


Visual Overview

Abstract
Idiosyncratic drug reactions (IDRs) range from relatively common, mild reactions to rarer, potentially life-threatening adverse effects that pose significant risks to both human health and successful drug discovery. Most frequently, IDRs target the liver, skin, and blood or bone marrow. Clinical data indicate that most IDRs are mediated by an adaptive immune response against drug-modified proteins, formed when chemically reactive species of a drug bind to self-proteins, making them appear foreign to the immune system. Although much emphasis has been placed on characterizing the clinical presentation of IDRs and noting implicated drugs, limited research has focused on the mechanisms preceding the manifestations of these severe responses. Therefore, we propose that to address the knowledge gap between drug administration and onset of a severe IDR, more research is required to understand IDR-initiating mechanisms; namely, the role of the innate immune response. In this review, we outline the immune processes involved from neoantigen formation to the result of the formation of the immunologic synapse and suggest that this framework be applied to IDR research. Using four drugs associated with severe IDRs as examples (amoxicillin, amodiaquine, clozapine, and nevirapine), we also summarize clinical and animal model data that are supportive of an early innate immune response. Finally, we discuss how understanding the early steps in innate immune activation in the development of an adaptive IDR will be fundamental in risk assessment during drug development.
Significance Statement Although there is some understanding that certain adaptive immune mechanisms are involved in the development of idiosyncratic drug reactions, the early phase of these immune responses remains largely uncharacterized. The presented framework refocuses the investigation of IDR pathogenesis from severe clinical manifestations to the initiating innate immune mechanisms that, in contrast, may be quite mild or clinically silent. A comprehensive understanding of these early influences on IDR onset is crucial for accurate risk prediction, IDR prevention, and therapeutic intervention.
I. Introduction
Idiosyncratic drug reactions (IDRs) represent a spectrum of unpredictable adverse drug reactions, ranging from mild, more common reactions to potentially life-threatening, less common reactions. IDRs can affect any organ, but a common target of IDRs is the liver. This can lead to liver failure and liver transplantation or death. IDRs may affect the skin and can range in presentation from a mild rash to toxic epidermal necrolysis (TEN), which has a high mortality rate and leaves survivors with permanent scars and often blindness. The bone marrow is also a common target, presenting as agranulocytosis, which can lead to sepsis and death. IDRs are responsible for a substantial burden on patient morbidity, mortality, and health care expenses, and because we cannot predict which drugs may cause IDRs, it also represents a risk to drug development (Suh et al., 2000; Pirmohamed et al., 2004; Breckenridge, 2015).
Although their mechanisms are still poorly understood, there is considerable evidence to suggest that IDRs are immune-mediated. Clinical features such as antidrug or antinuclear antibody detection, human leukocyte antigen (HLA) associations, delayed reaction onset with rapid onset during rechallenge, and involvement of lymphocytes, particularly cytotoxic T cells (identified by histology and by their activation in response to drug exposure in vitro) are all highly suggestive that IDRs are the result of aberrant activation of the adaptive immune response. It is likely that specific attributes of the adaptive immune system are what make IDRs idiosyncratic. For example, HLA associations alone often do not accurately predict the risk of developing IDRs. It is possible that the correct combination of HLA and T-cell receptor (TCR), which are randomly generated in each individual, is required to initiate the adaptive response that leads to the IDR. However, the events that lead up to this, i.e., the innate immune response that precedes antigen presentation, may not be idiosyncratic.
The postulation that an innate immune response is a necessary initiating mechanism in the progression to a serious IDR has been proposed by a number of groups (Cho and Uetrecht, 2017; Sawalha, 2018; Holman et al., 2019; Ali et al., 2020; Hastings et al., 2020; Yokoi and Oda, 2021). However, IDR research to date has predominantly focused on the role of the adaptive immune response and the clinical manifestations of these reactions during the IDR itself, but the events leading up to the clinical manifestation of the IDR remain largely uncharacterized. Thus, this review aims to encourage prospective research on the mechanisms that are involved during the time between commencement of drug administration and the onset of an adaptive IDR by providing an overview of the innate immune system and supporting evidence that drugs that cause IDRs can also induce an innate response. First, we provide a brief overview of the major classes of IDRs with reference to general characteristics, treatment strategies, and drugs frequently associated with the reactions. We then introduce fundamental principles in innate immunology, as well as mechanisms of adaptive immune activation, that may play a mechanistic role in the subclinical phase preceding the development of an IDR. This includes the cells and soluble mediators of the innate immune system in addition to mechanisms of antigen formation, antigen uptake, antigen presentation, and adaptive immune activation. Subsequently, using four archetypal IDR-associated drugs (amodiaquine, amoxicillin, clozapine, and nevirapine), we summarize the available clinical and animal model literature that is supportive of early immune involvement and activation. Patterns and differences among the data for these drugs will be discussed, and current knowledge gaps will also be emphasized. Lastly, we suggest the application of this research to relevant fields in toxicology.
II. Review of Types of Idiosyncratic Drug Reactions
IDRs have been extensively reviewed elsewhere (Uetrecht and Naisbitt, 2013; Böhm et al., 2018), and describing these reactions in considerable detail is not the purpose here. The main presentations of IDRs will be briefly described, with a focus on the clinical features and studies that illustrate the involvement of the immune system.
A. Idiosyncratic Drug-Induced Liver Injury
Between 1975 and 2007, of 47 drugs that were withdrawn from the market, 15 were withdrawn because of hepatotoxicity, highlighting the burden of this adverse event on patient safety and drug development (Stevens and Baker, 2009). The liver is likely such a common target of IDRs because of its role in drug metabolism. The LiverTox website (http://www.livertox.nih.gov/) identifies 12 different types of drug-induced liver injury based on clinical phenotype (Hoofnagle, 2013). Idiosyncratic drug-induced liver injury (IDILI) may occur unpredictably after drug administration. To broadly classify the type of IDILI, an R ratio is calculated using alanine aminotransferase (ALT) and alkaline phosphatase (ALP) levels, expressed as a multiple of the upper limit of normal: ALT/ALP ≤ 2 indicates cholestatic liver injury, ≥ 5 indicates hepatocellular liver injury, and intermediate values indicate a mixed phenotype. Particular HLA class II molecules may influence the pattern of liver injury (Andrade et al., 2004).
1. Hepatocellular Liver Injury
Hepatocellular liver injury is caused by hepatocyte death. The time to onset can vary widely, with 1–3 months being most common. The severity in presentation also varies, with mild and transient elevations in liver enzymes presenting more frequently than severe liver injury that may require liver transplantation or result in death (Uetrecht, 2019a). Symptoms can include allergic features such as fever or rash (Uetrecht and Naisbitt, 2013). Many drugs have been associated with causing hepatocellular IDILI, including various anti-infective agents (e.g., sulfonamides, minocycline, nitrofurantoin, rifampicin, isoniazid, nevirapine), troglitazone, lamotrigine, and diclofenac; immune checkpoint inhibitors are also emerging as a major cause of liver injury (Andrade et al., 2019; Uetrecht, 2019a; Shah et al., 2020).
Histologic examination has revealed the involvement of various cell types, although there is often a mononuclear infiltrate, and there may be eosinophils (Zimmerman, 1999). Eosinophilia in peripheral blood and liver biopsies was correlated with a better prognosis (Björnsson et al., 2007). Increases in CD8+ T cells [cytotoxic T cells (Tc cells)] and macrophages have been noted by immunohistochemical staining (Foureau et al., 2015). An immune response can be a response to injury rather than its cause; however, the major role of CD8+ T cells is to kill virus-infected cells and cancer cells, not to repair damage. In a mouse model, we showed that depletion of CD8+ T cells protected against amodiaquine-induced liver injury, suggesting that these cells do indeed mediate the injury (Mak and Uetrecht, 2015b). In patients treated with isoniazid who had a mild increase in liver enzymes, Th17 cells secreting interleukin (IL)-10 were also increased in peripheral blood (Metushi et al., 2014).
Various antibodies have also been detected in patients with IDILI. For instance, a number of cases of anti–cytochrome P450 antibodies have been reported for different drugs (Kullak-Ublick et al., 2017), which suggests that drug bioactivation is important in producing the immune response. A recent study found that anti-mitochondrial antibodies correlated with the severity of liver injury better than did anti-nuclear antibodies (Weber et al., 2020).
Most genetic associations with the risk of IDILI development have been related to HLA polymorphisms (Kaliyaperumal et al., 2018). In some cases, other associations have been found, such as an association with an IL-10-low producing phenotype that correlated with an absence of peripheral eosinophilia and more severe liver injury (Pachkoria et al., 2008), an association between increased risk of IDILI with a genetic variant linked to differential expression of interferon regulatory factor-6 in the context of interferon (IFN)-β treatment in multiple sclerosis (Kowalec et al., 2018), and an association between increased risk of IDILI and a missense variant of the gene encoding protein tyrosine phosphatase, nonreceptor type 22 gene (Cirulli et al., 2019).
2. Autoimmune Liver Injury
Certain drugs cause a syndrome that closely resembles autoimmune hepatitis with hypergammaglobulinemia and detectable serum autoantibodies including anti-nuclear antibodies and smooth muscle antibodies (de Boer et al., 2017). The histology also tends to be consistent with that of autoimmune hepatitis, such as interface hepatitis and hepatic rosette formation (Hennes et al., 2008). The onset of autoimmune IDILI is typically later, often after over a year of drug administration. Nitrofurantoin and minocycline are two of the most commonly implicated drugs (Björnsson et al., 2010).
3. Cholestatic Liver Injury
Cholestatic liver injury arises from problems within the biliary system. In some cases, cholestatic IDILI has been associated with a lower risk of death compared with hepatocellular IDILI (Andrade et al., 2005; Björnsson and Olsson, 2005), but in other cases, the mortality was found to be higher, although the cause of death was not often the liver injury itself (Chalasani et al., 2008). Such findings may depend upon the patient population, as cholestatic IDILI is more commonly observed in older patients (Lucena et al., 2009). In terms of the course of the liver injury, the recovery from cholestatic IDILI tends to be more prolonged than for hepatocellular IDILI, possibly because cholangiocytes regenerate more slowly than hepatocytes (Abboud and Kaplowitz, 2007). Cholestatic IDILI may also lead to ductal injury, such as vanishing bile duct syndrome (Hussaini and Farrington, 2007). Drugs associated with cholestatic drug–induced liver injury include various anti-infective agents (e.g., amoxicillin-clavulanate, flucloxacillin, penicillins) and oral contraceptives (Andrade et al., 2019).
Bile salt export pump (BSEP) inhibition has been identified as a possible mechanism that induces cholestatic IDILI. The rationale for this hypothesis is based upon the finding that genetic defects in BSEP activity cause liver failure with a cholestatic pattern (Jacquemin, 2012). Although correlations have been identified between in vitro BSEP inhibition and drugs that cause IDILI (Morgan et al., 2010), there has not been convincing evidence that this is the mechanism in vivo. Indeed, many of these drugs cause hepatocellular, rather than cholestatic, liver injury, so this is not consistent with the proposed mechanism. One group found that the in vitro results predict IDILI as well as the Biopharmaceutics Drug Disposition Classification System, but because this system is not based upon mechanistic hypotheses of liver injury, BSEP inhibition as a mechanism cannot be a reliable predictor of drug-induced liver injury (Chan and Benet, 2018). Additionally, although it is plausible that BSEP inhibition could lead to the accumulation of bile salts in the liver and induce cytotoxicity or cell stress, few clinical studies to examine bile salt levels in patient sera have been performed to further test this hypothesis.
Amoxicillin/clavulanic acid is most commonly associated with cholestatic IDILI, and multiple HLA associations have been identified in different ethnicities (Hautekeete et al., 1999; Lucena et al., 2011; Stephens et al., 2013). HLA associations have also been found for flucloxacillin (Daly et al., 2009; Nicoletti et al., 2019), and a polymorphism in BSEP 1331 has been found for cholestatic IDILI caused by estrogen (Meier et al., 2008).
B. Severe Cutaneous Adverse Reactions
Skin rash is a highly reported adverse effect likely because it is visible to the patient, even if it is not usually severe. Additionally, as a barrier between the host and the environment, the skin has high immune activity and contains a number of immune cells including macrophages, Langerhans cells, mast cells, and multiple lymphocytes (Sharma et al., 2019). Although the skin has very low cytochrome P450 activity relative to the liver (Rolsted et al., 2008), it contains other enzymes capable of xenobiotic metabolism, such as sulfotransferases and acetyltransferases, which can bioactivate drugs and generate covalently modified proteins (Baker et al., 1994; Dooley et al., 2000; Bhaiya et al., 2006; Luu-The et al., 2009). The focus of this section will be the severe cutaneous drug reactions, which can be life-threatening skin reactions with systemic involvement and fever.
1. Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis
Stevens-Johnson syndrome (SJS) and TEN are considered to be on the same spectrum of disease, wherein SJS is classified as involving ≤10% of total body surface area, TEN as ≥30% of body surface area, and SJS-TEN as intermediate involvement (Gerull et al., 2011). TEN is the most severe of the skin reactions and has a mortality rate of 30%. The onset usually ranges from about 1 to 3 weeks. Drugs with a high risk of causing SJS or TEN include antiepileptics (e.g., carbamazepine, lamotrigine, phenytoin, phenobarbital), antibiotics (e.g., trimethoprim-sulfamethoxazole, nevirapine), oxicam NSAIDs (e.g., meloxicam, piroxicam), allopurinol, and sulfasalazine (Mockenhaupt et al., 2008).
Full-thickness epidermal necrosis, keratinocyte apoptosis, and a mild mononuclear infiltrate characterize the histology (Uetrecht and Naisbitt, 2013). Involvement of various inflammatory mediators has been identified in the pathology of SJS/TEN, including tumor necrosis factor (TNF)-α (Paquet et al., 1994; Nassif et al., 2004b), soluble Fas ligand (Viard et al., 1998; Abe et al., 2003; Murata et al., 2008), granzyme B and perforin (Posadas et al., 2002; Nassif et al., 2004a), and granulysin (Chung et al., 2008). These mediators are highly suggestive of CD8+ T-cell involvement, and indeed these cells have been identified in patient blister fluid (Chung et al., 2008). In addition, CD8+ T cells from patients proliferate in response to culprit drugs in vitro (Nassif et al., 2004a; Hanafusa et al., 2012), although this is not always the case (Tang et al., 2012). Monocytes have also been identified in patient blister fluid (de Araujo et al., 2011; Tohyama and Hashimoto, 2012).
2. Drug Reaction with Eosinophilia and Systemic Symptoms
Drug reaction with eosinophilia and systemic symptoms (DRESS) was first identified as being caused by anticonvulsant medications and was originally referred to as anticonvulsant hypersensitivity syndrome (Shear and Spielberg, 1988), but this term is now seldom used (Bocquet et al., 1996; Uetrecht and Naisbitt, 2013). DRESS is characterized by rash, fever, and at least one additional symptom indicating organ involvement (lymph nodes, liver, kidney, lung, heart, thyroid, or blood) (Peyrière et al., 2006; Walsh and Creamer, 2011). However, the presentation of the syndrome is highly heterogeneous, and diagnosis can be quite difficult; for example, a rash is not always present, and if it is, it can vary in its histopathology (Ortonne et al., 2015). The onset of DRESS is typically 2–6 weeks, and its associated mortality rate is about 10% (Cacoub et al., 2011). Moreover, multiple human herpesviruses and other viruses have been found to be reactivated in patients experiencing DRESS (Kano et al., 2006). Drugs that have been associated with DRESS include antiepileptics (e.g., carbamazepine, phenytoin, lamotrigine), antibiotics (e.g., trimethoprim-sulfamethoxazole, minocycline), allopurinol, and abacavir (Behera et al., 2018).
3. Acute Generalized Exanthematous Pustulosis
Acute generalized exanthematous pustulosis (AGEP) presents as a sterile pustular rash on the trunk, face, axillae, upper extremities, and groin. Neutrophilia and eosinophilia may be present, and systemic symptoms are less common than with the other skin reactions but may occur (∼20% of cases) (Beylot et al., 1980; Sidoroff et al., 2001; Feldmeyer et al., 2016). The onset of AGEP is shorter than with other skin reactions and can be as short as less than a day or up to 23 days (Roujeau et al., 1991; Choi et al., 2010). Antibiotics (e.g., penicillins, sulfonamides, terbinafine) are the most common cause of AGEP, although other drugs have been implicated as well (e.g., hydroxychloroquine, diltiazem) (Sidoroff, 2012).
Both CD4+ T cells (helper T cells, Th cells) and CD8+ T cells have been identified in the dermis and epidermis of patients with AGEP, and neutrophils are observed in the pustules (Britschgi et al., 2001). These T cells were found to be drug-reactive and secreted IL-8 [C-X-C motif chemokine ligand (CXCL) 8]. Both CD4+ and CD8+ T-cell subsets appear to be activated to a cytotoxic killer phenotype, and perforin, granzyme B, and Fas ligand are involved in the tissue damage (Schmid et al., 2002). Variants in interleukin 36 receptor antagonist (IL-36RN) have been associated with the development of AGEP (Navarini et al., 2013), as well as HLA-A*31:01 (McCormack et al., 2011).
C. Idiosyncratic Drug-Induced Blood Dyscrasias
Several IDRs affect blood cells, possibly by enhancing their destruction or impairing their production and maturation. These blood reactions include agranulocytosis, hemolytic anemia, and thrombocytopenia.
1. Idiosyncratic Drug-Induced Agranulocytosis
Agranulocytosis is a deficiency of granulocytes in the peripheral blood, which is classically defined as a neutrophil count below 500 cells per microliter of blood (Andrès and Maloisel, 2008; Andrès et al., 2011). Agranulocytosis can be the result of a sequestering of neutrophils in tissue reservoirs, decreased production of neutrophils in the bone marrow (where there is an absence of neutrophil precursors beginning at the promyelocyte stage), and/or increased destruction of neutrophils or their precursors (Schwartzberg, 2006). Like other IDRs targeting blood and bone marrow, the time to onset of idiosyncratic drug-induced agranulocytosis (IDIAG) is usually delayed, typically between 1 and 3 months (Andrès et al., 2017). It can present clinically as septicemia, septic shock, and/or severe infection; however, often patients may remain relatively asymptomatic, highlighting the need for routine monitoring of neutrophil counts for high-risk drugs (Palmblad et al., 2016; Andrès et al., 2019). Drugs frequently associated with this IDR include antibiotics (e.g., cotrimoxazole and amoxicillin ± clavulanic acid), antithyroid drugs (e.g., carbimazole), psychotropics (e.g., clozapine and carbamazepine), antiviral agents (e.g., valganciclovir), antiaggregants (e.g., ticlopidine), analgesics (e.g., metamizole), disease-modifying antirheumatic drugs (e.g., sulfasalazine), and immune checkpoint inhibitors (e.g., nivolumab and ipilimumab) (Andrès and Mourot-Cottet, 2017; Boegeholz et al., 2020). Some risk factors have been identified, such as the presence of certain HLA haplotypes. For instance, several HLA-B haplotypes and HLA-DQB1 are associated with an increased risk of agranulocytosis with clozapine (Legge and Walters, 2019).
Rescue of neutrophil counts to baseline levels can usually be achieved by halting treatment with the suspected drug, and recovery can be assisted with the administration of granulocyte colony stimulating factor or granulocyte-macrophage-colony stimulating factor, thereby reducing the likelihood of infections or other fatal complications (Andersohn et al., 2007; Andrès and Mourot-Cottet, 2017). Although this treatment is useful for patients who have already developed agranulocytosis, it does not prevent the onset of this IDR. Overall, the underlying mechanism of IDIAG is not well understood, but preclinical and clinical research suggests that the reaction likely involves an immune component linked with the formation of reactive metabolites of the drug by myeloperoxidase (Johnston and Uetrecht, 2015).
2. Idiosyncratic Drug-Induced Hemolytic Anemia
Hemolytic anemia is characterized by the premature destruction of erythrocytes that can occur intra- or extravascularly. Patients may be asymptomatic or present with a variety of symptoms, including dyspnea, fatigue, hematuria, tachycardia, and jaundice. Management simply involves discontinuation of the implicated agent (Phillips and Henderson, 2018). There is considerable overlap between drugs that cause agranulocytosis or thrombocytopenia and hemolytic anemia, with reports of patients experiencing more than one hematologic IDR from a single drug (Garratty, 2012). Frequently implicated drugs include the antiarrhythmics (e.g., quinidine, procainamide), antibiotics (e.g., piperacillin, minocycline), the antihypertensive α-methyldopa, and the diuretic hydrochlorothiazide (Al Qahtani, 2018). The suggested mechanisms of this IDR include either drug-dependent or autoimmune antibodies (Gniadek et al., 2018), with some drug-dependent antibodies demonstrating potential selectivity for certain blood group antigens (Garratty, 2009).
3. Idiosyncratic Drug-Induced Thrombocytopenia
Thrombocytopenia is a deficiency in circulating platelets, typically characterized by a platelet count of less than 150,000 cells per microliter of blood, although patients may be asymptomatic until counts fall below 50,000 cells per microliter, at which point purpura may be observed (Gauer and Braun, 2012). With counts below 10,000 cells per microliter, spontaneous bleeding may occur; this constitutes a hematologic emergency (https://www.ncbi.nlm.nih.gov/books/NBK542208/). Typically, treatment involves discontinuation of the causative agent and allowing counts to recover without further intervention, although corticosteroids or platelet transfusions may be administered if the hemorrhage is life-threatening (Andrès et al., 2009). The most common drugs reported in association with immune thrombocytopenia include the anticoagulant heparin; the antimalarial quinine; the antiarrhythmic quinidine; the antibiotics rifampicin, cotrimoxazole, and penicillin; and several oral antidiabetic agents (Andrès et al., 2009). Depending on the offending drug, several mechanisms responsible for the decrease have been proposed, including myelosuppression or the expedited clearance of platelets caused by anti-platelet or anti-haptenated platelet antibodies or platelet-specific autoantibodies (Narayanan et al., 2019). One recent example is a case of moxifloxacin-induced thrombocytopenia, in which IgM and IgG antiplatelet antibodies were detected in serologic testing and were found to be enhanced in the presence of moxifloxacin, but not with pantoprazole or esomeprazole (Moore et al., 2020).
D. Other Idiosyncratic Drug Reactions
Although reactions targeting the liver, skin, and blood cells are among the most common IDRs, several other classes exist, including autoimmune reactions and kidney injury.
1. Idiosyncratic Drug-Induced Autoimmune Reactions
A number of drugs may cause organ-specific autoimmune-type reactions, such as autoimmune hemolytic anemia or autoimmune hepatitis, as described above. Frequently, drugs may cause more than one type of autoimmune reaction, although the pattern of reactions observed may be unique for different drugs (Uetrecht and Naisbitt, 2013). Drug-induced vasculitis is another example of a delayed-type autoimmune reaction, whereby patients may develop antineutrophilic cytoplasmic antibodies against a variety of cytoplasmic neutrophil antigens, including myeloperoxidase, lactoferrin, or granule proteins (Guzman and Balagula, 2020). Notably, myeloperoxidase can oxidize many drugs that are associated with autoimmune reactions, and this likely represents a key mechanistic step in the progression to IDRs (Hofstra and Uetrecht, 1993; Uetrecht, 2005). Drug-induced vasculitis may present with morbilliform eruptions but is also manifested by blood vessel wall inflammation and necrosis (Shavit et al., 2018). Medications from a variety of classes have been associated with rare cases of vasculitis, including TNF-α inhibitors such as etanercept (Shavit et al., 2018).
Conversely, the autoimmune reaction induced by hundreds of drugs and herbal medications presents with systemic lupus erythematosus-like clinical characteristics within the first few weeks to months of treatment (Solhjoo et al., 2020). Although the clinical manifestation of different drugs can vary considerably, a positive antinuclear antibody score usually is observed, with autoantibodies including anti-histone antibodies, anti-phospholipid antibodies, and anti-neutrophilic cytoplasmic antibodies. The necessity of both an innate and adaptive immune response in the onset of drug-induced autoimmunity has also been proposed (Sawalha, 2018). One of the earliest drugs reported to have a high incidence of drug-induced lupus during chronic treatment was procainamide, with the majority of patients presenting with anti-nuclear antibodies (Uetrecht and Woosley, 1981). Sulfasalazine, a disease-modifying antirheumatic drug, has also been associated with a significant number of autoimmune reactions (Atheymen et al., 2013), identified risk factors for which include HLA-DR4 and HLA-DR*03:01 (Gunnarsson et al., 2000). Resolution of drug-induced autoimmunity is commonly achieved by discontinuation of the implicated agent.
2. Idiosyncratic Drug-Induced Nephropathy
Drug-induced acute interstitial nephritis (DIAIN) is most prominent at the corticomedullary junction. Drug treatment accounts for between 70% and 90% of biopsy-confirmed acute interstitial nephritis (Nast, 2017), and it is the third most common reason for acute kidney injury in hospitalized patients (Raghavan and Shawar, 2017). Typically, symptoms of DIAIN are nonspecific (e.g., general fatigue, myalgia, and arthralgia) (Perazella, 2017), with approximately 50% of cases accompanied by cutaneous reactions (Raghavan and Eknoyan, 2014). The most accurate diagnosis of interstitial nephritis is achieved with a kidney biopsy, as blood tests are generally not useful and various imaging modalities (e.g., computed tomography scans, ultrasounds) and urinary tests (e.g., urine microscopy, eosinophiluria) do not provide highly sensitive and/or specific findings (Perazella, 2017). Key histopathological findings include focal to diffuse interstitial edema and an inflammatory infiltrate of T cells that is frequently accompanied by plasma cells and macrophages but infrequently may be accompanied by eosinophilia, depending upon the causative drug (Nast, 2017).
More than 250 drugs have been associated with the risk of DIAIN, including NSAIDs (e.g., diclofenac and naproxen), proton pump inhibitors (e.g., omeprazole and esomeprazole), and antibiotics (e.g., penicillins and sulfonamides) (Eddy, 2020), each presenting with differing histology. On average, NSAIDs induce less severe injury and rarely have infiltrating interstitial eosinophils, whereas eosinophils are observed in more than 80% of proton pump inhibitor–induced acute interstitial nephritis (AIN), which also appears to be a more severe reaction and often takes more than 6 months to resolve (Valluri et al., 2015). DRESS may also involve the kidney in approximately 10%–30% of cases caused by antibiotics (Eddy, 2020).
The onset of AIN frequently occurs within the first few weeks of treatment with antibiotics, although cases of NSAID-induced AIN have been reported after 6–18 months of treatment (Eddy, 2020). With proton pump inhibitors, the range of onset is 1–18 months, and in many cases of drug-induced AIN, the initiating mechanisms are unclear (Nast, 2017). A possible initiating mechanism includes the covalent binding of the drug or its metabolites to proteins in the kidney, as may occur with β-lactam or sulfonamide antibiotics (Raghavan and Shawar, 2017). Resolution of injury often occurs after removal of the offending agent and may be aided with corticosteroid treatment; however, in the elderly population, return to baseline kidney function may not be achieved in up to 50% of patients (Valluri et al., 2015). Moreover, some acute cases of tubulointerstitial nephritis may progress to chronic kidney disease with interstitial fibrosis and tubular atrophy (Perazella, 2017).
E. Rationale for Current Review
Throughout this section, it is clear that there is involvement of the adaptive immune system across IDRs affecting different organs. Delayed onset, multiple symptoms, and HLA-associated risk factors of severe IDRs are most consistent with an adaptive immune response. But cells of the adaptive immune system require activation from the innate immune system, and the following section outlines how drugs may cause activation of the innate immune system. Understanding this process is crucial in understanding the development of IDRs. Additionally, although the adaptive response appears to be idiosyncratic because of patient-specific factors, the innate response is unlikely to be idiosyncratic, as it is the body’s first and nonspecific line of defense after the detection of pathogens and other harmful stimuli. Therefore, this may represent a means of identifying drug candidates that carry the risk of causing IDRs during drug development and will be discussed in more detail below.
Ultimately, the goal of this review is to highlight the need for research on the initiating factors of IDRs to delineate the events that occur between the commencement of drug administration and the development of the IDR. To guide these investigations, we look to the fundamentals of immunology to describe how an immune response may develop in response to the administration of small-molecule drugs.
III. Innate Mechanisms Contributing to Adaptive Immune Activation
Overwhelmingly, the adaptive arm of the immune system has been the focus of IDR research, as this is the mechanism that is likely responsible for clinically significant IDRs. The adaptive immune response is likely also what makes IDRs idiosyncratic: individuals possess unique and dynamic TCR repertoires, formed through random somatic recombination events (Krangel, 2009), and without the major histocompatibility complex (MHC) presentation of drug-modified peptides to cognate TCRs, adaptive immune activation and subsequent IDR manifestation cannot occur (Usui and Naisbitt, 2017; Hwang et al., 2020).
However, a fundamental dogma of immunology is that an innate immune response is required to initiate an adaptive immune response, and although progression to a severe IDR may be uncommon, it is likely that a greater proportion of patients experience an innate immune response that resolves without intervention and without leading to a significant adaptive immune response. Therefore, a more comprehensive understanding of the subclinical early immune mechanisms preceding IDR onset is necessary to guide future strategies in disease management and prevention. Thus, from neoantigen formation to consequences of immunologic synapse formation, this section will provide a succinct overview of important principles in innate immunology as well as mechanisms of adaptive immune activation that are potentially involved preceding the development of an IDR. These concepts are summarized in Fig. 1. Admittedly, the innate immune response is much more complex and nuanced than can be adequately addressed here; however, these topics provide a basic framework to be considered when designing future mechanistic studies for drugs associated with the risk of IDRs. For those already familiar with the innate immune system, this section may be skimmed, skipped, and/or referred to when necessary in accompaniment with Section IV. Support for Immune Activation Using Model Drugs.

A working hypothesis of the early immune mechanisms involved in idiosyncratic drug reactions. First, drugs may bind to MHC molecules and alter the repertoire of peptides presented by the MHC molecules, known as the altered peptide hypothesis. More commonly, drugs or their reactive metabolites (generated by various enzymes) covalently bind to cellular proteins, generating drug-modified, or haptenated, proteins. These haptenated proteins may be transported to APCs via extracellular vesicles or endocytosis mechanisms, or may be generated by the APC itself. Additionally, protein modification leads to cell stress, damage, or death, which prompts the release of proinflammatory molecules such as DAMPs. These mediators result in the recruitment of effector innate immune cells such as neutrophils or other granulocytes, which may degranulate or release NETs, monocytes or macrophages (which may result in cytokine release), and/or ILCs. Another response induced in these cells may include activation of the inflammasome and the subsequent release of IL-1 cytokines. Within APCs, the drug-modified proteins are processed and presented in the context of MHC molecules. The recognition of DAMPs and cytokines by APCs induces the upregulated expression of costimulatory molecules and also causes inflammatory cytokine release by the APCs themselves, ultimately resulting in the activation of T cells.
A. Cells of the Innate Immune System
Initiation of any inflammatory response is dependent upon the recruitment and activation of innate effector cells. When this immune response is triggered by the detection of endogenous danger signals without the detection of pathogens or pathogen-associated molecular patterns, it is described as sterile inflammation (Chen and Nuñez, 2010). Although the types of innate immune cells that may participate in this type of inflammation are generally similar, the function of the sterile response is not to clear an infection but, ultimately, to repair the damage caused by chemical or physical insult; thus, the role of effector cells may vary considerably. Responding leukocytes include granulocytes (neutrophils, eosinophils, basophils, and mast cells), professional antigen-presenting cells (APCs: monocytes, macrophages, and dendritic cells), and innate lymphoid cells (ILC groups 1–3). Other immune cells, including platelets, megakaryocytes, and erythrocytes, and nonimmune cells, such as mesenchymal stem cells, fibroblasts, and hepatocytes, may also function during the immune response and are introduced briefly. Ultimately, since the function of the innate immune system is to provide a first line of defense against foreign or potentially harmful stimuli, including potential damage caused by binding of drug-reactive metabolites, activation of innate immune cells represents a more universal, non–patient-specific mechanism to be studied in the context of IDRs.
1. Granulocytes
a. Neutrophils
Neutrophils are essential for innate immunity, not only as phagocytes that engulf and destroy invading pathogens but also as rapid responders during sterile inflammation (McDonald et al., 2010; Lämmermann et al., 2013), and can even possess a reparative function (Wang et al., 2017). Moreover, in vitro, neutrophils have been demonstrated to function as APCs under inflammatory conditions, further highlighting the diverse roles of these cells (Mehrfeld et al., 2018).
Mature neutrophils, derived from common myeloid progenitors in the bone marrow, are the most abundant leukocyte present in the human circulation, although a large store of mature cells also exist in the bone marrow or transiently arrested within blood capillaries (Lawrence et al., 2018). After the detection of any of an extensive array of inflammatory stimuli [such as chemokines or damage-associated molecular patterns (DAMPs), discussed below], marginated neutrophils are released rapidly into the circulation and, through the process of chemotaxis, can migrate to the site of inflammation. Although once considered to be a single, short-lived population, significant neutrophil heterogeneity has been reported in the steady-state (Fine et al., 2019) as well as in the context of numerous inflammatory (Silvestre-Roig et al., 2016; Yang et al., 2017) and cancer models (Hellebrekers et al., 2018; Giese et al., 2019), with extended neutrophil life spans observed in the presence of inflammation (Filep and Ariel, 2020). Reparative and immunosuppressive phenotypes have also been described (Rosales, 2020).
Neutrophils contain several types of granules and secretory vesicles, the contents of which can be released in a stimulus-dependent manner, either intracellularly via fusion with a phagocytic vacuole or extracellularly via degranulation or exocytosis (Giese et al., 2019). In addition to the many enzymes, receptors, and cytokines released in granules and secretory vesicles, neutrophils are also able to generate reactive oxygen species (Sheshachalam et al., 2014; Winterbourn et al., 2016). Together, these components can mediate pathogen destruction, induce recruitment of additional inflammatory cells, or contribute to tissue injury or repair (Silvestre-Roig et al., 2019).
Moreover, after stimulation, neutrophils can release web-like structures called neutrophil extracellular traps (NETs), composed of histone-linked DNA fragments, cathepsin G, elastase, and myeloperoxidase (Brinkmann et al., 2004). Interestingly, NET release can occur in a lytic or nonlytic manner, meaning neutrophil lysis and subsequent cell death may or may not occur during the process (Castanheira and Kubes, 2019). Of note, the enzyme myeloperoxidase, which is present in both neutrophil granules and NETs, has also been shown to bioactivate a variety of drugs to reactive metabolites in vitro (Hofstra and Uetrecht, 1993) and to contribute to the covalent binding of drugs observed in vivo (Lobach and Uetrecht, 2014b). Whereas IDIAG is the result of a delayed, adaptive immune response, paradoxical neutrophilia has been reported in the first few weeks of treatment with drugs associated with this IDR, namely clozapine (Section IV. D. Clozapine). Overall, neutrophils are integral for coordination and resolution of an inflammatory response and for tissue repair, and they can also play a role in the generation of neoantigens through myeloperoxidase-mediated reactive metabolite formation.
b. Eosinophils
Eosinophils are among the rarest leukocytes in circulation in a healthy state, but their numbers can increase up to 20-fold during certain pathologic conditions (Klion et al., 2020). They are fundamental effector cells in innate immunity against a wide variety of pathogens but also contribute to acute and chronic inflammatory conditions including asthma, eczema, and different types of autoimmunity and can mediate both tissue damage and repair (Ferrari et al., 2020; Nagata et al., 2020). In addition to granular proteins, eosinophils synthesize more than 40 proinflammatory mediators, such as TNF-α, IL-1 family cytokines, IL-4, IL-6, IL-8, granulocyte-macrophage-colony stimulating factor, leukotrienes, and reactive oxygen species (Spencer et al., 2014; Melo and Weller, 2018). These mediators can be released via classic exocytosis; through eosinophil cytolysis, whereby intact granules are liberated directly into target tissues; or through piecemeal degranulation, whereby cytokines are selectively mobilized to vesicles from the main granules and are then released (Spencer et al., 2014). Much like neutrophils, eosinophils can release extracellular traps of DNA and DAMPs, although these networks are more resistant to degradation compared with NETs (Ueki et al., 2016). Overall, eosinophils are a hallmark of allergic inflammation, and as discussed in Section IV. Support for Immune Activation Using Model Drugs, eosinophilia is frequently reported during the initial weeks of clozapine therapy in patients, indicative of an innate immune response. More broadly speaking, eosinophilia is also seen in other IDRs, such as DRESS, AGEP, or liver injury.
c. Basophils
Although they are the rarest and weakest phagocytic granulocyte in circulation, basophils play a key role in tissue inflammation; namely, skin, lung, and gastrointestinal tract inflammatory responses that are commonly triggered by either an invading parasite or allergen (Schwartz et al., 2016). Basophils are activated by allergen-induced crosslinking of their IgE receptors (Knol, 2006). Indeed, the basophil activation test is used as a reliable diagnostic tool for identifying various allergens. In the context of drug allergy, however, the basophil activation test is not as sensitive as it is in identifying other types of allergens (Eberlein, 2020). Possibly, this could be because the covalent modification of proteins by drugs produces a range of antigens such that it is not accurately reproduced in vitro.
Basophils are a source of IL-13 and are known to constitutively express IL-4, which are cytokines necessary for B-cell stimulation and differentiation to plasma cells and also differentiation of naïve helper T cells to Th2 cells (Liang et al., 2011), thus representing an important bridge between the innate and adaptive immune responses. Basophil-derived IL-4 has also been shown to have an important function in alternatively activated (M2) macrophages, which are involved not only in type 2 immunity but also in tissue repair and physiologic homeostasis (Yamanishi and Karasuyama, 2016). Basophils can quickly migrate to inflamed tissues and are among the first responding cells during skin injury (Chhiba et al., 2017). Activated basophils release a variety of mediators stored in cytoplasmic granules, including the bioactive lipids leukotrienes and prostaglandins, histamine, chemokines, and other cytokines (Chirumbolo et al., 2018), and also present with transcriptional heterogeneity, depending upon the stimuli (Chhiba et al., 2017). Additional innate effector cells such as eosinophils and ILC2 have also been demonstrated to be recruited by basophils to inflammatory sites (Schwartz et al., 2016). Although basophils were once considered a redundant counterpart of tissue-resident mast cells, they have more recently been acknowledged to play many unique roles during the inflammatory response that extend beyond allergy and hypersensitivity reactions.
d. Mast cells
Mast cells share functional and morphologic characteristics with basophils but are considered sentinels of the innate immune system, and although they are found in most tissues of the body, terminally differentiated mast cells are typically not detected in circulation. Although mast cells have diffuse cytoplasmic granules comparable to basophils and other classic granulocytes, there has been considerable debate as to whether the progenitors of the mast cell lineage are more closely related to megakaryocyte/erythroid or granulocyte/macrophage progenitors. However, it appears that mast cells are derived independently from either group and only share the early common myeloid progenitor (Franco et al., 2010). Both positive and negative immunoregulatory roles have been ascribed to mast cells. They function as a first line of defense against pathogens, and they are particularly useful in degrading venoms and toxins (Dudeck et al., 2019). Additionally, they contribute to allergic inflammatory responses by recruiting additional innate cells to the site of inflammation and by activating adaptive immune cells, thus promoting chronic responses (Kubo, 2018; Olivera et al., 2018). Moreover, excessive and sustained activation of mast cells can cause anaphylaxis and tissue damage, respectively. These effector functions can be attributed to the release of mast cell secretory granules, which contain proteases, lysosomal enzymes, biogenic amines, and cytokines (TNF, IL-4, IL-5, IL-6, etc.), among numerous other constituents (Wernersson and Pejler, 2014). Mast cells are most abundant in areas exposed to high levels of antigen, including the skin, other connective tissues, and the gastrointestinal and respiratory tracts (Krystel-Whittemore et al., 2016), and their roles in innate immunity can vary depending on the local milieu of mediators.
2. Professional Antigen-Presenting Cells
Professional APCs are cells that possess constitutive or inducible expression of high levels of MHC II molecules, process antigen, and express costimulatory molecules to facilitate the development of adaptive immunity to specific antigens. Classically, dendritic cells, macrophages, and B cells are considered professional APCs. B cells will not be discussed here, aside from their antigen presentation function, which is briefly described later. Although it has been recognized that other cell types, referred to as atypical or nonprofessional APCs, can also express MHC II, there is little evidence that they can activate naïve T cells (Kambayashi and Laufer, 2014). APCs facilitate the surveying of antigen by CD4+ T cells to efficiently expand the small subset of T cells expressing the cognate T-cell receptors to respond to antigenic challenge.
a. Dendritic cells
Dendritic cells (DCs) received their name from their many branched cellular processes (Steinman and Cohn, 1973). DCs can be categorized as conventional or plasmacytoid, both of which arise from a committed dendritic cell precursor in the bone marrow. These then diverge, as conventional dendritic cell precursors leave the bone marrow and seed other organs, whereas plasmacytoid dendritic cell precursors remain. Conventional DCs (cDCs) are the predominant cell type responsible for T-cell activation, and the far less abundant plasmacytoid DCs are specialized in sensing viral RNA and DNA and can produce large amounts of interferons to drive the antiviral response (Sichien et al., 2017; Musumeci et al., 2019). Historically, cDCs have been further categorized as migratory or lymphoid-resident; however, more recent studies have resulted in the classification of cDCs as cDC1 and cDC2 based upon surface marker expression and transcriptomic analyses (Ziegler-Heitbrock et al., 2010; Guilliams et al., 2014, 2016). Langerhans cells were originally presumed to be DCs based upon their function, but based upon their ontogeny, they are resident macrophages (Doebel et al., 2017), highlighting the complexity in classifying these types of cells. Langerhans cells likely play an important role in mediating skin IDRs.
DCs are usually found in a resting or immature state and survey their environment by sampling antigen. Because they have both low surface expression and rapid turnover of MHC II molecules, they are unable to activate naïve T cells (Drutman et al., 2012). In an inflammatory milieu, DAMPs and pathogen-associated molecular patterns are present and engage various pattern recognition receptors on the DC surface, causing the DC to mature. The cells are then able to express cytokine and chemokine receptors, facilitating migration to lymph nodes. MHC II turnover decreases while expression increases, allowing for presentation of the peptides found in the inflammatory context (Cella et al., 1997). Additionally, costimulatory molecule expression and cytokine secretion are induced, and the combination of these changes is sufficient to induce naïve T-cell activation (Curtsinger and Mescher, 2010). Depending upon the stimuli received by the DC, it will secrete different cytokines and influence the differentiation of cognate T cells into different subsets of effector T cells (Blanco et al., 2008).
DCs may also be tolerogenic in certain cases. Thymic DC populations appear to be important in maintaining central tolerance during T-cell development (Lopes et al., 2015). Peripherally, a small proportion of DCs undergo maturation under homeostatic conditions and upregulate MHC II expression, but this maturation results in tolerance rather than naïve T-cell activation (Lutz and Schuler, 2002). Indeed, antigen presentation without DC priming resulted in antigen-specific tolerance (Probst et al., 2003). The vast heterogeneity of DCs, as well as the varied outcomes of maturation based on environmental influences, results in numerous functions for DCs.
b. Monocytes
Monocytes are cells of the myeloid lineage that are derived from the bone marrow and are released into circulation. Monocytes are phagocytic and scavenge apoptotic cells and toxic macromolecules in circulation. They also function as important orchestrators of inflammatory responses by producing cytokines after the detection of tissue damage or pathogens. Monocyte subsets exist across a spectrum of differentiation, initially taking on an “inflammatory” phenotype upon egress from the bone marrow and then taking on a “patrolling” phenotype over time because of transcriptional changes (Mildner et al., 2017). Under steady-state conditions, monocytes can enter tissue, return to circulation with minimal differentiation, and traffic to lymph nodes to present antigen to T cells (Jakubzick et al., 2013). Although monocytes may themselves function as APCs, upon entering inflamed tissue, they can also differentiate into macrophages or dendritic cells to propagate the inflammatory response (Jakubzick et al., 2017).
c. Macrophages
Macrophages are phagocytes that are usually found in tissues, and many have been named depending upon the organ in which they reside (e.g., Kupffer cells in the liver, microglia in the central nervous system, or osteoclasts in bones). The environment in which macrophages are found influences their phenotype and function; various tissue-resident macrophage populations express different surface proteins, and even within an organ may have different phenotypes (Hume et al., 2019). Macrophages survey the tissue in which they reside and phagocytose foreign molecules and debris.
Like DCs, macrophages express pattern recognition receptors, which can stimulate their activation. Macrophage activation states are highly complex and are often described as polarization to an “M1-like” or “M2-like” phenotype, correlating with Th1 and Th2 responses, respectively. As macrophage activation is a dynamic process, different macrophages in the same tissue likely express different mixtures of activation markers and perform different functions; this also evolves over time (Murray, 2017). For example, in response to IFN-γ, activated macrophages secrete proinflammatory cytokines (e.g., IL-1β, IL-6, IL-12). In response to IL-4, however, they can secrete insulin-like growth factor 1 and resistin-like molecule-α, which can stimulate fibroblast survival and promote extracellular matrix deposition, respectively (Mosser and Edwards, 2008).
Macrophages can participate in antigen presentation, but unlike DCs, they cannot activate naïve T cells. Their ability to present antigen can be influenced by their environment; for example, antigen presentation to T cells and CD40 expression were increased with the uptake of necrotic but not apoptotic cells (Barker et al., 2002). Crosspresentation of antigen by macrophages from dead tumor cells has been shown to be important in antitumor immunity (Asano et al., 2011). Macrophages have also been shown to present lipid antigens to invariant natural killer T cells (Barral et al., 2010).
Macrophages also have reparative functions and can secrete growth factors and anti-inflammatory mediators including IL-10 and TGF-β during tissue repair (Vannella and Wynn, 2017). Overall, macrophages play varied and dynamic roles in the steady-state and throughout the inflammatory response.
3. Innate Lymphoid Cells
Over the past decade, ILCs have come to be recognized as fundamental effectors of the innate immune response (Moro et al., 2010; Neill et al., 2010; Price et al., 2010), both in health and in disease states ranging from type 2 inflammatory conditions (e.g., atopic dermatitis and asthma) to autoimmune diseases (e.g., psoriasis and inflammatory bowel disease) (Ebbo et al., 2017; Kobayashi et al., 2020). Aside from natural killer (NK) cells, which are localized in secondary lymphoid organs, ILCs are generally under-represented in lymphoid tissues but are predominantly found in the liver, skin, intestine, lungs, adipose tissue, and mesenteric lymph nodes and are most prominent at mucosal barriers (Klose and Artis, 2016). At the most rudimentary level, ILCs can be classified as group 1, group 2, and group 3, with each group sharing similarities in cytokine production and transcriptional regulation with a particular T-cell subset (although ILC antigenic receptors do not undergo the genetic rearrangement that adaptive lymphocytes undergo) (Spits et al., 2013). Group 1 ILCs include both ILC1s (Th1-like) and natural killer cells (cytotoxic T cell-like) and are characterized by their production of IFN-γ and TNF (Spits et al., 2016), whereas group 2 ILCs are a single population (Th2-like) that produce amphiregulin, IL-4, IL-5, and IL-13 (Klose and Artis, 2016). Group 3 ILCs comprise three populations (Th17-like)—lymphoid tissue inducer cells, natural cytotoxicity receptor-negative cells, and natural cytotoxicity receptor-positive cells—that all secrete IL-22, but only the first two population also secrete IL-17A/IL-17F (Montaldo et al., 2015).
Additionally, some T-cell subsets are “preprogrammed” and behave like innate cells in that they can respond rapidly to a limited and conserved antigenic repertoire. These include invariant natural killer T cells, which differ most prominently from conventional T cells in that they recognize lipid-based antigens in the context of CD1d; mucosal-associated invariant T cells, subsets of γδ T cells; and certain memory T-cell subsets (Vivier et al., 2018).
Like many cells, ILCs demonstrate significant plasticity, and their functionality is dependent on their local microenvironment. Even within specific ILC groups, significant heterogeneity has been reported. For example, among natural killer cell subsets, some possess more cytolytic activity and contain high concentrations of granzyme and perforin, whereas others are more reactive to activation by proinflammatory mediators, and surface receptor expression varies between hepatic, intraepithelial, and other natural killer cell populations (Spits et al., 2016). Natural killer cells, as discussed in the next section, have also been shown to mediate the inflammatory response induced by amodiaquine, and they are likely to play fundamental roles in the early immune responses to other IDR-associated drugs, as well. Moreover, based on the emerging roles of various ILC subsets in inflammatory conditions, as well as their localization within organs most commonly associated with IDRs (e.g., the liver and skin), it is reasonable to question whether other members of the ILC family play a crucial part in the innate immune response to drugs associated with IDRs.
4. Other Innate Immune Cells
Other immune cells, such as megakaryocytes, their platelet derivatives, and erythrocytes, may also be important contributors during an innate immune response. Although the immunologic role of megakaryocytes is not as well defined, their expression is dependent on the demand for platelets, which can be upregulated during inflammatory conditions or infection, vascular damage, and tissue repair (Noetzli et al., 2019). Beyond a role in hemostasis, platelets have significant immunomodulatory potential: they can release both proinflammatory [e.g., CXCL4, chemokine (C-C motif) ligand (CCL) 5, histamine, epinephrine, and high mobility group box 1 (HMGB1)] and proresolving mediators and can form complexes with a variety of immune cells, including neutrophils and monocytes (Margraf and Zarbock, 2019). Platelets can release these mediators through microvesicles and exosomes (Heijnen et al., 1999). Likewise, erythrocytes contribute to innate immunity and are more than just oxygen carriers. Interestingly, these cells can bind a variety of chemokines, in turn, either preventing recruitment of effector cells such as neutrophils or possibly extending the half-life of these mediators to prolong the inflammatory response, referred to as the sink hypothesis and reservoir hypothesis, respectively (Anderson et al., 2018).
5. Nonimmune Cells
Although the focus of this section is to introduce the reader to cells of the innate immune system, it is also worth emphasizing that countless nonimmune cells can have inflammatory or immunoregulatory functions. Naturally, any damaged or dying cells can release DAMPs and proinflammatory mediators that can activate immune cells both locally and in circulation, resulting in recruitment to the location of injury and initiation of an innate immune response; this is not dependent on immune cell status. However, nonimmune cells can also secrete bioactive and chemotactic molecules in response to the detection of a stimulus, as well. Such cells include, but are not limited to, hepatocytes, mesenchymal stem cells, and fibroblasts.
a. Hepatocytes
As the predominant parenchymal cell in the liver, hepatocytes are well known for their role in metabolism, xenobiotic detoxification, and protein synthesis, but they are also critical players in innate immunity (Mehrfeld et al., 2018). The liver is highly vascularized, receiving 25% of total cardiac output (Eipel et al., 2010), and is also responsible for the production of up to 50% of the lymph collected by the thoracic duct (Ohtani and Ohtani, 2008). While not in direct contact with the sinusoidal blood flow, hepatocytes can extend filopodia through fenestrations in the adjacent endothelium to enable interactions with circulating leukocytes (Warren et al., 2006). Under steady-state conditions, hepatocytes only express MHC I (Mehrfeld et al., 2018), but they may express MHC II under inflammatory conditions (Herkel et al., 2003). Thus, hepatocytes may function as APCs with the potential to interact with both helper and cytotoxic T cells; indeed, hepatocytes have been shown to activate CD8+ T cells, although they did not promote survival (Bertolino et al., 1998). Like most of the cells discussed thus far, hepatocytes not only share the ability to target pathogens for destruction but can also secrete a variety of proinflammatory mediators, such as soluble CD14, soluble myeloid differentiation 2, IL-6, CCL2, and CXCL1 (Zhou et al., 2016). Moreover, among the proteins synthesized and secreted into the blood by hepatocytes are acute phase proteins, such as C-reactive protein, serum amyloid A, and serum amyloid P. The concentrations of these mediators can dramatically increase after the detection of inflammation, thus acting to amplify the immune response (Schrödl et al., 2016). As most reactive metabolite formation occurs in the liver, and the liver is the target of a large proportion of IDRs, hepatocytes are likely fundamental in the initiation of the innate immune response to drugs that cause IDILI (Uetrecht, 2019b; Ali et al., 2020; Hastings et al., 2020; Mosedale and Watkins, 2020; Yokoi and Oda, 2021).
b. Mesenchymal stem cells
Mesenchymal stem cells, also referred to as mesenchymal stromal cells, have been identified in various tissues and have the capacity to differentiate into chondrocytes, osteoblasts, and adipocytes (Dominici et al., 2006). Moreover, these multipotent stem cells help maintain the tissue microenvironment, under both normal and inflamed conditions, often promoting an immunosuppressive milieu via the release of growth factors and anti-inflammatory molecules (e.g., transforming growth factor-β, IL-1 receptor antagonist, IL-10, prostaglandin E2, etc.) after the recognition of proinflammatory stimuli (Wang et al., 2014). Exosomes have also been shown to contribute to the immunomodulatory capabilities of these cells, and even apoptotic mesenchymal stem cells maintain suppressive properties (Shi et al., 2018). Mesenchymal stem cells can also migrate to the site of tissue damage to participate in regeneration and can activate or suppress the activation of various innate cells, including neutrophils, macrophages, DCs, and mast cells (Le Blanc and Mougiakakos, 2012; Shi et al., 2018).
c. Fibroblasts
Although a key function of fibroblasts is to maintain connective tissue structural integrity, these sentinel cells also have the capacity to respond to pathogens and endogenous danger signals, to secrete inflammatory signals, and to initiate tissue repair (Hamada et al., 2019). For instance, intestinal immunity is shaped by the secretion of cytokines, chemokines, growth factors, and metalloproteinases by epithelial cells and myofibroblasts (Curciarello et al., 2019). These cells have also been shown to contribute to the persistence of inflammation, such as during rheumatoid arthritis, where synovial fibroblasts produce a variety of proinflammatory and matrix-degrading molecules (Frank-Bertoncelj et al., 2017).
B. Antigen Formation and Cell Damage
Multiple theories have been proposed to explain how drugs may cause IDRs; these are discussed in detail elsewhere (Cho and Uetrecht, 2017). We will present the hypotheses that are relevant to the discussion of innate immune system activation. It has long been understood that foreign peptides are recognized by the immune system. The hapten hypothesis and the altered peptide repertoire model describe distinct processes by which drug administration may ultimately result in the exposure of the immune system to novel peptides. These neoantigens may serve as targets for the immune response, potentially resulting in the development of an IDR.
More recently, it was also recognized that there needs to be a signal (e.g., a DAMP, discussed below) that damage is occurring. This is known as the danger hypothesis. This hypothesis most likely complements the hapten and altered peptide hypotheses. Similarly, in conjunction with the hapten hypothesis, it is plausible that sufficient covalent binding in the cell may result in cellular damage. The endoplasmic reticulum (ER) stress and unfolded protein response, as well as mitochondrial toxicity, may result from the generation of covalently modified proteins. These processes may also result in the release of DAMPs. Thus, these hypotheses likely work together to describe the initiating events in IDRs.
1. Hapten Hypothesis
Small-molecule drugs are too small to be detected by the immune system, which recognizes larger molecules, such as proteins (Erkes and Selvan, 2014). However, many drugs that cause IDRs have reactive metabolites. The hapten hypothesis posits that drugs are bioactivated to a reactive metabolite that then covalently binds to endogenous protein, thereby altering the protein and provoking an immune response (Landsteiner and Jacobs, 1935; Faulkner et al., 2014; Cho and Uetrecht, 2017). Although it is very difficult to prove that reactive metabolites cause IDRs, there are some cases in which they have been shown to be responsible. For example, penicillin hypersensitivity involves IgE, and IgE from hypersensitive patients has been shown to react to penicillin-modified protein (forming the basis of diagnostic skin tests) (Levine et al., 1967). There have also been studies of multiple drugs characterizing drug-protein adducts in patient samples, although these have not necessarily been causally linked to IDR onset. Additionally, nevirapine is another case in which the reactive metabolite was identified. Female brown Norway rats develop a rash when chronically administered nevirapine. The findings that 12-hydroxynevirapine sulfate was covalently bound in the skin and that topical application of a sulfotransferase prevented both the covalent binding and the rash demonstrates that this reactive metabolite was indeed responsible for causing the skin rash (Sharma et al., 2013).
2. p-i Concept
The pharmacological interaction of drugs with immune receptors concept (p-i concept) attributes IDRs to the activation of immune receptors, MHC and the TCR specifically, by direct, noncovalent interaction of the culprit drug (Pichler, 2002). This is based on the observation that drugs can activate lymphocytes from patients who have experienced an IDR to that drug in the absence of metabolism. However, in the case of nevirapine-induced skin rash, it has been shown that the rash is caused by a reactive metabolite, and yet cells from rats or humans who have a history of nevirapine-induced skin rash are activated by the parent drug (Chen et al., 2009). Thus, although direct activation of immune cells by parent drug may occur in IDR patients, this mechanism may not play a role in the initiation of the IDR.
3. Altered Peptide Repertoire Model
A mechanism related to the p-i concept is the altered peptide repertoire model, which describes the noncovalent binding of drug to the HLA molecule itself, thereby altering its conformation and the peptide repertoire that it is able to present. This is illustrated by abacavir, which has been shown to reversibly bind to the F pocket of the peptide-binding groove of HLA-B*57:01 and alter the repertoire of peptides that it can present (Illing et al., 2012; Norcross et al., 2012; Ostrov et al., 2012).
4. Danger Hypothesis
Although foreign peptide is a requirement for activation of the immune response, it is not usually sufficient; indeed, the body is exposed to non–self-proteins constantly from food sources and gut microflora, for example. It would be detrimental if the immune system were constantly activated as a result of these sources. The danger hypothesis recognizes that it is not necessarily the detection of an entity that appears foreign but, in fact, an entity that causes damage that activates the immune system (Matzinger, 1994). Cell damage causes the release of DAMPs, which signal to the immune system that there is likely a pathogen that needs to be eliminated. Very broadly speaking, cell damage may manifest as cell death, in which intracellular contents may be passively released and serve as DAMPs, or the cell may continue to survive, in which case DAMPs may be actively secreted.
Additionally, the type of cell death influences the types of DAMPs that are released. In apoptosis, cellular contents are not necessarily released into the extracellular milieu as membrane integrity is maintained; however, ATP is released in a controlled manner as a “find-me” signal (Elliott et al., 2009). In contrast, in cells dying by necrosis, cellular contents are released as cell death is uncontrolled. Necroptosis, a regulated form of necrosis mediated by death receptor activation, and pyroptosis, cell death regulated by inflammasome and caspase-1 activation, may similarly both result in the release of a number of DAMPs. Some DAMPs are released in the context of both types of programmed cell death (e.g., HMGB1, heat shock protein 90, ATP, IL-1α), whereas some have only been observed in necroptosis (e.g., S100A9, IL-33) or pyroptosis (e.g., ASC specks) to date (Frank and Vince, 2019).
5. Endoplasmic Reticulum Stress and the Unfolded Protein Response
The ER is the location of protein folding and post-translational modification in the cell. Disruption of this process can cause ER stress. A series of pathways, termed the unfolded protein response (UPR), maintain quality control of protein synthesis through sensing deficiencies in protein folding capacity. Proteins in their proper conformation proceed to the Golgi apparatus as the next step in the secretory pathway, whereas misfolded proteins are retained in the ER (Schröder and Kaufman, 2005). As cytochrome P450 enzymes tend to localize in the ER (Szczesna-Skorupa and Kemper, 1993), reactive metabolites can be formed in the ER and adduct to proteins, inducing the UPR.
Unfolded proteins take on a conformation of a higher free energy than that of their native conformations. A variety of chaperones can sense this increased free surface energy as hydrophobic residues are exposed to water. To maintain a balance with the folding capacity of the ER, the UPR employs two strategies: first, to increase folding capacity by inducing ER-resident molecule chaperones and foldases and by increasing the size of the ER, and second, to decrease the misfolded protein load by downregulating protein synthesis and by increasing clearance of misfolded protein by upregulating ER-associated degradation (Schröder and Kaufman, 2005).
Ultimately, if the unfolded protein burden remains too great, the UPR response can result in apoptosis. It has also been shown that chronic ER stress can lead to inflammation. For example, ER stress has been shown to induce nuclear factor of the κ light chain enhancer of B cells (NF-κB) activation (Deng et al., 2004), NLR family pyrin domain containing 3 (NLRP3) inflammasome activation (Menu et al., 2012), and DAMP secretion either freely (Andersohn et al., 2019) or packaged in extracellular vesicles (Collett et al., 2018).
Unfolded protein is not the only possible trigger of ER stress, although it is the most well studied. Aberrations in lipid homeostasis may also induce ER stress (Song and Malhi, 2019). Although, compared with proteins, changes to lipids have not been studied as extensively in the context of IDRs, this may be an interesting avenue to explore; for example, lipid-smooth ER inclusions were found in hepatocytes of brown Norway rats administered nevirapine (Sastry et al., 2018), which is also known to induce smooth ER hypertrophy (Sharma et al., 2012).
The absolute number of proteins modified by covalent binding of a drug is quite small (Evans et al., 2004), and compared with other sources of unfolded protein, drug modification of protein may not induce sufficient protein unfolding to trigger activation of the UPR. Additionally, a transcriptomic study of primary human hepatocytes predicted a suppression, rather than induction, of pathways related to the UPR (Terelius et al., 2016).
6. Mitochondrial Toxicity
Mitochondrial toxicity has been identified as an adverse effect of many medications. Its role in IDRs in particular, however, has been a matter of debate (Boelsterli and Lim, 2007; Cho and Uetrecht, 2017). The mechanisms underlying mitochondrial toxicity include inhibition of the electron transport chain, interference with mitochondrial transcription and translation, inhibition or uncoupling of ATP synthase, inhibition of enzymes in the citric acid cycle or mitochondrial transporters, and increased reactive oxygen species (ROS) production (Vuda and Kamath, 2016; Will et al., 2019). As these mechanisms have been extensively covered elsewhere, we will focus on how mitochondrial toxicity may result in inflammation.
Increased ROS production causes activation of redox-sensitive transcription factors such as NF-κB. It has also been shown that autophagy negatively regulates NLRP3 inflammasome activation, whereas increased ROS positively regulates NLRP3 inflammasome activation and inflammation, at least in part due to cytosolic localization of oxidized mitochondrial DNA (Nakahira et al., 2011; Zhou et al., 2011; Shimada et al., 2012). In addition to mitochondrial DNA, other mitochondrial-derived molecules can function as DAMPs, such as ATP, mitochondrial transcription factor A, N-formyl peptide, succinate, cardiolipin, and cytochrome-c (Nakahira et al., 2015; Grazioli and Pugin, 2018). Cytochrome-c release into the cytosol can induce apoptosis via inducing oligomerization of apoptosis-protease activating factor 1 and initiating caspase activation. Depending upon the context and the cleavage products of the caspases involved, this may result in apoptosis, but it may also result in cell differentiation and proliferation (Garrido et al., 2006). Thus, the causes and outcomes of mitochondrial toxicity are varied and complex.
In general, there is little direct evidence that drugs that cause IDRs do so by inducing mitochondrial damage. An exception is valproic acid–induced liver injury, however, which has been associated with variants in mitochondrial DNA polymerase γ (Stewart et al., 2010) and which may present with steatosis and lactic acidosis (Chaudhry et al., 2013; Farinelli et al., 2015; Pham et al., 2015). Acetaminophen also causes mitochondrial damage, but it does not cause IDRs (Jaeschke et al., 2019).
C. Mediators of Inflammation
Depending on the location and severity of the tissue injury, a variety of factors may be involved in the initiation and propagation of a sterile inflammatory response, including DAMPs, cytokines, and chemoattractants. The transcriptional regulation of many of these proinflammatory molecules by NF-κB is also an important consideration. Moreover, other body systems such as the microbiome and the nervous system have the potential to contribution to inflammation.
1. Damage-Associated Molecular Patterns
As discussed above, the injury or death of a cell may result in the release of intracellular contents. Once outside of their normal subcellular location, these components are referred to as danger signals or DAMPs, at which point they can initiate an inflammatory response (Medzhitov, 2008). DAMPs can be classified based on their normal location in or on the cell and include stimuli such as nuclear and mitochondrial DNA, RNA, ATP, S100, heat shock proteins, HMGB1, and extracellular matrix fragments (Chen and Nuñez, 2010; Zindel and Kubes, 2020). The detection of DAMPs then leads to effector cell recruitment and propagation of the sterile inflammatory response.
In the context of efferocytosis, DAMPs such as ATP, UTP, lysophosphatidylcholine, and sphingosine-1-phosphate, in addition to adhesion molecules and receptors such as intracellular adhesion molecule 3 and CX3C chemokine receptor, can act as chemotactic find-me signals, and concurrent with surface expression of eat-me signals such as phosphatidylserine, contribute to the removal of apoptotic cells by phagocytes (Westman et al., 2020).
The concept of danger signals in the initiation of IDRs has been proposed several times (Pirmohamed et al., 2002; Li and Uetrecht, 2010; Hassan and Fontana, 2019; Uetrecht, 2019b). Although reactive metabolites of drugs associated with IDRs can covalently bind to cellular proteins that may, in turn, cause cell damage or cell death, the release of DAMPs can also occur in response to a wide array of insults, such as UV irradiation, hemorrhagic shock, starvation, and other forms of injury or trauma (Schaefer, 2014). Since DAMPs are simply a mechanism by which the immune system is alerted that there is tissue injury, they are not idiosyncratic in their release. Therefore, it is possible that a similar pattern of DAMPs may be released after treatment with a drug associated with IDRs. This pattern of DAMPs could function as potential biomarkers during preclinical development by indicating that a drug candidate may carry the risk of causing IDRs; however, it is likely too nonspecific to be useful for clinical diagnosis of the early onset of an IDR. Thus, characterization of the specific DAMPs released after treatment with different drugs associated with IDRs is certainly an avenue for future research, as it may provide insight into the specific type and target of cell injury or death that is stimulating the observed innate immune response.
2. Cytokines, Chemokines, and Acute Phase Proteins
A wide range of classic soluble mediators have already been highlighted for various functions in innate immunity. To reiterate, cytokines such as TNF-α, IL-1β, IL-4, IL-6, and IL-18; chemokines such as CCL2, CCL5, CXCL1, CXCL2, and CXCL8; and acute phase proteins such as C-reactive protein, serum amyloid A, and serum amyloid P contribute to effector cell recruitment and activation and the propagation of the inflammatory response. These mediators are released in coordinated spatial and temporal responses, the patterns of which can influence the types and absolute numbers of innate leukocytes that are recruited to the site of injury. Not yet mentioned are the type I (i.e., α and β) and type II (γ) interferons, which act to potentiate proinflammatory signaling via enhanced cytokine production and antigen presentation, macrophage priming, and natural killer cell function, among numerous other effects (Kopitar-Jerala, 2017). Although additional proinflammatory molecules are elaborated on elsewhere (Turner et al., 2014; Akdis et al., 2016; Kapurniotu et al., 2019), the role of IL-1 family cytokines is worth emphasizing because of their fundamental importance in innate immunity.
a. Interleukin-1 cytokines and their activation
The IL-1 family of cytokines consists of 11 soluble mediators, including proinflammatory IL-1α, IL-1β, IL-18, IL-33, IL-36α, IL-36β, and IL-36γ, as well as several receptor antagonists and the anti-inflammatory cytokine IL-37 (Dinarello, 2018). IL-1 cytokines are first translated into inactive precursors (except for IL-1α), which then attain functional maturity after enzymatic cleavage in a process mediated predominantly by caspase-1 and the inflammasome (Mantovani et al., 2019). Fundamentally, the inflammasome is a multimeric protein complex that, when activated, leads to the maturation of caspase-1, the cleavage and release of mature IL-1 cytokines, and the induction of additional inflammatory effector mechanisms (Walsh et al., 2014). Several cytoplasmic pattern recognition receptors can assemble as independent inflammasomes, each responding to specific DAMPs or other stimuli. These pattern recognition receptors (PRRs) are expressed in multiple cells, including neutrophils, monocytes, macrophages, and dendritic cells, and play an important role in innate immunity (Sharma and Kanneganti, 2016).
The NLRP3 inflammasome may be the most relevant for the study of drug-induced immune activation, as it is activated by the widest array of stimulants, although several other inflammasomes may be involved (Schroder and Tschopp, 2010; Latz et al., 2013). Several drugs associated with serious IDRs have also been demonstrated to activate inflammasomes and increase IL-1β release in vitro (Kato and Uetrecht, 2017; Mak et al., 2018; Kato et al., 2019, 2020). Additionally, it is known that animals deficient in components of the inflammasome are resistant to contact hypersensitivity (Watanabe et al., 2007), a reaction that may parallel events in the early immune response to drugs that cause IDRs. Whether inflammasome activation occurs in humans treated with these drugs has yet to be clearly demonstrated.
3. Bioactive Lipids
In addition to inflammatory protein mediators, several classes of bioactive lipids exist and play various roles in inflammation, immunoregulation, and maintenance of tissue homeostasis (Chiurchiù and Maccarrone, 2016). The main types of proinflammatory lipids include classic eicosanoids (Dennis and Norris, 2015), certain endocannabinoids (Chiurchiù et al., 2015), lysoglycerophospholipids, and sphingolipids (El Alwani et al., 2006), and specialized proresolving lipid mediators are a relatively new class of lipids that terminate acute inflammation and drive resolution and tissue repair (Serhan, 2014). These molecules are all generated from ω-6 or ω-3 essential polyunsaturated fatty acids precursors (e.g., arachidonic acid) but demonstrate significant heterogeneity in structure and function after maturation (Das, 2018).
Classic eicosanoids (e.g., certain prostaglandins, prostacyclins, thromboxanes, leukotrienes, hydroxyeicosatetraenoids, and lipoxins) are typically considered highly proinflammatory mediators that are usually produced by innate cells such as neutrophils and monocytes within the first several hours of an inflammatory stimulus. Specifically, leukotrienes can act as chemoattractants for neutrophils, macrophages, and eosinophils (De Caterina and Zampolli, 2004), and prostaglandins can function to enhance proinflammatory cytokine gene transcription and release and can also amplify the innate response to DAMPs (Hirata and Narumiya, 2012). However, some eicosanoids can be immunosuppressive and promote immune tolerance in certain contexts (Obermajer et al., 2012; Wang et al., 2014). The endocannabinoids, such as 2-arachidonoylglycerol, are ubiquitously expressed molecules that have diverse immunomodulatory effects on monocytes/macrophages, dendritic cells, and granulocytes, and unsurprisingly, perturbations in endocannabinoid homeostasis have been shown to contribute to neuroinflammatory and autoimmune diseases (Chiurchiù et al., 2018). Lysoglycerophospholipids (e.g., lysophosphatidylcholine and lysophosphatidylinositol) and sphingolipids (e.g., ceramide 1-phosphate and sphingosine 1-phosphate) are key signaling molecules controlling inflammatory cascades, trafficking and activation of immune cells, cell survival, and apoptosis (Sevastou et al., 2013; Gomez-Muñoz et al., 2016).
NSAIDs are one class of drugs for which the potential role of bioactive lipids in the innate immune response is particularly relevant. Although NSAIDs are the most frequently used medications for the management of pain and inflammation, they are also associated with some of the highest incidence rates of drug hypersensitivity reactions (Conaghan, 2012). Reported reactions include urticaria and other cutaneous reactions, acute interstitial nephritis, and hepatotoxicity (Nast, 2017; Yamashita et al., 2017; Wöhrl, 2018). Mechanistically, NSAIDs inhibit the enzymes cyclooxygenase-1 and -2, blocking the synthesis of inflammatory prostanoids such as prostaglandin E2. It has even been postulated that an innate immune response contributes to the onset of NSAID-mediated adaptive IDRs, potentially through the activation of eosinophil and mast cell degranulation or through the shunting of arachidonic acid precursors to the production of other proinflammatory lipid mediators such as leukotrienes (Doña et al., 2020). Based on the fundamental roles of bioactive lipids in the initiation and propagation of an inflammatory response, future research to delineate key mediators in the early immune response to drugs that are associated with IDRs is necessary.
4. Pattern Recognition Receptors
As part of the innate immune system, pattern recognition receptors have evolved to recognize conserved molecular patterns of danger or invading pathogens. Thus, PRRs represent a key aspect of the innate immune system that is not likely to be idiosyncratic, as they are conserved, germline-encoded receptors that are not antigen-specific and respond to a structurally diverse range of molecules (Gong et al., 2020), in contrast to an individual’s randomly generated TCR repertoire. PRRs include Toll-like receptors (TLRs), nucleotide-binding oligomerization domain-like receptors (NLR), retinoic acid–inducible gene-1–like receptors, C-type lectin receptors, receptor for advanced glycation end products, and scavenger receptors (Gordon, 2002; Xie et al., 2008; Palm and Medzhitov, 2009; Takeuchi and Akira, 2010). Notably, DAMPs are largely recognized by PRRs. For example, HMGB1 can signal through TLR4 or receptor for advanced glycation end products (Lu et al., 2013). TLR signaling can result in NF-κB signaling (discussed below) and ultimately the production of proinflammatory cytokines (Vidya et al., 2018). PRR signaling may also result in cell death (Amarante-Mendes et al., 2018). If signaling through PRRs was directly responsible for the onset of IDRs, however, it is likely that these severe reactions would be observed in most, if not all, patients given a particular drug because of the conserved nature of these receptors, but this is not what is observed clinically. Thus, although likely a necessary first step for the development of an IDR, pattern recognition is not itself sufficient to cause an IDR. Again, we emphasize that this innate aspect of the immune response should occur in most patients taking drugs that are associated with IDRs if they cause cellular damage and is not idiosyncratic, but other downstream pathways contribute to the development of IDRs themselves.
5. Transcriptional Regulation of Inflammation
Several families of transcription factors are activated in response to inflammatory stimuli, such as signal transducers and activators of transcription, interferon regulatory factors, and most notably, NF-κB (Smale and Natoli, 2014; Irazoqui, 2020). The NF-κB family consists of several inducible transcription factors—NF-κB1 (p50), NF-κB2 (p52), RelA (p65), RelB, and c-Rel—that bind to κB enhancer DNA elements as dimers to modulate gene transcription (Liu et al., 2017). Activation can occur via the canonical pathway in response to ligand binding of various cytokine receptors, PRRs, and TNF receptors, or via the noncanonical pathway in response to ligand binding of a specific subset of TNF receptors (Cildir et al., 2016; Sun, 2017).
NF-κB signaling results in the upregulation of target genes related to cell adhesion, survival and proliferation, dendritic cell maturation, neutrophil recruitment, M1 macrophage polarization, and other inflammatory mediators that function to amplify the detected inflammatory response (Lambrou et al., 2020). Key proinflammatory cytokines and chemokines under NF-κB regulation include IL-6, IL-8, TNF-α, CCL2, CCL5, CXCL1, and CXCL2 (Liu et al., 2017). Moreover, activation of NF-κB is necessary for signal 1 (priming) in inflammasome activation, as transcription of inflammasome-related components such as pro-IL-1β, pro-IL-18, and NLRP3 is upregulated after NF-κB activation (Latz et al., 2013). If drugs associated with IDRs cause an inflammatory response that is characterized by elevated levels of NF-κB–regulated mediators, then this provides a starting point to determine which canonical or noncanonical receptors are activated after drug administration and may provide clues as to the types of cell damage or neoantigens formed (i.e., potential receptor ligands) with that drug.
6. Other Contributing Factors
In addition to the multifarious range of activation signals presented thus far, multiple junctures of interaction have been identified between the innate immune system and both the microbiome and the nervous system; however, these will only be introduced briefly.
a. Interaction with the microbiome.
Although most commonly associated with the gut, the human microbiome refers to the collection of genes of all microorganisms (e.g., archaea, bacteria, fungi, protists, viruses) that reside on or in all bodily tissues and fluids, including the biliary tract, respiratory tract, and skin (Marchesi and Ravel, 2015). To maintain a commensal relationship and prevent the initiation of an inappropriate immune response, extensive crosstalk between the microbiota and immune cells, particularly ILCs (Thaiss et al., 2016; Negi et al., 2019), must occur. For instance, it has been shown that germ-free mice have a significantly altered innate immune system, with attenuated myeloid cell development in the bone marrow (Khosravi et al., 2014). Although this is an extreme example that would not be particularly relevant to humans, it does highlight the potentially profound impact of the microbiome on innate immunity.
Commensals are necessary to educate the immune system and often promote tolerance (Grice and Segre, 2011), but how these microorganism interactions may shape the metabolism of and subsequent inflammatory response to drugs that cause IDRs has yet to be adequately investigated (Marchesi and Ravel, 2015). One notable exception, however, is immune checkpoint inhibitor–induced colitis. A recent investigation demonstrated that ipilimumab altered microbiome composition and the subsequent risk of colitis (Dubin et al., 2016). Countless drugs can target components of the microbiome, the most obvious being the antibiotics; therefore, understanding the reverse of this relationship will likely provide novel insights into patient-specific risk factors for IDRs.
b. Communication with the nervous system.
An important function of the nervous system is to interact with immune cells. Unsurprisingly, innate immune cells, including neutrophils, macrophages, and dendritic cells, express receptors for several neurotransmitters (e.g., α- and β-adrenergic and acetylcholinergic receptors), and neurons can express various pattern recognition and cytokine receptors, facilitating effective crosstalk between the systems (Chavan et al., 2017). Additionally, at peripheral sites of inflammation and tissue injury, both afferent and efferent neural circuits have been shown to have immunoregulatory functions (Pavlov and Tracey, 2015). As many drugs associated with IDRs have therapeutic effects in the CNS, including a multitude of anticonvulsant and antischizophrenic agents, it is necessary to consider how these psychotropics may influence the neuroimmune axis and the consequential immune activation.
D. Antigen Reception/Uptake by Antigen-Presenting Cells
There are multiple means by which APCs may obtain peptides or proteins and present them (Avalos and Ploegh, 2014; Roche and Furuta, 2015; Allen et al., 2016; Lindenbergh and Stoorvogel, 2018; Li and Hu, 2019). Antigen presented by APCs is most often thought to originate from within the cell itself or to be received via uptake from the extracellular environment by processes such as phagocytosis (Roche and Furuta, 2015; Kotsias et al., 2019). Based on the numerous HLA associations that have been identified as risk factors for different drugs and reactions (Usui and Naisbitt, 2017; Chen et al., 2018), this step is likely to be important in the pathogenesis of IDRs.
1. Presentation by Major Histocompatibility Complex I: Endogenous Protein, Crosspresentation
MHC I molecules are expressed by all nucleated cells, which allows for CD8+ T cells to survey host tissue for aberrations suggestive of intracellular pathogens or malignancy (Jongsma et al., 2019). Peptides originating from within the cell are usually presented in the context of MHC I (Neefjes et al., 2011). In what is considered the conventional processing route, proteins are digested by the proteasome to generate shorter peptide fragments, which are then translocated to the ER via the transporter associated with antigen processing for loading onto MHC I molecules after assembly of the peptide-loading complex (Vyas et al., 2008).
In some cases, proteins exogenous to the cell may be presented by MHC I, particularly in the case of DCs; this process is termed crosspresentation (Li and Hu, 2019). Peptide loading is described as following either the cytosolic pathway or the vacuolar pathway. The cytosolic pathway appears to require the proteasome for peptide processing, and peptide loading may occur in phagosomes or endosomes, whereas the vacuolar pathway is lysosome-dependent and both peptide processing and loading may occur in lysosomes (Embgenbroich and Burgdorf, 2018).
2. Presentation by Major Histocompatibility Complex II: Phagocytosis, Endocytosis, Macropinocytosis, Autophagy
Constitutive expression of MHC II is largely restricted to professional APCs, although myeloid cells, including eosinophils, neutrophils, and basophils, as well as ILCs, have been demonstrated to upregulate expression of MHC II in certain conditions (Kambayashi and Laufer, 2014). Peptides originating from exogenous sources are most often presented in the context of MHC II; however, endogenous peptides may also follow this pathway via autophagy (Duffy et al., 2017). Exogenous proteins may be acquired via different methods (Roche and Furuta, 2015). Phagocytosis is the internalization of pathogens or particulate antigens and is considered to be the most important mechanism of antigen uptake in vivo (Stuart and Ezekowitz, 2005). This process can be enhanced by opsonins, which are host proteins such as antibodies or complement that can coat foreign entities. Clathrin-mediated endocytosis is the internalizing of ligands complexed to surface receptors and soluble macromolecules (Mantegazza et al., 2013). Macropinocytosis is a nonspecific process during which uptake of extracellular fluid containing soluble antigens and macromolecules occurs (Liu and Roche, 2015). Proteins are then processed via the endocytic pathway to peptides in specialized late endosomes that are enriched with MHC II molecules for antigen presentation (Neefjes et al., 2011).
3. Crossdressing: Trogocytosis, Extracellular Vesicles, Nanotubes
In some cases, preformed MHC-peptide complexes may be transferred from the surface of a donor cell to a recipient cell; the process is referred to as crossdressing (Campana et al., 2015). Multiple mechanisms have been proposed to describe the transfer of these complexes. Trogocytosis refers to the phenomenon in which patches of the plasma membrane are rapidly transferred from one live cell to another upon cell-cell contact. In some cases, phagocytosis may not be possible if the target cell is too large; the phagocyte may instead ingest smaller pieces of the cell by “nibbling” at the membrane and potentially the cytoplasm (Dance, 2019).
Extracellular vesicles refer to either microvesicles, formed by plasma membrane budding, or exosomes, formed as intraluminal vesicles within endosomal multivesicular bodies and then released by fusion of the multivesicular body with the plasma membrane. Because these two types of vesicles are indistinguishable after their release, they are collectively termed extracellular vesicles (Groot Kormelink et al., 2018). In the context of IDRs, extracellular vesicles have been shown to be involved in the transport of drug-modified antigen to target cells, such as in the case of amoxicillin (Sánchez-Gómez et al., 2017; Ogese et al., 2019). Extracellular vesicles may also transfer proteins that can be processed by the recipient cell and presented by MHC molecules, or they may transfer the MHC-peptide complexes themselves. Additionally, extracellular vesicles derived from multiple cell types including B cells (Raposo et al., 1996) and DCs (Théry et al., 2002) have been shown to activate T cells themselves. Conversely, hepatocyte-derived exosomes have also been implicated in the promotion of immune tolerance in the liver, and dysregulation of this tolerogenic mechanism may be an important step in the onset of IDILI (Holman et al., 2019).
Tunneling nanotubules are intercellular structures that have been shown to mediate the exchange of MHC I molecules (Schiller et al., 2013). Such an exchange may be another means by which crossdressing can occur.
These varied methods of antigen acquisition, processing, and presentation describe different means by which antigen may be presented to the adaptive immune system. Understanding how antigen may reach both APCs and target T cells will aid in the understanding of the pathogenesis of IDRs.
E. Naïve Lymphocyte Activation by Antigen-Presenting Cells
Naïve T cells and B cells are activated by APCs once they receive sufficient activation signals (Mak et al., 2014). Classically, the three-signal model is used to describe this sequence of events: signal one refers to the binding involving the MHC molecule presenting the antigenic peptide of interest; signal two refers to costimulatory molecule engagement, which has been upregulated as a result of exposure to inflammatory conditions; and signal three refers to the cytokine help that permits the lymphocyte to survive and proliferate. This model describes the activation process for helper T-cell, cytotoxic T-cell, and B-cell activation, although there are some differences between the cell types.
1. Helper T Cells
Only mature DCs can activate naïve T cells. For Th cell activation, the first signal between these cells is the binding of cognate TCRs to MHC II-peptide complexes on the DC; a strong interaction over several hours results in the signaling cascades that induce cell polarization and forms the immunologic synapse. T-cell receptor engagement induces NF-κB signaling (Liu et al., 2017). This also induces CD40L and CD28 expression on the T-cell surface; CD40 engagement on the DC surface by CD40L upregulates expression of B7 molecules by the DC.
In most cases, costimulatory molecule engagement is required for T-cell activation, although in some cases, the MHC-peptide complex may deliver a strong enough signal to bypass the need for signal two (Wang et al., 2000). The B7 molecules on the DC surface interact with CD28 on the T-cell surface. This permits upregulation of cytokine receptors and induces CD4+ T-cell production of proinflammatory cytokines such as IL-2 and IFN-γ.
Signal three is delivered by APCs in the form of cytokine release. For CD4+ T cells, these cytokines include IL-1, TNF-α, and IL-6 (Pape et al., 1997; Joseph et al., 1998; Curtsinger et al., 1999; Ben-Sasson et al., 2009). This results in activation, proliferation, and differentiation to Th effector cells as well as licensed DCs. Licensed DCs may then proceed to activate naïve Tc cells.
2. Cytotoxic T Cells
Signal one is delivered to the Tc cell by engagement of MHC I on a licensed DC, the product of Th cell activation, to the T-cell receptor (Joffre et al., 2009). Signal two, or costimulation of Tc cells, is more dependent upon CD28 engagement, as B7 is already upregulated on the DC surface (Curtsinger et al., 2003). Finally, in signal three, the naïve Tc receives cytokine help, such as IL-12, from activated Th cells and APCs, thus allowing for proliferation and differentiation to precytotoxic lymphocytes (Curtsinger et al., 2003; Curtsinger and Mescher, 2010). These precytotoxic lymphocytes may then leave the lymph node and migrate to the site of inflammation, where signals such as IL-12, IFN-γ, and IL-6 induce differentiation to armed cytotoxic lymphocytes (Mescher et al., 2007). Protein synthesis for the contents of the cytotoxic granules is induced. Finally, engagement of the T-cell receptor by antigen presented on MHC I within the tissue induces targeted cell destruction by the cytotoxic Tc cell (Groscurth and Filgueira, 1998).
3. B Cells
Some antigens are considered to be T-independent in that the antigens themselves can stimulate the B cell to proliferate without T-cell help (Mond et al., 1995). Most antigens, however, are T-dependent and require the same three signals for B-cell activation (MacLennan et al., 1997).
Multiple antigens are required to bind to the B cell receptors on a single cell, termed the B-cell microcluster (Wan and Liu, 2012). This allows for the intracellular signaling cascades that prepare the B cell to receive T-cell help. An important distinction from the T-cell activation process is that the B cell can recognize whole antigen (Li et al., 2019).
Signal two is provided to B cells by activated Th cells: costimulatory signals are delivered by the Th cell, primarily by the interaction of CD40L, and the receptor, CD40, which is constitutively expressed on the B-cell surface (Banchereau et al., 1994). This induces the B cell to internalize the antigen engaged by its B-cell receptors, process the peptides, and present the peptides to the T cell. MHC II on the B cell is engaged by the T-cell receptor, which means that both the B and Th cells must recognize the same antigen, although not necessarily the same epitopes. This is known as linked recognition (Smith, 2012).
Finally, cytokines are also required as signal three for B-cell proliferation (Zubler and Kanagawa, 1982). The Th cell in contact with the B cell is usually the source of these cytokines. IL-4 is critical to induce the primed B cell to proliferate, while other cytokines support this process (Takatsu, 1997).
F. Fate of the Adaptive Immune Response
After the formation of the immunologic synapse, there are several potential outcomes with respect to the adaptive immune response that are dependent upon the strength of the signals received. At a fundamental level, the result of synapse formation may be 1) no adaptive immune activation (if signals are below the threshold of activation), 2) the promotion of tolerance via anergy or clonal deletion (if there is an engagement of coinhibitory molecules), or 3) the initiation of an adaptive immune response, resulting in T- and/or B-cell activation and effector cell maturation (after the successful formation of an immunologic synapse, complete with the engagement of the MHC-TCR complex and costimulatory receptors) (Finetti and Baldari, 2018). This spectrum of potential consequences likely explains why some individuals develop IDRs, whereas some develop mild reactions that resolve, and others may have no such adverse effects. Even if an individual has drug-modified proteins that have caused cell stress and have stimulated an innate immune response, it is unlikely that they will have the specific MHC molecule to present and/or the specific TCR clone to recognize the neoantigens in the correct conformation, or the interaction may not be strong enough to stimulate T-cell activation and expansion. There are likely other contributing factors to the idiosyncrasy of adaptive immune activation that have yet to be characterized; thus, severe IDRs remain difficult to predict.
IV. Support for Immune Activation Using Model Drugs
Hundreds of drugs have been reported to cause various severe IDRs (Mockenhaupt et al., 2008; Andrès et al., 2009, 2019; Chalasani et al., 2014; Hussaini and Farrington, 2014; Björnsson, 2016; Al Qahtani, 2018; Behera et al., 2018; De et al., 2018; Eddy, 2020; Solhjoo et al., 2020). Although different drugs are associated with different IDRs, and many drugs can cause more than one type of IDR, this section will summarize the available clinical and animal model literature demonstrating early immune involvement using four archetypal IDR-associated drugs: amodiaquine, amoxicillin, clozapine, and nevirapine (Fig. 2). Together, these IDR-associated drugs provide a representation of the majority of target organs, encompassing liver, skin, and blood reactions.

Chemical structures of the drugs associated with idiosyncratic drug reactions that are discussed in this review: amodiaquine, amoxicillin, nevirapine, and clozapine. Reactive species of each drug that have been demonstrated or proposed to contribute to the onset of the associated idiosyncratic reactions are shown in red: (A) amodiaquine can form a quinone imine, (B) amoxicillin is itself a reactive β-lactam, (C) nevirapine can form both a 12-hydroxy species and a quinone methide, and (D) clozapine can form a nitrenium ion.
Using an extensive combination of keywords related to the innate immune response, many of which were presented in Section III. Innate Mechanisms Contributing to Adaptive Immune Activation, we searched the available literature for each drug of interest. Reviewed studies were only included if the focus of the research was on early responses to drug treatment and not on the study of an IDR. In vitro studies were largely omitted to focus on the effect of drugs administered in vivo because of the complexity of the immune response, which is not adequately recapitulated using in vitro models. We also emphasize studies that focused on the healthy state, rather than disease or injury, to isolate the specific effects of the drug on the immune system.
Although the clinical manifestations of many IDRs have been well documented, research characterizing the mechanisms preceding these adaptive immune processes is limited, particularly for human data. Of course, such mechanistic studies are exceptionally difficult to undertake, as research in patients is usually limited to immune changes observed in blood samples; more detailed studies on organ effects cannot be performed. Additionally, the timing and duration of the innate immune response are likely to vary for different drugs, and the extensive patient monitoring required to capture such a response would be quite expensive, time-consuming, and generally impractical. Moreover, the characteristics of an early immune response can diverge greatly depending on the stimuli involved, and attempting to encapsulate all potential biomarkers of an innate response in clinical testing would be impossible. Therefore, in addition to data from patients, relevant studies investigating immune-related changes in experimental animal models are also discussed (refer to Supplemental Data for more detailed discussion of the individual studies). Although there are evident species differences and the immune response observed in animals may not be identical to that experienced by patients in the early weeks of drug treatment, such studies can provide important mechanistic insight into the general cells, pathways, and inflammatory mediators that may be involved in the immune response.
A. Amodiaquine
The 4-aminoquinolone amodiaquine was introduced as an alternative antimalarial medication to chloroquine. Although it is still in use in malaria-endemic areas, amodiaquine was withdrawn from most markets because of the occurrence of several serious IDRs, including agranulocytosis (Rouveix et al., 1989) and hepatotoxicity (Neftel et al., 1986).
Although the mechanisms of amodiaquine-induced IDRs are not completely understood, the bioactivation of amodiaquine in both the liver and immune cells has been extensively investigated, providing insights into the formation of neoantigens and potential immune activation. In the liver, amodiaquine is metabolized by CYP2C8 to N-desethylamodiaquine (Li et al., 2002). Both amodiaquine and N-desethylamodiaquine can be oxidized to a reactive quinone imine by cytochrome P450s in the liver and myeloperoxidase in neutrophils, leading to significant levels of covalent binding (Maggs et al., 1987, 1988; Clarke et al., 1990; Tingle et al., 1995; Naisbitt et al., 1997, 1998; Lobach and Uetrecht, 2014b) (Fig. 2A). The sites of reactive metabolite formation, i.e., CYP450 enzymes in the liver and myeloperoxidase in neutrophils and their precursors, are consistent with the pattern of IDRs caused by amodiaquine, i.e., liver injury and agranulocytosis.
Moreover, amodiaquine has been found to activate inflammasomes in vitro in a human acute monocytic leukemia cell line (THP-1 cells), with or without prior bioactivation of the drug by human hepatocarcinoma functional liver cell-4 cells (Kato and Uetrecht, 2017). In an impaired immune tolerance model, treatment of female programmed cell death protein 1 knockout (PD-1−/−) mice with anti–cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) antibodies and amodiaquine caused marked liver injury similar to IDILI in humans that was mediated by Tc cells (Mak et al., 2017).
These data support the role of early antigen formation in the progression to serious hepatotoxicity induced by amodiaquine, and further support of innate immune involvement is discussed below. Notably, no clinical studies reviewed reported any relevant data on early immune responses to amodiaquine, and thus, this section only highlights data obtained from rodent studies. This is likely reflective of discontinued amodiaquine use in many countries, although early clinical monitoring in areas actively using amodiaquine may reveal patterns of immune activation that could be leveraged to reduce progression to severe IDRs.
1. Data from Rodent and Human Studies
Several groups have investigated the impact of amodiaquine on hepatic structure and function. In general, studies characterizing the effects of amodiaquine monotherapy in the absence of a pre-existing disease have consistently demonstrated elevated ALT levels in the first few weeks of treatment, which then resolves (Clarke et al., 1990; Shimizu et al., 2009; Mak and Uetrecht, 2015a, 2019; Metushi et al., 2015; Liu et al., 2016).
Glutathione-depletion studies using buthionine sulphoximine (BSO) have been performed to evaluate the effect of detoxification of amodiaquine. However, the dosing paradigms used differ significantly. In one case, BSO (700 mg/kg intraperitoneally) was administered 1 hour before amodiaquine (180 mg/kg orally), and liver injury was greatly exacerbated in 6–48 hours compared with amodiaquine treatment alone (Shimizu et al., 2009). In contrast, BSO (4.4 g/l in drinking water), administered 1 week before amodiaquine (∼200 mg/kg per day in rodent meal), in addition to diethyl maleate (4 mmol/kg, intraperitoneally), administered 1 day before amodiaquine, prevented liver injury (Liu et al., 2016). It is important to note that the liver injury occurred acutely in the former model, which was likely due to the higher exposure of the mice to amodiaquine by bolus administration. Acute toxicity represents a different type of liver injury compared with what is observed clinically with patients with IDILI, as these drugs do not cause acute toxicity in humans at therapeutic doses. However, this does not preclude the fact that these drugs may cause a clinically silent immune response in patients.
Few studies have sought to characterize changes in inflammatory mediators with amodiaquine treatment. Amodiaquine monotherapy in female mice and male rats caused significant increases in numerous proinflammatory cytokines and chemokines beginning after 1 week of treatment (Metushi et al., 2015; Liu et al., 2016). Interestingly, the addition of amodiaquine was reported to attenuate increases in some inflammatory cytokines in models of acute tissue injury such as hepatitis or intracerebral hemorrhage (Yokoyama et al., 2007; Kinoshita et al., 2019). This could be due to the induction of tolerogenic mechanisms by amodiaquine, which prevents a pathogenic response to amodiaquine. This illustrates the complexity of immune responses.
Although the patterns observed are organ-specific with respect to timing and specific cell types, studies that investigated the effect of amodiaquine treatment on immune cells have consistently reported a decrease in leukocytes in the first several days to weeks of treatment, followed by an increase around a month of treatment (Clarke et al., 1990; Ajani et al., 2008; Mak and Uetrecht, 2015a, 2019; Metushi et al., 2015; Liu et al., 2016). In studies that undertook phenotyping of specific populations, NK cells were demonstrated to be the most important effector cell in response to amodiaquine treatment, with increased populations observed in the lymph node, spleen, and liver beginning after 1 week of treatment (Metushi et al., 2015; Liu et al., 2016; Mak and Uetrecht, 2019).
Only two studies, both using male rats, explored alterations in cell death pathways in response to amodiaquine treatment. Both reported an increase in apoptotic-related processes, either in the seminiferous tubules after 2 weeks (Niu et al., 2016) or in the liver after 5 weeks (Liu et al., 2016). These data suggest that amodiaquine-induced cell death may play a role in the activation of the immune response that ultimately results in severe IDRs. Covalent binding has been detected in several organs beyond the liver, including the kidney, spleen, and gut (Metushi et al., 2015), and thus, similar cell death effects may also occur elsewhere. Additional work is necessary to characterize the mechanisms preceding the onset of apoptosis and whether this occurs in other organs and, if so, at what time points.
Taken together, amodiaquine has been consistently shown to induce mild liver injury in rodent models that resolves spontaneously with continued treatment, and it has been shown that NK cells are important in mediating this injury. Whether the apoptosis that has been observed is induced by covalent binding of the drug itself or by the subsequent release of DAMPs and recruitment of NK cells or other immune cells remains to be determined. However, it is quite clear that amodiaquine induces an immune response that is not idiosyncratic.
B. Amoxicillin
Amoxicillin is a β-lactam antibiotic often used in the treatment of multiple bacterial infections. It is sometimes administered in combination with clavulanic acid, a β-lactamase inhibitor, to prevent the development of microbial resistance. Both of these agents are intrinsically reactive because of the β-lactam ring (Fig. 2B). Amoxicillin on its own is associated with different hypersensitivity reactions. Hypersensitivity reactions to β-lactam antibiotics can be classified as immediate or delayed. Immediate hypersensitivity reactions are IgE-mediated, involving basophil activation, and occur within 1 hour of drug administration, whereas delayed hypersensitivity, occurring over 1 hour after administration, tends to be T cell–mediated (Blanca et al., 2009).
The combination of amoxicillin and clavulanate is also associated with cholestatic IDILI (de Abajo et al., 2004). As amoxicillin alone is not associated with a high incidence of cholestatic IDILI (https://www.ncbi.nlm.nih.gov/books/NBK547854/), this suggests that clavulanate is the causative drug; however, as clavulanate is not used alone, there are no direct data to support this.
Covalent binding of amoxicillin to protein has been identified in in vitro studies, and some studies have also identified amoxicillin-modified proteins in exosomes, which may represent a means of transporting antigen to the immune system, as the exosomes were shown to activate naïve T cells in vitro in an HLA-A*02:01–dependent manner (Ogese et al., 2017, 2019; Sánchez-Gómez et al., 2017). Additionally, binding of amoxicillin and clavulanate to serum protein was identified in patients (Ariza et al., 2012; Meng et al., 2016).
1. Data from Rodent and Human Studies
There are few published studies on the immunomodulatory effects of amoxicillin in uninfected subjects. A study in rats administered amoxicillin/clavulanate for 30 mg/kg per day intraperitoneally (clavulanate dose not specified) for 14 days showed some signs of liver cell death, indicated by increased serum ALT and increased caspase expression in the liver. This appeared to have been caused by oxidative stress, as evidenced by increased malondialdehyde levels, cytochrome-c release, and increased ATPase activity (Oyebode et al., 2019). White blood cell counts were also elevated. However, another study in mice reported a decrease in white blood cell counts only at a higher dose of 500 mg/kg per day by oral gavage (amoxicillin only) for 28 days (Lebrec et al., 1994). Decreased thymus cellularity was also noted but was only significant at a dose of 100 mg/kg.
As mentioned, in humans, amoxicillin (Ariza et al., 2012) and clavulanate (Meng et al., 2016) have been identified covalently bound to serum albumin. However, treatment with oral amoxicillin (1 g)/clavulanate potassium (125 mg) twice daily for 5 days resulted in no change in cell counts of multiple leukocytes, intracellular TNF-α concentrations in monocytes, and intracellular TNF-α and IFN-γ concentrations in NK cells or CD8+ T cells stimulated ex vivo (Dufour et al., 2005).
Overall, these findings are perhaps not very surprising, as amoxicillin is used very frequently and is generally considered quite safe. The safety profile of amoxicillin illustrates the fact that covalent binding on its own is insufficient to cause IDRs. Liver toxicity was observed in the study of rats administered amoxicillin/clavulanate at 2 weeks, but such parameters were not measured in the clinical study. Although few innate parameters were measured in the clinical study, the general lack of changes reported suggests that there may not be a detectable systemic inflammatory response to amoxicillin and, possibly, that immune changes are localized to the liver.
C. Nevirapine
Nevirapine is a non-nucleoside reverse transcriptase inhibitor used in the treatment of HIV infections. Nevirapine is associated with skin reactions and IDILI (Popovic et al., 2010). It is noteworthy that, in some cases, such as perinatal transmission prophylaxis, it can be used as monotherapy; in others, it is administered in combination with other highly active antiretroviral therapy medications to avoid the development of resistance (Bardsley-Elliot and Perry, 2000). Thus, the effects of nevirapine treatment on immune parameters in clinical studies can be difficult to disentangle from the effect of other drugs when used in combination or from the background of HIV infection and subsequent effects of treatment efficacy (i.e., recovery of CD4+ T-cell counts).
As already mentioned, 12-hydroxynevirpine sulfate was found to covalently bind in the skin of female brown Norway rats and was determined to be responsible for the observed skin rash because the application of a topical sulfotransferase inhibitor prevented the development of the rash (Sharma et al., 2013). However, although this metabolite is responsible for skin rash, a quinone methide formed by cytochrome P450 is the major metabolite responsible for covalent binding in the liver (Sharma et al., 2012) (Fig. 2C).
In patients taking nevirapine, protein-nevirapine adducts have been detected in blood samples. A 12-hydroxynevirapine sulfate-His146 adduct was detected on human serum albumin from patients taking nevirapine, which was replicated in vitro by treatment of human serum albumin with 12-hydroxynevirapine sulfate (Meng et al., 2013). Nevirapine-derived adducts to the N-terminal valine of hemoglobin were also detected in patient samples (Caixas et al., 2012).
1. Data from Rodent and Human Studies
Generally, nevirapine administration caused an increase in serum ALT levels in male rats and mice (Adaramoye et al., 2012; Sharma et al., 2012; Awodele et al., 2015), but not in female brown Norway rats (Bekker et al., 2012; Brown et al., 2016), although this observation is likely due to the shorter duration of the latter studies (7 or 14 days). In a clinical study, nevirapine exposure was associated with reduced fibrosis, although again it is difficult to speculate upon the mechanism, as this was in the context of HIV and hepatitis C coinfection (Berenguer et al., 2008).
Histologic findings indicative of hepatocyte cell death were sometimes found in rats and mice (Adaramoye et al., 2012; Bekker et al., 2012; Sharma et al., 2012). Gene expression changes in the skin of female brown Norway rats 6 hours after 12-hydroxynevirapine treatment also appeared to indicate apoptosis or altered mitochondrial function (Zhang et al., 2013). In rodent studies, nevirapine caused an increase in malondialdehyde, although changes in antioxidant enzymes were not observed (Adaramoye et al., 2012; Awodele et al., 2015). Altogether, the studies in rodents appear to suggest effects on cell death and mitochondrial function.
The effects of nevirapine on mitochondrial function are less clear in clinical studies. One study showed that nevirapine (coadministered with stavudine and lamivudine) increased mitochondrial depolarization and lymphocyte apoptosis (Karamchand et al., 2008). In contrast, another study showed that switching to nevirapine from nucleoside reverse transcriptase inhibitors improved mitochondrial parameters, but this may simply indicate that nevirapine has less of an effect on mitochondria than nucleoside reverse transcriptase inhibitors, which are known to cause mitochondrial toxicity (Negredo et al., 2009). Infants treated with nevirapine were not shown to have significant mitochondrial toxicity (Jao et al., 2017). In terms of oxidative stress, a study measured plasma F2-isoprostane levels as a measure of lipid peroxidation and found that there was a trend toward decreased plasma F2-isoprostane levels with nevirapine treatment (Redhage et al., 2009). Altogether, mitochondrial function may be impaired or unchanged with nevirapine exposure, but any observed effects do not appear to be as substantial as with other antiretrovirals.
In general, nevirapine did not have a clear impact on blood cell counts in rodents; depending upon the timing and the dosing, nevirapine was found to decrease leukocytes (compared with controls, 6 mg/kg per day orally, 60 days) (Awodele et al., 2015) or increase lymphocytes and platelets (compared with reference range, 200 mg/kg per day by oral gavage, 21 days) (Bekker et al., 2012) in rats. A low dose of nevirapine acutely increased leukocyte emigration in rats (Orden et al., 2014). In the female brown Norway rat model of skin rash, nevirapine treatment appeared to induce macrophage infiltration in auricular lymph nodes that preceded T-cell recruitment (Popovic et al., 2006). Infants exposed to prophylactic nevirapine treatment had elevated monocyte counts and percentages and basophil counts at birth (Schramm et al., 2010).
In rodent models, nevirapine caused some changes in cytokine levels, although in most cases, these changes occurred 3 weeks or longer after initiation of drug treatment. Serum TNF-α was increased at 24 hours in one study (Bekker et al., 2012), but no changes were seen with IFN-γ up to 3 weeks (Popovic et al., 2006; Bekker et al., 2012). In clinical studies of HIV infection, nevirapine exposure was associated with decreased serum or plasma cytokines such as CCL3 and IL-8 (Shalekoff et al., 2009), IL-6 (Borges et al., 2015), and potentially soluble CD14 (Allavena et al., 2013). The latter two studies compared the effects of multiple drugs, so these results may speak to a nevirapine-specific effect rather than a broad antiviral treatment effect. Although there are not many clear or consistent changes in specific inflammatory markers, nevirapine does seem to have modulatory effects on inflammatory markers in general, which may even contribute to its efficacy.
Overall, nevirapine has been shown to cause mild liver injury in otherwise healthy rodents. In some cases, histologic findings of cell death may complement the observation of liver injury. There are conflicting results regarding mitochondrial toxicity, leukocyte changes, and cytokine changes. There is no convincing evidence that nevirapine mediates mitochondrial injury; if anything, it appears less likely to do so than other antiretrovirals used in the treatment of HIV. Nevirapine does appear to have effects on peripheral blood cells, although the differences in study duration, doses, and models used are problematic. It would be informative to determine whether changes observed in female brown Norway rats, particularly the macrophage recruitment to lymph nodes, are reproduced in other rodent models that do not develop a skin rash.
D. Clozapine
Clozapine, an atypical antipsychotic, has unique efficacy in the treatment of schizophrenia. However, it is infrequently prescribed because of the risks of IDIAG and, more rarely, IDILI (Wu Chou et al., 2014; Li et al., 2020). As mentioned, several HLA haplotypes have been associated with an increased risk of clozapine-induced agranulocytosis (Legge and Walters, 2019).
The initiating mechanisms of clozapine-induced IDRs are poorly understood but are hypothesized to involve an aberrant adaptive immune response against clozapine-modified proteins. Clozapine can be bioactivated by cytochromes P450 in the liver and myeloperoxidase in neutrophils and monocytes to a reactive nitrenium ion that covalently binds to cellular proteins in vitro and in vivo (Liu and Uetrecht, 1995; Maggs et al., 1995; Dragovic et al., 2013; Lobach and Uetrecht, 2014b) (Fig. 2D). If this neoantigen formation leads to significant cell damage or death, then the proinflammatory mediators and DAMPs released could stimulate an innate immune response that eventually leads to the onset of severe IDRs (Pirmohamed and Park, 1997; Johnston and Uetrecht, 2015).
Because of the rigorous hematologic monitoring required for patients starting clozapine, substantial evidence in support of an early innate immune response has been reported, as discussed below. Additionally, several animal models, predominantly albino rats, have been used to characterize immune changes throughout the first few weeks of clozapine treatment.
1. Data from Rodent and Human Studies
A review of the literature revealed more than 50 case reports and case series that noted the occurrence of an innate immune response with clozapine, most commonly evidenced by fever, eosinophilia, neutrophilia, leukocytosis, a left shift in blood cells, and increased C-reactive protein, ALT, ALP, and aspartate transaminase within the first 6 weeks of treatment (Lowe et al., 2007; Røge et al., 2012; Szota et al., 2013; Fonseka et al., 2016; Bellissima et al., 2018; Verdoux et al., 2019; de Leon et al., 2020). To avoid redundancy, those studies are not presented here.
Although short-term clozapine administration has been studied in close to 100 rodent studies, the focus of much of this work was to determine how clozapine alters disease and/or injury progression (e.g., in phencyclidine-induced schizophrenia) and not to characterize the effects of clozapine alone. Such models make it challenging to delineate a role for clozapine in the initiation of an innate immune response. Interestingly, many of these studies actually reported a protective effect of clozapine, often noting an attenuation of the disease model–induced inflammatory response. However, these disease models are physically or chemically induced, and such results may not reflect the true effects of clozapine monotherapy in patients.
Two male rat studies that evaluated clozapine effects in the liver demonstrated significant increases in injury (e.g., ALT increases, inflammatory cell infiltrates, increased liver weight) between 1 and 3 weeks of treatment (Jia et al., 2014; Zlatković et al., 2014). Significant covalent binding has also been demonstrated in the liver of clozapine-treated rats (Gardner et al., 2005; Ip and Uetrecht, 2008), and it is possible that the hepatic inflammation observed is in response to this haptenization.
The effects of clozapine on various other organs, including the brain, heart, and kidney, have been investigated using several rodent models. In studies characterizing the effects of clozapine in the absence of induced injury or disease, decreased splenic white pulp was observed in both female mice and male rats (Abdelrahman et al., 2014; Mohammed et al., 2020), and both ovarian and kidney injury were reported in rats (Khalaf et al., 2019; Mohammed et al., 2020). Moreover, significant cardiac inflammation and morphologic aberrations were observed during the first few weeks of treatment (Wang et al., 2008; Abdel-Wahab and Metwally, 2014; Abdel-Wahab et al., 2014; Nikolić-Kokić et al., 2018; Mohammed et al., 2020). This parallels what is observed clinically because, in addition to severe IDRs, clozapine has been associated with an increased risk of myocarditis in patients, which can present with fever, eosinophilia, and increased troponin levels, often during weeks 2 and 3 of treatment (Kilian et al., 1999; Ronaldson et al., 2010; Curto et al., 2015). This is clearly an innate immune response due to the acute onset and effector cells and mediators observed.
The potential for clozapine to trigger cell death has been explored in several organs, including the liver, heart, blood, and brain. The majority of rodent studies demonstrated evidence of apoptosis (e.g., increased terminal deoxynucleotide transferase dUTP nick-end labeling staining or caspase-3 activation) between weeks 1 and 4 of treatment (Wasti et al., 2006; Jarskog et al., 2007; Huang et al., 2012; Abdel-Wahab and Metwally, 2014; Abdel-Wahab et al., 2014; Jia et al., 2014; Zlatković et al., 2014; Hsu and Fu, 2016; Khalaf et al., 2019) using doses that would approximate therapeutic concentrations in patients (Lobach and Uetrecht, 2014a). One study also noted that clozapine induced autophagy within hours of administration (Kim et al., 2018), and others noted decreased proliferation within the first few weeks of treatment (Huang et al., 2012; Hsu and Fu, 2016; Khalaf et al., 2019) as well. Translocation of apoptosis-inducing factor was not observed in the striatum of clozapine-treated patients or in rodents after 1 month of treatment (Skoblenick et al., 2006), suggesting against the involvement of caspase-independent cell death with clozapine.
In various models of acute injury and disease, clozapine was not consistently found to attenuate changes in immune cell populations. Only a small number of studies investigated the effects of clozapine in healthy animals, almost all of which demonstrated induction of an immune response by clozapine in the first 3 weeks of treatment. Most commonly, an increase in neutrophils was reported in clozapine-treated male and female rats (Wasti et al., 2006; Abdel-Wahab and Metwally, 2014; Lobach and Uetrecht, 2014a; Ng et al., 2014) or rabbits (Iverson et al., 2010). In the only two mouse studies, clozapine caused not only a decrease in several leukocyte populations (Abdelrahman et al., 2014; Jiang et al., 2016) but also an increase in monocytes, suggesting that the immunomodulatory effects of clozapine may differ across rodent species. In clinical studies, however, clozapine demonstrated strong evidence of innate immune cell activation during the first several weeks of treatment. Depending on the patient population, studies reported an increased incidence of eosinophilia, neutrophilia, and/or leukocytosis that typically resolved with continued clozapine administration (Banov et al., 1993; Pollmächer et al., 1996; Chatterton, 1997; Tham and Dickson, 2002; Pui-yin Chung et al., 2008; Löffler et al., 2010). One small study also noted an increase in circulating CD34+ hemopoietic stem cells after 2 weeks of clozapine treatment (Löffler et al., 2010).
In rodent models in which immunomodulatory effects were investigated in the context of a disease or injury model, clozapine was frequently shown to attenuate the model-induced inflammation. Contrarily, few studies characterized the inflammatory mediator changes caused by clozapine alone. Of these studies, most demonstrated an increase in proinflammatory mediators in either rats or mice, including TNF-α, CXCL2, and heat shock protein 75 (Wang et al., 2008; Abdel-Wahab and Metwally, 2014; Abdel-Wahab et al., 2014; Lobach and Uetrecht, 2014a; Kedracka-Krok et al., 2016; Mohammed et al., 2020). Few studies have reported alterations in bioactive lipids in response to the drugs reviewed; however, dysregulated arachidonic acid signaling was also noted with clozapine treatment (Kim et al., 2012; Modi et al., 2013). Among the drugs investigated for this review, clozapine also provides the strongest support of innate immune activation in patients. All but one study reported an increased incidence of fever and/or increased serum levels of inflammatory mediators, such as TNF-α, soluble TNF receptor, soluble CD8, and soluble IL-2 receptor, most commonly occurring during the first month of treatment (Pollmächer et al., 1995, 1996, 1997; Maes et al., 1997, 2002; Hinze-Selch et al., 1998, 2000; Tham and Dickson, 2002; Pui-yin Chung et al., 2008; Kluge et al., 2009; Hung et al., 2017).
The research focused on the effects of amodiaquine, amoxicillin, or nevirapine on signal transduction pathways in rodent models is limited, although this is likely due to the preference for in vitro work in this area. Contrastingly, the effect of clozapine on a number of signaling pathways has been examined in rodents, although many of these observations have yet to be verified in subsequent studies. The reported effects of clozapine on the regulation of transcription vary greatly and depend on the timing of the studies, as well as the organs investigated. Clozapine-induced activation of hepatic and cardiac NF-κB was demonstrated in two rat models at 3 weeks (Abdel-Wahab and Metwally, 2014; Zlatković et al., 2014), but these changes were not observed in several brain regions in other models. Other studies have also characterized clozapine-induced activation of other signaling pathways, including the AMP-activated protein kinase (AMPK)-Unc-51–like kinase 1-Beclin1 pathway (Kim et al., 2018) and the protein kinase R–like ER kinase/eukaryotic translation initiation factor 2A ER stress axis (Weston-Green et al., 2018), although additional work is necessary to confirm the results of these reports. Notably, AMPK signaling has been shown to play a role in many biochemical pathways, including autophagy, mitochondrial biogenesis, and lipid metabolism (Hardie et al., 2016); thus, further investigation of clozapine’s impact on AMPK signaling and its potential role in inflammation should be undertaken.
Several rodent studies have also been conducted to evaluate changes in mitochondrial function due to clozapine. Clozapine often caused attenuated mitochondrial function or oxidative stress (e.g., increased malondialdehyde levels), which was noted most frequently in male rats after 3–4 weeks of treatment in various brain regions (Lara et al., 2001; La et al., 2006; Mehler-Wex et al., 2006; Streck et al., 2007; Bullock et al., 2008; Martins et al., 2008; Ji et al., 2009; Bishnoi et al., 2011; Zlatković et al., 2014; Cai et al., 2017), although cardiac-specific (Nikolić-Kokić et al., 2018) and ovarian-specific (Khalaf et al., 2019) aberrations were also reported. Additionally, another study reported increased markers of ER stress in the liver 1 hour after clozapine treatment (Lauressergues et al., 2012).
Although additional work is clearly needed to characterize the mechanisms underlying the findings discussed here, clozapine has frequently been shown to cause innate immune activation, both in patients and in various animal models. One avenue that should also be pursued moving forward is determining what initially triggers the immune response (e.g., triggers of myeloid cell recruitment) and, subsequently, whether inhibiting this immune response prevents progression to serious IDRs, effectively reducing the risks associated with clozapine use.
E. Summary
Overall, various early immune-related changes have been observed in animal models and human studies with the drugs presented here (Table 1). In rodents, the increased serum ALT observed with all drugs is indicative of liver damage. Additionally, the induction of apoptosis in many other organs was also observed with each of the drugs. A number of changes were described in various leukocyte populations, with some drugs causing increases in innate immune cells and, in fewer instances, some drugs causing decreases in leukocytes. In many cases, increases in proinflammatory cytokines were observed, and with clozapine, activated signal transduction pathways involved in proinflammatory signaling were also observed; this has not been studied in vivo for the other drugs examined. Markers of mitochondrial dysfunction were also reported after administration of amoxicillin, clozapine, and nevirapine.
An overview of the most commonly observed findings from rodent and human studies that investigated the effects of amodiaquine, amoxicillin, nevirapine, or clozapine on various innate immune parameters, excluding models of injury or disease
Refer to Supplemental Data for a comprehensive list of rodent and human studies (including models of injury or disease), including information on the dose, route of administration, study duration, and primary outcomes.
The study of the inflammation caused by amodiaquine is limited to rodent models. However, there is a clear indication of NK cell–mediated liver injury, which spontaneously resolves with continued treatment, in addition to other immune cell infiltrates detected in the liver, spleen, lymph node, and peripheral blood. The immune effects of amoxicillin ± clavulanic acid have not been studied in healthy subjects as extensively as some of the other drugs presented here. In general, the data do not suggest that amoxicillin causes an overt inflammatory response, but there are certainly changes that suggest some effects on mitochondria and leukocytes. Nevirapine treatment appears to cause liver damage but has variable effects on inflammatory mediators and immune cell counts. In rodents, clozapine shows the clearest pattern of a proinflammatory response of these drugs, which is not surprising, as it has been noted to induce fever, eosinophilia, neutrophilia, and increased proinflammatory cytokine release in patients.
Overall, the effects of each of these drugs are quite variable depending upon the models and doses used and the time points at which different parameters are evaluated. This highlights the complexity of the immune response and the potential differences that may be caused by different drugs, which likely depend upon the conditions in which their reactive metabolites are formed and which may also have a bearing on the types of IDRs that they cause. Studies that evaluate the time course of drug effects on inflammatory pathways are needed to better understand how drugs that cause IDRs induce innate immune responses.
V. Conclusions and Perspectives
Just as the drugs presented here are associated with different IDRs, they have also been demonstrated to cause a variety of immune-related effects during the first few weeks of treatment. These differences include the tissue localization of cellular dysfunction, injury, and death; the responding effector cells; the mechanisms contributing to inflammation; and the development of the immune response over time. These drug-specific observations emphasize the nuances and complexity underlying the activation and progression of an innate immune response. Based on the data presented here, it is clear that further research is necessary to expand our understanding of how drugs that are associated with severe IDRs can more frequently induce an early, transient immune response that typically resolves with continued treatment. Such research is fundamental to understanding the mechanisms of IDRs, and it is quite feasible to perform such research. Most drug metabolic pathways and immune responses share some similarities in animals and humans, and animal models are an important tool because they make it possible to perform controlled experiments and investigate organs such as the liver and spleen that could not be routinely looked at in patients. It is important to use doses in animals that would produce what would be a therapeutic level in humans because high doses are more likely to cause overt toxicity that is not involved in the mechanism of IDRs. However, even though most features are likely to be similar in rodents and humans, there are clearly important differences between animals and humans; therefore, it is essential to follow up the animal studies with studies in humans to make sure that the results in animals correspond to the immune response to drugs in humans.
The innate immune response caused by these IDR-associated drugs is likely to be mild in comparison with the overt injury induced by disease models and would easily be overlooked in studies not designed to capture these relatively subtle changes. Moreover, additional consideration should be given to how these innate immune responses resolve with persistent treatment, as this resolution/tolerogenic response, or lack thereof, may provide clues as to why certain individuals eventually develop severe IDRs while the majority do not. Although certain risk factors for different IDRs have been identified, such as particular HLA haplotypes, these factors only account for a small proportion of risk; for most drugs, it remains difficult to predict which individuals will develop a severe IDR. An individual’s T-cell receptor repertoire is likely to be a major factor, but it is much more difficult to study than HLA haplotypes. Many drugs, although highly efficacious in the treatment of their intended conditions, are therefore limited in their clinical use because of the risk of IDRs (including amodiaquine, clozapine, and nevirapine). Thus, a better understanding of the mechanisms contributing to the early immune response to these drugs may help predict and prevent or treat their associated IDRs, enabling the safer use of these agents. Although some work has been done in this area already, as reviewed here, the innate immune response has not been systematically studied across drugs that cause IDRs. More work is required to understand whether different drugs cause different responses, or whether there are certain commonalities in the immune changes caused by drugs that cause IDRs. Additionally, it will be important to test drugs that do not cause IDRs to ensure that they do not have the same effects. By identifying alterations in pathways that presage IDRs, these studies will identify potential biomarkers for drugs that can cause IDRs. These biomarkers could be used to develop a preclinical tool to screen drug candidates for the potential to cause serious IDRs. Such assays would facilitate the development of safer drugs and reduce the burden of IDRs on the drug discovery process. Additionally, understanding the specifics of the innate immune response to these drugs may reveal potential targets to inhibit to prevent the development of IDRs for drugs in clinical use. Altogether, although much work remains in this area, the study of the innate immune response is clearly important in improving drug safety.
Acknowledgments
We are very grateful to Glyneva Bradley-Ridout for assistance with conducting the literature review for Section IV. Support for Immune Activation Using Model Drugs.
Figures were produced using BioRender.com.
Authorship Contributions
Participated in research design: Sernoskie, Jee, Uetrecht.
Performed data analysis: Sernoskie, Jee.
Wrote or contributed to the writing of the manuscript: Sernoskie, Jee, Uetrecht.
Footnotes
↵1 S.C.S. and A.J. contributed equally to this work.
This work was supported by the Canadian Institutes of Health Research [Grant 142329]. S.C.S. was supported by an Ontario Graduate Scholarship, a Mitacs Research Training Award, and a University of Toronto Fellowship. A.J. was supported by a Natural Sciences and Engineering Research Council of Canada doctoral scholarship.
No author has an actual or perceived conflict of interest with the contents of this article.
↵
 This article has supplemental material available at pharmrev.aspetjournals.org.
This article has supplemental material available at pharmrev.aspetjournals.org.

Abbreviations
- AGEP
- acute generalized exanthematous pustulosis
- AIN
- acute interstitial nephritis
- ALP
- alkaline phosphatase
- ALT
- alanine aminotransferase
- AMPK
- AMP-activated protein kinase
- APC
- antigen-presenting cell
- BSEP
- bile salt export pump
- BSO
- buthionine sulphoximine
- CCL
- chemokine (C-C motif) ligand
- cDC
- conventional DC
- CXCL
- C-X-C motif chemokine ligand
- DAMP
- damage-associated molecular pattern
- DC
- dendritic cell
- DIAIN
- drug-induced acute interstitial nephritis
- DRESS
- drug reaction with eosinophilia and systemic symptoms
- ER
- endoplasmic reticulum
- HIV
- human immunodeficiency virus
- HLA
- human leukocyte antigen
- HMGB1
- high mobility group box 1
- IDIAG
- idiosyncratic drug-induced agranulocytosis
- IDILI
- idiosyncratic drug-induced liver injury
- IDR
- idiosyncratic drug reaction
- IFN
- interferon
- IL
- interleukin
- ILC
- innate lymphoid cell
- MHC
- major histocompatibility complex
- NET
- neutrophil extracellular trap
- NF-κB
- nuclear factor of the κ light chain enhancer of B cells
- NK
- natural killer
- NLR
- nucleotide-binding oligomerization domain-like receptor
- NLRP3
- NLR family pyrin domain containing 3
- NSAID
- nonsteroidal anti‐inflammatory drug
- PRR
- pattern recognition receptor
- Rel
- v-rel avian reticuloendotheliosis viral oncogene homolog
- ROS
- reactive oxygen species
- SJS
- Stevens-Johnson syndrome
- Tc cell
- cytotoxic T cell
- TCR
- T-cell receptor
- TEN
- toxic epidermal necrolysis
- Th cell
- helper T cell
- TLR
- Toll-like receptor
- TNF
- tumor necrosis factor
- UPR
- unfolded protein response
- Copyright © 2021 by The Author(s)
This is an open access article distributed under the CC BY Attribution 4.0 International license.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





{kind=link}
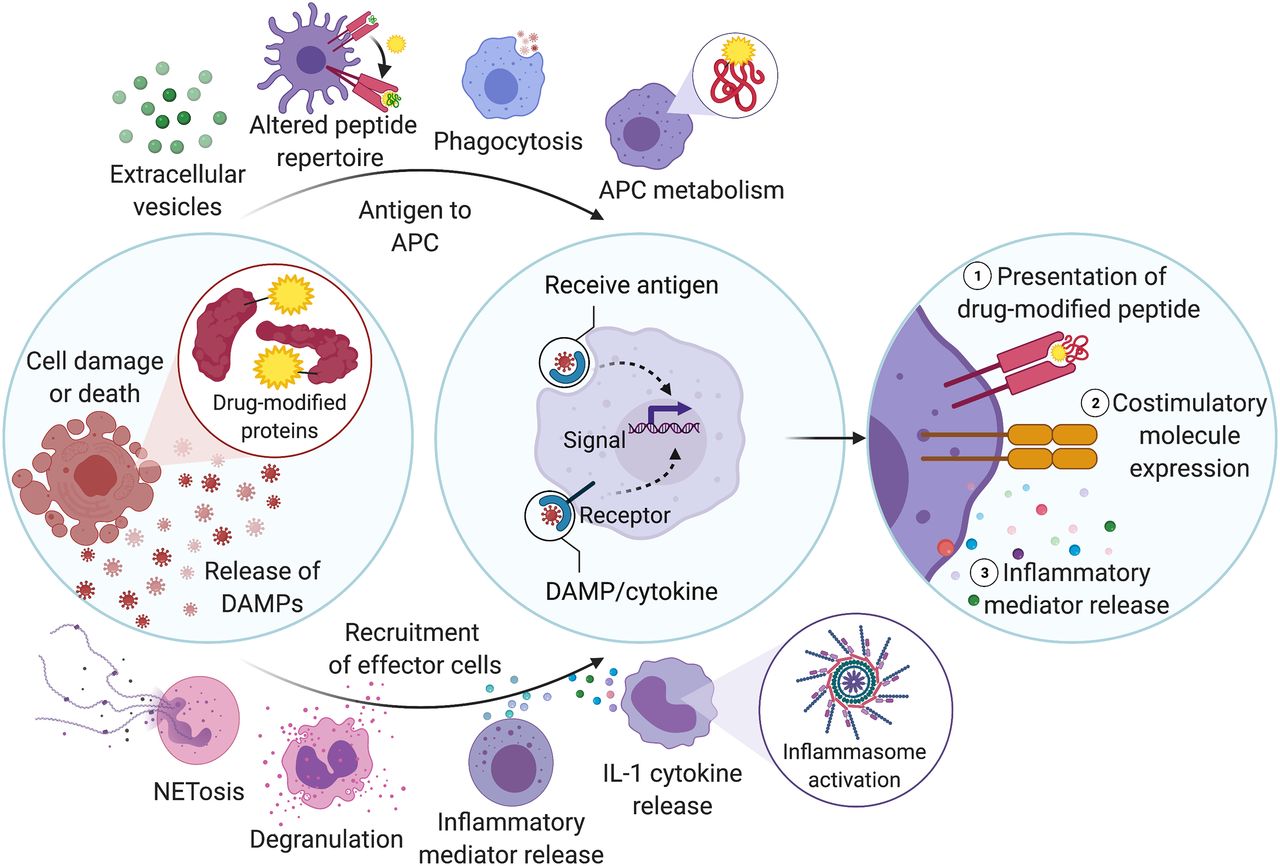{kind=link}
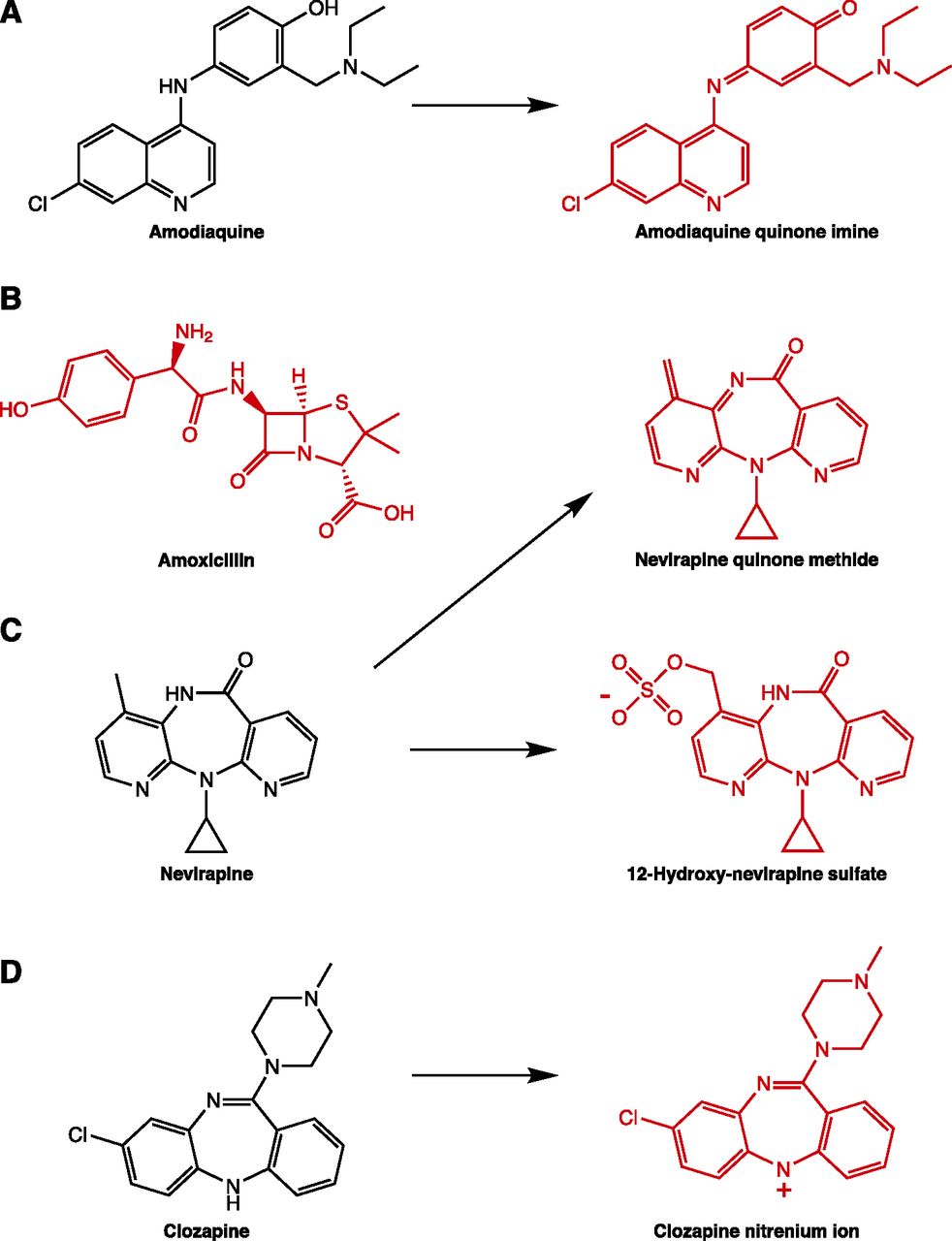{kind=link}
Jump to section
- Article
- Visual Overview
- Abstract
- I. Introduction
- II. Review of Types of Idiosyncratic Drug Reactions
- III. Innate Mechanisms Contributing to Adaptive Immune Activation
- IV. Support for Immune Activation Using Model Drugs
- V. Conclusions and Perspectives
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters
- PDF + SI