


Abstract
The phosphodiesterase 4 (PDE4) enzyme family plays a pivotal role in regulating levels of the second messenger cAMP. Consequently, PDE4 inhibitors have been investigated as a therapeutic strategy to enhance cAMP signaling in a broad range of diseases, including several types of cancers, as well as in various neurologic, dermatological, and inflammatory diseases. Despite their widespread therapeutic potential, the progression of PDE4 inhibitors into the clinic has been hampered because of their related relatively small therapeutic window, which increases the chance of producing adverse side effects. Interestingly, the PDE4 enzyme family consists of several subtypes and isoforms that can be modified post-translationally or can engage in specific protein-protein interactions to yield a variety of conformational states. Inhibition of specific PDE4 subtypes, isoforms, or conformational states may lead to more precise effects and hence improve the safety profile of PDE4 inhibition. In this review, we provide an overview of the variety of PDE4 isoforms and how their activity and inhibition is influenced by post-translational modifications and interactions with partner proteins. Furthermore, we describe the importance of screening potential PDE4 inhibitors in view of different PDE4 subtypes, isoforms, and conformational states rather than testing compounds directed toward a specific PDE4 catalytic domain. Lastly, potential mechanisms underlying PDE4-mediated adverse effects are outlined. In this review, we illustrate that PDE4 inhibitors retain their therapeutic potential in myriad diseases, but target identification should be more precise to establish selective inhibition of disease-affected PDE4 isoforms while avoiding isoforms involved in adverse effects.
Significance Statement Although the PDE4 enzyme family is a therapeutic target in an extensive range of disorders, clinical use of PDE4 inhibitors has been hindered because of the adverse side effects. This review elaborately shows that safer and more effective PDE4 targeting is possible by characterizing 1) which PDE4 subtypes and isoforms exist, 2) how PDE4 isoforms can adopt specific conformations upon post-translational modifications and protein-protein interactions, and 3) which PDE4 inhibitors can selectively bind specific PDE4 subtypes, isoforms, and/or conformations.
I. Introduction
Since the discovery of cAMP as a second messenger by Sutherland and Rall in 1958, its role in a wide variety of cellular processes, bodily functions, and pathologies has been thoroughly studied (Rall and Sutherland, 1958; Sutherland and Rall, 1958). Upon diverse extra- and intracellular cues, the second messenger cAMP is synthesized by adenylyl cyclases to relay signaling to adaptive changes in the cell. This notion indicates that cAMP is used as a single generic signaling molecule to convey and amplify information from different sources, a notion supported by the principle that evolution promotes utilizing the same machinery for different functions (Purvis and Lahav, 2013). Through precise regulation of the localization, abundance, and dynamics of cAMP, different signaling modes can be generated using the same generic molecule. Consequently, slight disturbances in cAMP regulation could promote pathophysiology in different cell types. Levels of cAMP are predominantly controlled through exclusive breakdown by the 3′,5′-cyclic nucleotide phosphodiesterase (PDE) enzyme family. This PDE enzyme family comprises 11 gene families (PDE1–11), which display different selectivity toward their substrates cAMP and cGMP. PDE4, PDE7, and PDE8 are cAMP-selective, and PDE5, PDE6, and PDE9 selectively degrade cGMP. The other gene families, PDE1, 2, 3, 10, and 11, can hydrolyze both cAMP and cGMP (Beavo, 1995; Bender and Beavo, 2006). PDE4 enzymes make up a majority of cAMP-selective PDEs in different organs and cell types (Lakics et al., 2010; Baillie et al., 2019). Hence, PDE4 enzymes are interesting pharmacological targets to specifically modulate cAMP signaling. Hence, inhibition of PDE4 has been and is clinically investigated as a therapeutic strategy in a multitude of disease areas, as also recently reviewed (Peng et al., 2020), including cognitive and affective disorders [e.g., Alzheimer disease (NCT03817684), fragile X syndrome (NCT03569631), schizophrenia (NCT02539550), depression (Hebenstreit et al., 1989), and substance dependence (NCT03489850)], autoimmune disorders [e.g., multiple sclerosis (NCT01982942) (Schepers et al., 2019), rheumatoid arthritis, atopic dermatitis, and Behçet syndrome (NCT02307513)], respiratory system diseases [e.g., chronic obstructive pulmonary disease and asthma (Lipworth, 2005)], dermatological conditions [e.g., psoriasis (NCT03022617)], and cancer [e.g., glioblastoma (NCT03782415)].
Although inhibition of PDE4 shows widespread therapeutic potential in preclinical research, the progression of PDE4 inhibitors into the clinic has been held back by severe adverse effects, including headaches, diarrhea, dizziness, nausea, and vomiting (Spina, 2008). In fact, only three PDE4 inhibitors made it to the market because of their limited or reduced adverse effects: roflumilast (Daliresp, Daxas), apremilast (Otezla), and crisaborole (Eucrisa) for chronic obstructive pulmonary disease, psoriasis, and moderate atopic dermatitis, respectively (Baillie et al., 2019). Interestingly, the PDE4 gene family consists of four paralogous genes, which, correspondingly, encode PDE4 subtypes (i.e., PDE4A–D). Each of these genes generates a variety of transcript variants that translate into different protein isoforms (e.g., PDE4D1–9). As these PDE4 subtypes and isoforms show tissue- and cell type–specific expression and intracellular compartmentalization patterns (Houslay, 2010) [reviewed in Baillie et al. (2019)], more selective inhibition could reduce the abovementioned adverse effects while maintaining treatment efficacy. An additional layer of complexity is added by the fact that PDE4 enzymes can adopt different conformational states as a result of various post-translational modifications and interactions with partner proteins. Consequently, this allows for more selective targeting, as PDE4 inhibitors will likely display different affinities toward different PDE4 subtypes, isoforms, and conformational states.
This review aims to provide an overview of the variety of PDE4 subtypes and isoforms and the mechanisms by which their cellular activity and inhibitor affinity is regulated through post-translational modifications and protein-protein interactions. Moreover, current advancements and strategies toward the development of PDE4 subtype- and/or conformation-specific compounds are discussed. Lastly, several mechanisms that potentially contribute to adverse side effect profiles of PDE4 inhibition are outlined to support the development of new, more specific, and safer PDE4-directed therapeutics.
II. Phosphodiesterase 4 Subtypes and Isoforms
Before the identification of the responsible enzymes in 1987, rolipram was shown to inhibit cAMP-specific PDE activity (Reeves et al., 1987). As this activity was distinct from three other types of PDE activity already known at the time, it was coined PDE IV. In retrospect, earlier studies had already identified rolipram-sensitive PDE activity to be present in rat brain material and to be involved in gastric secretion (Schwabe et al., 1976; Puurunen et al., 1978). A rat ortholog of the Drosophila cAMP-PDE enzyme, encoded by the dunce gene, was found to produce an enzyme that can be inhibited by rolipram (Davis et al., 1989; Swinnen et al., 1989a,b).
In mammals, four PDE4 genes can be distinguished, all of which show similar and evolutionarily conserved exon compositions, encoding the abovementioned PDE4 subtypes PDE4A, B, C, and D (Bolger et al., 1993, 1994; Milatovich et al., 1994; Johnson et al., 2010). Across species and among genes, there is particular sequence similarity in specific exons that encode the enzyme’s catalytic domain and two regulatory domains, upstream conserved region 1 (UCR1) and upstream conserved region 2 (UCR2). Next to their sensitivity to rolipram, PDE4 enzymes can be distinguished from other PDEs by the presence of these UCR1 and UCR2 domains. Apart from the UCR1, UCR2, and catalytic domains, the amino acid sequences of human PDE4 subtypes differ notably in the linker region 1 (between UCR1 and UCR2), linker region 2 (LR2) (between UCR2 and catalytic domain), and the C termini. These differences allow for subtype-specific modulation while maintaining core PDE4 regulation and functionality as discussed in section III. Phosphodiesterase 4 Modifications and Interactions.
Additional diversity is achieved at the gene level as each of the PDE4 genes contains alternative promoters that can generate distinct transcript variants by incorporating unique exons and through recursive splicing mechanisms (Sibley et al., 2015). Different promoters may contain distinct transcription response elements that allow for transcriptional regulation associated with a diversity of signaling pathways. For example, specific promoters of the PDE4D gene have been identified to contain regulatory elements for the transcription factors cAMP-response element binding protein (Vicini and Conti, 1997; D'Sa et al., 2002; Le Jeune et al., 2002), melanocyte inducing transcription factor (Khaled et al., 2010), or activating transcription factor 4 (Soda et al., 2013). These transcriptional control mechanisms allow for intricate transcriptional feedback loops that upregulate PDE4 expression to terminate cAMP signaling associated with particular cascades. The activity of certain promoters is thus regulated by the presence of the various transcription factors, and the accessibility of the promoter may also be subject to epigenetic regulation. Indeed, epigenetic alterations at the level of DNA (hydroxy)methylation and histone modifications of the PDE4D gene have been associated with changes in expression on specific transcript variants (Tilley and Maurice, 2005; Paes et al., 2021a). Although the exact responsiveness of the different PDE4 promoters remains to be explored further, prior findings already suggest that PDE4 transcription can be regulated in an intricately regulated manner that enables organ- and cell-specific expression patterns.
Depending on the location of the promoter, PDE4 mRNA transcripts will include the exons encoding both UCR1 and UCR2, only UCR2, a truncated UCR2, or only exons that encode a part of the catalytic domain (Nemoz et al., 1996; Johnston et al., 2004). Based on the presence of these UCRs, the protein products of these transcripts can be categorized as long, short, and supershort isoforms, respectively. Transcripts encoding catalytically inactive isoforms are called dead-short (Houslay, 2001). Based on deletion mutant studies, the UCR domains were found to regulate catalytic activity, showing differential enzymatic kinetics for the different isoform categories (Jacobitz et al., 1996; Saldou et al., 1998). Via alternative promoters and alternative splicing mechanisms, more than 20 human PDE4 transcript variants have been identified, allowing for tissue- and cell-specific expression regulation.
Figure 1 highlights the exon composition per human PDE4 transcript. As described below, certain PDE4 transcripts have been identified or characterized in humans or rodents only; Supplemental Table 1 provides an overview of which PDE4 transcript has been described per species. Human PDE4A encodes four long isoforms [PDE4A8 (Mackenzie et al., 2008); PDE4A4, which is named PDE4A5 in rodents (Bolger et al., 1993; Naro et al., 1996); PDE4A11 (Wallace et al., 2005); and PDE4A10 (Rena et al., 2001)]; one supershort isoform [PDE4A1 (Sullivan et al., 1998)]; and a dead-short, catalytically inactive isoform [PDE4A7 (Johnston et al., 2004)] (Supplemental Table 1).
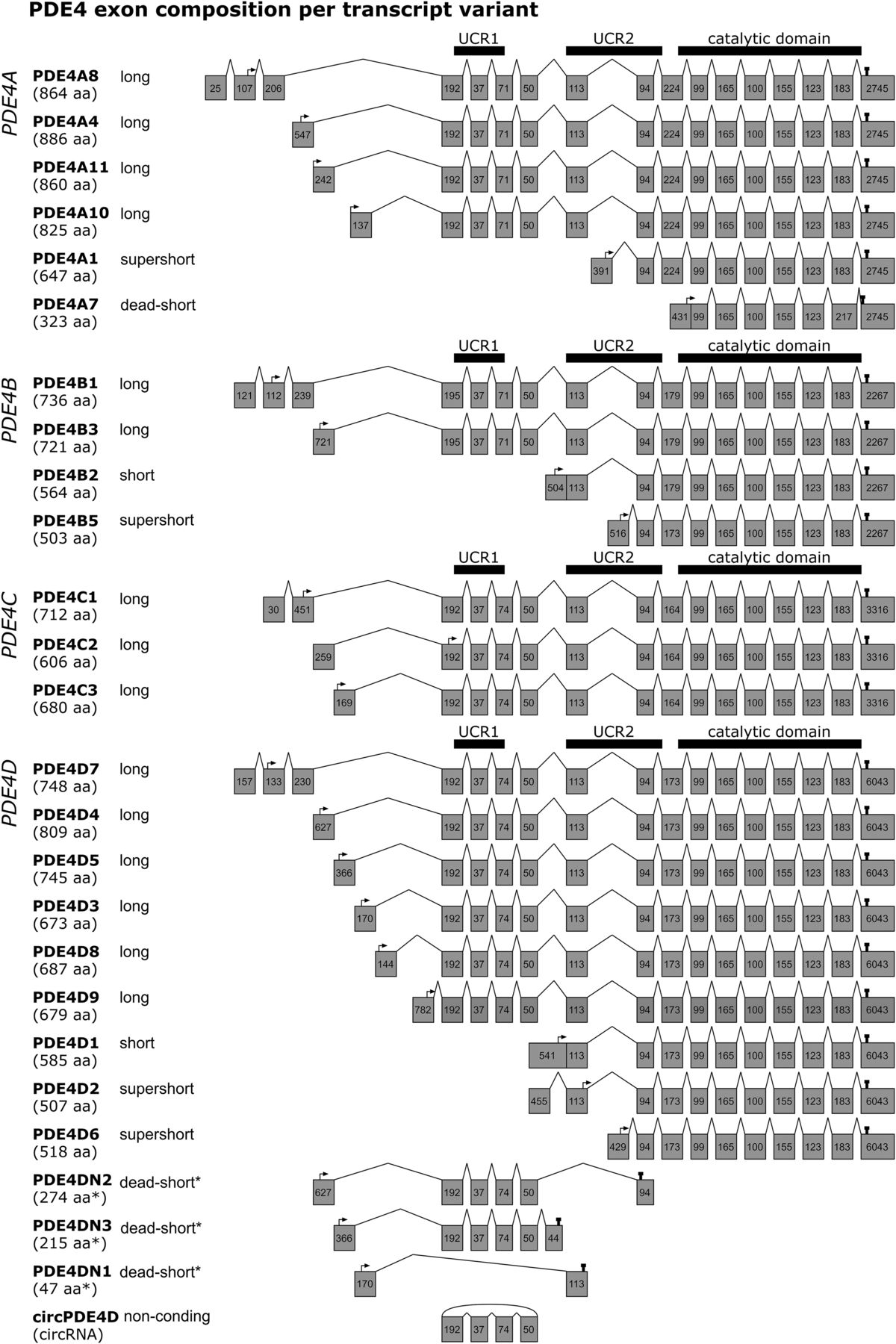
Exon composition per human PDE4 transcript variant. For each of the PDE4 genes (PDE4A–D), the exon composition is shown per transcript variant. Gray boxes depict exons and their nucleotide length. The protein isoform names and associated amino acid (aa) number per transcript are shown on the left. Start and stop codons are indicated by arrows and pins, respectively. The regions translating into UCR1, UCR2, and the catalytic domain are visualized by thick horizontal bars. *As the transcripts PDE4DN1–3 have only been identified on the mRNA level, amino acid lengths are isoform categories that are predictions based on in-frame stop codons. This figure was established through analysis and crossreferencing of online databases (NCBI: https://www.ncbi.nlm.nih.gov/gene and Ensembl: http://www.ensembl.org/index.html) and original cloning studies (see references to studies per transcript variant in section II. Phosphodiesterase 4 Subtypes and Isoforms). An overview indicating which transcripts have counterparts in rodents or are only found in rodents is provided in the Supplemental Material.
For human PDE4B, two long [PDE4B1 (Bolger et al., 1993) and PDE4B3 (Huston et al., 1997)], one short [PDE4B2 (McLaughlin et al., 1993)], and one supershort isoform [PDE4B5 (Cheung et al., 2007)] have been identified. In rodents, in addition, a long PDE4B4 isoform has been characterized that was suggested to have no functional equivalent in humans as a result of in-frame stop codons (Shepherd et al., 2003). However, PDE4B antibodies can clearly detect an 85-kDa PDE4B species in human brain tissue corresponding to rodent PDE4B4, but the exact sequence remains to be determined (Fatemi et al., 2008) (Supplemental Table 1).
The least well characterized PDE4 subtype is PDE4C, which comprises three long isoforms [PDE4C1 (Engels et al., 1995), PDE4C2 (Owens et al., 1997b), and PDE4C3 (Obernolte et al., 1997)], but likely generates additional variants through complex alternative splicing (Obernolte et al., 1997). Interestingly, despite relatively little insight into its function, several studies found that the DNA methylation signature of the PDE4C gene correlated with aging (Marquez-Ruiz et al., 2020).
Lastly, the human PDE4D gene produces the highest number of isoforms, i.e., six long isoforms [PDE4D3, PDE4D4, PDE4D5 (Bolger et al., 1997), PDE4D7, PDE4D8 (Wang et al., 2003), PDE4D9 (Gretarsdottir et al., 2003)], one short isoform [PDE4D1 (Bolger et al., 1997)], and two supershort isoforms [PDE4D2 (Bolger et al., 1997) and PDE4D6 (Wang et al., 2003)] (Supplemental Table 1).
Moreover, alternatively spliced transcripts have been described for PDE4D3, D4, and D5 that do not translate the catalytic domain because of in-frame stop codons caused by exon deletions or insertions (Fig. 1) (Miro et al., 2000). These variations of the “conventional” PDE4D3, D4, and D5 isoforms have been respectively coined PDE4DN1, PDE4DN2, and PDE4DN3 and can be categorized as dead-short forms based on the absence of the catalytic domain. For PDE4DN1, all exons encoding UCR1 are skipped, creating a transcript that encodes the unique N-terminal of PDE4D3 followed by 31 frame-shifted codons of the UCR2 (Fig. 1). Based on this sequence, PDE4DN1 may engage in similar protein-protein interactions as PDE4D3 would using its N-terminal residues, but the exact functional role of PDE4DN1 remains undetermined (see also section III. Phosphodiesterase 4 Modifications and Interactions and Fig. 2). In contrast to PDE4DN1, PDE4DN2 and PDE4DN3 do incorporate the UCR1-encoding exons in their transcripts (Fig. 1). Similar to PDE4DN1, the unique N termini, as also present in PDE4D4 and PDE4D5, may allow PDE4DN2 and PDE4DN3 to bind specific protein partners or putatively cause competitive binding for these binding sites with full-length PDE4D4 and PDE4D5, respectively. This competitive binding may subsequently induce altered distribution of full-length PDE4 forms causing distinct cellular cAMP dynamics. Intriguingly, the presence of UCR1 in these truncated forms may have functional consequences on full-length PDE4 forms. It has been demonstrated that a peptide fragment of the UCR1 can bind and activate full-length long PDE4 forms (Wang et al., 2015). PDE4DN2 and PDE4DN3, containing the same sequence as this peptide fragment, may exert similar actions and could biologically be relevant by providing an additional mechanism to elevate cellular PDE4 activity. The existence of these truncated forms at the protein level and their putative activating effects on full-length long PDE4D forms, however, remain to be validated.

Graphical representation of regulatory protein domains of PDE4 proteins and PDE4 amino acids or domains involved in inherent features of PDE4 enzymes, post-translational modifications and interactions with protein partners. Colored rectangles indicate regulatory domains. Inherent features of PDE4 enzymes and associated amino acids are also visualized in Supplemental Video. Those post-translational modifications and protein-protein interactions for which the involved PDE4 domains or specific amino acid residues have been identified are listed in this figure. If specific amino acids in involved regions have been identified to mediate the modification or interaction, these amino acids are indicated in bold and underlined. In case the specific amino acids are unidentified, the involved region is indicated by the region’s first and last amino acids connected by a line. Of note, for simplicity, PDE4D amino acids are shown for nonconserved amino acids involved in modifications or interactions that can occur in multiple PDE4 subtypes. Importantly, not all proteins or mechanisms in Figure 2 are listed in Table 1, and vice versa, as one of both aspects (i.e., either involved amino acids or effect of activity) may not have been revealed yet. The determination of amino acids involved in each of the modifications or interactions was based on the references as listed below: AKAP9 (Terrenoire et al., 2009); AKAP18δ (Stefan et al., 2007); Akt (Fang et al., 2015); AMPK, AMP-activated protein kinase (Sheppard et al., 2014); β-arrestin (Bolger et al., 2006; Baillie et al., 2007; Smith et al., 2007; Bolger, 2016); aII spectrin (Creighton et al., 2008); B55α PP2a subunit (Yun et al., 2019b); calcineurin (Zhu et al., 2010); CaMKII (Mika et al., 2015); caspase-3 (Huston et al., 2000); CDK5 (Plattner et al., 2015); C-term, C terminus; dimerization (Lee et al., 2002; Richter and Conti, 2002; 2004; Xie et al., 2014; Bolger et al., 2015; Cedervall et al., 2015); DISC1 (Millar et al., 2005; Cheung et al., 2007; Murdoch et al., 2007; Soda et al., 2013); ERK ( Hoffmann et al., 1999; Baillie et al., 2000; MacKenzie et al., 2000); HSP20 (Sin et al., 2011); integrin α5 (Yun et al., 2016, 2019b); JNK (Sharrocks et al., 2000; Bogoyevitch and Kobe, 2006; Zeke et al., 2015); LIS1 (Murdoch et al., 2011); LR1, linker region 1; Lyn (Beard et al., 2002); Lyn + Src (McPhee et al., 1999); mAKAP (Dodge et al., 2001; Carlisle Michel et al., 2004); Mdm2, mouse double minute 2 homolog (Li et al., 2009); membrane binding (Baillie et al., 2002); Mg2+ binding (Alvarez et al., 1995; Saldou et al., 1998; Laliberte et al., 2000; Liu et al., 2001); MK2 (MacKenzie et al., 2011; Bolger, 2016; Houslay et al., 2017; Houslay et al., 2019); myomegalin (Verde et al., 2001); N-term, N terminus; oxidative stress kinase (Hill et al., 2006; Bolger, 2016); p62 (SQSTM1, sequestosome 1) (Christian et al., 2010); PHD2 (Huo et al., 2012); phosphodegron motif (Zhu et al., 2010); PKA (Alvarez et al., 1995; Sette and Conti, 1996; Hoffmann et al., 1998; Beard et al., 2000; MacKenzie et al., 2002; Byrne et al., 2015; Bolger, 2016); p75NTR (Sachs et al., 2007; Houslay et al., 2019); RACK1 (Yarwood et al., 1999; Bolger et al., 2002; Bolger et al., 2006; Smith et al., 2007; Bird et al., 2010); Rheb (Kim et al., 2010; Meng et al., 2017); Shank2 (Lee et al., 2007); SIK1 (Kim et al., 2015); Src/Lyn/Fyn (O'Connell et al., 1996; Beard et al., 1999); SUMOylation (Li et al., 2010); ubiquitination (Li et al., 2009); UCR1-UCR2 interaction (Beard et al., 2000); XAP/aryl hydrocarbon receptor-interacting protein (AIP) (Bolger et al., 2003).
Importantly, the existence of these truncated PDE4DN1–3 forms has a practical consequence for quantitative polymerase chain reaction (qPCR) measurements. Isoform-specific PDE4D expression can be measured using qPCR primers that amplify part of the sequence of the isoform-unique exon (and the first UCR1 exon), but in the case of PDE4D4 and PDE4D5, the respective expression of PDE4DN2 and PDE4DN3 will also be detected by qPCR. Hence, PCR and gel electrophoresis should be performed in parallel using appropriate primers to determine whether expression changes are found for both the full-length and truncated transcript or for only one of these transcripts.
Recently, it has been described that PDE4D also encodes a highly stable, mainly cytoplasmic, circular RNA, circPDE4D, which is formed through circularization of exons 2–5 of the PDE4D gene (Fig. 1) (Wu et al., 2021). The expression of circPDE4D and linear PDE4D mRNA was found not to be correlated, indicating that distinct mechanisms produce these transcript types. Wu et al. (2021) identified regulatory regions in the flanking introns (i.e., upstream of exon 2 and downstream of exon 5) that are crucial for circPDE4D circularization. Through specific CRISPR-Cas9 editing, the authors were able to decrease circPDE4D expression and determine that QKI response elements are involved in circularization of the PDE4D pre-mRNA. QKI response elements bind the RNA-binding protein quaking (QKI), which has previously been shown to regulate the circularization of many pre-mRNAs (Conn et al., 2015). The functional role of circPDE4D remains largely to be determined, but Wu et al. (2021) identified that circPDE4D can bind specific microRNAs and thereby indirectly modulates the translation of mRNAs that otherwise would be degraded by the now-scavenged microRNA. In the same study, circPDE4D was found to be downregulated in osteoarthritic cartilage tissue, and intra-articular injection of circPDE4D could mitigate impairments in a mouse model of osteoarthritis.
As circPDE4D contains four exons that are also present in linear long-form PDE4D mRNA, it can be speculated that circPDE4D scavenges microRNAs that bind linear long-form PDE4D mRNA, and circPDE4D may therefore indirectly modulate PDE4D protein expression. Moreover, circRNAs can also directly regulate protein function, protein scaffolding, and protein localization, but these potential roles still remain to be determined for circPDE4D (Kristensen et al., 2019). These recent findings underline the complex transcriptional control of the PDE4D gene and warrant further investigation into non–protein-coding transcripts of the PDE4 gene family.
For murine Pde4d, the additional long PDE4D11 and supershort PDE4D10 isoforms have been described (Chandrasekaran et al., 2008; Lynex et al., 2008) (Supplemental Table 1). Furthermore, additional murine PDE4D transcripts have been identified that all encode supershort PDE4D2 but are generated through diverse exon incorporation, resulting in diverse 5′UTR sequences and lengths (Chandrasekaran et al., 2008) (Supplemental Table 1). Regarding 5′UTR and 3′UTR lengths, discrepancies exist regarding the reported transcript lengths when comparing NCBI Gene and Ensembl databases. 5′UTR lengths can differ depending on where the transcriptional machinery binds the DNA and initiates transcription, as exemplified for mouse PDE4D1 (McLaughlin et al., 1993). Similarly, the PDE4 transcript’s 3′UTR may differ in length, as it contains multiple polyadenylation sites (Sullivan et al., 1998; Wang et al., 2003). According to this variability at the 5′UTR and 3′UTR, certain gene data banks may show other lengths for the first unique and last common exons compared with those displayed in Fig. 1. As 5′UTR and 3′UTR sequences have been found to function as “zip codes” for specific intracellular transcript transport, it can be speculated that variation in these PDE4 transcript sequences may contribute to specific intracellular localization patterns for the different PDE4 isoforms (Andreassi and Riccio, 2009; Chin and Lecuyer, 2017). Wang et al. (2003) identified 10 putative consensus polyadenylation signals in the 3′UTR of PDE4D mRNA, which indicates that multiple transcripts that differ in their 3′UTR length can be generated depending on which polyadenylation site is used. Subsequently, differences in 3′UTR length may give rise to mRNA transcripts that exhibit different recognition motifs for several RNA-binding proteins (Di Liegro et al., 2014). For example, self-complementary sequences in mRNA can form secondary-structure hairpin loops that can be recognized and bound by RNA-binding proteins to regulate mRNA stability and transportation (Di Liegro et al., 2014). Depending on PDE4 3′UTR length, different secondary structures may be formed and bound by different transport proteins, which localize the transcript to distinct intracellular compartments. There, the specific PDE4 mRNA may be locally translated, after which the PDE4 protein isoform can be anchored to other proteins or membranes through its (isoform-specific) amino acids (see section III. Phosphodiesterase 4 Modifications and Interactions). Although the role of 5′UTR and 3′UTR sequence differences in mRNA transportation has not yet been explicitly demonstrated for PDE4 transcripts, these sequence differences could provide another mechanism through which PDE4 subtypes and isoforms can display a distinct cell type–specific and tissue type–specific intracellular distribution.
The intracellular localization of PDE4 isoforms is particularly regulated via isoform-specific N termini at the protein level, encoded by typical unique exon incorporation. These different N termini permit PDE4 isoforms to interact with protein partners or membranes in specific intracellular compartments (Houslay et al., 1995). In Aplysia, apPDE4 was found to require N-terminal residues for membrane binding, which indicates a conserved role for isoform-specific N termini in PDE4 localization (Jang et al., 2010). Likewise, different mammalian PDE4 subtypes (e.g., PDE4B and PDE4D) locate to different intracellular compartments (Blackman et al., 2011).
In addition to the scaffolding function, interactions with partner proteins can, as mentioned above, act upon the enzyme’s conformational state and hence affect its catalytic activity and inhibitor affinity. Similarly, PDE4 subtypes and isoforms are subject to post-translational modifications that differentially alter the conformation and activity of the enzyme.
To develop more efficacious and safer PDE4 inhibitors for different diseases, it is crucial to identify which PDE4 subtypes and isoforms should be targeted in disease-relevant tissues and cell types. Similarly, insight into which PDE4 subtypes and isoforms mediate adverse effects will determine which specific targets to avoid to improve the treatment’s safety profile. Additionally, understanding the conformational state of the isoform(s) involved in the compartmentalized signaling important to treat the disease at hand determines which compounds would be most potent. In the following section, an overview is provided on the different manners by which PDE4 activity and inhibitor affinity are influenced by post-translational modifications and interactions with partner proteins.
III. Phosphodiesterase 4 Modifications and Interactions
As mentioned before, catalytically active PDE4 isoforms can be categorized as long, short, or supershort based on the presence of UCR1 and UCR2 domains. Depending on the PDE4 subtype, isoform category, and unique N-terminal features, different post-translational modifiers and interaction partners can influence the conformation of the enzyme. These mechanisms allow for the dynamic regulation of the amount, localization, and activity of PDE4 enzymes to shape and respond to spatiotemporal cAMP signaling [as also recently reviewed for all PDE gene families (Baillie et al., 2019)]. The seminal work by Houslay and collaborators has provided a detailed understanding of which PDE4 amino acid residues are crucial for several modifications and interactions (Klussmann, 2016). Those post-translational modifications and protein-protein interactions for which involved PDE4 domains or specific amino acid residues have been identified are graphically represented in Fig. 2. These and other modifications and interactions, for which involved regions have not been determined, are discussed below in more detail. Furthermore, the known functional consequences of several modifications and interactions on PDE4 activity and inhibitor binding are elaborated upon in the following subsections and are summarized in Table 1.
Overview of modifications and interactions that influence PDE4 activity and the affinity of PDE4 inhibitors
A. Upstream Conserved Region 1-Upstream Conserved Region 2 Module, Dimerization, and Phosphorylation
PDE4 activity is profoundly regulated by its own UCR domains as, in long isoforms, the C-terminal of UCR1 forms a module with the N-terminal of UCR2, which autoinhibits its activity through the capping of the UCR2 α-helix NQVSE[F/Y]ISXTFLD across the catalytic domain (Kovala et al., 1997; Lim et al., 1999; Beard et al., 2000; Houslay and Adams, 2010) (Fig. 3; Table 1; Supplemental Video). Furthermore, the UCR1 and UCR2 domains enable long isoforms to homo- and heterodimerize, whereas short and supershort isoforms (lacking UCR1) exist as monomers (Xie et al., 2014). The UCR1-UCR2 module is disrupted upon serine phosphorylation in the conserved protein kinase A (PKA) consensus motif RRES in UCR1 of all long PDE4 isoforms. The liberation of this UCR1-UCR2 module attenuates the autoinhibitory effect causing enzyme activation (Hoffmann et al., 1998; Beard et al., 2000). As PKA is a direct downstream effector protein of cAMP, this modification serves as a negative feedback loop restoring cAMP levels through enhanced PDE4 activity. Although PKA phosphorylation can activate all long isoforms, the amplitude of PKA activation can differ among these isoforms as reported for long PDE4D isoforms (Richter et al., 2005). This indicates that additional regulatory mechanisms influence enzymatic activity. For example, the presence of additional PKA phosphorylation sites in PDE4D3 and PDE4D7, upon combined PKA phosphorylation, leads to differential effects in terms of catalytic activity (Sette and Conti, 1996; Collins et al., 2008; Byrne et al., 2015). Indeed, unique phosphorylation in the N-terminal PDE4D7, but not at the unique site in PDE4D3, induces an inhibitory effect on activity as opposed to PKA phosphorylation at the conserved UCR1 site (Byrne et al., 2015). Although PKA phosphorylation provides a negative feedback loop to restore cAMP levels, this regulation can be influenced by other protein interactors as well. For example, when PDE4D is bound by the protein coiled-coil and C2 domain-containing protein 1A (CC2D1), phosphorylation by PKA is prevented (Al-Tawashi et al., 2012; Al-Tawashi and Gehring, 2013). In addition to PKA, it was found that the same serine residue in UCR1 can be phosphorylated by Akt (Fang et al., 2015). Other conserved serine residues in the UCR1 also serve as phosphorylation sites for other kinases such as MK2, SIK1, CDK5, and AMP-activated protein kinase, which may modulate PDE4 activity by similarly affecting UCR1-UCR2 module formation/stabilization (MacKenzie et al., 2011; Sheppard et al., 2014; Kim et al., 2015; Plattner et al., 2015; Bolger, 2016; Houslay et al., 2019). Next to phosphorylation events, binding of phosphatidic acid or phosphatidylserine to the UCR1-UCR2 module increases PDE4 activity (Nemoz et al., 1997). The regulatory role of the UCR1-UCR2 module is further supported by the observation that several PDE4D mutations associated with the rare genetic disorder acrodysostosis localize to these regions, causing either increased or decreased PDE4D activity (Kaname et al., 2014; Gurney et al., 2015; Briet et al., 2017).

Tertiary structure of dimerized PDE4B demonstrating capping of UCR2 across the catalytic domain and helices 10 and 11. Crystal structure was derived from Protein Data Bank [PDB: 4WZI, (Cedervall et al., 2015)]. UCR1 and UCR2 regions of both monomers are indicated in purple and red, respectively. Catalytic domains are colored cyan and have the catalytic metals (i.e., Mg2+ and Zn2+) embedded as shown by the spheres. Wheat-colored helices form part of the linking region between catalytic domains and, nonmodeled, C termini. A three-dimensional representation of the image is provided in Supplemental Video.
B. Phosphorylation at Sites Other than Upstream Conserved Region 1
Through the use of multiple phosphorylation sites, PDE4 functionality can be modulated in a conditional manner, requiring multiphosphorylation, as has been reported for the role of PDE4D9 in mitosis (Sheppard et al., 2014). Next to the UCR1, the N terminus of the catalytic domain comprises phosphorylation sites for CaMKII and an “oxidative stress” or “switch” kinase. CaMKII phosphorylation is PDE4D-specific and induces enzyme activation in a manner distinct from PKA activation (Mika et al., 2015). In response to oxidative stress, an as of yet unidentified oxidative stress or switch kinase can increase PDE4 enzyme activity by switching the inhibitory effect of phosphorylation by extracellular signal–regulated kinase (ERK) to an activating effect (Hill et al., 2006; Bolger, 2016). Similarly, inhibition of PDE4D by ERK is diminished upon SUMOylation mediated by protein inhibitor of activated STAT protein gamma (PIASy), and this SUMOylation augments PKA activation of PDE4A and PDE4D (Fig. 2; Table 1) (Li et al., 2010).
Phosphorylation by ERK is established through the docking of ERK at the KIM and FQF motifs, which are located in catalytic domains and the C terminus, respectively (Fig. 2) (Houslay and Baillie, 2003). Upon docking of ERK, the consensus motif PXSP can be phosphorylated, which is present at the end of the catalytic domain in PDE4B, 4C, and 4D. Although PDE4A phosphorylation at this motif has been detected, this is unlikely induced by ERK, given the absence of the consensus motif, or it does not result in changed PDE4A activity (Baillie et al., 2000; Lario et al., 2001). Phosphorylation by ERK reduces the catalytic activity of long and supershort PDE4B, 4C, and 4D isoforms, presumably by intramolecular stabilization of the UCR2-capped configuration (Baillie et al., 2000; Houslay and Baillie, 2003). Conversely, short isoforms are activated upon ERK phosphorylation. However, removal of the UCR2 region in the short PDE4B2 form prevents an effect by phosphorylation by ERK, which supports the notion that UCR2 capping is required for ERK-mediated effects (Rocque et al., 1997b). Moreover, when ERK phosphorylation is paired with additional PKA or switch kinase phosphorylation, enzyme activity will increase in long isoforms as well (Hoffmann et al., 1999; Hill et al., 2006). Apart from these direct effects on PDE4 activity, phosphorylation by ERK may make PDE4 less prone to proteolysis (Lenhard et al., 1996). The PDE4D5 isoform may be minimally subjected to phosphorylation by ERK, as its unique N terminus binds the protein phosphatase B55α subunit, which dephosphorylates at the ERK site, reversing its inhibition (Yun et al., 2019). Furthermore, PDE4D5 specifically interacts with the proteins β-arrestin and RACK1, which can block the docking and phosphorylation by ERK, respectively (Table 1) (Bolger et al., 2006). Consequently, PDE4 activity can be increased upon these interactions, as reported for RACK1 (Yarwood et al., 1999; Bolger et al., 2002, 2006; Bird et al., 2010). Of note, binding of β-arrestin and RACK1 ablates dimerization, but they bind mutually exclusively as a result of overlapping binding residues in the PDE4D5 N terminus (Bolger, 2016). Additional mouse double minute 2 homolog–mediated ubiquitination putatively primes PDE4D5 to interact with β-arrestin rather than RACK1 (Li et al., 2009). Although β-arrestin preferentially binds PDE4D5, when bound to the RXFP1 rector, it prioritizes binding PDE4D3 (Halls and Cooper, 2010). These findings indicate that, because of the differential and combined interactions with partner partners, phosphorylation and binding events may be stimulated, prevented, or countered in an isoform-specific manner.
The ERK-associated KIM docking motif can also be used by c-Jun N-terminal kinase (JNK) (Sharrocks et al., 2000; Houslay and Adams, 2003). Intriguingly, the specific isoforms PDE4A8, PDE4B1, and PDE4D7 all contain an additional, evolutionary conserved docking site for JNK in their unique N terminus (Fig. 2). Previously, it has been demonstrated that PDE4B1 can indeed bind JNK, but it remains to be verified whether JNK effectively phosphorylates PDE4 and whether this changes PDE4 enzymatic activity (Zeke et al., 2015). A putative JNK phosphorylation site can be found in the accessible LLSTPAL motif in the catalytic domain, which corresponds with the heptapeptidic consensus sequence surrounding the JNK phosphorylation site (Bogoyevitch and Kobe, 2006).
C. Indirect Regulation of Phosphodiesterase 4 Activity and Interactions
Next to the aforementioned mechanisms, cellular PDE4 activity can be regulated through mechanisms distinct from phosphorylation. These regulatory mechanisms may be therapeutically relevant for the disease of interest, as exemplified by the disrupted in schizophrenia 1 (DISC1) protein. DISC1 is a PDE4 interaction partner and has been found to be a risk factor for the development of psychiatric diseases when mutated (Millar et al., 2005; Cheung et al., 2007; Soda et al., 2013). DISC1 can bind and inhibit both PDE4B and PDE4D through homologous amino acids, causing occlusion of the catalytic domain, and additional subtype-specific binding sites allow for a stronger interaction with PDE4B than with PDE4D (Fig. 2) (Murdoch et al., 2007). Consequently, upon cAMP elevation, DISC1 dissociates from PDE4D while retaining its interaction with PDE4B, resulting in functionally distinct roles for these PDE4 subtypes (Murdoch et al., 2007). Hence, mutations in either DISC1 or PDE4, or both, can impair or enhance this inhibitory action of DISC1, leading to aberrant PDE4 activity.
Through regulation of PDE4 mRNA and protein stability, cellular PDE4 activity can also be influenced. For example, cold-inducible RNA-binding protein, which acts as a cellular stress regulator, can stabilize PDE4B and PDE4D mRNA expression (Xie et al., 2020). Moreover, PDE4B mRNA was found to be stabilized by the common RNA modification N6-methyladenosine, providing another mechanism to regulate PDE4 translation and subsequent activity (Huang et al., 2020). At the protein level, PDE4 expression and activity were found to be decreased upon overexpression of the oxygen-sensing protein prolyl hydroxylase domain-containing protein 2 (PHD2) through hydroxylation of a specific site in the catalytic domain (Fig. 2) (Huo et al., 2012). PDE4D contains a phosphodegron motif in its C terminus, which upon dual phosphorylation by casein kinase I and glycogen synthase kinase 3β, induces PDE4D degradation (Fig. 2). However, through binding of the serine/threonine protein phosphatase calcineurin, PDE4D can be protected against phosphodegron-mediated degradation, which would stabilize PDE4 activity (Zhu et al., 2010). Notably, both calcineurin inhibitors and PDE4 inhibitors are used in the treatment of atopic dermatitis (Papier and Strowd, 2018). Based on the calcineurin-PDE4 interaction, these calcineurin inhibitors may, at least in part, be effective through stimulation of PDE4 degradation. Likewise, the small GTPase Rhebstabilizes PDE4D protein expression and dissociates upon cAMP elevation to activate the mTOR pathway (Kim et al., 2010; Meng et al., 2017). Lastly, PDE4B ubiquitination can be induced upon interaction with the E3 ubiquitin ligase SMAD ubiquitination regulatory factor 2 (Smurf2) (Cai et al., 2018). Thus, PDE4 activity is indirectly regulated through control of its degradation. Conversely, when these regulatory mechanisms are affected in a disease state (e.g., in case of DISC1 mutations), PDE4 activity can consequently become dysregulated.
D. Conformational States Impacting upon Phosphodiesterase 4 Activity and Inhibitor Binding
Upon modifications and interactions, PDE4 can exist in different conformational states. Historically, this has been recognized by PDE4 activity that could be distinguished based on different binding affinities for rolipram, a high-affinity rolipram binding site (HARBS) and low-affinity rolipram binding site (LARBS); see also section IV. Phosphodiesterase 4 Inhibitors below (Jacobitz et al., 1996; Rocque et al., 1997a,b; Souness and Rao, 1997). Multiple studies have demonstrated several factors that contribute to the existence of distinct conformers. For example, binding Mg2+ in the catalytic domain dose-dependently increases enzymatic activity but also increases the affinity to bind rolipram and other inhibitors (Table 1) (Wilson et al., 1994; Alvarez et al., 1995; Laliberte et al., 2000). Similarly, phosphorylation by PKA and RACK1 binding both increase PDE4 activity and induce an increase in rolipram affinity (MacKenzie et al., 2002; Zhang et al., 2018). However, the effect of phosphorylation by PKA on rolipram affinity can be different per long isoform (MacKenzie et al., 2002). Interestingly, increases in enzyme activity upon phosphorylation by PKA may actually be a consequence of an increase in Mg2+ sensitivity, which subsequently would facilitate cAMP catalysis as well as binding of certain inhibitors through water-mediated interactions (Saldou et al., 1998; Laliberte et al., 2000). Indeed, as proposed by Houslay and Adams (2003), modifications at the N terminus may be relayed via conformational changes to the catalytic domain to eventually influence enzyme activity. For example, upon PKA phosphorylation and possibly also other PDE4-activating phosphorylation events in the UCR1 (Fig. 2), the UCR1-UCR2 module and/or UCR2-catalytic domain interactions may be disrupted (Beard et al., 2000; Houslay and Adams, 2003). Subsequently, this altered conformation could change the orientation of helices 10 and 11 in the catalytic domain that flank the catalytic metals, thereby impacting the way these metals are held in place. More specifically, helices 10 and 11 form a tweezer-like structure (Fig. 4, dark blue helices) and are connected by a loop (Fig. 4, orange), which may interact with UCR2 and/or LR2 residues (Fig. 4). As such, N-terminal modifications and interactions can, directly or through modulation of this UCR2/LR2 stretch, alter the conformation of this connecting loop, thereby changing the way helices 10 and 11 stabilize catalytic metal binding. Consequently, catalytic activity and metal-mediated inhibitor binding will be influenced. Vice versa, inhibitor binding can impact the ability of PDE4 forms to engage in protein-protein interactions via inside-out signaling, which supports the notion that conformational changes are relayed between the catalytic domain and N-terminal regions. (Terry et al., 2003; Day et al., 2011). Interestingly, as the LR2 region is not conserved among PDE4 subtypes, its effect on metal coordination through interaction with the helix 10-11 connecting loop may be different per PDE4 subtype. Indeed, deletion of the UCR2 and LR2 regions significantly decreases the sensitivity of PDE4D, but not PDE4A or PDE4B, to bind Mg2+ (Saldou et al., 1998).

Tertiary structure of dimerized PDE4B demonstrating helices 10 and 11 and its connecting loop. One monomer is colored dark gray, and helices 10 and 11, flanking the catalytic metals, of the other monomer are now colored dark blue. The connecting loop between helices 10 and 11 is indicated in orange. The C terminus of UCR2 (red loose end in the middle) is connected to the N terminus of the catalytic domain (loose end in cyan in right-bottom corner) via LR2, which is not modeled but which would fold across the helix 10-11 connecting loop. Modifications and interactions in the N terminus are hypothesized to be relayed, via UCR2/LR2-associated changes, to the connecting loop of helices 10 and 11 to change catalytic metal ion coordination, which subsequently impacts cAMP catalysis and affinity toward certain metal-interacting inhibitors.
PDE4 enzymes exhibiting increased activity are not analogous to increased inhibitor affinity, as interactions that do not increase PDE4 activity also influence inhibitor binding. Binding of the proteins XAP2 and Lyn enhances the sensitivity to rolipram but has a negative or no effect on PDE4 activity, respectively (Table 1) (McPhee et al., 1999; Bolger et al., 2003). Furthermore, it has been postulated that the HARBS conformer is constituted of long isoforms since their dimerized state stabilizes HARBS (Richter and Conti, 2004). However, HARBS is not dependent on dimerization or the presence of UCR1 but rather is formed by inhibitor-UCR2 interactions, indicating that (super)short isoforms can also exhibit HARBS (Jacobitz et al., 1996). Indeed, truncated proteins similar to supershort forms that do possess the inhibitory UCR2 helix exhibit both HARBS and LARBS, whereas further deletion of the entire UCR2 only displays LARBS (Rocque et al., 1997a,b). These findings correspond to the fact that UCR2/LR2 residues may bind the connecting loop between helices 10 and 11 to regulate catalytic metal ion coordination. Absence of these residues would ablate the ability to change metal coordination and the consequent change in enzyme activity and metal-engaging inhibitor binding. Thus, both HARBS and LARBS comprise different states that depend on dimerization, degree of metal binding, interaction with partner proteins, and phosphorylation but which display similar affinities to inhibitors. Selective inhibition of PDE4 forms in HARBS or LARBS conformation has been shown to yield distinct biologic effects, although the exact PDE4 isoforms and/or conformation mediators remain to be specified. For example, inhibition of HARBS in the brain was found to produce antidepressant-like effects in rats (Zhang et al., 2006). Moreover, using PDE4 inhibitors that selectively bind HARBS or LARBS, Boomkamp et al. (2014) identified that inhibition of HARBS, but not LARBS, mediates myelination and neurite outgrowth in vitro.
The notion that inhibitors can display divergent affinities toward isoforms in different conformational states and that conformation-specific inhibition can exert different biologic effects has implications for drug discovery and development. Hence, it is crucial to understand which isoforms, in what conformational state, should be targeted in the disease of interest that determine the most potent inhibitor (see also section IV. Phosphodiesterase 4 Inhibitors).
E. Intracellular Phosphodiesterase 4 Localization and Anchoring
1. A-Kinase Anchoring Proteins
In addition to activity-altering interactions, PDE4 engages in protein-protein interactions that contribute to specific intracellular localization patterns. Accurate localization of PDE4 enzymes is crucial for the directed breakdown of compartmentalized cAMP. Many studies have indicated an essential role for A-kinase anchoring proteins (AKAP) in tethering signaling molecules regulating cAMP signaling, including PDE4 subtypes and isoforms [reviewed in, e.g., McConnachie et al. (2006), Wild and Dell'Acqua (2018), and Omar and Scott (2020)]. Through specific binding domains, the extensive collection of AKAPs allows the assembly of signaling modules consisting of PKA and PDE4 that are anchored to specific organelles, receptors [e.g., to the 5HT4b receptor (Weninger et al., 2014)], or cytoskeletal proteins (McConnachie et al., 2006). Important to note is that phosphorylation of the PDE4D3-specific PKA site facilitates its binding to mAKAP, which localizes to the ryanodine receptor (Carlisle Michel et al., 2004; Lehnart et al., 2005). Additionally, as PKA is bound to AKAPs as well, tethering PDE4D3 to mAKAP also promotes phosphorylation of the common PKA site, causing PDE4 activation near the ryanodine receptor (Dodge et al., 2001). Moreover, PDE4D3 can be brought into contact with IK calcium-activated potassium channels through selectively binding AKAP9 (Terrenoire et al., 2009; Terrin et al., 2012). Interestingly, the PDE4 residues involved in binding AKAP18δ are present in all isoforms, but tissue-specific expression causes only PDE4D3 and PDE4D9 to be associated with AKAP18δ (also known as AKAP7) in the kidney, highlighting the importance of studying cellular-, tissue-, or disease-relevant PDE4 expression (Stefan et al., 2007). Furthermore, PDE4 has been shown to associate with AKAP450 (also known as AKAP350, centrosome and Golgi localized PKN-associated protein (CG-NAP), Hyperion, or Yotiao) (Tasken et al., 2001; McCahill et al., 2005), AKAP95 (AKAP8) (Asirvatham et al., 2004; Clister et al., 2019), AKAP3 (does not bind PDE4D) (Bajpai et al., 2006), myeloid translocation gene on chromosome 16 (MTG16B) (Asirvatham et al., 2004), AKAP149 (Asirvatham et al., 2004), AKAP5 (also known as AKAP79) (Choi et al., 2011; Kocer et al., 2012), and Gravin (also called AKAP12, AKAP250, or Src-suppressed C kinase substrate (SSeCKS), which binds PDE4D3 and PDE4D5) (Willoughby et al., 2006). Although direct effects of AKAP binding on PDE4 activity remain largely undetermined, it is known that myomegalin (PDE4DIP) utilizes the same PDE4 residues that mediate UCR1-UCR2 interaction and dimerization and hence could influence the conformational state and activity of the bound PDE4 (Verde et al., 2001; Uys et al., 2011). Moreover, interactions between specific PDE4 forms and specific AKAPs may only occur in specific cell types or organs, as more specifically described elsewhere (Baillie et al., 2019; Zaccolo et al., 2021).
2. Not A-Kinase Anchoring Protein–Related
Independently of AKAPs, certain PDE4 isoforms can bind to specific structures through subtype- or isoform-specific amino acids. For example, the PDE4D4 and PDE4A4 isoforms have been found to localize to specific organelles and the plasma membrane through interactions with SRC homology 3 domains of the tyrosine kinases Lyn, Src, and Fyn via their N-terminal proline residues that possibly affect inhibitor binding (Table 1) (O'Connell et al., 1996; Beard et al., 1999, 2002; McPhee et al., 1999; Huston et al., 2000). Additionally, PDE4D4 is anchored to the cytoskeleton via interaction with αII spectrin (Creighton et al., 2008). The PDE4D-specific LR2 sequence allows interaction with integrin α5, which brings PDE4D in vicinity to a phosphatase that rectifies ERK-induced phosphorylation (Fig. 2) (Yun et al., 2016; Yun et al., 2019). The supershort PDE4A1 is membrane-bound via specific residues in its N terminus (Shakur et al., 1993; Baillie et al., 2002), and similar observations have been made for the short PDE4B2 isoform (Lobban et al., 1994). Likewise, PDE4A4 is associated with the membrane, but upon cleavage by caspase-3, it is redistributed within the membrane (Huston et al., 2000). Hence, post-translational modifications can alter intracellular distribution and compartmentalization, which is also supported by the fact that phosphorylation events can translocate membrane-associated PDE4 to the cytosol (Liu and Maurice, 1999). Through interactions in UCR1 and UCR2, mainly long PDE4D forms have been found to interact with Shank2 (Lee et al., 2007). As Shank2 also binds cystic fibrosis transmembrane conductance regulator (CFTR), this may explain the association of PDE4 enzymes with CFTR (Lee et al., 2007; Blanchard et al., 2014). Lastly, PDE4 enzymes can be intracellularly recruited to the p75NTR and neuropilin receptors, which are involved in neuronal growth (Sachs et al., 2007; Ge et al., 2015; Houslay et al., 2019). Vice versa, PDE4 enzymes can modulate cellular signaling cascades through scaffolding other molecules away from their usual binding partners. This is exemplified by the fact that PDE4 can recruit lissencephaly-1 (LIS1), causing it to disassociate from dynein. Subsequently, dynein function will be impaired, causing changes in microtubule transport and directed cell migration (Murdoch et al., 2011). As many interactions between PDE4 and partner proteins rely on isoform-specific amino acid stretches, these interactions can localize PDE4 isoforms to distinct subcellular compartments. Consequently, different PDE4 isoforms may regulate spatially distinct cAMP signaling cascades, and isoform-specific PDE4 inhibition would enable more precise modulation of these different cAMP cascades.
IV. Phosphodiesterase 4 Inhibitors
As an important cellular regulator of cAMP levels and associated intracellular signal transduction, PDE4 has been investigated as a therapeutic target in a wide variety of disorders. Based on promising observations upon treatment with nonselective PDE inhibitors and the prototypical PDE4-selective inhibitor rolipram, PDE4 inhibitors have been developed with improved potency. Despite increased potency, clinical progression of PDE4 inhibitors has been hampered primarily because of the occurrence of severe adverse side effects (see also below in section V. Adverse Effects of Phosphodiesterase 4 Inhibition). Therefore, to improve treatment efficacy and safety, PDE4 inhibitor specificity next to potency may have to be considered. As discussed above, the diversity in PDE4 isoforms that display distinct enzymatic properties, the amino acid differences among subtypes, and isoform-specific N termini allow for complex activity regulation by post-translational modifications and interactions with partner proteins. Eventually, this regulatory control can influence the enzyme’s activity through conformational changes. These specific interactions, nonconserved amino acids, and distinct conformational states can provide the opportunity to target PDE4 activity more specifically at the subtype and isoform level.
This section will outline several aspects that should be considered when determining PDE4 inhibitor specificity toward subtypes or isoforms by providing an extensive overview of inhibitor screening literature. Furthermore, this section will summarize which PDE4 inhibitors have been developed to show more specific targeting of subtypes, isoform, and/or conformations.
A. Phosphodiesterase 4 Inhibitors and High-Affinity and Low-Affinity Rolipram Binding Sites
As mentioned above, HARBS and LARBS represent different conformational states that exhibit different inhibitor affinities and can be influenced through several mechanisms (e.g., metal binding, phosphorylation, or binding of partner proteins). Based on the observation that preferential inhibition of HARBS was correlated with adverse effects, efforts were made to develop PDE4 inhibitors with reduced HARBS binding (Barnette et al., 1995; Duplantier et al., 1996). Selective inhibition of HARBS or LARBS can produce distinct cellular effects, which provides additional support for improved therapeutic potential of conformation-specific inhibitors (Zhang et al., 2006; Boomkamp et al., 2014). However, HARBS and LARBS conformations can occur in all PDE4 subtypes, and HARBS seems to require a part of the UCR2 domain, suggesting that both long and short PDE4 isoforms can exert HARBS (Souness and Rao, 1997). Thus, although HARBS and LARBS can be preferentially bound by certain inhibitors, selective binding of these conformations does not directly allow for PDE4 subtype or isoform selectivity. However, as described in subsection III.D., HARBS and LARBS conformations may, in part, be a consequence of differences in the orientation of two helices that flank the catalytic domain and that coordinate the catalytic metals (Houslay and Adams, 2003). The positioning of these helices is likely impacted by LR2 residues, which are nonconserved among PDE4 subtypes. Associated with these PDE4 subtype-specific LR2 residues, changes in metal orientation between HARBS and LARBS may be different among the PDE4 subtypes, which is supported by subtype-specific sensitivities to Mg2+ depending on whether LR2 is present or not (Saldou et al., 1998). Hence, although highly speculative, it may be possible to alter PDE4 activity subtype-selectively by means of allosteric modulation in the LR2 region, which subsequently modifies enzyme activity through changes in metal orientation. In support of this hypothesis, the kinases CaMKII and the oxidative stress kinase both phosphorylate residues in the LR2 region and have been shown to modulate PDE4 activity (Fig. 2; Table 1) (Hill et al., 2006; Mika et al., 2015).
As PDE4 subtypes and isoforms regulate distinct cellular and behavioral functions, PDE4 subtype-selective inhibition, rather than preferential binding of HARBS or LARBS, may show superior treatment efficacy compared with nonselective PDE4 inhibition (Blackman et al., 2011; Zhang et al., 2017).
B. Determining Phosphodiesterase 4 Inhibitor Subtype and Isoform Selectivity in Assays
1. The Influence of Phosphodiesterase 4 Construct and Assay Type on Phosphodiesterase 4 Inhibitor Screenings
Given the importance of subtype-, isoform-, or conformation-specific inhibition for treatment efficacy and safety, it is crucial to assess these properties of PDE4 inhibitors. Initial high-throughput screenings often used the catalytic domain of a single PDE4 subtype only to determine the ability of a compound to inhibit PDE4 activity. Since these catalytic domain constructs may not express UCR2 domains, these enzymes do not resemble cellular PDE4 activity, as UCR2-mediated autoinhibition or capping is not possible. Furthermore, the lack of the UCR2 alters the Mg2+ sensitivity of the enzyme and could therefore skew screening results, as both enzyme activity and Mg2+-mediated inhibitor binding may be impacted (Saldou et al., 1998). Hence, compound screening for PDE4 inhibitory activity using the catalytic domain of a single PDE4 subtype only will provide limited insight into the compound’s potential subtype selectivity. Subtype selectivity should therefore be assessed by using constructs of these different PDE4 subtypes and constituting a full-length protein rather than the catalytic domain only. Linked to this, as catalytic domains show large similarity among PDE4 subtypes, selectivity can more likely be achieved through interactions with amino acid residues outside of the catalytic domain (Wang et al., 2007a). Thus, to determine potential subtype selectivity of PDE4 inhibitors, the use of full-length PDE4 constructs will provide a better understanding compared with using only PDE4 catalytic domains.
PDE4 inhibitors can be tested in both cellular assays and assays utilizing purified PDE4, but affinity values cannot be directly compared across studies, as they are dependent on the used assay. For example, affinity values derived from assays using purified PDE4 versus overexpressed PDE4 constructs in a cellular assay can show an 80-fold difference (Wunder et al., 2013). Moreover, other studies showed that inhibitor affinity to isolated PDE4 enzymes does not reflect its potency in intact tissue (Harris et al., 1989). Furthermore, differences exist regarding which cell lines and heterologous expression systems are used for cellular assays or purified protein assays, respectively, which prevents comparison across studies that use different methodologies. Although assays using purified protein can provide a detailed understanding on the affinity and potency of an inhibitor, these assays do not reflect dynamic changes in (subtype-specific) PDE4 conformational states and associated affinity changes that would occur in a cellular environment. Hence, when assessing potential subtype specificity of PDE4 inhibitors, affinity values can only be compared within studies, and the most accurate approximation of in vivo PDE4 inhibition can be assessed by using full-length enzymes that contain all regulatory elements.
Table 2 provides a comprehensive overview of studies in which affinities of PDE4 inhibitors toward different PDE4 subtypes were assessed. As mentioned, affinity values, indicated as nanomolar concentrations that cause half-maximum inhibition (IC50), cannot be compared across studies, as different assays may have been used. For each subtype construct, it is indicated whether a particular isoform was used and whether the construct entailed the full-length protein or was truncated. From this overview, it becomes evident that several studies have assessed compound selectivity for PDE4 subtypes by using constructs of different isoform categories (i.e., long, short, or supershort) per subtype. Several studies indicate that inhibitors can exhibit different affinities toward PDE4 subtypes. For example, cilomilast seems to show slightly higher affinity toward PDE4D, irrespective of whether truncated supershort or long isoforms are used (Huang et al., 2007; Asaka et al., 2010). Interestingly, many compounds show notably lower affinity toward PDE4C, whereas other compounds (e.g., UFM24) preferentially bind PDE4C (Tsai et al., 2017). As PDE4C shows the least homology with the other subtypes, PDE4C-specific residues therefore may either promote or impede inhibitor binding, yielding PDE4C-specific or PDE4C-aversive binding, respectively.
PDE4 inhibitors tested for their selectivity against different PDE4 subtypes
Importantly, subtype selectivity may seemingly be a result of or biased by preferred binding to an isoform of particular length rather than specificity to the PDE4 subtype. Since isoforms of different lengths exhibit different properties as a result of the presence or absence of regulatory domains (e.g., long forms can dimerize via UCR1-UCR2 interactions), isoform categories of the same PDE4 subtype may be preferentially bound by certain inhibitors. To reveal potential isoform-specific binding, Table 3 presents an overview of compounds that were tested, in the same assay, for their affinity toward PDE4 constructs of different lengths of the same PDE4 subtype. Various compounds show increased affinity toward the long isoforms compared with its catalytic domain only, in which the regulatory UCR1, UCR2, and C-terminal domains are deleted (Wunder et al., 2013). As these regulatory domains convey effects of post-translational modifications and interactions with partner proteins, these modifications and interactions themselves may exert an effect on inhibitor affinity within the same PDE4 isoform, as explained earlier. Indeed, for example, PKA phosphorylation has been shown to profoundly impact inhibitor affinity (Hoffmann et al., 1998), which supports the notion that assays using purified, nonphosphorylated enzyme cannot be compared with cellular assays in which PDE4 activity is dynamically regulated (Wunder et al., 2013). As certain modifications or interactions can only occur in specific PDE4 subtypes or isoforms to induce conformational changes (Fig. 2), these conformations actually provide other means of achieving more specific PDE4 inhibition. Therefore, it is crucial to understand how different conformational states, caused by modifications or interactions, impact inhibitor affinity.
PDE4 inhibitors tested for their selectivity against different PDE4 isoforms or construct lengths of the same PDE4 subtype
Several studies have investigated these effects of conformational state on PDE4 inhibitor affinity, and these findings are summarized in Table 4. By means of mutations that mimic phosphorylation by PKA and ablate phosphorylation by ERK, the effects of PKA phosphorylation per se on inhibitor affinity can be simulated (Hoffmann et al., 1998; Burgin et al., 2010). This phosphorylation-mimicking mutation profoundly increases the affinity of rolipram and BPN14770 to bind the long-form PDE4D7 (Zhang et al., 2018) (see Table 4), indicating that inhibitors can preferentially bind PKA-phosphorylated PDE4 states. Mechanistically, increased inhibitor affinity upon phosphorylation by PKA could be due to a more favorable positioning of UCR2, increased Mg2+ sensitivity to aid inhibitor binding, or a combination of both. Interestingly, although PDE4 phosphorylation by PKA also increases enzymatic activity as a biologic feedback loop, this feedback mechanism would actually facilitate or strengthen binding of these inhibitors. Since PKA-phosphorylated PDE4 displays higher enzymatic activity, preferential inhibition of this activated state may actually produce more potent effects on cAMP levels in an in vitro or in vivo setting. Conversely, as inhibitors may bind only the phosphorylated fraction of PDE4 forms, inhibition of nonphosphorylated PDE4 may be minimal, yielding only a partial inhibition of total PDE4 activity. Therefore, it is essential to consider the potency of PDE4 inhibitors in addition to their affinity when determining which inhibitor, with its affinity profile against different PDE4 subtypes, isoforms, and conformations, shows the most therapeutic benefit in the disease of interest.
PDE4 inhibitors tested for their selectivity against different PDE4 conformational states or PDE4 isoforms in different cellular fractions
In addition to specific PDE4 phosphorylation, PDE4 isoforms can show different affinities toward inhibitors depending on whether these isoforms are located in the cytosol or have complexed with other cellular structures, as elaborately investigated by Houslay and collaborators (Bolger et al., 1997; Huston et al., 1996, 1997; Rena et al., 2001; Wallace et al., 2005). By subcellular fractionation, both cytosolic and particulate fractions of PDE4 forms can be separated, which can show differential affinities to inhibitors in an isoform-dependent manner (Table 4). Per cellular fraction, PDE4 isoforms may engage in different protein-protein interactions, which subsequently can alter their conformation, leading to divergent effects on inhibitor affinity. Thus, based on its specific subcellular localization and local interactions or modifications, the same PDE4 isoform can adopt different conformations that may be preferentially bound by certain inhibitors (e.g., the different affinities of rolipram toward particulate and cytosolic PDE4A4; Table 4). As such, these inhibitors can modulate PDE4 activity with different potency at the subcellular level.
2. Optimizing Phosphodiesterase 4 Inhibitor Screenings
The involvement of PDE4 in various cellular processes makes these enzymes attractive pharmacological targets, but it simultaneously makes nonspecific PDE4 inhibition prone to modulating unwanted biologic mechanisms. Hence, to optimize the efficacy and safety of PDE4 inhibitors, it is crucial to specify which PDE4 subtypes or isoforms are involved in the (disease-affected) cellular functions that are to be modified. In this subsection, we highlight how the choice of assay, target specification, and the use of “toolbox” compounds that apply distinct binding mechanisms can guide drug development toward safe and efficacious PDE4 subtype/isoform inhibition.
Firstly, considering that PDE4 enzymes can dynamically adopt several conformational states that can show different affinities to inhibitors, screening assays using purified PDE4 constructs are limited in predicting the inhibitory potential of a compound, as they assess affinities against a static, rather than a dynamic, enzyme. Hence, cell-based screening assays will more accurately indicate the PDE4 inhibitory potential of compounds when other influencing factors, including phosphorylation events, interactor proteins, and biologic feedback mechanisms, are present. When using cell-based assays, we argue it is essential to assess a PDE4-regulated phenotypical or physiologic readout that is relevant to a healthy or pathologic process of interest rather than an overall change in cAMP levels. Thus, the quality (i.e., cAMP elevation at the desired intracellular location) of PDE4 inhibition is more important than the quantity (i.e., profound cAMP elevation but not intracellularly confined) to achieve efficacious treatment while minimizing side effects. The use of cell-based assays that focus on a phenotypical or physiologic readout may also lead to discovery of efficacious PDE4 inhibitors with additional activity on other relevant targets, which would be undetectable in assays using solely purified PDE4 enzymes.
Using a cell-based, possibly disease-relevant, assay, experiments can be conducted to specify which PDE4 subtypes and isoforms regulate the chosen phenotypical or physiologic readout. By means of genetic knockout (e.g., using CRISPR-Cas9) or short-hairpin RNA–mediated knockdown, it can be specified which PDE4 subtypes or individual PDE4 isoforms regulate the biologic readout process. Conveniently, the typical PDE4 gene structures allow for targeting of isoforms selectively, as they each contain (parts of) a unique exon that can be targeted at the DNA level (e.g., using CRISPR-Cas9) or the transcript level (e.g., using short-hairpin RNA) (Fig. 1). Validation of the role of specific subtypes or isoforms in the chosen readout can be performed by overexpressing the subtype/isoform to assess whether it induces an opposite effect on the readout compared with subtype/isoform knockout. The involvement of specific isoforms in the process of interest can also be corroborated using a dominant-negative approach in which a catalytically inactive PDE4 form is being overexpressed. Subsequently, overexpressed inactive PDE4 isoforms will (partly) displace endogenous, active PDE4 isoforms, inducing local decreases in PDE4 activity. Functional roles of specific PDE4 isoforms have already been successfully identified using this approach (Perry et al., 2002; Campbell et al., 2017; Bolger et al., 2020). Although dominant-negative PDE4 forms can isoform-specifically displace endogenous forms, their overexpression may also cause excessive scaffolding of PDE4 interaction proteins that could alter cellular signaling. Hence, depending on the biologic mechanism of interest, validation of the role of specific PDE4 subtypes and isoforms may be best supported by a combination of the abovementioned strategies. Upon target specification and validation, drug design is suggested to be conducted in a structure-based manner. In case the functionally relevant PDE4 forms belong to a specific subtype, subtype-specific differences in PDE4 structure may be exploited to develop PDE4 subtype-selective inhibitors, as described in section C. Mechanisms for Phosphodiesterase 4 Subtype Selectivity: Interactions with Regulatory Domains.
Since there already exist PDE4 inhibitors that preferentially bind certain subtypes, isoforms, and/or conformations (Tables 2–4), these compounds can be used as toolbox compounds to determine which binding mechanism induces the most prominent effect on the assay readout. Parallel insights from the target validation approaches and use of toolbox compounds would eventually provide the insight and understanding to develop PDE4 inhibitors that apply a particular binding mode to selectively bind and inhibit the most relevant PDE4 forms to subsequently efficaciously attenuate a biologic dysfunction of interest.
C. Mechanisms for Phosphodiesterase 4 Subtype Selectivity: Interactions with Regulatory Domains
1. Interactions with the Upstream Conserved Region 2
Although the different subtypes possess highly similar catalytic domains, subtle amino acid differences exist in regulatory regions (i.e., UCR2 and C terminus) that can be positioned across the catalytic pocket. When the UCR2 is capped, certain residues can interact with inhibitors as their side chains extend into the catalytic domain (Fig. 3; Supplemental Video). UCR2 capping is postulated to occur via intermolecular actions in long, dimerized PDE4 isoforms in which the UCR2 of one monomer folds across the catalytic domain of the other monomer (Cedervall et al., 2015). As the UCR2 also is autoinhibitory in monomeric, short PDE4D1, intramolecular UCR2 capping may also occur (Kovala et al., 1997). Irrespective of whether UCR2 capping occurs in trans (intermolecularly) or in cis (intramolecularly), inhibitors can engage in interactions with UCR2 residues. Intriguingly, in primates, a polymorphism has occurred in PDE4D leading to the expression of a phenylalanine instead of tyrosine in the UCR2 region. This subtype-specific difference has been successfully exploited, and validated by mutation studies, to generate PDE4D-selective inhibitors (e.g., BPN14770) that interact with the PDE4D-specific phenylalanine in the UCR2 region (Table 4) (Burgin et al., 2010). Interestingly, Gurney et al. (2019) have shown that certain compounds that use UCR2 for binding, like BPN14770, behave as partial inhibitors. It is hypothesized that, in dimerized PDE4, through inhibitor-UCR2 interactions at one monomer, the other UCR2 cannot effectively trans-cap the other monomer. Since this UCR2-capping is involved in compound binding, inhibitor binding at this monomer will be reduced, resulting in overall partial inhibition. Conversely to binding the UCR2 phenylalanine, inhibitors can preferentially interact with the tyrosine residue in PDE4A-C, producing PDE4D-sparing actions, as reported for ABI-4 (PF‐06266047) (Hedde et al., 2017). Notably, several classes of PDE4 inhibitors have been found to stabilize a UCR2-capped state, as elaborately indicated by Day et al. (2011). Although PDE4 modifications and protein-protein interactions can influence inhibitor affinity by influencing UCR2-capping, stabilization of UCR2 capping by certain inhibitors can, conversely, also alter the conformation of regulatory domains that affect PDE4 modifications and interactions (Terry et al., 2003). In the case of PDE4A4, stabilization of UCR2 capping by inhibitor binding causes its intracellular redistribution, demonstrating that inhibitor binding can induce additional cellular changes next to elevation of local cAMP levels (Day et al., 2011). This use of UCR2 by certain compounds makes their affinity also dependent on post-translational modifications and interactions with partner proteins as they can influence the UCR2 capping state, as explained before.
2. Interactions with the C Terminus
Similar to the use of UCR2 residues to achieve subtype selectivity, amino acid differences in the C terminus can be employed to achieve subtype-specific inhibitor binding. Through interactions with residues unique to the PDE4B C terminus, PDE4B selectivity has been achieved for the compounds A33 and a tetrahydrobenzothiophene inhibitor (see Table 4) (Kranz et al., 2009; Naganuma et al., 2009; Fox et al., 2014). The C terminus is capped across the catalytic domain in an intramolecular manner, as the linker region between the C terminus and the catalytic domain is too short to achieve capping the other monomer in a PDE4 dimer. Inhibitors that employ C terminus residues may therefore preferentially bind capped over uncapped states producing a degree of conformation-dependent binding. As both UCR2 and C terminus capping are dependent on multiple cellular events, including phosphorylation or interactions with partner proteins, conformation-dependent inhibitors may bind PDE4 in a selective spatial and/or temporal manner.
D. Stereoisomerism and Metabolites of Phosphodiesterase 4 Inhibitors
In 1983, it had already been described that the enantiomer (R)-rolipram and racemic rolipram are more potent in increasing cerebral cAMP levels than (S)-rolipram (Wachtel, 1983a,b; Schneider, 1984). Accordingly, several studies have indicated that (R)-rolipram shows higher PDE4 affinity than (S)-rolipram (Torphy et al., 1992; Barnette et al., 1996; Laliberte et al., 2000). This suggests that stereoisomerism of PDE4 inhibitors can influence the inhibitor’s affinity depending on whether a racemic mixture or purified enantiomer is tested. Both (R)-rolipram and (S)-rolipram seem to exhibit similar binding modes in PDE4D2 crystal structures, but increased affinity of (R)-rolipram may be conveyed via other isoforms and/or conformational states than those captured by the reported crystal structures (Huai et al., 2003). Indeed, rolipram can adopt a slightly different conformation in crystals that include UCR2 domains (Cedervall et al., 2015). For another set of enantiomeric inhibitors (L-869298 and L-869299), stereochemistry does change the binding mode in a Mg2+-interacting manner, which is concurrent with differences in affinity (Huai et al., 2006). Next to the aforementioned enantiomeric inhibitors, also for the PDE4D-selective inhibitor GEBR32a, different affinities are reported for its enantiomers (Table 3) (Cavalloro et al., 2020). Hence, in the case of racemic inhibitors, it has to be considered that enantiomers can exhibit different binding modes and affinities for specific PDE4 conformations. Consequently, certain enantiomers may display favorable pharmacological properties superior to its racemic mixture. Enantiomer-specific effects can provide important insights into the molecular binding modes crucial for efficacy and can subsequently facilitate pharmacophore determination and inhibitor optimization.
Similar to the use of racemic PDE4 inhibitor mixtures, metabolism of administered PDE4 inhibitors can produce multiple metabolites that each show differences in their binding mode and affinity. For example, the PDE4 inhibitor roflumilast is metabolized into roflumilast N-oxide, which shows distinct subtype selectivity compared with roflumilast itself (Tables 2 and 3) (Claveau et al., 2004; Huang et al., 2007; Wunder et al., 2013). The inhibitory potential of inhibitor metabolites on PDE4 activity should therefore be taken into account as combined actions of PDE4 inhibitors, and its metabolites may cause certain PDE4 subtypes, isoforms, or conformations to be more potently inhibited in vivo, resulting in a more favorable or unfavorable pharmacological profile.
E. Modulators of Phosphodiesterase 4 Activity
Next to PDE4 inhibitors, molecules have been described that influence PDE4 activity in a noninhibiting manner. For example, atropine, a muscarinic acetylcholine receptor antagonist, has been described to allosterically inhibit PDE4 while also potentiating rolipram binding (Perera et al., 2017). These effects correspond to those of modulation by several intracellular factors as discussed above in section III. Phosphodiesterase 4 Modifications and Interactions. In contrast to allosterically inhibiting molecules, PDE4 activity can also be stimulated through allosteric binding. For example, early studies by the Conti and coworkers showed that binding of antibodies targeting the UCR2 autoinhibitory domain increased PDE4 activity similar to phosphorylation by PKA (Conti et al., 1995; Lim et al., 1999).
More recently, also small molecules have been described that increase PDE4 activity through allosteric binding (Omar et al., 2019). More specifically, these compounds bind and activate long, dimerized PDE4 and thereby mimic the activating actions of PKA. This mechanistic similarity of activation is further supported by the inability of these small molecules to increase activity in PKA-mimicking PDE4 mutants. Hence, these small molecule allosteric activators have therapeutic utility in disorders in which cAMP signaling is aberrantly increased. For example, in autosomal dominant polycystic kidney disease, elevated cAMP levels promote the formation of cysts. Omar et al. (2019) have demonstrated that the use of allosteric activators can successfully diminish cyst formation in cell models of autosomal dominant polycystic kidney disease . It can be speculated that allosteric PDE4 activators also have therapeutic applicability in other disorders displaying elevated cAMP signaling, such as the genetic condition McCune-Albright syndrome, in which gain-of-function Gs α subunit mutations lead to exaggerated cAMP synthesis (Levine, 1999; Innamorati et al., 2018). Hence, PDE4 enzymes have therapeutic utility as pharmacological targets in both conditions displaying reduced and conditions displaying heightened levels of cAMP.
Lastly, PDE4 peptide fragments have been developed that can either stimulate PDE4 activity (Wang et al., 2015) or reduce activation by scavenging the PDE4-activating phosphorylation by Cdk5 (Plattner et al., 2015).
V. Adverse Effects of Phosphodiesterase 4 Inhibition
Although PDE4 inhibition shows therapeutic potential in a variety of disease areas, clinical exploitation of PDE4 inhibitors has often been held back by dose limitations caused by adverse side effects. Side effects upon PDE4 inhibitor administration are mainly gastrointestinal in nature (i.e., nausea, vomiting and diarrhea), but headaches and dizziness have also been reported (Compton et al., 2001). These side effects may be overcome by more precise targeting of PDE4 subtypes, isoforms, or conformational states. Accordingly, subtype-selective PDE4 inhibitors have been developed that exploit subtle sequence differences and distinct conformational states that may result from different PDE4 lengths and specific interactions and modifications (Burgin et al., 2010; Fox et al., 2014; Hedde et al., 2017). While this allows for more precise targeting, insight into which PDE4 subtypes or isoforms mediate unwanted effects is therefore crucial to understand which subtypes/conformations not to target. This section will discuss PDE4 expression and functionality in brain and bodily areas, and potential molecular mechanisms related to the different PDE4-mediated side effects. In addition, an overview is provided of those PDE4 inhibitors that have been tested for possible adverse side effects.
A. Hypothermia
Several preclinical studies using mice, rats, and guinea pigs have demonstrated that PDE4 inhibitors can induce hypothermia (Wachtel, 1983a,b,c; McDonough et al., 2020b). These effects seem to be regulated via PDE4-mediated dopamine signaling in the hypothalamus, which is supported by the fact that PDE4 inhibitors that enter the brain with difficulty have less profound effects on body temperature (McDonough et al., 2020b). Although not explicitly described as an adverse side effect in humans, hypothermia may contribute to uncomfortable feelings during treatment with PDE4 inhibitors.
B. Dizziness
PDE4 inhibition has also been reported to cause dizziness (Blokland et al., 2019). In cochlear and vestibular nuclei in the brainstem, PDE4D was found to be expressed higher than other PDE4 subtypes (Iwahashi et al., 1996; Perez-Torres et al., 2000). More specifically, PDE4D1, PDE4D2, and PDE4D3 mRNA was expressed in the rat dorsal cochlear and vestibular nuclei (Miro et al., 2002b). As the cochlear and vestibular nuclei relay signaling from the inner ear, altered cAMP modulation by PDE4 inhibition in these areas may provoke feelings of dizziness and, through subsequent signaling to other brains stem areas, promote nausea and emesis. Interestingly, in the vestibular apparatus in the inner ear itself, PDE4D is expressed, and rolipram induces endolymphatic hydrops in the mouse, which is associated with feelings of dizziness (Nakashima et al., 2016; Degerman et al., 2017).
C. Gastrointestinal
1. Diarrhea
Concerning effects of PDE4 inhibition on the gastrointestinal system, diarrhea has been reported as an adverse effect. Through PKA-dependent activation of the CFTR, PDE4 inhibition may cause diarrhea by elevating intestinal Cl− secretion (Chao et al., 1994). Specifically, PDE4D may mediate this effect upon PDE4 inhibition, as PDE4D has been found to be recruited to CFTR via binding the scaffolding protein Shank2 (Lee et al., 2007). Moreover, PDE4 inhibition can facilitate 5HT4 receptor–mediated acetylcholine release, causing contraction of large intestinal circular smooth muscle (Pauwelyn et al., 2018). Specifically, the PDE4D3 and PDE4D5 isoforms have been found to associate with 5HT4(b) receptors and may, therefore, be involved in gastrointestinal effects caused by PDE4(D) inhibition (Weninger et al., 2014). Despite the fact that diarrhea has been reported as a PDE4-mediated adverse event, it is striking that roflumilast has actually been found to exhibit antidiarrheal effects in mice, which may be associated with antispasmodic actions of roflumilast in the jejunum (Rehman et al., 2020).
2. Nausea and Emesis
Regarding nausea and emesis, PDE4 actions within both the central nervous and gastrointestinal systems seem to be involved. In an early study, intravenous administration of rolipram and Ro20-1724 was found to increase gastric acid and pepsin secretion in anesthetized rats (Puurunen et al., 1978). Correspondingly, Lamontagne et al. (2001) suggested that, in the stomach, pepsinogen-releasing chief cells primarily express the PDE4D subtype, whereas acid-releasing parietal cells expressed PDE4A. Moreover, binding of specifically HARBS configurations in gastric glands showed strong correlation with the degree of gastric acid secretion (Barnette et al., 1995). Interestingly, gastric transit was found to be more strongly inhibited by roflumilast than by a selective PDE4B inhibitor, which may suggest PDE4B inhibition contributes relatively little to gastric side effects (Suzuki et al., 2013). Recently, it was shown that nonselective PDE4 inhibition induces gastroparesis (i.e., delayed gastric transit) in mice (McDonough et al., 2020a). More importantly, this study showed that genetic ablation of any of the four PDE4 subtypes does not affect gastroparesis or protect against inhibitor-induced gastroparesis, which suggests that two or more PDE4 subtypes contribute to PDE4-mediated gastroparesis. As gastroparesis is strongly associated with nausea and emesis in humans, this physiologic effect of PDE4 inhibition warrants further investigation as a possible predictive measure for the side effect profile of PDE4 inhibitors (Grover et al., 2019).
Although PDE4 inhibition causes profound local gastric effects, emesis is eventually effectuated through signaling in brainstem nuclei such as the nodose ganglion, area postrema (AP), and nucleus tractus solitarius (NTS) (Miller and Leslie, 1994; du Sert et al., 2012). In the nodose ganglion of the squirrel monkey, cell bodies of gastric vagal nerve fibers can be found in which predominantly PDE4D is present compared with lower expression levels of PDE4C or long PDE4A forms (Lamontagne et al., 2001). Similarly, the AP and NTS were found to mainly express PDE4D and, to a lesser extent, PDE4B in mouse (Cherry and Davis, 1999), rat (Iwahashi et al., 1996; Takahashi et al., 1999; Perez-Torres et al., 2000), and human brain (Mori et al., 2010). PDE4D expression was also found in medulla oblongata neurons that are innervated by substance P, which has been reported to be involved in emetic responses upon PDE4 inhibition (Robichaud et al., 1999; Lamontagne et al., 2001). At the PDE4D transcript variant level, mRNA of PDE4D1 and PDE4D4 showed high expression in the rat AP compared with lower levels of PDE4D2 and PDE4D5 and absence of PDE4D3. Furthermore, low levels of PDE4D2, PDE4D4, and PDE4D5 were detected in the NTS (Miro et al., 2002b). Strikingly, in another study by Miro et al. (2002a), no PDE4D4 mRNA was detected in the rat AP. Human data regarding PDE4D mRNA content in the AP also suggest that PDE4D4 is not abundantly present, and the highest expression was found for PDE4D3 and PDE4D9 (Vanmierlo et al., 2019).
In addition to localization studies showing high expression of PDE4D in emesis-related brainstem areas, mechanistically, PDE4D also seems to mediate emetic responses. In a study by Robichaud et al., (2002b), PDE4D, but not PDE4B, knockout mice displayed reduced sleeping time under xylazine/ketamine-induced anesthesia, a measure of emetic-like behavior. Furthermore, the PDE4 inhibitor PMNPQ did reduce anesthesia duration in wild-type and PDE4B-deficient mice but not in PDE4D-deficient mice, which again indicates an involvement of PDE4D in emetic-like behavior. Xylazine/ketamine-induced anesthesia assesses emetic-like behavior in nonvomiting species through α2-adrenergic receptor antagonism (Robichaud et al., 2001; Robichaud et al., 2002a; Nelissen et al., 2019). The involvement of PDE4D in this mechanism corresponds with the expression of both α2 adrenoceptor and PDE4D mRNA in the NTS (Scheinin et al., 1994; Lamontagne et al., 2001). However, PDE4A and PDE4B also seem to mediate α2-dependent signaling, at least in retinal rod bipolar cells (Dong et al., 2014). PDE4 inhibitors may enhance neuronal firing in both the AP and NTS of rats, as higher expression of the neuronal activity marker Fos was observed in these regions upon administration of rolipram or PMNPQ in rats (Bureau et al., 2006). Accordingly, AP neurons could be excited by cAMP, which would be elevated upon inhibitor administration (Carpenter et al., 1988). Notably, AP neurons exhibit a hyperpolarization-activated cation current (Ih) that acts as pacemaker current and is found to be activated through stimulation of cAMP signaling (Funahashi et al., 2003). Consequently, PDE4 inhibition may increase the frequency of this pacemaker current, leading to an increased firing rate. Actually, the influence of PDE4 on pacemaker currents has already been demonstrated in the sinoatrial node of the heart (St Clair et al., 2017). The xylazine/ketamine-induced anesthesia test suggests that PDE4 inhibition alters AP activity through antagonizing α2-adenergic receptor signaling (Nelissen et al., 2019). Accordingly, α2-adrenergic receptor stimulation was shown to inhibit cAMP and close hyperpolarized cyclic nucleotide–gated channels (Wang et al., 2007b). Vice versa, PDE4 inhibition could enhance cAMP, open hyperpolarized cyclic nucleotide–gated channels, increase hyperpolarization-activated currents (Ih), and increase the firing rate of AP neurons, leading to an emetic response. Anesthesia induced by injecting an α2-adrenergic agonist into the locus coeruleus, which innervates the AP, of rats could be reversed by rolipram. However, pretreatment of a PKA inhibitor blocked the effects of rolipram, suggesting emetogenic effects by PDE4 inhibitors are mediated through actions requiring cAMP-PKA signaling (Correa-Sales et al., 1992). Moreover, phosphorylation of PKA and ERK, downstream kinases of cAMP, was found to be highest during peak emesis in AP neurons (Zhong and Darmani, 2017).
These findings support the notion that PDE4-mediated cAMP signaling and its downstream effectors contribute to emetic behavior. Still, the exact mechanism underlying these adverse side effects caused by PDE4 inhibitors remains to be resolved. Since the AP may in part be outside of the blood-brain barrier, emetic effects can be dependent on both peripheral and central actions (Miller and Leslie, 1994). Based on the gene deletion and localization studies discussed above, PDE4D seems to be involved in central and peripheral actions, leading to the induction of emesis. However, emetic(-like) effects cannot be attributed entirely to PDE4D, as the PDE4D-selective inhibitors GEBR32a, V11294A, and BPN14770 appear to be well tolerated in animals and/or healthy volunteers [NCT02648672] [NCT02840279] (Rogers and Giembycz, 1998; Gale et al., 2002, 2003; Sutcliffe et al., 2014; Ricciarelli et al., 2017). It is likely that multiple PDE4 subtypes and isoforms in different bodily tissues contribute collectively to PDE4-associated side effects, as also suggested by Richter and colleagues (McDonough et al., 2020a). Hence, subtype- or isoform-selective PDE4 inhibitor may yield less severe side effects. As PDE4 subtypes and isoforms have nonredundant roles and show specific intracellular localization, certain subtypes or isoforms may be specifically involved in mechanisms that eventually evoke unwanted effects. The potency of the inhibitor to inhibit these particular isoforms would then explain their potency to induce adverse effects. However, which exact PDE4 subtypes and isoforms mediate these processes remains to be determined. Another explanation may lie in the differential affinities of the compound to different PDE4 conformational states. Indeed, correlations have been found between the degree of binding HARBS and emetic effects (Duplantier et al., 1996; Christensen et al., 1998; Hirose et al., 2007). Inhibitors that preferentially target LARBS or show no preference would therefore be safer, which is supported by the relatively low emetogenicity of cilomilast and roflumilast (Davis et al., 2009). In contrast, the PDE4D-selective inhibitor BPN14770 preferentially binds HARBS but appears to be well tolerated in humans [NCT02648672] [NCT02840279] (https://tetratherapeutics.com/wp-content/uploads/2016/11/FINAL-Tetra-Phase-1-121616-FINAL.pdf). Although BPN14770 can bind HARBS, its relatively low emetogenicity may be explained by its PDE4D-selective inhibition and/or the fact that it acts as a partial inhibitor [see section IV. Phosphodiesterase 4 Inhibitors (Burgin et al., 2010)]. Thus, the occurrence of adverse effects may not be fully attributed to preferential binding of HARBS or LARBS, or to PDE4D-selective inhibition, but rather is more complex, involving inhibition of specific PDE4 subtypes and/or isoforms in different central and peripheral tissues.
Thus, it has been shown that it is possible to develop PDE4 inhibitors with safer pharmacological profile compared with the prototypical rolipram. In Table 5, an overview is provided of several compounds that have been tested for their ability to elicit PDE4-associated side effects compared with rolipram. Because of discrepancies among studies regarding the type of test, species, and administration routes used, results can mainly be compared within studies using the same procedures. Table 5 indicates that multiple PDE4 inhibitors induce emetic and emetic-like effects at doses more than 30-fold of those induced by rolipram (e.g., CT-2450 and D159687), whereas some compounds require lower doses than rolipram (e.g., PMNPQ and D157140). Since adverse side effects are often assessed in nonprimate species (e.g., mice, rats, dogs, and ferrets), it should be considered that these tests do not accurately reflect the potential emetic effects of PDE4 inhibitors that exploit the primate PDE4D-specific phenylalanine in the UCR2 region. In addition, rodents cannot vomit and therefore require an indirect emesis test like the xylazine/ketamine test or pica feeding test. Despite the fact that several compounds can be used safely at higher doses than rolipram, their therapeutic potential is also dependent on the doses required for the desired therapeutic effect. As such, the eventual therapeutic windows of these compounds, i.e., the range of doses that elicit therapeutic, yet no adverse, effects may still be similar to those of rolipram for a specific disease indication. Nevertheless, a wide variety of PDE4 inhibitors have been developed with improved affinities to specific subtypes and/or conformational states (Tables 2–4) and minimized emetogenicity compared with rolipram (Table 5). Thus, to establish safer and more efficacious PDE4 inhibition for a disease, it should be determined which PDE4 subtypes, isoforms, or conformations have to be targeted to determine which inhibitor displays the broadest therapeutic window.
PDE4 inhibitors tested for their potential to induce emesis-like behavior (rodents) or emesis compared with rolipram
D. Strategies to Minimize Phosphodiesterase 4–Mediated Adverse Side Effects
Although PDE4 inhibition can produce several adverse side effects, as described above, multiple strategies can be exploited to minimize or prevent these unwanted responses while retaining efficacy. Most notably, more selective inhibition of PDE4 forms may be pursued to improve treatment efficacy and/or avoid inhibiting PDE4 forms mediating adverse side effects. Regardless, a mechanistic understanding of which PDE4 subtypes, isoforms, and/or conformational states mediate adverse effects will aid in the development of new-generation PDE4 inhibitors that then should actually avoid potently inhibiting these forms. Several PDE4 inhibitors already exist that preferentially bind nonconserved amino acids and different conformational states (see Table 4). Moreover, partial inhibitors may provide a safer pharmacological profile compared with full inhibitors through distinct potencies for different PDE4 conformations and/or subtypes (Gurney et al., 2011). In case PDE4 inhibition is desired in peripheral organs rather than the central nervous system, PDE4 inhibitors with limited brain penetrance may minimize the occurrence of side effects that are mediated through central actions (Aoki et al., 2001a). Likewise, depending on the target organ, a particular route of administration can be chosen to minimize systemic exposure (e.g., intranasal or topical administration, or via inhalation) (Tralau-Stewart et al., 2011). More precise targeting of PDE4 inhibitors has also been achieved by using inhibitor-antibody conjugates, which are internalized specifically by cells that express the antibody’s antigen (Yu et al., 2016). Systemic exposure can also be minimized by using PDE4 inhibitors that are quickly degraded, as recently reported (Larsen et al., 2020).
Finally, combination treatments may reduce the severity of PDE4-mediated side effects. For example, it has been described that diarrhea caused by high doses of roflumilast can be attenuated when paired with a cyclooxygenase inhibitor (Peter et al., 2011). Furthermore, inhibition of different PDE families (e.g., PDE4 and PDE5 or PDE2 and PDE4) can produce synergistic effects that enable the use of lower doses, with reduced chances of adverse effects, to produce the same treatment effect (Bollen et al., 2015; Gulisano et al., 2018; Paes et al., 2021b). Lastly, the treatment of certain diseases in which cAMP elevation is desired may benefit from dual PDE4/PDE7 inhibitors, which may subsequently reduce PDE4-selective inhibition and its associated side effects (Sharma et al., 2019; Ručilová et al., 2021).
VI. Outlook
As important regulators of cAMP signaling in the entire body, the PDE4 enzyme family presents an interesting and promising pharmacological target in a broad range of disease areas. Accordingly, PDE4 inhibition has preclinically shown therapeutic potential for many diseases, yet its nonselective inhibition is associated with severe adverse effects, which has seriously hampered its translation to the clinic. To increase efficacy and avoid adverse effects, more selective targeting is required. This is actually possible, as PDE4 enzymes consists of multiple similar, but different, subtypes that each comprise different isoforms. In this review it is summarized how these subtypes and isoforms can, through PDE4-inherent regulation, post-translational modifications, and interactions with other proteins, adopt different conformational states to which PDE4 inhibitors can selectively bind. Since PDE4 subtypes and isoforms are expressed in specific tissue, cell type, and intracellular expression patterns, selective PDE4 subtype/isoform inhibition will enable a more directed modulation of cAMP signaling in the target organ of interest. To further improve PDE4 inhibitor treatment efficacy and safety, it should also be investigated which PDE4 subtypes and isoforms contribute most to the organ’s disease-specific aberrant cAMP signaling. Additionally, upon PDE4 specification, the conformational state of these enzymes in their disease context would have to be determined to choose or develop the most efficacious inhibitors (Fig. 5). Consequently, it is crucial to determine preferential binding of PDE4 inhibitors to specific subtypes, isoforms, and conformations rather than testing their affinity toward PDE4 catalytic domains only. Moreover, a deeper insight into the mechanisms underlying PDE4-mediated unwanted effects in organs is also warranted to facilitate the development and use of safe PDE4 inhibitors to treat diseases. In conclusion, specification and subsequent selective inhibition of PDE4 subtypes, isoforms, or conformations grants the opportunity to effectively and safely modulate aberrant cAMP signaling in myriad diseases. This will increase the chance of success of more PDE4 inhibitors reaching the patient eventually.
PDE4 as a pharmacological target in a variety of disease areas. In a broad range of diseases, PDE4 inhibition shows therapeutic potential. Nonselective PDE4 inhibition is associated with adverse effects, but the existence of PDE4 subtypes (PDE4A–D) and associated isoforms allows for more specific targeting. PDE4 subtypes and isoforms can undergo conformational changes upon post-translational modifications and interactions with partner proteins. PDE4 inhibitors can display different affinities toward PDE4 subtypes, isoforms, and conformations. Disease-specific determination of which PDE4 isoform(s) should be inhibited will facilitate the development of safe and efficacious PDE4 inhibitors. This figure was created with BioRender.com. P, phosphorylation; SUMO, SUMOylation.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Paes, Schepers, Rombaut, van den Hove, Vanmierlo, Prickaerts.
Footnotes
This work was supported by Internationale Stichting Alzheimer Onderzoek/Alzheimer Nederland [Grant WE.03-2016-07].
Tim Vanmierlo and Jos Prickaerts have a proprietary interest in selective PDE4D inhibitors for the treatment of neurodegenerative disorders. The other authors declare that they have no competing interests.
↵
 This article has supplemental material available at pharmrev.aspetjournals.org.
This article has supplemental material available at pharmrev.aspetjournals.org.

Abbreviations
- AKAP
- A-kinase anchoring protein
- AP
- area postrema
- CaMKII
- calcium/calmodulin-dependent kinase II
- CDK5
- cyclin-dependent kinase 5
- CFTR
- cystic fibrosis transmembrane conductance regulator
- circPDE4D
- circular PDE4D
- DISC1
- disrupted in schizophrenia 1
- ERK
- extracellular signal–regulated kinase
- Fyn
- proto-oncogene tyrosine-protein kinase Fyn
- HARBS
- high-affinity rolipram binding site
- 5HT4
- 5-hydroxytryptamine 4
- JNK
- c-Jun N-terminal kinase
- LARBS
- low-affinity rolipram binding site
- LR2
- linker region 2
- Lyn
- Lck/Yes novel tyrosine kinase
- MK2
- MAPK-activated protein kinase 2
- NCBI
- National Center for Biotechnology Information
- NTS
- nucleus tractus solitaries
- PDE
- phosphodiesterase
- PHD2
- prolyl hydroxylase domain-containing protein 2
- PKA
- protein kinase A
- p75NTR
- p75 neurotrophin receptor
- QKI
- quaking
- qPCR
- quantitative polymerase chain reaction
- RACK1
- receptor of activated protein C kinase 1
- Rheb
- RAS homolog enriched in brain
- Shank2
- SH3 and multiple ankyrin repeat domains protein 2
- SIK1
- salt-inducible kinase 1
- Src
- proto-oncogene tyrosine-protein kinase
- UCR1
- upstream conserved region 1
- UCR2
- upstream conserved region 2
- 3′UTR
- 3′ untranslated region
- 5′UTR
- 5′ untranslated region
- XAP
- HBV X-associated protein 2.
- Copyright © 2021 by The American Society for Pharmacology and Experimental Therapeutics





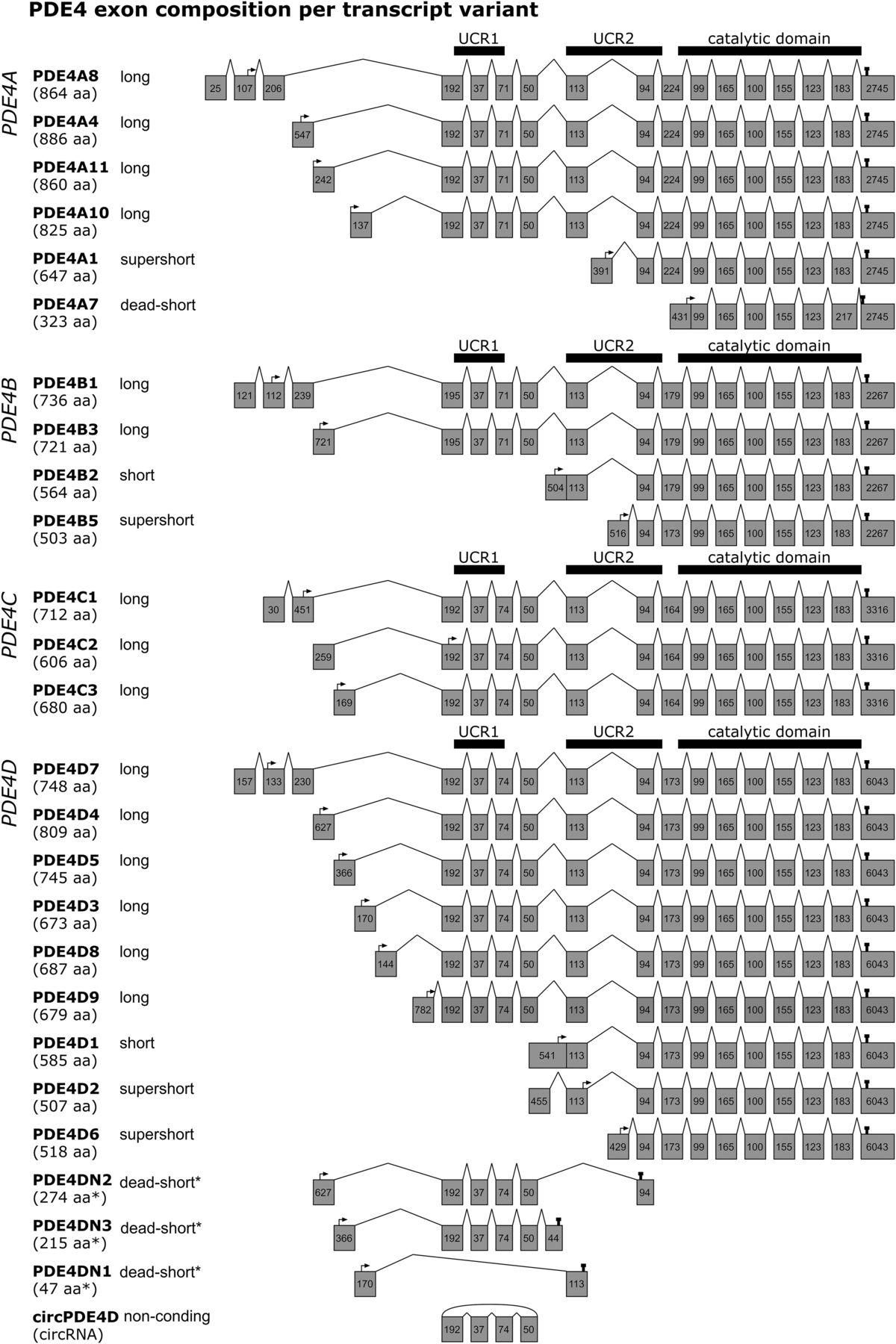{kind=link}
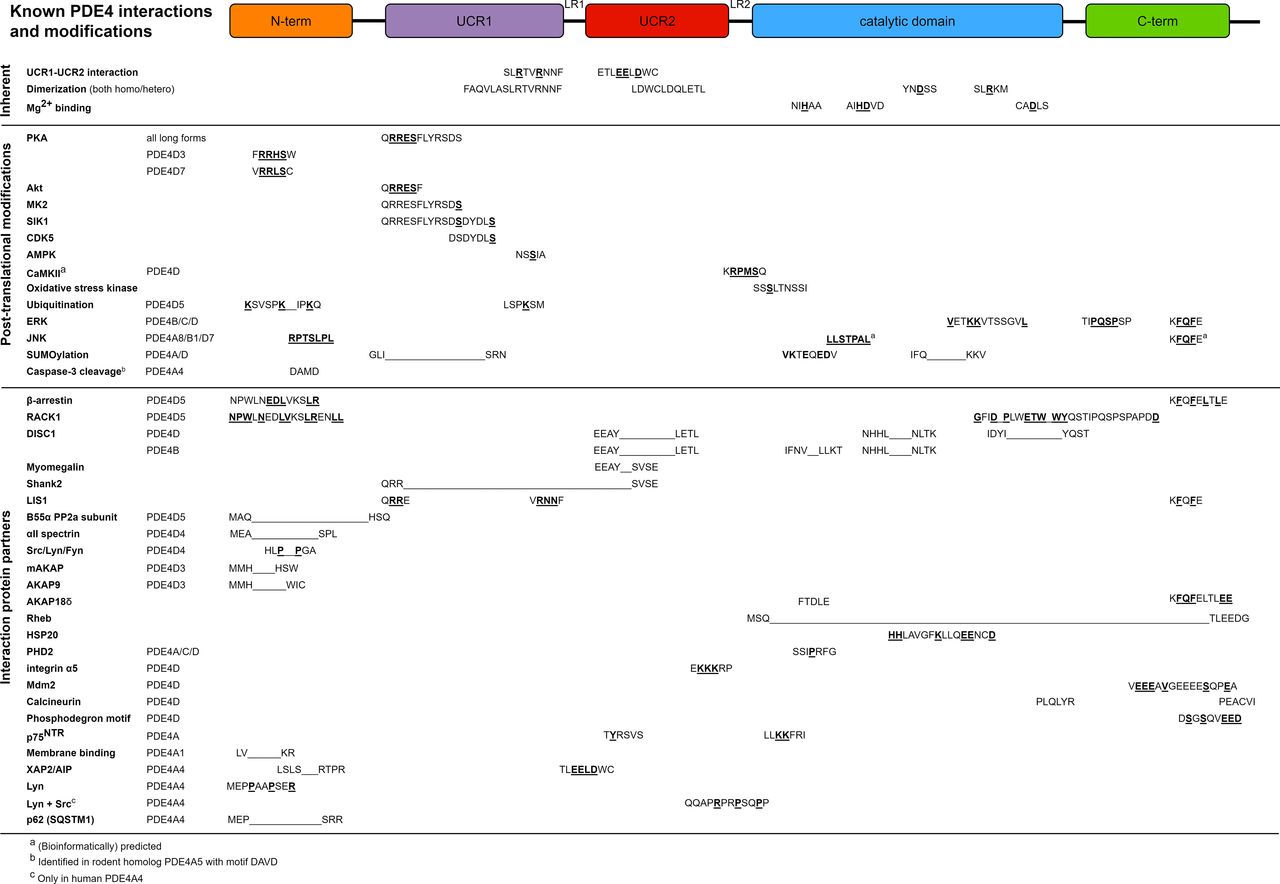{kind=link}
{kind=link}
{kind=link}
{kind=link}