


Abstract
G protein–coupled receptors (GPCRs) constitute the largest family of membrane proteins in the human genome and are important therapeutic targets. During the last decade, the number of atomic-resolution structures of GPCRs has increased rapidly, providing insights into drug binding at the molecular level. These breakthroughs have created excitement regarding the potential of using structural information in ligand design and initiated a new era of rational drug discovery for GPCRs. The molecular docking method is now widely applied to model the three-dimensional structures of GPCR-ligand complexes and screen for chemical probes in large compound libraries. In this review article, we first summarize the current structural coverage of the GPCR superfamily and the understanding of receptor-ligand interactions at atomic resolution. We then present the general workflow of structure-based virtual screening and strategies to discover GPCR ligands in chemical libraries. We assess the state of the art of this research field by summarizing prospective applications of virtual screening based on experimental structures. Strategies to identify compounds with specific efficacy and selectivity profiles are discussed, illustrating the opportunities and limitations of the molecular docking method. Our overview shows that structure-based virtual screening can discover novel leads and will be essential in pursuing the next generation of GPCR drugs.
Significance Statement Extraordinary advances in the structural biology of G protein–coupled receptors have revealed the molecular details of ligand recognition by this large family of therapeutic targets, providing novel avenues for rational drug design. Structure-based docking is an efficient computational approach to identify novel chemical probes from large compound libraries, which has the potential to accelerate the development of drug candidates.
I. Introduction to G Protein–Coupled Receptors: Pharmacology and Structural Biology
A. G Protein–Coupled Receptor Pharmacology and Therapeutic Relevance
G protein–coupled receptors (GPCRs) constitute the largest family of membrane proteins and transduce signals from the extracellular matrix into the cell (Fig. 1). About 350 of the 800 GPCRs encoded by the human genome are nonsensory receptors (http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=694) that are activated by diffusible ligands, and this group is the main focus of this review. GPCRs are activated by remarkably diverse ligand types, ranging from small molecules to peptides and proteins (Alexander et al., 2019). The binding of the endogenous ligand to the orthosteric site triggers a conformational change in the receptor (Fig. 2), which leads to activation of intracellular proteins (e.g., heterotrimeric G proteins or β-arrestins) that regulate multiple signaling pathways (Hilger et al., 2018). GPCRs are widely expressed in the body and play important roles in cellular communication. The GPCR signaling machinery is involved in numerous physiologic processes, which stimulated the development of chemical probes to study receptor function and numerous drug candidates (Vass et al., 2018). Currently, 475 drugs are known to target ∼100 of the nonsensory GPCRs, which modulate cellular signaling pathways associated with a large number of therapeutic indications (Hauser et al., 2017). In fact, 34% of marketed drugs bind to GPCRs. Over 142 agents targeting another 19 members of the GPCR family are currently in clinical development (Congreve et al., 2020). The importance of GPCRs as drug targets is the result of the diversity of cellular processes that are mediated by receptor signaling and of the many types of ligands that can interact with different druggable binding sites. GPCRs can adopt a spectrum of states, ranging from inactive conformations that are not compatible with coupling to intracellular G proteins or β-arrestins to active conformations that trigger one or both of these effectors. Ligands can stabilize GPCR conformations by interacting with the orthosteric binding site or by targeting allosteric sites that influence the effect of orthosteric ligands or G protein/β-arrestin coupling (Wingler and Lefkowitz, 2020). Orthosteric ligands have traditionally been classified as agonists, inverse agonists, or antagonists (Fig. 2). Full agonists activate a GPCR to the same extent as the endogenous agonist. Partial agonists also activate a GPCR, but they only have partial efficacy at the receptor compared with the endogenous agonist. Inverse agonists block the basal activity that a GPCR has in the absence of an agonist, whereas neutral antagonists compete with the endogenous ligands but do not affect basal signaling. Biased ligands can have different effects on signaling pathways, e.g., activate G protein or G protein–independent signaling pathways (Wingler and Lefkowitz, 2020). Allosteric ligands modulate receptors by targeting other pockets than the orthosteric site. Allosteric agonists can stabilize an active receptor conformation even in the absence of an orthosteric agonist, and allosteric antagonists block the activation by an orthosteric agonist (Schwartz and Holst, 2007). Positive allosteric modulators (PAMs) enhance the effect (affinity and/or efficacy) of the orthosteric ligand, whereas negative allosteric modulators (NAMs) have the opposite effect. Unlike PAMs and NAMs, neutral allosteric ligands bind to an allosteric site without affecting the effect of the orthosteric ligand. An ago-allosteric modulator both acts as an agonist and modulates the effect of the orthosteric ligand (Christopoulos et al., 2014; Changeux and Christopoulos, 2016).
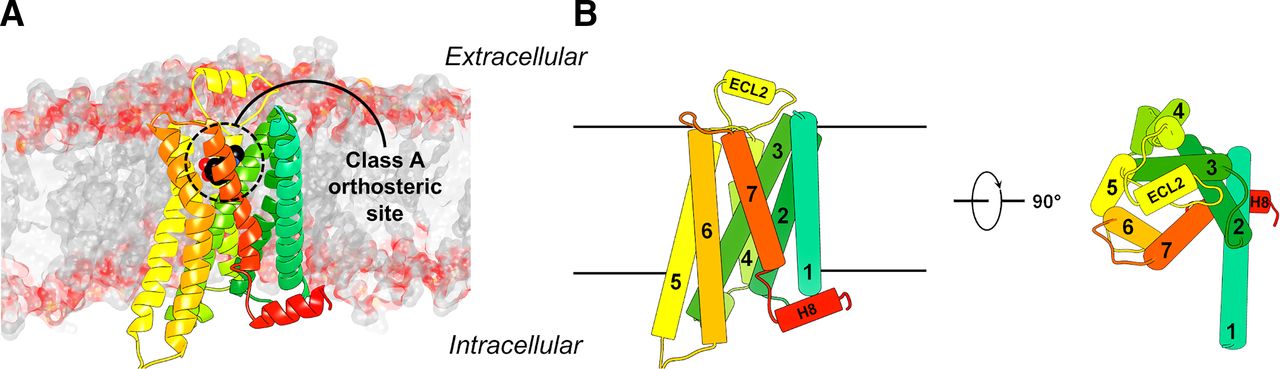
(A) Structure of a class A GPCR embedded in a lipid bilayer. The crystal structure of the β2 adrenergic receptor (Protein Data Bank code: 4LDO) is shown as a cartoon with different colors for the seven helices. The atoms of the orthosteric ligand are shown as spheres. (B) Schematic structure of a class A GPCR.

Overview of the effect of GPCR ligands on receptor response and structural aspects thereof. (A) Effect of GPCR ligands with different modalities on receptor response. Adapted from Ferguson and Feldman (2014). (B) Structures of the β1 adrenergic receptor (β1R, gray cartoon) capturing different states and ligand modalities. From left to right: β1R in complex with the inverse agonist (of the G protein pathway) carvedilol in red (Warne et al., 2012), the agonist isoprenaline (green) and G protein (orange surface) (Su et al., 2020), and the arrestin-biased agonist timolol (light green) and β-arrestin (purple surface) (Lee et al., 2020). The inlays highlight changes in TM5 and TM6 in response to agonist binding and receptor activation. Max, maximum.
GPCRs are divided into six classes based on sequence similarity, of which class A, B, C, and F receptors are found in humans (Fredriksson et al., 2003; Munk et al., 2016). GPCRs consist of seven transmembrane helices (TM1–7) connected by three extracellular loops (ECLs) and three intracellular loops and an intracellular C-terminal helix 8 (H8). The ∼300 nonsensory class A receptors (http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=694) typically have a relatively short and disordered N-terminal region. The orthosteric agonist binding sites of class A GPCRs are located in an extracellularly accessible binding pocket formed by the TM region, N terminus, and extracellular loops (Fig. 1). For class A GPCRs, the Ballesteros-Weinstein numbering scheme for the TM helices facilitates the comparison of different family members (Ballesteros and Weinstein, 1995). Residues in a TM helix (X) are numbered relative to the most conserved amino acid, which is defined as ×.50. In this review, the Ballesteros-Weinstein number of a residue is shown in superscript for class A GPCRs. Class B receptors are divided into secretin (B1) and adhesion (B2) subclasses. The 15 secretin receptors (http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=694) contain a large structured extracellular domain (ECD) that binds the largest portion of their peptide ligands, whereas the N-terminal portion of these ligands interacts with the TM region. The 33 adhesion (B2) GPCRs contain a variety of different large N-terminal domains that are involved in receptor signaling. The 22 class C GPCRs also bind endogenous agonists in large N-terminal domains and form hetero- or homodimers that are required for receptor signaling. Class F has 11 members (10 frizzled receptors and smoothened) that also contain a large N-terminal domain, and activation is controlled by other proteins as well as small-molecule ligands such as cholesterol (Kolakowski, 1994; Lagerstrom and Schioth, 2008; Munk et al., 2016; Pándy-Szekeres et al., 2018).
B. Structural Coverage of the G Protein–Coupled Receptor Superfamily
The first atomic-resolution GPCR structure was the crystal structure of bovine rhodopsin, which was solved in 2000 (Palczewski et al., 2000). It took almost another decade until the first diffusible ligand-bound structures were published: the β2 adrenergic (Cherezov et al., 2007), β1 adrenergic (Warne et al., 2008), and A2A adenosine (Jaakola et al., 2008) receptors. GPCR structure determination was enabled by the development of complementary approaches to stabilize receptors in detergent solution during purification, including receptor thermostabilization by site-directed mutagenesis, the introduction of fusion proteins to facilitate crystal packing during crystallization in lipidic cubic phase, and the use of high-affinity ligands ranging from small molecules to antibodies (Piscitelli et al., 2015; Erlandson et al., 2018). These approaches have revolutionized GPCR structural biology and resulted in the determination of >500 receptor crystal structures. Since 2017, cryo-electron microscopy (cryo-EM) is a complementary technique to solve GPCR structures. Cryo-EM has provided access to active-state conformations coupled to G proteins and large multidomain GPCRs such as class B and C receptors (Garcia-Nafria and Tate, 2020), resulting in 119 structures of 42 different receptors. At the end of March 2021, crystal or cryo-EM structures were available for 93 GPCRs (Fig. 3). The structures cover receptors from all human GPCR classes (A, B1, B2, C, and F) bound to a wide range of compounds (e.g., small molecules, peptides, and proteins) with different pharmacological modes of action, in active and inactive conformational states, and in complex with G proteins or arrestins. This increase of structural information has greatly enhanced the understanding of ligand binding and receptor signaling. Active and inactive structures have shown that agonists induce conserved conformational changes in the intracellular region, which is mainly characterized by an outward movement of TM6. The extracellular region shows more sequence variation, which allows recognition of different molecules. The structural changes in the orthosteric binding site upon activation are surprisingly subtle in most cases (Fig. 2) (Venkatakrishnan et al., 2016; Weis and Kobilka, 2018).

Structural coverage of the GPCR family and successful applications of these structures in molecular docking screens. All receptors with at least one experimental structure are shown on the phylogenetic tree as either circles or stars. Stars indicate receptors for which a successful structure-based virtual screen has been published. The stars and circles are colored according to the modality of the ligand present in the experimental structure (red, agonist or PAM; blue, antagonist or NAM; gold, both modality types). The GPCR tree was produced with the GPCR tree mapper (Kooistra and de Graaf, unpublished) and is based on Fredriksson et al. (2003).
C. The Atlas of G Protein–Coupled Receptor Binding Sites
The 338 determined structures of unique GPCR-ligand complexes reveal distinct binding pockets, ranging from cavities in the ECDs and TM region to pockets at the receptor-membrane interface and in the intracellular G protein binding site. Figures 4 and 5 summarize the different ligand binding sites observed in experimentally determined GPCR structures. The structures and relevant references used in the analysis of GPCR binding sites are summarized in Supplemental Table 1.

Summary of experimental structures of (unique) GPCR-ligand complexes. aAllosteric agonist. bPositive allosteric modulator. cAllosteric antagonist. dNegative allosteric modulator. eMajor pocket. fMinor pocket. gExtracellular vestibule. hIntracellular pocket. iExtracellular domain. jDeep transmembrane pocket.

Binding mode diversity in the TM domain of GPCRs. The binding sites are broadly categorized into the major and minor pockets, main pockets, deep pockets, extrahelical pockets, and the ECV. For each receptor class, all the small-molecule ligands are shown as colored sticks, and a representative receptor structure is shown as a transparent gray cartoon. Only the TM domain and H8 are shown (extracellular domains are not shown). The Protein Data Bank codes for the representative structures are 4BVN, 6X18, 7C7S, and 6O3C for class A, B1, C, and F, respectively.
Most of the ligands identified in experimental structures bind to a common extracellular binding cavity, which comprises residues from the TM helices and the three ECLs (Figs. 4 and 5). This site is the orthosteric pocket of class A and B1 GPCRs and an allosteric pocket in class C and F receptors. The extracellular binding site can be divided into a major pocket (between TM3 and 7), a minor pocket (between TM1 and 3, and TM7), and an extracellular vestibule (ECV; between the N terminus, the ECLs, and the ends of TM1–7). Most of the small-molecule ligands occupying this site are primarily anchored in the major pocket. For example, this type of binding site has been observed in structures of adrenergic (α2A, α2B, α2c, β1, and β2), dopamine (D1, D2, and D3), histamine (H1), muscarinic (M1, M2, M3, M4, and M5), serotonin (5-HT1B, 5-HT2A, and 5-HT2B), adenosine (A1 and A2A), neuropeptide Y (NPY1), neurotensin (NTS1), opioid (DOR and MOR), neurokinin (NK1), chemokine (CXCR4), free fatty acid (FFA1), leukotriene (CysLT1 and CysLT2), prostanoid (DP2), P2Y (P2Y1 and P2Y12), opsin (Rho), melatonin (MT1 and MT2), metabotropic glutamate (mGlu1 and mGlu5), and the frizzled (SMO) receptors. In a few GPCR structures, the minor pocket is primarily involved in ligand recognition, which has been observed in structures of proteinase-activated (PAR1 and PAR2), chemokine (CCR2, CCR6, CXCR2, and CXCR4), leukotriene (BLT1), prostanoid (EP3, EP4, and thromboxane A2), succinate (SUCNR1), GPR52 (a class A orphan), and glucagon (GLP-1) receptors. In many GPCR crystal structures, receptor-ligand contacts involve the major binding pocket in combination with the minor binding pocket. The binding modes of such bitopic compounds have been observed in structures of several GPCRs, e.g., adrenergic (β1 and β2), dopamine (D2 and D4), serotonin (5-HT1B, 5-HT2A, 5-HT2B, and 5-HT2C), melanocortin (MC4), oxytocin (OT), adenosine (A1 and A2A), angiotensin (AT1 and AT2), apelin (APJ), complement peptide (C5a1), endothelin (ETB), formyl peptide (FPR2), opioid (KOR, MOR, and NOP), orexin (OX1 and OX2), lysophospholipid (LPA1 and S1P1), cannabinoid (CB1 and CB2), platelet-activating factor, bile acid (GPBAR), corticotropin-releasing factor (CRF1 and CRF2), glucagon (GLP-1R, GCGR, SCTR, and GHRH), adhesion (ADGRG), and frizzled (SMO) receptors. The major and minor pockets are typically accessible from the extracellular side of the membrane, but some ligands of the free fatty acid receptor (FFA1) access the site from the membrane bilayer (Supplemental Table 1).
Several GPCR ligands interact with binding sites outside the minor and major pockets (Figs. 4 and 5). In structures of class B, C, and F receptors, binding sites in the ECDs have been observed in calcitonin (CTR, CGRP, AM1, and AM2), metabotropic γ-aminobutyric acid (GABAB1 and GABAB2), metabotropic glutamate (mGlu5), and frizzled (SMO) receptors. A few small-molecule ligands were found to target only the ECV, including PAM-bound muscarinic receptors (M1 and M2). Recently, structures of nonpeptide agonists targeting the interface of the TM domain and the ECV of the glucagon-like peptide-1 receptor have been determined. In the corticotropin-releasing factor receptor 1 (CRF1), a NAM locks the receptor into an inactive conformation by targeting a distinct ligand binding pocket that is deeply buried in the core of the TM region and formed by TM3, TM5, and TM6. NAMs binding to a different deep buried pocket formed by TM2–3 and TM5–7 have been identified in metabotropic glutamate receptors (mGlu5). Similarly, a few compounds are anchored in the major/minor binding sites and extend deeply into the TM region in class A, C, and F receptors. The most prominent example among class A GPCRs is the leukotriene (BLT1) receptor structure, in which the ligand extends into a buried pocket and interacts with conserved residue Asp2.50. In class C and F receptors, compounds with such binding modes have been observed for metabotropic γ-aminobutyric acid (GABAB1 and GABAB2), metabotropic glutamate (mGlu5), and Frizzled (SMO) receptors (Supplemental Table 1).
In recent years, binding sites located in the extrahelical TM interface with the membrane bilayer have been identified in structures of class A, B, and C receptors (Figs. 4 and 5). For class A GPCRs, an extrahelical binding pocket located in the intracellular half of TM3–5 accommodates agonists/PAMs of the free fatty acid FFA1, β2 adrenergic, and D1 dopamine receptors as well as antagonists/NAMs of the complement peptide C5a1 receptor, demonstrating that this region is an allosteric hub that can be targeted to stabilize either active and inactive receptor conformations. Additional extrahelical binding sites of NAMs/antagonists involve TM1–3 in the P2Y1 receptor, TM2–4 in the cannabinoid CB1 receptor, and TM2–4 in PAR2. Several extrahelical binding sites have also been observed in class B and C receptors. In a glucagon receptor GLP-1 structure, a PAM bound at the TM1–2 interface was observed to interact directly with the orthosteric peptide agonist. The intracellular half of TM5–7 in glucagon receptor GCGR and GLP-1 structures accommodates allosteric antagonists/NAMs locking TM6 in an inactive conformation. In metabotropic γ-aminobutyric acid (GABAB2) receptor structures, PAMs bind to an extrahelical pocket at the intracellular tip of TM6, which stabilizes the heterodimer interface that is necessary for signaling in class C GPCRs (Supplemental Table 1).
Ligands that bind in the G protein binding site have been identified in class A GPCRs (Figs. 4 and 5). The allosteric antagonist observed in the intracellular binding pockets of β2 adrenergic and chemokine (CCR2, CCR7, CCR9, and CXCR2) receptor structures partially overlap with the G protein binding pocket formed by TM1–3, TM6–7, and H8 (Supplemental Table 1).
II. Molecular Docking: Methodology and Virtual Screening Workflow
The potential of structure-based design was recognized early (Beddell et al., 1976). Rational ligand design using atomic-resolution protein structures has become an important component of the drug discovery process (Sledz and Caflisch, 2018). Molecular docking is a central method in structure-based ligand design and is used both to screen for ligands in chemical libraries and to model protein-ligand interactions in lead optimization (Kitchen et al., 2004; Shoichet, 2004). Based on a protein structure, molecular docking algorithms aim to predict both the binding mode of a ligand and the affinity of the complex. If this would be possible, the technique would revolutionize target-based drug discovery. Instead of screening chemical libraries experimentally by high-throughput screening (HTS), the compounds with the highest affinities would be rapidly identified. Virtual screening can also score compounds that have not yet been synthesized, which makes it possible to consider orders-of-magnitude-larger libraries than empirical screening and reduce synthesis costs (Shoichet et al., 2002). However, docking methods need to sacrifice accuracy for speed to screen large chemical libraries. Currently, docking scoring functions are unable to predict ligand affinities with high accuracy (Warren et al., 2006). Docking should be viewed as a method to filter out unlikely ligands from libraries and thereby enrich active compounds among the top ranked. A vast majority of the predicted ligands should be expected to be inactive, and a hit rate of 5%–10% (i.e., the percentage of active compounds among those evaluated experimentally) is a successful result. Despite the high false-positive rate, molecular docking screening has proven able to identify novel ligands of important targets and is now an established tool that is complementary to other lead discovery methods such as HTS (Irwin and Shoichet, 2016). In this section, we introduce the molecular docking method and describe the practical steps of a structure-based virtual screen.
A. The Molecular Docking Method
Molecular docking predicts the orientation and conformation of a ligand in a receptor binding site and can be divided into two steps: sampling and scoring. In the sampling step, a large number of ligand poses are generated on the receptor surface, resulting in thousands of possible complexes. The most probable structures of the complex, as well as ligand affinity, are predicted in the scoring step. The binding affinity is approximated using simplified scoring functions that are either physics-based or calibrated on experimental data (Kitchen et al., 2004). One of the first docking programs enabling computational prediction of protein-ligand complexes was DOCK, which was developed in the eighties (Kuntz et al., 1982). Today, there are a large number of docking programs with different sampling algorithms and scoring functions. Several widely used docking programs are listed in Table 1.
List of commonly used docking programs
To enable screens of large chemical libraries, docking programs make many approximations in both the sampling and scoring steps. Protein flexibility is generally disregarded completely. The ligand is treated as flexible, but only a limited conformational ensemble can be explored and search space is constrained to a predefined binding site volume. The docking scoring function predicts ligand affinity from a single structure of the complex, and the description of the energy terms contributing to ligand binding are only approximate. Terms capturing receptor-ligand interactions (e.g., hydrogen bonding and van der Waals interactions) are the main components of all scoring functions. Several solvent (e.g., binding site desolvation) and entropic (e.g., from restricting ligand and binding site flexibility in the complex) contributions to ligand binding are not accurately described or neglected. The development of methods to overcome the weaknesses of the docking method remains an active research field, which has been reviewed elsewhere (Leach et al., 2006; Pantsar and Poso, 2018; Salmaso and Moro, 2018; Zhenin et al., 2018). For example, predictions can be improved by using consensus techniques (Feher, 2006), by applying more rigorous scoring methods to a subset of the screened library (Zhong et al., 2010), or through filters such as interaction fingerprints (Deng et al., 2004; Marcou and Rognan, 2007). Binding site plasticity can at least be partially considered by treating side chains as flexible (Ravindranath et al., 2015) or by docking to an ensemble of receptor conformations (Totrov and Abagyan, 2008). Major efforts have also been made to capture contributions from binding site waters. Water molecules identified as important for ligand recognition can be treated as part of the binding site (Verdonk et al., 2005; Huang and Shoichet, 2008), and accounting for energy contribution from water displacement can improve predictions (Abel et al., 2008; Balius et al., 2017).
B. Structure-Based Virtual Screening Workflow
The key steps of a molecular docking screen are 1) design of a chemical library, 2) selection and preparation of the receptor structure, 3) assessment of docking performance, 4) docking screen and compound selection, 5) experimental evaluation, and 6) hit-to-lead optimization. In this section, we describe how to perform a prospective virtual screen and important considerations in each step (Fig. 6).

Workflow of a prospective molecular docking screen. In the first step (1), relevant receptor structures are identified. If experimental structures are not available, computationally derived models can be explored. Retrospective docking screens based on available structures of complexes and known ligands (redocking and ligand enrichment calculations) are used to optimize virtual screening performance. In the second step (2), a chemical library is selected, and the compounds are screened by docking. In the third step (3), top-ranked compounds from the docking screen are analyzed based on the predicted binding modes, novelty, and diversity. A set of compounds are then selected for experimental evaluation. In the fourth step (4), compounds are evaluated in experimental assays. The hit-to-lead optimization step (5) is often iterative and involves testing of several compound sets guided by SAR.

GPCR structures that have been used in prospective molecular docking screens. Each receptor is shown in surface representation with the target pocket colored orange (orthosteric) or green (allosteric), respectively. The co-crystallized ligands are shown as sticks.

Hit rates from molecular docking screens of chemical libraries using crystal structures of GPCRs. Docking was primarily performed to the orthosteric site with the goal to find ligands, i.e., there was generally no clearly defined goal regarding the properties of the compounds (circles). A few studies explored strategies to identify agonists (squares), allosteric modulators (triangles), multitarget ligands (diamonds), and selective ligands (crosses). Hit rates are based on the definition of actives in each individual study.

Comparison of models and crystal structures of GPCR-ligand complexes. Top-scoring models from the GPCR Dock assessments, which challenged researchers to predict both the receptor structure and ligand binding mode, are shown. The receptor is shown as a cartoon, and the ligand is shown as sticks. The predicted and experimentally determined structures are colored green and gray, respectively. Each model was aligned to the relevant crystal structure (Protein Data Bank codes: D3R/eticlopride: 3PBL; 5-HT1BR/ergotamine: 4IAR, 5-HT2BR/ergotamine: 4IB4, A2AR/ZM241385: 3EML, SMO/SANT-1: 4N4W, SMO/LY-2940680: 4JKV, CXCR4/IT1t: 3ODU). The coordinates of the models were either obtained from the GPCR Dock assessment or directly from the participating researchers.
1. Design of a Chemical Library
The screening library defines the region of chemical space that will be explored by the docking screen. Typical sources of screening libraries are in-house collections and commercially available compounds. Examples of online resources for commercial chemical libraries are shown in Table 2. Several million compounds are currently available in-stock, e.g., in the ZINC database (Irwin et al., 2020), and can be delivered immediately by chemical suppliers. Make-on-demand libraries with readily synthesizable molecules provide additional coverage of chemical space. Alternatively, virtual libraries can be generated in-house based on available building blocks. Commercial make-on-demand libraries from chemical suppliers currently reach >20 billion compounds, which can be custom-synthesized and delivered within 1 month, e.g., the Enamine REAL Space database (Hoffmann and Gastreich, 2019; Grygorenko et al., 2020).
Compound databases for virtual screening and hit-to-lead optimization
The design of a chemical library depends on several factors. The ultimate goal of many virtual screening campaigns is to identify a high-affinity compound with drug-like properties, e.g., as defined by Lipinski’s rule of 5 (Lipinski et al., 2001). The choice of chemical library determines the path to identify the ligand. Screening libraries are typically generated based on a set of physicochemical properties, e.g., the calculated octanol/water partition coefficient (log P) and molecular weight. Subsets of chemical libraries with fragment- and lead-like properties are common starting points for ligand discovery (Hann and Oprea, 2004; Keserű et al., 2016). Fragment-based drug discovery initially focuses on compounds that are approximately half the size of a drug (mol. wt. <250 Da). Screens of fragment libraries result in higher hit rates (∼2%–8%) than HTS, but the identified ligands will usually have weak affinities, and optimization is required to obtain a potent lead (Erlanson et al., 2016; Keserű et al., 2016). Libraries with lead-like compounds (mol. wt. ∼250–350 Da) are closer to the final drug-like molecule (mol. wt. <500 Da) in property space, and high-affinity ligands can emerge directly from a screen, but at the expense of hit rate and chemical space coverage (Teague et al., 1999). In addition to physicochemical property filters, the library can be tailored for a specific target by using information about known ligands and the binding site. This can be accomplished using ligand-based virtual screening methods (e.g., pharmacophore, quantitative structure-activity relationship, or chemical similarity methods), which have been reviewed elsewhere (Cherkasov et al., 2014; Muratov et al., 2020). Target-focused libraries (Harris et al., 2011) can increase screening hit rates and reduce the computational cost of the docking screen but may reduce the chances of identifying novel chemotypes. Another important step in library design is the removal of molecules that are likely to cause experimental artifacts. Chemotypes that lead to false positives because of assay interference or colloidal aggregation (McGovern et al., 2002; Baell and Holloway, 2010) have been identified and can to some extent be filtered out using cheminformatics tools. Useful webservers for identifying such compounds are listed in Table 3. The ZINC database provides pregenerated fragment and lead-like chemical libraries from several suppliers (Irwin et al., 2020).
Web-based services that predict frequent hitters
2. Selection and Preparation of the Receptor Structure
The receptor structure must be carefully prepared for a docking screen based on the goal of the study (e.g., ligand mode of action and selectivity profile). The quality of the receptor structure (e.g., resolution), the conformational state (active or inactive), and pharmacological properties of bound ligands should be considered. In some cases, the receptor structure needs to be refined by adding missing atoms (e.g., unresolved side chains, loop regions, and hydrogens) and reverting sequence modifications made to facilitate structure determination (e.g., mutations). If crystallographic waters are present in the receptor structure, the roles of these in ligand recognition should be analyzed in detail. In high-resolution structures, an average of four to five ordered water molecules interact with the ligand (Lu et al., 2007), and particular attention should be given to those that bridge receptor-ligand interactions. Prior to the virtual screen, docking performance of structures prepared in the presence and absence of binding site waters can be compared in retrospective tests (Huang and Shoichet, 2008). Protonation states of ionizable side chains (e.g., histidines), which can substantially affect the docking results, need to be assigned carefully. Assignment of protonation states can be guided by computational prediction of side chain pKa values (Bas et al., 2008). In some cases, energy minimization or sampling of low-energy conformations of the receptor with molecular dynamics (MD) or Monte Carlo simulations may also be relevant. The binding site is typically defined based on the ligand present in the experimental structure, but any surface on the protein can in principle be screened. If experimental structures are not available, computational methods can be used to create a binding site model. As a large number of GPCR structures are now available, receptor models can be created based on these as templates using homology modeling, e.g., with the program MODELLER (Webb and Sali, 2016). Several online resources for GPCR homology modeling are shown in Table 4. The GPCR database (https://www.gpcrdb.org) provides precalculated models of receptors in different activation states (Pándy-Szekeres et al., 2018).
Webservers for homology modeling of GPCRs
3. Assessment of Docking Performance
The performance of the docking protocol should be evaluated in several retrospective tests to minimize the number of false positives from the prospective virtual screening campaign. In a redocking calculation, the ability of the docking algorithm to reproduce the experimental binding mode of a ligand is evaluated, which can be quantified based on the root-mean-square deviation (RMSD). A redocking RMSD of <2 Å that captures key polar interactions is considered successful (Vieth et al., 1998). Another important test is to evaluate if actives are likely to be enriched among the top-ranked compounds from the docking screen (Huang et al., 2006). Such retrospective evaluations can be performed if there is a set of known ligands of the target. Sets of experimentally verified actives and nonbinders (decoys) of many GPCRs can be collected from the ChEMBL (Mendez et al., 2019) or the PubChem BioAssay databases (Wang et al., 2012). As there are generally too few experimentally confirmed nonbinders, presumed decoys from the Directory of Useful Decoys-Enhanced (DUD-E) database (http://dude.docking.org/generate) can be used (Mysinger et al., 2012a). By retrospectively evaluating different receptor structures and parameters, the docking protocol with the greatest ability to enrich ligands can be identified.
4. Docking Screen and Compound Selection
The speed of docking algorithms makes it possible to screen large chemical libraries efficiently. Even if several hundred million compounds are considered, which would take years to complete on a desktop computer, a screen can be performed in a few days on a high-performance computing cluster, which is available at most research institutions today (Lyu et al., 2019; Gorgulla et al., 2020). The result of a docking screen is a list of compounds ranked by score and their predicted binding modes. The next step is to select the best candidates for experimental evaluation from a top-ranked set of compounds. As one of the main objectives of structure-based virtual screens is to discover novel chemotypes, compounds that are too similar to known ligands can be excluded based on 2D similarity. The top-ranked compounds can also be clustered to facilitate the selection of a diverse set to test experimentally. The final set of compounds is rarely selected solely based on computational scoring. Instead, the binding modes for the set of top-ranked compounds remaining after the filtering steps are visually inspected (Fischer et al., 2021). The binding mode and interactions with the receptor are analyzed in detail, allowing the scientist to exclude compounds that are top-ranked because of weaknesses of the docking method or errors in library preparation. Common problems in the preparation of chemical libraries are the assignment of unlikely tautomerization and protonation states. Weaknesses in the scoring function can involve the balance between different binding energy contributions. The impact of certain interactions (e.g., hydrogen bonds) may be exaggerated, and several important energy terms contributing to binding are neglected, e.g., ligand strain energy (Tirado-Rives and Jorgensen, 2006). The number of compounds to select for experimental evaluation depends on the expected hit rate. If the target is expected to be challenging, it may be necessary to test a large number of compounds to find a ligand. For GPCRs, high hit rates of >25% have been achieved for several targets (de Graaf et al., 2011). In such cases, relatively small sets of only 10–20 compounds will result in the discovery of several ligands.
5. Experimental Evaluation of Predicted Ligands
The therapeutic relevance of GPCRs has led to the development of a large number of screening assays to identify ligands, which have been reviewed elsewhere (Zhang and Xie, 2012; Jacobson, 2015). Binding assays (e.g., using radiolabeled ligands) are common to evaluate the affinity of a compound for the orthosteric site and identify ligands with different efficacy profiles (e.g., agonists, antagonists, and inverse agonists) in a single experiment. If weak ligands are expected (e.g., in fragment screens), a sensitive biophysical screening method such as surface plasmon resonance (SPR) or nuclear magnetic resonance (NMR) can be advantageous (Congreve et al., 2011; Aristotelous et al., 2013; Shepherd et al., 2014). Compounds are initially tested at a single concentration to identify the most active, which is followed by full dose-response curves for the best hits to determine affinities. To assess the pharmacological properties of discovered ligands, binding assays can be complemented by functional experiments measuring activation of G protein–dependent and –independent signaling pathways. As compounds may not have the same activity in different pathways, investigation of effects in multiple assays is necessary to fully understand their properties (Smith et al., 2018). Finally, controls should be made to exclude false positives in the assays (McGovern et al., 2002; Baell and Holloway, 2010) and to verify compound identity and purity.
6. Hit-to-Lead Optimization
Hit-to-lead optimization will not be described in detail, as it is a complex phase of ligand discovery that aims to identify compounds with improved target affinity, efficacy, and selectivity, as well as absorption, distribution, metabolism, excretion, and toxicity properties (Hughes et al., 2011). The first step after discovering ligands from a virtual screen is to obtain structure-activity relationships (SAR). Analogs with a similar chemical structure as the hits can be identified in commercial chemical libraries or be synthesized in-house. Examples of online resources for identifying analogs are shown in Table 2.
III. Success Stories: Discovery of G Protein–Coupled Receptor Ligands by Molecular Docking Screening
In this section, structure-based docking screens using experimental GPCR structures are summarized, and we also highlight representative studies based on homology models. GPCRs targeted in structure-based virtual screens using experimental structures are shown in Figures 3 and 7. Statistics from these virtual screens are presented in Table 5. We identified 62 successful virtual screens for 22 unique targets belonging to 14 different receptor families (Fig. 3; Table 5). In all of these studies, at least one receptor crystal structure was used in the screen. The targets were class A GPCRs, except in the case of SMO, which belongs to class F. Commercially available chemical libraries with lead-like compounds were primarily used. The sizes of these libraries were typically in the order of a few million molecules, but they also reached >100 million in three cases targeting melatonin, dopamine, and cysteinyl leukotriene receptors. Several studies focused on docking of fragment libraries to adenosine, dopamine, histamine, melatonin, chemokine, and neurotensin receptor structures. Docking was primarily performed to the orthosteric site with the goal to find ligands, i.e., there was generally no clearly defined goal regarding the efficacy or selectivity of the compounds. However, in a few studies on the adenosine, adrenergic, and muscarinic receptors, attempts were made to bias the screening results toward the discovery of agonists. Several strategies to identify subtype-selective ligands were also explored (adenosine, chemokine, dopamine, muscarinic, opioid, and serotonin receptors), and docking to several structures allowed the identification of multitarget ligands (adenosine, dopamine, and chemokine receptors). Four screens focused on docking to allosteric binding pockets (adrenergic, dopamine, free fatty acid, and muscarinic receptors). All these virtual screens successfully identified ligands, but there was a large variation in hit rate and size of the screened libraries (Fig. 8).
Summary of molecular docking screening results using experimental GPCR structures of the target. Screens focused on hit optimization were omitted.
A. Adenosine Receptors
There are four adenosine receptor subtypes (A1, A2A, A2B, and A3), which are activated in response to organ or tissue stress. These GPCRs have been explored as therapeutic targets for a wide range of conditions, e.g., cancer, cardiovascular, and neurodegenerative diseases (Chen et al., 2013b). The first crystal structure of the A2A adenosine receptor (A2AR) was determined in 2008 (Jaakola et al., 2008) and revealed the binding mode of an antagonist in the major pocket. Subsequently, a large number of structures of the A2A (Jacobson et al., 2020) and A1 subtypes (Cheng et al., 2017; Glukhova et al., 2017; Draper-Joyce et al., 2018) in complex with agonists and antagonists were determined (Figs. 3 and 4; Supplemental Table 1). Several structure-based ligand design studies have been performed, and these primarily focused on the A2AR (Table 5).
Shortly after the release of the first A2AR crystal structure, two research groups screened commercial chemical libraries against the orthosteric binding site (Carlsson et al., 2010; Katritch et al., 2010). Katritch et al. (2010) optimized the docking performance by refining side chains and inclusion of crystal waters, followed by a virtual screen of 4.3 million drug-like compounds. Compound selection was guided by clustering of a top-ranked subset using 2D similarity. From the set of 56 selected compounds, 23 had Ki values ranging from 32 nM to 2.9 μM (hit rate = 41%). Carlsson et al. (2010) screened 1.4 million commercially available compounds using molecular docking. From the top-ranked 500 compounds, a set of 20 were selected and evaluated in binding assays. Seven of the predicted ligands were experimentally confirmed (hit rate = 35%). The most potent compound was an antagonist and had a Ki value of 200 nM. Subsequently, virtual screens have been carried out by other research groups, leading to the discovery of additional antagonist scaffolds (Table 5) (Lenselink et al., 2016a; Tian et al., 2017; Ballante et al., 2020). In all these studies, the ligands were predicted to primarily occupy the major binding pocket and form hydrogen bonds to Asn2536.55.
Wei et al. (2020) screened 1.5 million commercially available compounds to identify A1R antagonists by combining ligand-based methods with molecular docking. Ligand-based techniques (i.e., random forest classification and pharmacophore models) were initially used to identify compounds similar to A1R antagonists, which reduced the database from 1.5 million to 19,000 compounds. This set was then docked to an A1R structure in an inactive conformation and evaluated using three different scoring methods. The 1139 top-ranked compounds were clustered and visually inspected, which led to the selection of 22 compounds. In total, 18 of these were confirmed to be A1R ligands in binding assays (hit rate = 82%). Compound 15 showed the highest affinity (pKi = 7.13) and was >1000-fold selective for the A1R over the A2A subtype. This ligand was predicted to form key interactions with Asn2546.55 and Phe171EL2. Six compounds were evaluated in functional assays, and all of these were A1R antagonists. Compound 15 was similar to a known antagonist scaffold, but several other ligands represented novel chemotypes.
All hits from the first docking screens targeting the A2AR that were tested for functional activity were antagonists, which agreed with the fact that the crystal structure had been determined in an inactive conformation. When structures of complexes with agonists were determined, Rodriguez et al. (2015) performed a docking screen of 6.7 million compounds to investigate whether novel agonists could be identified. A set of compounds that formed interactions similar to agonists (e.g., with Ser2777.42 or His2787.43) and had a better ranking in active than inactive structures was prioritized for experimental testing. Of the 20 tested compounds, nine were ligands and the best compound (17) had an affinity of 16 nM (hit rate = 45%). However, functional assays showed that none of the discovered compounds activated G protein signaling via the A2AR.
Chen et al. (2013a) explored fragment libraries to identify A2AR ligands. A library with 328,000 commercially available compounds was docked to the orthosteric site of the A2AR in an inactive conformation. In total, 22 fragments were evaluated in binding assays, and 14 showed activity with Ki values between 2 and 240 μM. The fragment hit rate of 64% was substantially higher than that obtained using traditional fragment screening approaches for the same target (Chen et al., 2012). Fragment-to-lead optimization was focused on two of the discovered ligands and was based on the identification of similar compounds from commercial libraries in combination with docking and MD simulations, leading to a 3-fold improvement of affinity (compound 12c, Ki = 2.1 μM).
Two studies explored target-focused virtual libraries to design A2AR agonists. In contrast to the docking screens described above, commercial chemical libraries were not used. Instead, commercial building blocks that could be used to synthesize compounds were first identified. A virtual library of the products that could be obtained based on these building blocks was then generated and screened by molecular docking. Rodriguez et al. (2016) designed a virtual library with 6784 compounds to identify adenosine-based agonists scaffolds with novel bases. This library was docked to an A2AR crystal structure and homology models of the A1 and A3 subtypes. Thirteen compounds were synthesized, and two of these were ligands with submicromolar affinities for the A2AR, which were similar to adenosine. Among the hits for the other two subtypes, there were several atypical adenosine receptor agonist scaffolds (e.g., compound 14, Ki = 359 nM for A1R). In the second study, Tosh et al. (2012) focused on the design of 5′-substituted adenosine-based agonists. A computationally generated library based on 2000 building blocks was docked to an A2AR crystal structure to guide the identification of new substituents. In total, 16 predicted analogs were then synthesized and evaluated experimentally, leading to the identification of several high-affinity agonists.
Jaiteh et al. (2018) and Kampen et al. (2021) investigated whether multitarget compounds could be predicted by molecular docking screening. In the first study, the A2AR and monoamine oxidase B (MAO-B), which are relevant for the development of drugs against Parkinson’s disease (Meissner et al., 2011), were selected as targets. Docking screens of fragments (0.8 million compounds) and lead-like molecules (4.6 million compounds) to crystal structures of both targets were carried out, and a consensus score was used to predict 24 dual-target ligands. Experimental evaluation of these identified four compounds with activity at both targets (dual-target hit rate = 17%). Structure-guided optimization based on commercial chemical libraries resulted in two dual-target scaffolds, which antagonized the A2AR and inhibited MAO-B with submicromolar activities. The best compound (3) had a Ki value of 19 nM for the A2AR and inhibited MAO-B with an IC50 of 100 nM. In the second study, Kampen et al. (2021) used crystal structures of the A2AR and a homology model of the D2R to design dual-target ligands relevant for the development of antiparkinson drugs. A focused virtual library (10,535 compounds) was designed based on a dopamine receptor agonist and the binding site structures of both targets. Docking screens guided selection of 10 compounds binding to the orthosteric and secondary binding pockets, which were synthesized and tested experimentally. Three compounds with affinity for both the A2AR and D2R were identified (dual-target hit rate = 30%). Structure-guided optimization of affinity and efficacy led to the discovery of a series of potent dual-target compounds that antagonized the A2AR and activated the D2R. The best compound (Ki values of 160 and 370 nM at the A2AR and D2R, respectively) was also demonstrated to exert a therapeutic effect in a rat model of Parkinson’s disease.
Prior to the release of the first A2AR crystal structure, the modeling community was challenged to predict the receptor structure and binding mode of the antagonist ZM241385 (Michino et al., 2009). The goal of the GPCR Dock assessment was to evaluate whether structures of receptors and complexes with drugs could be modeled accurately. Analysis of the results of GPCR Dock showed that prediction of drug binding using homology models was very challenging, but a few groups were able to generate good models (Fig. 9). The ligand RMSD of the best model was 2.8 Å, and 45% of the receptor-ligand contacts were captured. After the A2AR crystal structure was published, several virtual screening studies have used homology models of other adenosine receptor subtypes to identify ligands in chemical libraries (Kolb et al., 2012; Ranganathan et al., 2015). As there is strong conservation of residues in the binding site, good models of the A1 and A3 subtypes based on the A2AR structure could be obtained. In a docking screen performed by Kolb et al. (2012), a model of the A1R was used to identify several ligands (hit rate = 21%). Ranganathan et al. (2015) used models of both the A1R and A3 adenosine receptor to identify A3-selective ligands using virtual screening of a fragment library (hit rate = 38%), followed by optimization of these to potent antagonists.
The development of the A2AR antagonist HTL-1071 illustrates how GPCR structures can guide drug discovery. Structure-based virtual screening using A2AR homology models led to the discovery of starting points for the design of potent antagonists (Langmead et al., 2012). The development of thermostabilized mutants of the A2AR enabled mapping of ligand binding modes by combining molecular modeling, mutagenesis, and biophysical screening (Zhukov et al., 2011). The predicted binding mode of one of the chromone-based ligands was later confirmed by crystallography (Fig. 10A) (Langmead et al., 2012; Mason et al., 2013; Jespers et al., 2020). A discovered 1,2,4-triazine antagonist scaffold was further optimized, leading to the identification of HTL-1071, which is being explored for the treatment of cancer in clinical trials by AstraZeneca (Congreve et al., 2012).

Experimentally confirmed binding models of ligands discovered from prospective molecular docking screens. (A) A2AR in complex with a compound discovered by Langmead et al. (2012) (Mason et al., 2013). Protein Data Bank (PDB) code of crystal structure: 6ZDR (Jespers et al., 2020). (B) β2R in complex with compound identified by Kolb et al. (2009). PDB code of crystal structure: 3NY9 (Wacker et al., 2010). The receptor is shown as a cartoon. The ligand and selected amino acid side chains are shown as sticks. The predicted and experimental binding mode of the ligand is colored green and gray, respectively.
B. Adrenergic Receptors (Adrenoceptors)
The adrenoceptors are classified into α and β adrenergic receptors, which consist of six and three subtypes, respectively. Experimental structures (Figs. 3 and 4; Supplemental Table 1) are currently available for five of the adrenergic receptors (α2A, α2B, α2C, β1, and β2). Several structure-based virtual screens focusing on the β2 adrenergic receptor (β2R) have been carried out, which illustrate strategies to identify agonists and antagonists from different types of chemical libraries (Table 5).
The β2R is a key target for drugs used in the treatment of asthma and cardiovascular diseases (Wang et al., 2018; Wendell et al., 2020). This receptor was also the focus of the first successful docking screen against a GPCR crystal structure. Currently, there are >60 determined structures of the adrenergic receptors, including structures in complex with different ligand types (agonists, inverse agonists, antagonists, and allosteric modulators) and with both G protein and β-arrestin (Figs. 3 and 4; Supplemental Table 1). Shortly after the release of the first structure of the receptor in an inactive conformation with an inverse agonist bound to the major pocket (Cherezov et al., 2007), two research groups docked chemical libraries to the orthosteric site and prioritized 150 (Sabio et al., 2008) and 25 (Kolb et al., 2009) of the top-ranked compounds for experimental testing, respectively. Both screens resulted in high hit rates of 21% (31 ligands) and 24% (six ligands), respectively. These results can be compared with a random selection from a chemical library, which was evaluated by Sabio et al. (2008) and resulted in a hit rate of only 0.3% (one hit out of 320 tested compounds). Several compounds discovered from the virtual screens had submicromolar affinities and represented novel chemotypes. The most potent compound (1) identified by Kolb et al. (2009) had a Ki value of 9 nM, and the predicted binding mode was also confirmed by a high-resolution crystal structure (Fig. 10B) (Wacker et al., 2010). Similar to many other β2R ligands, this compound formed a salt bridge with Asp1133.32 and extended toward TM5 with an aromatic moiety. In a follow-up study, similarity and substructure searches in combination with docking were used to identify analogs, which provided SAR and identified several additional high-affinity ligands (Schmidt et al., 2017). A large number of crystal structures of the β2R in inactive conformations are now available, and docking screens of lead-like libraries using alternative binding site conformations identified additional antagonist scaffolds (Table 5). For example, Scharf et al. (2020) predicted 27 ligands, and one of these showed activity, corresponding to a hit rate of 4%.
An interesting observation from the first docking screens was that the hits were inverse agonists, which was proposed to be a result of the fact that the used structure was determined in an inactive conformation (Kolb et al., 2009). The determination of β2R structures in active conformations (Rasmussen et al., 2011) allowed the investigation of whether agonists could be identified from docking screens. Several docking campaigns with the aim to identify β2R agonists were performed. Weiss et al. (2013) demonstrated that the active receptor structure, in contrast to the inactive, enriched agonists well and could distinguish agonists from inverse agonists. A library of 3.1 million compounds was docked to β2R structures determined in active and inactive states. Two criteria were used to select agonists among the top-ranked compounds from the docking to the active conformation. Compounds had to be predicted to bind stronger to the active state and form polar interactions similar to agonists (e.g., with serine residues in TM5). Out of the 22 compounds that were predicted to be agonists, six activated the β2R, corresponding to a hit rate of 27%. The four most potent ligands resembled catechol-based β2R agonists, whereas the two remaining compounds represented novel scaffolds. An alternative approach to identify β2R agonists was used by Kooistra et al. (2016). In this study, an energy-based docking scoring function was combined with a structural interaction fingerprint similarity scoring method to selectively identify agonists. Retrospective virtual screens based on multiple crystal structures allowed for the selection of optimal combinations of receptor-agonists interaction fingerprints, receptor conformations, and scoring cutoffs for the identification of β2R agonists. A prospective screen of ∼0.1 million compounds led to experimental testing of 63 compounds, and 41% of these showed activity. The most potent compound (48) was a partial agonist (Emax = 60%) with an EC50 of 37 nM and was similar to known β2R agonists. Most of the other hits were novel partial agonists with micromolar activity. More recently, Scharf et al. (2019) docked 3.8 million compounds to two active β2R conformations. Compounds were primarily selected based on high ranking in one or both screens. Of the 19 compounds that were evaluated experimentally, eight (42%) were β2R agonists. All the discovered agonists were based on an N-methylphenylethanolamine (halostachine) scaffold, which is a substructure of adrenaline.
One of the first applications of fragment-based drug discovery to GPCRs was a screen of a fragment library against a thermostabilized form of the β1 adrenergic receptor (β1R) (Christopher et al., 2013). The starting point was an experimental screen of a fragment library with 650 compounds using SPR, which yielded two promising hits with affinities of 5.6 and 16 μM. Analysis of β1R crystal structures and docking guided the selection of analogs, and several of these were experimentally validated as high-affinity ligands. Crystal structures were solved for complexes with two optimized ligands. The compound with the highest affinity (19, Ki = 67 nM) formed a salt bridge with Asp1213.32 and had an indole moiety interacting with Ser2115.42.
Chevillard et al. (2018) developed a computational fragment-growing approach to optimize β2R ligands based on the design of focused virtual libraries. The study was initiated from five fragments that were predicted to bind to the major binding site of the β2R and form salt-bridge interactions with key residue Asp1133.32. Guided by the models of these complexes, the authors identified a secondary pocket where the fragments could be grown into by reacting them with commercial building blocks. The set of building blocks was then docked to the secondary binding pocket to identify those that could be connected to the fragment ligands. The most promising products were then docked to the binding site to guide the selection of compounds. This protocol identified eight synthetically accessible products, leading to four ligands with up to 40-fold improved experimental affinities compared with the initial fragments (compound 11, Ki = 520 nM).
Chevillard et al. (2019) developed a computational protocol to identify ligands in a virtual library by docking to β2R crystal structures determined in active and inactive conformations. The docking was carried out in several steps and included ∼77,000 compounds that could be synthesized from commercial building blocks using two different reactions. A set of 240 compounds were selected for synthesis, and 127 of these were successfully obtained. Binding affinities for the β2R stabilized in active and inactive conformations by nanobodies were determined for 15 compounds. Of these, 12 displayed KD values <200 μM (hit rate = 9%). Additional docking and synthesis resulted in a second set of 30 compounds, which led to a 40-fold improvement of affinity (0.5 μM, compound redA05B51) for the β2R stabilized in an active state.
One structure-based screen focusing on the identification of allosteric modulators of the β2R was carried out (Liu et al., 2020). A commercial chemical library was docked to a pocket in the extracellular vestibule that is occupied by part of the agonist salmeterol. One of the hits from the screen acted as a weak NAM and was further optimized for activity. However, the determination of a crystal structure with one of the optimized NAMs revealed that the compound did not bind to the pocket predicted by the docking screen. Instead, the compound was found to occupy a completely novel site in the TM region that faces the membrane (Fig. 11).
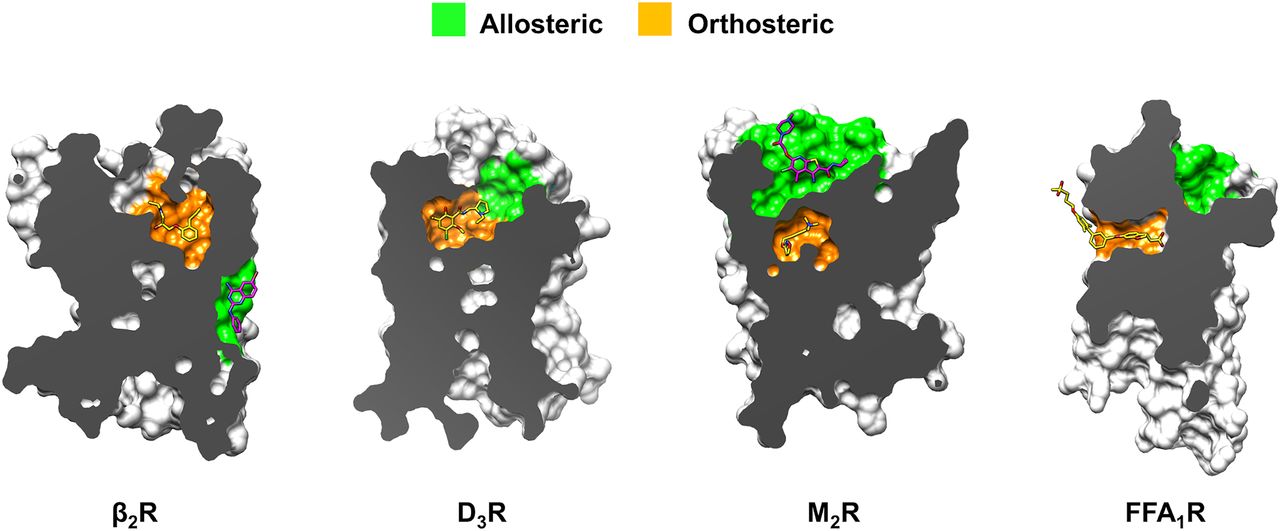
Targets of molecular docking screens for allosteric modulators. Receptors are shown in surface representation with orthosteric and allosteric sites colored orange and green, respectively. Structure-based docking screens successfully discovered ligands of allosteric pockets identified in crystal structures of the D3R, M2R, and FFA1R (pockets shown in green). Screens for allosteric modulators of the β2R unexpectedly identified a NAM binding to an extrahelical pocket, but the docking was carried out to a pocket located in the ECV (Fig. 7).
Docking screens using homology models of adrenergic receptors have also been carried out. Szőllősi et al. (2015) performed a parallel computational and experimental fragment screen of a library with 3071 compounds. The binding site structure of the α2C adrenergic receptor was modeled based on a β2R crystal structure. Docking to the homology model was able to identify five of the 16 experimentally confirmed ligands in the library among the 30 top-ranked compounds, corresponding to a 13-fold better result than expected from random selection.
C. Chemokine Receptors
Chemokine receptors are important regulators of inflammation and immune cell behavior. This group is composed of 23 GPCRs, which represent promising drug targets for immune-related pathologies, e.g., multiple sclerosis, rheumatoid arthritis, human immunodeficiency virus-1 infection, and cancer (Zhao et al., 2019). In 2010, the first crystal structure of a chemokine receptor was determined, revealing a peptide and small molecule bound to the orthosteric site (Wu et al., 2010). Recently, several additional chemokine receptor structures have been solved in complex with both orthosteric and allosteric ligands (Figs. 3 and 4; Supplemental Table 1). Structure-based virtual screens have primarily focused on the chemokine receptor CXCR4 using fragment and lead-like libraries, and explored the prediction of ligand selectivity and polypharmacology (Table 5).
Mysinger et al. (2012b) docked a library of 4.2 million lead-like compounds to the orthosteric site of a CXCR4 crystal structure in an inactive conformation. The top-ranked compounds were visually inspected, and molecules that interacted with Glu2887.39 were prioritized for experimental evaluation. In total, 23 top-ranked compounds were selected and assayed. Of these, four were antagonists in functional assays with IC50 values ranging from 55 to 77 μM (hit rate = 17%). The performance of docking to the CXCR4 crystal structure was compared with that of a virtual screen against a homology model, which was performed prior to the release of the experimental structure. A set of 24 compounds was selected based on a docking screen of 3.3 million compounds. Of these, one compound was active and had an IC50 value of 107 μM, corresponding to a hit rate of 4%.
Adlere et al. (2019) used a structure-based virtual screening approach to identify fragment-like molecules forming ionic interactions with Asp972.63 and Glu2887.39 in the minor binding pocket of CXCR4. A focused fragment library composed of 52,500 compounds that fulfilled a pharmacophore based on two known CXCR4 antagonists was screened using docking and structural interaction fingerprints. The top-ranked 200 compounds were visually inspected, and 23 fragments with low chemical similarity to known CXCR4 ligands were purchased and tested in binding experiments. Four hits showed >50% inhibition of endogenous chemokine binding at 63 μM (hit rate = 17%). One hit was used as a starting point for ligand optimization and SAR exploration, resulting in CXCR4 antagonists (compounds 13, 22, and 28) with submicromolar affinities (IC50) and micromolar functional potencies. Structural information and three-dimensional quantitative structure-activity relationship studies enabled the identification of important interactions and ligand binding mode features that could explain affinity and antagonist efficacy.
Mishra et al. (2016) screened a commercial GPCR-focused library (13,000 compounds) using both ligand- and structure-based approaches to search for CXCR4 ligands. The structure-based screen was performed using two different docking programs. Based on docking scores and favorable ligand-protein interactions, nine diverse compounds were evaluated experimentally. In addition to the structure-based predictions, six compounds were selected from the ligand-based screen. Two antagonists and one agonist were identified from the structure-based screen (hit rate = 33%), whereas one antagonist and one agonist were confirmed from the ligand-based screen. The antagonists showed IC50 values ranging from 0.3 to 3 µM. Exploration of chemical space around one discovered agonist (NUCC-390) allowed to identify additional CXCR4 ligands and rationalize differences between agonist and antagonist ligands.
Schmidt et al. (2015) performed docking screens to identify dual binders of CXCR3 and CXCR4 as well as subtype-selective ligands for each target. Simultaneous modulation of CXCR3 and CXCR4 represents a promising strategy to maximize the pharmacological effect. A commercial chemical library with 2.4 million lead-like molecules was screened against a homology model of CXCR3 and a crystal structure of CXCR4. The top-ranked 500 compounds were visually inspected, and a dual-target ranking protocol was applied to identify scaffolds with the potential to interact with both targets. In total, 17 compounds were selected for experimental evaluation. Of these, seven and six were predicted to be CXCR3- and CXCR4-selective, respectively, and the remaining four were predicted to be dual-target modulators. Four out of seven compounds were confirmed as CXCR3-selective ligands (hit rate = 57%) that inhibited G protein activation (KB = 12 nM for the most potent compound, 3) with no detectable activity at CXCR4. Three out of six compounds were confirmed selective for CXCR4 (hit rate = 50%, KB = 66 nM for the most potent compound, 9). Out of the four compounds predicted as dual binders, two modulated both receptors (hit rate = 50%). Six additional compounds were evaluated to explore the SAR of a dual-target scaffold. All six analogs were confirmed to be dual binders, with KB values ranging from 2 to 300 nM.
In the GPCR Dock 2010 assessment, the modeling community was challenged to blindly predict the structure of CXCR4 in complex with a small-molecule ligand and a peptide (Kufareva et al., 2011). At this time, the best available templates had 25% sequence identity to CXCR4, making the homology modeling process challenging. Only a few research groups were able to identify the right binding site of the small-molecule ligand. The best model only captured 36% of the receptor-ligand contacts with a ligand RMSD of 4.88 Å (Fig. 9).
D. Dopamine Receptors
Dopamine receptors are drug targets for the treatment of neuropsychiatric and neurodegenerative disorders, e.g., schizophrenia and Parkinson’s disease (Missale et al., 1998). There are five receptor subtypes and experimental structures of both D1-like (D1R) and D2-like receptors (D2-, D3-, and D4R) (Figs. 3 and 4; Supplemental Table 1). Docking screens against three receptor subtypes illustrate different strategies to identify orthosteric and allosteric GPCR ligands by virtual screening of chemical libraries of varying size and composition (Table 5).
Carlsson et al. (2011) docked a library with 3.6 million commercially available lead-like compounds to the orthosteric site of the D3R crystal structure determined in an inactive conformation. Among the top-ranked compounds, 25 ligands were predicted and experimentally evaluated in binding assays. Five compounds had binding affinities better than 10 μM (hit rate = 20%), and all of these were confirmed to be antagonists. The discovered ligands were predicted to bind in the same pocket as the cocrystallized ligand and to establish a salt bridge with Asp1103.32. The compound with the highest affinity had a Ki of 300 nM (compound 28). A major finding of this study was the comparison of these results to a screen against a D3R homology model based on adrenergic receptor crystal structures. As this screen was carried out prior to the release of the D3R crystal structure, the study could compare the ability of GPCR homology models and crystal structures to identify ligands. The homology model screen resulted in a comparable hit rate (23%) and yielded ligands with affinities similar to those discovered using the crystal structure.
Lane et al. (2013) used docking screens of commercial chemical libraries to discover orthosteric and allosteric ligands of the D3R. The receptor structure was first optimized for virtual screening guided by docking of actives and decoys. In this step, small variations of backbone and side chain rotamers were considered, leading to improved enrichment of actives in retrospective screens. A library with 4.1 million drug-like compounds was first docked to the orthosteric site. Experimental validation studies identified 14 ligands (hit rate = 56%) with binding affinities ranging from 76 nM to 3.8 μM. Most of these compounds were antagonists and predicted to be bitopic ligands, i.e., targeted both the major and minor pockets. The second screen was focused on identifying allosteric ligands binding primarily to the minor pocket (Fig. 11). The screening library was docked to a structural model of the D3R in complex with dopamine, which had been modeled in the orthosteric site. In this case, eight novel ligands were discovered (hit rate = 32%). The ligands displayed a variety of functional activity profiles, including noncompetitive allosteric modulation.
Several studies have explored fragment-based approaches to identify D3R ligands based on docking to crystal structures (Vass et al., 2014a,b; Egyed et al., 2021). In the first study, a D3R crystal structure and a homology model of the D2R subtype were used to design selective ligands. Fragment libraries were docked to the major and minor pockets to identify fragment-sized compounds that could be linked to create a drug-like ligand. Three linked compounds were synthesized and evaluated experimentally in binding assays. One of these bitopic ligands had subnanomolar affinity for the D3R (Ki = 0.67 nM, compound 3) and 55-fold D2/D3 selectivity. Selectivity was predicted to be achieved by targeting the D3R-specific residue Tyr361.49 (Leu411.39 in the D2R) with a sulfonylurea moiety (Vass et al., 2014a). In the second study, a fragment library with ∼13,000 compounds was docked to the orthosteric site of a D3R crystal structure and to an ensemble of 27 MD simulation snapshots generated using the same structure. Ligands were predicted separately from the crystal structure (50 compounds) and MD ensemble (56 compounds) screens. In total, 25 compounds showed significant binding, and Ki values were determined for eight hits from the screen (0.17–2.8 μM). The hit rate from the screen using MD snapshots (32%) was higher than that from the screen against the crystal structure (18%). Encouragingly, the sets of hits from the two screens were only partially overlapping, suggesting that using ensembles of receptor structures can increase the diversity of hits from virtual screening (Vass et al., 2014b).
Three docking screens for D4R ligands have been carried out using crystal structures, which illustrate different approaches to design the screening library (Wang et al., 2017; Lyu et al., 2019; Ballante et al., 2020). In the first study, Wang et al. (2017) docked a library containing 0.6 million cationic lead-like compounds, which hence was a target-focused library based on the observations that the vast majority of dopamine receptor ligands are positively charged. Top-ranked compounds that occupied the major pocket and extended into a minor binding pocket formed by TM1 and TM2, which was not present in the closely related D3R crystal structure, were selected to identify subtype-selective ligands. In total, 10 top-ranked compounds were experimentally evaluated, and two of these were confirmed to be submicromolar ligands (hit rate = 20%). Despite the fact that the docking was carried out using a structure determined in an inactive conformation, the two hits showed agonist activity. In a second step, one scaffold was optimized for selectivity by identifying 102 analogs in commercial chemical libraries. This led to the discovery of a compound with a Ki of 3 nM, which was strongly selective for D4 over the D2 subtype and had an unusual arrestin-biased signaling profile. In a second study on the D4R, Ballante et al. (2020) carried out structure-based virtual screens with the aim to identify ligands with target selectivity, but a different approach to design the chemical library was used. Publicly available HTS bioactivity data were first analyzed to identify compounds that had been screened extensively (in >100 HTS assays) but had never shown biological activity. The idea was that if active compounds could be identified in this set of “dark chemical matter” (Wassermann et al., 2015), these ligands would have excellent selectivity profiles. The dark chemical matter was docked to a D4R crystal structure. Among the 18 top-ranked compounds that were tested experimentally, two had submicromolar affinities (hit rate = 11%). Both discovered ligands showed some selectivity for the D4R over the D3 subtype (4, Ki = 0.42 μM for D4R, 26-fold selective). More importantly, these D4R ligands were experimentally confirmed to lack activity at hundreds of off-targets by HTS and hence represent favorable starting points for the development of drugs with improved safety profiles. In the third study using D4R crystal structures, Lyu et al. (2019) performed a structure-based virtual screen of 138 million compounds, a library that was orders of magnitude larger than those carried out previously. This advance was enabled by access to a make-on-demand library enumerated from available building blocks by the chemical supplier Enamine (Grygorenko et al., 2020). To compare the results of selecting compounds automatically (by docking score) and manually (by visual inspection), 114 (docking score) and 124 (visual inspection) compounds were selected for experimental testing from the top 1000 ranking compound clusters. From these sets, 23% and 26% were confirmed to be ligands with Ki values better than 10 μM. The hit rates were hence similar for automated and manual compound selection, but there was a shift toward higher ligand affinity in the set selected by visual inspection. To assess whether docking enriched active compounds, the activities of 549 compounds with scores ranging from high to low were measured. Hit rates varied from 22%–26% among the highest-ranking molecules to 0% for the set of compounds with the worst scores, indicating a good docking performance for this target. Notably, the most potent discovered agonist was a diastereomeric mixture (compound ZINC621433144). All four individual diastereomers were synthesized, which uncovered that one of the pure stereoisomers had a very potent EC50 of 180 pM. The best hits from the three studies focusing on the D4R contained a positively charged amine moiety and were predicted to form a salt bridge to Asp1153.32, which is conserved in all dopamine receptors.
Several studies used homology models in structure-based virtual screening and modeling of receptor-drug complexes. As in the case of the A2AR and CXCR4, the modeling community was challenged in the GPCR Dock assessment to predict the structure of the D3R (Kufareva et al., 2011). Several research groups were able to predict the binding mode of the cocrystallized ligand eticlopride with high accuracy (Fig. 9). The best model had a ligand RMSD of only 0.96 Å and captured 58% of the receptor-ligand contacts. Three studies used homology models of the D2R subtype to identify ligands. Kaczor et al. (2016) used a D2R homology model based on a crystal structure of the D3 subtype in a virtual screen of a library with 6.5 million compounds. Of the 21 selected compounds, 10 showed activity (hit rate = 48%) in a binding assay (58 nM to 24 μM), and seven high-affinity compounds were confirmed to be antagonists. The second and third virtual screens focused on the discovery of D2R agonists using homology models. Weiss et al. (2013) docked commercially available fragment and lead-like libraries (3.1 million compounds) to a homology model of the D2R based on an active conformation of the β2R. In total, 15 compounds were selected for experimental testing using similar criteria as used in the docking screen for β2R agonists in the same study (formation of key interactions and better ranking for the active compared with an inactive model of the receptor). Two of the active compounds were agonists, and the remaining one was an inverse agonist of the G protein pathway, corresponding to a ligand hit rate of 20%. In the third study, Männel et al. (2017) constructed a focused virtual library based on a known D2R ligand scaffold (phenylpiperazine) linked to a moiety that could extend into secondary binding pockets. Molecular docking of the 13,000 virtual products to a D2R homology model based on a D3R crystal structure identified 18 compounds, which were subsequently synthesized. Of these, 16 displayed activity in functional assays measuring either G protein or β arrestin–mediated signaling, and a few ligands showed some preference for one of these pathways. Such a biased signaling profile could have advantages in drug development for neurodegenerative and neuropsychiatric diseases, as one pathway could be responsible for the therapeutic effect, whereas others can be associated with adverse effects.
E. Free Fatty Acid Receptors
Free fatty acid receptors are activated by short- to long-chain fatty acids, and these GPCRs have received attention for their involvement in inflammation and diabetes (Stoddart et al., 2008). The free fatty acid receptor 1 (FFA1R) plays a key role in glucose homeostasis and mediates insulin secretion. Receptor activation by long-chain free fatty acids enhances insulin secretion, and FFA1R is a target for the treatment of type 2 diabetes. Crystal structures of FFA1R (Figs. 3 and 4; Supplemental Table 1) in complex with different ligands (Srivastava et al., 2014; Lu et al., 2017) revealed two different binding sites: a hydrophobic interhelical site between TM3 and TM4 and an extrahelical site between TM3, TM4, and TM5. A third possible binding pocket between the extracellular poles of TM1, TM2, and TM7 was also predicted, but no ligand was bound to this site in the experimental structures. Lückmann et al. (2019) carried out MD simulations of the FFA1R, which suggested that stabilizing the solvent-exposed extracellular pocket (Fig. 11) could influence receptor signaling. A database with 13 million commercially available compounds (Table 5) was then docked to this site in a stepwise fashion. After an initial screen against a single FFA1R structure, the 100,000 top-scored compounds were docked to four simulation snapshots. Docking scores combined with physicochemical property filters for drug-like compounds guided the selection of 99 compounds for experimental evaluation. One screening hit (compound 1) displayed a similar potency and efficacy as the endogenous ligand (oleic acid) but was unable to displace TAK-875 from the receptor (Fig. 11). As predicted, these results supported that the ligand was able to bind to a different site than TAK-875. Additional experiments showed that the compound acted as an agonist and PAM of oleic acid. Site-directed mutagenesis indicated that the ligand interacted with Lys2597.36, Ala662.64, and Tyr121.39 in the predicted extracellular pocket. These results suggested that the compound stabilizes the extracellular site and thereby promotes an active-like conformation of the receptor. Further studies were performed on 96 commercially available analogs, and two of these had a pharmacological profile similar to the screening hit. Custom-made synthesis led to the discovery of two compounds (4 and 5) with improved potency and efficacy (EC50 ≈ 1 µM).
F. Histamine Receptors
The histamine receptor family comprises four receptors (H1–H4R) and is implicated in allergic inflammation, gastric acid secretion, neurotransmission, and immunomodulation (Tiligada and Ennis, 2020). In 2011, the first crystal structure of a histamine GPCR was published (Figs. 3 and 4; Supplemental Table 1), and several docking screens were subsequently carried out (Table 5). The crystal structure showed the inverse agonist doxepin binding deep in the major pocket (Shimamura et al., 2011). A docking screen using this crystal structure was first published by de Graaf et al. (2011). A protocol combining protein-ligand interaction fingerprint scoring and a conventional energy-based scoring function was first developed by docking of known H1R ligands and decoy molecules. A focused library with ∼0.1 million commercially available fragments was then docked to the orthosteric site. The results were filtered to identify compounds forming a salt bridge with Asp1073.32 and scored using the developed screening protocol. The remaining compounds were clustered and visually inspected, resulting in the selection of 26 molecules for experimental validation. Remarkably, 19 of the compounds were found to be H1R antagonists or inverse agonists (hit rate = 73%) with affinities ranging from 6 nM to 11 µM. In a follow-up study, Kooistra et al. (2016) revisited the previous docking study and further evaluated their approach. In addition to the combined approach, two sets of compounds were selected using either the energy-based scoring method or the protein-ligand interaction fingerprint scoring while maintaining all other steps of the virtual screening approach. The energy-based approach identified 15 new H1R ligands out of a set of 33 compounds (hit rate = 45%). For the fingerprint approach, 20 new H1R ligands were identified in a set of 33 compounds (hit rate = 61%). The combined scoring approach hence resulted in the highest hit rate (73%). One of the hits from these screens was used as a starting point for evaluating the SAR for binding to the H1R using analysis of water networks in the binding site and mutagenesis (Kuhne et al., 2016). By synthesizing 23 analogs of the hit and experimentally evaluating their affinity to the H1R, the contribution to ligand binding from three different regions of the major pocket were systematically explored (the amine binding region near Asp1073.32 and two aromatic regions). Displacement of ordered water molecules from the amine binding region was found to explain the SAR for closely related compounds. Egyed et al. (2021) developed a fragment-based approach to optimize the selectivity profile of an H1R antagonist by identifying compounds binding to a secondary binding pocket using molecular docking. By covalently linking the orthosteric antagonist to fragments predicted to bind in the secondary pocket, ligands with selectivity for the H1R over the M1R were identified.
Virtual screens have also been carried out using homology models of the other histamine receptor subtypes (Kiss et al., 2014; Vass et al., 2014b; Istyastono et al., 2015; Kiss and Keserű, 2016; Schaller et al., 2019). Both Vass et al. (2014b) and Istyastono et al. (2015) used homology models of the H4R in virtual screening of fragment libraries. Vass et al. (2014b) performed docking of a library with ∼13,000 fragments to an H4R homology model (based on the H1R crystal structure) and MD-refined structures. Out of the 85 compounds that were tested experimentally, 15 showed significant binding (hit rate = 18%). Istyastono et al. (2015) used H4R homology models based on β2R and H1R crystal structures, resulting in the discovery of nine ligands (hit rate = 24%).
G. Leukotriene Receptors
Leukotriene receptors are GPCRs involved in inflammation (Back et al., 2011), which are classified into two main types based on their recognition of either leukotriene B4 or the cysteinyl leukotrienes. The two cysteinyl leukotriene receptors (CysLT1R and CysLT2R) are activated by a set of leukotrienes (LTC4, LTD4, and LTE4) (Back et al., 2011), which are lipid mediators that are released through the 5-lipoxygenase pathway during allergy-induced inflammation (White, 1999). These GPCRs are also involved in pathways mediating allergic asthma (Athari, 2019), and antagonists were identified as a promising therapy for this condition (Trinh et al., 2019). Experimental structures (Hori et al., 2018; Gusach et al., 2019; Luginina et al., 2019) are available of both receptor types (Figs. 3 and 4; Supplemental Table 1), and a virtual screening was performed for the CysLT1R and CysLT2R (Table 5). The first crystal structures of CysLTRs were determined in 2019 and revealed a unique binding mode of antagonists (Gusach et al., 2019; Luginina et al., 2019). The ligands are deeply buried in the TM region and stabilize a gap between TM4 and TM5 facing the lipid membrane. This gap may serve as an entry point to the orthosteric binding site.
Sadybekov et al. (2020) performed a prospective docking screen against both CysLT1R and CysLT2R. Conformational ensembles of the receptors were first prepared starting from the crystal structures, and the predictive abilities of these models were benchmarked by measuring the enrichment of antagonists over decoys. Binding site water molecules were considered, and these were found to improve the discrimination between actives and decoys. A total of 115 million drug- and lead-like compounds were docked to the crystal structures and the optimized models using a flexible receptor docking algorithm. The top-ranked 20,000 molecules from this screen were then docked again using more extensive conformational sampling. Subsequently, the 2000 best-ranked compounds from the crystal structures and the two optimized models were merged, filtered based on their interaction patterns in the binding site, and clustered by chemical similarity. Based on structural novelty and diversity, 155 compounds were selected, and 139 of these were successfully synthesized by a chemical supplier. None of these compounds activated the receptors in a functional assay measuring inositol-1-phosphate (IP1) production. Five compounds acted as antagonists of CysLT1R signaling induced by LTD4 in a dose-dependent manner, and two of these yielded full inhibition of the agonist-mediated response (Ki = 0.22–1.03 µM). One compound fully inhibited the agonist effect at CysLT2R (Ki = 6.46 µM). Functional potencies were determined for the best compound (BRI-12359), resulting in affinities (KB) of 0.34 µM and 105 µM at CysLT1R and CysLT2R, respectively. BRI-12359 also displayed inverse agonism at the CysLT2R mutant L129Q, which is constitutively active and linked to uveal melanoma (Nell et al., 2021). Analysis of the docking pose in the CysLT1R pocket showed that BRI-12359 occupied the upper part of the binding site and established a hydrogen bond with Y832.64.
H. Melatonin Receptors
The two melatonin receptors (MT1R and MT2R) regulate the circadian rhythms and are drug targets for the treatment of insomnia, depression, and cancer (Liu et al., 2016). The first crystal structures of the MT1 and MT2 receptors in complex with agonists (Figs. 3 and 4; Supplemental Table 1) were published in 2019 (Johansson et al., 2019; Stauch et al., 2019) and have been used in two virtual screens (Table 5).
Stein et al. (2020) performed a virtual screening campaign by docking of >150 million commercially available make-on-demand compounds to the orthosteric site of an MT1R structure. The top-scoring 300,000 compounds were clustered by topological similarity, and 10,000 clusters were visually inspected. In compound selection, molecules that were able to interact with the key residues Asn1624.60 and Gln181EL2 were favored. In total, 38 compounds were selected and tested experimentally; 15 molecules (hit rate = 39%) were active in functional assays at MT1R, and four of these were MT1 selective agonists. There were both agonists and inverse agonists among the hits with activities of 1 nM and 1.2 µM, respectively, for the best compounds. In total, 12 ligands were selected for further optimization by searching for analogs in the make-on-demand library. Several thousand commercially available analogs were docked into the MT1R structure, followed by the synthesis and testing of 131 compounds. Of these, 94 compounds were active at one or both the MT receptors. Three subtype-selective compounds (two MT1 selective inverse agonists and one selective MT2 agonist, respectively) were further evaluated in vivo. Phase-shift experiments showed that the two MT1 selective inverse agonists advanced the mouse circadian clock by 1.3–1.5 hours (a typical effect of the endogenous agonist melatonin) when administrated to mice at dusk. This agonist-like effect was absent in MT1- but not in MT2-knockout mice, thus revealing an unknown role of the MT1R.
Patel et al. (2020) docked a library of 8.4 million commercially available compounds to the orthosteric pockets of both the MT1 and MT2 receptors. Prior to the screen, the MT1 and MT2 receptor structures were analyzed to optimize the docking performance. The focused screening library was constructed by selecting commercially available fragment-like compounds from the ZINC database with physicochemical properties similar to known ligands. The 5000 top-ranked compounds for each receptor were selected and docked with an increased computational sampling to both the MT1 and MT2 receptors. From each receptor screen, the top 500 compounds were considered, and compound selection was guided by chemical clustering and docking scores. In total, 62 compounds (23 from MT1R, 25 from MT2R, and 14 from both) were finally prioritized for binding and functional experiments based on their structural novelty, chemical diversity, and interactions with the receptors. Out of the 62 compounds, 11 activated one or both subtypes with submicromolar potencies in Gi/o signaling assays. Of these, 10 displayed Ki values <10 µM, and six were MT2-selective. The most potent compound (28, a melatonin derivative) showed an EC50 of 0.04 nM at both MT1 and MT2. Compound 21, which was the most potent new chemotype, displayed a 30-fold selectivity for the MT2R (EC50 = 0.36 nM and 12 nM for MT2 and MT1 receptors, respectively). The identified hits were also evaluated for functional selectivity. Compounds 21 and 28 displayed reduced β-arrestin recruitment at the MT2R compared with melatonin. Conversely, compound 37 showed no Gi signaling but marked β-arrestin–mediated activity at the MT1R. These compounds hence displayed signaling bias, and the predicted binding modes provided hypotheses regarding the structural basis of the observed functional selectivity, e.g., interactions with Asn4.60 and Tyr7.38.
I. Muscarinic (Acetylcholine) Receptors
There are five muscarinic (acetylcholine) receptors (MRs) in humans, and these are drug targets for a large number of conditions, e.g., Alzheimer’s disease, diabetes, and asthma (Kruse et al., 2014). However, as the MRs share very high sequence similarity in the orthosteric site, the development of subtype-selective ligands has been challenging. The determination of crystal structures of all MR subtypes (Figs. 3 and 4; Supplemental Table 1) provided the opportunity to use molecular docking to screen for selective ligands, which has been explored in three virtual screens by targeting either orthosteric or allosteric binding sites (Table 5).
Kruse et al. (2013) focused on structure-based discovery of orthosteric MR ligands. The crystal structures of the M2R and M3R confirmed that the orthosteric sites are very similar and only differ by a single amino acid (Phe and Leu in position 5.33 of the M2R and M3R, respectively). A commercial chemical library with 3.1 million compounds was first docked to the M3R, and 18 top-ranked compounds that formed interactions with key binding site residues (Asp1033.32, Asn4046.52, and Trp4006.48) were prioritized for experimental evaluation. Of these, 11 showed significant binding to the M2R (Ki values <50 μM), corresponding to a hit rate of 61%, and the best compound had an affinity of 390 nM (compound 1). A second docking screen of the same library was then carried out for the M3R. In this case, compounds that were top-ranked for the M3R and had a lower rank in the M2R screen were extracted to bias the screening result toward subtype selectivity. A set of 16 compounds that formed interactions with nonconserved Leu2255.33 were selected, and eight of these were shown to bind to the M3R with affinities ranging from 780 nM to 64 μM (hit rate = 50%). Although a large fraction of the hits showed some selectivity for the M3R, the differences in affinities were small, and the best compound reached a selectivity ratio of only 6-fold. A majority of the discovered compounds did not show agonist activity, which was consistent with the fact that the docking screens were performed using receptors crystallized in an inactive state. One compound (16) was a partial M3R agonist (EC50 = 5.2 μM, Emax = 65%), but it showed no detectable activity for the M2 subtype.
Korczynska et al. (2018) explored an alternative approach to identify selective ligands by docking to an allosteric site in an M2R crystal structure. An allosteric pocket was identified at the extracellular entrance of the orthosteric site, which was separated from the cocrystallized antagonist by a layer of aromatic residues (Fig. 11). In contrast to the orthosteric site, the sequence conservation of the ECV is relatively low, and ∼50% of the residues in this pocket differ between the M2 and M3 subtypes. A structure-based virtual screen of 4.6 million compounds was performed. The effects of 13 top-ranked compounds were evaluated in binding assays, and three of these showed a significant allosteric effect (hit rate = 23%). One compound (‘589) was found to increase the affinity of the antagonist N-methyl scopolamine by 22%, corresponding to PAM activity, and had an affinity of 4 μM for the allosteric site. Structure-based optimization of this ligand using docking of commercially available analogs resulted in the identification of a compound (‘628) that had an affinity of 1.1 μM and also showed PAM activity in functional assays. The optimized modulator was also tested at the other four MRs and showed either no or very weak activity in all cases, demonstrating that the compound was subtype selective.
Fish et al. (2017) searched for new M2R agonist scaffolds using the active-state structure of M2R in complex with the agonist iperoxo. Based on the binding mode of iperoxo, a set of 19 aromatic compounds were designed to establish a hydrogen bond interaction with Asn4046.52 and a salt bridge with Asp1033.32. Prior to performing the biological experiments, this set of molecules was then docked to active and inactive M2R conformations. Based on hydrogen bonding with Asn4046.52 and docking scores, the compounds were classified as agonists or nonagonists. These predictions were then compared with the results from functional assays. The compounds displayed affinities in the mid to low micromolar range at M1R, M2R, and M3R, with two of them reaching nanomolar Ki values. One compound (3) was correctly predicted to be an agonist (Ki = 14 µM, Emax = 75%), and 16 compounds were correctly classified as M2R antagonists or inverse agonists. Compound 17 was a high-affinity antagonist (Ki = 0.14 µM, Emax = −14%) but had been predicted to be an agonist, and compound 2 was an agonist but was expected to be an antagonist. Further optimization identified compound 22, which displayed improved affinity but lower efficacy (Ki = 1 µM, Emax = 44%) compared with 3. Finally, a docking screen using a fragment library with 2.2 million compounds was also performed against the M2R active-state crystal structure. The top-ranked 1000 fragments were inspected, and 10 compounds were prioritized for experiments among those showing interactions with Asn4046.52 and Asp1033.32, which were expected to be key interactions for receptor activation. Of these, seven fragments (hit rate = 70%) had Ki values <50 µM, and the best affinity was 6 µM (compound 29). Three compounds were shown to be agonists in functional assays, with EC50 values in the low micromolar range. Compound 29 was the most potent agonist, with EC50 and Emax values of 9.9 µM and 74%, respectively.
J. Neurotensin Receptors
There are two neurotensin receptors, and several crystal and cryo-EM structures of the NTS1 subtype (Figs. 3 and 4; Supplemental Table 1), which is a target for central nervous system diseases, have been determined (White et al., 2012; Egloff et al., 2014; Krumm et al., 2015, 2016; Kato et al., 2019; Yin et al., 2019; Huang et al., 2020). Ranganathan et al. (2017) performed structure-based screening of fragment- and lead-like chemical libraries (Table 5) with the goal to discover novel NTS1R ligands targeting the orthosteric site, which recognizes the C-terminal end of the peptide agonist. The commercially available fragment- and lead-like libraries contained 0.5 and 1.8 million compounds, respectively. Each library was docked to the orthosteric site, and 25 fragment-like and 27 lead-like compounds were selected for experimental evaluation by SPR. The experimental binding affinities of the eight identified fragment-like ligands (hit rate = 32%, KD = 0.2–0.3 mM) were lower than for the five lead-like ligands (hit rate = 19%, KD 1.2–42 μM), but the hits covered complementary chemical space. The discovered ligands were predicted to be anchored by interactions with Arg3276.54, Tyr1463.29, and Tyr3477.31. Structure-guided optimization of hits by identification of analogs in commercial chemical libraries led to the discovery of small-molecule agonists with low micromolar to submicromolar affinity.
K. Opioid Receptors
The opioid receptor family is composed of the µ, δ, and k receptors (MOR, DOR, and KOR, respectively), and the opioid-related nociceptin receptor (NOP). Opioid receptors are therapeutic targets for pain treatment (Stein, 2016; Manglik, 2020). All three opioid receptors mediate analgesia, but drug interactions result in different side effects depending on their regional expression and functional activity in central and peripheral systems (Valentino and Volkow, 2018). MORs are associated with respiratory depression, addiction, sedation, tolerance, constipation, itch, and nausea. DORs mediate respiratory depression, constipation, dependence, and convulsions. KORs cause dysphoria, sedation, and diuresis (Stein, 2016). For these reasons, there is a major interest in the development of improved analgesic drugs. Crystal structures of the MOR, DOR, KOR, and NOP have been determined (Granier et al., 2012; Manglik et al., 2012; Thompson et al., 2012; Wu et al., 2012) (Figs. 3 and 4; Supplemental Table 1) and contributed to several successful structure-based ligand discovery campaigns (Table 5).
Negri et al. (2013) docked a library containing 4.5 million commercially available lead-like molecules to an inactive KOR crystal structure. The top-ranked compounds were visually inspected, and a set of 22 were selected based on identifying contacts similar to the cocrystallized ligand (e.g., with Asp1383.32), chemotype novelty, and interactions with KOR-specific residues. Experimental validation led to the identification of four ligands that displayed significant affinities in a binding assay. Out of these, the racemic mixture of MCKK-17 activated GαoB signaling, and the two stereoisomers of this compound were synthesized. The S-stereoisomer (MCKK-17S) was identified as a full and subtype-selective KOR agonist with micromolar potency (EC50 = 7.2 μM).
Zheng et al. (2017) performed a structure-based virtual screening campaign for KOR ligands. In addition to a KOR crystal structure, two modified receptor structures, which were optimized for ligand enrichment, were also used. A total of 4.5 million commercially available compounds were docked into the three different KOR models. Based on chemical diversity, novelty, and predicted binding affinity, 43 compounds were prioritized for experimental testing, and 14 had Ki values <10 µM (hit rate = 33%). The most active compound had an affinity of 0.2 µM (compound 28). Hit-to-lead optimization was attempted for six chemotypes by combining similarity searches in commercial chemical databases and docking. In total, 40 analogs were tested experimentally, resulting in 11 ligands with submicromolar affinities that represented four different scaffolds. The compound with the highest affinity (64, Ki = 0.09 µM) was an antagonist of both G protein and β-arrestin–mediated signaling.
Manglik et al. (2016) screened >3 million commercially available lead-like compounds with the goal to identify MOR agonists. Compounds were docked to an inactive MOR structure, and ligands were prioritized based on their ability to interact with key residue Asp1473.32 and other residues considered important for receptor affinity and specificity. In total, 23 molecules from the top-scored 2500 compounds were prioritized for experimental evaluation. Seven compounds showed micromolar activity (hit rate = 30%), and the highest affinity was 2.3 µM. A similarity search based on the three most potent compounds identified 500 commercially available analogs, and 15 of these were evaluated experimentally. Seven analogs had Ki values between 42 nM and 4.7 µM, and the most potent agonist showed robust activation of Gi/o and low arrestin recruitment. Structure-guided optimization of this compound led to the identification of PZM21, a potent and selective Gi-biased MOR agonist (EC50 = 4.6 nM, Ki = 1.1 nM). In tests on mice, PZM21 induced longer and robust analgesia with less respiratory depression and constipation than morphine.
In a study by Weiss et al. (2018) focusing on identifying KOR-selective ligands, crystal structures of both the KOR and MOR were used in a docking screen of 3 million lead-like compounds. Compounds that ranked in the top 1% for the KOR structure were further investigated and ranked according to the ratio of their ranks from the KOR and MOR screens. The compounds with the highest rank ratio were visually inspected, and 22 of these were purchased and experimentally evaluated. Nine compounds showed affinity for the KOR (hit rate = 41%), with two displaying >18-fold selectivity for this receptor subtype. However, there were also compounds among the hits that showed similar selectivity for the MOR, i.e., the opposite selectivity than predicted. These results highlighted the difficulty in utilizing structure-based virtual screening to identify selective compounds for closely related receptors.
L. Orexin Receptors
The orexin signaling system is involved in a plethora of behavioral functions, including regulation of the sleep-wake cycle (Scammell and Saper, 2007). It consists of two GPCRs, the orexin receptor subtypes 1 (OX1R) and 2 (OX2R), which are activated by the orexin A and B neuropeptides (de Lecea et al., 1998; Sakurai et al., 1998). In 2014, the Food and Drug Administration approved the nonselective orexin receptor antagonist suvorexant for the treatment of insomnia (Winrow and Renger, 2014). The sequence identity between the two receptors is 64%, with nearly identical binding sites, which makes it challenging to develop subtype-selective compounds. Crystal structures of the OX1R (Yin et al., 2016; Hellmann et al., 2020; Rappas et al., 2020) and OX2R (Yin et al., 2015; Suno et al., 2018; Rappas et al., 2020) with diverse antagonist chemotypes have been determined (Figs. 3 and 4; Supplemental Table 1) and were used in two studies with the goal to identify novel and selective ligands (Table 5).
Gunera et al. (2020) performed a docking screen against a crystal structure of OX2R using lead- and drug-like sets from the ZINC database with ∼11 million compounds. Two docking programs were used in combination with four scoring functions. Visual inspection of 6500 top-ranked molecules led to the selection of 43 commercially available compounds, which were experimentally tested at both the OX1 and OX2 receptors. Eleven compounds showed measurable pKi values at the OX2R (hit rate = 26%), and the highest affinity ligand had a pKi of 5.55 (compound P33). The discovered compounds showed similar affinities for both receptors. In total, 54 analogs were selected based on docking scores, predicted binding poses, and commercial availability, and 16 of these showed a detectable affinity for the OX2R with a highest pKi of 6.18 (compound F33.3). F33.3 was predicted to bind with a suvorexant-like pose and establish a hydrogen bond with Asn3246.55. Functional assays for 19 discovered ligands revealed antagonist activity at both OX1R and OX2R. As the hits from the screen showed similar activity at both subtypes, a second virtual screen was carried out with the goal to identify OX2-selective ligands. In this case, results from docking screens against both the OX1R and OX2R structures were considered, and 25 OX2-selective ligands were predicted. Experimental evaluation resulted in a hit rate of 24% (six ligands) and similar affinities as the hits from the single-target screen (pKi = 5.80 for the best compound). However, despite the fact that the docking screen used structures of both subtypes, only a weak subtype selectivity (5-fold) was achieved.
An excellent example of how crystal structures can facilitate the identification of selective compounds is the study by Hellmann et al. (2020), which used OX1 and OX2 receptor structures to design a subtype-selective analog of the drug suvorexant. Suvorexant shows similar affinity for both subtypes, and crystal structures revealed that only two amino acids differed in the binding sites (Ser1032.61 and Ala1273.33 of OX1R are exchanged for Thr1112.61 and Thr1353.33 in the OX2R). Molecular docking calculations guided the design of suvorexant analogs with modifications on the homopiperazine moiety of suvorexant. By moving a methyl to a different position on the homopiperazine and optimizing the size of this substituent, a pocket that is created by the smaller side chain in position 3.33 could be filled in the OX1R binding site. In the OX2R, substituents in this position would clash with Thr1353.33. Based on this observation, a set of compounds with small hydrophobic substituents were synthesized and evaluated experimentally. The most selective compound (JH112) retained affinity for the OX1R (Ki = 0.72 nM), but there was a large loss of affinity at the OX2R (Ki = 54 nM) compared with suvorexant. As predicted, the compound was hence OX1R-selective with a 75-fold higher affinity for this subtype. A crystal structure of OX1R in complex with JH112 confirmed the predicted binding mode and interactions of the designed substituent.
M. Serotonin (5-Hydroxytryptamine) Receptors
The 5-Hydroxytryptamine (5-HT) receptors are targets of several therapeutics, including antipsychotics and antimigraine drugs. The large number of receptor subtypes recognizing serotonin makes it difficult to identify selective ligands. Experimental structures of four serotonin receptor subtypes (5-HT1BR, 5-HT2AR, 5-HT2BR, and 5-HT2CR) are currently available (Figs. 3 and 4; Supplemental Table 1) (Liu et al., 2013; Wacker et al., 2013; Wang et al., 2013; Wacker et al., 2017; García-Nafría et al., 2018; McCorvy et al., 2018; Peng et al., 2018; Yin et al., 2018; Kimura et al., 2019; Kim et al., 2020), which guided three virtual screening campaigns focusing on subtype-selective ligands (Table 5).
Rodriguez et al. (2014) aimed to identify ligands that were selective for the 5-HT1BR over the 5-HT2B subtype. A target-focused library with 1.3 million commercially available compounds, which all had a positively charged nitrogen moiety, was docked to 5-HT1B and 5-HT2B receptor crystal structures. The rankings of the compounds in these two screens were then compared. A set of 500 compounds that were top-ranked for the 5-HT1BR and ranked substantially lower (>100,000) for the 5-HT2B subtype were inspected visually. Of these, 22 compounds were experimentally evaluated in binding assays, leading to the discovery of 11 ligands with affinities <10 μM (hit rate = 50%). Nine compounds showed selectivity for the 5-HT1B over the 5-HT2B subtype, and the fold difference in affinity was >10 in four cases. Three 5-HT1BR-selective compounds were also tested in functional assays, and all of them activated the G protein pathway, which agreed with the fact that the 5-HT1BR had been crystallized in complex with an agonist. The most promising agonist had an affinity of 300 nM for the 5-HT1BR and >300-fold subtype selectivity.
Rataj et al. (2018) attempted to identify ligands that were selective for the 5-HT2BR using a combination of ligand- and structure-based techniques. A machine learning method based on 2D fingerprints was first trained to identify 5-HT1B and 5-HT2B ligands and subtype selectivity using compounds with known activity. This method was then used to predict 5-HT2B-selective ligands in a library with 4.8 million commercially available compounds. The resulting focused library (24,849 compounds) was docked to several 5-HT1B and 5-HT2B receptor crystal structures. The docking results were filtered based on interactions with conserved residue Asp3.32 in the orthosteric site, binding site waters, and nonconserved residues in a secondary binding pocket to favor the identification of selective compounds. A set of 231 compounds fulfilled the interaction criteria and also had a better ranking in the 5-HT2B docking screen than in that carried out for the 5-HT1B subtype. Nine of these were selected for experimental evaluation based on novelty and visual inspection. Three compounds showed significant activity in binding assays (hit rate = 33%), and as predicted, all of these were selective for the 5-HT2B subtype. The best compound (8) displayed an affinity of 0.3 nM for the 5-HT1BR, and it was nearly 10,000-fold selective.
Egyed et al. (2021) used a fragment-based approach to optimize the selectivity of lysergic acid diethylamide (LSD) for the 5-HT2B receptor. LSD binds to the orthosteric site of both the 5-HT1B and 5-HT2B subtypes, but affinity is 10-fold higher for the former receptor. The goal of the study was to design a bitopic ligand based on LSD that would increase selectivity for the 5-HT2BR. A set of 119 fragments were docked to a secondary pocket of the 5-HT2BR with LSD placed in the orthosteric site. Top-scoring fragments were then linked to LSD via an amide bond, and the resulting compounds were then docked again, which confirmed that the LSD scaffold and the fragments maintained their binding modes. Two of these bitopic compounds were synthesized, and binding assays confirmed the predicted selectivity profile in both cases. Compound 5 had an affinity of 0.2 µM for the 5-HT2B receptor and was >60-fold selective over the 5-HT1B subtype. The selectivity profile was further evaluated by testing at five other aminergic GPCRs (adrenergic, serotonin, and dopamine receptors), and compound 5 again showed the highest affinity for the 5-HT2BR.
The GPCR Dock 2013 assessment demonstrated that access to crystal structures of other aminergic GPCRs enabled accurate prediction of the 5-HT1B and 5-HT2B receptor structures using homology modeling (Kufareva et al., 2014). The overall binding mode of the ligand ergotamine was well predicted (RMSD from the crystal pose of 1.51 and 1.05 Å, respectively), and ∼50% of the ligand contacts were captured in the best models (Fig. 9). Several virtual screening studies have used homology models of serotonin receptors (Lin et al., 2012; Weiss et al., 2018). Weiss et al. (2018) attempted to use a structure-based approach to identify a multitarget compound binding to the D2R and the 5-HT2AR, but without any affinity for H1R. Compounds with such a polypharmacological profile could be useful in the development of efficient antipsychotics with reduced side effects (Roth et al., 2004). As there were no experimental structures of the D2R and 5-HT2AR at this point, homology models were created for each receptor based on a D3R crystal structure, and an H1R crystal structure was used as an antitarget. In total, 3 million commercially available lead-like compounds were docked into the two models and the H1R structure. Molecules that were top-ranked in the D2R and 5-HT2AR screens and did not show similarity to top-ranked compounds for the H1R or known H1R ligands were visually inspected. Out of 28 selected compounds, 17 showed activity at the 5-HT2AR (hit rate = 61%), 10 showed activity for D2R (hit rate = 36%), and eight compounds showed activity for both receptors (hit rate = 29%). However, 16 of the compounds also showed affinity for the antitarget (hit rate = 57%). A second virtual screen with the same aim, but with a modified docking protocol and a new structural model of the H1R, was then performed. Experimental testing of 20 predicted compounds yielded similar results as the first screen, i.e., high hit rates for both the targets and antitarget. Hence, although high hit rates were obtained for the two targets, it was difficult to avoid interactions with the closely related H1R.
N. Smoothened
Smoothened (SMO) was the first class F GPCR to be crystallized and is a target of cancer drugs (e.g., vismodegib). Crystal structures revealed that several SMO antagonists bind in buried TM pockets that partially overlap with the orthosteric site of class A GPCRs (Figs. 3 and 4; Supplemental Table 1). These complexes revealed that certain SMO antagonists are ineffective against tumors because binding site mutations disrupt receptor-ligand interactions (Wang et al., 2013, 2014; Weierstall et al., 2014; Byrne et al., 2016; Zhang et al., 2017; Qi et al., 2019). Two structure-based virtual screens have been carried out with the goal to identify novel antagonists binding in the TM region (Table 5).
Lacroix et al. (2016) performed the first docking screen for SMO ligands using a structure of the complex with the antagonist LY2940680 (taladegib). The docking protocol was optimized by docking of actives and decoys, followed by a screen of 3.2 million lead-like compounds. The top 0.2% of the ranked list (∼6400 compounds) was visually inspected, and 21 of these were selected for experimental evaluation. The selected compounds were predicted to interact with at least two residues known to be important for antagonist binding (Asn219, Asp473, Arg400, Lys394, Glu518, or Asp384) and to explore different subpockets in the TM region. Four compounds were experimentally confirmed to inhibit signaling mediated by SMO with IC50 values ranging from 5.3 to 34.4 μM (hit rate = 19%). A set of 231 analogs of the hits were identified in commercial chemical libraries and docked to SMO. Of these compounds, 46 were experimentally tested, which led to the identification of several inhibitors in the micromolar range. One of these ligands (45b, IC50 = 3.1 μM) was also tested against the drug-resistant SMO mutant Asp473His and showed only a 3-fold loss of activity, which can be compared with a 100-fold reduction for vismodegib.
Lu et al. (2018) developed a structure-based virtual screening protocol to discover SMO antagonists. Four SMO structures were first evaluated based on redocking and ligand enrichment calculations, and two binding site conformations were used in the prospective screen. A library with >1 million compounds was first docked and scored in each structure. The 100,000 top-ranked compounds were then extracted and docked in two steps using increasingly advanced scoring functions. The 1000 top-ranked compounds for each structure were filtered to identify molecules with drug-like properties and clustered. From the remaining set, 21 compounds were selected for experimental evaluation. The compounds were tested in a functional assay measuring inhibition of hedgehog signaling. Six compounds had IC50 values <10 μM (hit rate = 29%). The most potent compound (20) displayed an IC50 of 47 nM, which is similar to marketed drugs targeting SMO. Twelve analogs of compound 20 were identified by similarity and substructure searches in commercial chemical libraries. Several of these were active, but they were not more potent than the initial screening hit. MD simulations and binding free-energy calculations with the molecular mechanics/generalized Born surface area (MM/GBSA) method were used to interpret SAR, which identified Asn219, Val386, Ser387, Tyr394, Arg400, and Phe484 as key residues for SMO antagonism.
Prior to the release of the first SMO crystal structures, the GPCR Dock 2013 assessment evaluated whether this receptor in complex with ligands could be modeled accurately. At that time, there were only templates with very low sequence identity available (<15% in the TM region). The results of the assessment showed that it was not possible to predict the complexes with the two ligands (LY-2940680 and SANT-1) with high accuracy (Fig. 9). Although the binding site of the ligands was identified, only a small fraction of the receptor-ligand contacts (<10%) were captured, and the RMSDs from the crystal poses were high (4.42 and 4.31 Å, respectively) (Kufareva et al., 2014).
IV. Opportunities and Limitations: What Can Molecular Docking Do for You?
A. Can Docking Screens Discover G Protein–Coupled Receptor Ligands?
Yes, the large number of successful prospective virtual screens using crystal structures clearly shows that molecular docking can identify GPCR ligands (Fig. 8; Table 5). Novel ligand chemotypes that can serve as starting points for drug discovery were identified in most of these screens, and with high affinities in several cases. Based on the available results, there are some trends regarding what can be expected from a prospective docking screen targeting the orthosteric binding site. The hit rates and ligand affinities will primarily depend on the nature of the binding pocket (Fig. 12). The highest hit rates and ligand affinities were obtained for well defined and buried binding pockets with a few key polar receptor-ligand interactions. Such sites are likely to bind small-molecule ligands with high affinity, and chemical libraries generally contain a large number of compounds endowed with the required pharmacophore features. As a consequence, a majority of the most successful docking screens focused on receptors having such binding pockets (e.g., adenosine, adrenergic, muscarinic, dopamine, histamine, serotonin, and melatonin receptors). Conversely, there are fewer published virtual screening studies for peptide- and protein-binding GPCRs (e.g., neurotensin, orexin, and CXCR4 receptors). In these cases, the orthosteric sites are more complex with either shallow, open, and/or larger pockets than receptors that recognize small molecules. Such binding site features will make it more challenging to identify a drug-like ligand. In addition, the docking algorithms can be expected to perform worse for these types of binding sites compared with buried pockets. Encouragingly, ligands have been discovered even for the peptide- and protein-binding GPCRs (e.g., NTS1R and OX2R). However, the hit rates have generally been lower, and the ligand activities have been weaker (Figs. 8 and 12). Even if molecular docking clearly can discover ligands of diverse GPCRs, it should of course not be assumed that all virtual screens are successful. There are likely many docking screens that did not result in any hits, which remain unpublished.
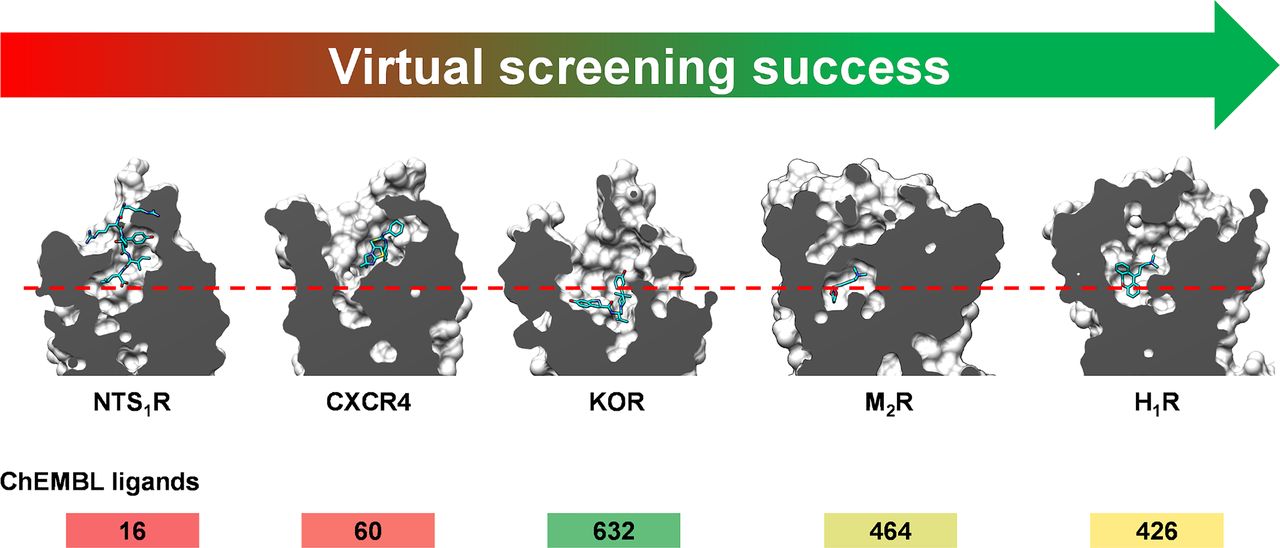
Virtual screening success depends on the nature of the receptor binding sites. High hit rates and ligand affinities/activities were obtained for well defined and buried binding pockets with a few key polar receptor-ligand interactions, which include small-molecule binding receptors such as M2R and H1R. Peptide and protein-binding GPCRs (e.g., NTS1R and CXCR4) have more complex orthosteric sites with either shallow, open, and/or larger pockets than receptors that recognize small molecules. Such binding sites are generally more challenging, which is also reflected by the number of known ligands in the ChEMBL database (activity ≤ 10 µM and mol. wt. ≤ 350). The dashed red line illustrates the difference in pocket depth between the two types of binding sites (shallow: NTS1R and CXCR4; buried and/or deep: M2R, H1R, and KOR).
Allosteric modulators of GPCRs have great potential as drugs (Conn et al., 2009). Although several structures of GPCRs in complex with allosteric modulators are available (Lu and Zhang, 2019; Wakefield et al., 2019), only a few virtual screens for such ligands have been performed (Lane et al., 2013; Korczynska et al., 2018; Lückmann et al., 2019; Liu et al., 2020). The most challenging aspect of such studies is to identify the site to target. Four docking screens focusing on allosteric pockets located in the minor pocket or ECV have been carried out (i.e., β2R, FFA1R, D3R, and M2R) and all of these identified ligands. Whereas extracellular, intracellular, and intrahelical sites are straightforward to screen with molecular docking, extrahelical pockets are more challenging to target because standard docking algorithms have not been parameterized to consider lipid-exposed sites. An illustrative example is the binding site of the P2Y1 receptor antagonist BPTU, which was unexpectedly shown to bind to an extrahelical site (Zhang et al., 2015a).
Several strategies can be used to increase the chances of screening success. Retrospective calculations to ensure that known ligands are enriched by the receptor structure are widely used. In this step, several receptor structures can be considered to identify the best performing binding site conformation (Rodriguez et al., 2015; Kooistra et al., 2016; Scharf et al., 2020). The receptor structure can also be optimized computationally before carrying out the screen to identify additional binding site conformations (Katritch et al., 2010; Lane et al., 2013; Vass et al., 2014b; Zheng et al., 2017; Lückmann et al., 2019; Patel et al., 2020; Sadybekov et al., 2020). The inclusion of water molecules in the binding site can improve the docking performance (Katritch et al., 2010; Lenselink et al., 2014). Some studies also use combinations of several scoring functions to further improve the predictions from the virtual screen (Vass et al., 2016; Gunera et al., 2020; Wei et al., 2020). In the vast majority of prospective virtual screens, a set of top-ranked compounds, which can range from hundreds to thousands of complexes, are inspected in the last step to identify the most promising candidates. Compounds forming similar interactions as known ligands are often prioritized for testing, and those that score well due to deficiencies of the docking method are excluded. The composition of the chemical library can be tailored to increase the chances of success. Depending on the target and screening assay, fragment-, lead-, or drug-like compound libraries may be most relevant (Table 5). Target-focused libraries were also generated by matching the expected formal charge of the ligands (de Graaf et al., 2011; Wang et al., 2017; Adlere et al., 2019) or using pharmacophore models (Wei et al., 2020).
B. Can Docking Screens Predict G Protein–Coupled Receptor Ligands with a Specific Functional Effect?
Prospective docking screens have identified orthosteric agonists, antagonists, as well as inverse agonists of GPCRs. However, is it possible to bias the screening results toward the discovery of ligands with a specific efficacy profile? Prediction of the functional effect of a ligand, e.g., receptor activation of G protein signaling, represents another level of difficulty compared with simply identifying compounds with affinity for the target. The propensity of a GPCR to activate in response to ligand binding is target-dependent. However, the prediction of agonists can generally be expected to be more difficult than the identification of antagonists because the stabilization of an active conformation requires the formation of several specific interactions (Fig. 2).
A ligand typically has some affinity for both active and inactive receptor conformations, and the relative binding strength to the different states determines the observed functional effect (Staus et al., 2016). For this reason, it is not surprising that agonists have emerged from docking screens against inactive receptor conformations (Kruse et al., 2013; Manglik et al., 2016; Wang et al., 2017; Lyu et al., 2019), and conversely, antagonists and inverse agonists were identified using active receptor structures (Rodriguez et al., 2015; Stein et al., 2020). Additional steps can be introduced to bias a screen toward the discovery of agonists. One strategy is to compare docking results obtained with the inactive and active receptor conformations. This approach was used successfully in prospective screens to identify β2R agonists (Weiss et al., 2013) but failed for the A2AR (Rodriguez et al., 2015). The binding site conformations of the active and inactive state are generally very similar, but there are often clear differences between what interactions the agonists and antagonists make (Lebon et al., 2011; Rasmussen et al., 2011). In screens for ligands with a specific functional effect, compounds forming (or lacking) key interactions can be prioritized in visual inspection of predicted complexes (Fish et al., 2017) or by automated filtering for contacts such as hydrogen bonds (Weiss et al., 2013). A more advanced approach is to use interaction fingerprints, which has the potential to screen for a specific functional effect through the selection of an appropriate reference fingerprint (Kooistra et al., 2015, 2016). As ligand interactions that trigger activation are generally not transferrable between receptors recognizing different types of endogenous ligands, the prospects of success in docking screens for agonists will be dependent on how well the structural basis of activation is understood. An encouraging result from the docking screens is that biased signaling has been observed for a number of discovered ligands (Weiss et al., 2013; Manglik et al., 2016; Wang et al., 2017; Patel et al., 2020), but the structural basis of functional selectivity is still not entirely clear.
The composition of the screening library also plays an important role in screens for ligands with a specific efficacy profile. If the goal is to identify agonists, the endogenous ligand and synthetic agonists can guide the selection of a suitable library. If the known agonists are small and have low molecular complexity (e.g., adrenaline or melatonin), fragment libraries are likely to contain potent agonists, and such sets can be prioritized for screening. For example, fragment-sized agonists were discovered by docking to both the adrenergic (Weiss et al., 2013; Kooistra et al., 2016) and melatonin (Patel et al., 2020) receptors. If the target receptor recognizes more complex endogenous compounds (e.g., larger peptides/proteins, or complex chemistry, e.g., nucleotides), drug-like molecules or focused libraries that capture features required for activation may be necessary to identify agonists. One example is the prospective docking screen for agonists of the A2AR. Although the same strategy allowed to identify β2AR agonists, none of the discovered high-affinity ligands activated the A2AR. One potential explanation for this result was a bias toward A2AR antagonist chemotypes in the library for this target and the complex interaction network formed by the ribose moiety of the agonist (Rodriguez et al., 2015). In such cases, focused virtual libraries with more complex compounds, which may not be present in the commercial libraries, can be screened, and this strategy resulted in the discovery of several adenosine receptor agonists (Rodriguez et al., 2016).
C. Can Docking Screens Predict Selectivity and Polypharmacology?
Subtype-selective GPCR ligands are difficult to predict using molecular docking screening because of limitations of available structural data and the docking method. First, atomic-resolution structures may not be available for all the relevant subtypes. In addition, each experimentally determined structure only represents one of many accessible receptor conformations, which is a major limitation if the aim is to predict ligand selectivity. Second, docking scoring functions have not been developed to compare affinities for different binding sites.
Several strategies to identify selective GPCR ligands using structure-based virtual screening have been explored. In screens for subtype-selective ligands, the least computationally expensive approach is to dock a library to the target receptor and select compounds that form interactions with binding site residues that are not conserved in the antitarget (Negri et al., 2013; Wang et al., 2017). The advantage of this method is that the structure of the antitarget is not required. An alternative method is to perform docking screens against both the target and antitarget, followed by the identification of compounds that score better for the target (Rodriguez et al., 2014; Weiss et al., 2018). Known selective ligands can also be docked to gain knowledge regarding the structural basis of selectivity (Katritch et al., 2011; Ranganathan et al., 2015). However, as most docking programs either do not consider binding site flexibility or have very limited representation of induced fit, it is difficult to make accurate predictions. Even if a compound has an unfavorable docking score for the antitarget structure, in reality the binding pocket can rearrange and still bind the ligand with high affinity. For these reasons, it is not surprising that docking screens were unable to identify subtype-selective ligands, e.g., for the muscarinic (Kruse et al., 2013), orexin (Gunera et al., 2020), and opioid (Weiss et al., 2018) receptors. However, from a pragmatic point of view, the computational cost of filtering for specific interactions or performing docking to an additional structure is relatively small, and there are also examples of successful predictions of subtype-selective ligands using this approach, e.g., for the serotonin (Rodriguez et al., 2014) and chemokine (Schmidt et al., 2015) receptors. The approach is most likely to succeed if there is a well defined pocket or interactions that are accessible in the target, but not in the antitarget. To further improve prediction accuracy, multiple conformations of the antitarget can be considered to account for receptor flexibility (Ranganathan et al., 2015).
A screening library can be designed to favor the discovery of selective ligands. One successful study focusing on the serotonin receptors used machine learning to identify compounds with features similar to selective ligands, and the resulting library was docked to the receptor structure (Rataj et al., 2018). Ligand-based approaches can also be used to remove compounds from the library that are similar to ligands of the antitarget, reducing the probability of selecting such chemotypes (Weiss et al., 2018). As demonstrated by Ballante et al. (2020), public HTS data can be used to identify compounds that have been experimentally observed to lack activity at a large number of targets. Such a library of molecules was used to identify A2AR and D4R ligands with reduced off-target activity. An alternative to screening lead- or drug-like compounds is to start with a fragment library. A fragment screen against the target is first carried out to identify many starting points for optimization. In a second step, the identified fragments are optimized to achieve potency and selectivity (Vass et al., 2014a; Ranganathan et al., 2015).
There is an increasing interest in compounds that can modulate several GPCRs relevant for the same disease (Anighoro et al., 2014). Polypharmacology could lead to synergistic therapeutic effects, and multitarget activity is an essential property of many antipsychotic drugs (Roth et al., 2004). The prospects of virtual screening success will depend primarily on how closely related the targets are and whether they recognize similar compounds. Four structure-based virtual screening studies have used molecular docking to identify multitarget ligands. The first two studies focused on closely related chemokine (Schmidt et al., 2015) and aminergic (Weiss et al., 2018) receptors and successfully identified dual-target ligands. In these studies, crystal structures were not available for all targets, and homology models were used. Jaiteh et al. (2018) and Kampen et al. (2021) focused on GPCR and enzyme targets with disparate binding sites (A2AR and either monoamine oxidase B or D2R) and identified several ligands with potent dual-target activity. As new GPCR structures become available, it will be possible to screen large panels of targets and antitargets to identify leads with tailored selectivity profiles. At this point, the size of the screening library may be a limiting factor because the number of compounds that fulfill the desired activity profile will decrease as the number of targets and antitargets increases.
D. Can Docking Guide Optimization of Screening Hits for Affinity?
Hits from virtual screens often have weak activity and need to be optimized to become useful chemical probes or lead candidates. How can molecular docking contribute to the hit-to-lead optimization process? First, it is well established that docking scoring functions are rarely able to rank congeneric ligands by affinity (Warren et al., 2006). Instead, docking should be viewed as a tool to generate ideas regarding what analogs to test. In a few cases, crystal structures of ligands have been used to guide structure-based ligand optimization (Hellmann et al., 2020), but typically the binding mode generated by docking is the only available model. The predicted complex can be helpful to identify key interactions with the receptor and provide insights into what regions of the binding site are not occupied. This knowledge can be an important guide in deciding what part of the chemical structure to modify. The predicted complex should first be analyzed in detail to assess whether alternative binding modes are possible. In this case, testing compounds that are predicted to be inactive based on the model of the complex can also be informative to judge its accuracy (Kolb et al., 2009; Rodriguez et al., 2014). The most common and straightforward approach to obtain initial SAR is to identify similar compounds in commercial chemical libraries, which often has led to the identification of ligands with improved activity (Manglik et al., 2016; Ranganathan et al., 2017; Jaiteh et al., 2018). Considering that there are now billions of commercially available make-on-demand compounds, the prospects of performing rapid optimization of hits have improved substantially in recent years (Lyu et al., 2019; Grygorenko et al., 2020).
In the hit-to-lead optimization process, other useful structure-based methods can complement the docking calculations. One example is the analysis of binding site hydration networks to identify ordered waters that can be displaced to gain ligand affinity, which has been applied to several GPCRs (Higgs et al., 2010; Mason et al., 2013; Sabbadin et al., 2014; Kuhne et al., 2016). To make more quantitative predictions, more rigorous computational methods can also be applied. MD simulations and free-energy calculations are substantially more computationally demanding methods but should in theory be able to predict relative binding affinities with higher accuracy than docking scoring functions. MD simulations have already played an important role in understanding the mechanism of GPCR activation (Latorraca et al., 2017), and recent reviews summarize the state of the art of binding affinity prediction methods (Cournia et al., 2017). Increasing computational power and more automated tools to prepare such calculations make it possible to use free-energy simulations in prospective ligand discovery studies, and there are already a few examples of applications to GPCRs (Lenselink et al., 2016b; Matricon et al., 2017; Deflorian et al., 2020; Jespers et al., 2020; Egyed et al., 2021).
E. Is an Experimentally Determined G Protein–Coupled Receptor Structure Required to Perform a Virtual Screen, or Can a Homology Model Be Used?
Community-wide assessments of GPCR structure prediction demonstrated that homology model accuracy is strongly dependent on access to a structure of a closely related receptor. In order for a homology model to be useful in virtual screening, the binding site structure must be well predicted. The three GPCR Dock assessments, which challenged modelers to blindly predict the structures of the A2AR, D3R, CXCR4, 5-HT1BR, 5-HT2BR, and SMO, provide some guidelines regarding when to consider using homology models in virtual screening (Michino et al., 2009; Kufareva et al., 2011, 2014). If only distant templates (TM sequence identity <30%–35%) were available, it was difficult to generate good models of the binding site. In such cases, it is unlikely that the model will be useful in virtual screening. Good binding site models were obtained if structures of closely related receptors (TM sequence identity >35%–40%) were available and if the target and template recognize similar compounds. Based on benchmarking studies and prospective screens, the same virtual screening success rates with a homology model as with a crystal structure can be expected at this level of model quality (Carlsson et al., 2011; Jaiteh et al., 2020). However, as several details of the binding site structure (e.g., loop regions) may not be modeled accurately, prediction of the efficacy or selectivity of the compounds will be more difficult compared with using a high-resolution experimental structure (Rodriguez et al., 2014; Weiss et al., 2018). Virtual screening performance of homology models can be enhanced when known actives are available. Models generated with different methods and/or templates can then be evaluated by calculating the ligand enrichment, and the structure with the optimal performance can be used in the prospective virtual screen (Lim et al., 2018; Costanzi et al., 2019; Jaiteh et al., 2020). Most of the successful virtual screens based on homology models used a structure of a closely related receptor as template (Carlsson et al., 2011; Kolb et al., 2012; Vass et al., 2014b; Lam et al., 2015; Ranganathan et al., 2015; Szőllősi et al., 2015; Kaczor et al., 2016; Weiss et al., 2018). If only distant templates are available, the binding site model will generally be poor, and the chances of virtual screening success are lower. For example, virtual screening using homology models of the A2AR (Langmead et al., 2012) and CXCR4 (Mysinger et al., 2012b) based on distant templates resulted in hit rates that were >4-fold lower than subsequent screens based on crystal structures (Carlsson et al., 2010; Mysinger et al., 2012b). Notable examples of docking screens that identified ligands using homology models based on distant templates are the discovery of modulators of orphan receptors MAS related GPR family member X2 (MRGPRX2), GPR65, and GPR68 (Huang et al., 2015; Lansu et al., 2017).
V. Conclusions
The rapidly increasing structural coverage of the GPCR family provides ample opportunities to use virtual screening to discover ligands of these therapeutically important targets. Our summary of successful molecular docking screens shows that this approach can contribute to the discovery of novel leads for a wide range of GPCRs. The chances of identifying GPCR ligands by molecular docking are not only dependent on the screening approach itself but also heavily influenced by the nature of the binding site and the composition of the chemical library. In favorable cases, the efficacy and selectivity profiles of the ligands can also be predicted, but there is a clear need for more accurate computational methods in this area. The application of virtual screening to the large number of GPCR structures that have recently been solved has the potential to contribute to the discovery of chemical probes, which can yield insights into the biological functions of GPCRs and facilitate drug discovery.
Acknowledgments
The authors thank past and present members of Jens Carlsson’s research group for helpful discussions. The authors also thank Peter Kolb, Jonathan Mason, and Stefano Costanzi for providing coordinates of predicted ligand binding modes.
Authorship Contributions
Participated in research design: Ballante, Carlsson.
Performed data analysis: Ballante, Kooistra, Kampen, de Graaf, Carlsson.
Contributed to the writing of the manuscript: Ballante, Kooistra, Kampen, de Graaf, Carlsson.
Footnotes
J.C. has received funding from the European Research Council under the European Union’s Horizon 2020 Research and Innovation Programme [Grant Agreement 715052]. The work was also supported by grants from the Swedish Research Council [2017-04676], the Swedish strategic research programme eSSENCE, and the Science for Life Laboratory to J.C., F.B., A.J.K., S.K., and C.d.G. participate in the European COST Action CA18133 ERNEST (European Research Network on Signal Transduction). This article/publication is based upon work from COST Action ERNEST (CA18133), supported by COST (European Cooperation in Science and Technology, www.cost.eu).
No author has a conflict or perceived conflict of interest with the contents of this article.
↵
 This article has supplemental material available at pharmrev.aspetjournals.org.
This article has supplemental material available at pharmrev.aspetjournals.org.Participated in research design: Ballante, Carlsson.
Performed data analysis: Ballante, Kooistra, Kampen, de Graaf, Carlsson.
Contributed to the writing of the manuscript: Ballante, Kooistra, Kampen, de Graaf, Carlsson.

Abbreviations
- 2D
- two-dimensional
- A1R, A1
- adenosine receptor
- A2AR, A2A
- adenosine receptor
- BLT1
- leukotriene B4 receptor 1
- CCR
- chemokine (C-C motif) receptor
- cryo-EM
- cryo-electron microscopy
- CXCR4
- chemokine (C-X-C motif) receptor 4
- CysLT1R
- cysteinyl leukotriene receptor 1
- CysLT2R
- cysteinyl leukotriene receptor 2
- DOR
- δ-opioid receptor
- D2R
- D2 dopamine receptor
- D3R
- D3 dopamine receptor
- D4R
- D4 dopamine receptor
- ECD
- extracellular domain
- ECL
- extracellular loop
- ECV
- extracellular vestibule
- Emax
- maximal effect
- EP
- prostaglandin E receptor
- FFA1R
- free fatty acid receptor 1
- GLP
- glucagon-like peptide receptor
- GPCR
- G protein–coupled receptor
- GPR
- G protein-coupled receptor
- H8
- C-terminal helix 8
- H1R
- H1 histamine receptor
- H4R
- H4 histamine receptor
- 5-HT2AR
- 5-hydroxytryptamine receptor 2A
- 5-HT1BR
- 5-hydroxytryptamine receptor 1B
- 5-HT2BR
- 5-hydroxytryptamine receptor 2B
- HTS
- high-throughput screening
- KOR
- κ-opioid receptor
- LSD
- lysergic acid diethylamide
- LT
- leukotriene
- MAO-B
- monoamine oxidase B
- MD
- molecular dynamics
- mGlu1
- metabotropic glutamate receptor 1
- mGlu5
- metabotropic glutamate receptor 5
- MOR
- μ-opioid receptor
- M2R
- M2 muscarinic (acetylcholine) receptor
- M3R
- M3 muscarinic (acetylcholine) receptor
- MR
- muscarinic (acetylcholine) receptor
- MT1R
- MT1 melatonin receptor
- MT2R
- MT2 melatonin receptor
- NAM
- negative allosteric modulator
- NOP
- Nociceptin/Orphanin FQ receptor
- NTS1R
- neurotensin receptor 1
- OX1R
- OX1 orexin receptor
- OX2R
- OX2 orexin receptor
- PAM
- positive allosteric modulator
- PAR2
- proteinase-activated receptor 2
- P2Y1
- P2Y1 receptor
- β1R
- β1 adrenergic receptor
- β2R
- β2 adrenergic receptor
- RMSD
- root-mean-square deviation
- SAR
- structure-activity relationship
- SMO
- smoothened
- SPR
- surface plasmon resonance
- TM
- transmembrane (helix)
- ZINC
- ZINC is not commercial
- Copyright © 2021 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.
References
In this issue





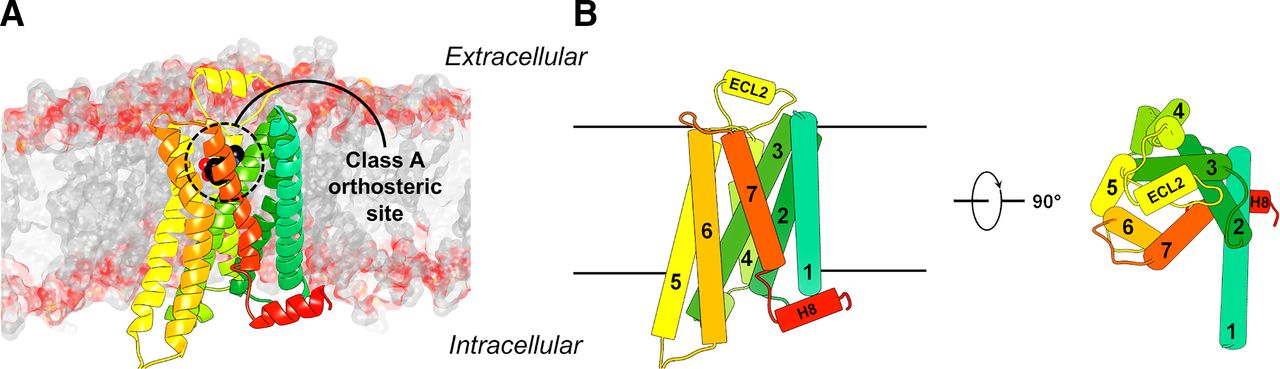{kind=link}
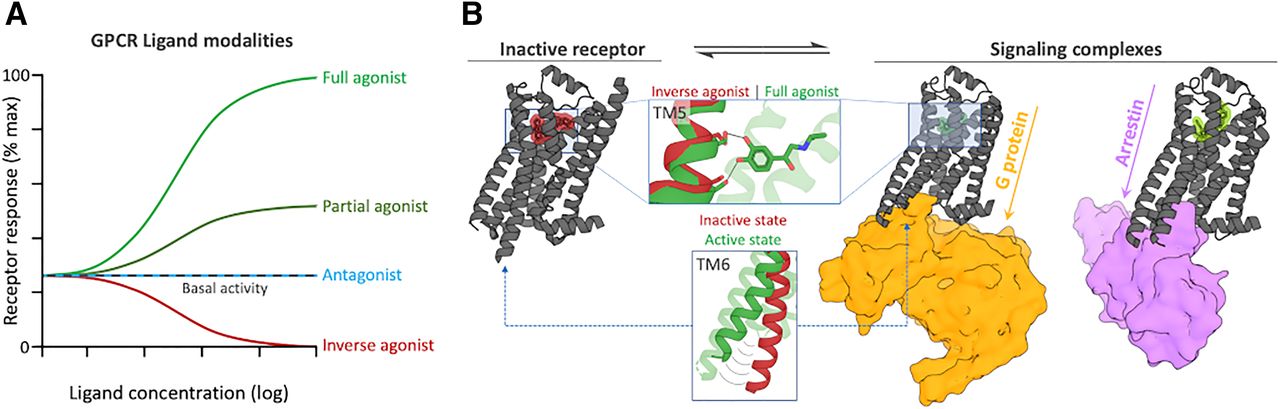{kind=link}
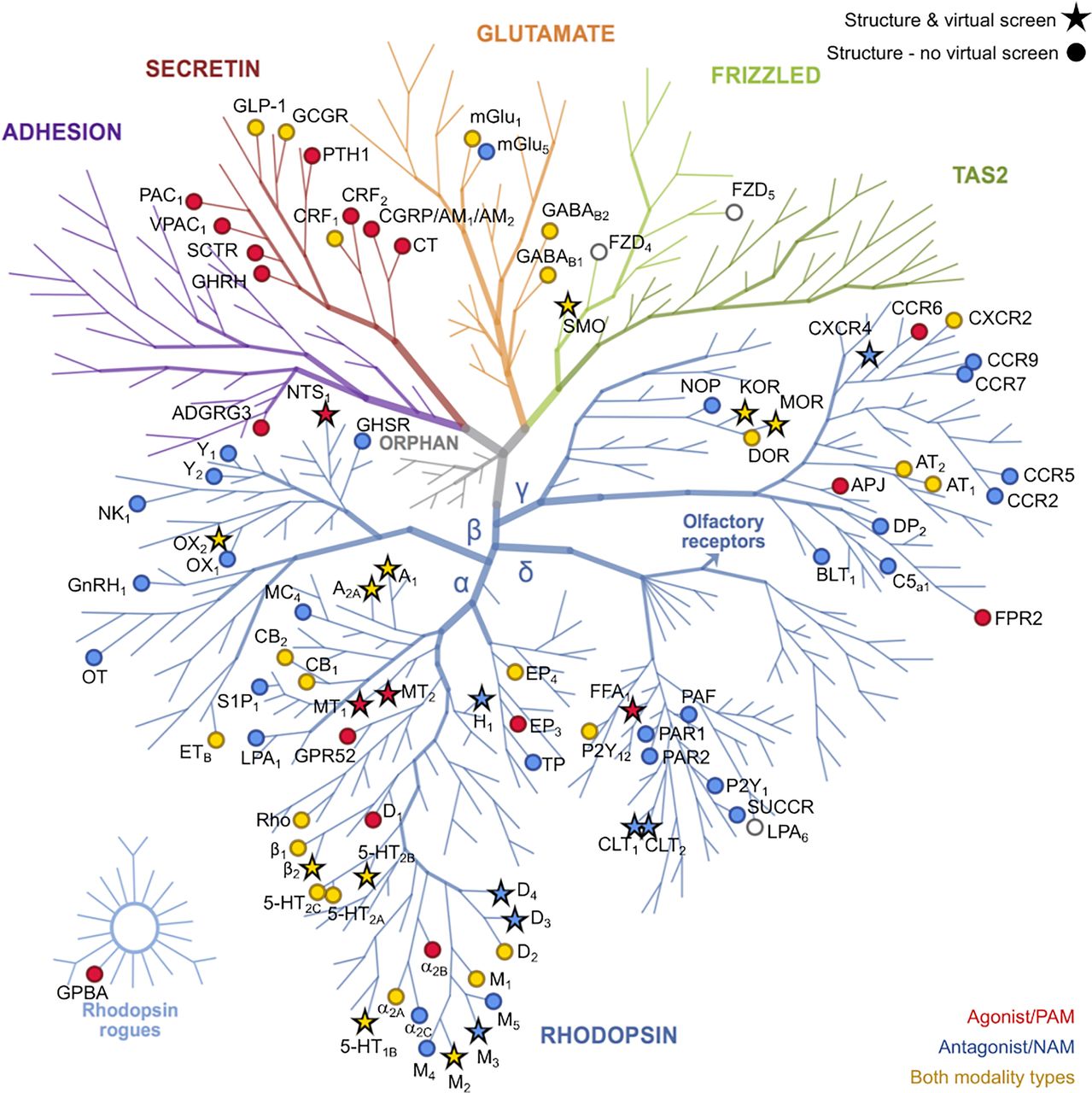{kind=link}
{kind=link}
{kind=link}
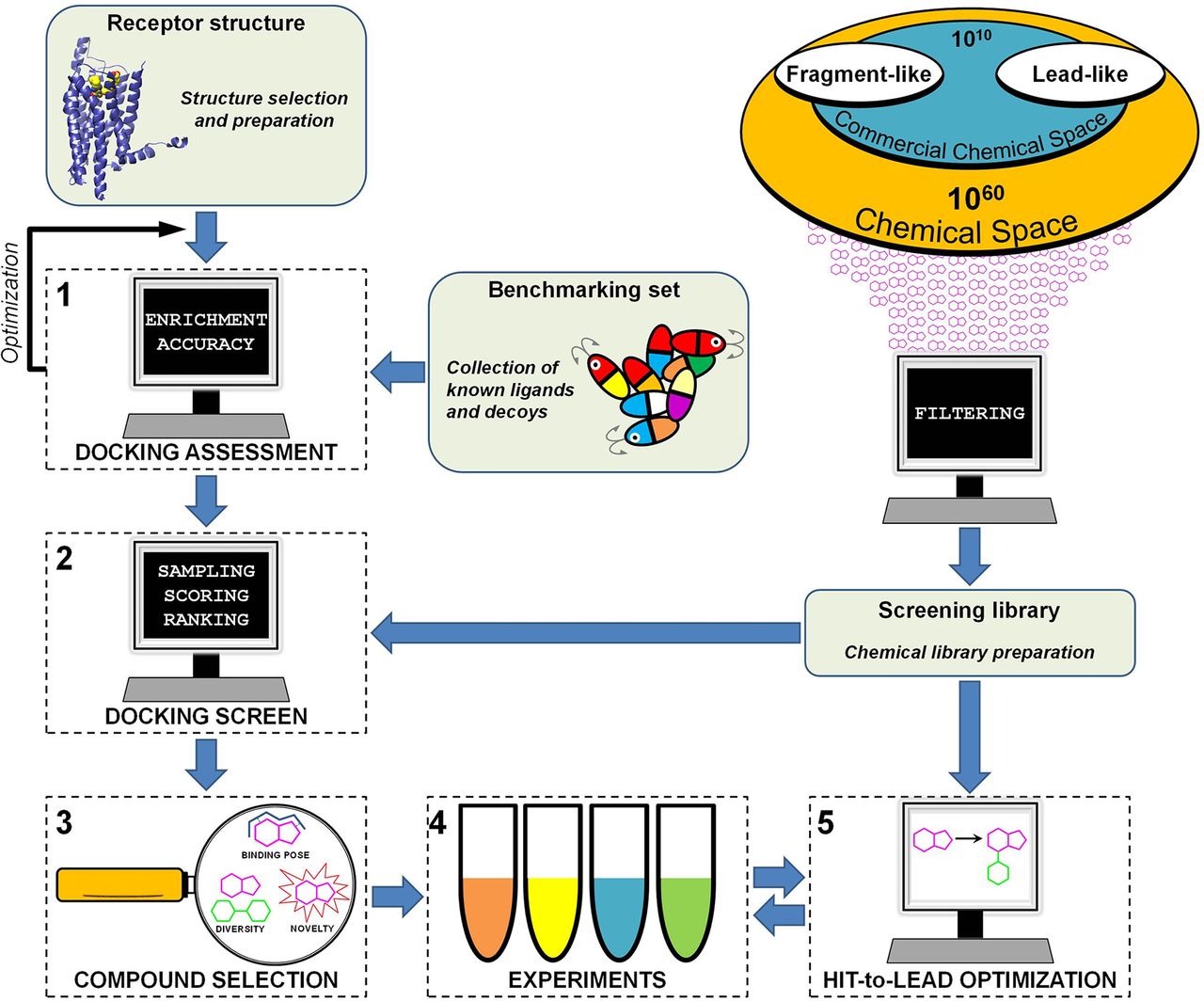{kind=link}
{kind=link}
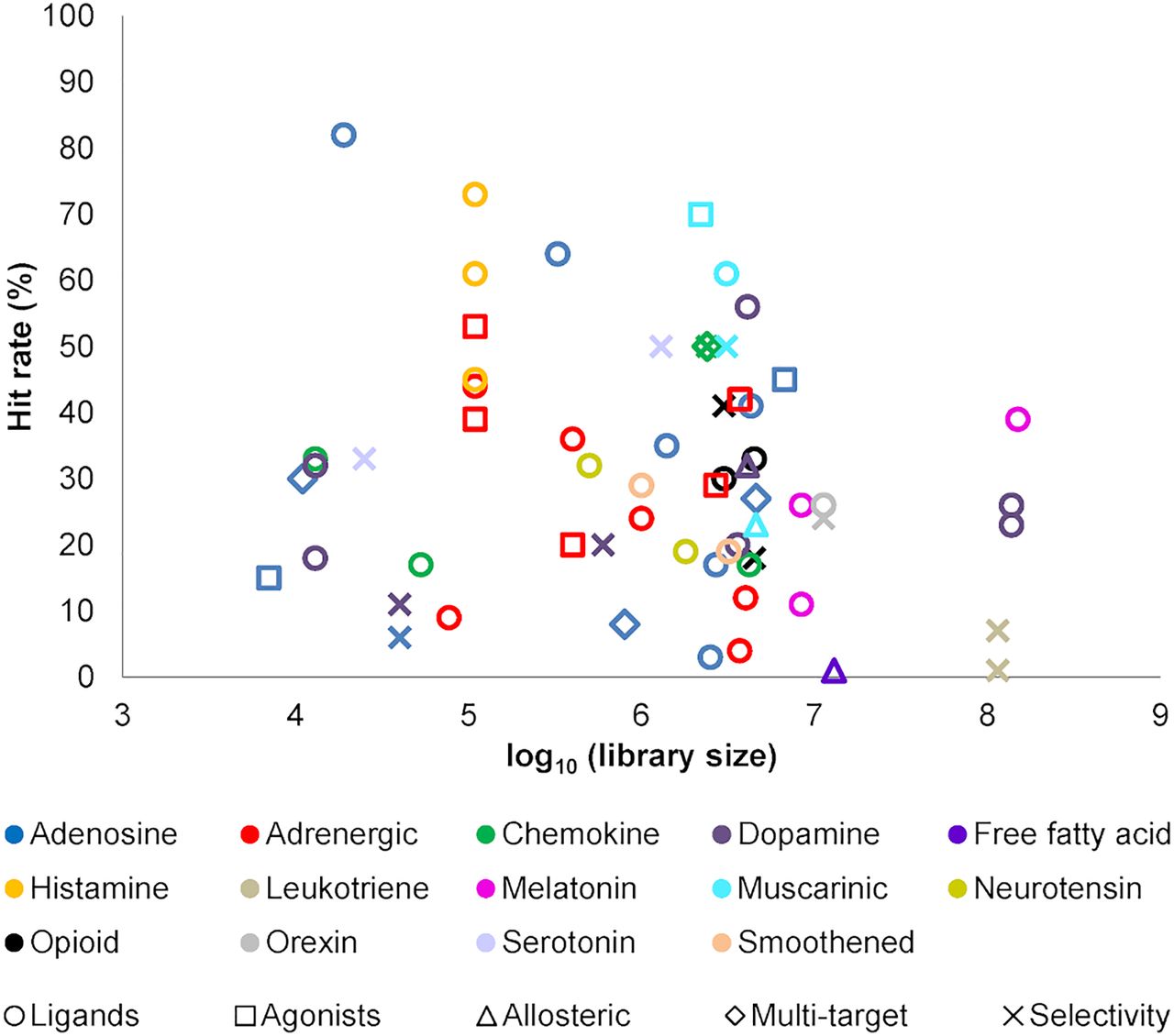{kind=link}
{kind=link}
{kind=link}
{kind=link}
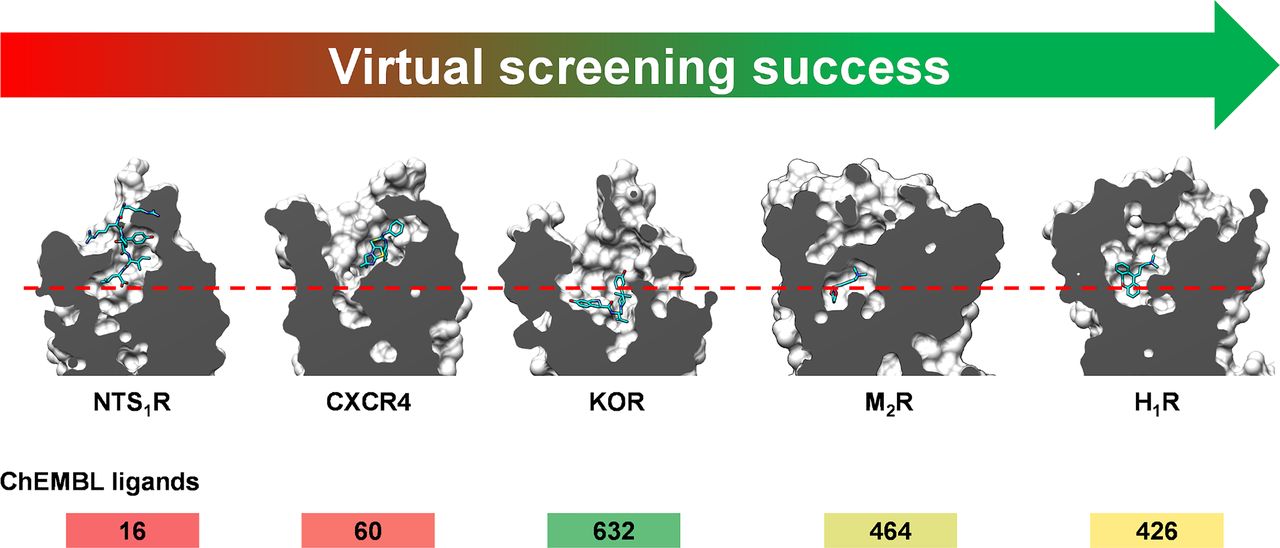{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction to G Protein–Coupled Receptors: Pharmacology and Structural Biology
- II. Molecular Docking: Methodology and Virtual Screening Workflow
- III. Success Stories: Discovery of G Protein–Coupled Receptor Ligands by Molecular Docking Screening
- IV. Opportunities and Limitations: What Can Molecular Docking Do for You?
- V. Conclusions
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters
- PDF + SI