK because of their demonstrated behavioral effects. However, it is possible that additional HNKs have similar biologic activities, or may exert additional effects, and future studies should aim to elucidate the full breadth of HNK-induced actions.
After ketamine administration, HNKs are rapidly formed via liver metabolism and can be detected in circulation within minutes of dosing in animals (Leung and Baillie, 1986; Zarate et al., 2012; Zhao et al., 2012; Moaddel et al., 2015; Zanos et al., 2016, 2019a; Pham et al., 2018; Yamaguchi et al., 2018; Farmer et al., 2020; Tůma et al., 2020) and at the end of ketamine infusion in humans (Zarate et al., 2012; Zhao et al., 2012), with quantifiable levels observed at the earliest time points studied (Leung and Baillie, 1986; Zanos et al., 2016, 2019a; Yamaguchi et al., 2018; Tůma et al., 2020) and (2R,6R;2S,6S)-HNK being the predominant HNK metabolite. Although the time course of elimination of HNKs is variable between species and individual HNK stereoisomers, some HNKs, including (2R,6R)-HNK, can be detected at low concentrations hours to days after dosing (Lankveld et al., 2006; Zarate et al., 2012; Zhao et al., 2012; Moaddel et al., 2016; Sandbaumhüter et al., 2016; Theurillat et al., 2016; Zanos et al., 2016, 2019a; Grunebaum et al., 2019).
The peak brain concentrations of HNKs, and specifically of (2R,6R)-HNK, achieved in humans after an antidepressant infusion of ketamine and in rodents after doses of ketamine and (2R,6R)-HNK that produce antidepressant-relevant behavioral effects, are of particular interest for evaluating the pharmacodynamic actions that may underlie these behaviors (see Table 1). After a typical antidepressant dose of ketamine in humans, the peak concentration of (2R,6R;2S,6S)-HNK in the brain was estimated to be ∼40 nM (≤37.8 ± 14.3 nM, mean ± S.D.) (Shaffer et al., 2019). In mice, (2R,6R)- and (2S,6S)-HNK reach peak concentrations between 1.54 and 2.46 µmol/kg after a dose of ketamine (10 mg/kg, i.p.) frequently reported to induce antidepressant-relevant behavioral effects (Zanos et al., 2016, 2019a). Finally, a dose of (2R,6R)-HNK commonly reported to induce antidepressant-relevant behaviors in mice (10 mg/kg, i.p.) results in concentrations of ∼10–20 µmol/kg in brain tissue (10.66–18.70 µmol/kg) (Zanos et al., 2016; Pham et al., 2018; Yamaguchi et al., 2018; Lumsden et al., 2019), ∼8 µM in the extracellular hippocampal space (7.57 ± 2.13 µM, mean ± S.E.M.) (Lumsden et al., 2019), and 18.40 µM in the CSF (Yamaguchi et al., 2018).
Some investigators have hypothesized that (2R,6R)-HNK’s mechanism of action centers around NMDAR inhibition, based upon the presumption that the parent compound, ketamine, acted solely in this manner. However, (2R,6R)-HNK does not displace radioligand binding to NMDARs or functionally inhibit NMDAR activity at antidepressant-relevant concentrations (Moaddel et al., 2013; Zanos et al., 2016; Morris et al., 2017; Suzuki et al., 2017; Lumsden et al., 2019), as described above (Table 2). In addition, (2R,6R)-HNK–induced potentiation of glutamatergic transmission is not occluded by NMDAR inhibition (Riggs et al., 2019).
Although some data indicate that high concentrations of (2R,6R)-HNK (50 µM) partially inhibit miniature NMDAR-mediated currents (Suzuki et al., 2017), there is evidence to suggest that NMDAR blockade does not underlie the behavioral effects of (2R,6R)-HNK. (2R,6R)-HNK blocks NMDAR-mediated synaptic transmission with an IC50 of 211.9 µM (Lumsden et al., 2019), which is a concentration that is substantially higher than the concentrations achieved after antidepressant-relevant dosing of ketamine or (2R,6R)-HNK in vivo (noted in Table 1) or those concentrations necessary to exert a range of antidepressant-relevant biologic effects in vitro, including synaptic potentiation (EC50 = 3.3 µM) (Riggs et al., 2019), BDNF release (10 nM) (Fukumoto et al., 2019), structural plasticity (0.5–1 µM) (Cavalleri et al., 2018; Shaffer et al., 2019), or intracellular signaling, for instance, via cAMP (10 µM) (Wray et al., 2019). As such, it is important to consider actions beyond NMDAR inhibition to fully understand the actions of HNKs.
A growing number of studies have begun to highlight potential sites of action of HNKs, as inferred from changes in various molecular, cellular, synaptic, and structural processes. Collectively, the literature suggests that HNKs rapidly facilitate glutamatergic synaptic transmission via enhanced glutamate release (Zanos et al., 2016; Ho et al., 2018; Pham et al., 2018; Riggs et al., 2019; Shaffer et al., 2019; Wray et al., 2019), resulting in subsequent activation of downstream signaling pathways, including the BDNF/TrkB and mTOR pathways (Paul et al., 2014; Zanos et al., 2016; Fred et al., 2019; Fukumoto et al., 2019; Lumsden et al., 2019; Anderzhanova et al., 2020), which can contribute to the observed postsynaptic increases in AMPAR expression induced by (2R,6R)-HNK at later time points (Zanos et al., 2016; Ho et al., 2018; Shaffer et al., 2019). These putative mechanisms of action are summarized in Fig. 2. In addition to glutamate, HNKs also increase extracellular levels of serotonin and norepinephrine (Pham et al., 2018; Ago et al., 2019), although no direct effects on these neurotransmitters or the monoaminergic systems, such as dopamine, have been detected (Can et al., 2016; Ago et al., 2019). Thus, the observed actions on monoamines are likely indirect effects of HNKs mediated via glutamatergic actions.

Putative synaptic mechanisms of (2R,6R)- and (2S,6S)-hydroxynorketamine. (2R,6R)- HNK acts on the presynaptic terminal to increase glutamate release, possibly via signaling mechanisms convergent with mGlu2, whereby (2R,6R)-HNK disinhibits the mGlu2-induced cAMP release, or via another glutamate release mechanism. Subsequent to enhanced glutamate release, AMPAR activation leads to enhanced BDNF release, TrkB activation, and subsequent activation of plasticity-relevant signal cascades, including an increase in protein kinase B (AKT), extracellular signal–related kinases (ERK)/mitogen-activated proteain kinases (MAPK), and mTORC1 pathway activity. These signaling cascades result in protein synthesis, including increased AMPAR expression, and synaptogenesis, ultimately promoting enhanced synaptic strength. (2R,6R)-HNK may also disrupt TrkB/AP-2 interactions, thereby inhibiting TrkB endocytosis and enhancing TrkB stability at the synapse. Additionally, (2S,6S)-HNK has moderate affinity to inhibit NMDARs and may act to increase intracellular signal cascades via an NMDAR inhibition–dependent pathway, including inhibition of eEF2 signaling in addition to increased AKT, ERK/MAPK, and mTORC1 signaling.
Data from the literature suggest that (2R,6R)-HNK dynamically engages the coordinated activity of several brain regions, including the hippocampus and prefrontal cortex, and support the hypothesis that the acute synaptic effects of (2R,6R)-HNK can evoke network-wide excitation, as systemically administered (2R,6R)-HNK (10 mg/kg, i.p.) leads to a rapid increase in electrocorticographic high-frequency γ oscillations (30–80 Hz) in mice (Zanos et al., 2016, 2019b).
To date, several independent research groups have demonstrated robust behavioral effects of (2R,6R)-HNK in a variety of rodent tests thought to predict antidepressant efficacy (Nelson and Trainor, 2007; Zanos et al., 2016, 2019b; Chou et al., 2018; Highland et al., 2019; Fukumoto et al., 2019; Lumsden et al., 2019; Chen et al., 2020; Chou, 2020; Ko et al., 2020; Rahman et al., 2020). Albeit to a lesser extent, (2S,6S)-HNK has also demonstrated similar antidepressant-relevant actions (Zanos et al., 2016; Yokoyama et al., 2020). Notably, rodent studies indicate an innocuous side effect profile for (2R,6R)-HNK (Zanos et al., 2016; Highland et al., 2019; Lilius et al., 2018), and animal studies have also demonstrated its favorable oral bioavailability (Highland et al., 2019). Together, these characteristics suggest that (2R,6R)-HNK may be a favorable antidepressant drug, although its antidepressant effectiveness awaits testing in human clinical trials.
In addition to antidepressant-relevant actions, (2R,6R)- and (2S,6S)-HNK have been reported to exert stress-protective effects in rodents (Chen et al., 2020), and several studies demonstrated effectiveness of HNKs administered 24 hours to a week prior to testing (e.g., 5–125 mg/kg, i.p.) (Zanos et al., 2016, 2019b; Highland et al., 2019; Fukumoto et al., 2019; Lumsden et al., 2019; Chen et al., 2020). These findings may suggest that the therapeutic applications for HNKs may extend beyond the reversal of depressive symptoms to the prophylactic prevention of stress-related disorders, which could include preventing the stress-induced onset or exacerbation of depression, anxiety, or post-traumatic stress disorder, although these potential applications require further study. Additional psychiatric applications of interest include those in which the parent compound, ketamine, has shown promise, including obsessive-compulsive disorder (Rodriguez et al., 2013). In addition to the treatment of mood and psychiatric disorders, HNKs may have therapeutic applications in the treatment of neuroinflammation (Ho et al., 2019; Rahman et al., 2020), as well as chronic pain, or could offer other analgesic uses, as there is preclinical evidence of analgesic actions of (2R,6R)-HNK (Kroin et al., 2019).
Altogether, although a growing body of evidence has demonstrated clinically relevant effects of (2R,6R)- and (2S,6S)-HNK for treatment of depression, future studies are needed to fully elucidate the mechanisms by which these HNKs exert their biologic effects and to determine whether the observed effects translate into clinical efficacy in humans. Additionally, the activities of multiple HNK stereoisomers remain largely unknown. Thus, further studies are needed to fully understand the full range of activities of all HNKs.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Highland, Zanos, Riggs, Georgiou, Clark, Morris, Moaddel, Thomas, Zarate, Pereira, Gould.
Footnotes
This work was supported by National Institutes of Health (NIH) National Institute of Mental Health [Grant R01-MH107615] and US Department of Veterans Affairs Merit Awards [1I01BX004062 and 101BX003631-01A1] (to T.D.G.); NIH National Institute of Allergy and Infectious Disease [Grant R21-RAI145211A] (to T.D.G. and S.M.C.); and by the National Institute of Aging (R.M.), National Institute of Mental Health (C.A.Z.), and National Center for Advancing Translational Sciences (C.J.T. and P.J.M.) NIH intramural research programs.
The following authors declare competing financial interests: R.M. and C.A.Z. are listed as co-inventors on a patent for the use of (2R,6R)-hydroxynorketamine, (S)-dehydronorketamine, and other stereoisomeric dehydro- and hydroxylated metabolites of (R,S)-ketamine in the treatment of depression and neuropathic pain. P.Z., R.M., P.M., C.T., C.A.Z., and T.G. are listed as co-inventors on a patent application for the use of (2R,6R)-hydroxynorketamine and (2S,6S)-hydroxynorketamine in the treatment of depression, anxiety, anhedonia, suicidal ideation, and post-traumatic stress disorders. R.M., P.M., C.A.Z., and C.T. have assigned their patent rights to the United States government but will share a percentage of any royalties that may be received by the government. P.Z. and T.G. have assigned their patent rights to the University of Maryland Baltimore but will share a percentage of any royalties that may be received by the University of Maryland Baltimore. T.D.G. has received research funding from Allergan and Roche Pharmaceuticals and has served as a consultant for FSV7, LLC, during the preceding 3 years. All other authors declare no competing interests.
Abbreviations
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- AMPAR
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- AP-2
- activator protein 2
- AUC
- area under the curve
- BDNF
- brain-derived neurotrophic factor
- CA1
- hippocampal cornu ammonis region 1
- CRPS1
- complex regional pain syndrome 1
- CSF
- cerebrospinal fluid
- eEF2
- eukaryotic elongation factor 2
- ERα
- estrogen receptor α
- fEPSP
- field excitatory postsynaptic potential
- GluA
- glutamate ionotropic receptor AMPA type subunit
- GluN
- glutamate ionotropic receptor NMDA type subunit
- HK
- hydroxyketamine
- HNK
- hydroxynorketamine
- iPSC
- induced pluripotent stem cell
- mEPSC
- miniature excitatory postsynaptic current
- malus
- metabotropic glutamate receptor
- mTOR
- mammalian target of rapamycin
- mTORC1
- mammalian target of rapamycin complex 1
- NMDA
- N-methyl-d-aspartate
- NMDAR
- N-methyl-d-aspartate receptor
- p4E-BP1
- phosphorylated eukaryotic initiation factor 4E binding protein 1
- pERK
- phosphorylated extracellular signal–related kinase
- TrkB
- tropomyosin receptor kinase B
- U.S. Government work not protected by U.S. copyright





{kind=link}
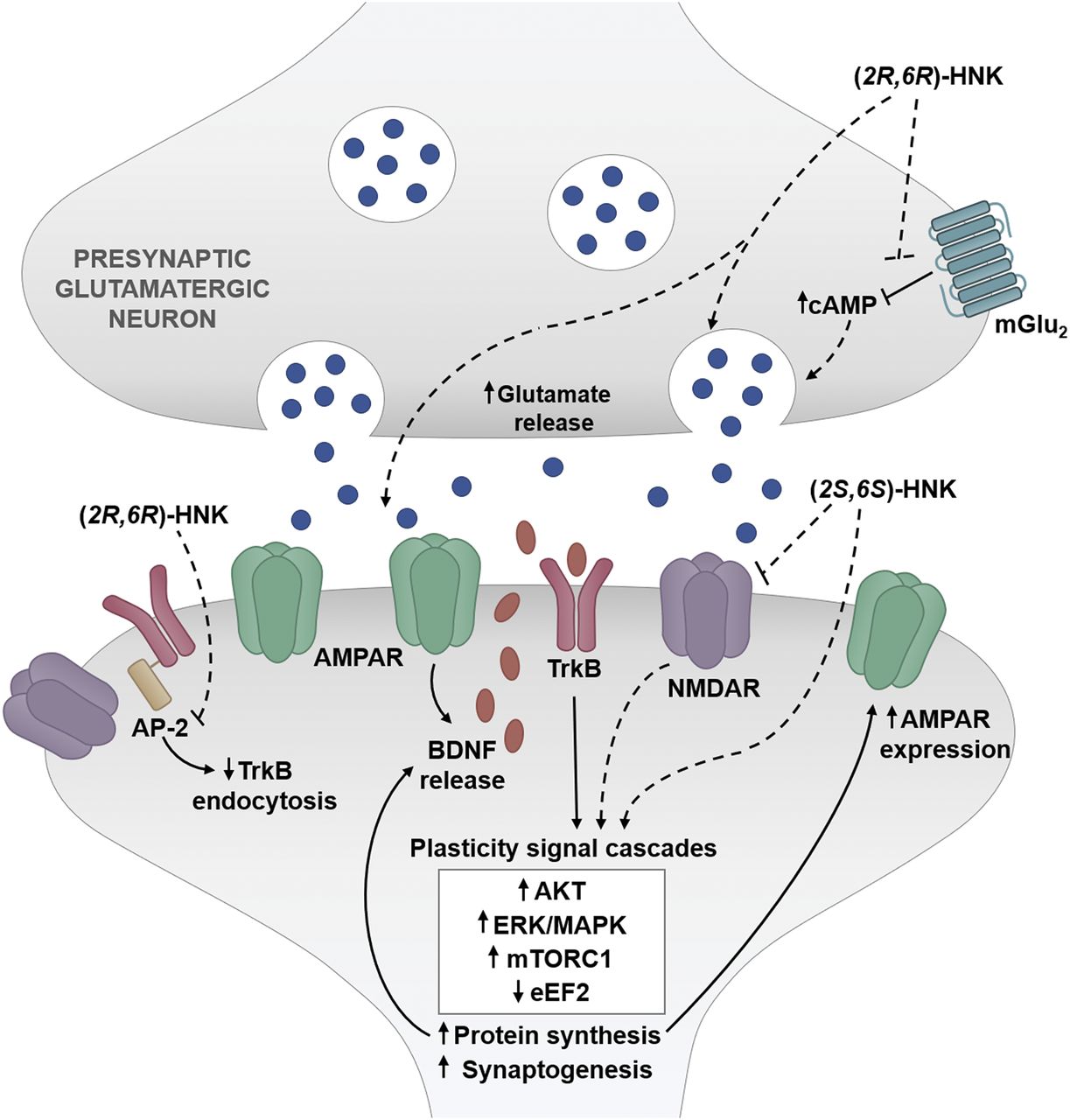{kind=link}