


Abstract
The purpose of this review is to summarize essential pharmacological, pharmaceutical, and clinical aspects in the field of orally inhaled therapies that may help scientists seeking to develop new products. After general comments on the rationale for inhaled therapies for respiratory disease, the focus is on products approved approximately over the last half a century. The organization of these sections reflects the key pharmacological categories. Products for asthma and chronic obstructive pulmonary disease include β-2 receptor agonists, muscarinic acetylcholine receptor antagonists, glucocorticosteroids, and cromones as well as their combinations. The antiviral and antibacterial inhaled products to treat respiratory tract infections are then presented. Two “mucoactive” products—dornase α and mannitol, which are both approved for patients with cystic fibrosis—are reviewed. These are followed by sections on inhaled prostacyclins for pulmonary arterial hypertension and the challenging field of aerosol surfactant inhalation delivery, especially for prematurely born infants on ventilation support. The approved products for systemic delivery via the lungs for diseases of the central nervous system and insulin for diabetes are also discussed. New technologies for drug delivery by inhalation are analyzed, with the emphasis on those that would likely yield significant improvements over the technologies in current use or would expand the range of drugs and diseases treatable by this route of administration.
Significance Statement This review of the key aspects of approved orally inhaled drug products for a variety of respiratory diseases and for systemic administration should be helpful in making judicious decisions about the development of new or improved inhaled drugs. These aspects include the choices of the active ingredients, formulations, delivery systems suitable for the target patient populations, and, to some extent, meaningful safety and efficacy endpoints in clinical trials.
I. Introduction
Over the last 50 years, we have witnessed the emergence of modern oral inhalation treatments of respiratory diseases. These became an essential and often the predominant pharmacotherapeutic intervention for the affected patients. Although the origins of therapeutic inhalations can be traced to ancient times, the modern products reflect half a century of transformational advances in the quality of the active ingredients, formulations, and delivery devices. The focus of this review is the products developed and approved during this period. To capture the essence of knowledge and experience accumulated during this process that resulted in the approval and general acceptance of these therapies, the contributions to this review were written by active participants in the development of many of the approved products.
The rationale for the inhaled administration of medicines is provided in Section II, and this is followed by presentation of typical criteria, processes, and methods for selection of drugs for this route of administration in Section III. The most common use of inhaled medicines is for asthma and chronic obstructive pulmonary disease (COPD).
The key pharmacological categories of products for these diseases as well as their combinations are presented in Sections IV–VIII. The respiratory tract infections therapies using anti-infectives are discussed in Section IX. There has been only one inhaled protein so far approved for a respiratory disease—dornase α (recombinant human DNase) for cystic fibrosis (Section X). Mannitol, another “mucoactive” drug for cystic fibrosis, is covered in Section XI. Inhaled prostacyclins are so far the only class of inhaled medicines approved for pulmonary arterial hypertension (Section XII). The challenging treatment of ventilator-supported, prematurely born infants with lung surfactants is discussed in Section XIII.
Approved orally inhaled aerosol products (i.e., excluding inhalation gases) for systemic delivery of drugs are reviewed (Section XIV). As the reason for such therapies is very different from the use of this route for respiratory diseases, the choice of suitable candidate drugs, formulations, and devices requires a somewhat different approach.
Future advances are necessary to enable drug delivery by inhalation to a wider range of patients and for administration of a greater variety of therapeutic options. The review builds on the successes and discusses the shortcomings of the past to offer general guidance on the suitability of therapeutic candidates for inhalation delivery. This review also presents novel technologies that may be enabling or an improvement over the formulations and devices in common use today (Section XV).
The readers are referred to excellent references on the history of inhalation medicines (Clark, 1995a; Sanders, 2007; Stein and Thiel, 2017). There are also comprehensive reviews of delivery technologies available (Dhand, 2018; de Boer et al., 2018).
Indeed, in this review we often make use of the extensive literature, including prior reviews, of the topics covered in this manuscript. Our primary goal is to aid in decisions about the future development and approval of inhaled therapies rather than providing an exhaustive review of all the elements of basic sciences, engineering, and clinical research required in this process. For example, because of the scarcity of validated animal models of respiratory diseases to study the efficacy of potential candidates for their treatments by inhalation, we have deliberately avoided this topic, as no general guidance can be given (Sécher et al., 2020). In contrast, the requirements for preclinical toxicology testing of inhaled therapies generally follow the strict regulatory guidelines. Publications on these topics are available (e.g., Tepper et al., 2016), but the interested reader should consult also the current regulatory guidelines pertinent to the particular type of product under consideration (e.g., small molecules, biologics, or nicotine-based products that at present require a somewhat different battery of preclinical toxicology tests vs. drug products that use previously approved drugs that generally have somewhat abbreviated toxicology requirements compared with new chemical entities).
It would have been much beyond the scope of this review to attempt to capture many of the exciting developments in orally inhaled therapies, such as new experimental therapies in preclinical and clinical testing stages.
II. Why Inhaled Medications?
Humans need to breathe air containing oxygen continuously to stay alive. The respiratory tract is the only organ of the human body in which we have limited ability to protect it against the constant barrage of insults from the environment. Unlike clean water, which is taken for granted as being an essential item for each individual, the control of air quality is not viewed in the same fashion so far (Boyd, 2019).
The human respiratory tract has a complex, multilayer system protecting against unwanted visitors: naso- and oro-pharynx together with the branching airways act as filters particularly for the larger particles and are further aided by the mucociliary clearance system to remove the deposited particles (Gonda, 1997). Macrophages and other endocytotic cells are also capable of removal and digestion of potentially toxic entrants that succeed to penetrate the earlier barriers. When the protective mechanisms in the respiratory tract are overloaded, unprepared for a new challenge, or somehow compromised, respiratory diseases may follow.
The respiratory tract has a highly developed immune system connected to the lymphatics and blood circulation. It also has some metabolic capacity to remove toxic materials (Holt et al., 2008; Hartl et al., 2018). However, the rapid spread of the SARS-CoV-2 virus primarily transmitted via inhalation is one of the starkest recent examples of just how vulnerable our respiratory tract is. Respiratory diseases have been on the rise both in absolute numbers of affected individuals as well as a proportion of the overall disease burden for a multitude of reasons, including aging population, air pollution, and ease of international travel in the case of respiratory infections in particular (Forum of International Respiratory Societies, 2017).
Since the human lung comprises only a relatively low mass of the body—typically 1 kg in an adult—treating respiratory disease via oral or intravenous delivery may not be very efficient and may require systemic doses that cause side effects at off-target locations unless there is a highly favorable partition between blood and lung tissues. Indeed, achieving adequate concentrations of large molecules, such as proteins, in the airway lumen may be impossible by any other route than inhalation because of poor permeability of the endothelial and epithelial barriers to macromolecules.
In general, inhaled medications can be rapidly and “noninvasively” delivered to the respiratory tract and may have beneficial therapeutic effects there with doses that cause low exposure to the rest of the body to minimize systemic adverse effects. Indeed, a lot of knowledge presented in subsequent sections has been accumulated on the “ideal” characteristics of drugs delivered by inhalation.
Inhalation for the treatment of respiratory diseases as the preferred route of administration is therefore used for one or more of the following reasons:
1) The therapeutic agent cannot reach adequate concentrations in the lung given systemically. An example of this would be the protein drug recombinant human DNase (dornase α) used as inhalation for the treatment of cystic fibrosis (Section X).
2) There is significant systemic toxicity of the therapeutic agents when given at doses that reach therapeutic concentrations in the lung. Examples are aminoglycoside antibiotics (Section IX), in which the efficacious inhaled doses are significantly lower than the equipotent doses that would have to be given systemically but would cause serious systemic side effects administered chronically.
3) The drug has poor oral bioavailability and therefore would need to be injected to exert its effect in the lung. Inhaled aminoglycosides (Section IX), cromones (Section VIII), and lung surfactant (Section XIII) are such examples.
4) A rapid onset of action is needed such as, for example, with short-acting bronchodilators (Section IV).
There is potentially another reason—if the systemic delivery to achieve therapeutic effects in the lung would require doses that would not be economically viable, such as for macromolecules (e.g., peptides, proteins, RNA and DNA therapies) that often show poor transport efficiency between the blood and lung compartments.
Of course, careful consideration needs to be given prior to embarking on the development of an inhaled product (Section III), especially vis a vis the oral ingestion route, which is preferred by most people, or convenient long-acting injectable forms that might be a more attractive alternative for some patients.
As described in the following sections of this review, there are technical and biologic constraints on the development of orally inhaled products (Section III). Some of the challenges are precisely because of the innate barriers to entry of “foreign” particles and their disposition after landing in the respiratory tract. Others have to do with both nonspecific and specific causes of irritation and long-term toxicity of certain types of molecules in the respiratory tract. For example, cough is a common defense mechanism and a frequent side effect of inhaled drugs.
Inhalation products are more complicated to use than oral or transdermal dosage forms. Therefore, judicious approach needs to be taken before selecting the inhalation route over other alternatives. Different types of devices have different instructions for use that can be confusing especially for patients on multiple different inhalation devices. Lack of compliance with proper use of inhalation devices, including the correct mode of breathing, has been a challenge for decades, with overall nonadherence of ∼50%. The flip side is that this is also a great opportunity for development of new inhalation systems that make them much less reliant on the person’s dexterity to use them through innovative formulations and devices (Section XV), including those with electronics and web connectivity (Gonda, 2019a).
Of special note here is the use of the inhalation route deliberately for administration of drugs for systemic effects (Section XVI). Here, the choice of the inhalation route needs to have a higher level of scrutiny, as the competitive landscape is generally much greater, and the ratio of the concentrations between the respiratory tract and the rest of the body becomes a disadvantage if pulmonary toxicity risks outweigh the benefits of noninvasive delivery or better bioavailability than the oral route. Nevertheless, there have been historically undeniable successes, most notably the use of inhalation anesthetics.
The sections on the approved orally inhaled products for various respiratory diseases in this review serve as examples of therapies wherein the test of time proved them to be valuable medicines.
Given the large and growing burden of respiratory diseases around the world, there has never been a greater need to develop new or improved inhaled therapies when the inhalation administration makes most sense for the target population of patients.
III. Critical Attributes of Orally Inhaled Drug Products for Treatment of Respiratory Diseases
A. Introduction
As described in the previous section, inhaled drug products may have significant benefits for the treatment of respiratory diseases in comparison with systemically administered drugs. Since desirable drug and drug-product attributes of topically active inhaled drugs differ in many aspects from those of oral drug products, these benefits are best realized when a drug molecule is specifically designed for the inhaled route (e.g., Bonn and Perry, 2021). However, there are several examples of drugs that were successfully “repurposed” for inhalation to improve efficacy and safety when treating respiratory conditions (see, for example, inhaled anti-infectives, Section IX). There are also examples of drugs (often biomolecules) in which low oral bioavailability necessitated inhalation delivery (e.g., Sections VIII and X).
This section focuses on desirable drug and drug-product attributes of orally inhaled drug products intended for the treatment of local respiratory diseases. Emphasis is placed on those attributes for which the desired properties are either different from those for “ideal” orally ingested drugs or in which characterization of the attributes requires specific techniques or considerations. In addition, strategies for optimizing inhaled drugs and drug products as well as techniques for characterizing critical drug and drug product attributes are briefly discussed. In this context, specific emphasis is placed on emerging techniques, such as clinically relevant test methods for the characterization of inhaled drug product performance as well as the use of computational tools to aid optimization of drug and drug-product performance.
B. Critical Drug Attributes and Points to Consider during Drug Optimization
The definition of a candidate drug target profile may be considered the first step on the path to a new inhaled drug. The actual targets and to some extent the strategies for achieving target properties are by necessity depending not only on the specific reason for selecting inhalation in the first place (Section II) but also strongly depending on therapeutic target, drug classes, and intended patient population. It is also wise at this stage to consider adding targets which address any limitations posed by available drug delivery systems. Such limitations may be related to insufficient moisture protection, need for compatibility with critical excipients, and packaging material (de Boer and Thalberg 2021a,b,c).
As outlined by Bonn and Perry (2021), designing an inhaled compound for topical treatment aims to maximize local exposure while minimizing systemic exposure. To achieve this, an inhaled drug should preferably have attributes providing: 1) retention in lung as to optimize local exposure and to provide a reasonable dosing regimen, 2) high potency to limit the need for high therapeutic doses, and 3) low oral bioavailability to reduce systemic exposure (Table 1). These properties and the principles by which they are achieved are often in direct contrast to attributes characterizing orally administered drugs (Lipinski et al., 2001). For instance, novel inhaled drugs [e.g., vilanterol (Procopiou et al., 2010) and fluticasone furoate (Biggadike et al., 2008)] are purposely designed with metabolic liabilities severely reducing oral bioavailability to limit systemic exposure from the swallowed dose. Therefore, inhaled drugs possess a somewhat different and wider distributed physicochemical property space than that proposed for oral drugs by Lipinski et al. (2001).
Critical drug attributes: inhaled drugs for treatment of respiratory diseases
The most important of the key attributes listed in Table 1 and probably the most challenging to build into a novel inhaled drug is lung retention. As shown in Table 1, lung retention can be achieved by a variety of mechanisms. Some of these, chiefly slow dissolution and strong tissue interactions, tend to reduce free drug concentrations favoring highly potent drugs. Novel β2-agonists have, for example, EC50 values in the nanomolar range (Procopiou et al., 2010). Low permeability could also enhance the impact of low solubility and tissue interactions. For example, Shaw et al. (2016) speculated that low permeability in combination with tissue interactions caused by basic amines was the key factor enabling lung retention for a series of platelet-derived growth factor receptor inhibitors. Interestingly, low permeability also results in very long residence time for large molecules, such as proteins, antibodies, and DNA- and RNA-based drugs (Adjei and Gupta, 1997). In addition, permeability may potentially be decreased with appropriate selection of counter-ions forming noncovalent ion pairs increasing polar surface area (Dutton et al., 2020). Ultimately, selection of a strategy for optimization of lung retention is intimately dependent on the available chemotypes and the location of the target. For instance, low permeability retains the drug in lumen and may thus favor drugs like the antimicrobials, whereas it may be detrimental for intracellular targets like the glucocorticoid receptor.
During lead optimization, in vitro tools to screen candidate drug compounds for target affinity, metabolism, and toxicity are akin to those applied for systemically active drugs. However, preclinical animal models designed to assess local efficacy and PK as well as local safety differ in that they normally require the drug to be administered topically. Hence, local concentrations in airways depend not only on total (body burden) dose administered but also on the distribution of drug. As discussed in a recent review (Sécher et al., 2020), no animal models perfectly mimic human disease, physiology, anatomy, and, indeed, any relevant clinical drug administration technique. This could result in differences in drug deposition profiles as well as species differences in rate of dissolution, mucociliary clearance, and permeation (Sécher et al., 2020). For instance, whereas inhalation may give the most clinically relevant drug distribution to the pulmonary region in humans, only a fraction [in rats typically 10% (Phillips, 2017)] of the inhaled dose reaches the lung. The remainder deposits in the rat nose region and will be absorbed from the nose or swallowed and thus available for gastrointestinal absorption into the blood circulation. Administration directly to the lung of animals avoids this problem (e.g., by instillation into the trachea) but can create other difficulties associated with a lung distribution pattern that is less clinically relevant (Phillips, 2017). In addition, some techniques, such as intratracheal instillation, require sedation of the animal, which may alter the functionality of absorptive and nonabsorptive clearance processes in lung (e.g., Wollmer et al., 1990). Naturally, this limits their predictiveness and suggests that the choice of disease model and species as well as administration technique must be carefully considered.
The disposition of poorly soluble drugs is generally sensitive to the state of the drug (crystalline, amorphous, or solubilized), their specific surface area (particle size), and the dose over solubility ratio [dose number (Amidon et al., 1995)]. Hence, high local solid drug concentrations in the respiratory tract of, for example, rodents at lung doses greatly exceeding equivalent human doses may cause artifacts not expected to be present in humans at clinical doses wherein the drug could have ample fluid in the lung to dissolve. Taken together, these challenges require careful design of the lead optimization and preclinical safety assessment programs and the test models used to enable successful translation of preclinical data into the clinical setting.
Finally, a successful inhaled therapeutic requires not only that the drug be tolerable, safe, and effective but also that it be compatible with a suitable delivery system. This is normally evaluated prior to selecting a candidate drug for clinical development. At this stage, it is important to consider the target patient population (e.g., their inspiratory flow rates and lung volumes), the predicted therapeutic dose, and commercial constraints and opportunities as well as available device and formulation platforms (e.g., intellectual property and barriers and opportunities and in-house capabilities). For instance, a drug that is to be administered in a dry powder–type inhalation device should preferably be compatible with the available formulations and processes (e.g., particle size reduction). This could include considering dose limitations as well as the factors affecting chemical and physical stability of the solid drug (e.g., excipient compatibility, melting/glass transition temperature, impact of humidity) (Shetty et al., 2020).
In conclusion, critical drug-attribute targets are different for drugs designed for typical treatment via inhalation compared with drugs designed for systemic treatment. High potency and effect duration as well as topical safety and tolerability are obtained through optimization of drug and formulation attributes governing high lung retention, optimal target interactions, and low oral bioavailability. These properties are preclinically evaluated in in vitro and in-animal models after local administration. Translation of preclinical results into a clinical setting must therefore consider differences between preclinical and clinical inhalation devices and formulations.
Ideally, formulations used in a preclinical test program should be similar to those intended for the clinical program. However, this is generally not possible for more complex formulations given that limited substance amounts are available at this stage. Then a scientifically sound bridging strategy should be evolved to ensure that the data obtained with different formulations during the development are valid for development in humans. Furthermore, compatibility between a candidate drug and the prospective commercial inhalation device should be carefully considered, and since the performance of an inhalation device will likely be dependent on the detailed properties of the formulation, an early decision on the device-formulation combination is encouraged.
C. Critical Drug Product Attributes and Points to Consider during Development
The clinical development phase starts once a suitable candidate drug molecule has been selected based on appropriate pharmacological activity, prepared in a form suitable for inhalation and found to be tolerable and safe in animals. Clearly, it needs to be amenable at this stage for inhalation delivery to humans. The clinical program is currently often a protracted process starting with assessing drug tolerability in man via assessment of therapeutic effect, dose response, and dosing regimen in patients to large safety studies in patients. During this process, clinically suitable test products must be developed and tested, ultimately resulting in the final commercial product. The latter also involves tests and studies aimed at understanding patient use and patient compliance with intended product use (FDA, 2016). Therefore, the specific target population of patients must be defined (e.g., adult patients with severe asthma). A key difference in the overall regulatory strategy for orally inhaled versus orally ingested products, such as tablets, is that currently there is no universally agreed methodology to bridge between the formulation and devices used at different stages in the development of inhaled products.
What they have in common with other dosage forms is that inhaled products have attributes and specification limits that control the quantity and quality of the dose to the patient, including its chemical and physical properties during the delivery and the product shelf-life. However, as shown in Table 2, inhaled products have specific critical requirements with respect to the assessment of dose and performance attributes. Common to all inhaler products is that they generate an aerosol that is at least to some extent capable to reach the lung and thus become available at the therapeutic target site. The lung dose and the dose deposition pattern that determines the therapeutically relevant dose depend on the aerodynamic particle size distribution (APSD) of the aerosol and the patient inhalation profile. The APSD intimately depends on the interactions between the formulation, the inhalation device, and, for most products, the inhalation maneuver of the subject. Hence, controlling dose encompasses not only an assessment of the administered dose (the body burden dose) but also characterization and control of the therapeutically available dose. The latter is unique for inhalation and is in practice controlled by specifications on APSD (as measured by a cascade impactor at standardized conditions). Hence, as alluded to above, changes to any product component (device and formulation) could potentially change the therapeutic dose even if the body burden dose is maintained.
Critical drug product attributes: inhaled drugs for treatment of respiratory diseases
Furthermore, as outlined in a recent review by Amini and Hochhaus (2021), there is increased evidence that the local and systemic exposure after inhalation of poorly soluble drugs (such as some of the glucocorticosteroids) is dependent not only on the therapeutic dose but also on the dissolution rate of the active drug. Methods to assess the dissolution rate of an inhaled aerosol are not yet standardized and must be developed fit for purpose.
The selection of an appropriate inhalation platform for a commercial product is a complex undertaking balancing clinical considerations (patient characteristics and needs), commercial aspects (e.g., cost, development risk/time, and marketing considerations), availability of technology (intellectual property and in-house competence), regional regulatory requirements, and, naturally, the drug product properties (e.g., dose size and stability).
Inhaled product delivery technologies can roughly be divided into dry powder inhalers (DPIs), pressurized metered dose inhalers (pMDIs), and nebulized or soft-mist aqueous formulations (de Boer and Thalberg 2021a,b,c).
Breath-actuated DPIs are widely used for treatment of respiratory disorders. The developments of DPIs were driven partly as user-friendly alternatives to chlorofluorocarbon (CFC)- and hydrofluoroalkane (HFA)-propellant–based pMDIs and to overcome some of the difficulties with coordination of pMDI actuation and inspiration. The three main DPI systems are capsule-based premetered single-dose devices, multiunit dose inhalers (factory-metered), and multiple-dose inhalers wherein the patients activate an in-built mechanism that meters out a dose of powder formulation from a reservoir of powder (Usmani, 2019). Aerolizer (e.g., Breezehaler, Novartis) and RS-01 Monodose, Plastiape’s low-resistance Cyclohaler (Plastiape), are some of several refinements of the original capsule-based Cyclohaler (Pharmachemie). The original Diskhaler or the “double disk” Ellipta inhaler devices (Glaxo) are widely used factory-filled multidose inhalers as well as a number of later developed generics of Diskhaler. Turbuhaler (AstraZeneca), the first widely used multidose reservoir inhaler device in the middle of the 1980s, has been followed by the development of several other devices of this class, each with its more or less unique technical solutions (e.g., EasyHaler, Orion).
Metered dose inhalers (MDIs) (e.g., Ventoline Evohaler, Glaxo), although in principle technically the same as their first appearance on the market in 1956, seem still to be considered as the first line of treatment of major respiratory disorders, such as asthma and COPD (Report Linker/PR Newswire, 2020). The small size and convenience to use are factors influencing the wide use of these devices as well as the relatively low cost and widespread availability of medications. The conventional pMDIs are, however, regarded as relatively inefficient in delivering the drugs to the lungs, partly because of patient errors. Basically, these drawbacks remain, although many modifications have been made to this class of devices, from improving the valve mechanism and its materials to coating the inside of the canister as well as adapting the propellants to current environmental requirements. Mouthpiece extensions, such as spacers and valved holding chambers like, for example, AEROCHAMBER PLUS FLOW-VU Chamber (Trudell Medical), were introduced to eliminate discoordination at the press-breathe sequential procedure, including their use for pediatric population. Furthermore, the breath-actuated inhalers, for example, Autohaler (3M Pharmaceuticals) inhalation device, were introduced for the same reason but with the advantage of being in a portable size like the conventional pMDIs. After AutoHaler, various technological innovations have been made with the aim of improving patient compliance during the press-breathe sequence, either controlling the inhaled flow rate or by allowing the inhaled air to flow past the triggering mechanism only when the MDI is depressed.
Jet nebulizers are the most common type of large-volume aqueous aerosol generators driven by compressed air used worldwide, and among currently marketed devices the majority are constructed with jet nozzles consisting of coaxial tubes. Some of these devices are breath-enhanced nebulizers (BENs), such as, for example, the PARI LC PLUS (PARI GmbH); these reduce the aerosol output during the expiratory period and increase it during inspiration. From the outer tube, the solution or suspension is sucked from a reservoir via underpressure created by the central compressed air nozzle for subsequent aerosolization. Further improvements for more consistent dose delivery and reducing waste of formulation are the breath-actuated nebulizers (BANs), such as AeroEclipse, Trudell Medical International (Suggett et al., 2014).
Later came the low-velocity sprays known as soft mist inhalers (SMIs). These devices represent a class of multidose inhaler devices containing liquid formulations similar to those in nebulizers. A variety of principles are used like ultrasonics, vibrating meshes, and several other novel approaches. Many of these devices are able to achieve very high lung deposition. A portable soft mist inhaler (Respimat, Boehringer Ingelheim) was launched in Europe in 2004 for asthma and COPD to replace CFC pMDI or DPI products for the same drugs (Newman, 2006).
Within these three categories exist significant choices in terms of the ways the drug can be processed and formulated and device design selected for the intended use (de Boer and Thalberg 2021a,b,c). To balance the development risk, some companies indeed prefer to use simple off-the-shelf delivery systems and well understood formulation technologies used in approved products, provided they reasonably satisfy the medical need and commercial drivers. Conversely, technical formulation and device innovation are driven by necessity (e.g., improvement of patient requirement for precise regional lung dosing, such as for inhaled insulin or competitive commercial pressures). Looking forward to new therapeutic areas and novel drug classes (e.g., biomolecules), technical innovations could become more important to enable meeting the nontechnical drivers for their development. However, looking at the plethora of approved and successfully commercialized orally inhaled drug products, a new drug for the inhalation route could probably use existing formulation technologies and devices, bearing in mind the importance of the nontechnical development drivers mentioned above.
D. Mechanistic Computational Models to Support Drug Product Development
The application of statistical Quantitative Structure-Activity Relationship models to understand molecular drivers of pulmonary disposition is well established (see, Edwards et al., 2016). However, Quantitative Structure-Activity Relationship models are less applicable for understanding product performance, as this must combine information on drug molecular attributes, drug material properties (solubility), and drug-product attributes (such as dose deposition and dissolution rate) as well as subject physiology (specific to, for example., age and disease state). Understanding of product performance in support of translational science during clinical development is thus better served by the application of mechanistic physiology-based pharmacokinetic (PBPK) models.
In the recent decade, significant progress of computational models to support and derisk clinical development of orally inhaled medication has been achieved (Bäckman et al., 2018). To a large extent this happened also thanks to the recent development of in vitro and ex vivo methods to improve the predictability of the attributes required to inform the models. Today, for example, clinically relevant aerodynamic particle size distributions of delivered doses can be measured by adding to the traditional quality-control methods of realistic mouth-throat replicas in combination with realistic breath profiles (Olsson et al., 2013, 2021). Likewise, methods to measure dissolution kinetics of the fraction of the delivered dose that is assumed to deposit in the lung have been developed (Price et al., 2018, Bäckman and Olsson, 2020). Using isolated perfused lungs and other ex vivo methods, improved estimates of permeability and binding to the lung tissues have been achieved (Bäckström et al., 2016; Eriksson et al., 2018; Bäckström and Fridén, 2021; Enlo-Scott et al., 2021). In parallel, computational models that focus on mechanistic relationships between measurable product attributes and biology continue to be advanced (Himstedt et al., 2021). Such PBPK models have the advantage over regressed statistical models that the parameters of the model reflect physical properties that are quantitatively linked to local and systemic exposure via mechanistic relationships derived from independent knowledge. Ideally, in the future such models would contain no fitted parameters, although the complexity of the respiratory tract, including intersubject variation and disease impact should not be underestimated.
PBPK modeling for orally inhaled drugs is a relatively young discipline with only few published applications. Most literature is focused on a single drug or drug class and is of semiempirical nature (i.e., not all processes are mechanistically described) (see Himstedt et al., 2021 and references therein). Of the more generally applicable models that are publicly available, the Mimetikos Preludium PBPK model (Emmace Consulting AB, Lund, Sweden) (Olsson and Bäckman, 2018) is specific for targeting orally and nasally inhaled drugs. It is semimechanistic in that it provides a fully mechanistic description of all pulmonary PK processes, whereas the systemic disposition is represented by an empirical compartmental PK model. The GastroPlus Pulmonary Compartmental Absorption and Transit module (Simulations Plus, Lancaster CA) is another commercially available generic PBPK model for oral inhalation. Systemic disposition may be modeled either as empirical PK compartments or as a fully mechanistic model. The deposition model is the now outdated ICRP66 model, but the deposition fractions in the various lung regions may be manually specified, as, for example, estimated from the MPPD software [Applied Research Associates, Inc. (ARA), Albuquerque, NM] (Miller et al., 2016). The very comprehensive open-source PBPK suite with PK-Sim and MoBi (Open Systems Pharmacology Suite, Bayer AG, Leverkusen, Germany) unfortunately does not specifically include modules for inhaled administrations. However, the MoBi software allows the user to implement such modules (Eriksson et al., 2020). Additionally, some pharmaceutical companies have in-house PBPK software, for example. Lung-Sim at AstraZeneca (Tehler et al., 2018), PulmoSim at Pfizer, and the “lung platform” at Merck & Co (Cabal et al., 2016).
Attributes required to inform a PBPK model may for simplicity be divided into three categories (for example, Bäckman and Olsson, 2018):
1) Batch- and product-specific attributes (e.g., delivered dose, aerodynamic particle size distribution, dissolution kinetics, inhalation flow profile, and bolus dose profile). Whether an attribute should be considered batch- or product-specific depends on the impact of batch-to-batch variability on product performance.
2) Drug-specific attributes that depend on molecular properties that are manifested for the drug in solution (e.g., permeability, tissue binding, system pharmacokinetics).
3) Population-specific attributes (i.e., how, for example, sex, age, and disease modify the attributes of the two preceding categories).
From a PK perspective, the most therapeutically influential product performance attributes are the lung dose and the release rate. Initial lung dose may be derived from delivered dose and aerodynamic particle size distribution through lung deposition modeling (Miller et al., 2016; Olsson and Bäckman, 2018; Hofmann, 2020). The initial lung dose is eliminated by competition between absorptive and nonabsorptive (e.g., mucociliary) clearance mechanisms. Release rate may be derived from experimental dissolution curves and modeled according to Noyes-Whitney type functions to derive parameters that can be employed in the PBPK model, such as the distribution of initial specific surface area available for dissolution (Bäckman and Olsson, 2020). Note that effective therapeutic lung dose as well as the release rate are potentially affected by each pathway leading to and including plasma clearance and that the kinetics of these pathways are interdependent, leading to a system that in terms of the governing parameters is inherently nonlinear but that may display linearity when one process is dominant.
The influence of population-specific attributes is less well explored. Bäckman and Olsson (2016) used PBPK modeling to understand the underlying causes for reduced systemic exposure of fluticasone propionate in asthmatics (compared with healthy subjects) (Brutsche et al., 2000; Harrison and Tattersfield, 2003) and found that this could be explained by increased nonabsorptive clearance in asthmatics as a result of higher central deposition due to airway constriction. Although the examples of the use of PBPK modeling to understand age-related differences (e.g., children vs. adults) is not observed in literature, no technical limitations exist, as the PBPK models are applicable to any lung physiology and morphometry specified.
There are several areas in the development of orally inhaled medication that could benefit from application of PBPK models. For example, to judge bioequivalence (BE) between a test and a reference product (note that test could be an innovator development as well as a generic product) normally requires a PK-BE study, which is an expensive endeavor prone to failure until eventual success (e.g., Lähelmä et al., 2015). Using a relevant PBPK model, investigators may be able to assess the probability of success and take appropriate action (proceed with the clinical study or continue product development with the benefit of mechanistically derived hypotheses). Ultimately, the outcome of a validated PBPK model may in the future suffice to declare PK-BE or not. Another example is when the target is not a reference product but a desired product performance profile.
Here, a relevant PBPK model may through sensitivity and what-if analyses provide valuable insights regarding the effects of device and formulation changes on product performance. Furthermore, a relevant PBPK model can facilitate the planning of clinical studies, for example, by predicting at what dose level a potential nonlinear transition between permeability and dissolution limited absorption would take place. A final example is that a PBPK model may facilitate the interpretation of unexpected findings in a PK study by providing a structured framework for investigations.
We will limit ourselves to highlight just two examples of successful applications of physiologically based modeling. The first example shows how subtle differences in dissolution kinetics of fluticasone propionate for different strengths of Advair Diskus translate to differences in systemic PK, demonstrating that systemic PK is capable of reflecting upstream local processes (Bäckman and Olsson, 2020). The second example shows how local free concentrations of salbutamol in the lung subepithelium are predictive of the extent and time course of the pharmacodynamic response (bronchodilatation) after inhaled and oral delivery, demonstrating the validity of PBPK model predictions of free drug concentrations in lung tissue (Boger and Fridén, 2019).
E. Conclusions
Critical drug attribute targets are different for drugs designed for typical treatment via inhalation compared with drugs designed for systemic treatment. High-potency effect duration as well as topical safety and tolerability are obtained through optimization of drug and formulation attributes governing high lung retention, optimal target interactions, and low oral bioavailability. Translation of preclinical results into a clinical setting must consider differences between preclinical and clinical inhalation devices and formulations as well as physiologic and anatomic difference between species. Furthermore, compatibility between a candidate drug and the prospective commercial inhalation device should be carefully considered, and since the performance of an inhalation device will likely be dependent on the detailed properties of the formulation, an early decision on the device-formulation combination is encouraged. Emerging mechanistic computer-based PBPK models show potential to synthesize the influence of drug attributes (e.g., permeability and tissue interactions), product attributes (e.g., dose, dose deposition, and dissolution), and population-specific attributes (e.g., age, disease) into a qualified understanding of in vitro–in vivo relationships that could support translational science and product design and thus derisk the clinical development process.
IV. β-2 Receptor Agonists
A. Introduction
Bronchodilators have been a mainstay treatment of symptoms from obstructive airways diseases since ancient times (Chu and Drazen, 2005). Acute symptomatic relief was achieved by the ingestion or smoking of plant materials that had xanthine or anticholinergic properties. However, such therapies require high doses to achieve benefit, leading to many adverse effects.
The use of the inhaled route to treat asthma was based on both the history of using that route and the desire to have high local effects without systemic adverse effects (Stein and Thiel, 2017). In 1929, the first report was made of the successful use of inhaled adrenal extract to treat asthma (Camps, 1929). This led to the use of adrenaline delivered from atomizers for some 30 years until the development of the β adrenoceptor agonist isoprenaline delivered by a chlorofluorocarbon-driven MDI. Subsequently, molecules with selectivity to the β2 adrenoceptor and muscarinic receptors have been developed.
B. Classes of Inhaled Bronchodilators
There are two classes of inhaled bronchodilators: muscarinic acetylcholine receptor antagonists (which will be discussed elsewhere—Section V) and β2 adrenoceptor agonists, which are discussed in this section.
β2 Agonists are available in two subclasses. The first group is molecules with a rapid onset of therapeutic benefit, but they have a short duration of benefit typically only up to 4 hours [known as short-acting β2 adrenoceptor agonists (SABAs)]. Currently available molecules are listed in Table 3. The second group is molecules that have a duration of therapeutic benefit of at least 12 hours [known as long-acting β2 adrenoceptor agonists (LABAs)], which are listed in Table 4.
Products, inhalers, and characteristics of short-acting β2 adrenoceptor agonists
Products, inhalers, and characteristics of long-acting β2 adrenoceptor agonists
C. Pharmacology
β2 Adrenoceptors are found on almost all cell types and are found throughout the lung. There is consensus that their main therapeutic action is via stimulating the airway smooth-muscle β2 adrenoceptors, thus causing relaxation of those cells leading to bronchodilatation. The possibility that their therapeutic benefit is contributed to by action on other cell types within the lung has been explored (Barnes, 1996). Although these effects can be shown in vitro and in disease models, so far no human clinical data has confirmed such contribution.
β2 Adrenoceptor agonists achieve their therapeutic effect by binding to the active site of β2 adrenoceptors. This binding activates cyclic AMP via a well described G-protein mechanism. Adrenaline is not specific for this receptor, as it has affinity for both α and all β adrenoceptor receptors. Isoprenaline is selective for all the β adrenoceptor subtypes. All the other compounds developed are selective for the β2 adrenoceptor, and some also show partial agonism to that receptor. All the selective β2 molecules are more potent than the original β agonist isoprenaline. However, selectivity and potency are less important in a topically acting product (i.e., when given by the inhaled route), as low potency can be overcome by creating high local concentration, which can avoid systemic exposure.
The duration of therapeutic effect depends upon the residency time at the receptor or within the tissues where the receptors lie. To achieve reversal of bronchoconstriction, the effective concentration needs to be reached rapidly; however, for prolonged bronchodilatation the levels must be maintained for the length of time desired to make the label claim. Therefore, some molecules (e.g., salmeterol) show a sustained activity in vivo and in vitro, whereas others (e.g., salbutamol) show neither or only show sustained effects when given topically in vivo (e.g., formoterol). Inherently short-acting molecules can gain increased therapeutic duration by increasing the dose, but this may also increase systemic exposure (e.g., salbutamol) and therefore adverse effects, although some molecules (e.g., formoterol) can be dosed topically to get an extended duration of therapeutic benefit without such increase in systemic effects (Lofdahl and Svedmyr, 1989).
If the site of action for these products is the airway smooth muscle, then the anatomic site of action is in airways that can contribute to airway obstruction, thus from the third division of the bronchial tree to the bronchioles. So, delivery of the product to larger or smaller airways will not contribute to therapeutic benefit but may add to systemic exposure (Usmani et al., 2005).
D. Therapeutic Use
The SABAs are used exclusively to treat acute episodes of breathlessness, although they are also effective prophylactically if taken before exercise in patients who have symptoms triggered by exercise (Godfrey and König, 1976). Despite being available since the 1960s they are still universally used because they have rapid onset of bronchodilatation and are very well known and trusted by the medical community and patients. In addition, they are easy to make in large quantities and cost-effective for this indication.
The LABAs are used for maintenance therapy taken regularly once or twice a day mostly in fixed combination therapy (discussed in Section VII). Although some have a relatively fast onset of therapeutic benefit (formoterol), they are less cost effective as a rescue medication, being no more effective than salbutamol (Lofdahl and Svedmyr, 1989). For asthma, the guidelines recommend their use in patients uncontrolled on inhaled glucocorticoids (Global Strategy for Asthma Management and Prevention, www.ginasthma.org), and in COPD they are recommended as first-line regular controller treatments (GOLD, Global Strategy for the Diagnosis, Management and Prevention of Chronic Obstructive Pulmonary Disease 2020 Report, www.goldcopd.org). Taken as recommended in the label, there is no clinical difference in the measured clinical outcomes (lung function or exacerbation control) between the once- and twice-a-day products, and although there is a theoretical compliance advantage of the once-daily dosing, this has not been shown in clinical trials to date.
The objective of the inhaler, which is a device/formulation combination, is to deliver the active ingredient to the anatomic site of action in a formulation that allows enough of the molecule to reach the receptors on the airway smooth muscle. The solubility of the molecule in the airway lining fluid will influence the behavior of the molecule. With a soluble drug, all particles will dissolve in the airway lining fluid and are thus readily available to the receptor. However, with an insoluble drug there is a need to have smaller particles so that the drug can dissolve in the lining fluid. To ensure that these small particles are delivered and stay in the correct part of the lung, they can be delivered via binding to carrier particles in the formulation, such as larger lactose particles.
All devices used to deliver orally inhaled medicines have an operational range of product mass that can be delivered efficiently to the lung. This puts limits on the choice of product, as potency and physical properties of a molecule as the device/formulation combination will determine how much drug can be delivered in an acceptable manner to patients. Patients need to have an easy-to-follow dosing regimen with as few doses as possible and a frequency that is easily remembered, ideally one dose once or twice a day for maintenance use and one dose as required for rescue use. This means that there is a limit to which molecules can be successfully formulated for the device types available for inhaled drugs.
All the products on the market have been selected and therapeutic dose has been chosen by showing that maximal bronchodilatation is achieved without systemic effect. So, they are differentiated by their onset of therapeutic benefit or frequency of dosing. There are a number of different devices and even formulations of drugs within the same type of device. These differences may affect the dose to the lung and therefore the prescribed dose, but there is little evidence that these change the fundamental safety/efficacy ration of the drug being delivered.
A number of these molecules are no longer patent-protected. However, few substitutable generics have come to the market. This is due to the exacting requirements of a match in in vitro testing as well as in PK and clinical endpoint. A number of nonsubstitutable copies have been developed using the regulations for line extension (device change), so-called 505 b(2) in the United States; they are less exacting but can lead to different doses if the delivery is not matched.
E. Aerosol Delivery
There are two primary types of inhalation devices for the delivery of β-2 agonist bronchodilators for asthma and COPD: MDIs and DPIs. Other methods include soft mist inhalers and nebulizers. Many commonly prescribed drugs are available in multiple delivery formats, as a number of technologies exist today to allow them to be formulated in a range of delivery devices. Decisions on which formulation/device (combination product) to develop are often driven by commercial considerations.
The most widely used SABAs (e.g., salbutamol) are predominantly delivered by MDI. LABAs are typically now only provided in combination with an inhaled glucocorticoid or anticholinergic (see Section VII). For both short- and long-acting β-agonists, solution formulations have been developed for use in nebulized therapy. These are typically used in hospital settings for patients who are severely ill.
An MDI is a complex pressurized device designed to produce a fine mist of medication for inhalation directly to the airways. These products were first developed over 60 years ago (Stein and Thiele, 2017) and are particularly suited to administration of therapy when respiratory function is compromised. Historically the MDI used CFCs as propellants, but in the past decade the CFCs have been replaced by hydrofluorocarbons [hydrofluorocarbons (HFCs) or HFAs], which have a lower environmental impact. The MDI remains popular because it is generally less expensive than powder inhalers.
DPIs are devices that deliver powdered medication (active ingredient alone or mixed with excipient, typically lactose) without the need for a propellant. There are many different devices that deliver powder medication. Most are available exclusively from a single pharmaceutical company that has patented the device. Most commonly used respiratory drugs (i.e., β agonists, including salbutamol) have been formulated successfully for DPIs and are now widely available.
DPIs fall into two categories: single-dose and multidose. Single-dose DPIs, which have been in use for more than 60 years, use a capsule containing one dose inserted into the device. The capsule is opened within the device, and the powder is inhaled. The capsule must be discarded after use and a new capsule inserted for the next dose. They are inexpensive but may be susceptible to humidity. Multidose DPIs, which have been in use for more than 20 years, typically contain enough doses for at least 1 month’s treatment. There are two types of multidose DPI, one with individual doses in which the metering is conducted during manufacture and the second that loads a measured amount of medicine for inhalation from a reservoir in the device.
A number of new chemical entities (drugs) over the past 15 years have been commercialized as DPI products as companies are finding that it is not cost-effective to develop both DPI and MDI formulations for these new treatments.
Nebulizers are devices that are filled with drug dissolved or suspended in aqueous solution, which is converted to inhalable droplets using compressed air, ultrasonics, or vibrating mesh. In principle, any formulation could be used with any nebulizer; however, this leads to widely different outputs (aerosol particle size) and dose to the patient. This has come under greater regulatory scrutiny in recent years, and a nebulizer is likely to be recommended for a particular formulation based upon data provided in the product submission to regulators. A nebulizer can take several minutes to deliver the required dose, and because of the long administration time, they are relatively inconvenient to use.
Small portable devices termed SMIs that produce aerosols of respirable diameter from aqueous formulations have been developed in recent years (Leiner et al., 2019). One (Respimat) is now widely available in a number of countries delivering a range of therapeutic entities. These new-generation devices produce an aerosol through mechanisms different from those described for nebulizers. The mechanisms include collision of two jets of liquid to produce an aerosol or forcing liquid through tiny micron-sized holes or vibrating mesh/plate or other novel mechanisms (e.g., electro-hydrodynamic effects). They can be distinguished from nebulizers in that they endeavor to deliver a complete dose within one or two breaths. The combination of improved efficiency and smaller aerosol particle size from these devices ensure that the aerosol they generate can be deposited deeply into the lungs.
Although there are now a wide range of delivery systems available to deliver most of the therapeutic agents to the lung, there is likely to be further change in the range of devices available in the coming years. Environmental pressure on the existing hydrofluorocarbon propellants used in MDIs (HFC-134a and HFC-227ea) through the Kagali Amendment to the Montreal Protocol (Pritchard, 2020) will likely lead to further reformulation work. Any inhalation propellant must be safe for human use and meet several other criteria relating to safety and efficacy before becoming widely available.
F. Conclusions
Rapidly acting SABAs will remain the treatment of choice for rescue use for the foreseeable future. LABAs have a place in the chronic maintenance treatment of both asthma and COPD but are not currently recommended as monotherapy.
V. Muscarinic Acetylcholine Receptor Antagonists
A. Introduction
Cholinergic transmission is the dominant neural pathway in control of airway tone in healthy persons (tonic activity) and in COPD (dominant role), and it is an important pathway in asthma (Matera and Cazzola, 2016). Thus, muscarinic acetylcholine receptor (mAChR) antagonists play an important role in the therapy of obstructive pulmonary diseases.
Considering the general physiologic importance of cholinergic neurotransmission and receptors, the therapeutic use of agonists or antagonists up till now is surprisingly limited: mAChR antagonists are registered for premedication in anesthesia and ophthalmology, treatment of gastrointestinal spasms and peptic ulcer, overactive bladder, duodenal ulcer, exocrine gland hypersecretion (drooling associated with chronic neurologic conditions), and treatment of the obstructive pulmonary diseases, COPD and asthma. Chronic use is rather limited except for pulmonary diseases. This constraint is explained by the difficulty to selectively target an organ or tissue. Even mAChR subtype–selective agents offer limited opportunities to solve this problem, as one subtype is typically orchestrating various effects in multiple organs. In contrast, topical application in combination with topical selectivity was highly successful to develop efficacious, selective, and safe medications for obstructive pulmonary diseases. Nevertheless, intense research and development efforts for subtype-selective mAChR antagonists are ongoing, especially in neurology (Moran et al., 2019).
In this section, muscarinic effects and mAChR subtypes involved and therapeutic interventions in all organ systems will be summarized with special emphasis on the available drugs, formulations, and devices for oral inhalation in lung disease.
B. Cholinergic Neurotransmission and Receptors in Different Organs and in the Lungs (Table 5)
Acetylcholine (ACh), the classic vagal transmitter of the parasympathetic nervous system, can signal through two distinct classes of receptors: ligand-gated cation channels termed nicotinic ACh receptors and G-protein–coupled mAChRs. Both receptor classes play important roles in the central and peripheral nervous system (Eglen, 2012; Moran et al., 2019). Activation of nicotinic ACh receptors typically promotes a net influx of positively charged ions resulting in membrane depolarization and generation of action potentials in postsynaptic cells serving neurotransmission. ACh via mAChRs acts as “classic” neurotransmitter for ganglionic transmission and neuroeffector junctions and beyond these can mediate hormonal effects. The physiologic role is exerted via five mAChR subtypes in different organs. Therefore, stimulation or inhibition of muscarinic receptors may result in both desired therapeutic effects but also unwanted adverse reactions depending on subtype selectivity of the mAChR modulator, distribution to the organ/compartment in question, and dose. As for therapy of obstructive pulmonary diseases, M3 over M1 selectivity and especially over M2 is the preferred option.
C. Pharmacology of Inhaled Muscarinic Receptor Antagonists for Treatment of Obstructive Pulmonary Diseases (Table 6)
Atropine and related anticholinergic alkaloids are widespread in the plant kingdom and have been used in healing arts for centuries. Inhaled atropine (asthma cigarettes) has been used as a bronchodilator in obstructive pulmonary disease until the mid-1970s. The problem with atropine—even if inhaled—was its lipophilicity, causing rapid and complete oral as well as pulmonary absorption and distribution across membranes throughout the body, including the central nervous system. As a consequence, peripheral and central adverse reactions were frequent already at bronchodilating doses. This improved dramatically with the chemical synthesis of quaternary derivatives of atropine, which have low lipid solubility, in consequence are poorly absorbed in the oropharynx and intestine, do not pass the blood-brain barrier, and distribute slowly after inhalation.
The antimuscarinic agents available today for treatment of lung diseases differ by mAChR subtype selectivity, potentially inverse agonisms at constitutively active mAChRs (Casarosa et al., 2010; Salmon et al., 2013; Babu and Morjaria, 2017; Hedge et al., 2018), duration of action, onset of action, pharmacokinetics and clearance, formulation, and inhaler device. The quaternary amine atropine derivative ipratropium (bromide) introduced in Europe in 1974 initially in a CFC-driven MDI and still used today modified to a hydrofluoroalkane-driven device (HFA-MDI) was shown to be an effective and safe bronchodilator. Affinity to mAChR subtypes (M1, M2, and M3) is similar, and kinetic subtype selectivity may not contribute because the dissociation from all subtypes is rapid. Ipratropium owes its lung selectivity to the inhaled topical administration and the quaternary structure as explained before (Gross et al., 1988; Cazzola et al., 2012).
Tiotropium (bromide), the most potent long-acting muscarinic antagonist, is characterized by very slow dissociation from mAChRs, qualifying for a once-daily treatment regimen. It showed kinetic receptor subtype selectivity M3 over M1 and especially over M2. Steady state, as evidenced by plasma levels, and maximal respiratory effects are reached after a few days of treatment. The quaternary structure supports lung selectivity after inhalation, as explained for ipratropium. Once absorbed into the systemic circulation, tiotropium is predominantly eliminated via renal excretion.
Glycopyrronium (bromide) is of special interest for this review because it is in use for a number of indications by different routes of administration: as bromide in inhaled, oral, and parenteral formulations and as tosilate in a cream for topical use (Chabicovsky et al., 2019). The compound showed a slow dissociation from mAChRs in line with a once- or twice-daily treatment regimen. It is a highly potent antagonist with kinetic receptor subtype selectivity M3 over M1 and especially over M2. The quaternary structure again supports lung selectivity after inhalation. Glycopyrronium is predominantly cleared renally.
Administration of an oral solution, which is indicated for drooling at a median dose of 2 mg (glycopyrrolate) for an adult patient, resulted in maximum plasma levels (Cmax) of 0.318 ng/ml and an area under the curve (AUC0-inf) of 1.81 ng*h/ml. An inhaled dose of 50 µg once daily indicated for COPD resulted in Cmax of 0.166 ng/ml and AUC0-24h of 0.464 ng*h/ml. The 2- to 3-fold higher systemic exposure to glycopyrronium for the oral indication compared with the treatment of COPD by inhalation results in a higher frequency of typical antimuscarinic adverse drug reactions: “dry mouth” 40% versus up to 10% and “urinary retention” 15% versus up to 1% (Prescribing Information Cuvposa/FDA, package leaflet Seebri Breezhaler/UK). The examples demonstrate the advantage of inhaled administration at a fully efficacious dose (in the airways) with a favorable level of adverse reactions (systemic or local, not lungs) compared with a carefully titrated oral dose with a higher frequency of adverse reactions, of course, in different indications and patient populations. Much higher drug exposure in the lung compartments in comparison with systemic compartments is outlined below (safety profile).
Aclidinium (bromide) showed a slow dissociation from mAChRs in line with a once- or twice-daily treatment regimen. It is a highly potent antagonist with some kinetic receptor subtype selectivity M3 over M1 and over M2. Again, the quaternary structure supports lung selectivity after inhalation. An advantage is its rapid hydrolytic inactivation reducing the potential for systemic adverse events but may also be the reason that twice-daily inhalation allows a lower total daily dose compared with once daily.
Umeclidinium (bromide), a highly potent muscarinic antagonist, qualifies for once-daily posology by slow dissociation from mAChRs. It showed high kinetic receptor subtype selectivity M3 over M2 (M1 not published). The quaternary structure supports lung selectivity after inhalation. The compound is predominantly cleared by hepatic metabolism.
Revefenacin, which is also a highly potent mAChR antagonist, is characterized by slow dissociation from M3 receptors, qualifying for once-daily administration. Although revefenacin is not a charged quaternary structure, the inhalational route of administration and very high kinetic receptor subtype selectivity M3 over M2 (M1 not published) are major factors to provide selectivity for antimuscarinic effects in the lungs.
It is instructive to illustrate the need for the inhaled route to achieve adequate pulmonary selectivity for lung diseases for this class of compounds by mentioning an example of a failed development: the oral antimuscarinic bronchodilator OrM3 (Merck). The compound is highly selective for M3 receptors (120-fold based on Ki values). In a phase 2 study, forced expiratory volume in 1 second (FEV1) change from baseline at the highest dose of 4 mg p.o. was superior to placebo, but only two-thirds of the response to the standard dose of ipratropium. In contrast, systemic adverse drug reactions were much higher with OrM3 versus ipratropium or placebo (most frequent adverse reaction dry mouth: 46.3% versus 0.5% versus 1.5%; similarly higher frequency for OrM3 for constipation and urinary retention at 10% level). Although M2-antagonistic effects, such as tachycardia, were not observed the M3-blockade mediated adverse reactions were not acceptable (Lu et al., 2006).
D. Therapeutic Use of Inhaled Muscarinic Acetylcholine Receptor Antagonists (Table 6)
With a duration of action of about 6 hours, ipratropium MDI bronchodilates longer than atropine or SABAs but still needs 3–4 administrations per day and thus qualifies as a short-acting muscarinic antagonist (SAMA). Treatment-related adverse events are infrequent. In chronic treatment of patients with COPD ipratropium was inferior to long-acting muscarinic antagonist (LAMA) (Hansel and Barnes, 2002) or LABA treatment (Cazzola and Page, 2014). For this reason, the place of SAMA is for acute reliever therapy in COPD and asthma.
All available LAMAs are qualified by large and long-term studies in patients with COPD documenting efficacy in improving lung function and symptoms, reduction of dyspnea, improvement in health-related quality of life (HRQoL), reduction of exacerbations of COPD (except revefenacin), and documentation of adequate safety. There are only subtle differences in the profiles (Matera and Cazzola, 2016).
As COPD does not affect pediatric patients, regulatory authorities have granted a waiver for pediatric development. In asthma and cystic fibrosis, pediatric indications had to be considered.
Tiotropium (by DPI) was primarily registered as a first-line treatment in COPD and was shown to provide improvement in lung function in comparison with placebo, ipratropium, and the LABA salmeterol using once-daily inhalation. Tiotropium was shown to improve symptoms of COPD, dyspnea, and HRQoL versus placebo. In long-term randomized controlled studies for up to 4 years, tiotropium was shown to reduce exacerbations of COPD and even mortality (supportive evidence, Kesten et al., 2009) with a low frequency of treatment-related adverse events. Tiotropium by SMI (an aqueous-based formulation delivered by a multiple-dose soft mist inhaler Respimat) was shown to be equivalent to HandiHaler (dry powder inhaler) in large studies in COPD and is the only LAMA fully profiled and registered in asthma as an add-on treatment of patients still symptomatic (with exacerbations) on inhaled glucocorticosteroid (GCS) or LABA-GCS treatment. The development program included studies in pediatric cohorts from 1 to 5 years (only safety assessed and confirmed), 6 to 11 years, and 12 to 17 years resulting in approval for moderate to severe asthma in the age group ≥6 years in the European Union and United States. The health status in patients with cystic fibrosis, including pediatric patients, was not improved by tiotropium treatment, so the indication is not registered [Spiriva Respimat (tiotropium bromide) inhalation 2019 FDA prescribing information, Spiriva Respimat Fachinformation 2020]. To be noted, the nominal dose in pediatric asthma of all age groups was not different from the adult dose. If needed, children used the Aerochamber Plus Flow-Vu with or without face mask. The systemic exposure was comparable in children ≥6 years and adults.
Glycopyrronium in COPD (50 µg bromide once daily or 15.6 µg twice daily by DPI) is similarly qualified as for efficacy and safety, with a duration of action slightly shorter than tiotropium when administered once daily. The drug provides a rapid onset of action, which is discussed as an advantage for symptomatic patients (Cazzola and Page, 2014).
Acclidinium in COPD is administered twice daily by a multidose DPI, and maximum bronchodilation is already seen with the first dose. Efficacy and safety results are similar to the other LAMAs. A point of discussion is a potential advantage (lung function in the evening/night) or disadvantage (convenience/compliance) of twice-daily versus once-daily administration. Controlled studies to this aspect are missing (Cazzola and Page, 2014).
Umeclidinium inhaled once daily by a multidose DPI in COPD again qualifies as a very long-acting LAMA by similar favorable outcome data (Ismaila et al., 2015).
Revefenacin, which is provided as solution in a unit dose vial (UDV) for once-daily inhalation by nebulizer in COPD, is the only nonquaternary LAMA. Although based on a smaller database, it is similarly profiled for efficacy, in other words, improvement in lung-function and HRQoL, reduction in breathlessness and dyspnea, and safety (based on a review by Antoniu et al., 2020). The compound offers a fast onset of action discussed as an advantage in symptomatic patients. The revefenacin database includes one 52-week phase III comparison with tiotropium DPI in typical patients with moderate to very severe COPD, showing a numerically smaller lung-function improvement (trough FEV1) at the end of the treatment period and no difference in other outcomes. However, in a 28-day phase IIIb, which recruited patients with COPD and suboptimal peak inspiratory flow rate (<60 l/min), nebulized revefenacin was superior to tiotropium DPI in improving trough FEV1 (significant in the subgroup with very low peak inspiratory flow rate (33–45 l/min) independent of COPD severity (Antoniu et al., 2020). To be noted, no similar comparisons were made with LAMAs in other devices (e.g., MDIs or SMIs) that also do not require the patient’s breathing effort to disperse the particles into fine aerosols.
E. Safety Profile
Drug delivery by inhalation is a key factor to expose target mAChRs in the airways and avoid systemic adverse reactions. Lung compartment pharmacokinetics typically cannot be directly assessed in man. To fill the gap, Hendrickx et al. (2018) used a translational model to predict pulmonary pharmacokinetics and efficacy in man for inhaled bronchodilators. Their compartmental model allows translation between animal species and predicted drug concentrations in human lungs and correlates with forced expiratory volume change. Inhaled antimuscarinic and β2-adrenergic drugs were studied. Simulated lung concentrations were 102 times (ipratropium) to 103 times (tiotropium, glycopyrronium) higher than plasma concentrations. Bartels et al. (2013) used a similar population pharmacokinetic modeling approach to analyze glycopyrronium exposure with comparable results and explain the large difference by slow absorption of the inhaled and deposited dose from the lungs followed by rapid systemic clearance.
The LAMAs discussed here have a favorable safety profile with no substantial differences (Tashkin, 2015).
The most frequent adverse reactions, as can be derived from Table 5, are dry mouth and taste disturbances (direct mucosal exposition), blurred vision (especially by directly exposing the face), urinary retention, constipation, and tachycardia (rare) (Williams and Rubin, 2018). Labeling (warnings and precautions) of inhaled mAChR antagonists point to special sensitivities (careful benefit to risk evaluation needed) as follows: narrow-angle glaucoma, prostatic hyperplasia, bladder-neck obstruction, myocardial infarction within the last 6 months, serious heart rhythm disorders, and reduced renal function (creatinine-clearance <50 ml/min) for antimuscarinics with predominant renal clearance. Of course, direct spraying into the eyes has to be avoided because of the risk to induce narrow-angle glaucoma. One remaining concern is based on potentially vulnerable subgroups: The safety database was mostly generated from randomized controlled trials, which typically exclude patients who are unstable (e.g., of a high cardiovascular risk group) who may show a different efficacy and safety profile (Rogliani et al., 2019).
Role of muscarinic acetylcholine receptor subtypes in different organs
Table compiled based on reviews: Racké et al., 2006, Giglio and Tobin, 2009, Eglen, 2012, Matera and Cazzola, 2016, Moran et al., 2019.
F. Aerosol Delivery
Inhaled mAChR antagonists for treatment in COPD and asthma target muscarinic receptors in the respiratory tract expressed on several cell types. The respiratory tract is innervated by the vagus nerve consisting of preganglionic cholinergic fibers in airway walls and postganglionic fibers, which innervate airway smooth muscle and submucosal glands. M3 ACh receptors are localized to smooth muscle in all airways; however, the density is much higher in larger airways and directly activated by ACh release from vagal nerve fibers (Barnes, 2004). In addition, the total cross-sectional area of the conducting airways increases exponentially starting with the fifth or sixth generation, so airflow resistance and in consequence airflow obstruction is dominated by large airways (Santus et al., 2020). Thus, following “classical” considerations, a formulation/device should predominantly target the larger airways [respirable particles, mean mass aerodynamic diameter (MMAD) <5 µm] and avoid deposition in the oropharyngeal tract prone to induce local adverse reactions, which should be followed by intestinal absorption contributing to systemic adverse reactions.
Whether a muscarinic antagonist should be targeted to the small airways too by a higher proportion of fine particles (MMAD < 3.1 µm) or even extrafine particles (MMAD < 2 µm), is not clear at this point. There is no doubt that inflammation followed by pathologic changes can severely affect the small airways in pulmonary diseases (Santus et al., 2020). Although there is no vagal innervation in small airways, the current hypothesis is that non-neurogenic ACh released from epithelial cells can activate M3 receptors on smooth muscle (Matera et al., 2020). In vitro and animal studies suggest anti-inflammatory and antiremodeling effects of M3 antagonists (Matera and Cazzola, 2016). A single-dose study in patients with COPD showed acute improvements in tests specific for small airways (lung pressure-volume curve, single-breath N2-washout test: similar improvements are reported for tiotropium and indacaterol; Pecchiari et al., 2017). Confirmatory clinical studies comparing the same LAMA administered by a standard-sized versus small-sized particles generating device/formulation are not yet available (Lavorini et al., 2017). Currently available clinical studies typically do not include endpoints with specific sensitivity to small airways. The characteristics of devices available with SAMAs or LAMAs (MDI, SMI, single-dose and multiple-dose DPI, nebulizer/ UDV) are described in Sections IV and VII.
The available formulations for the SAMA ipratropium and the LAMAs are summarized in Table 6.
Products, inhalers, and characteristics of muscarinic acetylcholine receptor antagonists
Table compiled based on reviews: Eglen, 2012, Cazzola et al., 2012, Matera and Cazzola, 2016, Williams and Rubin, 2018, Chabicovsky et al., 2019, Prescribing information USA and EU countries.
Clinical use of mono-product SAMA and LAMAs is by now rather limited because fixed dose combinations of SAMA-SABA, LAMA-LABA, or LAMA-LABA-GCS (inhaled glucocorticosteroids) play a dominant role (Section VII). As a recent example, an American Thoracic Society clinical practice guideline recommends using LABA-LAMA combination therapy over LABA or LAMA monotherapy in all patients with symptomatic COPD (Gartman et al., 2021).
G. Conclusions
Like many other drugs, the history of inhaled anticholinergic drugs can be traced back to a “natural product”—in this case atropine. It is noteworthy that some of the authors of this review remember the days when the delivery system of this drug for patients with asthma was cigarettes!
Inhaled anticholinergics or muscarinic acetycholine receptor antagonists achieve high therapeutic ratios due to the combination of targeted delivery to the lungs followed by slow systemic absorption, poor oral bioavailability, and selective receptor binding. Their therapeutic benefits in asthma and COPD are derived by binding to the receptors in the airways; it is uncertain at present whether additional deposition in the small airways would provide added benefit. The patients today have a wide choice of delivery systems for the monotherapy products as well the combination products that include nebulizers, MDIs, DPIs, and SMIs—a long way from “asthma cigarettes.”
VI. Glucocorticoids
A. Introduction
Extract of adrenal gland was first used to treat patients with asthma in 1900 and shown to be efficacious (Solis- Cohen, 1900). By the 1950s cortisol had been identified, synthesized, and used to treat asthma (Chu and Drazen, 2005). Systemic cortisol and other similar molecules became part of the acute severe asthma treatment, and oral prednisolone became part of the maintenance of chronic severe asthma. The use of these drugs was limited by the many and serious side effects. In 1972, the first inhaled glucocorticoid beclomethasone dipropionate was available in a chlorofluorocarbon-driven MDI. Currently an inhaled glucocorticoid alone or in combination with a bronchodilator is recommended in all national treatment guidelines for all but the mildest asthmatic patients. The Table 7 lists the available inhaled products containing glucocorticoids.
Products, inhalers, and characteristics of inhaled glucocorticoids
B. Pharmacology
All glucocorticoids bind to the same intracellular receptor known as the NR3C1 receptor. When the drug binds to the receptor the complex enters the nucleus by an active transport mechanism. In the nucleus it has two functions: first to bind to the DNA responsive elements to activate gene transcription, so called transactivation. This leads to protein production, and some have anti-inflammatory actions. Second, the complex can also bind to other transcription factors and prevent them from upregulating inflammatory proteins with so-called transrepression.
Both the transactivation and transrepression actions of the glucocorticoid lead to the therapeutic benefit and to the side-effect profile that is common to all molecules. The only differentiating factors for the molecules are their potency and specificity for the NR3C1 receptor. However, as the molecules are delivered locally by devices with limited dosing capacity range, there is a limit to the acceptable potency range of commercially available molecules (e.g., 100–2000 µg). Also, the major side effects are not due to the local concentrations, therefore, lack of systemic availability is likely to be a more important factor and will limit the dose range available.
These mechanisms are found in all cells with a nucleus, so any beneficial effect on an inflammatory process will be balanced by a potentially unwanted effect of another cell type either in the same anatomic location or further afield if the drug enters local or systemic circulation.
C. Therapeutic Use
As the therapeutic effects require either transcription of new proteins or the suppression of the production of inflammatory proteins, the onset of action is over 4–6 hours. The duration effect from a single dose can be detected for 24 hours. This contrasts with the plasma pharmacokinetics that show maximum concentrations within 2 hours and rapid distribution/elimination thereafter (Vathenen et al., 1991; Rohatagi et al., 2004). It has been calculated that the lung residence time (absorption phase) is shorter than the clinical effect time, as could be expected by their indirect mechanism of action. Inhaled glucocorticoids are therefore used for maintenance therapy to prevent exacerbations and reduce symptoms of asthma and COPD. In addition, taking higher-than-usual doses can be used to treat acute exacerbations not requiring other medical intervention instead of using oral corticoids.
In asthma guidelines they are recommended as first-line controller therapy, and in COPD they are recommended for use when regular bronchodilator therapy fails to control the patient. Most are prescribed once or twice a day, meaning the drug-specific regimen decided by the clinical program used for registration rather than definitive clinical studies supporting the dosing schedule. Taken as recommended in the label there is no clinical difference between the once- and twice-a-day products, although there is a theoretical compliance advantage of the once-daily dosing, this has not been shown in clinical trials to date.
Although it is appreciated by clinicians treating patients with obstructive airways disease that some patients are better treated by higher doses of inhaled glucocorticoids than others, there is little evidence of a dose response in clinical trials. However, trials in patients requiring oral glucocorticoid treatment have shown a dose response to inhaled glucocorticoids drugs (Noonan et al., 1995) which indicates that a very homogeneous population is required to show the effect. Probably in a naive patient population with individual titration of the dose of glucocorticoids in each patient, it may be possible to demonstrate a clear dose response; however, such studies are not practical in view of the widespread use of the products. Because of the difficulty in demonstrating a dose response, it has proven difficult to clinically differentiate between products, and there is a tendency for an individual product to have a limited range of doses available. Therefore, like bronchodilators (Section IV) the choice of inhaled glucocorticoid may come down to the cost and device type available.
The purpose of the device formulation is to deliver the molecule in a form that can allow access to the site of inflammation. There is some evidence that inflammation can be found in the very small peripheral airways, but it is likely that the main site is in the same generation of airways that responds to bronchodilators. Studies have suggested that “ultra” fine particle aerosols that reach the peripheral airways may be more effective than normal-size particles, but as they increase the dose delivered to the lung it is difficult to assess those claims (El Baou et al., 2017). Thus, the effective particle size range is limited to that of the bronchodilators. Given the difficulty in showing any dose response in efficacy, the role of clinical trials with different devices/formulations is to examine the relative systemic availability from the device and patient attributes of the devices rather than study the differences between products. There are a number of different devices and even formulations of drugs within the same type of device. These differences may affect the dose to the lung and therefore the prescribed dose, but there is little evidence that these change the fundamental safety/efficacy ratio of the drug being delivered. One nonlung potential differentiator between formulation and device combinations is the side effect of local candidiasis in the mouth and throat, and optimization of the product could potentially reduce this side effect.
A number of these molecules are no longer patent-protected. However, few substitutable generics have come to the market. This is due to the exacting requirements of a match in in vitro testing as well as in PK and clinical endpoint. A number of nonsubstitutable copies have been developed using the regulations for line extension (device change), so-called 505 b(2); in the United States these are less exacting but can lead to different doses if the delivery is not matched.
D. Aerosol Delivery
There are two primary types of inhalation devices for the delivery of inhaled glucocorticoids (ICSs), the MDI, and DPIs. Many glucocorticoids are available in both formats, although only a limited number are available as a suspension for nebulization. This, in part, is due to the technical challenges for development of aqueous suspension formulations that are readily redispersable for nebulization.
Because of the physicochemical properties of beclomethasone dipropionate, it was not possible to develop a suspension HFA MDI formulation, thus a solution MDI was developed. This in turn allowed the opportunity to develop a product that had a “superfine” aerosol output, with the ability to provide deeper lung deposition. Other ICSs have also been developed as solution MDIs, including ciclesonide and flunisolide (Stein and Thiel, 2017).
Most ICSs are available in multiple delivery formats (MDI and DPI), as technologies exist today to allow them to be formulated in a range of delivery devices. Decisions on which formulation/device (combination product) to develop are often driven by commercial considerations.
There are some limits to the delivery of high-powder-load DPIs using lactose carrier formulations (in low-resistance passive devices) because of the cohesive nature of the formulation when the active levels are around 10% or greater. These formulations present challenges to powder flow and dispersion.
To mitigate the potential for local candidiasis in the mouth and throat and to facilitate inhaled glucocorticoids delivery in the pediatric population, spacer devices were developed to be used in conjunction with MDIs (Dolovich et al., 1983). In addition, several integrated spacer devices (with MDIs) were developed historically (e.g., Azmacort, now discontinued in the United States) that facilitated lower throat deposition.
E. Conclusions
Glucocorticoids will remain primary maintenance therapy for asthma and have a secondary role in COPD for the foreseeable future. Unfortunately, some patients remain uncontrolled on them alone or even in combination with bronchodilators. This could be due to dose limitation, the disease process, patient compliance, or a combination of these factors. Technology may improve lung targeting and improve the percentage of patients fully controlled, but it is likely that there will be a role for noninhaled therapy in this uncontrolled group.
VII. Combination Products
A. Combination Rationale
As presented in Sections IV–VI, there are three major classes of inhaled drugs for asthma and COPD: the bronchodilators β2-adrenoceptor (β2-AdR) agonists and mAChR antagonists and the anti-inflammatory GCSs (also called interchangeably here glucocorticoids). Within the bronchodilator classes, there are important distinctions between the short-acting rescue therapies (short-acting β2-AdR agonists, SABA, and short-acting mAChR antagonists, SAMA) versus the long acting members of these classes LABA and LAMA that are used generally for maintenance therapy.
Both asthma and COPD like many other conditions are managed in a progressive fashion. First a drug of one pharmacological class is used (often a short-acting bronchodilator, SABA) on demand. If satisfactory control is not achieved, then a drug of a second class (typically an inhaled GCS in asthma or LAMA in COPD) is added and eventually other classes of drugs until control is achieved. As reviewed in Sections IV and V, the bronchodilators β2-AdR agonists and mAChR antagonists have different points of interaction with airway smooth-muscle tone: mAChRs appear to be more prominent in the central airways, whereas β2-AdRs have a higher expression level in peripheral airways. So, a combination should provide optimized bronchodilation in all regions of the lungs. Concomitant administration of these two classes of bronchodilators by pMDIs or by nebulizers showed early on additive bronchodilator effects in several large trials (Cazzola et al., 2012). Therefore, additive bronchodilation or protection against constrictive stimuli can be achieved by adding well tolerated doses of agents of the two classes. In essence, the intended therapeutic effects are additive, whereas most adverse drug reactions of the two classes are different, so not additive. This combination rationale applies to short/rapidly acting agents (SABA/SAMA) as well as long-acting agents (LABA/LAMA).
Inflammation and airway tone play dominant roles in asthma and COPD, indicating a major role of both the anti-inflammatory ICSs and bronchodilator treatments.
Without the combination products, patients may have to use several different individual “monotherapy” inhalers and sometimes even at different times of the day. Such polypharmacy may therefore increase the risk of incorrect use, as the techniques for correct use of the devices may be quite different. In fact, several studies have reported that 50% to 60% of patients with asthma or COPD cannot use their inhalers well enough to benefit from treatment (Lavorini et al., 2014).
The additive effects of different classes of therapy from separate inhalers were shown to carry over to combination inhalers.
Poor compliance with therapy is often thought to be a cause of uncontrolled disease in patients with asthma and COPD (GINA, 2021; GOLD, 2020). Once it was realized that several different treatments were required to treat all but the mildest patients, physicians became concerned that patients would rely on faster-acting bronchodilators (β2-AdR agonists) at the expense of the slower-onset controller medicines (ICS). This led to a push in the mid-1990s to develop combination inhalers containing both bronchodilators and GCS, which should theoretically improve patient compliance, as they simplify the patients` drug regimen who need to learn to use different types of inhalers correctly. Thus, there were convincing arguments to develop fixed-dose combination inhalers.
Subsequently, combinations of two bronchodilators—an LABA and a GCS and triple combinations of two bronchodilators and a GCS—have been brought to the market. There are now a range of combination products available to treat patients with asthma and COPD. These are listed in Tables 8–11.
Products, inhalers, and characteristics of short-acting combination therapies
Prescribing Information: www.accessdata.fda.gov/scripts/cder/daf/ (USA), www.fachinfo.de/(EU, Germany), www.medicines.org.uk/emc/(UK).
Products, inhalers and characteristics of ICS-LABA combination therapies
Prescribing Information: www.accessdata.fda.gov/scripts/cder/daf/(USA), www.fachinfo.de/(EU, Germany), www.medicines.org.uk/emc/(UK).
B. Therapeutic Use
The pharmacology of the components of the combinations are discussed in the single-entity Sections IV–VI.
Except for the rapid and short-acting combinations [ipratropium + salbutamol (or fenoterol)] and in some markets the budesonide/formoterol combination, which also has a rescue indication, the combination therapies are used as maintenance treatment for improvement of airflow and relief of symptoms, and some combinations have shown a reduction of exacerbations in COPD (LAMA-LABA, GCS-LABA, and GCS-LABA-LAMA) and asthma (GCS-LABA). One of the GCS-LABA-LAMA combinations (Enerzair Breezhaler) is registered for prevention of symptoms and exacerbations in asthma in the EU.
LAMA-LABA combinations are registered in COPD only, so they are not an option for pediatric patients. GCS-LABA combinations are typically registered in COPD and asthma. Most combinations have been profiled for patients with asthma ≥12 years of age. Only few have been investigated in infants ≥5–11 years of age, and for this age group only the lowest dose is recommended (Table 10). Most GCS-LABA-LABA combinations too are registered in COPD only (Trelegy Ellipta, Trimbow, Bretztri Aeroshere). Enerzair Breezhaler is registered in asthma; however, pediatric clinical data are not reported.
Products, inhalers and characteristics of LAMA-LABA combination therapies
Assessed by transition dyspnea index (TDI), (c) by St.Georges Respiratory Questionnaire (SGRQ), (d) moderate + severe exacerbations. Prescribing Information: www.accessdata.fda.gov/scripts/cder/daf/ (USA), www.fachinfo.de/(EU, Germany), www.medicines.org.uk/emc/(UK).
Products, inhalers, and characteristics of ICS-LABA-LAMA combination therapies
Prescribing Information: www.accessdata.fda.gov/scripts/cder/daf/ (USA), www.fachinfo.de/(EU, Germany), www.medicines.org.uk/emc/(UK).
Interestingly, it appears that no dose-finding studies were necessary for the combinations, and the doses used were set by those of the individual drug components. Nevertheless, some combinations have confirmed the appropriateness of the doses with phase II dose-ranging studies (e.g., Spiolto Respimat).
The international guidelines for asthma recommend the use of the LABA-GCS combination at step 3 and the LABA-GCS-LAMA combination at step 5 of the treatment guidelines (GINA, 2021). For COPD, the guidelines reserve all combinations for the more severe patients who not adequately controlled with LAMA or LAMA-LABA or GCS-LABA treatment (GOLD, 2020). Taken as recommended in the label, there is no clinical difference between the once- and twice-a-day products, although there is a theoretical compliance advantage of the once-daily dosing. However, this has not been shown in clinical trials to date.
The actual choice between different combination products may come down to patient and doctor preference of the delivery systems used. There are several different devices and even formulations of drugs within the same type of device. These differences may affect the dose to the lung and therefore the deposited dose, but there is little evidence that these change the fundamental safety/efficacy ratio of the drug being delivered.
A number of these drug molecules are no longer patent-protected. However, few substitutable generics have come to the market. This is due to the exacting requirements of a match in in vitro testing as well as in pharmacokinetic and clinical endpoints. A number of nonsubstitutable versions of the innovators’ products have been developed using the so-called 505 b(2) regulation in the United States, and they are less exacting but can lead to different doses if the delivery is not matched (e.g., Teva AirDuo RespiClick).
C. Aerosol Delivery
Combining drugs in metered dose inhaler suspension formulations has been known since the early 1960s (Medihaler Duo: isoproterenol and phenylephrine) (Stein and Thiel, 2017). As shown in Tables 8–11, there are now a range of therapeutic combinations delivered either via an MDI or DPI. There are, however, limitations in delivering some drug combinations particularly via DPIs.
The combination of formoterol and budesonide was successfully developed in a multidose reservoir powder inhaler (Turbuhaler/Flexhaler – Loof et al., 2008); however, this product was not registered in the United States. Instead, the company developed an HFA (hydrofluoroalkane) MDI formulation that is now widely prescribed and accepted by patients and payers.
Although both premetered (capsule) and reservoir DPI devices have been developed containing two drugs and have demonstrated acceptable dose consistency, there are technical challenges to achieve consistent dose delivery, particularly when the amounts of drug delivered cover a wide range (Canonica et al., 2015) or there is a need to deliver triple drug combinations.
To overcome these challenges, companies have deployed differing technical approaches. A novel multidose DPI was developed (Ellipta, Grant et al., 2015) that contains two premetered drug-containing strips. This allows drugs to be formulated separately (two drugs in one strip, one in the other) and can achieve the required dose consistency and stability through the life of the product.
An alternate approach is to formulate the drug in a complex matrix within an MDI. By using cosuspension technology, the sources of dose variability can be overcome (Doty et al., 2018). This technology has been successfully deployed in commercial combination inhalers.
The change from CFC MDIs to HFA-powered MDIs was also a transition to solution-based aerosol generation, which allowed a higher proportion of extrafine particles (MMAD < 2 µm). These inhalers deposit a higher proportion of the drugs in the small airways (<2 mm in diameter). There is no doubt of the importance of inflammation and pathologic changes in the small airways in asthma and COPD, and a number of studies claim superiority of extrafine GCS, GCS-β2-AdR-agonist combinations, and triple combinations versus the “conventional” formulations in asthma and COPD (Santus et al., 2020). In contrast, a systematic review and meta-analysis (El Baou et al., 2017) support the viewpoint of no difference between extrafine and standard aerosols in asthma. However, as a note of caution in interpreting these data, it should be considered that the endpoints assessed in most clinical studies to date are not sensitive to small airway changes.
As an alternative to MDI or DPI delivery, a range of molecules (including combinations) is available in an aqueous multidose soft mist inhaler (Respimat) that aerosolizes a metered dose of aqueous drug(s) solution into a slower-velocity fine mist for inhalation with one deep inspiration. The device is powered by the energy of a compressed spring (see also Section IV). In some countries these products are now available as reusable refills with the delivery mechanism being used for multiple cartridges (up to six) for 1 month supply each (Lavorini et al., 2014).
D. Conclusions
Complex diseases, such as asthma and COPD, benefit from polypharmacy to increase control at a reduced adverse effect load so the development of combination products was a logical next step. These combination products will remain the main maintenance therapy for moderate and severe patients for the foreseeable future.
VIII. Cromones–Sodium Cromoglycate and Nedocromil
A. Introduction
Like many drugs developed in the 1950s and 1960s, inhaled cromones were developed after discoveries based on plant extracts. Sodium cromoglycate (SCG), the first approved cromone, was developed from a Middle Eastern extract known as Khellin. The “antispasmatic” action of Khellin was well known (Mustapha, 1879) with typical doses being in the tens to hundreds of milligrams delivered orally or by intramuscular injection. Kennedy and Stock (1952) investigated its bronchodilator action in patients who were asthmatic. Oral dosing was from 50 to 400 mg daily. They concluded “that the results are sufficiently encouraging to warrant further work on the action and clinical use of Khellin as a bronchodilator.”
B. Sodium Cromoglycate
In early 1963, using Khellin derivatives as a starting point, a new cromone, later to be known as disodium cromoglycate, was synthesized (Howell, 2005).
SCG with a pKa of 2 is almost completely ionized at physiologic pH and is thus poorly absorbed from the gastrointestinal tract (Murphy, 1988). Thus, it was initially developed as an inhaled dry powder using lactose blend technology and delivered via a dry powder inhaler. The Spinhaler had originally been designed to deliver proteolytic enzymes but was ideal for the task of delivering high doses of cromolyn powder directly to the lungs (Howell, 2005). Because SCG needed to be delivered prophylactically to protect against allergens and because of its lack of immediate bronchodilator effect, it was initially developed in combination with low-dose bronchodilator isoprenaline (Holgate 1996). The product introduction was met with some enthusiasm, as treatment of asthma in children was very limited at the time, and inhaled isoprenaline alone was not approved for use in children. Silverman and Godfrey initially investigated the effect of long-term daily treatment with this combination product (Silverman, 1972). This trial was successful in that 71% of the children on active treatment were still well controlled after 1 year compared with 24% in the placebo group.
These reports developed a considerable interest in both acute and regular daily treatment with SCG given by inhalation directly to the target organ, the lung. Studies were carried out to investigate the acute effect on the bronchoconstriction induced by inhaled allergen (Howell and Altounyan, 1967) and exercise (Davies, 1968; Read and Rebuck, 1969) and to investigate long-term control of asthma (Kennedy, 1969; Read and Rebuck, 1969; Jones and Blackhall, 1970). When SCG was inhaled from the Spinhaler, no delay was required between administration and the start of exercise because of the immediate onset of its protective action (Fig. 1) (Silverman and Andrea, 1972). The major advantages of SCG were its inhibition of the late response to inhaled allergens (Dahl and Mölgaard Henriksen, 1980), the lack of tolerance to its protective effect when used daily, and its safety profile driven by its poor oral bioavailability and its inability to partition into cells and interfere with intercellular function (Murphy, 1988).

Mean values for % change in peak expiratory flow rate (P.E.F.R.) from baseline before, during, and after exercise with no drug in the presence of placebo and after inhaling 10 mg of disodium cromoglycate (DSCG) from a capsule 20 minutes before exercise, at the start of exercise, and immediately at the end of exercise. (Silverman and Andrea 1972).
The mechanism of action of SCG was not clear at the time, but it was known that it did not have any direct action on bronchial smooth muscle (Orr and Cox, 1969), and it was ineffective against challenge by histamine (Jenkins and Breslin 1987). However, it was reported to inhibit adenosine-induced extravasation, suggesting it may be a functional antagonist of tachykinins (Tamaoki et al.,1999; Yamawaki et al., 1997). Although it was demonstrated to have an action on “C” fiber nerves (Dixon et al., 1980), its primary effect was thought to be in inhibiting the release of mediators, probably histamine, from mast cells (Riley and West, 1953; Flint et al., 1985; Leung et al., 1986) that are present in the pulmonary epithelium and free in the bronchial lumen (Wasserman, 1984). The mechanism for this inhibition was proposed as preventing interaction between IgE and the mast cell, the crucial event that caused degranulation and release of histamine (Cox, 1967). It was considered that the asthmatics most likely to benefit from treatment with inhalation of SCG were those with positive skin reactions to common aero allergens (Pepys et al., 1968).

Duration of the effect of sodium cromoglycate in suppressing exercise-induced asthma in three groups of patients selected by the time course of their response. Exercise was performed every 2 hours. The drug appeared to exert a relatively long effect in five subjects (A), a relatively short effect in five subjects (B), and no effect in four subjects (C). (Godfrey, Silverman and Anderson 1973). DSCG, disodium cromoglycate; P.E.F.R., peak expiratory flow rate.
As bronchial challenge testing with inhaled allergens in the clinic was impractical and potentially unsafe, exercise was used to investigate the acute protective effect of SCG and its duration of action (Silverman and Andrea, 1972; Silverman et al., 1972). The initial exercise studies were carried out in a small number of adults, and SCG was effective in both skin-test-positive and skin-test-negative subjects with exercise-induced bronchoconstriction (Silverman and Turner-Warwick, 1972). Pediatricians were keen to investigate the protective effect of SCG, 20 mg delivered by inhalation from the Spinhaler, on children with asthma provoked by exercise (Silverman et al., 1972). At the time it was shown that exercise by running was more potent than cycling or swimming (Anderson et al., 1971), so running on a treadmill was chosen as the challenge to provoke an asthma attack. In brief, a third of the group had excellent protection from an asthma attack provoked by exercise, one-third received some effect, and in one-third there was no protective effect (Fig. 2) (Godfrey et al., 1973, Silverman et al., 1973). The results in 80 children for four tests performed at 2-hour intervals after 20 mg of SCG illustrate the relatively short duration of its protective effect (Fig. 3) (Anderson et al., 1975). Some years later, important studies were reported on the duration of the protective effect and the effect of varying doses (Fig. 4) (Tullett et al., 1985) on exercise and hyperventilation with cold air (Juniper et al., 1987).

The duration of action of sodium cromoglycate in 80 patients assessed by repeating exercise tests every 2 hours after a single dose of drug or placebo before the first test on each of 2 separate days. The drug had a progressively diminishing effect in suppressing the postexercise fall in peak expiratory flow. (Anderson et al., 1975).
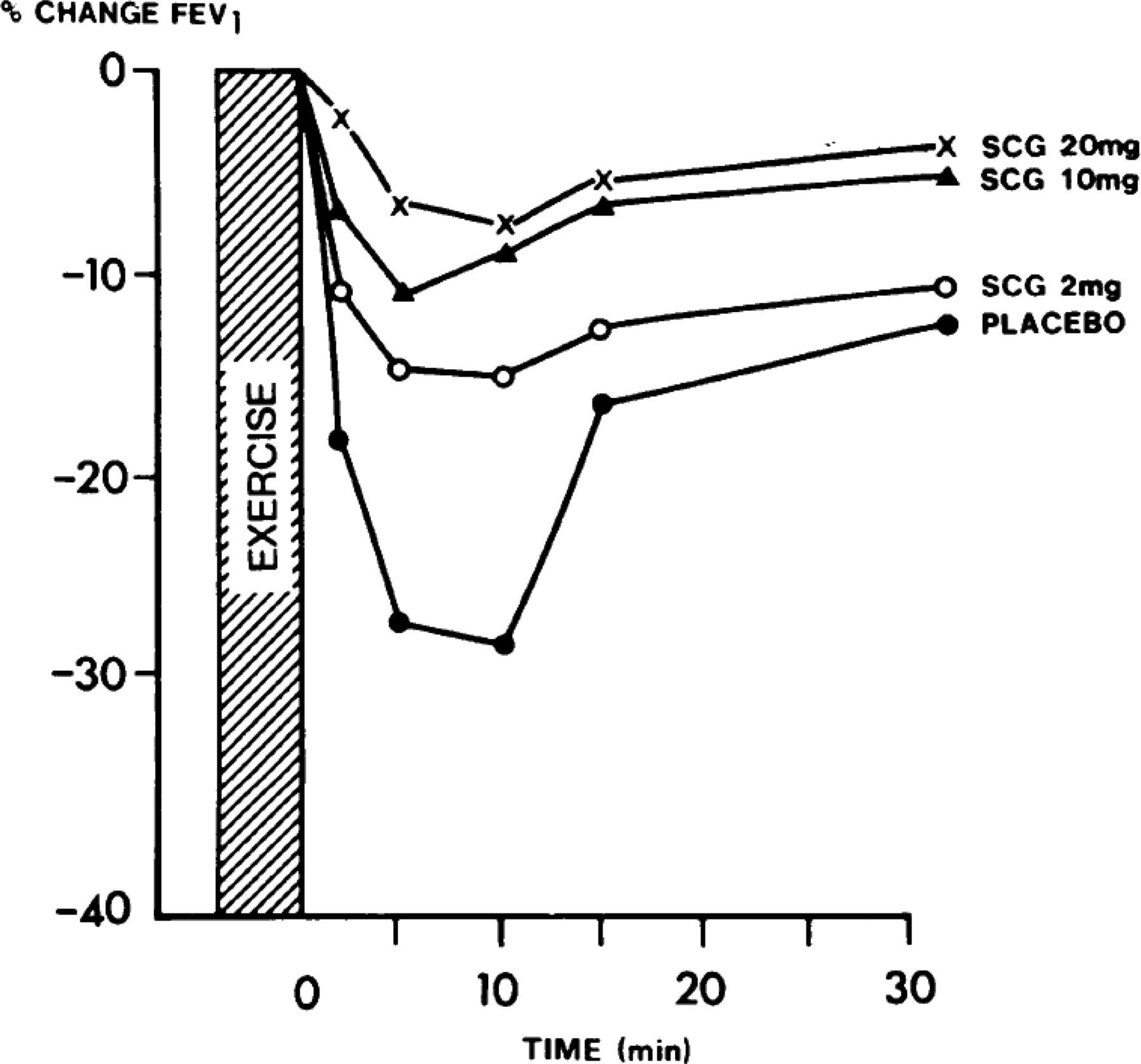
Mean percentage change in FEV1 (from baseline after administration of drug) over 30 minutes after exercise with different doses of sodium cromoglycate aerosol. (Tullett et al., 1985).

Mean percentage (%) fall in FEV1 in 12 children (6.5–13.5 years) with asthma after 6 minutes of running exercise at the baseline test and 30 minutes after inhaling a placebo, 4 mg of nedocromil sodium and 10 mg of sodium cromoglycate delivered either directly from a metered dose inhaler or via a Fisonair spacer in a randomized, double-blind crossover design (Comis et al., 1993).
Subsequently it was shown in a long-term study that inhaled SCG resulted in 65% of the children being well controlled after 3–5 years of treatment (Godfrey et al., 1975). The children most likely to benefit from treatment with SCG were those with mild-to-moderate persistent asthma with the drug appearing to play no role in controlling children with severe asthma.
Over the years SCG was developed in all of the now popular inhalation modalities (Table 12). Sodium cromoglycate with low-dose isoprenaline as a lactose blend formulation was the first, and this was followed by SCG alone in both blend and pelletized formulations. At the time both inhalation powder blends (Newman and Jenkins, 1968) and pelletized powder formulations (Gunning and Hartley, 1975) were new technologies, but they subsequently became the mainstay of dry powder inhaler development. Dry powder formulations were followed by a 1% aqueous nebulizer solution and two pMDI at doses of 1 mg and 5 mg SCG per actuation. Intal 5 (Intal being the brand name of SCG) was then updated as a CFC-free pMDI using the HFA227 as a new propellant. Although these product forms spanned a wide range of nominal doses, it should be remembered that the delivery efficiencies of the differing modalities also vary. In scintigraphy studies, delivery of the 20-mg nominal dose using the Spinhaler resulted in 1.1–3.4 mg depositing in the lung depending on the inhaled flowrate (Newman et al., 1994). Using similar imaging techniques, the 5 mg pMDI (2 actuations per dose) delivered 0.9 mg and 1.1 mg to the lungs when used with and without a small volume spacer, respectively (Newman et al., 1991), indicating that despite the wide range in nominal doses lung doses are similar (Table 12). Pharmacokinetic studies using urine collection in children estimated lung deposition from the 20-mg nebulizer solution was approximately 0.3 mg.
Major product forms of sodium cromoglycate and nedocromil sodium
C. Nedocromil
Nedocromil, a second-generation cromone, was developed during the 1970s and came to market in the 1980s. It had a similar profile to SCG in terms of protection against exercise-induced bronchoconstriction. (Fig. 5). (Albazzaz et al., 1989; Speelberg et al., 1992; Comis et al., 1993; de Benedictis et al., 1995; Oseid et al., 1995; Kelly et al., 2001; Spooner et al., 2002). However, in vitro it exhibited far greater potency, up to 200 times that of SCG, in inhibiting release of mediators, such as histamine, leukotriene C4, and prostaglandin D2. Nedocromil’s safety and effectiveness was born out in bronchial challenge tests with exercise and with sulphur dioxide (Fig. 6) (Barnes, 1993). Further, its recommended dose of 4 mg (two times 2 mg per actuation) twice daily from a pMDI showed efficacy in the treatment of reversible obstructive airway disease (Gonzalez and Brogden, 1987). In a pharmacokinetic study using urinary excretion, the estimated lung dose from the 4 mg pMDI was approximately 0.3 mg (Aswania et al., 1998), indicating that lung doses achieved with nedocromil were similar to that of SCG despite its higher in vitro potency. The initial CFC pMDI was replaced in 2003 with a CFC-free pMDI using HFA227 (Table 12).

The effect of nedocromil sodium (Ned) on sulfur dioxide–induced bronchoconstriction and dyspnea measured by visual analog scale in six asthmatic subjects (Barnes, 1993 modified from Dixon et al., 1987).
Albazzaz et al. (1989) performed a dose-response study in exercise-induced asthma involving 10 patients and three concentrations of nebulizer solutions, 0.5%, 1%, and 2%. Aerosols were generated by a Wright Nebulizer and involved tidal breathing for 5 minutes, giving a delivery of approximately 1 ml that was similar to the delivery from a 2-ml ampoule using a conventional nebulizer. All solution concentrations significantly improved FEV1 compared with placebo, and there was no difference in response between the concentrations, indicating that the 0.5% solution generated an effective dose. A 0.5% solution was subsequently developed as the commercial product form (Table 12).
D. Conclusions
Although the prophylactic use of cromones in allergic and exercise-induced bronchoconstriction was extremely successful, their relatively short duration of action requiring four-times-a-day dosing eventually limited their use in comparison with the twice-a-day and once-a-day bronchodilators and glucocorticoids. Early findings of benefits of inhibition of certain types of cough using cromones (Hargreaves and Benson, 1995) have not resulted in additional approvals for this indication. More recent studies of alternative pathways that are affected by cromones (Sinniah et al., 2017) do not seem to have appeared to result in expanded clinical use for either asthma or other respiratory diseases. Nevertheless, the use of cromones despite their limitations is likely to continue in some patient populations because of the very low incidence and intensity of side effects.
IX. Anti-Infectives
A. Introduction
Lower respiratory tract infections (e.g., tuberculosis, influenza, pneumonia, measles, severe acute respiratory syndrome) represent some of the greatest threats to global health, with more than 4 million deaths recorded worldwide in 2012 (Forum of International Respiratory Societies, 2017; WHO Global Tuberculosis Report, 2020). With the rapid increase in antimicrobial resistance (Marston et al., 2016) and emerging respiratory pathogens, such as SARS-CoV-2 (Cevik et al., 2020), the need for novel therapeutics and new therapies that target these pathogens within the respiratory tract is of critical importance.
Administration of anti-infectives by oral inhalation targets the drug directly to the site of the infection in the lungs resulting in high local drug concentrations that are often not attainable by oral or parenteral therapy, reductions in systemic drug concentrations, and reductions in drug exposure to gut microbiota (Woods and Rahman, 2018; Weers, 2015; Bassetti et al., 2020; Cipolla et al., 2016a). Regional deposition within the respiratory tract is also of importance, as the site of the infection (e.g., the airway lumen, the lung parenchyma, or intracellular in pulmonary macrophages) also influences the design and development of the inhaled drug product.
A key challenge with inhaled anti-infectives is that they typically have low potency, necessitating administration of nominal doses between ∼10 mg and 6000 mg (Table 13). This limits the choice of delivery systems to nebulizers and in some cases dry powder inhalers. It also imposes constraints on formulation design. The high doses of drug and excipient delivered to the pulmonary epithelium also impose unique challenges from a drug delivery perspective. These include local safety and tolerability concerns (e.g., postinhalation cough, throat irritation, bronchospasm; Sahakipijarn et al., 2020) as well as concerns with respect to device portability, administration time, and daily treatment burden (Sawicki et al., 2009).
Inhaled anti-infective products
The physicochemical properties of the anti-infective (e.g., lipophilicity, aqueous solubility, stability in airway surface liquid) affect the suitability of the drug for inhalation, with a key requirement being to achieve and typically also maintain high drug concentrations within the respiratory tract. Indeed, dramatic improvements in efficacy have been observed with inhaled anti-infectives by optimizing their pharmacokinetic/pharmacodynamic profile. Hence, the development of sustained release formulations (e.g., liposomes, microparticles) may enable superior treatment of lower respiratory tract infections (Cipolla et al., 2016a; Zhang et al., 2018; Bassetti et al., 2020). The concentrations of drug should not only exceed the minimum inhibitory concentration for the susceptible target organism(s) but preferably also prevent the growth of the less susceptible single-step mutants to avoid development of antimicrobial resistance; thus, the anti-infective drug should exceed also the mutant prevention concentration, MPC (Blondeau et al., 2004).
The current marketed products developed to treat lower respiratory tract infections are summarized below.
It is noted that so far, there has been a remarkably small number of inhaled anti-infectives approved vis a vis the potential addressable healthcare problem. The only exception is the treatment of chronic respiratory tract infections with Pseudomonas aeruginosa in cystic fibrosis in which multiple products have been approved.
B. Tobramycin
1. Tobramycin Inhalation Solutions
The first inhaled antibiotic to receive marketing authorization was tobramycin inhalation (TOBI) solution. Tobramycin is an aminoglycoside antibiotic produced by Streptomyces tenebrarius, with activity against gram-negative bacteria (Bulitta et al., 2015). TOBI is indicated for the management of chronic P. aeruginosa infections in patients with cystic fibrosis (CF) above the age of 6 (TOBI Technical Monograph, 1998). The drug is administered twice daily according to a 1-month-on/1-month-off treatment regimen designed to limit the development of antimicrobial resistance (Ramsey et al., 1999).
TOBI contains 300 mg of tobramycin in 5 ml of sodium chloride (2.25 mg/ml) at pH 6.0 (TOBI Technical Monograph, 1998). The acidic pH enables the TOBI solution to remain stable without the addition of preservatives (Alothman et al., 2002). This was an important finding, as marketed parenteral formulations of tobramycin containing preservatives caused bronchospasm in children when inhaled. The sterile TOBI solution is hypoosmolar, with an osmolality between 158–183 milliosmoles (mOsm) per kg. TOBI has a permeant anion (chloride) concentration of ∼75 mM (TOBI Technical Monograph, 1998). The presence of small-hydrated anions like chloride that are able to permeate and depolarize the nerve membrane are effective in suppressing the cough reflex (Boggs and Bartlett, 1982; Godden et al., 1986). The pH, osmolality, and chloride ion concentration fall within the ranges deemed critical for airway tolerability and safety (Weber et al., 1997; Chang et al., 2020). Although tobramycin is relatively stable in aqueous solution and serum, the cationic drug can be inactivated when bound to negatively charged mucins and DNA fragments in sputum (Hunt et al., 1995).
Compared with parenteral administration, pulmonary administration of tobramycin with a jet nebulizer (mass median aerodynamic diameter, MMAD ∼4 µm) enables significantly higher doses of drug to be administered to the bronchial airways while minimizing plasma levels of drug. The total lung dose of TOBI using the approved jet nebulizers determined by gamma scintigraphy is ∼15% of the dose placed in the nebulizer (Lenney et al., 2011) (some of the dose deposits in the mouth, but the oral bioavailability of tobramycin is insignificant). The improved lung targeting afforded by TOBI leads to reductions in P. aeruginosa sputum density, improvements in lung function, and reductions in pulmonary exacerbations, inflammatory markers, and the incidence of systemic adverse events (e.g., ototoxicity and nephrotoxicity) (Ramsey et al., 1999). TOBI has contributed to the significant increase in life expectancy of patients with CF (Sawicki et al., 2012).
Twice-daily administration of TOBI with a jet nebulizer places a significant burden on the patient with an administration time of 15–20 minutes per dose plus additional time required for nebulizer cleaning and disinfection (Sawicki et al., 2009).
As a result, second-generation formulations of inhaled tobramycin have focused on reducing treatment burden and improving treatment convenience. This includes the development of more concentrated solutions of tobramycin for nebulization (Bethkis, Vantobra) coupled with the use of vibrating mesh nebulizers (e.g., e-Flow, PARI) providing more rapid delivery (Mazurek et al., 2014; Sands et al., 2014) and the advancement of a novel dry powder formulation product, TOBI Podhaler (Geller et al., 2011).
2. Tobramycin Inhalation Powder/TOBI Podhaler
Owing to the high dose requirements of inhaled tobramycin, traditional dry powder formulation technologies (e.g., lactose blends, spheronized particles) are not suitable for inhaled antibiotics (Geller et al., 2011; Weers, 2015). The dry powder formulation in TOBI Podhaler is prepared by spray drying an emulsion-based feedstock to create small porous particles (PulmoSphere technology; Weers and Tarara, 2014). The core-shell particles are comprised of a core of amorphous tobramycin sulfate coated with a 2:1 molar ratio of distearoylphosphatidylcholine to calcium chloride. The spray-dried particles have a median geometric diameter of 1.7–2.7 µm and an MMAD <4 µm (Geller et al., 2011). The engineered particles provide highly efficient pulmonary delivery (∼60% of nominal dose in an in vitro anatomic throat model) that is independent of patient inspiratory flow rate (Haynes et al., 2016). The nominal dose of tobramycin in the TOBI Podhaler is 112 mg administered in four capsules according to the same treatment regimen as nebulized TOBI (Geller et al., 2011). The dry powder formulation showed comparable peak levels of tobramycin in serum to the nebulized drug product (i.e., ∼1 µg/ml). This is significantly less than the systemic drug levels observed with intravenous tobramycin (i.e., 10–12 µg/ml) that are associated with oto- and nephro-toxicity, while providing significantly higher concentrations of drug in sputum (Geller et al., 2011).
The dry powder formulation enables reductions in administration time from 15–20 minutes to 4–6 minutes while eliminating the time needed for cleaning and disinfection (Konstan et al., 2011a). The portable dry powder inhaler, room temperature storage, and lack of power source requirements also allow for patients to go “wireless,” enabling more convenient delivery outside of the home (Geller et al., 2011).
TOBI Podhaler is well tolerated by most patients. The safety profile of TOBI Podhaler is similar to nebulized TOBI with respiratory disorders (e.g., cough, lung disorder, dyspnea, oropharyngeal pain, dysphonia, and hemoptysis) being the primary source of adverse events.
C. Aztreonam Lysinate/CAYSTON
Aztreonam is a monobactam antibiotic with good activity against a range of gram-negative bacteria, including Escherichia coli, P. aeruginosa, and K. pneumoniae (Brogden and Heel, 1986).
Cayston (aztreonam lysinate solution for inhalation) is comprised of a 75-mg nebulized dose administered to patients with CF three times daily with the Altera nebulizer, a variant of the PARI e-Flow vibrating mesh nebulizer (Elborn and Henig, 2010; Plosker, 2010). Cayston is indicated to improve respiratory symptoms in patients with CF with P. aeruginosa.
Aztreonam lysinate is packaged as a lyophilized powder that is reconstituted in 1.0 ml of 0.17% saline. The nebulized solution is administered over a period of 2–3 minutes. The nebulized droplets have an MMAD of 3.8 µm, with about 30 mg of the 75-mg nebulized dose deposited in the lungs (Elborn and Henig, 2010). The lysinate salt replaced the arginate salt used in parenteral formulations due to concerns regarding the role of arginine in nitric oxide production in the lungs (Sapienza et al., 1998).
Safety and efficacy of Cayston in the treatment of P. aeruginosa infections in patients with CF was demonstrated across multiple clinical studies (Elborn and Henig, 2010) for up to nine 28-day-on/28-day-off cycles (Oermann et al., 2010). In each cycle, Cayston showed a mean improvement in FEV1 followed by a return to baseline in the off cycle.
It is of note that Cayston is hyperosmolar with an osmolality of ∼550 milliosmoles/kg (Weers, 2015). Sudden alterations in the composition (e.g., pH, osmolality) of the periciliary fluid lining the respiratory epithelium may induce changes in ciliary beat frequency and induce protective reflexes, such as cough and apnea (Godden et al.,1986; Sahakijpijarn et al., 2020). To prevent adverse pulmonary effects, especially bronchoconstriction, patients are pretreated with bronchodilators. It is possible that the olecular nature of this drug also contributes to its irritation potential.
D. Levofloxacin/QUINSAIR
Levofloxacin is a broad-spectrum fluoroquinolone antibiotic with activity against both gram-negative and gram-positive organisms (Davis and Bryson, 1994). The Quinsair drug product is comprised of a solution of levofloxacin complexed with magnesium chloride. Complexation increases the water solubility of the drug from 17 mg/ml to 100 mg/ml (Ross and Riley, 1992) while reducing the bitter taste and improving the pharmacokinetic profile. The 240-mg Quinsair dose (2.4 ml) is administered twice daily to patients with CF over about 5 minutes with the Zirela nebulizer system (a variant of the e-Flow vibrating mesh nebulizer). The nebulized droplets have an MMAD of ∼4 µm. Quinsair is indicated for the treatment of patients with CF with chronic infections with P. aeruginosa and is approved in the EU and Canada but has not yet been approved by the FDA.
Significant reductions in P. aeruginosa sputum density and improvements in lung function were observed for Quinsair in a placebo-controlled phase III study (Flume et al., 2016). Quinsair was noninferior to TOBI in terms of improvements in lung function (Elborn et al., 2015). In addition to its antibacterial activity, Quinsair shows evidence of immunomodulatory effects (i.e., decreases in the proinflammatory cytokines IL-6 and IL-8; Tsivkovskii et al., 2011). Like other inhaled antibiotics, prominent adverse events include cough and dysgeusia.
E. Colistimethate Sodium/COLOBREATHE
Colistin is a polypeptide antibiotic from the polymyxin family isolated from the soil bacterium Bacillus colistinus (Li et al., 2005). Its importance has increased over the last decade because of it being one of the few remaining options for treatment of multidrug-resistant organisms. It is available in two forms, as colistin sulfate and as the prodrug colistimethate sodium (Conole and Keating, 2014). There is evidence that the prodrug may be better tolerated (Westerman et al., 2004). Colistimethate has very low absorption from the gastrointestinal tract (EMA, 2016).
Parenteral formulations of colistin (e.g., Coly-Mycin) have been nebulized, particularly in Europe, for more than 2 decades. More recently, a dry powder formulation of colistimethate has been advanced but has not yet been approved by FDA (Colobreathe).
Colobreathe (colistimethate powder for inhalation) is a dry powder formulation of neat micronized colistimethate (Schwarz, 2015). The 125-mg dose is administered twice daily; at each administration event, the contents of a single capsule are inhaled using the portable Turbospin dry powder inhaler over approximately three breaths. Approximately 12% of the nominal dose is delivered into the lungs without pretreatment with a bronchodilator (Su et al., 2014). Patients on Colobreathe who also take a short-acting inhaled bronchodilator are advised to take it prior to the dose of inhaled colistimethate.
Colobreathe was noninferior to TOBI for chronic suppressive treatment of P. aeruginosa infections in patients with CF while significantly decreasing the administration time to 1–2 minutes (Schuster et al., 2013).
Like other inhaled antibiotics for the treatment of CF, the incidence of respiratory adverse events, including cough, are elevated relative to parenteral formulations.
The development of Cayston, Quinsair, Colobreathe, and other classes of antibiotics is critical for effective chronic suppressive treatment of P. aeruginosa infections and minimization of the development of drug resistance in patients with CF. The availability of multiple antibiotics enables the development of alternative treatment regimens, including the potential use of different antibiotics in alternating 28-day or 14-day treatment cycles.
F. Amikacin/ARIKAYCE
Amikacin sulfate is a semisynthetic aminoglycoside antibiotic derived from kanamycin A, a natural antibiotic isolated from Streptomyces kanamyceticus. Amikacin is active against a broad spectrum of gram-negative and some gram-positive microorganisms, including nontuberculous mycobacteria (NTM). Amikacin’s polycationic structure allows it to associate with lipopolysaccharide, phospholipids, and anionic proteins on the surface of the bacteria, and following active transport across the cell membrane and binding to the 30S bacterial ribosome subunit, it interferes with bacterial protein synthesis (Ramirez and Tolmasky, 2017). Amikacin has very low oral bioavailability.
The application of amikacin is primarily limited by the side effects of nephrotoxicity and ototoxicity, which are common for the aminoglycoside class of antibiotics (Prayle et al., 2010). These observations provide motivation to treat lung infections by inhalation delivery, which increases the pulmonary amikacin concentration and reduces systemic exposure (Weers, 2015) in addition to replacing injections. Utilizing an inhaled sustained release amikacin formulation like ARIKAYCE provides additional features over inhaled amikacin alone by modifying its disposition in the lung and increasing its uptake into pulmonary macrophages, which may harbor NTM while further reducing its systemic exposure (Shirley, 2019).
ARIKAYCE encapsulates 590 mg of amikacin (as the sulfate salt) in small liposomes (<300 nm) with a high drug-to-lipid ratio of ∼1.5. These liposomes are composed of two biocompatible lipids, dipalmitoyl phosphatidylcholine (DPPC) and cholesterol in a 2:1 weight ratio. The 8.4 ml of ARIKAYCE is delivered by oral inhalation once daily via an optimized eFlow Technology Nebulizer (Lamira Nebulizer System, PARI Pharma GmbH, Munich, Germany), achieving a lung dose of ∼43% (relative to the initial dose in the nebulizer) in patients with NTM lung disease (Olivier et al., 2016). Many liposomal formulations are susceptible to disruption during nebulization (Cipolla et al., 2013), including ARIKAYCE. However, a feature of the ARIKAYCE composition is the consistent generation of ∼30% free amikacin and 70% encapsulated amikacin after mesh nebulization across both the respirable aerosol size range and over the course of the aerosol treatment (Li et al., 2008).
In preclinical development, inhaled ARIKAYCE demonstrated a reduction in systemic amikacin exposure in rats and an ability to penetrate P. aeruginosa biofilms (Meers et al., 2008) and later NTM biofilms (Zhang et al., 2018). Additionally, after inhalation delivery to rats, a 6-fold increase was observed in pulmonary macrophage uptake for liposomal amikacin compared with amikacin alone (Zhang et al., 2018).
After demonstrating a reduction in systemic exposure of amikacin in healthy subjects (Weers et al., 2009) and evaluation first in patients with CF with P. aeruginosa lung infections (Clancy et al., 2013; Bilton et al., 2020) and then in non-CF bronchiectasis, ARIKAYCE made a successful pivot to treating patients with refractory pulmonary NTM infections (Griffith et al., 2018). In a randomized, controlled phase III trial, when added to guideline-based therapy, ARIKAYCE improved culture conversion from 8.9% for the active comparator to 29.0% (P < 0.001).
Once-daily oral inhalation of ARIKAYCE was approved by EMA in 2020 (EMA ARIKAYCE label) and FDA in 2018 under the limited population pathway for antibacterial and antifungal drugs and is indicated “for the treatment of refractory Mycobacterium avium complex lung disease as part of a combination antibacterial drug regimen in adults” (US FDA ARIKAYCE, 2018), and this therapy is further supported by the recent NTM pulmonary lung disease treatment guideline (Daley et al., 2020).
G. Neuraminidase Inhibitors
Influenza enters the human body mainly via inhalation or by oral ingestion. The disease symptoms are typically initially in the respiratory tract, and therefore drug inhalation that targets the respiratory tract for prophylaxis and early intervention is a logical path for administration.
The viral enzyme neuraminidase (also known as sialidase) has a critical role in the infectious spreading of influenza viruses A and B. The understanding of the structural basis of the mechanism of action of inhibitors of this enzyme and refinements in the drug design resulted in several drugs to treat these infections, including the two inhaled drugs Relenza and Inavir (Von Itzstein, 2007).
1. Zanamivir/RELENZA
Relenza, which contains influenza virus neuraminidase inhibitor zanamivir, is indicated for the treatment of acute influenza type A and B infections in patients aged 7 years and older who have been symptomatic for no more than 2 days. It is also indicated for prophylaxis in patients aged 5 years and older (GlaxoSmithKline, 2018).
The mechanism of action of this drug as a member of the class of sialidase inhibitors (see above) was described in detail by Von Itzstein (2007).
The clinical development leading to approval was reviewed by Elliott (2001), and the key aspects of the development leading to approval are included in the package insert (GlaxoSmithKline, 2018) and summarized below.
a. Device, formulation, deposition, and disposition in humans
The Relenza drug product is manufactured as a dry powder formulation. Two inhalations, each containing 5 mg of zanamivir in 20 mg of the carrier lactose, constitute a single dose administered from the blister-based Diskhaler device. Two 10 mg doses are recommended to be taken twice a day.
The early evaluation of this drug-device combination resulted in estimated total bronchial and lung deposition of about 13.2% of the dose in the blister, with the majority of the dose content (77.6%) depositing in the oropharynx (Cass et al., 1999b). The concentration of zanamivir in the respiratory tract after inhalation is estimated to significantly exceed the inhibitory concentrations for influenza virus. Although there is significant oropharyngeal deposition, the systemic exposure is low because of the low oral bioavailability (Elliott, 2001), with only 5% of the unchanged drug appearing in urine in 8 hours postinhalation (Cass et al., 1999a). These early data are similar to the information provided in the package insert (GlaxoSmithKline, 2018), with 4–17% of the inhaled dose absorbed and serum half-life of 2.5–5.1 hours.
b. Pivotal efficacy trials
Treatment of adult patients with early symptoms of the flu with inhaled zanamivir versus placebo in controlled trials suggested a reduction in the median time to improvement of symptoms of about 1–1.5 days, although statistical significance was not reached in these trials. Similar results were observed in the pediatric population (GlaxoSmithKline, 2018).
Two trials with slightly different designs were conducted in postexposure prophylaxis in family households with one infected person initially. Both of these trials demonstrated that 19.0% versus 4.1% of the households receiving placebo versus zanamivir developed flu symptoms in at least one additional family member (GlaxoSmithKline, 2018).
Relenza was also tested as prophylaxis in community-acquired seasonal flu. In the first trial, 86% of the community remained unvaccinated. The incidence of symptomatic laboratory confirmed influenza was 6.1% and 2.0% in the placebo and zanamivir groups, respectively. In a second trial in which the majority of the community (67%) was vaccinated, the incidence of symptomatic laboratory confirmed influenza was 1.4% versus 0.2% in the placebo and zanamivir groups, respectively (GlaxoSmithKline, 2018).
Relenza appears to be a safe and effective medication for pre-exposure and postexposure prophylaxis of influenza, with trends suggesting efficacy in subjects with early symptoms of the disease. The high local concentration in the respiratory tract combined with its low systemic exposure (Cass et al., 1999a,b) is likely to improve the therapeutic index versus oral drugs with a similar mechanism of action. This could be the reason for lower incidence of drug-related side effects of Relenza compared with the orally administered neuraminidase inhibitor Tamiflu (oseltamivir), but this may be also the result of different molecular structures with zanamivir containing features that make it more similar to sialic acid – the natural substrate for sialidase (Shie and Fang, 2019). However, Relenza needs to be used with care in patients with chronic airway disease to prevent bronchospasm; pretreatment with bronchodilators in this population is recommended (GlaxoSmithKline, 2018).
Relenza was studied in pediatric population (ages 5–16) for prophylaxis of influenza, and no differences were found between the safety and efficacy in this group compared with adults. Similarly, no differences were found in the adolescent patients compared with adults when Relenza was used for the treatment of influenza. However, development of adequate inspiratory flow rate with the Diskus dry powder inhaler was a challenge for some younger children with influenza. Children and adolescents may be at higher risk of neuropsychiatric disorders (GlaxoSmithKline, 2018).
2. Laninamivir/INAVIR
Inavir (Daiichi Sankyo) contains an ester prodrug of the neuraminidase inhibitor laninamivir. It is marketed in Japan as a dry powder formulation blend of 40 mg of the drug with lactose and administered from the TwinCap dry powder inhaler (Pharmaceutical and Food Safety Bureau Ministry of Health, Labor and Welfare Japan, 2010). Adults require two devices and eight inhalations from four capsules, whereas children take four inhalations from two capsules in a single device (Ikematsu and Kawai, 2011). The multiple inhalations are presumably required to empty the high formulation dose in each capsule.
After inhalation, the prodrug is retained in the lung for a long period of time and is metabolically converted into its active form. Studies in human volunteers taking a single inhaled dose of the dry powder formulation showed that the laninamivir concentration in the airway epithelial lining fluid exceeded the 50% inhibitory concentration against viral neuraminidases for at least the duration of the last observation of 240 hours (Ishizuka et al., 2012). The lung concentrations of both the prodrug and the drug measured in the bronchoalveolar lavage and in alveolar macrophages are markedly higher than the plasma concentrations of these molecules.
Inavir therefore affords a single dose administration by inhalation that should be effective for at least 10 days. It is an interesting example of the combination of inhalation with the prodrug approach wherein the prodrug has favorable characteristics because of its relatively low solubility, long retention in the lung, and use of local metabolism for conversion into the active drug moiety.
a. Comparative efficacy
Inhaled laninamivir octanoate (Inavir) showed clinical efficacy comparable to oral oseltamivir (Tamiflu) and inhaled zanamivir (Relenza) against the H1N1 pandemic influenza strain from 2009, seasonal H3N2 influenza, and influenza B viruses (Ikematsu and Kawai, 2011). The key differentiating factor is a single-dose administration for Inavir.
b. Pediatric population safety
As with Relenza, younger subjects are more likely to exhibit neuropsychiatric side effects. It is advisable to provide adult supervision to teenagers receiving Inavir (Nakano et al., 2013).
H. Ribavirin/VIRAZOLE
Respiratory syncytial virus (RSV) is a most common cause of lower respiratory tract infections in children and elderly people. Its journey in humans typically starts by replication in the upper airways (naso- and oro-pharynx), which is followed by a spread into the lower respiratory tract. The infection can cause severe disease, and it can be particularly dangerous in high-risk populations, such as the elderly and immunocompromised subjects (Schweitzer & Justice, 2020).
The use of the inhalation route for the prevention and treatment of RSV seems intuitively obvious, yet the only product so far developed and approved for this purpose, nebulized ribavirin, is restricted to the most severe populations of hospitalized children and immunocompromised adults (Nicholson and Munoz, 2018).
Ribavirin is a synthetic nucleoside with antiviral activity with unknown mechanism of action, potentially substituting for natural nucleosides in the viral metabolism. VIRAZOLE (Ribavirin for Inhalation) is supplied as 6 g of lyophilized ribavirin, which is reconstituted in 300 ml of water. It is administered by a small-droplet-size nebulizer Spag-2 (MMAD = 1–2 µm; Newth and Clark, 1989; Walsh et al., 2016). The recommended treatment regimen is continuous aerosol administration for 12–18 hours per day for 3–7 days. It can also be used in ventilated patients (Bausch Health, 2020).
1. Pivotal Clinical Trials
Two placebo-controlled trials in hospitalized unventilated infants with lower respiratory tract RSV infections showed improvement of symptoms and in one of them a significant reduction of viral load in the respiratory secretions. In a controlled study in ventilated infants, Virazol significantly decreased the duration of mechanical ventilation (4.9 vs. 9.9 days, P = 0.01) and duration of required supplemental oxygen (8.7 vs. 13.5 days, P = 0.01) (Bausch Health, 2020).
Virazol may cause sudden decrease of respiratory function. It is detected in plasma at micromolar concentrations, but its bioavailability has not been determined (Bausch Health, 2020).
I. Pentamidine/NEBUPENT
Pneumocystic pneumonia is an alveolar infection caused by Pneumocystic jiroveci, previously called P. carinii. It is spread through air by people, but it usually only affects patients who are immunocompromised, such as those with AIDS/human immunodeficiency virus infections, patients with cancer, or organ transplant patients on immunosuppressive drugs (CDC, 2020). There is no inhaled drug approved for the treatment of P. jiroveci pneumonia (PJP). NebuPent is the only approved inhaled drug indicated for prophylaxis against P. jiroveci in high-risk patients with human immunodeficiency virus. Its active ingredient is pentamidine isethionate, which interferes with nuclear metabolism in microbes. However, its exact mechanism of action is unknown. It shows in vitro activity against P. jiroveci.
1. Device, formulation, deposition, and disposition in humans
Inhaled pentamidine isethionate has a long half-life in the epithelial lining fluid in the lung in humans (Conte and Golden, 1988).
A single dose of NebuPent contains 300 mg pentamidine isethionate; it is supplied as a lyophilized powder to be reconstituted with 6 ml sterile water and then administered with the Marquest Respigard II nebulizer every 4 weeks (FDA, 2020—Nebupent package insert). The MMAD and geometric standard deviation (GSD) of the aerosol are 0.76 µm and 1.9, respectively; the distribution of these aerosols in patients is similar to radioactive gas (Smaldone et al., 1991).
Although there was some interindividual variability in lung deposition of pentamidine, the bronchoalveolar lavage concentrations correlated with the deposited doses estimated from gamma scintigraphy (Smaldone et el. 1991). In patients with AIDS, a single inhaled dose of 300 mg of the drug resulted in mean bronchoalveolar lavage fluid concentration of 23.2 ng/ml 18–24 hours postadministration. A similar intravenous dose (4 mg/g) in the same population resulted in mean bronchoalveolar lavage drug concentration 18–24 hours postadministration of 2.6 ng/ml. The peak plasma levels postinhalation are substantially lower than the maximum plasma concentrations after intravenous pentamidine isethionate (FDA, 2020 and references therein), suggesting that inhalation delivery achieves very good persistent specificity for the lung compared with systemic administration.
2. Pivotal clinical trials
In a controlled clinical trial in subjects with a previous history of PJP, a significantly better protective effect (P < 0.01) against PJP with the 300-mg every-4-week dosage regimen was found compared with the 30-mg every-2-week dosage regimen. The 300-mg dose regimen reduced the risk of developing PJP by 50% to 70% compared with the 30-mg regimen. Cough and bronchoconstriction can affect patients taking NebuPent, and pretreatment with a bronchodilator minimizes these side effects (FDA, 2020 and references therein).
J. Conclusions
Approved inhaled anti-infectives are good examples of drugs that are administered by inhalation typically because the doses that would be required to achieve adequate therapeutic activity via systemic administration would cause systemic toxicity. Several of these drugs have very low oral bioavailability, which makes inhalation superior both because of its noninvasive nature and absence of potentially harmful systemic exposure of the portion of the dose that ends up in the gastrointestinal tract.
A fundamental limitation of the inhaled route approach to treating respiratory diseases in general and lung infections in particular is that the aerosols can only reach ventilated areas. Poorly ventilated locations in the respiratory tract or those that are completely obstructed are therefore likely to be “resistant” to this approach.
As several of the inhaled anti-infectives are used as chronic therapy, there is concern that patients may become refractory to the treatment through development of microbial organisms resistant to the drug in question.
Several long-term studies investigated this question in cystic fibrosis and found no evidence of clinically significant changes in therapeutic effects (Quon et al., 2014). Multidrug-resistant bacteria were no more common in the groups treated with inhaled tobramycin or aztreonam than in the placebo groups. The absence of clinical impact despite the changes in the susceptibility of the microbes to the antibiotics is most likely the result of very high antibiotic concentration in the lungs after the inhalation treatment. Furthermore, the practice of alternating 1 month on, 1 month off treatment may also help to prevent the loss or reduction of therapeutic effect, as the susceptibility changes are reversible. However, in trials with inhaled tobramycin and aztreonam, the “opportunistic” fungal infections, such as Candida albicans and related species, were more common in the inhaled antibiotic groups.
Similar investigations were conducted as a part of two phase III clinical trials in patients with non–cystic fibrosis bronchiectasis and chronic infections with P. aeruginosa treated with an inhaled ciprofloxacin formulation combining liposome-encapsulated and free drug (Vandenvanter et al., 2019). Although there were some losses in susceptibility of P. aeruginosa to ciprofloxacin, this had no impact on the key clinical goal of reduction of pulmonary exacerbations. Importantly, no evidence of emergence of opportunistic bacterial infections was observed in the drug treatment groups.
In summary, there does not appear to be evidence of risk to the individual patients of developing clinically important resistance to inhaled antibiotics, at least in the most frequently studied infection with P. aeruginosa. In some patients, “opportunistic” fungal infections may appear. There is also interest to assess the risk of transmission of resistant microorganisms within patient groups (e.g., in the cystic fibrosis community) (Denton et al., 2002). Such findings need to be considered with care, as the definition of “resistance” is based on concentrations achievable with systemic administration. Development of meaningful criteria for “resistance” or “susceptibility” appropriate for inhaled antibiotics is desirable (Kidd et al., 2018).
The history of inhaled antibiotics and especially the decades of experience with these in cystic fibrosis with multiple approved products provide compelling evidence that this therapeutic approach may be useful for the treatment of a wide range of respiratory infections.
X. Recombinant Human Deoxyribonuclease
A. Introduction
Recombinant human deoxyribonuclease (Pulmozyme rhDNase) was approved in 1993 for the management of CF and was one of the most rapid development programs in the modern pharmaceutical era with approval coming less than 5 years after product conception (Shak, 1995; Gonda, 1996; Shire, 1996; Pressler, 2008; Shire and Scherer, 2014; Lazarus and Wagener, 2019). The rationale for the development of rhDNase was the observation that many patients with CF had viscous purulent secretions in their airways, a result of recurrent bacterial infections and the process now called NETosis in which the infiltrating host neutrophils release DNA to form neutrophil extracellular traps in an effort to entangle pathogens and combat the infection. The presence of extracellular DNA in the lung secretions of patients with CF increases their viscoelasticity and makes their clearance difficult.
The decision to deliver rhDNase as an inhaled aerosol was because more conventional routes of protein delivery (e.g., intravenous or subcutaneous injection) were unlikely to achieve an adequate dose in the airway lumen. After inhalation, the rhDNase depositing in the airways would be in close proximity to the high-molecular-weight DNA, enabling it to be cleaved into shorter fragments. The resulting reduced viscoelasticity of the purulent secretions would allow for improved clearance (Shak et al., 1990; Lazarus and Wagener, 2019).
The ability of deoxyribonuclease of animal origin to reduce dramatically the viscosity of lung secretions has been known for a long time; however, it was found that using it for inhalation therapy in humans was associated with adverse reactions, and eventually such products were withdrawn from the US market. The reasons for these adverse reactions were not fully elucidated, but they could been allergic reactions to this molecule being a nonhuman protein and also presence of contaminants (Lazarus and Wagener, 2019).
At the time that rhDNase was under development at Genentech, Inc., no recombinant human biologic had previously been approved for delivery by inhalation. However, formulation of biologics as stable aqueous solutions for injection was reasonably well understood. To minimize development time and risk, approved nebulizer-compressor systems utilizing aqueous formulations to generate aerosol droplets were selected for development (Cipolla et al., 1994a) [Genentech did initiate a second-generation pharmaceutical development of an inhalable dry powder formulation of rhDNase, but this product did not advance to human testing (Chan et al., 1997)].
The initial selection of the doses was based on multiple constraints that included 1) concentration-dependent stability in simple aqueous solutions, 2) convenient volume for nebulization—this was selected to be 2.5 ml, 3) preclinical inhalation toxicology data, and 4) estimates of efficacious concentrations in sputum. As these were still the early days of the biotechnology industry, the cost of production of rhDNase was also a consideration.
The ultimate choice for the commercial product was based on a dose-ranging phase 2 trial in which doses of 0.6 and 10 mg of rhDNase in 2.5 ml of aqueous formulations were delivered twice daily with a jet nebulizer driven by a compressor for 10 days to patients with cystic fibrosis (Gonda, 1996; Lazarus and Wagener, 2019). Based on these results, 2.5 ml of a 1 mg/ml rhDNase solution was selected for late-stage development and approval.
B. Formulation Development
rhDNase is a glycosylated 260-amino-acid protein with a molecular weight of ∼30 kDa and a predicted isoelectric point of 4.5 (Shire, 1996). Assays were developed to evaluate the potential to observe degradation of rhDNase, and those included monitoring for protein aggregation by size-exclusion chromatography and gel electrophoresis, deamidation of an active site asparagine residue at position 74 using tentacle ion-exchange chromatography, and activity in an assay that measured the release of an intercalated methyl green dye during DNase digestion (Cipolla et al., 1994a,b; Shire, 1996; Shire and Scherer, 2014).
Additionally, the secondary and tertiary structures of rhDNase were assessed by far and near UV circular dichroism, respectively.
Because inhalation of a protein therapeutic was novel, the guiding principle of rhDNase formulation development was to minimize the use of excipients that could potentially be toxic in the respiratory tract or induce cough or bronchoconstriction upon inhalation (Beasley et al., 1988; Cipolla et al., 1994a; Shire and Scherer, 2014). A buffer is typically desirable to retain the formulation pH within the optimum range during nebulization and over the product’s shelf life. However, some buffers (e.g., citrate) could provoke cough (Chang et al., 2020. Fortunately, since the rhDNase protein itself was found to provide adequate buffering capacity, it was decided to not use a buffer. The optimum pH of 6.3 ± 0.7 was selected to balance the competing concerns between safety and tolerability and increased deamidation of the Asn 74 residue at more alkaline pH and the potential for rhDNase to precipitate at more acidic pH (Cipolla et al., 1994a). Sodium chloride at 150 mM was added to create an iso-osmotic formulation (Cipolla et al., 1994a) given that bronchospasm had been observed upon inhalation of both hypo-osmotic and hyperosmotic solutions (Beasley et al., 1988). Calcium was added to the formulation at a 33-fold molar excess to rhDNase (1 mM), as the binding of calcium was essential to stabilize the conformation of the protein (Shire, 1996). After investigations with a variety of formulations, 1 mg/ml rhDNase in 150 mM NaCl, 1 mM CaCl2, pH 6.3 with 2.5 ml filled into a plastic ampoule using “blow-fill-seal” technology was selected. This format was easier for the patient to dispense the liquid volume into a nebulizer than using traditional glass vials (Cipolla et al., 1994a,b; Shire, 1996; Shire and Scherer, 2014).
C. Nebulization
The primary question that needed to be addressed was whether the rhDNase formulation was stable to the nebulization process and the resulting exposure to shear and air-liquid interface inherent in droplet formation (Cipolla et al., 1994a, Cipolla and Gonda, 1994c, Cipolla et al., 1994d). Degraded protein would not be effective at cleaving the DNA in the sputum. Even more concerning, formation of protein aggregates, as had been reported for nebulization of other proteins, was linked to an increased potential for immunogenicity in patients (Shire and Scherer, 2014).
Both the US and European regulatory authorities were concerned about continuous access and ability to select the appropriate nebulization system for patients in their territories. It was therefore necessary to develop testing methodologies and criteria to evaluate the performance of various nebulization systems (Gonda 1996). For example, a method had to be developed to quantitatively collect the rhDNase aerosol for stability evaluation without introducing artifactual degradation in the collection apparatus, which can occur upon drying on the collection surface of the glass fiber filters routinely used to capture aerosols (Cipolla and Gonda, 1994c). The use of a prewetted sintered glass frit resulted in recovery of up to 98% of the rhDNase placed in the nebulizer, providing assurance that if degradation was occurring during nebulization, it would be detected (Fig. 7). The rhDNase in the collected aerosol as well as that remaining in the nebulizer that had been exposed to the complete duration of the nebulization process were evaluated using the stability assays noted above. No degradation was observed after jet nebulization of the 1 and 4 mg/ml rhDNase formulations using 16 different combinations of nebulizers and compressors or compressed oxygen (Cipolla et al., 1994d). In contrast, one ultrasonic nebulizer that gradually heated the formulation up to 58°C by the end of nebulization resulted in the formation of soluble aggregates of rhDNase due to thermal denaturation. Thus, jet nebulizers were selected for rhDNase delivery in the clinical setting.

Methodology to collect biologic aerosols with retention of protein integrity (with permission from Cipolla and Gonda, 1994c).
The second focus of the nebulization characterization studies was to quantify the emitted aerosol and its droplet size distribution to enable an estimate of the rhDNase dose that would be delivered to the lungs (Cipolla et al., 1994d). Each combination of jet nebulizer and compressor was characterized for its nebulizer efficiency or emitted dose (the proportion of the rhDNase placed in the nebulizer that left the nebulizer as aerosol), the MMAD, the GSD, and the respirable fraction (the proportion of rhDNase mass in droplets between 1 and 6 µm). The density and viscosity of the placebo and rhDNase solutions over the 0.25 to 4 mg/ml concentration range were comparable, leading to similar nebulizer performance in terms of nebulization time, emitted dose, and aerosol droplet size distribution.
However, the choice of nebulizer and compressor led to a wide variation in the in vitro performance, which could translate into differences in the pulmonary deposition and distribution of the therapeutic dose. Calculation of the lung dose was made by multiplying the emitted dose times the respirable fraction divided by two (to account for the patient only inhaling half of the time during continuous nebulization). This quantity varied widely from 5.2% to 24.7% in the tested systems (Cipolla et al., 1994d).
To rapidly screen the large number of available systems in the United States and particularly Europe, a laser diffraction method was developed to estimate the aerodynamic size distribution (Cipolla et al., 1994d) and subsequently qualified (Clark, 1995b). The performance characteristics for the three nebulizer systems approved in the US label provided an in vitro estimate of lung dose of 11.7%–14.2% of the loaded dose, or 290–360 µg rhDNase (Table 14). The European authorities accepted the in vitro performance using laser diffraction as an adequate demonstration of comparable performance based on the proposed criteria. For inclusion on the US label, FDA additionally requested a clinical trial showing clinical comparability of the nebulizer systems (Fiel et al., 1995), which was successfully executed.
Characterization of inhalation systems used to deliver rhDNase in the clinical setting
D. Clinical Evaluation
The jet nebulizer and compressor system used in the two phase I clinical trials was the Marquest Acorn II nebulizer using the DeVilbiss Pulmo-Aide compressor (Aitken et al., 1992; Hubbard et al., 1992). In a Phase I nonrandomized repeat-dose escalation study, Aitken et al. (1992) reported that rhDNase was well tolerated in 12 healthy subjects and 14 patients with CF with improvements observed in lung function and dyspnea score for the adult CF patients after 10 days of three-times-a-day dosing rhDNase (2, 6, or 10 mg nebulizer loaded dose). In a placebo- controlled crossover investigational study in 16 adult patients with CF, Hubbard et al. (1992) demonstrated that BID rhDNase (10 mg nebulizer loaded dose) improved lung function (FVC and FEV1) by 10%–20% compared with placebo (n = 11) throughout the 6-day treatment period.
The Marquest Acorn II nebulizer using the DeVilbiss Pulmo-Aide compressor was also used in two Phase II clinical trials (Ramsey et al., 1993; Ranasinha et al., 1993). In a placebo-controlled trial, Ramsey et al. (1993) evaluated 0.6, 2.5, and 10 mg rhDNase (all in a 2.5 ml volume) BID for 10 days in 181 children and adults with CF. No major adverse events were noted, and rhDNase was well tolerated. Improvements in FVC and FEV1 against placebo were observed for all three doses of rhDNase, but there was no benefit of the 10-mg over the 2.5-mg BID dose. In the second placebo-controlled phase II trial in 71 patients with CF, only the 2.5-mg BID dose (2.5 ml of 1 mg/ml rhNase) was evaluated, and it was well tolerated over the 10-day period (Ranasinha et al., 1993). An increase in FEV1 of 13.3% from baseline was observed compared with a decline of 0.2% for placebo.
These positive results for the 2.5-mg BID dose in phase II led to a 24-week randomized, double-blind, placebo-controlled phase III design comparing QD and BID 2.5 mg rhDNase (Fuchs et al., 1994) in patients with CF with FEV1 >40% predicted. The Acorn II nebulizer was replaced by the Hudson T Up-Draft II nebulizer, but the compressor was not changed. In the 968 children and adults with CF, the risk of exacerbations was significantly reduced for the QD and BID rhDNase by 28% and 37%, respectively, compared with placebo. Statistically significant improvements in FEV1 and FVC were also observed for both QD and BID rhDNase compared with placebo. This clinical evidence combined with acceptable nonclinical safety (Green, 1994) resulted in FDA approval of once-daily 2.5 mg rhDNase for the management of CF using an approved nebulizer with the caveat that some patients may benefit from twice-daily rhDNase.
E. Postapproval Studies
Pulmozyme rhDNase has continued to be evaluated in various segments of the CF population and continues to demonstrate benefit including those with severe pulmonary disease (FVC <40% predicted; Shah et al., 1995; McCoy et al., 1996), children aged 6–10 with mild disease (FVC >85%; Quan et al., 2001; Robinson, 2002), and effect on the rate of pulmonary colonization (Frederiksen et al., 2006). Additional studies have reviewed longitudinal data from CF registries (Konstan and Ratjen, 2012), including a comprehensive safety evaluation of Pulmozyme rhDNase from the Epidemiologic Registry of Cystic Fibrosis indicating that children under age 5 tolerated rhDNase as well as older patients (McKenzie et al., 2007). Additionally, in the Epidemiologic Study of CF, 2-year treatment with Pulmozyme rhDNase was associated with a reduction in the rate of decline in FEV1 (Konstan et al., 2011b).
In contrast, Pulmozyme rhDNase has not shown promising outcomes so far in indications outside CF (Lazarus and Wagener, 2019).
Since jet nebulizers with compressors take a relatively long time to deliver a dose, other potential delivery systems for rhDNase have been explored. In particular, the in vitro evaluation of rhDNase using the Pari eFlow vibrating mesh nebulizer demonstrated more rapid and efficient delivery than the labeled jet nebulizers (Scherer et al., 2011). One concern with reusable vibrating mesh systems was the potential for clogging of the holes in the mesh, which may increase the nebulization time and possibly contaminate or alter protein formulations (Rottier et al., 2009). However, Scherer et al. (2011) found that although the average nebulization time did increase modestly, rhDNase retained its integrity and activity. The eFlow was modified (eRapid) to provide comparable lung dose to the jet nebulizers. (Shire and Scherer, 2014). In a randomized crossover trial in 87 patients with CF comparing delivery of rhDNase by the Pari eRapid to the Pari LC Plus (Sawicki et al., 2015), comparable safety and efficacy were observed for both systems. This data allowed the Pari eRapid to be added to the Pulmozyme rhDNase label as an approved nebulizer.
F. Conclusions
Prior to the approval of Pulmozyme rhDNase in 1993, CF received very little attention and half of patients with CF did not reach adulthood. That dynamic has dramatically changed, first with the approval of rhDNase, then with inhaled antibiotics, and finally with the oral CFTR correctors so that most patients now thrive well into adulthood. And there is hope that gene therapy will one day provide a cure for CF. Pulmozyme rhDNase has now been on the market for almost 3 decades without any competing biosimilars gaining approval. The only significant advancement has been the addition of the Pari eFlow delivery system to the product label, which improves the convenience for the patient by reducing the administration time. Even with the new oral CFTR correctors, Pulmozyme rhDNase remains a mainstay of therapy in CF.
XI. Mannitol
A. Introduction
Mannitol as a therapeutic medication is delivered by inhalation as a dry powder. Although the exact mechanisms contributing to its therapeutic effects are unknown, inhaled mannitol increases the hydration of the airway surface liquid, improves the physical properties of abnormal mucus, and contributes to increased mucus clearance of the retained secretions through mucociliary activity and cough.
Mannitol (C6H14°6, molecular weight 182.2) is a naturally occurring nonionic sugar alcohol that is neither absorbed nor transported across the gastrointestinal epithelium. It has very low toxicity and is widely used as an excipient in tablets. Mannitol is an osmotic agent and is also given intravenously as a treatment to relieve cerebral edema.
In healthy airways, the volume of the periciliary fluid layer (PCL) in the first 10 generations of human airways is small, likely less than 1 ml (Anderson et al., 1989; Anderson et al., 2018). Thus, only small doses of inhaled mannitol are needed to create an osmotic gradient for water to move across the airway epithelium into the airway lumen of these airways. This event causes a relatively rapid (30 minutes) and marked increase in mucociliary clearance (Daviskas et al., 1997). Although the hyperosmolarity of the PCL created by the inhaled mannitol is only transient, the increased hydration of the airway surface liquid (PCL and mucus layer) is maintained while the mannitol is present (Daviskas and Anderson, 2016). Mannitol is cleared from the airways by the mucociliary escalator or slowly by way of the paracellular pathway of the airway epithelium.
Mannitol given by inhalation being an osmotic stimulus usually provokes cough, as hyperosmolarity is well known to stimulate the sensory nerves (Lowry et al., 1988; Jia and Lee 2007; Bonvini et al., 2015). Patients who have excessive secretions rely on cough to clear secretions. A major advantage of inhaled mannitol is that it makes cough effective in disease by increasing the hydration and by changing the physical properties of mucus favorably (Daviskas et al., 2010a). Coughing, when effective, is considered beneficial, as it promotes clearance in patients with excessive secretions.
In the mid-1990s a dry powder preparation of mannitol suitable for inhalation and deposition in the lower airways was developed. The properties of the inhalable particles of mannitol obtained by spray drying depend on the composition of the spray dried mannitol solution, the nature of the manufacturing equipment, and the conditions during the manufacturing process (Tang et al., 2008; Hulse et al., 2009; Littringer et al., 2013). As mannitol can exist in multiple crystalline and amorphous forms, precise control of the parameters during the manufacturing is required. The development of Bronchitol also had to consider that to preserve the product functionality, the desired properties of the mannitol particles need to remain unaltered during the product lifetime, including the storage and use by the patients.
The spray-dried mannitol dry powder formulation was investigated for its potential to increase mucociliary clearance in asthmatic and healthy subjects (Daviskas et al., 1997). The favorable results suggested that the mannitol dry powder could be used to enhance the clearance of excessive secretions from the airways of patients with respiratory disease, such as cystic fibrosis (Robinson et al., 1999) and bronchiectasis (Daviskas et al., 2001).
B. Impact of Mannitol Inhalation in Subjects with Abnormal Mucus and Mucociliary Clearance
Excessive production and secretion of mucus is a feature of several respiratory diseases, including bronchiectasis and cystic fibrosis. If the mucus is not cleared, the patients suffer from chronic cough, persistent airway inflammation, recurrent infective exacerbations, and poor quality of life, and they have an increased risk of morbidity and mortality. Importantly, they have reduced lung function that is deteriorating at an abnormally high rate (King et al., 2005; Martinez-Garcia et al., 2007; Breuer et al., 2018).
A key cause of the failure of mucus to clear adequately from the airways in diseases, such as cystic fibrosis, is likely to be due to its state of insufficient hydration. This poor hydration of the mucus results from an imbalance between volume of mucus and availability of water for its hydration when it reaches the airway surface (Boucher, 2004). This imbalance causes the mucus to become thick and sticky. The thickened and sticky mucus makes normal ciliary function difficult to achieve, cough is ineffective, and, as a result, the mucus becomes stagnated. By forming mucus plugs, stagnated mucus may cause obstruction to airflow. These events contribute to chronic inflammation, recurrent infections, decrease in lung function, and development and progression of airway disease (King et al., 2005; Caudri et al., 2018). Both viscosity and elasticity of mucus as well as its interfacial tension (surface tension) determine the effectiveness of its transport up the airways by cilia and by cough, and they are both affected by the state of hydration of the mucus and need to be optimally balanced (Daviskas and Rubin, 2013). Mannitol given by inhalation as a dry powder has been shown to reduce both the viscosity and elasticity of mucus in patients with asthma and bronchiectasis (Daviskas et al., 2007; Daviskas et al., 2010b; Daviskas and Rubin, 2013). In addition, it was shown to reduce the surface tension of mucus (primarily responsible for effective clearance by cough), and this correlated negatively with the increase in FEV1 in response to mannitol in patients with cystic fibrosis (Daviskas et al., 2010c; Daviskas and Rubin 2013). The initial stimulus for improved mucociliary clearance is likely the osmotic force created by mannitol deposited in the airways acting to increase the paracellular movement of water. The result is an increase in the availability of water at the airway surface.
The greater availability of water leads to increased hydration of the mucus, allowing it to achieve the appropriate physical properties to permit its transport by cilia and by cough (Robinson et al., 1999; Daviskas et al., 2008, 2010b,,c, 2017; Daviskas and Rubin, 2013).
Importantly, daily treatment with inhaled dry mannitol powder is associated with clinical benefits in patients with non-CF bronchiectasis and cystic fibrosis manifested in a positive impact on lung function (Bilton et al., 2011, 2013, 2014; Aitken M et al., 2012) described in greater detail below.
C. Development of the Dry Powder Mannitol Inhaler for Cystic Fibrosis and Bronchiectasis
The advantage of mannitol over other saccharides is that mannitol particles are of a crystalline nature that makes them physically stable when prepared by spray drying (Chew and Chan 1999; Glover et al., 2006, 2008). The benefits in using a dry powder compared with a nebulized aerosol are portability and ease of delivery.
Mannitol is stable as a dry powder resisting absorption of water even at high relative humidity. The powder is provided in capsules ready for delivery from a dry powder inhaler (Anderson et al., 1997).
In vitro studies established the relationship between particle size of mannitol powder, inhaler device efficiency, and inhalation flow rate, confirming that powder of spherical particles with a median particle size of around 3 microns was superior to powders with a large size of 5 or 7 microns (Chew and Chan 1999).
In the early studies, various inhaler devices that had been well characterized and were commercially available were used for inhalation and delivery of the mannitol powder (Anderson et al., 1997). These included the Halermatic (Fisons Pharmaceuticals), the Inhalator (Boehringer Ingelheim), and the low resistance inhaler device the Dinkihaler (Rhone Poulenc Rorer). All these devices had been used to deliver mannitol for studies in asthmatics. The subjects were required to inhale at 50–120 l/min from the Halermatic, >28 l/min from the Inhalator, and 80–120 l/min from the Dinkihaler (Chew and Chan 1999).
To investigate the therapeutic benefit of using dry powder mannitol by inhalation, several clinical trials were conducted in patients with non-CF bronchiectasis (Daviskas et al., 2004, 2005) and cystic fibrosis (Jaques et al., 2008; Teper et al., 2011). In these clinical trials, the Osmohaler (RS-01 Plastiape Italy) was used to deliver the mannitol for inhalation. A minor modification was made to increase inspiratory resistance, as this was shown to decrease cough and improve emptying of the capsule.
In the initial studies in patients with adult cystic fibrosis, an improvement in lung function assessed by FEV1 occurred after 2 weeks of treatment with mannitol and was retained for 26–52 weeks while continuing with regular clearance of secretions. The improvement in FEV1 in response to treatment with mannitol was irrespective of concomitant treatment with dornase alfa, another inhaled “mucoactive agent” frequently used in patients with cystic fibrosis. (Jaques et al., 2008; Bilton et al., 2011; Aitken et al., 2012).
Two large multicenter clinical trials were carried out in 317 adult patients with cystic fibrosis who received 400 mg or control (50 mg mannitol) twice daily for 26 weeks (Bilton et al., 2011; Aitken et al., 2012). These phase III trials were of a double-blind, randomized, controlled, parallel group design. The results were pooled and analyzed together (Bilton et al., 2013). There was a significant improvement in both absolute (99.5 ml) and a relative change in % predicted normal FEV1 of 4.72% (P < 0.001) compared with control [Fig. 8 (Flume et al., 2015)].

Change in FEV1 from baseline sustained during double-blind phase of the studies (intention to treat) (Flume et al. 2015). CI, confidence interval.
A phase II study randomized placebo-controlled crossover study was reported in 92 cystic fibrosis children aged 6–17 years (mean age 12 years), with a baseline of 72.2% predicted (De Boeck et al., 2017). The subjects received a dose of 400 mg or a matching placebo every 12 hours for 8 weeks followed by 8 weeks of washout and 8 weeks of the alternate treatment. The investigators reported the absolute change (i.e., milliliters of FEV1, expressed as a % of the predicted FEV1). This value was +3.59% (P < 0.004) over the baseline value for FEV1 after treatment with mannitol compared with +0.17% after treatment with placebo [i.e., a difference of +3.42% of the % predicted FEV1 (P < 0.004)]. The relative change in FEV1% predicted FEV1 was +5.72% after mannitol and +0.75% after placebo, and a difference of +4.97% was also highly significant (P = 0.005). The relative change in Forced Expiratory Flow over the middle one half of FVC (FEF 25-75) was 10.52% (P = 0.013). Almost twice as many subjects in the group receiving mannitol as in the placebo group had a relative improvement in % predicted FEV1 of ≥5% (51.7% vs. 28.7%) and ≥10% (32.2% vs. 19.5%) (De Boeck et al., 2017).
Inhaled mannitol could potentially provoke airway narrowing in patients with hyperresponsive airways (Anderson SD et al., 1997; Brannan et al., 2005). Although bronchospasm after inhalation of an osmotic stimulus is usually mild, short-lived, and responds well to treatment with a bronchodilator (Rodwell and Anderson, 1996; Anderson et al., 1997; Briffa et al., 2011), it was prudent in the clinical trials to pretreat the patients with a short-acting bronchodilator before the inhalation of mannitol. In addition, and prior to initiation of the long-term treatment with mannitol in the trials, patients had an assessment with a dose of inhaled mannitol to identify then exclude those with airway hyperresponsiveness. The percentage of CF subjects who had significant airway narrowing in response to the mannitol dose and were excluded from participating in the first and second phase III trials was 7% and 4% respectively (Bilton et al., 2011, 2013; Aitken et al., 2012). In addition, very few patients screened and dosed as described above reported bronchospasm during the treatment period in the clinical trials (Jaques et al., 2008; Bilton et al., 2011; Aitken et al., 2012).
D. Conclusions
Inhaled mannitol has regulatory approval for the treatment of patients with cystic fibrosis to facilitate clearance of mucus and improve their lung function. In the final approved product (Bronchitol) for cystic fibrosis, the twice-daily dose of 400-mg precision spray-dried form of mannitol is inhaled from 10 capsules (40 mg each) with a close to full inspiration at an inspiratory flow of >45–60 l/min or more followed by a 5-second breath hold using the OsmohalerTM (RS-01 Plastiape Italy) dry powder inhaler with modified inspiratory resistance.
In all markets, patients are required to have a tolerance test prior to being prescribed Bronchitol to ensure that they are not hyper-responsive to mannitol and are pretreated with a short-acting bronchodilator prior to taking each dose of Bronchitol.
XII. Prostacyclins
A. Introduction
Pulmonary arterial hypertension (PAH) is a chronic disease characterized by proliferation and remodeling of vascular endothelial and smooth muscle cells in the small pulmonary arteries and arterioles (Farber and Loscalzo, 2004). This leads to decreases in the lumen of the vessels, resulting in progressive increases in pulmonary vascular resistance, elevations in pulmonary artery pressure, right heart failure, and, eventually, death. The most commonly reported symptoms for patients with PAH are dyspnea, physical fatigue, and a low exercise capacity.
B. Pharmacology
Current therapies for PAH aim to alleviate vasoconstriction, vascular endothelial cell proliferation, smooth muscle cell proliferation, and endothelial dysfunction within pulmonary arteries (Humbert et al., 2004; Frumkin, 2012). The three principal molecular pathways targeted for PAH therapeutics are the prostacyclin, endothelin, and nitric oxide pathways (Frumkin, 2012; Ghofrani and Humbert, 2014). PAH is associated with reduced pulmonary levels of prostacyclin as a result of underexpression of endothelial prostacyclin synthase and an imbalance with endogenous thromboxane levels that promote vasoconstriction. Prostacyclin analogs (e.g., epoprostenol, treprostinil, iloprost) are potent vasodilators that also impact tissue remodeling by inhibiting smooth muscle cell growth. Endothelin receptor antagonists (e.g., bosentan, ambrisentan, macitentan), block the effect of endothelin, a potent endogenous vasoconstrictor and mitogen, at smooth muscle cell receptors. Phosphodiesterase type 5 (PDE) inhibitors (e.g., sildenafil, tadalafil) competitively inhibit cGMP hydrolysis by PDE5 enzymes on the nitric oxide pathway. This results in accumulation of cGMP and relaxation of vascular smooth muscle. Soluble guanylate cyclase stimulators (e.g., riociguat) achieve the same end result on the nitric oxide pathway by increasing synthesis of cGMP in smooth muscle cells at a rate that exceeds hydrolysis by PDE5 enzymes.
C. Inhaled Vasodilators
Pulmonary administration enables noninvasive, targeted delivery of vasodilators directly to the site of action in the lungs, thereby enhancing pulmonary selectivity while potentially reducing adverse events related to off-target delivery. Compared with oral administration, inhaled vasodilators may have a more rapid onset of action and reduced variability in dose delivery that occur as a result of first-pass effects (Hill et al., 2015). Inhaled therapeutics may also lead to improvements in ventilation-perfusion matching, resulting in improvements in oxygenation, especially in those patients who have concomitant lung disease (Hill et al., 2015).
Currently, nebulized prostacyclin analogs are prescribed as an add-on therapy for patients who have not reached their improvement goals but who have not deteriorated to the point of requiring infusion treatment (Hill et al., 2015). Inhaled prostacyclin analogs are also used in more severe patients who are unable to manage or tolerate parenteral therapy, which requires an infusion pump and carries the risk of infections and infusion-site reactions (Poms and Kingman, 2011).
Prostacyclin-related side effects are observed with all routes of administration, including inhalation (Poms and Kingman, 2011). These include nausea, flushing, diarrhea, jaw discomfort, musculoskeletal pain, headache, rash, and thrombocytopenia. Side effects can be effectively mitigated by titrating the dose, and some (e.g., headache) may resolve over time.
D. Iloprost/VENTAVIS
The first inhaled therapeutic approved for the treatment of PAH was Ventavis (nebulized iloprost) (Olschewski et al., 2002). Iloprost has a very short duration of action (about 30 minutes), necessitating frequent administration (6–9 times daily) (Olschewski et al., 2002). Ventavis is currently administered with the I-neb adaptive aerosol delivery system (Philips/Respironics, Murrysville, PA) (Dhand, 2010). The I-neb is based on a vibrating mesh nebulizer platform coupled with Adaptive Aerosol Delivery (AAD) technology. Software in the AAD system analyzes the patient’s breathing pattern and system analyzes the patient’s breathing pattern and adapts the pulse of medication for delivery only during patient inspiration. The low dead volume and lack of drug release during expiration leads to an exhaled fraction of less than 1%. The daily treatment burden, including the time required to gather the supplies, prepare the nebulizer, administer the dose, and clean the nebulizer, was determined clinically and found to be about 2 hours per day (Chen et al., 2013).
The follow-up product is now available in some geographies under the name Breelib (Gessler et al., 2017). It incorporates similar breath-control electronics to I-neb in a small, portable, and faster mesh nebulizer that is a customized version of the Fox inhaler (Fox Nebulizer, 2021).
E. Treprostinil/TYVASO
A second inhaled prostacyclin (Tyvaso, inhaled treprostinil) was later developed that had an extended duration of action (terminal elimination half-life ∼4.5 hours) (Channick et al., 2012). This decreased the number of treatments to four times daily while significantly reducing treatment burden to 39 minutes per day (Chen et al., 2013). The impact of the reduced treatment burden with Tyvaso was reflected in improved scores on the Treatment Satisfaction Questionnaire for Medication (Chen et al., 2013).
Despite the improvements in treatment convenience with Tyvaso, there remain perceived disadvantages with current inhaled vasodilator therapies (Hill et al., 2015). These include: 1) delivery systems that are cumbersome and time consuming to use; 2) treatment regimens that are cumbersome with administration 4 to 9 times daily; 3) prostacyclin-related side effects; 4) irritant effects on airways (cough, bronchospasm, throat irritation), which are described as mild to intractable resulting in discontinuation (Poms and Kingman, 2011); and 5) treatments that are very costly. All of these factors can have a negative impact on patient adherence to therapy and ultimately on the effectiveness of the product.
Portable inhalers (e.g., dry powder inhalers, metered dose inhalers, smart mist inhalers) may provide an alternative option to further improve patient convenience and treatment satisfaction for inhaled vasodilator therapies (Voswinckel et al., 2009; Hannon et al., 2016; Feldman et al., 2020). Indeed, numerous inhaled vasodilators administered with portable dry powder inhalers are currently in clinical development, including vasodilators targeting both the prostacyclin (e.g., treprostinil) and nitric oxide (e.g., vardenafil, riociguat) pathways.
F. Conclusions
Inhaled vasodilators are indicated as a maintenance therapy for the treatment of PAH. Compared with oral delivery of vasodilators, pulmonary administration leads to a more rapid onset of action, increased pulmonary selectivity, improved dose consistency, and improved oxygenation. New formulations of prostacyclins and other vasodilators are being developed in portable dry powder inhalers because of the improved convenience and decreased treatment burden.
XIII. Lung Surfactant
A. Introduction
Respiratory distress syndrome (RDS) is a serious disorder of pulmonary insufficiency that occurs mainly in premature infants (gestation age less than 35 weeks). RDS is caused by surfactant deficiency and immature, structurally underdeveloped lungs. Pulmonary surfactant lowers the surface tension at the air-liquid alveolar interface, thus preventing alveolar collapse and the results of atelectasis (Walti and Monset-Couchard, 1998). Not all premature babies develop RDS, and reported incidence is 92% at 24–25 weeks, 88% at 26–27 weeks, 76% at 28–29 weeks, and 57% at 30–31 weeks gestation age (GA) (Sweet et al., 2013).
B. Bolus Surfactant Delivery of Approved Products
Currently, RDS is treated or prevented by administration of supplemental exogenous surfactants, which were discovered and extensively studied in the late 1980s and early 1990s. Two types of surfactant have seen clinical use (Walti and Monset-Couchard 1998). Those isolated from animal lungs are of bovine (Infasurf, Survanta, Alveofact, BLES) and porcine (Curosurf) origin. Those with synthetic origin are Exosurf and Surfaxin. Both synthetic and natural origin surfactants have been approved as safe and effective in several territories. Their safety and efficacy with this delivery mode were demonstrated in randomized controlled studies.
The widespread use of exogenous surfactant resulted in decreased mortality and reduction of adverse events of barotrauma, volutrauma, and neurotrauma (Carlo, 2012; Dargaville, 2015)—see Table 15.
Surfactants approved for ETT instillation in North America and Europe
All current surfactants are approved for delivery only as a bolus instillation via endotracheal tubing (ETT) to ventilated infants. However, inserting an ETT for ventilatory support or surfactant administration is not a benign procedure and risks oxygen desaturation, bradycardia, ETT misplacement, infection, and airway trauma (Jorch et al., 1997). To mitigate the issues, less invasive techniques have been investigated, such as Intubate-SURfactant- Extubete (INSURE) (Kanmaz et al., 2013), Less Invasive Surfactant Administration, and similar techniques, and are being implemented in clinical practice (Göpel et al., 2015).
Rapid bolus delivery of liquid to the lungs also causes issues, such as transient bradycardia and decreased blood pressure. Nevertheless, at present, the rapid bolus technique remains the recommended method of surfactant administration (Nouraeyan et al., 2014).
Mechanical ventilation can in itself result in adverse events (e.g., barotrauma and volutrauma), and clinicians have continued to study the best surfactant preparation and the best way to ventilate the preterm lung to minimize barotrauma. Although bolus surfactant administration has been shown to benefit preterm infants with RDS (Sinha et al., 2008), some preterm infants can be supported with nasal continuous positive airway pressure (nCPAP) alone. However, noninvasive ventilatory support without delivery of surfactant does not effectively address the underlying cause of RDS-surfactant deficiency, which results in the need for intubation/cannulation and surfactant administration (Carlo, 2012; Dargaville, 2015; Dargaville et al., 2016).
C. Aerosolized Surfactant Delivery
Because of major drawbacks, bolus instillation, and the associated need for mechanical ventilation as well as new risks created by less invasive surfactant administration techniques, researchers have turned to a new approach to RDS treatment and prevention (More et al., 2014). This approach is aerosolized surfactant administered via noninvasive nCPAP ventilatory support.
Aerosolized surfactant is an attractive alternative to bolus surfactant instillation, as it eliminates the risks associated with placement and management of endotracheal tubes and catheters in the infant trachea and the risks arising from instillation of large volumes of liquid delivered directly into the lungs (Mazela et al., 2007; Shah, 2011).
In the past 2 decades, five studies have been published using aerosolized surfactant (Table 16). These involved 2 surfactants of natural origin, bovine or porcine, and 2 synthetics. Clinicians’ interest is also evident in six review papers (Mazela et al., 2007; Shah, 2011; Pillow and Minocchieri, 2012; Trevisanuto and Marchetto, 2013; More et al., 2014; Sardesai et al., 2017).
Overview of studies with aerosolized surfactant
The experimental published work indicates feasibility and safety of this new delivery route, but, except for two publications, efficacy of the tested products was questionable. The authors reported issues regarding the type of surfactant, aerosol generation technique, deposition of medication in the airway, and dosing strategies. Of the clinical studies reported to date in preterm infants (Jorch et al., 1997; Arroe et al., 1998; Berggren et al., 2000; Finer et al., 2010; Minocchieri et al., 2019), two have shown promising results. Jorch et al. (1997) showed improvement in the arterial to alveolar oxygen tension ratio, and Minocchieri et al. (2019) showed a reduction in the need for intubation and instillation in 20- to 34-week-olds (odds ratio 0.53).
The delivery of aerosolized surfactant for RDS in premature infants was first investigated by Jorch et al. (1997) for the delivery of nebulized Alveofact and was demonstrated to be safe with noticeable improvements in oxygenation and alveolar ventilation. Endotracheal intubation and mechanical ventilation were avoided in 70% of the patients. To achieve these effects, 150 mg/kg was delivered once or twice, resulting in 300 mg/kg surfactant delivered in some patients. Observed side effects of nebulizing surfactant (Alveofact) were increased secretions after administration. Overall, the study demonstrated clinical feasibility and rapid improvement of respiratory parameters, with some adverse events without any apparent relationship to the nebulization of surfactant.
Arroe et al. (1998) aerosolized a synthetic surfactant, Exosurf. The goal of this pilot study (n = 22) was to estimate whether inhalation of aerosolized Exosurf at different dosages would improve systemic oxygenation in infants with mild-to-moderate RDS treated with nCPAP. Exosurf was administered as 1, 2, 4, or 8 vials with two inhalations 6 hours apart using a closed nCPAP system and nebulizer (Sidestream 45). The mean GA was 30 5/7 ± 3 weeks. Two infants were intubated and mechanically ventilated during the study period, one shortly after the end of the first inhalation and one during the second period of inhalation. Both infants had a pneumothorax. Six infants were intubated more than 2 hours after the end of the second inhalation. The overall effect was not statistically significantly different from zero (+0.02, 95% CI −0.02 to +0.06). The relation between the arterial to alveolar partial oxygen pressure ratio (a/A-ratio) response and the postnatal age were close to significance (P = 0.05), with a better response being associated with a higher postnatal age. The study concluded that all infants needed oxygen and nCPAP and had no other likely cause for respiratory distress than RDS. The inhalation did not improve the a/A-ratio and a relation between the a/A-ratio response and the dose of surfactant, GA, and a/A-ratio before the first inhalation was not found.
In a study of 32 infants randomized 1:1, Berggren et al. (2000) studied aerosolized Curosurf delivered via nCPAP versus an nCPAP alone. The study was conducted using an Aiolos nebulizer (Karlstad, Sweden). The authors noted that no beneficial effects of aerosolized surfactant were demonstrated during the trial, which was in contradiction with their animal feasibility data. They concluded that the lack of efficacy could be a reflection of the differences in administration techniques. In this study, aerosolized surfactant in premature infants with RDS showed no beneficial effects, suggesting that, in spite of the precautions taken to enhance delivery, the amount of surfactant retained in the lungs was too low to compensate for the underlying surfactant deficiency and counterbalance the presence of surfactant inhibitors in the airspaces. Given the results of this study, the authors concluded that further work is needed to optimize delivery of aerosolized surfactant to the neonatal lung in clinical practice.
Finer et al. (2010) evaluated the effects of an inhaled aerosolized synthetic surfactant, Aerosurf (a peptide-containing synthetic surfactant), delivered with an Aeroneb Pro vibrating mesh nebulizer (Aerogen Ltd). The study included 17 infants (GA 28–32 weeks; no control group) and two dosing regimens—up to four doses of Aerosurf 20 mg/ml over 48 hours with each treatment separated by at least 3 hours or Aerosurf 20 mg/ml up to four doses with each treatment separated by at least 1 hour. The study assessed feasibility and safety. In this proof-of-concept study, researchers encountered several technical difficulties with the viscosity and delivery of this surfactant. The study showed that Aerosurf could be safely administered via nCPAP in preterm infants at risk for RDS and that it may provide an alternative to surfactant administration via an ETT. However, because of the lack of a control group, the researchers were unable to conclude whether Aerosurf was effective in reducing the requirement for a high fraction oxygen content in the inspired air oxygen (FiO2).
A recent review article from Sardesai et al. (2017) summarized the current treatment options and provided clinician’s perception of the future in treating premature neonates with RDS. Similar to previous papers, the authors concluded that administering surfactant via aerosol is a promising therapeutic option worth investigating.
Most recently, Minnochieri et al. (2019) reported on the use of aerosolized Curosurf in preterm infants ranging from 29 0/7 to 33 6/7 weeks GA. They aerosolized Curosurf using a modified PARI e-Flow nebulizer, giving an initial nominal dose of 200 mg/kg followed by a second dose of 100 mg/kg after 12 hours if needed. Sixty-four patients were placed on nCPAP, with 32 receiving aerosolized surfactant. Their primary endpoint was the need for intubation and mechanical ventilation at 72 hours. Twenty-two of 32 patients required intubation in the nCPAP alone group, and 11 of 32 required intubation in the aerosol group. The mean intubation risk ratio for the aerosol surfactant group was 0.526 with a 95% confidence range of 0.292–0.950. The authors concluded that early nebulized surfactant may reduce the need for intubation in the first 3 days of life compared with nCPAP alone in infants born between 29 and 33 GA with mild RDS, but that confirmation requires further adequately powered studies.
It is evident from these most recent trials (Finer et al., 2010 and Minocchieri et al., 2019) that technical problems with efficient aerosolized surfactant delivery still remain. Given the need to deliver the surfactant to the air exchange areas (i.e., alveoli) through the narrow airways of prematurely born neonates, these reviews highlight the need for a dedicated approach. The choice of the aerosolization equipment and its suitability to deliver the individual surfactants with their specific physical characteristics and the need for coordination of the aerosol delivery with the infant’s breathing as well as reduced dilution of aerosol to achieve sufficient output rate to facilitate acceptable treatment times need to be addressed. In a recent modeling study (Clark, 2021), it was also suggested that breath-synchronized delivery be accompanied by aerosol delivery early in an inspiration followed by sufficient “chase air” to clear the anatomic dead space. In preterm infants, the anatomic dead space can represent 10%–20% of the tidal volume, and for maximum efficiency aerosol delivery should thus be restricted to the first 80% of each breath.
D. Conclusions
Exogenous surfactant delivery via intubation and instillation has proven effective and likely represents the greatest advance in neonatal care in the last 30 years. However, instillation of a large surfactant bolus has its risks, and less invasive techniques are being adopted. Aerosolized surfactant, the ultimate in noninvasive delivery, shows considerable promise once the technical issues have been properly resolved, and it may well be the next major advance in the treatment of infant RDS.
XIV. Drugs Delivered by Inhalation for Systemic Therapies
A. Introduction
Although the main focus of this review is the treatment of respiratory diseases, there are opportunities to use inhalation delivery to target diseases outside the pulmonary space. Several reviews on systemic delivery via oral inhalation can be consulted for further details (Adjei and Gupta, 1997; Patton et al., 2004; Gonda, 2006; Cipolla, 2016b). We also refer the reader to comprehensive reviews of therapies with inhaled gases (Gentile, 2011; Miller et al., 2020).
The respiratory tract is an easily accessible portal for delivery of certain drugs to the systemic circulation and thus to other organs or targets within the body. There may be one or more reasons to explore this route of delivery for targets outside the respiratory tract:
1) It offers noninvasive delivery versus injectable treatments.
2) Compared with the gastrointestinal tract, there are relatively modest enzymatic and metabolic activities to deactivate small molecules, peptides, and proteins in the respiratory tract. Furthermore, there is no “first-pass” metabolism via the liver.
3) The lung has a large and permeable surface area, particularly in the alveolated regions, that enable facile absorption of most small molecules.
The ideal properties of drugs for inhalation delivery for the treatment of respiratory diseases discussed in Section II (i.e., achievement of high concentrations and long residence time in the respiratory tract and minimizing systemic exposure) are practically the opposite of what is required for systemic delivery via this route, perhaps with one exception when respiratory tract delivery would be used to generate a biologic response (e.g., immune reaction) that does not require systemic absorption. Furthermore, although diseases for which most respiratory drugs have been developed (e.g., asthma, COPD, CF) primarily affect the airways, systemic delivery via the lung generally benefits from delivery deeper into the lung (Colthorpe et al., 1992, 1995). The distal alveolated regions provide a vastly greater surface area than the airways and generally possess more permeable membranes; importantly, they do not have the competing mucociliary clearance mechanism.
It is of interest that the inhalation of products not regulated as medicines that contain addictive and substance-dependent drugs, in particular tobacco and cannabis particles and vapor, constitute by far the largest inhaled “drug” market. The key reasons for utilization of the inhalation route for these substances are the same as for some of the therapeutic agents inhaled for systemic efficacy, i.e., that noninvasive delivery achieves rapidly high drug concentrations in the pulmonary arteries supplying blood to the brain. This also represents one of the reasons for the speed and efficacy of anesthetic gases.
B. Inhaled Therapies for the Diseases of the Central Nervous System
“Nipping it in the bud” is often the most effective way to deal with central nervous system problems, such as episodic pain or craving for the nicotine in cigarettes. For this purpose, the rapid entry of inhaled drugs, especially small molecules delivered into the alveolated regions of the lung, can be used successfully to elicit efficacious responses. In addition to inhaled nicotine replacement therapies, two additional products are currently approved.
1. Inhaled Nicotine Replacement Therapy Products
There has been a significant global effort to replace cigarettes and other products that generate aerosolized nicotine in the form of tobacco smoke with pharmaceutical products containing “clean” nicotine. It has been recognized that dependence on nicotine is for many people a chronic condition, and therefore the goal of modern nicotine replacement therapy (NRT) strategies is to eliminate or at least reduce the use of tobacco smoking. A key element of this approach is to reduce craving for such products.
Although in terms of benefit versus risk and cost, NRT in its various noninhaled forms (patches, lozenges, gums, mouth sprays, and films) is one of the most effective healthcare interventions, its efficacy as a percentage of long-term quitters is low: A Center for Disease Control (2017) study concluded that: “In 2015, approximately two thirds of cigarette smokers were interested in quitting… fewer than one in 10 smokers overall quit successfully in the past year. Approximately three in five adults who had ever smoked had quit” (http://dx.doi.org/10.15585/mmwr.mm6552a1).
Although there is undoubtedly an attractive benefit/risk ratio for smokers using these therapies, the quit rates even with NRT products are relatively modest (Surgeon General (2020)).
It has been speculated that the reason for this low efficacy is that none of these therapies emulate the experience and “ritual” with tobacco smoke inhalation. However, the more plausible explanation is that the reduction of craving is related to the speed of penetration of nicotine into the brain as evidenced by the intensity and duration of craving reduction from high early arterial peaks using an experimental nicotine inhaler, consistent with an earlier study with cigarettes (Cipolla et al., 2008; Gonda et al., 2009).
More recent evidence to support the need for fast nicotine pharmacokinetics to provide an effective smoking cessation tool comes from comparisons of various types of nicotine vaping devices. The products that have the shortest time (Tmax) to high peaks (Cmax) show the largest reduction in craving for cigarettes. Indeed, the total (t = 0 to infinity) areas under the nicotine plasma concentration curve bear very little relationship to efficacy if the Cmax and Tmax are significantly different between products (Cipolla and Gonda, 2015).
The development of a “clean cigarette” (i.e., an inhalation product that would be an effective NRT) has encountered many challenges related to tolerability and efficacy. Cigarette smoke because of its very small particles and the smokers’ habit of deep inhalation followed by breath-holding results in a very effective alveolar delivery system, causing almost instantaneous entry of nicotine via the pulmonary arterial circulation into the brain. Nicotine is also quite irritant, and deposition higher up in the respiratory tract is in many people associated with severe cough (even cigarette smoke has that effect initially, but most smokers quickly develop tolerance to this irritation). A satisfactory therapeutic “clean cigarette” therefore needs to achieve high arterial nicotine concentrations quickly without excessive deposition in upper and larger airways that could cause poor tolerability.
2. Nicotrol Inhaler (Nicorette Inhalator, Nicorette Inhaler)
We include this inhaled therapy here, as it was originally developed as a nicotine aerosol product for lung delivery, but it was later discovered that only a very small fraction of the dose reaches the lung, and most of the nicotine is absorbed from the upper airways (Bergstrom et al., 1995; Lunell et al., 1996, 2000).
The name of this product and exact composition and use may vary somewhat from country to country.
Nicotine in this inhaler is stored in liquid form in a cartridge containing a porous plug, with a total of 10 mg nicotine per plug and also some menthol. During use, about 4 mg nicotine is released from the plug over 20 minutes of inhalations repeated every 15 seconds. The amount absorbed is only about 2 mg (the rest is presumably exhaled). The maximum arterial plasma concentration of nicotine is ∼6 ng/ml at ∼15 minute postinhalation. This peak is substantially lower and later than that from a typical cigarette (>40 ng/ml at ∼5 minutes) (Nicotrol Inhaler Package Insert, 2019).
a. Pivotal clinical evidence and indication
In two placebo-controlled pivotal trials in healthy smokers (n = 445 total) that consisted of treatment of 3–6 months using from 4–20 cartridges per day dosing as needed, at 6 months the Nicotrol groups achieved statistically significant quitting of tobacco smoking in 20%–21% versus 6%–11% in the placebo groups; however, at 12 months, this difference was reduced to 11%–13% in the Nicotrol groups versus 5%–10% in the placebo groups and was not statistically significant. Nicotrol Inhaler also demonstrated statistically significant reduction in the urge to smoke compared with placebo (Nicotrol Inhaler Package Insert, 2019).
In studies in tobacco smokers, it is often difficult to distinguish between the adverse long-term effects of smoking versus the effects of the treatment, especially if the latter is with “inhaled” nicotine. Nevertheless, compared with placebo, the Nicotrol Inhaler was associated with a greater proportion of irritation in the mouth and throat (66% vs. 42% and 40% vs. 18%, respectively, vs. placebo). Coughing and rhinitis had higher rates in the Nicotrol group (32% vs. 12%, 23% vs. 16%, respectively, vs. placebo). Dyspepsia was also higher in the Nicotrol group (9% vs. 3% for placebo), but withdrawal symptoms were similar in the two groups (Nicotrol Inhaler Package Insert, 2019).
As for most other NRTs, the Nicotrol Inhaler is indicated to assist tobacco smoking cessation by reduction of nicotine withdrawal symptoms. The recommended dosage is 6–16 cartridges per day for the initial period of up to 12 weeks, with gradual reduction in the following period of up to 12 weeks. The goal of the therapy is abstinence from tobacco smoking.
3. Voke/Nicotine 0.45-mg Inhaler
This nicotine inhaler therapy available in the United Kingdom was approved as a “hybrid medicine” via comparability against the already authorized Nicorette Inhalator (see previous product review) (MHRA, 2014).
It is formulated as a solution of nicotine (0.056%) in the propellant HFA134a also containing propylene glycol, ethanol, saccharin, and levomenthol.
Each Voke inhaler device—a “pack”—has 20 × 0.45–mg doses in a pressurized metered-dose-inhaler–like canister that charges each dose into a “stick” with the looks of a cigarette. Except for the first dose, the delivered dose per stick is 0.43 mg. The device is used dosing as needed via multiple inhalations from each stick charge, with a maximum recommended daily dose of two packs (i.e., 40 stick charges) (medicines.org.uk, 2019).
a. Clinical development and therapeutic indication
Initially, a number of prototype devices and formulations were tested in crossover trials versus the Nicorette Inhalator (Nicotrol). The “to be marketed” final product was tested in a randomized crossover clinical study in 24 healthy smokers. Each participant was advised to inhale the entire contents of one stick from the Voke Inhaler taking one inhalation every 15 seconds until the device was empty, but taking no longer than 4 minutes. In the reference period, the participants were advised to use the Nicorette Inhaler in line with the manufacturer’s prescribing information to take 4 inhalations every minute for 20 minutes, with the maximum dose achieved with deep inhalations (MHRA, 2014).
The mean venous peak nicotine concentration and area under the curve for the Voke inhaler were both less than 50% of those for the Nicorette Inhalator, whereas the Tmax for the former was 21.0 versus 36.3 minutes for the Nicorette. Interestingly, the reduction of the craving for cigarettes using the visual analog scale was not only earlier but also greater throughout the study for the Voke product than for the Nicorette. These findings are consistent with the explanation that it is the attainment of early Cmax (i.e., shorter Tmax) in the arterial blood nicotine levels that is important for the craving reduction timing and magnitude (Gonda et al., 2009). The mechanistic explanation for the differences in the PK between the two products is likely due to the difference between the predominantly large airway and esophagus absorption of nicotine vapor produced by the Nicorette Inhalator (Lunell et al., 1996) versus the faster pulmonary absorption of the nicotine aerosol generated by the Voke, which deposits in the pulmonary spaces (MHRA, 2014).
Voke is used to relieve or prevent cravings and nicotine withdrawal symptoms associated with tobacco dependence. It is indicated to aid smokers wishing to quit smoking and represents a safer alternative to smoking for smokers unable to quit and those around them.
4. Loxapine/ADASUVE
Acute agitation is a serious episodic problem in some people with psychiatric disease. If not treated quickly, it can exacerbate further. Rapid intervention is therefore preferred but oral (ingested) treatments are generally too slow, and the subject may not be amenable to be treated with injection.
An inhaled form of the antipsychotic drug loxapine was therefore developed and approved for this purpose. The mechanism of action of this drug is unknown. It has been used in France via intramuscular injection to deal with acute agitations (Spyker et al., 2015).
Adasuve is a single-dose disposable inhaler containing 10 mg of loxapine powder. Inhalation through the device triggers rapid heating of a thin film of the drug powder causing evaporation and subsequent condensation into fine particles suitable for alveaolar delivery with an MMAD of ∼2–3 µm and a narrow size distribution (Dinh et al., 2011; Adasuve Package Insert, 2012).
a. Essential clinical trial evidence
In healthy volunteers, inhalation from Adasuve resulted in maximum plasma concentrations in ∼2 minutes followed by a half-life of 7.6 hours ((Adasuve Package Insert, 2012).
Using an instrument to assess the level of agitation, Adasuve showed superiority over placebo in the change in this instrument at 2 hours postadministration. The effect of Adasuve was apparent 10 minutes postadministration.
Because of the findings of bronchospasm (Gross et al., 2014), Adasuve can be only used in the United States in registered healthcare institutions that have the capability to rescue patients who may develop acute bronchospasm when administered Adasuve. This product is also contraindicated in patients taking asthma or COPD medication or have history of these diseases. Increased mortality was found in elderly patients with senile dementia (Adasuve Package Insert, 2012).
5. Inhaled Levodopa/INBRIJA
L-dopa (levodopa) is the precursor of the neurotransmitter dopamine. Oral administration of L-dopa is a key part of the treatment of Parkinson Disease (PD), whose underlying pathology includes degeneration of dopaminergic neurons. Because premature conversion of L-dopa to dopamine in the blood can cause nausea and vomiting, it is used in combination with carbi-dopa to retain more of it as a prodrug prior to reaching the brain.
So called “off” periods of poor motor control in PD may be the result of inadequate L-dopa blood concentrations. In addition to the general interference with absorption of L-dopa from food interactions and first-pass metabolism, oral administration of L-dopa may be less effective particularly in advanced PD as a result of delays in gastrointestinal transit. The purpose of developing an inhaled L-dopa therapy was therefore to provide reliable fast remedy during these “off-periods” (Hauser et al., 2019).
The Inbrija formulation is a dry powder made of porous particles containing L-dopa, sodium chloride, and DPPC. A dose of Inbrija is 84 mg of L-dopa contained in 2 capsules; the contents are inhaled from the Inbrija Inhaler. Inhalation of Inbrija results in maximum plasma concentrations of L-dopa in ∼0.5 hours (Inbrija Package Insert, 2018).
a. Essential clinical evidence
In a 12-week randomized placebo-controlled study, a dose of the study medication (82 mg of L-dopa or placebo) could be inhaled up to 5× a day during the off period. Subjects with chronic respiratory diseases within the last 5 years were excluded. The average observed number of doses was about two. Using the change at 12 weeks between the predose off state and 30 minutes postdose in the Unified Parkinson’s Disease Rating Scale Part III (motor syndrome) score as the primary endpoint, Inbrija was superior to placebo. A greater proportion of the subjects on Inbrija compared with placebo remained in the on state at 60 minutes postdose as well. As frequently observed with other inhaled therapies, the most common adverse effect of Inbrija versus its placebo was cough (15% vs. 2%, respectively) (Inbrija Package Insert, 2018).
In another study of subjects with moderate or mild asthma on a stable treatment of that condition, 60% of those taking Inbrija versus 0% on placebo reported cough. Ten subjects (40%) on Inbrija had a significant postdose bronchoconstriction event, and four of those also had such bronchoconstriction postinhalation of placebo (Inbrija Package Insert, 2018).
Inbrija is indicated for the intermittent treatment of the off episodes for patients with PD treated with oral carbidopa/levodopa. It is not recommended for subjects with chronic lung diseases.
C. Insulin
1. Introduction
People with type I diabetes are dependent on regular doses of exogenous insulin. In the large and growing population of type II diabetes subjects, many patients, especially those with the more severe form of the disease, need insulin therapy.
The interest in noninvasive delivery of insulin and specifically by inhalation is almost as old as insulin discovery itself (Gaensslein, 1925). Insulin is a polypeptide, and despite many attempts to deliver it noninvasively by various routes of administration, for many decades the only form of insulin therapy was injections. The earlier inhalation attempts in this area failed both because of poor understanding of the requirements for efficient and reproducible delivery as well as absence of technologies capable of providing the required performance to achieve these goals (Kohler, 1992).
A few companies emerged in the 1990s that began developing modern inhalation delivery systems that would be capable to address the challenges of pulmonary delivery of insulin. These included precision delivery required by the narrow therapeutic index (to balance the potential occurrence of hyperglycemia or hypoglycemia) and substantial alveolar deposition to maximize the systemic bioavailability of the lung dose. These two requirements were addressed by the development of technologies to achieve inhaled products with small aerodynamic diameters and, in some cases, by controlling the inspiratory flow rate. Furthermore, all these emerging products included instruction to enhance alveolar delivery by first exhaling and then taking a slow deep inhalation of the insulin aerosols followed by breath-holding. Since the variability of pulmonary deposition is largely the product of oropharyngeal deposition, the small aerodynamic diameters, especially those associated with slow inspiratory flow rate to minimize inertial impaction in the upper airways, also suppressed the variability in delivery because of this source.
The substantial challenge with the choice of excipients to stabilize insulin during manufacture, storage, and use and to satisfy tolerability and safety criteria also needed to be dealt with. Last but not least, insulin therapy by injection is relatively inexpensive, and therefore the economic factors were a consideration as well.
2. Exubera
The first approved inhaled insulin was Exubera. Its pharmaceutical development was reviewed by Stevenson and Bennett (2014). It was a combination product utilizing an amorphous spray-dried formulation containing insulin, sodium citrate, mannitol, glycine, and sodium hydroxide packaged in blisters. Two product strengths were developed: 1 and 3 mg insulin (1.7 and 5.1 mg nominal powder fill weight, respectively) to enable flexibility in dosing for postprandial control of glucose.
To achieve adequate precision of delivery with reduced dependence on the patient’s dexterity and breathing effort, the Exubera device had a translucent holding chamber in which the cloud of aerosol was formed by compressed air provided from a pump integral in the device. The patients were instructed to take a slow deep inhalation to empty the aerosol from the chamber, continue to take a full breath and then briefly hold their breath to avoid exhalation of the therapy. The energy of the compressed air caused deagglomeration of the powder formulation into particles with an MMAD of ∼3 µm, which were inhaled until the aerosol from the chamber disappeared. The inspiratory flow rate was limited by the critical orifice in the device.
a. Key clinical evidence leading to approval
The bioavailability of Exubera compared with subcutaneous regular insulin was ∼10% (Stevenson and Bennett, 2014). It achieved earlier Cmax in blood than subcutaneous injection of regular insulin and resembled more the fast-acting subcutaneous insulin analog lispro. The intrasubject variability in glucose control was similar to subcutaneous injections (Exubera US Package Insert, 2006).
The primary efficacy was reduction in hemoglobin A1C (HbA1C). In type I diabetes, Exubera showed comparable reduction in HbA1C to subcutaneous injection insulin. In type II diabetes, Exubera either alone or in combination with oral hypoglycemic agents showed better control of HbA1C than oral therapies alone.
Furthermore, Exubera was generally superior in the clinical trials in the reduction of fasting glucose levels. The most common nonrespiratory side effects were hypoglycemia, chest pain, dry mouth, and otitis media in type I diabetes. The most common respiratory adverse effect that was consistently observed for Exubera was cough in patients with both type I (21.9%) and type II (29.5%), compared with 3.7%–10.2% in patients receiving insulin injection or oral hypoglycemic drugs (Exubera US Package Insert, 2006). It is possible that the use of citrate in the formulation was the cause (Chang et al., 2020).
There were generally greater losses in lung function (FEV1 and diffusing capacity for carbon monoxide) in the Exubera groups compared with comparators (Exubera US Package Insert, 2006).
b. Postapproval developments
The sales of Exubera were vastly lower than the bombastic predictions for many years prior to its approval. The marketing partner for Exubera Pfizer decided to return the product to its licensor and original developer Nektar. Soon after, the two other major pharmaceutical companies developing inhaled insulin terminated their programs.
An even more damaging event for inhaled insulin was the press release by Pfizer that lung cancer occurred in more patients on Exubera than in subjects who were taking other diabetes medicines. It should be noted that all subjects diagnosed with lung cancer had a history of smoking. As the number of lung cancer cases was within the range of general population statistics, it was unclear whether Exubera in particular or inhaled insulin in general was causing the lung cancer (Heinemann, 2008).
These events had a major negative impact on the inhalation research and development and especially on inhaled biologics (Gonda, 2019b).
3. Afrezza
Although big pharmaceutical companies pulled out from the development of inhaled insulins, Mannkind Corporation continued in their mission to provide patients with diabetes with an inhaled insulin therapy.
Afrezza is an approved drug-device combination comprising a dry powder formulation containing regular human recombinant insulin adsorbed on particles made out of fumaryl diketopiperazine and polysorbate 80. These excipients appear to be the reason why this formulation results in ultrafast absorption from the lung. There are two strengths cartridges: four-unit or eight-unit cartridges (0.35 mg and 0.7 mg insulin, respectively). The powder is administered via a passive breath-powered inhaler. The instructions for use ask the patient to first exhale and take a deep breath from the device, and this is followed by a breath hold. The inspiratory flow rate is limited by the high resistance of the device (Leone-Bay et al., 2010).
After inhalation, the plasma pharmacokinetics is more rapid than that of the fast-acting subcutaneous insulin analog lispro, but this is not reflected in faster effect on glucose reduction. The pharmacokinetic and pharmacodynamic profiles are consistent with the indication (Afrezza Package Insert, 2014) as a rapid-acting insulin to be administered at the beginning of the meal and indicated to improve glycemic control in adult patients with diabetes mellitus.
a. Key clinical evidence leading to approval (Afrezza Package Insert, 2014; Goldberg and Wong, 2015)
In a study in patients with poorly controlled type I diabetes, Afrezza used at mealtimes met the prespecified noninferiority criteria for reduction of HbA1c versus insulin aspart injections. In patients with type II diabetes poorly controlled with oral antidiabetic drugs (OADs), Afrezza plus OAD was superior to OAD + Afrezza placebo in terms of reduction of HbA1c.
Acute bronchospasm has been reported in patients with asthma and COPD using Afrezza, and it is therefore contraindicated for the patients with diabetes with these pulmonary comorbidities. The safety and efficacy of Afrezza has not been established in smokers.
Hypoglycemia as well as severe hypoglycemia were more common in the Afrezza group of patients with type II diabetes compared with placebo. Cough and throat irritation were more common in the Afrezza-treated patients compared with the control groups, although the differences were smaller when the carrier particles for insulin were used as the control, suggesting that the carrier itself may be also the causative factor. As with Exubera, a greater decline in FEV1 was observed in the patients using Afrezza compared with the control groups.
D. Conclusions
Approved inhaled therapies for the treatment of respiratory diseases have been developed to achieve adequate therapeutic concentrations in the respiratory tract with doses that are safe and well tolerated both locally and systemically. The targeting specificity in this situation is enhanced by inhaled delivery. This is not the case with the use of the inhalation route for systemic administration. In these situations, the respiratory tract is exposed to the full dose intended for systemic administration, and in the case of drugs with incomplete absorption from the lung, a greater dose. Addition of excipients necessary for appropriate pharmaceutical performance, including storage stability, further increases the potential risk for adverse respiratory reactions.
If the key advantages of the inhalation route, namely rapid onset of action, noninvasive method of administration, and, in the case of poor absorption from oral administration, superior bioavailablity, do not outweigh the risks of adverse respiratory reactions, then it is questionable whether an attractive balance of benefit versus risk for the patient can be achieved.
Reviews analyzing the various factors that contributed to the troublesome history of inhaled insulin products have been published (Heinemann, 2008; Oleck et al., 2016) together with “lessons learned” to avoid the pitfalls inherent in the development and marketing of Exubera when considering future inhaled products delivering biologics (Gonda, 2019b) as well as small molecules for respiratory disease and for systemic effects (Cipolla, 2016b).
It is noteworthy that most of the inhaled therapies for systemic administration result in respiratory adverse reactions, in particular cough and bronchoconstriction. This not only often leads to exclusion from the approved label of subjects with known existing respiratory comorbidities but also creates a risk for subjects who may have an undiagnosed respiratory disease. It is an area that deserves closer attention to minimize the frequency and severity of such adverse events (Chang et al., 2020).
XV. New Developments in Inhalation Technology
A. Introduction
As outlined in Section III, there may be various “drivers” for introduction of innovation into inhalation medicines. New and often very different types of active ingredients—small molecules, proteins, and DNA and RNA constructs as well as cell therapies or new patient population and targets within their respiratory tract—are such examples. But they could also be improvements in formulations and devices of existing therapies to make them safer, more effective, or easier for the patients to use—a very successful example of that is the combination products of already approved drugs for asthma (Section VII).
B. Improved Lung Targeting and Dose Consistency
The target of most pharmaceutical aerosol products is the lower respiratory tract (i.e., the lungs), with off-target deposition in the upper respiratory tract (URT) considered undesirable. Nonetheless, most marketed formulations deposit a large percentage of the delivered dose (50%–90%) in the mouth and throat, where it can result in both local adverse events (e.g., opportunistic infections, dysphonia, throat irritation, cough), and systemic adverse events for drugs that are orally bioavailable. Off-target delivery also leads to increases in the nominal dose, which can be problematic for those drugs that are expensive to manufacture (e.g., biologics) or for drugs with low potency (e.g., inhaled antibiotics). Improved lung targeting may overcome or minimize these issues while also significantly reducing variability in drug delivery (Tayab and Hochhaus, 2005).
In the new millennium there has been a step change in the total lung dose (TLD) that can be achieved with pharmaceutical aerosols. The TLD with portable inhalers has increased from 10%–30% of the nominal dose to 40%–70% with some recently marketed products (Pitcairn et al., 2005; Haynes et al., 2016), with new studies suggesting that TLD values exceeding 90% may be possible (Ung et al., 2016; Weers et al., 2019b; Bass et al., 2021).
Although there is currently significant effort being paid to reducing dosing variability associated with nonadherence, the large variability in TLD resulting from differences in the anatomic features of the URT is largely ignored. The mean coefficient of variation on measures of TLD is about 30%–50% for formulations that deposit 10%–30% of their dose in the lungs, decreasing to 10%–20% when the TLD exceeds ∼40% (Stahlhofen et al., 1989; Borgström et al., 2006; Cipolla et al., 2010). For aerosols that effectively bypass deposition in the URT through the control of their aerodynamic size and velocity, the variability resulting from oropharyngeal filtering of particles should approach 0% (Gonda, 1992).
Dry powder formulations that more effectively bypass deposition in the URT also typically have a decreased flow rate dependence in TLD. In this regard, spray-dried formulations with improved lung targeting typically have much lower flow-rate dependencies than do lactose blends and spheronized particles (Weers and Clark, 2017).
Not only have new delivery systems been advanced with improved targeting to the lungs, but there have also been advances in targeting of drug regionally within the lungs (e.g., to the small airways) (Usmani, 2012; Leach et al., 2016; Virchow et al., 2018). There is growing evidence that the small airways contribute to the pathophysiologic and clinical expression of asthma and COPD (Usmani, 2012; Carr et al., 2017; Lavorini et al., 2017). Indeed, the small airways represent the major site of obstruction in COPD and may precede the development of emphysema (McDonough et al., 2011; Stockley et al., 2017). Advances in the development of extrafine pMDI formulations with particle sizes of ∼1.0 μm have enabled increased delivery into the small airways (Usmani, 2012; Leach et al., 2016). New dry powder formulation strategies (e.g., excipient enhanced growth) may enable high-efficiency delivery to the airways while minimizing alveolar deposition and particle exhalation (Hindle and Longest, 2010; Bass et al., 2021).
C. The Emergence of “Bottom-up” Particle Engineering Technologies
Effective drug delivery to the lungs typically requires the production of fine micronized particles with a geometric size less than 5 µm. Currently, most dry powder inhalers and suspension-based pMDIs use fine crystalline drug particles that are produced by “top-down” manufacturing processes, wherein large nonrespirable drug particles are milled to the desired size (Midoux et al., 1999; Kluge et al., 2012). Top-down methods (e.g., jet milling, high-pressure homogenization) produce irregular polydisperse particles with limited control of the surface properties of the particles. Indeed, the milling process often results in modifications to the particle surface (e.g., electrostatic charging or the development of high energy sites and amorphous domains) (Ward and Schultz, 1995). These surface modifications can lead to unacceptable physical and chemical stability and inconsistency in aerosol performance. As a result, milled particles often undergo a conditioning step to enable more consistent dose delivery (Brodka-Pfeiffer et al., 2003; Müller et al., 2015).
Fine micronized particles exhibit strong interparticle cohesive forces that result in poor powder flow. As a result, micronized drug particles are often blended with coarse lactose carrier particles or spheronized into larger agglomerates. Owing to the large size, drug must be dispersed from the carrier or agglomerate to be delivered into the lungs.
The adhesive properties between the drug and carrier in lactose blends can be modified by the addition of a force control agent (e.g., magnesium stearate), thereby enabling improved lung targeting with these formulations (Begat et al., 2005).
The new millennium has seen the emergence of bottom-up processing methods that enable greater control of the micromeritic properties of the particles, including their size, density, surface composition, surface morphology, and physical form of the drug substance. Bottom-up processing methods include spray drying (Vehring, 2008; Weers and Tarara, 2014; Weers, 2019; Vehring et al., 2020), spray freeze drying (Maa et al., 1999; Rogers et al., 2002), various supercritical fluid-processing methods (Sun, 2015; Hadiwinoto et al., 2018), and lithography or “printing” of highly uniform particles with a consistent shape (Garcia et al., 2012).
Arguably the most advanced of these particle creation technologies with multiple products having received market authorization is spray drying. By controlling the feedstock composition and drying parameters, it is possible to control the Peclet Number (a dimensionless number describing the ratio of the rates of the competing processes of diffusion of the solute in the spray-dried solution and evaporation rate of the solvent), which enables the creation of core-shell particles (Vehring et al., 2020). In these engineered particles, the drug substance is present in the core of the particle with excipients (e.g., buffers, glass-formers, common ions) that ensure physical and chemical stability of the drug substance, and the shell is comprised of a hydrophobic excipient that controls the micromeritic properties (e.g., density, surface roughness, surface energy, environmental robustness) of the powder. Various formats have been developed for incorporating drug into spray-dried formulations. These formats enable control of the physical form of the drug substance (e.g., amorphous or crystalline) in the spray-dried drug product (Weers and Tarara, 2014; Weers et al., 2019a).
The improved control of interparticle cohesive forces enables acceptable powder flow and dose delivery to the lungs without the need to be blended with coarse lactose carrier particles. This enables nominal doses of drugs that are more than three orders of magnitude larger than current asthma drugs to be delivered with a portable inhaler (Geller et al., 2011). Dry powder formulations offer a reduced treatment burden and improved convenience relative to jet nebulizers for the delivery of inhaled antibiotics (Geller et al., 2011; Weers, 2015).
Bottom-up processing methods enable effective mixing of drug with excipients (e.g., buffers and glass-forming excipients) in an amorphous glass, enabling dry powder formulations of macromolecules with long-term physical and chemical stability at room temperature (White et al., 2005; Sadrzadeh et al., 2010; Vehring et al., 2020).
As discussed above, effectively bypassing deposition in the URT also significantly reduces variability in lung delivery, which results from anatomic variability in the soft tissues of the mouth and throat between subjects (Stahlhofen et al., 1989; Borgström et al., 2006). Dry powder formulations prepared by spray-drying also exhibit large reductions in flow rate dependence in lung delivery compared with spheronized particle and lactose blend formulations (Weers and Clark, 2017).
Cosuspensions of micronized drug with small porous lipid particles improve suspension stability in HFA propellants, reducing the potential for variability in shake-pause-fire testing (Vehring et al., 2012). These cosuspensions also exhibit no coformulation effects and enable uniform delivery of nominal doses less than 1 mg.
D. Improvements in Delivery of Aqueous Aerosols
Nebulizers, as a refinement of atomizers that generate coarse aerosols by virtue of simple liquid break-up, first appeared in the mid-1800s (Nikander and Sanders, 2010; Stein and Thiel, 2017). Jet nebulizers have changed little since that time. In modern jet nebulizers, a stream of compressed gas is used to entrain solution and generate droplets, which are directed at impingement baffles designed to capture large droplets and allow respirable droplets to exit and be inhaled. Jet nebulizers have many disadvantages: high dead volume due to capture of solution on the baffles; high shear stress due to continued recirculation of the large droplets through the atomizer; and wasted aerosol during exhalation due to the continuous nature of their operation and a limited ability to combine high delivery rates and short delivery times with fine droplet sizes. Although still popular, these disadvantages have been the target of the next generation of advanced jet nebulizers. These advanced devices use vents, valves, and chambers to help synchronize inhalation with aerosol generation to improve delivery efficiency (Newman, 2009).
Ultrasonic nebulizers have been developed in two main forms. Transducers produce aerosol directly using capillary waves and cavitation and generally operate in the Mhz region, whereas ultrasonic mesh nebulizers use transducers to vibrate a multiple aperture mesh in the high kHz range (Carvalho and McConville, 2016). The former of these has the disadvantage of inducing both temperature and shear stress into the solution, which can denature biologics (Cipolla et al., 1994d).
Vibrating mesh nebulizers were introduced in 1993 (Dhand, 2002; Pritchard et al., 2018). This technology does not require recirculation of solution and generates a respirable aerosol in a single pass through a mesh consisting of many small apertures (Dhand, 2002). The mesh is typically vibrated at over 100 kHz and contains apertures of around 3-µm diameter, which produces aerosols with MMADs of around 5 µm. Their output is typically around 0.5 ml/min but is limited by number of holes per unit area and the available surface area of the mesh. Recent improvements in mesh architecture driven by a new manufacturing technique known as Photo Defined Aperture Plates have allowed manufacture of meshes with smaller hole diameters and high aperture densities and have resulted in an ability to generate aerosols with MMADs of around 2 µm while maintaining acceptable delivery rates (Fink et al., 2016) (note: Delivery rate is proportional to droplet volume and droplet volume scales as the cube of the diameter; hence the hole density, number of droplets generated per vibration, must increase as the inverse of the cube of the droplet diameter to maintain delivery rates). Mesh nebulizers also have a rapid onset of aerosol generation, allowing true synchronization of delivery with inspiration.
Although the base technologies of nebulization have been around for many years, with the advent of mesh technology nebulizer devices continuing to become more sophisticated, there are offerings, such as more accurate control of droplet size, true breath synchronization, matching delivery to inspiratory profiles, and helping instruct patients to inhale correctly. Adaptive Aerosol Delivery (Hardaker and Hatley, 2010) synchronizes aerosol delivery with a patient’s inhalation. More recently, a branded version of the Fox (Fox Nebulizer, 2017), a small hand-held mesh nebulizer, Breelib (Bayer), was introduced for delivery of iloprost. It assists in controlling the patient’s inspiratory flow rate, the inspiratory volume, and the timing of aerosol delivery during the inspiration to enhance consistency, efficiency, and rate of dosing (Gessler et al., 2017). Fink et al. (2017) reported on an innovative use of mesh technology in a micronebulizer designed to deliver insulin. The device incorporated lights to help instruct the patient on how to inhale and a restrictive airflow to ensure the patients inhaled at a low (5–10 l/min) flow rate. Scintigraphy studies indicated that this combination resulted in both high total and peripheral lung deposition.
The “final frontier” for nebulizer technology is the efficient and reproducible delivery of aerosols in the critical care environment (Clark et al., 2016). Here mesh nebulizers come into their own, as they do not introduce additional air into a ventilation circuit. In addition, use of the Photo Defined Aperture Plate architecture facilitates aerosol sizes capable of passing through the nasal airways of neonates (Clark, 2021; Section XIII of this issue) and coupled to breath synchronization can potentially facilitate efficient reproducible delivery of drugs directly to a neonate’s lungs.
Small hand-held aerosol inhalers that form droplets by break-up of jets formed by mechanical extrusion of liquids through orifices are examples of SMIs (Leiner et al., 2019). In the only example so far successfully commercialized (Respimat) (Dalby et al., 2004), two impinging jets collide to generate respirable droplet aerosol. The AERx system was a unit dose inhaler in which each dosage form had its own disposable nozzle assembly through which the liquid was extruded under mechanical pressure. The electric version of the system used a programmable motion piston for the liquid extrusion with real-time visual feedback to guide the patients into the correct breathing maneuver; the device also had a miniature heater to aid evaporation of the solvent to minimize droplet size (Schuster et al., 1997). Later development of precision laser micromachining enabled manufacture of nozzles in the submicron regions to enable the formation of small (2–3 µm) droplets without the need for a heater, and the energy for the extrusion, synchronization of the aerosol generation with inspiration, and the control of inspiratory flow rate were all achieved purely through mechanical means (Cipolla et al., 2008; Cipolla and Gonda, 2015). In 2007, de Boer et al. (2017) described a multiple nozzle system, Medspray, that used small apertures of 1.5–2.5 µm diameter to produce sprays with MMADs in the respirable range. The Medspray device has a similar format to the popular pMDI, although like most SMIs, the spray duration is much longer (several seconds vs. 100 milliseconds), and the plume is less dynamic (several ms−1 vs. 30–50 milliseconds−1). SMIs are promising, however, and just as with all pharmaceutical inhalation dosage forms, they have limitations (Leiner et al., 2019). Generally, the volume of solution that can be delivered in an inhalation is limited to a few tens of microliters, and the range of solution properties limits the concentrations that can be atomized (Carvalho and McConville, 2016). Thus, their application is limited to low-dose/high-potency molecules.
E. Sustained Release in the Lungs
Being able to control the clearance of drug from the lungs is of critical importance (Tayab and Hochhaus, 2005). Once-daily administration of drugs leads to improvements in patient adherence relative to drugs dosed more frequently (Izquierdo et al., 2016). Moreover, maintaining drug concentrations at the site of action in the lungs may improve their therapeutic index (Tayab and Hochhaus, 2005). Indeed, the advantage of sustaining concentrations of drug in the lungs to optimize pharmacokinetic/pharmacodynamic metrics has been demonstrated for inhaled corticosteroids and antibiotics (Tayab and Hochhaus, 2005; Weers et al., 2019b).
Achieving sustained release within the lungs is challenging because of the multiple clearance pathways and the need to avoid accumulation of excipient within the lungs (Smyth, 2011). Traditionally, sustaining drug within the lungs has been accomplished through molecular engineering, as is evidenced by the evolution of β-agonists from the original adrenaline to short-acting, rescue medications like albuterol to long-acting molecules like formoterol or salmeterol that require twice-daily dosing to once-daily therapeutics like oladaterol, vilanterol, and indacaterol.
Increasing the residence time in the lungs can also be achieved by controlling the dissolution of the drug substance. This can be done by using the neutral form of the drug or by inclusion of design features in the drug substance that limit dissolution (Daley-Yates, 2015; McShane et al., 2018). Treatment of pulmonary arterial hypertension with inhaled prostanoid therapy currently requires four or more administrations per day, and the rapid systemic uptake can lead to increased side effects. Several approaches, including nanoparticle (Garcia et al., 2012) or liposome-encapsulation (Kan et al., 2020), to modify the release profile have demonstrated promise in preclinical development and the use of a prodrug strategy to delay dissolution and release of the active drug in the lung that has advanced into the clinic either alone (Chapman et al., 2020) or combined with a lipid-nanoparticle approach (Leifer et al., 2018).
More recently, the first formulation approaches to achieve sustained release have been advanced into late-stage development, and one has been approved (Arikayce, Insmed Corp.). Arikayce is a liposomal formulation of amikacin approved for the treatment of Mycobacterium avium complex lung disease as part of a combination antibacterial drug regimen in adults who have limited or no alternative treatment options (Zhang et al., 2018). Liposomes have the advantage that the excipient is rapidly cleared by a natural catabolism process.
Penetration of the nanosized particles into mucus or biofilms may provide an additional therapeutic benefit (Meers et al., 2008). A liposomal formulation of ciprofloxacin comprising both free and encapsulated drug has also been advanced into late-stage clinical development for the treatment of bronchiectasis (Cipolla et al., 2016a).
Particle size and shape may also be leveraged to control particle clearance by evading macrophage clearance (Champion and Mitragotri, 2006). Large porous particles with a geometric size between 5 and 30 μm may be too large to be phagocytosed (Edwards et al., 1997), whereas nanoparticles that are smaller than bacteria may not be recognized (Kawaguchi et al., 1986).
Other formulation technologies are also being explored, including polymeric microparticles and semisolid and solid lipid nanoparticles (Smyth, 2011; Cipolla et al., 2014). Interestingly, in a mouse model, the slow clearance (half-life of 10–11 hours) of semisolid lipid nanoparticles was not changed by the inflammation (Patel et al., 2016).
Although polymer-based systems have been used extensively for controlling release in oral and parenteral formulations, they have yet to achieve adoption in pulmonary formulations because of concerns about excipient clearance and toxicity in the lung. Work continues in this area.
Increasing the residence time of particles in the lungs may enable targeting of drug to pulmonary macrophages to enable intracellular treatment of infections, such as tuberculosis and nontuberculosis mycobacteria. The inclusion of specific molecules (e.g., antibodies, mannan, phosptidylserine) into the surface of the liposomes or microparticles may make the particles “tastier” to macrophages (Bot et al., 2001). More work is needed to better understand the implications of increasing the residence time of particles in the airways from a safety perspective (Gonda, 1988; Weers et al., 2019a; Sahakijpijarn et al., 2020).
F. Improved High Dose Delivery
A large percentage of the respiratory drugs on the market today have been developed for the treatment of asthma and COPD, with comparatively few drugs developed for other indications. Asthma/COPD therapeutics are generally highly potent with nominal doses for bronchodilators and inhaled glucocorticosteroids in the range from 10 µg to 500 µg. Not surprisingly, the technologies developed for these indications are not well suited for delivering doses in the range of 10–100 mg (Geller et al., 2011).
High dose delivery has traditionally been accomplished using jet nebulizers (e.g., for inhaled antibiotics including TOBI). Disadvantages of jet nebulizers include a high daily treatment burden related to the long administration times and the added time needed for cleaning and disinfection and dose preparation. Recently, advances in delivery technologies have enabled more rapid delivery with vibrating mesh nebulizers (see the segment of this section Improvements in Delivery of Aqueous Aerosols above) as well as the development of the first high-dose dry powder formulations as discussed in this section above.
Vibrating mesh nebulizers (e.g., PARI e-Flow, Aerogen Solo, Phillips Respironics Innospire) provide improved portability and more efficient delivery into the lungs with significantly reduced administration times compared with jet nebulizers (Martin and Finlay, 2015; Ari and Fink, 2020). Vibrating mesh nebulizers may also cause less degradation of biologics than jet nebulizers and require much less drug in inhaled toxicology studies, making them a favored device in these applications (Liang et al., 2020).
Dry powder formulations with nominal doses of ∼100 mg have been approved utilizing neat drug (Schwarz, 2015), small porous particle (Geller et al., 2011), and large porous particle (Paik, 2020) technologies. Various other formulation technologies may hold promise for high dose delivery, including comicronization or mechanofusion of drug and force control agents like leucine or magnesium stearate (Begat et al., 2009). To reduce the burden of treatment of the patient, it is important that the dose be delivered in a single receptacle, if possible. Maximizing pulmonary delivery for a dry powder from a given sized powder receptacle (e.g., a capsule), depends on mass of powder that can be filled into the receptacle, the drug loading in the formulation, and the efficiency of the aerosol in delivering drug into the lungs (Weers and Miller, 2020). Alternative delivery systems with larger-sized receptacles have also been advanced (Young et al., 2014; Parumasivam et al., 2017).
G. Electronic Enhancements of Inhalers
“Electronic,” “intelligent,” or “smart” inhalers date back to at least the 1980s (Howard et al., 2014; Kikidis et al., 2016), but their potential scope has greatly increased with universal access to high-speed internet.
Medical inhalers are typically used daily and in many instances multiple times a day. With built-in or add-on device electronics, they can collect and process information about the users pertinent to their health, which can be then communicated (Dundon et al., 2020). By “connecting” them to telecommunication systems, such devices can act as the interface between the user and other stakeholders in the user’s personal and overall healthcare systems (Gonda, 2019a).
The benefit for the patient can be manifold. Firstly, adherence with the instructions for correct use can be monitored and analyzed, and the patient can be provided with guidance to improve their technique or compliance. Specifically, the delivery technique can be improved, for example, by sensing the subject’s breathing pattern and actuating the dose at the optimum time during the inspiration, as has already been discussed above with electronically aided nebulizers and the AERx soft mist inhaler.
A variety of sensors can be incorporated within or associated with these devices, including those based on analyses of acoustic signals and airflow rates.
Real-time feedback to the patient (e.g., visual feedback to guide the user to inhale at the desired inspiratory flow rate) and postdosing feedback (e.g., “comments” to shake a suspension metered dose inhaler prior to the inhalation, to exhale prior to inhalation, and to increase the duration of breath-holding) can be provided using either the intelligence built into the device or some external device, such as a smartphone connected to the internet. Such information can be very useful for healthcare providers to assess whether failure of the treatment is due to poor compliance with instructions for use or is the result of a true lack of response to the particular drug (Gonda, 2019a and references therein).
In asthma, the contextual information related to acute exacerbation is very important. For example, the environmental triggers of such events could be the quantity of air pollutants or exposure to high altitude. Linking the local information about these potential triggers using the geographical location of the user, monitoring the frequency of use of short-acting rescue medications like bronchodilators, and, ultimately, emergency phone calls by the patient can be very useful to inform about the ways to prevent or reduce occurrence of such crisis episodes (Williams et al., 2019).
The first FDA-approved “smart” add-on device was most likely the Nebulizer Chronolog (Kikidis et al., 2016) recording the timing of the patient’s use of inhalation therapy (Howard et al., 2014; Kikidis et al., 2016) to check compliance with the dosing regimen.
An example of a recently approved “add-on” technology with a focus on the improvement of asthma patient adherence to therapy is the Hailie (Adherium Ltd). This is an internet-connected device for inhalers that captures medication use data and can provide real-time feedback to patients and their physicians via an app.
Perhaps the most comprehensive asthma disease management “connected add-on” device for inhalation therapy with pMDI asthma inhalers was the SmartMist (Aradigm Corporation) approved by FDA in 1996. The device recognized from the barcode the nature of the inhaler, reminded the user of the last time they were using that medication, and recorded the date and time of the new dosage event. It only actuated the metered dose inhaler if the correct inspiratory flow rate was achieved early during inspiration and then provided visual guidance for maintenance of the flow rate in a preprogrammed range and encouraged the subject to take a full breath. Moreover, the device also had the capability to measure lung function. The information from SmartMist could be downloaded into a computer to track adherence over time and effect of compliance on lung function (Gonda et al., 1998).
Although the sensor devices can be added as a separate item to already approved inhalers, there have been recent approvals of inhalers in which the connectivity is built into the approval process or, indeed, is an integral part of the device.
Teva obtained approval for three “digital” dry powder inhalers containing albuterol, fluticasone propionate, or a combination of the latter with salmeterol (https://www.businesswire.com/news/home/20200921005170/en/).
All three inhalers are based on the Digihaler technology that contains Bluetooth connectivity via phone to a mobile app. The products may be used to inform the patients or their parents if the patient is a child and their healthcare providers about how often the devices have been used, measure inspiratory flow rates during dose administration, and determine whether inhalation technique may need improvement.
The Enerzair Breezhaler (Novartis) (European Medicines Agency, 2020) is a dry powder inhaler containing the triple combination of indacaterol, glycopyrronium bromide, and mometasone for the treatment of poorly controlled asthma. It comes with an optional electronic sensor developed by Propeller Health that records the patient’s use of the medication and then sends this data to the patient’s smartphone or other mobile device.
H. Conclusions
New disease indications and weaknesses of approved therapies, such as existence of significant numbers of patients who are refractory to their current medications, will most likely drive product innovation in the inhalation field. The ability to deliver higher doses in a convenient manner opens the door for less potent drugs provided that the high doses do not cause tolerability or safety issues as well as unacceptable costs. The technologies driving higher efficiency of delivery also tend to result in less intrasubject and intersubject variability and better lung targeting.
The miniaturization of electronics and universal access to high-speed internet enables collection of information about the inhaler use from their users. When they are employed during the new product development, there should be a relatively smooth transition to postapproval use. The collected information can be in principle integrated into large databases in which it can be analyzed and used for better healthcare for the participating individuals and the population in general. Most of the current applications are focused primarily on monitoring and improving adherence to the dosage regimens and the user technique, but it is likely that we will see in the future a greater integration of various disease management tools, including respiratory diagnostics with these connected inhalers, similar to the development of “smart” insulin pens used for the management of diabetes in conjunction with continuous glucose monitoring (Jendle et al., 2021).
XVI. Current Clinical Developments, Thoughts on Future Opportunities, and Challenges
Although modern inhaled medicines made many valuable contributions to the care of patients with respiratory diseases as described in this review, there are still significant opportunities to make improvements in the indications wherein products are already available as well as to expand to many other areas where there is underserved medical need.
Development of future inhaled therapies will hopefully benefit both from the experience of the advances as well as the failures of the past. The selection of suitable candidates can be guided by historical precedents based on the properties of approved inhalation products (Strong et al., 2018) using the principles described in various sections of the review, especially Section III.
The receptor specificity may be the first consideration in the early design of a new compound to be delivered by inhalation (as detailed, for example, in Section V). Potency of the drug is a key consideration because of the limitation on the amount of material that for tolerability, safety, and practical reasons can be delivered by inhalation. This goes hand in hand with the duration of action, as a short duration may necessitate impractically frequent administration or excessively high less-frequent doses to retain sufficient activity.
The selection criteria will favor drugs that have high “lung selectivity” characterized by receptor binding in the respiratory tract. Binding to other respiratory materials that can act as the reservoir for the active ingredient in the vicinity of the target may also be beneficial. Minimum absorption of the drug into the systemic circulation or rapid systemic clearance to minimize undesired biologic effects is naturally desirable.
From a practical perspective, for the transition from basic research to first in-human study, the “no observed adverse effect level” (i.e., the maximum concentration or dose of drug at which adverse local and systemic side effects in animal models are absent) (Tepper et al., 2016) has to afford a safety margin for first dose in humans that is likely to result in beneficial pharmacological effects for the target patient population. The latter may be hard to determine in the absence of any efficacy studies in humans and, more often than not, lack of validated in vitro or animal models of human respiratory diseases for the particular indications of interest. Such large uncertainty over the efficacious doses in humans often remains until at least the phase 2 efficacy studies. Finding the right balance between efficacious doses that do not cause unacceptable adverse reactions is central in preparations for phase 3 studies.
The choice of the animal species is therefore important because one of the key purposes of preclinical toxicology is to look for toxicity signals that should be monitored in humans. No animal species is a perfect model of the human respiratory tract and especially the respiratory tract of humans with lung disease.
Rats are a particularly sensitive animal favored for that reason by regulatory authorities for inhalation toxicology studies; their respiratory clearance mechanisms have a limited capacity with a threshold generally viewed as being lower than in other species, including humans. This may result in artifacts with poorly soluble drugs or formulations in rat inhalation toxicology studies caused by the overload of their normal clearance mechanisms, which then provokes an inflammatory reaction and may end up with fibrosis and ultimately cancer (Oberdörster, 2002). The use of additional or alternative models that can provide toxicity signals relevant to humans is therefore advisable.
Another example of the need for extra caution in the choice of the animal models and their interpretation is the testing of toxicity of inhaled recombinant human proteins or biologics, such as DNA and mRNA, wherein the differences in the structure of human and animal proteins may cause the animal’s immune system to recognize the new therapeutic protein as “foreign” and cause an adverse effect in the animal (Green, 1994).
Of course, animals cannot be trained to inhale the test articles from the devices that are used by humans! Instead, special aerosol generation and exposure systems have been developed. The estimated lung doses in animals are then used to determine the doses deemed to be safe for humans using a mixture of experimental data and theoretical deposition values and species-dependent multipliers to allow for adequate safety margins for dosing humans (Tepper et al., 2016). However, the scientific shortcomings of these animal models (Sécher et al., 2020) are being increasingly recognized not just for toxicology but even more so for efficacy modeling. Initiatives are being taken to supplement and replace these by in vitro and in silico alternatives (Movia and Prina-Mello, 2020).
Changes of formulations and devices during the clinical development raise questions about the validity of the prior findings and the possibility of having to do clinical comparability studies before advancing to the next stage of development. It is therefore preferable to make the choices of the device and the formulation relatively early in clinical development. The final configuration of the product used in the pivotal trials is needed to support regulatory approvals.
To increase the overall probability of success of the product in the commercial phase, many other considerations are therefore taken into account prior to entry into late-stage development, such as 1) the evidence that the pharmaceutical formulation is likely to have sufficient long term stability, 2) security of the supply chain for the drug and other formulation ingredients with adequate quality attributes, and 3) manufacturability of the formulation on commercial scale. Similar considerations apply to the device choice. The regulatory status of the formulation ingredients as well as the device are scrutinized to avoid the risk that a regulatory failure of a component could result in the overall failure of the product.
The intellectual property consideration and the overall cost of manufacturing of the drug product versus the socioeconomic value it is to provide are also important points in the competitive commercial environment for pharmaceutical products. A guide for the points to consider is in Table 17.
Points to Consider in Inhaled Drug Development
The probabilities of success from a new therapeutic idea to an approved product are still disappointingly low for respiratory medicines: Even once the product enters phase 1, the failure rate is over 90%, and it remains over 30% from phase 3 to approval (BIO, 2016). Although the discussion above provides some important considerations used in the decision making, this review would be incomplete without mentioning the critical role of individuals and teams involved in this long and complex process of new product development that, in addition to scientific competence, requires much perseverance and personal career risk taking because even after many years of research and development, the product could fail to be approved. This process is long with many serious obstacles and the need to make hard decisions in the face of great uncertainties. Although the history of discovery, development, and approval of inhaled cromones (Howell, 2005) is one of the few that has been published, there are probably many more that may not be very dissimilar.
Ultimately, the success of a product will depend on its acceptance and use by the target population of patients. Continuous investigations of the ability of the patients to use and maintain their product correctly according to the instructions for use is therefore an essential part of inhalation drug product development.
Nonadherence with prescribed dosage regimens and its healthcare consequences impact all routes of administration, but with inhalation there are additional causes associated with the incorrect technique to use the medications (Lavorini et al., 2014). Improvements in drugs, formulations, and devices to address these weaknesses are underway. We already have capability for real-time feedback using internet-connected devices that can “nip the problem in the bud” and also collect information to link the level of daily adherence to therapeutic outcomes (Section XV; Gonda, 2019a).
Both acute and severe respiratory infections remain a major burden on global health (Forum of International Medical Societies, 2017). Tuberculosis is an example of a severe chronic lung infection wherein intuitively inhaled medicines could overcome the toxicity associated with systemic delivery of antituberculosis drugs. The success of inhaled liposomal amikacin in a related disease—nontuberculous mycobacterial lung infections (Section IX) —would suggest that this may be possible.
Idiopathic pulmonary fibrosis (IPF), hereditary emphysema due to the α-1 antitrypsin deficiency and non–cystic fibrosis bronchiectasis are just a few examples of less common but severe respiratory diseases wherein development of inhaled therapies has been initiated, but no products have been approved yet.
There was a promising wave of development of inhaled biologics in the 1990s, but the field stagnated since then. There are no fundamental reasons why biologics could not be successfully delivered by inhalation (Gonda, 2019b), as demonstrated by dornase α for cystic fibrosis (Section X). Several biologics are being tested now in clinical trials (Table 18).
Examples of novel inhaled therapies in clinical developmenta
DNA and RNA delivery to the respiratory tract may be optimal therapies for some disorders (Alton, 2019). Respiratory delivery of bacteriophages, viruses, and microbes to restore a healthy microbiome is stretching the limits of our current knowledge, but isolated data in humans in the case of a severe necrotizing infection with resistant bacteria is quite promising (Maddocks et al., 2019).
An overview of innovative inhaled therapies in recent clinical development is presented in Table 18 based on a search in July 2021 in the two largest clinical trials registers, the United States and EU. It is apparent that many of these development use drugs already approved by other routes of administration either for the same or other indications. This “repurposing” or “repositioning” of therapies significantly reduces the systemic toxicity risk. The development is very much accelerated, as most if not all of the basic research is already completed (Cipolla and Gonda, 2015; Chan and Cipolla, 2018). An even greater risk reduction and acceleration is achieved, of course, if a drug is already approved for use via the inhalation route, and the novelty is its use for a new indication. An important recent example is the treatment of early stages of COVID-19 with inhaled budesonide (Ramakrishnan et al., 2021).
Indeed, the SARS-CoV-2 prompted an impressive effort in research and development of inhaled therapies for prophylaxis and treatment of COVID-19, as the initial infection starts usually in the respiratory tract and may end up with predominantly severe respiratory problems (e.g., pneumonia and RDS).
A number of inhaled formulations of anti-infectives are being tested in humans, such as itraconazole, teicoplanin, voriconazole, and a new RSV polymerase inhibitor, PC786.
For IPF, one of the established oral treatments, pirfenidone (Esbriet), is in clinical development as an inhaled formulation (AP01). TD-1058 is a novel inhaled approach for this indication.
The rare diseases CF and primary ciliary dyskinesia (PCD) are targeted by a number of new therapies by inhalation: epithelial sodium channel inhibition (ENaC), neutrophil elastase inhibition, and mucus modifying agents (Oligo-G, SNSP113).
We also note advances in the obstructive lung diseases asthma and COPD with innovative inhaled treatments typically targeting subgroups with severe disease. Inhaled capsaicin and a novel nociceptor-targeting approach (NOC-100) are investigated as chronic and acute cough treatments.
For treatment of PAH, clinical stage research using new mechanisms of action, such as guanylate cyclase stimulation and platelet-derived growth factor receptor antagonism, is underway.
New products using inhalation route for systemic effects are also being explored, for example, conversion to sinus rhythm in recent onset atrial fibrillation using an inhaled formulation of the antiarrhythmic flecainide. Inhaled formulations of cannabinoids are studied in cancer and fibromyalgia pain and a serotonin receptor antagonist for relief from nausea and vomiting syndromes.
We note the fascinating innovative therapeutic repurposing of the asthma drug sodium cromoglycate (Section VIII) for systemic delivery via inhalation for the treatment of postischemic stroke, amyotrophic lateral sclerosis, and early Alzheimer disease. The rationale for these studies is based on presumed qualities as a mast cell stabilizer, neuroinflammatory microglia modulator, and inhibitor of β-oligomerization.
The need to have technologies that can be used to make accessible, affordable, and easily used products for these purposes for large numbers of subjects with varied needs and capabilities certainly provides a fertile ground for innovations in inhaled medicines.
Acknowledgments
The authors would like to thank Jacqueline Perry [the American Society for Pharmacology and Experimental Therapeutics (ASPET)] for her excellent assistance with the manuscript preparation and submission and the reviewers for their helpful comments.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Anderson, Atkins, Bäckman, Cipolla, Clark, Daviskas, Disse, Entcheva-Dimitrov, Fuller, Gonda, Lundbäck, Olsson, Weers.
Footnotes
- Received June 18, 2020.
- Accepted August 6, 2021.
This paper received no external funding.
Anderson previously received 13.2% share of the royalties received by the Sydney Local Health District (the owner of the intellectual property) for the use of mannitol to promote mucociliary clearance, and she is the inventor on the patent. She also owns shares of Pharmaxis Ltd worth less than US $5,000 that she purchased herself. Cipolla is a paid employee of Insmed Inc. Two of Insmed’s products were discussed in these sections, including amikacin liposome inhalation suspension (ALIS, ARIKAYCE) and inhaled treprostinil palmitil. Daviskas received 16% share of the royalties received by the Sydney Local Health District (about US $10,000 per annum). She also owns shares of Pharmaxis worth less than US $3,000 that she purchased herself. Disse received compensation for consultant services from Boehringer Ingelheim, Actelion and Sanofi-Aventis in recent years. Olsson is owner of Mimetikos AB mentioned in the text.
Abbreviations
- ACh
- acetylcholine
- AIDS
- acquired immunodeficiency syndrome
- APSD
- aerodynamic particle size distribution
- AUC
- area under the curve
- BE
- bioequivalence
- BID
- twice-a-day dosing
- CF
- cystic fibrosis
- CFC
- chlorofluorocarbon
- COPD
- chronic obstructive pulmonary disease
- COVID-19
- disease caused by the infection with the SARS-CoV-2 virus
- DPI
- dry powder inhaler
- DPPC
- dipalmitoyl phosphatidylcholine (1,2-dipalmitoyl-sn-glycero-3-phosphocholine)
- EMA
- European Medicines Agency
- ENaC
- epithelial sodium channel
- ETT
- endotracheal tubing
- EU
- European Union
- FDA
- Food and Drug Administration
- FEV1
- forced expiratory volume in 1 second
- FiO2
- fraction oxygen content in the inspired air oxygen
- FVC
- forced vital capacity
- GA
- gestational age
- GINA
- Global Initiative for Asthma
- GCS
- glucocorticosteroid
- GOLD
- Global Initiative for Chronic Obstructive Lung Disease
- GSD
- geometric standard deviation
- HFA
- hydrofluoroalkane
- HFC
- hydrofluorocarbon
- HRQoL
- health-related quality of life
- ICS
- inhaled corticosteroid (or inhaled glucocorticosteroid)
- IL
- interleukin
- IPF
- idiopathic pulmonary fibrosis
- LABA
- long-acting β2 adrenoceptor agonist
- LAMA
- long-acting muscarinic antagonist
- L-dopa
- levodopa
- mAChR
- muscarinic acetylcholine receptor
- MDI
- metered dose inhaler
- MHRA
- Medicines and Health Care products Regulatory Agency
- MMAD
- mass median aerodynamic diameter
- nCPAP
- nasal continuous positive airway pressure
- NRT
- nicotine replacement therapy
- NTM
- nontuberculous mycobacteria
- OAD
- oral antidiabetic drug
- PAH
- pulmonary arterial hypertension
- PBPK
- physiology-based pharmacokinetic
- PCD
- primary ciliary dyskinesia
- PCL
- periciliary fluid layer
- PD
- Parkinson disease
- PDE5
- phosphodiesterase type 5
- PJP
- P. jiroveci pneumonia
- PK
- pharmacokinetics
- pMDI
- pressurized metered dose inhaler
- p.o.
- per os
- QD
- every day
- rhDNase
- recombinant human DNase dornase α
- RDS
- respiratory distress syndrome
- RSV
- respiratory syncytial virus
- SABA
- short-acting β2 adrenoceptor agonist
- SAMA
- short-acting muscarinic antagonist
- SARS-CoV-2
- severe acute respiratory syndrome coronavirus 2
- SCG
- sodium cromoglycate
- SMI
- soft mist inhaler
- TLD
- total lung dose
- Tmax
- time at which the Cmax occurs
- TOBI
- tobramycin inhalation
- UDV
- unit dose vial
- URT
- upper respiratory tract.
- Copyright © 2022 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Visual Overview
- Abstract
- I. Introduction
- II. Why Inhaled Medications?
- III. Critical Attributes of Orally Inhaled Drug Products for Treatment of Respiratory Diseases
- IV. β-2 Receptor Agonists
- V. Muscarinic Acetylcholine Receptor Antagonists
- VI. Glucocorticoids
- VII. Combination Products
- VIII. Cromones–Sodium Cromoglycate and Nedocromil
- IX. Anti-Infectives
- X. Recombinant Human Deoxyribonuclease
- XI. Mannitol
- XII. Prostacyclins
- XIII. Lung Surfactant
- XIV. Drugs Delivered by Inhalation for Systemic Therapies
- XV. New Developments in Inhalation Technology
- XVI. Current Clinical Developments, Thoughts on Future Opportunities, and Challenges
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters