


Abstract
Despite nearly 30 years of development and recent highlights of nitric oxide (NO) donors and NO delivery systems in anticancer therapy, the limited understanding of exogenous NO’s effects on the immune system has prevented their advancement into clinical use. In particular, the effects of exogenously delivered NO differing from that of endogenous NO has obscured how the potential and functions of NO in anticancer therapy may be estimated and exploited despite the accumulating evidence of NO’s cancer therapy–potentiating effects on the immune system. After introducing their fundamentals and characteristics, this review discusses the current mechanistic understanding of NO donors and delivery systems in modulating the immunogenicity of cancer cells as well as the differentiation and functions of innate and adaptive immune cells. Lastly, the potential for the complex modulatory effects of NO with the immune system to be leveraged for therapeutic applications is discussed in the context of recent advancements in the implementation of NO delivery systems for anticancer immunotherapy applications.
SIGNIFICANCE STATEMENT Despite a 30-year history and recent highlights of nitric oxide (NO) donors and delivery systems as anticancer therapeutics, their clinical translation has been limited. Increasing evidence of the complex interactions between NO and the immune system has revealed both the potential and hurdles in their clinical translation. This review summarizes the effects of exogenous NO on cancer and immune cells in vitro and elaborates these effects in the context of recent reports exploiting NO delivery systems in vivo in cancer therapy applications.
I. Introduction
Nitric oxide (NO) is an endogenous gaseous molecule that plays a myriad of biologic and pathophysiological functions involved in cardiovascular homeostasis, neurotransmission, angiogenesis, immune response, and apoptosis (Fukumura et al., 2006; Carpenter and Schoenfisch, 2012). Inspired by its in vivo functions (Fukumura et al., 2006; Carpenter and Schoenfisch, 2012) as well as its biocompatibility (Fukumura et al., 2006; Kim et al., 2014) originating from its role as an endogenous signaling molecule and rapid degradation into nontoxic ions after reaction, there has been continuous effort toward the development of anticancer drugs that leverage NO donors (Hrabie and Keefer, 2002; Wang et al., 2002) that are defined as “NO releasing small molecules or functional groups” (Kim et al., 2014). The most widely explored paradigm of NO donors in anticancer therapy seeks to exert NO’s cytotoxic effects on cancer cells by achieving burst intracellular release of NO. For example, O2-(2,4-dinitrophenyl) 1-[(4-ethoxycarbonyl)piperazin-1-yl]diazen-1-ium-1,2-diolate) (JS-K) has been intensively explored for this purpose (Ren et al., 2003; Shami et al., 2003; Shami et al., 2006; Udupi et al., 2006; Kiziltepe et al., 2007; Chakrapani, Goodblatt et al., 2008; Chakrapani, Kalathur et al., 2008; Simeone et al., 2008; Kitagaki et al., 2009; Liu et al., 2017; Liu, Huang et al., 2018; Zhao et al., 2019) because it allows the burst release of NO in response to the high intracellular redox environment of cancer cells. In addition, several NO delivery systems have been developed with the goal of delivering a high concentration of NO donors to the tumor microenvironment to elicit NO mediated cytotoxicity as an anticancer therapeutic approach (Kumar et al., 2010; Park, Im et al., 2019; Kim, Suh et al., 2022). Although various NO donors and delivery systems have demonstrated anticancer effects in vitro as well as in several in vivo xenograft tumor models (Ren et al., 2003; Shami et al., 2003; Shami et al., 2006; Udupi et al., 2006; Kiziltepe et al., 2007; Chakrapani, Goodblatt et al., 2008; Chakrapani, Kalathur et al., 2008; Simeone et al., 2008; Kitagaki et al., 2009; Kumar et al., 2010; Stevens et al., 2010; Xu et al., 2015; Fan et al., 2017; Liu et al., 2017; Liu, Huang et al., 2018; Zhao et al., 2019), their clinical translation is currently limited due to observations that at concentrations lower than the threshold that elicits cytotoxicity, NO can also accelerate tumor progression (Mocellin et al., 2007; Kim et al., 2014; Kim et al., 2017). In addition to its cytotoxic functions, NO also has drug sensitizing effects (Mocellin et al., 2007; Fan et al., 2015; Kim et al., 2017; Deng et al., 2018; Hu et al., 2018; Pramanick et al., 2018; Wang et al., 2019; Ding et al., 2019; Feng et al., 2019; Wang et al., 2019; Zhang et al., 2018; Gao et al., 2020), which have accelerated the rapid progress of NO donors or delivery systems for use in combination with other therapeutic modalities including chemotherapy, radiation therapy, photothermal therapy, and photodynamic therapy (Mocellin et al., 2007; Fan et al., 2015; Kim et al., 2017; Deng et al., 2018; Hu et al., 2018; Pramanick et al., 2018; Wang et al., 2019; Ding et al., 2019; Feng et al., 2019; Wang et al., 2019; Zhang et al., 2018; Gao et al., 2020). For example, NO donor RRx-001 [N-(bromoacetyl)-3,3-dinitroazetidine] was evaluated for its potential as a radiosensitizer in clinical trials (Kim et al., 2016; Oronsky, Scicinski, Cabrales et al., 2016; Oronsky, Scicinski, Ning et al., 2016). However, sensitizing effects as well as cytotoxic functions of NO donors and delivery systems have been investigated almost entirely at an in vitro level or in immune-deficient mouse models. As a result, NO’s effects on the immune system have been underexplored.
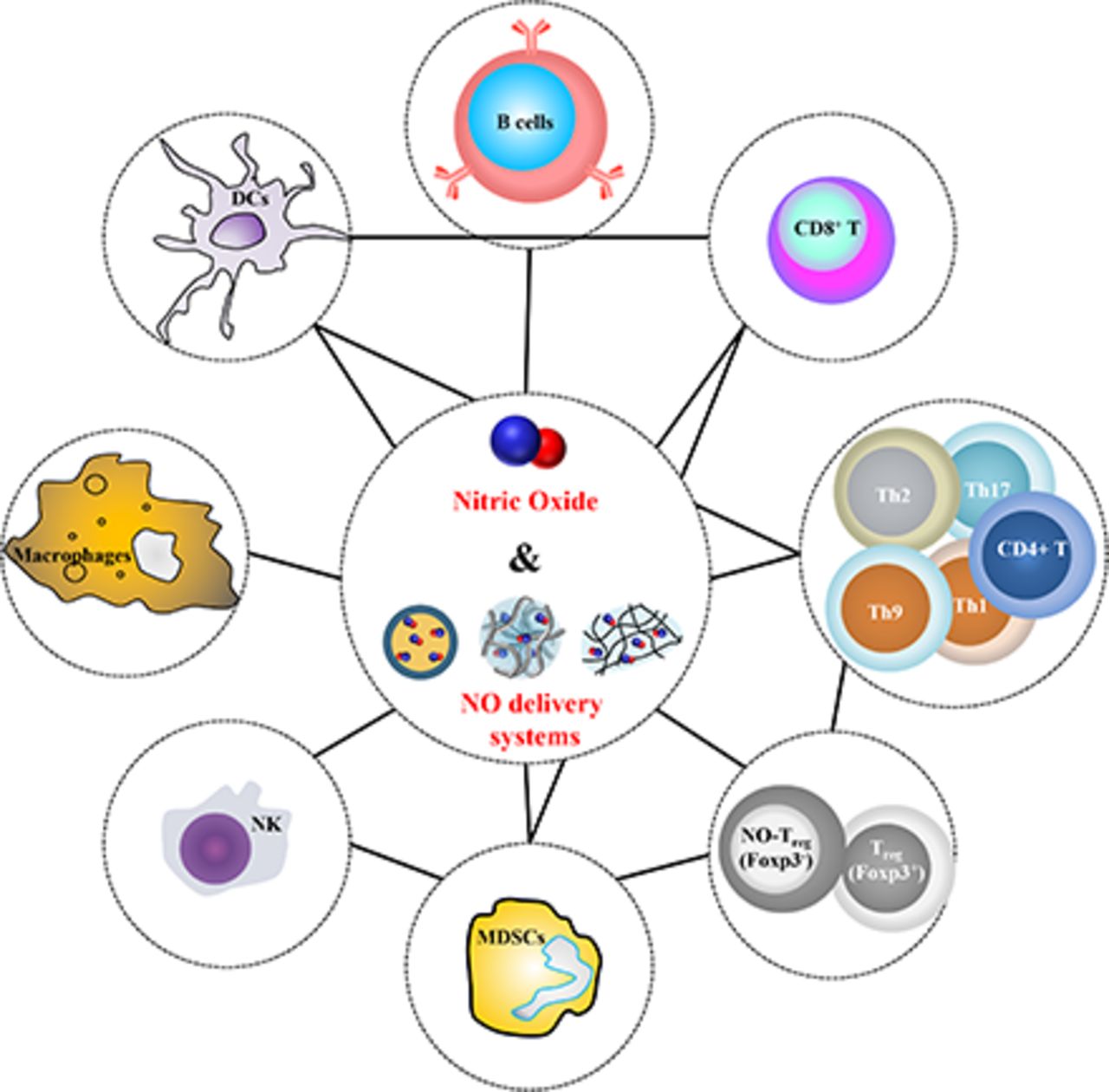
A substantial body of literature has reported endogenous NO’s pivotal role in various immunologic mechanisms. However, the effects of exogenously delivered NO on tumor progression have not summarily been presented. In this review, major NO donor and delivery system classes are described. A synthesis of the existing literature landscape of NO’s known direct effects on cancer cells and immune cells, including dendritic cells (DCs), macrophages, B cells, CD8+ T cells, CD4+ T cells, regulatory T cells (Tregs), natural killer (NK) cells, and myeloid-derived suppressor cells (MDSCs), based on in vitro studies is then presented. Finally, contemporary studies developing NO delivery systems are summarized, revealing the complex immune networks regulated by NO and its capacity to potentiate anticancer immunotherapy.
II. NO-donors
Numerous NO donors have been developed for medicinal purposes (Hrabie and Keefer, 2002; Wang et al., 2002). The characteristics of such NO donors, which vary in their chemistry and mechanism of NO donation, are herein presented.
A. Nitrates/Nitrites
The oldest class of NO donors is comprised of organic nitrates and nitrites that have a generic structure RONO2 and RONO, respectively. Representative examples include glyceryl trinitrate and amyl nitrite, which have been pharmacologically used to treat angina for more than a century. Organic nitrates/nitrites can be metabolized by reacting with cellular cysteine and glutathione (GSH) (Needleman and Krantz, 1965; Needleman et al., 1971; Ignarro and Gruetter, 1980; Horowitz et al., 1983; Wang et al., 2002; Thatcher et al., 2004). Although decomposition of organic nitrates/nitrites to release NO was reported to be associated with the enzymatic reaction with cytochrome P450 and/or glutathione S-transferase (GST) (Schroder, 1992; Seth and Fung, 1993; McDonald and Bennett, 1993; Meyer et al., 1994; Kenkare et al., 1994; Wang et al., 2002; Thatcher et al., 2004), it is currently believed to be associated with mitochondrial aldehyde dehydrogenase-2, independent of cytochrome P450 (Wenzel et al., 2007; Mayer and Beretta, 2008; Daiber and Munzel, 2015; Neubauer et al., 2015; Lopez et al., 2021). Organic nitrates and nitrites are synthesized by reacting alcohols with nitric acid and nitrous acid, respectively (Wang et al., 2002; Omar et al., 2012). Because of their easy and simple chemistry, nitrates/nitrites have been used to develop hybrid prodrugs to release NO and nonsteroidal anti-inflammatory drugs (NSAID) (Thatcher et al., 2004; Dunlap et al., 2008), as well as incorporated into NO delivery systems.
B. Amidoximes/Oximes
Amidoximes and oximes, which have the generic structure R1C(= N-OH)R2, mimic the N-hydroxy-L-arginine, which is a kind of amidoxime produced as an intermediate of enzymatic reactions between endogenous NO synthases (NOS) and L-arginine substrates (Blatt, 1938; Tavakol and Arshadi, 2009; Belmar et al., 2013; Bohle et al., 2013; Novikov and Bolotin, 2017; Sahyoun et al., 2019).
Amidoximes are synthesized by reacting nucleophile hydroxylamine with nitriles, thioamide, amidine hydrochloride, hydrazide imide, iminoether, or imidoylbenzo-triazole (Tiemann, 1884; Warburton, 1966; Bjoklund and Coburn 1980; Katritzky et al., 2006; Ranjbar-Karimi et al., 2018; Sahyoun et al., 2018; Sahyoun et al., 2019), by reacting oximinoether and hydroximic acid chlorides with ammonia (Lossen W, 1889; Sahyoun et al., 2019), or by reducing nitrosolic acids with hydrogen sulfide (Wieland and Bauer, 1906; Sahyoun et al., 2019). Oximes on the other hand are prepared via condensation of aldehydes or ketones with hydroxylamine (Sahyoun et al., 2019) or by introducing hydroxyimino group into the diene moiety using nitrite under acidic conditions (Wang et al., 2002). Amidoximes and oximes can be decomposed to release NO via an oxidation process with singlet oxygens (Öcal and Erden, 2001) and enzymatic reaction with cytochrome P450 enzymes, the reduced form of NADP, and O2 (Andronik-Lion et al., 1992; Boucher et al., 1992; Mantyla et al., 2004). However, NOS does not contribute to the oxidation process of amidoximes and oximes except the L-arginine and N-hydroxy-L-arginine (Moali et al., 2000; Caro et al., 2001). Oximes that have been explored in biomedical applications include NOR-1, NOR-3, and NOR-4.
C. S-nitrosothiols
S-nitrosothiols are classified as analogs of O-nitroso compounds or organic nitrites, which have the generic structure R-S-N = O (RSNO), an electronegative oxygen and sulfur, the latter of which is slightly more electrically negative than nitrogen (Hogg, 2002). Nitrosation makes α-proton and α-carbon downfield shifted in 1H and 13C nuclear magnetic resonance spectra. S-nitrosothiols also exhibit characteristic UV-visible absorbance in 255 to 261 nm (π→π* transition), 330 to 350 nm (n0→π* transition), and 550 to 600 nm (nN→π* transition). The third band determines the color of S-nitrosothiol compounds from red to green (Wang et al., 2002; Zhang et al., 2017). In infrared spectra, N-O vibration and N-S vibration appear in 1430 to 1710 cm−1 and 610 to 685 cm−1 or 1000 to 1170 cm−1, respectively (Zhang et al., 2017).
Formation and metabolism of S-nitrosothiol regulate numerous physiologic functions. As examples, serum albumin is one of the major forms of S-nitrosothiol in blood plasma, and erythrocytes also can contain a S-nitrosothiol modified hemoglobin. These S-nitrosothiols in blood not only maintain vascular tone homeostasis and exert important vasodilation functions with the help of GSH to be reversely converted to S-nitrosoglutathione (GSNO) (Hogg, 2002), but also transfer S-nitrosothiol functional groups to the cells via cystine-mediated transnitrosation (Zhang and Hogg, 2004). On the other hand, GSNO is one of the major intracellular NO donors because GSH is highly abundant in cells, and a thiol in GSH with a simple structure is thermodynamically easier to be reacted with nitronium cation (NO+) produced by nitrosative biologic reactions than thiols in proteins with larger molecular weight and tertiary structures (Massa et al., 2021). Nevertheless, close interactions of NO+, GSNO, and other NO donors with proteins allow S-nitrosylation of proteins (Massa et al., 2021). This post-translational modification plays an important role in signal transduction with the help of dinitrosyl iron complexes–mediated transnitrosation, which modulates the activity and functions of proteins (Konorev et al., 2000; Hogg, 2002; Bosworth et al., 2009; Kevil and Patel, 2010).
S-nitrosothiols can be synthesized via nitrosation by reacting thiol compounds with N-oxides (NOCl, N2O4, N2O3, NO2, HNO2) (Zhang et al., 2017). For example, GSNO can be synthesized by reacting GSH with sodium nitrite (NaNO2) in acidic aqueous conditions (Hart 1985). This aqueous nitrosation method has been the most widely used in synthesizing RSNO and S-nitrosothiol conjugated NO delivery systems, which is beneficial in preventing the unwanted N-nitrosation but is limited for use with material systems that undergo hydrolysis in acidic conditions (Wang et al., 2002; Damodaran et al., 2012; Kim et al., 2014; Zhang et al., 2017). On the other hand, the nonaqueous nitrosation method utilizing tert-butyl nitrite in organic solvents not only can prevent the hydrolysis of synthetic substrates but also restrain spontaneous decomposition of SNO groups during synthesis and purification in aqueous conditions.
The decomposition of SNO groups results in the release of NO, which is dependent on temperature, light, heavy metal ions, and redox conditions. GSH, ascorbic acid, and heavy metal ions including Cu+, Fe2+, Hg2+, and Ag+ induce catalytic release of NO by reducing the RSNO (Wang et al., 2002). Indeed, intracellular concentration of GSH or ascorbic acid leads to a burst NO release by acting as a reducing agent or nucleophile (Wang et al., 2002; Kim et al., 2020). Initial rates of decomposition of RSNO are dependent on its initial concentration because RSH and RS˙ produced during decomposition of RSNO contribute to the autocatalytic reaction (de Oliveira et al., 2002). Likewise, Cu+ ions act as a reducing agent to accelerate the decomposition of RSNO (Stamler and Toone, 2002), however, whether nitrosothiols on proteins and peptides are susceptible to copper-catalyzed decomposition remains unclear since free copper is only very lowly abundant in most tissues (Stamler and Toone, 2002). Accordingly, copper-containing biomaterials have been developed to accelerate NO release from endogenous GSNO (Hwang and Meyerhoff, 2008; Zhou et al., 2021). It should be noted that RSNO is not stable at room temperature, as it undergoes thermal decomposition with homolytic cleavage of S-N bond with 20 to 30 kcal mol−1 of homolytic bond dissociation energy to release NO˙ (Bartberger et al., 2001; Wang et al., 2002). In addition, RSNO exhibits photolytic release of NO under UV light due to the absorption band as previously described (Wang et al., 2002; Marazzi et al., 2012). Although their low thermal- and photostability remain to be addressed, S-nitrosothiols have been widely employed in (pre)clinical applications and mechanistic studies due to the ease of this chemical modification on any type of thiol-containing substance (Kim et al., 2014). In addition to NO delivery systems, S-nitrosothiol prodrugs based on α-S-nitroso-α-phenyl acetic acid have been developed to improve its stability for practical therapeutic applications (Lu et al., 2007).
D. Metal Nitrosyl Complexes
Metal nitrosyl complexes have a generic coordination structure of M-NO where NO binds as a ligand to metals (M) including Fe, Ru, Co, Ni, Cu, Cr, MO, etc. (Wang et al., 2002; Wright and Hayton, 2012). An example of metal nitrosyl complexes is sodium nitroprusside (SNP; Na2[Fe(CN)5NO]) which has been used clinically for the treatment of angina for decades. SNP spontaneously releases NO, which can be accelerated under light irradiation and by reductive conditions as well as changes in oxygen levels, pH, and temperature (Wang et al., 2002; Rose and Mascharak, 2008; Fry and Mascharak, 2011; Xiang et al., 2017). However, reduction of SNP results in the release of cyanide, which is cytotoxic (Butler and Megson, 2002; Wang et al., 2002; Rose and Mascharak, 2008; Fry and Mascharak, 2011; Xiang et al., 2017). Other metal nitrosyl examples include Roussin’s black salt (RBS; [Fe4S3(NO)7]−) and Roussin’s red salt (RRS; [Fe2S2(NO)4]2−), a class of iron-sulfur cluster nitrosyls mimicking iron-sulfur clusters in endogenous enzymes (Wang et al., 2002). RBS and RRS spontaneously release NO, which can be accelerated by light exposure, free thiols, and high temperature (Bourassa et al., 1997; Butler and Megson, 2002; Wang et al., 2002; Rose and Mascharak, 2008). Another example is dinitrosyl iron complexes (DNICs) that contain two NO ligands [Fe(NO)2L2]x+ where L is a ligand that can be -SR, -NR2, or -OR (Butler and Megson, 2002; Wang et al., 2002). DNICs also spontaneously release NO, which can be increased by light exposure, free thiols, acids, and elevated temperature (Wang et al., 2002).
Metal nitrosyl complexes have been actively explored in the development of photoresponsive NO delivery systems owing to their distinctive photolytic NO release behaviors via dπ(M) → π*(NO) transitions (Mascharak, 2012; Ford, 2013; Kim et al., 2014). However, most metal nitrosyl complexes exhibit photolytic NO release in response to the UV and/or visible light that are toxic and/or not appropriate to penetrate to tissues (Mascharak, 2012; Ford, 2013; Xiang et al., 2017). Accordingly, there has been demand for NO donors that are responsive to near-infrared (NIR) light that is relatively biosafe and can penetrate tissues at greater depths relevant to (pre)clinical applications. The wavelength of light required to induce photolytic NO release from metal nitrosyl complexes is dependent on the coordinated ligands and transition metal of the donor (Rose and Mascharak, 2008; Fry and Mascharak, 2011; Xiang et al., 2017). Therefore, several metal nitrosyl complexes have been developed to enable metal nitrosyl complexes to release NO in responsive to the longer wavelength of light by using various polydentate ligands and transition metals to tightly bind NO and ligands (Wecksler et al., 2004; Eroy-Reveles et al., 2008; Rose and Mascharak, 2008; Akl et al., 2016; Xiang et al., 2017). In addition, these novel metal nitrosyl complexes show improved stability of metal nitrosyl complexes during storage and the prevention of ancillary ligand loss to induce nonspecific cytotoxicity during their decomposition (Rose et al., 2008; Kim et al., 2014; Akl et al., 2016; Xiang et al., 2017).
E. Sydnonimines
Sydnonimines are meso-ionic heterocyclic compounds, that include molsidomine and 3-morphorlinosydnonimine (SIN-1). In particular, molsidomine that is used as an oral anti-anginal drug is a prodrug that undergoes deacetylation and ring cleavage to release SIN-1 via enzymatic and nonenzymatic reaction (Wang et al., 2002; Granik, 2010). SIN-1 spontaneously decomposes to release NO to exert its biologic effects by consuming oxygen. In addition, light irradiation can improve the release of NO from SIN-1 in an oxygen-dependent manner (Ullrich et al., 1997). Because of the simultaneous generation of superoxide anion (O2-˙) capable of reacting NO during decomposition of SIN-1, sydnonimines are also peroxynitrite (ONOO−) donors. Inspired by therapeutic activity of SIN-1 and clinical use of molsidomine, several SIN-1 prodrugs have been developed by conjugating 5-imines of SIN-1 with substrates responsive to enzymatic degradation or reductive conditions (Cai et al., 2005; Kim, Suh et al., 2022). These SIN-1 prodrugs show high storage stability and exhibit a stimuli-responsive NO release behaviors, which are beneficial characteristics for clinical applications.
F. Diazeniumdiolates
Diazeniumdiolates, the abbreviated name of diazen-1-ium-1,2-diolates and frequently referred to as NONOates, have a structure of X-N(O) = N-O− with a zwitterionic electropositive diazen group (N = N) and two electronegative oxygen atoms (Hrabie and Keefer, 2002; Kim et al., 2014). A diazeniumdiolate conjugated to the carbon (X = C) is called as C-diazeniumdiolate, whereas its conjugation to nitrogen (X = N) is categorized as N-diazeniumdiolate. N-diazeniumdiolate is a synthetic compound not found in nature, while C-diazeniumdiolate is (Hrabie and Keefer, 2002). Acidified nitrite similar with the aqueous nitrosation method for S-nitrosothiols is generally employed to synthesize C-diazeniumdiolate on primary amine or oximes. However, NO (g) under high pressure and strong basic conditions can produce C-diazeniumdiolate as well as N-diazeniumdiolate. O1- or O2-protected diazeniumdiolates can also be developed when the first or second position of oxygen is protected via alkylation, respectively. While O1-protected diazeniumdiolates are only found in C-diazeniumdiolates, O2-protected diazeniumdiolates are available in both C- and N-diazeniumdiolate. Despite N-diazeniumdiolate being less stable than C-diazeniumdiolate, only N-diazeniumdiolates have been widely used in the development of NO donors and delivery systems because most C-diazeniumdiolates do not release NO but instead produce nitrous oxide. In addition, N-diazeniumdiolates on primary amines are much less stable than those on secondary amines (Drago and Karstetter, 1961; Hrabie and Keefer, 2002). Accordingly, herein only the N-diazeniumdiolates formed on secondary amines are discussed in detail.
N-diazeniumdiolates are generally synthesized by reacting secondary amine-containing materials with NO (g) under high-pressure (5 − 10 atm) and strong basic conditions using sodium methoxide (NaOCH3) (Hrabie and Keefer 2002; Besson et al., 2009; Nguyen et al., 2010; Kim et al., 2011; Hong et al., 2013). Electron-withdrawing N-diazeniumdiolates make α-proton and α-carbon downfield shifted in 1H and 13C nuclear magnetic resonance spectra (Kim et al., 2011), and N1s and C1s peaks in the X-ray photoelectron spectroscopy (Hong et al., 2013). N-diazeniumdiolates also exhibit a characteristic UV-visible absorbance in 220 to 250 nm (Hrabie and Keefer 2002; Besson et al., 2009; Kim et al., 2011). In the infrared spectra, N-N, N-O, and N = N stretches and NO vibrations appear in 1000 to 1070 cm−1, 1480 to 1540 cm−1, 1390 to 1410 cm−1, 1620 to 1640 cm−1, and 1735 cm−1, respectively (Keefer et al., 2001; Hrabie and Keefer 2002; Besson et al., 2009; Nguyen et al., 2010; Hong et al., 2013). The decomposition of N-diazeniumdiolates is initiated by protonation of secondary amines bearing a diazeniumdiolates group, which follows acid catalyzed first-order kinetics (Davies et al., 2001; Hrabie and Keefer 2002). The NO release kinetics of N-diazeniumdiolates are significantly affected by the compound’s molecular structure and functional groups that influence the hydrogen bonding and hydrophobicity with the diazeniumdiolates (Zhang et al., 2003; Horstmann et al., 2002; Kim et al., 2011; Lu et al., 2011).
The low stability of bare N-diazeniumdiolates during storage and in vivo limits their practical application. In addition to the NO delivery systems that will be discussed in the next section, various O2-substituted N-diazeniumdiolates have been developed to address this issue. O2 positions of N-diazeniumdiolates can be protected with the alkyl or aryl groups by using alkyl or aryl halides, alkyl sulfates, and epoxides (Saavedra et al., 1992; Makings and Tsien, 1994; Saavedra et al., 2001; Hrabie and Keefer 2002). The O2-substituted N-diazeniumdiolates are not only stable under physiologic conditions but also exbibit stimuli-responsive NO release behavior if the protecting groups are substrates for specific stimuli including enzymes, redox conditions, etc. Examples include β-lactamase-responsive cephalosporin-3′-diazeniumdiolates (Yepuri et al., 2013), glycosidase-responsive glycosylated diazeniumdiolates (Wu et al., 2001; Valdez et al., 2008), esterase-responsive O2-acetoxymethylated diazeniumdiolates (Saavedra et al., 2000), and NSAID (Velázquez et al., 2007); UV-responsive O2-benzyl derivatives-substituted diazeniumdiolates (Bushan et al., 2002; Ruane et al., 2002; Pavlos et al., 2004); and GST- and GSH-responsive O2-2,4-dinitrophenyl diazeniumdiolates (Saavedra et al., 2001; Chakrapani, Kalathur et al., 2008). Representative O2-substituted N-diazeniumdiolates include JS-K with O2-2,4-dinitrophenyl substitution on the diazeniumdiolate group, which has been widely explored as an anticancer agent (Ren et al., 2003; Shami et al., 2003; Shami et al., 2006; Udupi et al., 2006; Kiziltepe et al., 2007; Chakrapani, Goodbatt et al., 2008; Chakrapani, Kalathur et al., 2008; Simeone et al., 2008; Kitagaki et al., 2009; Liu et al., 2017; Liu, Huang et al., 2018; Zhao et al., 2019).
III. Design of NO Delivery Systems
NO delivery systems aim to deliver NO to tissues and cells of interest at bioactive doses in a controlled release manner, which can be accomplished by incorporating NO donors into drug delivery systems (DDSs). NO donors and DDSs have their own intrinsic physicochemical properties, so their combinations facilitate the development of various and diverse NO delivery systems (Riccio and Schoenfisch, 2012; Kim et al., 2014; Kim et al., 2017; Kim et al., 2017; Yang, Zelikin et al., 2018; Wu et al., 2021; Yang et al., 2021). Fundamentally, NO delivery systems are designed not only to prevent the unintended decomposition of NO donors during storage and in vivo outside of the intended tissue or cell target by protecting the direct exposure of NO donors to environments that can trigger NO release, which include aqueous environments, low pH, oxygen, enzymes, and/or light but also to exhibit stimuli-responsive NO-releasing behaviors at the target sites. Therefore, NO release kinetics of NO donors vary with NO delivery system, which differ widely from those of bare NO donors. Therefore, instead of introducing all examples of specific NO delivery systems, the fundamentals in designing NO delivery systems based on the general physicochemical properties of NO donor and DDS combinations is the focus of the ensuing discussion. It is important note that strategies that indirectly increase endogenous NO without utilizing NO donors will not be covered in this discussion, such as NOS gene delivery (Chen et al., 2002; Cooney et al., 2007; Sharif et al., 2012), arginine substrate delivery (Kudo and Nagasaki, 2015; Cao et al., 2016; Fan et al., 2017; Jiang et al., 2018; Wan et al., 2018; Wang et al., 2019; Ma et al., 2020; Tao et al., 2022), copper catalysis delivery (Hwang and Meyerhoff, 2008; Zhou et al., 2021), approaches that interfere with endogenous NO production mechanisms (Sharma et al., 2013; Guo et al., 2015; Zhang, Lai et al., 2019; Costa et al., 2021), and so on. In addition to NO donors discussed in the previous section, NO delivery systems covered in this section will provide a backdrop for the subsequent discussion of their effects on immune system elaborated in subsequent sections.
A. Physical Loading of NO Donors
Physical loading of NO donors to DDSs can be achieved via simple mixing, which however requires the consideration of physicochemical properties of DDSs and NO donors, such as their hydrophobicity/hydrophilicity and electric charge.
Micelles, liposomes, polymersomes, mesoporous silica nanoparticles (MSN), and polymeric nanoprecipitations are widely used DDSs for physical loading of various drugs, which is generally achieved by hydrophobic or hydrophilic interactions between the DDS matrix and the agent to be incorporated. Accordingly, the hydrophobicity/hydrophilicity of NO donors should be first considered when selecting DDSs for physical loading of NO donors in the design of NO delivery systems. Hydrophobic NO donors include nitrate/nitrite–NSAID, NOR-3, SIN-1 prodrug, and O2-substituted diazeniumdiolates. However, other hydrophilic NO donors can be chemically modified to exhibit hydrophobicity as needed (Huang X et al., 2019). Micelles comprised of amphiphilic block copolymers have a self-assembled hydrophobic core and hydrophilic shell, which allow the hydrophobic NO donors to be loaded into the hydrophobic core (Kumar et al., 2010; Kaur et al., 2013; Pramanick et al., 2018; Kang et al., 2019). Liposomes and polymersomes comprised of phospholipids or amphiphilic polymers consist of an aqueous core and bilayer lipids or polymers, which allow both hydrophilic (Dinh et al., 2005; Tai et al., 2010; Suchyta and Schoenfisch, 2015; Suchyta and Schoenfisch, 2017; Yoshikawa et al., 2019) and hydrophobic NO donors (Nakanishi et al., 2015) to be loaded into either the aqueous core or hydrophobic bilayers, respectively. MSN also exhibit uniform mesoporous channels that can act as a reservoir for hydrophobic NO donors (Li, Song et al., 2020). Nanoprecipitation is a traditional method to physically encapsulate drugs into nanoparticles (NPs) via emulsion methods, which is used in the physical loading of NO donors as well. In detail, oil-in-water emulsion facilitates the load of hydrophobic NO donors into polymeric particles by sequentially emulsifying hydrophobic NO donors and/or hydrophobic polymers in organic phases into hydrophilic polymers or amphiphilic copolymers in aqueous solutions. In contrast, water-in-oil-in-water emulsions enable the loading of hydrophilic NO donors into polymeric particles by sequentially emulsifying hydrophilic NO donors in aqueous solvents into hydrophobic polymers or amphiphilic copolymers in organic solvents, followed by hydrophilic polymer addition in an aqueous solvent (Yang, Hwang et al., 2018).
Charged NO donors including diazeniumdiolates, SNP, RBS, and RRS can also be loaded into DDSs via electrostatic adsorption. In particular, negatively charged RBS and RRS have been widely explored to develop photoresponsive NO DDSs relevant to in vivo applications by adsorbing to positively charged amine functionalized upconversion NPs (Garcia et al., 2012; Tan et al., 2017; Zhao, Hu et al., 2020) and quantum dots (Tan et al., 2014) that are excited by NIR wavelength light that is capable of penetrating into deep tissues, and then the resulting emitted light induces the decomposition of NO donors that are incorporated into the DDSs.
In addition to nano-particulate systems, hydrogels have been widely used to achieve the sustained release of NO donors and NO by simple physical mixing any types of NO donors into the polymeric hydrogel matrix (Shishido et al., 2003; Halpenny et al., 2009; Pelegrino et al., 2018; Zahid et al., 2019; Liu et al., 2020; Santos et al., 2021; Kim, Francis et al., 2022). The release kinetics of NO donors and NO from hydrogels are dependent on not only hydrogel stability but also hydrophobic, electrostatic, and/or hydrogen-bonding interactions between NO donors and polymer matrix that may influence free diffusion.
B. Chemical Conjugation of NO Donors
There are two fundamental strategies that are employed for the chemical conjugation of NO donors to DDSs in the development of NO delivery systems. First, NO donors with functional groups facile to further chemical reactions can be conjugated to DDSs or materials comprising DDSs. For example, a hydroxyl group of hydrophobic nitrobenzene to release NO in response to UV light exposure was conjugated to a block copolymer, which self-assembled into the core of micelles (Naoki et al., 2010). Another example includes a redox responsive SIN-1 prodrug with a thiol group, which were conjugated to thiols in DDSs including albumin, 4-arm poly(ethylene glycol) thiols (4-arm PEG-SH), and silica NPs-SH via a disulfide bond (Kim, Suh et al., 2022). However, this strategy is limited to NO donors that are stable and have reactive functional groups for chemical conjugations. On the other hand, another approach is the direct formation of NO donors on the precursors in DDSs and materials comprising DDSs, which account for the majority of developed NO delivery systems and are discussed in the following text.
Various polymers with thiols or secondary amines have been investigated in the formation of S-nitrosothiols or N-diazeniumdiolates, which have been explored for controlled NO release in a form of unstructured polymers (Park et al., 2013; Lu et al., 2014; Ahonen et al., 2019; Maloney et al., 2021), coating materials (Zhang et al., 2002; Reynolds et al., 2006; DeRosa et al., 2007; Wan et al., 2009; Reynolds et al., 2010), or hydrogels/scaffolds (Lipke and West, 2005; Kim et al., 2011; Damodaran et al., 2012; Schanuel et al., 2015; Yao et al., 2015; Hasan et al., 2021). In particular, block copolymers bearing thiols and secondary amines have been exploited for the development of NO delivering micelles and polymersomes. NO donors on hydrophilic segments of block copolymers are exposed to aqueous environments in the self-assembled structures of micelles and polymersomes (Song et al., 2014; Fan et al., 2021). As S-nitrosothiols and N-diazeniumdiolates are polar functional groups that can spontaneously release NO in aqueous solutions, however, they are generally designed to be protected in hydrophobic core or inner layer of micelles or polymersomes to prevent unintended decomposition during storage and in vivo delivery. It can be generally achieved by forming NO donors in the hydrophobic blocks of polymers, by surrounding the NO donors with hydrophobic moieties, or by forming the NO donors at the end of hydrophobic segments in chemical polymer structures (Duong et al., 2013; Gao et al., 2015; Yu et al., 2015; Schudel et al., 2018; Hou et al., 2019; Park, Im et al., 2019; Wu et al., 2020). Interestingly, block copolymers with a hydrophilic segment and a hydrophilic segment containing secondary amines were reported to be self-assembled into a micelle after the formation of N-diazeniumdiolates on the hydrophilic segment containing secondary amines despite the hydrophilic and polar nature of N-diazeniumdiolates (Jo et al., 2009). However, the mechanism behind the hydrophilic-to-hydrophobic conversion in the formation of N-diazeniumdiolates has remained unclear. In addition to synthetic polymers, proteins as natural polymers can be also used for the development of NO delivery systems. Representative examples of protein-based NO delivery systems include albumin-SNO synthesized by converting a thiol in albumin to S-nitrosothiol, which has been explored as a NO delivery system for antitumor therapy (Katayama et al., 2010; Ishima et al., 2013) owing to the albumin’s favorable circulation times that enable tumor accumulation and active tumor targeting capability (Frei, 2011; Hoogenboezem and Duvall, 2018).
Several inorganic metal NPs including gold NPs and superparamagnetic NPs have been under development as a platform for NO delivery systems by modifying the surface of NPs with materials containing precursors for NO donors via self-assembled monolayer chemistry, followed by the formation of NO donors (Rothrock et al., 2005; Polizzi et al., 2007; Duong et al., 2014; Santos et al., 2016). Nevertheless, the most extensively explored inorganic NO delivery systems are silica materials based on well-defined silane chemistry, such as silica NPs, MSN, and xerogels. In brief, S-nitrosothiols or N-diazeniumdiolates can be functionalized on thiols- or amines-functionalized silane-based DDSs that were prepared via a co-condensation of thiolalkoxysilane or aminoalkoxysilane with alkoxysilanes or via a condensation of thiolalkoxysilane or aminoalkoxysilane on the as-prepared silane-based DDSs (Nablo et al., 2001; Zhang et al., 2002; Marxer et al., 2003; Nablo et al., 2005; Hetrick et al., 2007; Shin et al., 2007; Riccio et al., 2009; Carpenter et al., 2011; Riccio et al., 2011; Carpenter et al., 2012; Riccio et al., 2012). In addition, aminoalkoxysilane can be functionalized with N-diazeniumdiolates or O2-protected N-diazeniumdiolates in advance and then condensed with alkoxysilanes to prepare silane-based NO delivery systems (Hetrick et al., 2008; Shin and Schoenfisch 2008; Hetrick et al., 2009; Stevens et al., 2010; Carpenter et al., 2013; Storm and Schoenfisch, 2013). Furthermore, silane-based NO delivery systems with S-nitrosothiols or N-diazeniumdiolates can be covered with other polymers (Nablo and Schoenfisch 2004; Nablo and Schoenfisch 2005) or calcium phosphate (Choi et al., 2016) or further modified with superhydrophobic fluorine (Storm et al., 2014), which achieve sustained and/or pH-responsive NO release.
NO coating strategies, which result in the modification of materials, nanoparticles, vessel stents, etc., with NO releasing moieties (e.g., NO donors), allow the surface of materials to release NO. Layer-by-layer methods have been employed as one NO coating strategy that is achieved by alternating deposition of positively charged materials containing secondary amine groups and negatively charged materials, followed by formation of N-diazeniumdiolates (Park et al., 2019a; Park et al., 2019b; Tanum et al., 2019). The zwitterionic negative charge of N-diazeniumdiolates contributes to the stabilization of the layers. In addition, hydrogels (Lipke and West, 2005; Kim et al., 2011; Damodaran et al., 2012; Schanuel et al., 2015; Yao et al., 2015; Hasan et al., 2021) and xerogels (Nablo et al., 2001; Marxer et al., 2003; Riccio et al., 2009; Riccio et al., 2012; Storm and Schoenfisch, 2013) chemically conjugating NO donors also exhibit sustained release of NO owing to the reduced diffusion rates in the polymeric matrix, such as hydrogels physically loaded with NO donors (Shishido et al., 2003; Halpenny et al., 2009; Pelegrino et al., 2018; Zahid et al., 2019; Liu et al., 2020; Santos et al., 2021; Kim, Francis et al., 2022). Polycatecholamine-diazeniumdiolates have also emerged as a versatile NO coating strategy (Hong et al., 2013; Park et al., 2016; Adnan et al., 2018; Sadrearhami Z et al., 2019). Polycatechnolamines are synthesized by oxidative self-polymerization of catecholamines including dopamine, norepinephrine, and so on, which can be formed on the surface of any material substrate including metals, polymers, semiconductors, and ceramics, regardless of shape and size (Lee et al., 2007). Techniques for polycatecholamine–diazeniumdiolates were developed by simply forming N-diazeniumdiolates on the secondary amines of polycatecholamine films on substrate materials. The advantages of polycatecholamine–diazeniumdiolates over hydrogels and xerogels were reported to include material-independent coating ability and thin thickness (nm vs. μm-mm) (Hong et al., 2013).
C. Considerations on the Immunologic Effects of NO Donors and Their Therapeutic Realization Using NO Delivery Systems
Since the cellular and microenvironmental balances of enzymes, singlet oxygen, O2, and GSH levels decisively regulate a tissue’s immune microenvironment (Ghezzi 2011; Gostner et al., 2013; Noman et al., 2015; Chen et al., 2016), their consumption or usage in the decomposition of NO donors, which vary widely in their decomposition mechanism ranging from consuming or using aldehyde dehydrogenase-2, singlet oxygens, cytochrome P450, the reduced form of NADP, O2, temperature, light, heavy metal ions, GSH, GST, ascorbic acid, or esterase, etc., as previously discussed, also can affect the overall biologic outcomes in addition to the effects of NO itself. Furthermore, even with the similar or same DDS and NO donors, engineering inter- and intramolecular bonding and association between NO donors and materials comprising NO delivery systems can govern kinetics of NO release (Shin et al., 2007; Riccio et al., 2009; Kim et al., 2011; Hong et al., 2013), followed by various concentration- and releasing kinetic-dependent mechanisms of NO (Fukumura et al., 2006; Carpenter and Schoenfisch, 2012; Kim et al., 2014). In particular, numerous materials used in the NO delivery systems also elicit an immune response on their own. As examples, silica, which is widely used for NO releasing xerogels, NPs, and MSNs, have been reported to act as an adjuvant, whose efficacy and pathway are dependent on size, morphology, and surface functional groups (Vallhov et al., 2007; Heidegger et al., 2016; Li et al., 2016; Abbaraju et al., 2017; Nguyen et al., 2019; Zhao, Jiang et al., 2020). How new mechanistic insights into NO’s immune regulatory roles that have been largely generated using NO donors can be realized for therapeutic ends using NO delivery systems more amenable to clinical translation should therefore be carefully considered.
IV. Direct Effects of NO on the Immunogenicity of Cancer Cells In Vitro
Given the myriad of physiologic roles of NO, NO donors and delivery systems have been widely explored for their potential as therapeutics. Herein, activities of NO as potentiating cancer therapy that are relevant to immunotherapy, directly in modulating cancer cell immunogenicity, are discussed while NO’s effects on the immune system will be discussed in subsequent sections.
Even without considering its complex effects on the immune system, NO itself has seemingly opposing effects on modulating the proliferation and viability of cancer cells (Mocellin et al., 2007; Kim et al., 2014). High NO concentrations can induce cancer cell apoptosis and necrosis by directly damaging DNA, activating caspases-1 and -10, phosphorylating p53, inhibiting DNA repair mechanisms associated with poly(ADP-ribose) polymerase and DNA-dependent protein kinase, downregulating antiapoptotic protein survivin, and increasing Fas (CD95/APO-1) receptor expression (Ren et al., 2003; Shami et al., 2003; Shami et al., 2006; Udupi et al., 2006; Kiziltepe et al., 2007; Mocellin et al., 2007; Chakrapani, Goodblatt et al., 2008; Chakrapani, Kalathur et al., 2008; Simeone et al., 2008; Kitagaki et al., 2009; Kumar et al., 2010; Stevens et al., 2010; Singh and Gupta 2011; Xu et al., 2015; Fan et al., 2017; Kim et al., 2017; Liu et al., 2017; Liu, Huang et al., 2018; Zhao et al., 2019). Seemingly in complete opposition to this, NO can also augment the proliferation of cancer cells by stabilizing hypoxia inducible factor 1 subunit alpha, activating matrix metalloproteinases, upregulating vascular endothelial growth factor, increasing antiapoptotic proteins including B-cell lymphoma-2 and cyclooxygenase-2, and scavenging cytotoxic radical species (Ren et al., 2003; Shami et al., 2003; Shami et al., 2006; Udupi et al., 2006; Kiziltepe et al., 2007; Mocellin et al., 2007; Chakrapani, Goodblatt et al., 2008; Chakrapani, Kalathur et al., 2008; Simeone et al., 2008; Kitagaki et al., 2009; Kumar et al., 2010; Stevens et al., 2010; Singh and Gupta 2011; Xu et al., 2015; Fan et al., 2017; Kim et al., 2017; Liu et al., 2017; Liu, Xiao et al., 2018; Zhao et al., 2019). NO has also been shown to synergize with chemotherapy-, radiotherapy-, photothermal therapy– and photodynamic therapy–mediated apoptosis and necrosis effects by affecting multidrug-resistance, hypoxia, autophagy, balance of reactive oxygen species (ROS), and so on (Matthews et al., 2001; Frederiksen et al., 2003; Konovalova et al., 2003; Evig et al., 2004; Riganti et al., 2005; Bonavida et al., 2006; Hirst and Robson, 2007; Mocellin et al., 2007; Bonavida et al., 2008; Bratasz et al., 2008; Huerta et al., 2008; Bonavida and Garban, 2015; Fan et al., 2015; Kim et al., 2017; Deng et al., 2018; Hu et al., 2018; Pramanick et al., 2018; Wan et al., 2018; Ding et al., 2019; Feng et al., 2019; Hays and Bonavida, 2019a; Wang et al., 2019; Zhang, Jin et al., 2019; Bonavida B, 2020; Gao et al., 2020; Pieretti et al., 2020). Taking into account the numerous research and review papers discussing those actions and mechanisms (Ren et al., 2003; Shami et al., 2003; Shami et al., 2006; Udupi et al., 2006; Kiziltepe et al., 2007; Mocellin et al., 2007; Bonavida et al., 2008; Chakrapani, Goodblatt et al., 2008; Chakarapani, Kalathur et al., 2008; Simeone et al., 2008; Kitagaki et al., 2009; Kumar et al., 2010; Stevens et al., 2010; Singh and Gupta, 2011; Kim et al., 2014; Fan et al., 2015; Xu et al., 2015; Fan et al., 2017; Liu et al., 2017; Deng et al., 2018; Hu et al., 2018; Kim et al., 2017; Liu, Xiao et al., 2018; Pramanick et al., 2018; Wan et al., 2018; Ding et al., 2019; Feng et al., 2019; Park, Im et al., 2019; Wang et al., 2019; Zhang et al., 2018; Zhao et al., 2019; Gao et al., 2020), in this section, we focus on the effects of NO donors and delivery systems on the immunogenicity (Mahanty et al., 2015) of cancer cells (Table 1).
Influences of NO donors on cancer cells in vitro
A. NO Effects on Ligands of Immune Checkpoints: CD155 and Programmed Cell Death Ligand 1
Immune checkpoints play crucial regulatory functions in the homeostasis of immune system and the prevention of autoimmunity. However, immune checkpoints are also exploited by tumors to evade immune surveillance (Pardoll, 2012). In this regard, modulation or blockade of immune checkpoints and their ligands provide alternative ways to subvert the evasion of cancer cells of immune surveillance, leading to more robust tumor control by cancer immunotherapies (Shin and Ribas, 2015; Burugu et al., 2018; Marin-Acevedo et al., 2018). Despite the expression of various immune checkpoint ligands by cancer cells, such as galectin-9, glucocorticoid-induced tumor necrosis factor (TNF) receptor family-related protein ligands, CD112/CD155, and programmed cell death ligand 1 (PD-L1) (Shin and Ribas, 2015; Burugu et al., 2018; Marin-Acevedo et al., 2018), it has not been well investigated whether, which, or how immune checkpoint ligands on cancer cells are affected by exogenous NO. In this section, how exogenous NO modulates the expression of T cell immunoreceptor with Ig and ITIM domains (TIGIT) and PD-L1 on cancer cells is discussed.
The effects of exogenous NO on TIGIT pathways have been explored in multiple myeloma (MM) cell lines. MM is characterized by abnormal growth of plasma cells in bone marrow, exhibiting various symptoms, such as abnormal proteins from plasma cells, low blood cells, weak immunity, accelerated osteoclasts, and kidney damage (Stoopler et al., 2007), which express the poliovirus receptor (PVR/CD155) that is a ligand of both DNAX accessory molecule-1 and TIGIT (Shin and Ribas, 2015; Zhu et al., 2016; Gao et al., 2017; Burugu et al., 2018; Marin-Acevedo et al., 2018; Stamm et al., 2018; Harjunpää and Guillerey, 2020). TIGIT is an immune checkpoint that acts similar with cytotoxic T-lymphocyte-associated protein-4 (CTLA-4). In brief, the activation of cytotoxic T cells and NK cells is inhibited when TIGIT on T cells or NK cells binds to PVR/CD155 on antigen presenting cells (APCs), while binding of CD226 on T cells or NK cells to PVR/CD155 on APCs leads to the opposite effect (Shin and Ribas, 2015; Zhu et al., 2016; Gao et al., 2017; Burugu et al., 2018; Marin-Acevedo et al., 2018; Stamm et al., 2018; Harjunpää and Guillerey, 2020). In addition, the interaction of DNAX accessory molecule-1 on NK or T cells with PVR/CD155 on cancer cells exerts cytotoxicity via cytotoxic cytokines (Chan et al., 2010) such as interleukin (IL)-2, IL-12, or IL-21. Accordingly, antibodies antagonizing TIGIT have been developed (Gao et al., 2017; Harjunpaa and Guillerey, 2020) and chemotherapeutic drugs that up-regulate PVR/CD155 ligands on MM cells have been explored (Soriani et al., 2009; Chan et al., 2010; Fionda et al., 2013). Interestingly, chemotherapeutic drugs upregulating PVR/CD155 ligands are associated with DNA damage responses (Soriani et al., 2009; Fionda et al., 2013; Fionda et al., 2015) and NO is a radical species capable of exerting genotoxicity. In this regard, the effects of NO donors on the expression of PVR/CD155 ligands followed by NK cell–mediated immunotherapy were investigated (Fionda et al., 2015). Fionda et al. (2015) demonstrated that NO donors (200 µM) including diethylenetriamine NO adduct (DETA-NONOate), NO aspirin (NCX4040), and JS-K induce the upregulation of PVR/CD155 on six different MM cell lines including SKO-007(J3), U266, OPM-2, RPMI-8226, ARK, and LP1 via a DNA damage response pathway, which enhanced the susceptibility of those MM cells to NK cells to induce cytotoxicity in vitro (Fionda et al., 2015) (Fig. 1). Since other cancer cells including melanoma cells also express PVR/CD155 (Mahnke and Enk, 2016; Zhu et al., 2016; Gao et al., 2017; Stamm et al., 2018; Harjunpaa and Guillerey, 2019), the demonstrated potential of NO donors on modulating tumor expression of PVR/CD155 could be explored for its effects in other indications.

Direct effects of exogenous NO on cancer cell immunogenicity. Exogenous NO can suppress PD-L1 expression by cancer cells, which enhances the efficacy of anti-tumor CD8+ T cell immunity. It can also upregulate the PVR/CD155 expression by cancer cells, which induces NK cell responses. It can also stimulate the ATP release as well as CRT expression, which recruits APCs. These immunogenic effects on cancer cells mediated by the application of exogenous NO are dependent on the types of cancer cells and NO donors/delivery systems, as well as the dose of NO donors/delivery systems.
Exogenous NO modulates PD-1/PD-L1 pathways as well. Summarizing the major mechanism of PD-1/PD-L1 simplistically, the binding of PD-1 on T cells to PD-L1 on APCs or cancer cells inhibits the antitumor functions of cytotoxic CD8+ T cells [cytotoxic T lymphocytes (CTLs)] (Pardoll, 2012; Shin and Ribas, 2015; Burugu et al., 2018; Marin-Acevedo et al., 2018). As a result, various antagonistic antibodies for PD-1 and PD-L1 have been developed and approved by the US Food and Drug Administration (FDA) for anticancer immunotherapy by allowing CD8+ T cells or cancer cells to be more activated or susceptible to CTLs, respectively (Pardoll, 2012; Shin and Ribas, 2015; Burugu et al., 2018; Marin-Acevedo et al., 2018). In addition, several strategies have been explored to downregulate PD-L1 on cancer cells by exploiting small interfering RNA, microRNA, CRISPR, and CRISPR-associated protein 9 gene-editing techniques or hypoxia-regulating NPs (Zou et al., 2018; Guan et al., 2019; Phung et al., 2019; He et al., 2020). Although it is well-known that PD-L1 expression on cancer cells is governed by hypoxia (Sun et al., 2018; Lequeux et al., 2019) that is regulated by NO via ubiquitous transcription factor Yin Yang 1 and hypoxia-inducible factor 1 subunit alpha (Mateo et al., 2003; Bonavida et al., 2008; Dai et al., 2018; Hays and Bonavida, 2019a,b), the effects of NO on PD-L1 expression began to be investigated only very recently (Sung et al., 2019; Kiriyama et al., 2020). Sung et al. (2019) revealed that NO releasing NPs (>5 µM) regulated the activity of transcription factor SP1 in vitro, which led to the suppression of the PD-L1 expression on hepatocellular carcinoma cells in vitro in a dose-dependent manner (Sung et al., 2019). However, the opposite results were also reported, with DETA-NONOate (>200 µM) upregulating PD-L1 expression on A172 glioblastoma cells in vitro via c-Jun N-terminal kinase pathways (Kiriyama et al., 2020). These results imply that the expression of PD-L1 can depend on the cancer cell type and the concentration and kind of NO donor or delivery system (Fig. 1).
B. NO Effects on Immunogenic Cell Death: Calreticulin, ATP, and High Mobility Group Protein B1
Immunogenic cell death (ICD) is a process in which dying apoptotic cells expose and release damage-associated molecular patterns including calreticulin (CRT), ATP, and high mobility group protein B1 (HMGB1), which activate anticancer immunity (Krysko et al., 2012). ICD is induced by simultaneous actions of ROS and endoplasmic reticulum stress (Krysko et al., 2012). Although NO not only regulates intracellular ROS levels but also modulates the functions of endoplasmic reticulum (Fukumura et al., 2006; Gotoh and Mori, 2006; Mocellin et al., 2007; Carpenter and Schoenfisch, 2012), there have been few reports to investigate the effects of NO donors and delivery systems on ICD by cancer cells.
CRT is called as an “eat-me” signal to be recognized by phagocytosis receptors of CD91 positive APCs (Gotoh and Mori, 2006). Doxorubicin (DOX) is an anticancer chemotherapeutic drug well-known to induce CRT on cancer cells (Gotoh and Mori, 2006). Interestingly, DOX-mediated CRT exposure was not observed in inducible NOS (iNOS) knockout human colon cancer cells lines (HT29iNOS− cells) in contrast to normal HT29 cells (De Boo et al., 2009). In addition, treatment with a NO donor (SNP; 100 µM) per se upregulated the externalization of CRT on HT29iNOS− cells in vitro, while treatment with NO scavenger (2-phenyl-4,4,5,5,-tetramethylimidazoline-1-oxyl 3-oxide) diminished the effects of SNP on CRT exposure (De Boo et al., 2009). In particular, SNP significantly improved the phagocytosis of HT29iNOS− cells by DCs and proliferation of allogenic lymphocytes in a mixed lymphocyte reaction (MLR) (De Boo et al., 2009). An additional study demonstrated that a NO donor [S-nitrosopenicillamine, (SNAP); 100 µM) facilitated the externalization of CRT in both normal and drug-resistant HT29 cells (HT29-dx cells) via soluble cyclic guanosine monophosphate (cGMP) (Kopecka et al., 2011). Similar results were also reported with the increased externalization of CRT on Jurket cell lines (human leukemic T-cell lymphoblast) that were treated with NO donor S-nitroso-L-cysteine ethyl ester (600 µM) (Tarr et al., 2010). In contrast with those results utilizing very high concentrations of NO (>10 µM) sufficient to induce apoptosis of cancer cells, low concentrations of a NO donor (GSNO; ∼0.1 µM), which is not enough to exert cytotoxicity but sufficient to facilitate the chemosensitization, exhibited negligible effects on the CRT exposure in B16F10 murine melanoma cells (Kim et al., 2020). However, the low concentrations of nitrosothiols (∼0.1 µM) formulated in paclitaxel (PTX)-delivery NPs (pPTX/pCD-pSNO) enhanced the CRT externalization compared with PTX-delivery NPs (pPTX/pCD-pSH) without NO donors (Kim et al., 2020). These results imply that NO donors and delivery systems improved the immunogenicity of cancer cells by upregulating the externalization of CRT when their concentrations are enough to induce apoptosis or when they are combined with other ICD-inducing drugs or DDSs (Fig. 1).
HMGB1 attracts immune cells to stimulate the production of proinflammatory cytokines (Krysko et al., 2012). To date, the effects of NO donors and delivery systems on the release of HMGB1 are not fully established. A study demonstrated that SNP (2 mM) suppressed the expression of HMGB1 in lysates of papillary thyroid cancer cells obtained from patients with tyroid cancer (Mardente et al., 2010). However, DETA-NONOate (500 µM or 1–10 µM) significantly increased the release of HMGB1 from noncancer cells, such as rat insulinoma (RINm5F cell lines) or murine macrophages (RAW 264.7 cell lines), respectively (Jiang and Pisetsky, 2006; Steer et al., 2006). Accordingly, further investigations are required with different types and concentrations of NO donors and delivery systems to clarify the effects and mechanisms of NO on the HMGB1-mediated ICDs by various cancer cells.
ATP is a representative “find-me” signal released from apoptotic cells, which facilitates the P2Y2- and P2X7-mediated phagocytosis by APCs (Martins et al., 2014; Elliott et al., 2009; Krysko et al., 2012). Although there have been several reports exploring the effects of NO on red blood cells and astrocytes, it is unclear whether NO promotes or inhibits the release of ATP due to diversity in experimental conditions, such as oxygen level and types and concentrations of NO donors (Bal-Price et al., 2002; Olearczyk, Ellsworth, et al., 2004; Olearczyk, Stephenson et al., 2004; Cao et al., 2009). Likewise, the effects of NO donors and delivery systems on ATP release from cancer cells have not been clearly evaluated (Fig. 1). Our group recently investigated the effects of low NO donor concentration (∼0.1 µM) on ATP release from B16F10 murine melanoma cells (Kim et al., 2020). Like CRT exposure, the low concentration of the NO donor (GSNO; ∼0.1 µM) exhibited negligible effects on ATP release, while pPTX/pCD-pSNO enhanced the ATP release compared with pPTX/pCD-pSH (Kim et al., 2020). There are two representative mechanisms in the release of ATP; caspase activation and autophagy (Martins et al., 2014). However, pPTX/pCD-pSNO failed to enhance autophagy process compared with pPTX/pCD-pSH, whereas free PTX and PTX/pCD-pSH led to enhanced autophagy compared with PBS and GSNO (Kim et al., 2020). These results imply that the modulation of ATP by low concentrations of NO in PTX codelivery is not associated with autophagy mechanisms (Kim et al., 2020). On the other hand, high concentrations of JS-K (1–5 µM), which are enough to elicit cytotoxicity to cancer cells, improved the autophagy in A2780 and SKOV3 ovarian cancer cell lines (Liu et al., 2019) and MDA-MB-453 breast cancer cell lines (McMurtry et al., 2011) in vitro. However, these results cannot guarantee that NO induces autophagy in a concentration-dependent manner because there have been reports of oppositive effects, demonstrating that different types and concentrations of NO donors and NOS inhibitors differentially suppress autophagy in various cell types (Sarkar et al., 2011; Benavides et al., 2013; Shen et al., 2014; Zhang, Jin et al., 2019). In addition, whether NO mediated changes in autophagy indeed regulate ATP release also need to be further investigated.
In addition to the ligands of immune checkpoints and ICD mechanisms as previously discussed, it was reported that NO enhances the degradation of antiapoptotic survivin in cancer cells, which sensitizes cancer cells to DC-induced cell death mediated by Fas–Fas ligand–independent as well as dependent pathways (Huang et al., 2005). These reports imply the potential presence of additional mechanisms that exogenous NO has on the immunogenicity of cancer cells.
In conclusion, NO donors and NO delivery systems have potential to improve the immunogenicity of cancer cells, although the types and dose are required to elicit these favorable effects remain to be further optimized.
V. Direct Effects of NO on Immune Cells In Vitro
Endogenous NO synthesized via an enzymatic reaction with endogenous NOSs and L-arginine substrates modulates the functions and populations of multiple immune cell subtypes in autocrine, paracrine, and endocrine manners, expertly summarized by Bogdan (2001, 2015). However, in several cells, exogenously delivered NO can act differently from endogenously generated NO (Gansauge et al., 1997; Nakano et al., 2000). Moreover, the immune system functions through the coordinated effects among various immune cells that govern tumor progression (Burkholder et al., 2014; King et al., 2017; Navarro-Tableros et al., 2018). These complexities make it difficult to understand the mechanism behind the physiologic outcomes in NO-based anticancer therapy. Accordingly, in this section, we discuss how NO donors and delivery systems directly regulate immune cells in vitro (Table 2 and 3).
Influences of NO donors on dendritic cells and B cells in vitro
Functions of NO donors on T cells and NK cells in vitro
A. Dendritic Cells
DCs are a specialized APC subtype that play pivotal roles in shaping innate and adaptive immune response (Wculek et al., 2020). Endogenous NO is also reported to directly modulate the functions of DCs as represented by TNF- and iNOS-producing DCs (Tip-DCs) to be involved in the innate immune response, priming of T cells, and modulation of CD8+ T cell activity (Shimamura et al., 2002; Serbina et al., 2003; Nicolas et al., 2007; Serbina et al., 2008; Marigo et al., 2016; Xue et al., 2018). Functions of Tip-DCs are reported to be significantly modulated by NO, which have emphasized the crucial roles of NO in the actions of DCs (Shimamura et al., 2002; Serbina et al., 2003; Nicolas et al., 2007; Serbina et al., 2008; Marigo et al., 2016; Xue et al., 2018). Likewise, there have been significant efforts to investigate the effects of exogenous NO donors on DCs. DC-mediated antitumor immunity is comprised of a cascade pathway: differentiation of immature DCs, antigen uptake by immature DCs in peripheral tissues, migration of the maturating DCs into secondary lymphoid organs, and antigen presentation of DCs to T cells (Palucka and Banchereau, 2012). Accordingly, the effects of NO on each aspect of DC-regulated immune response are discussed stepwise in the following text.
NO donors such as SNP (50 nM), SIN-1 (50 µM), DEA-NO (50 µM), and DETA-NONOate (50 µM) were reported to differentiate hematopoietic stem cells and monocytes into DCs (Fernández-Ruiz et al., 2004; Tiribuzi et al., 2013). Interestingly, DCs expanded by NO donors (SNAP; 500 nM) seem to consist of IL-6–, IL-12–, and TNF-producing effector DCs and IL-10– and PD-1–expressing regulatory DCs (Si et al., 2016). An iNOS-inhibitor [L-N6-(1-iminoethyl)lysine; 40 µM] selectively induced the differentiation of effector DCs, as contrasted with opposite results observed for NO donor-treated (SNAP; 500 µM) DCs in vitro (Si et al., 2016).
DCs matured by TNF-α have a suppressed endocytic ability via intracellular accumulation of the lipid messenger ceramide (Sallusto et al., 1996). However, the DETA-NONOate (100 µM) and SNAP (200 µM) significantly improved the endocytic ability of TNF-α-treated DCs by inhibiting intracellular accumulation of ceramide via the cGMP pathway (Paolucci et al., 2000). Uptaken antigens are digested and processed by DCs and are then presented on the DC cell surface. Interestingly, a NO donor [3-(2-hydroxy-2-nitroso-1-propylhydrazino)-1-propanamine; 20 µM] reduced the presentation of antigens to the major histocompatibility complex class I by DCs when a protein antigen to be digested was loaded, whereas it had no effect on antigen presentation compared with control when a peptide antigen was used (Siddiqui et al., 2011). These results suggested that NO hampers the intracellular antigen processing step by inhibiting proteolysis (Siddiqui et al., 2011).
During antigen processing, DCs undergo maturation and migrate into the secondary lymphoid organs where they initiate the adaptive immune response. GSNO (1–50 µM) and DETA-NONOate (50 µM) have been shown to exert negligible effects on the maturation of DCs, respectively (Paolucci et al., 2003; Kim et al., 2020). However, NO donors showed unpredictable results when they are treated with other agents or DDSs to induce the maturation of DCs. While DETA-NONOate (50 µM) and S-nitrosothiol (1–50 µM) improved the maturation of TNF-α- or pPTX/pCD-SH–treated DCs (Paolucci et al., 2003; Kim et al., 2020), SNAP (100–1000 µM), dipropylenetriamine NONOate (600 µM), or N-(-4-ethyl-2-hydroxyimino-5-nitrohex-3-enyl)pyridine-3-carboxamide (NOR-4; 25–100 µM) suppressed the expression of IL-6, IL-12, and TNF-α in DCs matured by lipopolysaccharide (LPS; 100 or 500 µg/mL) (Xiong et al., 2004; Giordano et al., 2006; Obregon et al., 2015; Si et al., 2016).
The effects of NO donors on the migration of DCs have been also investigated (Giordano et al., 2006). NOR-4 (25–100 µM) was reported to improve LPS-matured DC responses to CCL19 in a dose-dependent manner. NOR-4 slightly enhanced CCR7 expression in LPS-treated DCs, implying that NOR-4–mediated enhanced migration of mature DCs toward CCL19 was not simply associated with the increase of CCR7 but achieved by a mechanism unrelated to receptor expression. LPS promoted expression and activity of cGMP kinase that phosphorylates vasodilator-stimulated phosphoprotein, which disrupted focal adhesion and inhibited cell migration. Interestingly, NOR-4 reversed the effects of LPS on cGMP kinase, which facilitated the enhanced migration of DCs toward CCL19. Furthermore, NOR-4 increased the expression of CXCR4 in LPS-treated DCs, which were correlated to the enhanced migration of the matured DCs toward CXCL12. The enhanced migration toward CCL19 and CXCL12 by NOR-4 was not observed in the immature DCs, suggesting the requirement of maturation status in exogenous NO-mediated enhancement of DC migration toward secondary lymphoid tissues.
Mature and activated DCs that migrate into secondary lymphoid tissues prime and instruct T cells to manifest antigen-specific adaptive immune response. NO donors (GSNO, 1–20 µM, and DETA-NONOate, 50 µM) themselves have been shown to exhibit negligible effects or slightly enhance the proliferation of CD3+ or CD8+ T cells in MLR using DCs or DC-containing splenocytes as stimulating cells (Paolucci et al., 2003; Obregon et al., 2015; Kim et al., 2020). However, the proliferation of T cells was significantly improved when cells were cotreated with NO donors in addition to PTX or TNF-α (Paolucci et al., 2003; Obregon et al., 2015; Kim et al., 2020).
Overall, exogenous NO has complex and seemingly opposing effects on DCs (Table 2). Delivered NO appears to promote the differentiation into the DCs, improve the endocytic ability of mature DCs, improve the migration of mature DCs toward secondary lymphoid tissues, and induce negligible proliferation of primed T cells in vitro (Fig. 2). In addition, T cell activation and proliferation induction by DCs can be improved when NO donors are used in combination with other agents including TNF-α, PTX, and PTX-containing DDS in vitro. However, exogenously supplied NO seems to also expand regulatory DCs, suppress the intracellular antigen processing of DCs, and exert negligible effects on DC activation, that can only marginally enhance T cell proliferation in vitro (Fig. 2). Accordingly, further investigations and optimizations are required to preferentially exploit the DC-stimulatory functions of NO delivery for antitumor immunotherapy.

Direct effects of exogenous NO on DCs. Exogenous NO improves differentiation of hematopoietic stem cells and monocytes to DCs. It enhances the endocytic functions of DCs, while hampering processing of intracellular antigen. Although exogenous NO itself has negligible effects on or inhibits the maturation of DCs, it can improve the effects of DC stimulating agents. It can also promote CCL19-mediated migration of DCs into the secondary lymphoid tissues. Although exogenous NO itself has an insignificant effect on or slightly enhances T cell proliferation, it can improve the efficacy of DC-stimulating agents on induction of T cell proliferation. These exogenous NO effects on DCs can be dependent on DC state as well as the types and dose of NO donors/delivery systems.
B. Macrophages
Macrophages not only serve as T cell instructors but also play an important role in innate immunity in vivo by taking up pathogens and apoptotic cells (Chaperot et al., 2000; Hume, 2008; Ferenbach and Hughes, 2008; Gottschalk and Kurts, 2015). Macrophages are largely subcategorized into M1 and M2, based on their polarization states (Ley, 2017; Russell et al., 2019). Classically, M1 is defined as CD11b+F4/80+MHCII+ macrophages to produce NO via an enzymatic reaction using L-arginine and iNOS, while M2 is defined as CD11b+F4/80+CD206+ macrophages that avidly consume L-arginine by arginase-1 (Ley, 2017; Russell et al., 2019; Nath and Kashfi, 2020). The proinflammatory M1 secrets high levels of cytokines including IL-12, IL-1β, and IL-23 to foster polarization of T lymphocytes to a T helper (Th) cell 1 type as well as DC maturation, whereas anti-inflammatory M2 produces IL-4, IL-10, and TGF-β (Ley, 2017; Russell et al., 2019; Nath and Kashfi, 2020). Tumor-associated macrophages are believed to accelerate the growth, angiogenesis, and metastasis of tumors, as well as contribute to the acquired resistance to chemotherapeutic drugs and immune checkpoint blockade by certain cancers (Ley, 2017; Lin et al., 2019; Russell et al., 2019; Nath and Kashfi, 2020). Because M2 is the dominant phenotype of tumor-associated macrophages, there have been continued efforts to reprogram M2 macrophages toward an M1 phenotype (Zanganeh et al., 2016; Rodell et al., 2018). Despite the potential antitumor effects of M1 macrophages (Zanganeh et al., 2016; Pang et al., 2018; Rodell et al., 2018), the functions of NO produced by M1 macrophages are debated as NO released from iNOS of M1 macrophages has been reported to not only exert direct cytotoxicity but also suppress the proliferation and activation of T cells (Bingisser et al., 1998; Lu et al., 2015; van der Veen et al., 2000).
In line with the pivotal roles of endogenous NO in macrophage-mediated cancer progression and response to therapy, the effects of exogenous NO on the polarization of macrophages have been investigated (Lu et al., 2015; Lee et al., 2020) (Fig. 3A). The solution containing NO generated by microwave plasma generator (NO2− 316.7 µM + NO3− 24.2 µM) induced M1 polarization with increased iNOS and decreased arginase-1 in RAW 264.7 macrophages (Lee et al., 2020). However, SNAP (100–1000 µM) suppressed iNOS expression by RAW 264.7 macrophages polarized to M1 by interferon (IFN)-γ (Lu et al., 2015). These results imply a potential feedback loop associated with NO in macrophage polarization.

Direct effects of exogenous NO on macrophages and B cells. (A) Exogenous NO promotes M1 polarization over M2, while the cotreatment with IFN-γ exerts the opposite effect. (B) Exogenous NO impairs the intracellular antigen processing functions of B cells by interfering with lysozyme activity.
C. B Cells
B cells are not only APCs but also specialized immunoglobulin (Ig)-producing cells (Nelson, 2010; Tsou et al., 2016; Yuen et al., 2016; Sarvaria et al., 2017; Wennhold et al., 2019; Petitprez et al., 2020). Moreover, B cells promote antigen presentation by other APCs (Kurt-Jones et al., 1988), which in turn modulate T cell priming and response to immune checkpoint blockade therapy (Candolfi et al., 2011; Wang et al., 2016; Griss et al., 2019; Cabrita et al., 2020; Helmink et al., 2020; Petitprez et al., 2020). In addition to their pivotal roles in adaptive immunity, B cells also have innate functions as B cells activated by IFN-α or TLR agonist (CpG-containing oligodeoxynucleotides) can directly elicit cytotoxicity against cancer cells (Kemp et al., 2004). Owing to their versatile functions, adoptive immunotherapies with activated and primed B cells have been under development, with demonstrated antitumor efficacy (Li et al., 2009; Ren et al., 2014; Wennhold et al., 2019).
B cells produce NO, implying a role for NO in B cell immune functions (Mannick et al., 1994; Zhao et al., 1998; Bogdan, 2001; Jayasekera et al., 2006; Tumurkhuu et al., 2010; Saini et al., 2014; Bogdan 2015). As with DCs, exogenous NO (N-[4-[1-(3-aminopropyl)-2-hydroxy-2-nitrosohydrazino]butyl]1,3-propanediamine; 100–200 µM) impaired intracellular antigen processing functions of B cells in vitro (Lemaire et al., 2009) (Fig. 3B). These results are correlated with in vivo reports of a negligible effect of GSNO on B cell populations (Kim et al., 2020) and higher production of antibody in iNOS-deficient mice than wild-type mice against influenza virus infection (Jayasekera et al., 2006). Beyond what is known with respect to effects on intracellular antigen processing (Table 2), further investigations are required to elucidate the effects of exogenous NO on the functions and differentiation of B cells.
D. T Cells
CD8+ T cells are one of the central effector cells in adoptive immune response to elicit antigen-specific cytotoxicity, and in many respects represent the holy grail in anticancer immunotherapy (Pardoll, 2012; Shin and Ribas, 2015; Burugu et al., 2018; Marin-Acevedo et al., 2018; Waldman et al., 2020). Although the differentiation, activation, and priming of CD8+ T cells is mainly shaped by the APCs with which they interact, CD4+ T cells including Th and Tregs also influence the fate of CTLs by interacting with various immune cells and secreting cytokines and chemokines (Pardoll, 2012; Shin and Ribas, 2015; Burugu et al., 2018; Marin-Acevedo et al., 2018; Waldman et al., 2020). To improve the activation and differentiation of CD8+ T cells, various antagonistic immune checkpoints blockade approaches have been developed with great clinical success and FDA approval (Pardoll, 2012; Shin and Ribas, 2015; Burugu et al., 2018; Marin-Acevedo et al., 2018; Waldman et al., 2020). In addition, numerous adoptive T cell transfer therapies utilizing chimeric antigen receptor T cells have been under clinical investigation worldwide, resulting in the FDA approval of KYMRIAH and YESCARTA for treatment of B cell lymphoma (June et al., 2018; Waldman et al., 2020).
NO is also one of the key molecules in the function and differentiation of T cells via signal transduction and post-translational modification via S-nitrosylation (Garcia-Ortiz and Serrador, 2018). T cells also express NOSs to produce endogenous NO to modulate T cell metabolism and CD4+ T cell differentiation, implying the pivotal role of NO in T phenotype (Garcia-Ortiz and Serrador, 2018). Interestingly, NO released from Tip-DCs improves the activity and proliferation of CD8+ T cell in vivo, while NO generated from cancer cells is immunosuppressive against T cells (Shimamura et al., 2002; Serbina et al., 2003; Nicolas et al., 2007; Serbina et al., 2008; Marigo et al., 2016; Garcia-Ortiz and Serrador, 2018; Xue et al., 2018). These results imply yet again seemingly paradoxical functions of NO in T cell–mediated immune response.
The direct in vitro effects of exogenous NO on T cells have been widely investigated (Table 3). GSNO (100–500 µM), SNP (1 mM), and N-ethyl-2-(1-ethyl-2-hydroxy-2-nitrosohydrazino)ethanamine (100 µM) inhibited the proliferation of SupT1 (human T cell lymphoma), ST4 (human T cell lymphoma), PF382 (human T acute lymphoblastic leukemia), and Jurkat (human acute lymphoblastic line) cells (Allione et al., 1999; Henson et al., 1999). Treatment of SIN-1 (1–10 mM) resulted in tyrosine nitrosylation of the T cell receptor (TCR)–CD8 complex, which inhibited the proliferation of antigen-specific CD8+ T cell by impairing the binding of peptide major histocompatibility complex (pMHC) to CD8+ T cells, an effect that occurred without affecting T cell expression of CD8 and TCR (Nagaraj et al., 2007) (Fig. 4A). In addition, DETA-NONOate and SNAP (50–200 µM) inhibited the proliferation of CD4+ T cells in a MLR assay using DCs as APC (Markowitz et al., 2017) (Fig. 4A).

Direct effects of exogenous NO on T cells. (A) Exogenous NO nitrosylates the TCR of T cells, which suppress the proliferation of T cells in response to antigen recognition. (B) Low concentrations of exogenous NO promotes the differentiation of CD4+ T cells to a Th1 type, while high concentrations suppresses it. (C) Exogenous NO enhances the production of IL-4 from activated Th2 cells. (D) Exogenous NO suppresses Th17 differentiation from CD4+ T cells partially by expansion of NO Tregs. In addition, exogenous NO suppresses Treg differentiation from CD4+ T cells and improves the proliferation of Th9 type cells.
NO donors govern the differentiation of CD4+ T cells in a concentration-dependent manner. Low SNAP [1–0 µM (Niedbala et al., 1999) 10–100 µM (Lee et al., 2011)] and DETA-NONOate [10–100 µM (Lee et al., 2011); 5–10 µM (Niedbala et al., 2002)] concentration enhanced the differentiation of CD4+ T cells toward a Th1 phenotype, while higher SNAP [50–500 µM (Niedbala et al., 1999) 10–100 µM (van der Veen et al., 1999)] or DETA-NONOate [10–100 µM (Niedbala et al., 2002)] suppressed Th1 differentiation (Niedbala et al., 1999; Niedbala et al., 2002) as well as the proliferation of Th1 and Th2 cells (van der Veen et al., 1999) (Fig. 4B). On the other hand, SNP (1–100 µM) and SNAP (20–200 µM) increased the production of IL-4 from Th2 clones and EL4 cells activated by concanavalin A or O-tetradecanoylphorbol 13-acetate/A23187 (Chang et al., 1997) (Fig. 4C). The polarization and functions of Th17 cells were also found to be controlled by NO donors (Niedbala et al., 2011; Obermajer et al., 2013; Yang et al., 2013). Physiologic concentrations of DETA-NONOates (10–25 µM), which are comparable to the NO level produced by MDSCs induced Th17 differentiation with insignificant effects on CD4+ T cell proliferation, whereas higher concentrations (100–200 µM) suppressed the proliferation of CD4+ T cells (Obermajer et al., 2013) (Fig. 4D). This is in line with reports that DETA-NONOate (50–200 µM) decreases the size and proliferation of preestablished Th17 cell pools with the suppressed production of IL-17 and IL-22 in dose-dependent manner (Niedbala et al., 2011) (Fig. 4D). Likewise, SNAP (10–200 µM) suppressed the differentiation of Th17 from naïve CD4+ T cells isolated from wild type as well as iNOS knockout mice (Yang et al., 2013). DETA-NONOate (100 µM), which suppressed Th17 cells, resulted in the enhanced differentiation of Th9 cells that are associated with anti-inflammatory responses to infection and allergy (Niedbala et al., 2014) (Fig. 4D). Interestingly, DETA-NONOate (50–200 µM) induced the differentiation of NO Tregs defined as CD4+CD25+Foxp3− converted from CD4+CD25−Foxp3− when exposed to NO (Niedbala et al., 2007), while SNAP [10–100 µM (Lee et al., 2011)] and DETA-NONOate [10–100 µM (Lee et al., 2011), 25 µM (Brahmachari and Pahan, 2009)] significantly suppressed the differentiation of normal Tregs (CD4+CD25+Foxp3+) (Fig. 4D). The NO Tregs inhibited the proliferation of effector CD4+CD25− T cells (Niedbala et al., 2007) and the polarization of Th17 without interfering the differentiation and functions of Th1 (Niedbala et al., 2013) (Fig. 4D). These NO Tregs are distinguished from normal Tregs to suppress the polarization of both Th1 and Th17 cells (Niedbala et al., 2013) (Fig. 4D). In addition, peroxynitrite (ONOO−; 0.1–2.4 mM), a major product by superoxide and NO, inhibited the CCL-2 mediated T cell migration by suppressing the expression of CXCR4 (Kasic et al., 2011).
Overall, very high concentrations (>100 µM) of exogenous NO appear to inhibit the differentiation, proliferation, and migration of T cells, whereas low NO concentrations help the differentiation of CD4+ T cells into Th1, Th2, Th17, and NO Treg cells.
E. Natural Killer Cells
NK cells are mainly responsible for the innate immunity by utilizing granzyme B and perforin (Guillerey et al., 2016). Although NK cells lack antigen specificity, their rapid cytotoxicity against target cells have promoted the progress and development of genetic engineering and surface modification of NK cells for the treatment of various cancers (Kim et al., 2019; Rezvani et al., 2017). Endogenous NO production has been reported to be closely linked to the function and activation of NK cells (Cifone et al., 1994; Cifone et al., 2001; Lamas et al., 2012). Depletion of arginine also led to the suppression of proliferation, activation, and cytotoxic functions of NK cells (Lamas et al., 2012). Arginine supply increased the cytotoxic ability of NK cells, while NOS inhibitors suppressed it (Cifone et al., 1994; Cifone et al., 2001). In addition, the enhanced production of endogenous NO was accompanied with the activation of NK cells by IL-2 (Cifone et al., 1994). In contrast to endogenous NO, exogenously supplied NO inhibited the functions of NK cells via nitrosylation of functional components of NK cells, similar to effects seen in T cells (Stiff et al., 2018). SNAP (100 µM) treatment nitrosylated the tyrosine residues in NK cells, which impaired Fc receptor-mediated NK cell functions in vitro, such as production of IFN-γ and antibody dependent cellular cytotoxicity of NK cells on the cancer cells (Stiff et al., 2018) (Table 3). Further investigations are required to understand the roles of exogenous NO on the functions and differentiation of NK cells.
F. Myeloid-Derived Suppressor Cells
MDSCs play a central role regulating CD4+ T, CD8+ T, NK cells, and macrophage functions and are generally considered to promote tumor growth and metastasis (Burkholder et al., 2014; Kumar et al., 2016). Importantly, the functions of MDSCs are highly dependent on the NO produced by NOSs (Liao et al., 2014; Hirano et al., 2015; Yang et al., 2016). In detail, NO released from MDSCs nitrosylates the tyrosines in TCR, which suppresses the expansion of antigen specific CD4+ and CD8+ T cells in vivo (Nagaraj et al., 2007; Nagaraj et al., 2010; Nagaraj et al., 2013; Markowitz et al., 2017). In addition, iNOS is expressed by various cancer cells, which directly provokes the recruitment and expansion of MDSCs in tumor microenvironment by modulating vascular endothelial growth factor secretion in tumor and upregulating STAT3 and ROS in MDSCs (Jayaraman et al., 2012; Kim et al., 2020). That is, NO from tumors recruits and expands MDSCs, which in turn fosters a high reactive nitrogen species environment in the tumor to accelerate further infiltration of MDSCs (Jayaraman et al., 2012; Kim et al., 2020). The resultant iNOS-dependent positive feedback loop seems to result in nitrosylation of the chemokine CCL2 within tumors, which impedes the infiltration of T cells with antitumor functions into the tumor microenvironment (Molon et al., 2011; Kim et al., 2020). Considering that MDSCs promote the expansion of Tregs through CD40–CD40 ligand interactions, NO-mediated expansion of MDSCs may contribute to increasing local Treg populations (Pan et al., 2010). MDSCs furthermore restrain the Fc receptor–mediated functions of NK cells in antibody-coated target cells via iNOS expression (Stiff et al., 2018).
Several in vitro results discussed in the sections of T cells and NK cells are in line with the immunosuppressive functions of NO produced by MDSCs in vivo (Nagaraj et al., 2007; Kasic et al., 2011; Obermajer et al., 2013; Yang et al., 2013; Markowitz et al., 2017; Stiff et al., 2018). In particular, the immunosuppressive functions of MDSCs and exogenous NO donors were reversed in iNOS knockout animals (Nagaraj et al., 2007) and when used in combination with NOS inhibitors in wild-type animals (Yang et al., 2013; Markowitz et al., 2017; Stiff et al., 2018), emphasizing the role that exogenously delivered NO plays in mimicking the immunosuppressive functions of NO that is endogenously produced by MDSCs. Unfortunately, the direct effects of exogenous NO donors and delivery systems on the functions, differentiation, and expansion of MDSCs have not been well investigated in vitro.
VI. Recent Advances in Antitumor Immunotherapy Leveraging NO-Delivery Systems In Vivo
Although traditional strategies using NO donors and delivery systems focus on exerting NO’s cytotoxicity directly on cancer cells, which exhibit powerful anticancer effects against various cancer cells in vitro, there have been conflicting reports of whether these strategies really work (Shami et al., 2003; Kiziltepe et al., 2007; Duan et al., 2012; Fan et al., 2017; Yin et al., 2017; Deng et al., 2018; Park, Im et al., 2019; Zhao et al., 2019; You et al., 2020) or not (Feng et al., 2018; Ding et al., 2019; Li et al., 2019; Zhang, Lai et al., 2019; Zhang Jin et al., 2019; Chen et al., 2020) even in complex xenograft tumor models that use immune-deficient animals. Likewise, using NO donors and delivery systems without any help of other therapeutic agents has produced conflicting reports in allograft tumor models using conventional mouse models (Fan et al., 2015; Liu, Xiao et al., 2018; Studenovsky et al., 2018; Dong et al., 2019; Feng et al., 2019; Kang et al., 2019; An et al., 2020; Kim et al., 2020). S-nitrosothiol conjugated upconversion NPs (Fan et al., 2015), SNP (Feng et al., 2019), 1,3-bis-(2,4,6-trimethylphenyl)imidazolylidene NO-loaded micelles (Kang et al., 2019), GSNO containing zeolitic imidazolate framework with cancer cell membrane (An et al., 2020), nitrate-conjugated polymers (Studenovsky et al., 2018), nitrate-conjugated bovine serum albumin–protected gold nanocluster (Liu et al., 2018), and S-nitrosothiol–conjugated mesoporous silica NPs (Dong et al., 2019) exhibited negligible antitumor effects on murine breast 4T1 (Fan et al., 2015; Liu, Xiao et al., 2018; Dong et al., 2019; Feng et al., 2019; Kang et al., 2019; An et al., 2020), murine melanoma B16F10 (Kim et al., 2020), and EL4 murine T-cell lymphoma (Studenovsky et al., 2018) tumor models. Intratumoral GSNO treatment (570 µg/kg, one-time injection) even led to increased B16F10 tumor growth compared with saline treatment (Kim et al., 2020). In contrast, there are several reports demonstrating therapeutic effects of NO in vivo (Sung et al., 2019; Lee et al., 2020; Li, Ji et al., 2020). A solution containing NO generated by microwave plasma generator (Lee et al., 2020) and an implantable and wireless powered NO release system that stimulates more NO release from GSNO under wirelessly powered light-emitting diode to irradiate 335 or 545 nm (Li, Ji et al., 2020), elicited antitumor effects in allograft tumor-bearing normal mouse, specifically in B16F10-bearing C57BL/6 and luciferase expressing CT26 murine colorectal carcinoma cells (CT26-luc)-bearing Balb/C mouse, respectively. It is unclear how a solution containing NO generated by microwave plasma generator (Lee et al., 2020) and wireless-powered NO release system (Li et al., 2020) can lead to the antitumor effects as contrasted with other reports (Fan et al., 2015; Liu, Xiao et al., 2018; Studenovsky et al., 2018; Dong et al., 2019; Feng et al., 2019; Kang et al., 2019; An et al., 2020; Kim et al., 2020). These conflicting claims may be ascribed to the different kinds of NO donors/delivery systems, dose, administration routes, and tumor types used in those studies. Indeed, very high concentrations of NO were frequently administrated or implanted into and near the tumor with a solution containing NO generated by microwave plasma generator (Lee et al., 2020) or implantable and wireless-powered NO release system (Li, Ji et al., 2020), which might exert efficient direct cytotoxicity to tumor cells regardless of the complex interactions with the in vivo immune system. These reports suggest the potential of intratumoral administration of NO delivery systems to deliver very high concentrations of NO in the treatment of primary tumors.
As discussed, NO has both immune stimulatory and suppressive functions, which are highly dependent on NO concentration. Accordingly, it is natural to try to exploit immune-stimulatory functions more selectively by minutely controlling NO concentration. Sung et al. (2019) hypothesized that low NO dose could normalize the abnormal tumor vessel as NO plays a pivotal role in blood vessel homeostasis (Kim et al., 2011; Carpenter and Schoenfisch, 2012; Godo and Shimokawa, 2017) and vasodilation (Carpenter and Schoenfisch, 2012; Deepagan et al., 2018; An et al., 2020), while high doses of NO can eradicate the tumor by destroying the vessel of tumor as high concentration of NO can induce cytotoxicity to cells including blood vessel endothelial cells as well as cancer cells (Mocellin et al., 2007; Kim et al., 2017). Indeed, a very high dose of DNIC [Fe(μ-SEt)2(NO)4] containing lipid-PLGA hybrid NPs (DNIC equivalent to 0.5–1 mg/kg, six times intravenous injection) facilitated the suppression of tumor (murine hepatocellular carcinoma HCA-1 tumor model) growth with decreased mean vessel density in the tumor (Sung et al., 2019). However, CD4+ and CD8+ T cells were not expanded in the tumor. On the other hand, a low dose of DNIC (0.1 mg/kg, 6 times intravenous injection) NPs significantly enhanced perfusion and functional perfused vessel. Interestingly, the low dose of DNIC NPs selectively polarized M1 (F4/80+CD86+) over M2 (F4/80+CD206+), which significantly expanded CD4+ and CD8+ T cells in the tumor. In particular, DNIC NPs suppressed the PD-L1 expression on HCA-1 tumor cells in a dose-dependent manner in vivo. As a result, a low dose of DNIC NPs allowed the efficient T cell–mediated antitumor effects in the codelivery of a vaccine despite the insignificant antitumor effects of a vaccine or low dose of DNIC NPs used alone. Furthermore, a low dose of DNIC NPs suppressed the distal metastasis with negligible effects on the primary tumor.
Our group shed light on the issue of how high-dose NO suppresses the expansion of T cells (Kim et al., 2020) using a B16F10-OVA dual mouse tumor model. Melanomas were established by implanting B16F10-OVA cells in the left dorsal skin as a primary (1°) tumor on day 0 and in the right dorsal skin as a secondary (2°) tumor to investigate both the local and systemic anticancer effects as well as immune response as a result of NO donor treatment. One-time intratumoral administration of GSNO (570 µg/kg) into the 1° tumor on day 7 increased 1° tumor size without affecting the size of the 2° tumor. MDSCs numbers were found to be significantly increased in both the 1° and 2° tumors as well as the lymph nodes draining each tumor seven days after one-time treatment of the primary tumor by intratumoral administration of GSNO (570 µg/kg). However, the number of total CD8+, antigen-experienced CD8+ (PD-1+ CD8+), and antigen-specific CD8+ (tetramer+ CD8+) T cells were negligibly changed in both the 1° and 2° tumor, spleen, and both tumor-draining lymph nodes despite increased levels of activated and expanded DCs in the 2° tumor and spleen, ratios of M1 (CD11b+F4/80+CD86+) to M2 (CD11b+F4/80+CD206+) macrophages in the spleen and lymph nodes draining the 2° tumor (2° dLNs), and NK cells (CD3-NK1.1+) in 2° dLNs. These results suggest that NO treatment induced the expansion of MDSCs (Nagaraj et al., 2007; Nagaraj et al., 2010; Jayaraman et al., 2012; Nagaraj et al., 2013; Markowitz et al., 2017), which contribute to preventing the instruction (Nagaraj et al., 2007; Nagaraj et al., 2010; Nagaraj et al., 2013; Markowitz et al., 2017) and infiltration (Molon et al., 2011) of CD8+ T cell, and M1 macrophage- and NK cell–mediated (Stiff et al., 2018) antitumor response. Taken together, both studies suggest that high concentrations of NO prevent the development of robust antitumor immunity despite the presence of expanded and activated DCs, as well as M1-polarized macrophages.
The development of combined NO delivery systems with other therapeutic methodologies including chemotherapy, radiation therapy, photothermal therapy, photodynamic therapy, and immunotherapy can be an alternative to overcome the previously discussed dose-associated problems of NO delivery systems (Fan et al., 2015; Liu, Xiao et al., 2018; Studenovsky et al., 2018; Dong et al., 2019; Feng et al., 2019; Kang et al., 2019; Sung et al., 2019; An et al., 2020; Kim et al., 2020) because therapeutic effects of combined NO delivery systems are primarily not dependent on NO but rather on other therapeutic methodologies including chemotherapy, radiation therapy, photothermal therapy, and photodynamic therapy. S-nitrosothiol–conjugated upconversion NP enhanced the therapeutic effects of radiation therapy (Fan et al., 2015), and docetaxel- and SNP-containing mesoporous Prussian blue NPs improved the therapeutic effects of docetaxel and photothermal therapy (Feng et al., 2019). The enhanced perfusion by a low dose of DNIC NPs facilitated the improved penetration of DOX and tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) protein to the tumor, which enhanced the therapeutic effects of DOX and TRAIL although the low dose of DNIC NPs did not elicit antitumor effects by themselves (Sung et al., 2019). 1,3-Bis-(2,4,6-trimethylphenyl)imidazolylidene nitric oxide and DOX-loaded micelles enabled to enhance the therapeutic effects of DOX by promoting vasodilation of tumor blood vessels (Kang et al., 2019). GSNO and chlorine e6-containing zeolitic imidazolate framework with cancer cell membrane exhibited the enhanced therapeutic effects compared with control groups (An et al., 2020). Nitrate-conjugated polymers promoted the antitumor effects of DOX-conjugated polymers (Studenovsky et al., 2018) and PTX-containing nitrate conjugated bovine serum albumin–protected gold nanocluster facilitated to encourage the antitumor effects of chemo and photothermal therapy (Liu, Xiao et al., 2018). DOX-loaded and S-nitrosothiol–conjugated mesoporous silica NPs showed higher antitumor effects than each DOX-loaded or S-nitrosothiol–conjugated one (Dong et al., 2019). Despite the therapeutic potential of combined NO delivery systems in allograft tumor models, most did not evaluate the associated immune response through immune phenotyping studies (Fan et al., 2015; Liu, Xiao et al., 2018; Studenovsky et al., 2018; Dong et al., 2019; Feng et al., 2019; Kang et al., 2019; Sung et al., 2019; An et al., 2020).
Our group demonstrated the potent immune response achieved through the combination of chemo- and NO-mediated cancer therapy by employing a B16F10-OVA dual tumor model (Kim et al., 2020). Interestingly, intratumoral administration of free PTX + free GSNO and pPTX/pCD-pSNO into the 1° tumor (PTX contents equivalent to 10 mg/kg PTX and NO contents equivalent to 570 µg/kg GSNO) showed similar antitumor effects on the 1° tumor compared with free PTX and pPTX/pCD-pSH, respectively. On the other hand, the intratumoral administration of free GSNO, free PTX+free GSNO, and pPTX/pCD-pSNO into the 1° tumor exhibited negligible antitumor effects on the 2° tumor compared with saline, while free PTX and pPTX/pCD-pSH improved the growth of 2° tumor. These results imply that NO mitigates systemic immune suppression induced by chemotherapeutic PTX but does not enhance its cytotoxic effects. Specifically, in response to pPTX/pCD-pSNO administration into the 1° tumor, MDSCs were expanded in the 1° tumor and its draining lymph nodes (1° dLN), and antigen-specific CD8+ T cells (tetramer+ CD8+ T) were significantly suppressed in the 1° dLN despite the activation (CD86+) and expansion of DCs (CD11b+CD11c+) in the 1° dLN, spleen, and 2° tumor, and expansion of NK cells (CD3-NK1.1+) in the dLN and M1 (CD11b+F4/80+CD86+) in spleen and 2° tumor.
Binding of CD28 on T cells to CD80/CD86 on APCs results in activation of T cells, while that of CTLA-4 on T cells to CD80/CD86 dampens the activations of T cells (Pardoll, 2012; Shin and Ribas, 2015; Burugu et al., 2018; Marin-Acevedo et al., 2018). As CTLA-4 has much higher affinity to CD80/CD86 than CD28, the antagonistic monoclonal antibodies to CTLA-4 facilitates the activation and expansion of antigen-specific CD8+ T cells (Pardoll, 2012; Shin and Ribas, 2015; Burugu et al., 2018; Marin-Acevedo et al., 2018). Based on the observed locoregional expansion of CD86-activated DCs resulting from administration of pPTX/pCD-pSNO into the 1° tumor (Kim et al., 2020), our group exploited antagonistic aCTLA-4 to overcome the attenuated expansion and antigen-presentation of CD8+ T cell by activated DCs and M1 macrophages (Kim et al., 2020). In so doing, intratumoral administration in the 1° tumor of dual melanoma-bearing animals with pPTX/pCD-pSNO in combination with aCTLA-4 showed improved control of both 1° and 2° (abscopal) tumors, which led to prolonged animal survival, whereas aCTLA-4 alone elicited marginal and negligible therapeutic benefits.
Inspired by reports that NO controls transcription factor AP-1 (Tabuchi et al., 1994) that potentially modulates metabolism of CTLA-4 (Valk et al., 2008), we further unveiled a mechanism associated with immunosuppressive functions of exogenous NO (Kim, Francis et al., 2022). Subcutaneous administration of GSNO (570 μg/kg) into naïve mice was found to upregulate the extra- and intracellular expression of CTLA-4 by F4/80−cDCs (CD11b+CD11c+F4/80−), CD11c− macrophages (CD11b+CD11c−F4/80+), and MDSCs in lymph nodes draining the injection site. It is important note that CTLA-4 can be expressed on cancer cells, DCs, and MDSCs in addition to T cells, which contributes to the suppression of T cell priming, differentiation, and function (Liu et al., 2009; Halpert et al., 2016; Chen et al., 2017; Hargadon, 2020; Kim, Francis et al., 2022). Considering that GSNO expanded and activated DCs as well (Kim et al., 2020; Kim, Francis et al., 2022), these results are interestingly in line with previous reports that NO donors expanding both effector and regulatory DCs (Si et al., 2016). However, intratumor administration of GSNO (570 μg/kg) in the B16F10-OVA dual tumor model resulted in the slight acceleration of both treated (1°) and untreated abscopal (2°) tumor growth, despite no effects on tumor cell proliferation and immunogenicity including CRT, CTLA-4, PD-1, and PD-L1 by tumor cells themselves in vitro and in vivo, respectively. These results imply that protumoral effects of GSNO may be attributed to the immunosuppression mediated by CTLA-4 expressing F4/80−cDCs, CD11c− macrophages, and MDSCs, which are also associated with the suppressed expansion, activation, and priming of CD8+ T cell despite the presence of activated and expanded DCs that result from the administration of GSNO. Accordingly, our group hypothesized that the cotreatment of aCTLA-4 could unleash the functions of DCs activated by GSNO if immunosuppressive functions of CTLA-4–expressing immune cells induced by GSNO were antagonistically suppressed (Fig. 5). In testing this hypothesis by coadministration of GSNO intratumorally into the 1° tumor and aCTLA-4 intraperitoneally, growth of both 1° and 2° tumors was observed that corresponded with significant expansions of total, activated CD25+ and LAG3+, antigen-experienced PD-1+, antigen-specific tetramer+ CD8+ T cells, NK cells, and NK T cells in the blood. Given their potent immunotherapeutic synergies, we sought to explore the potential for sustained corelease of NO and aCTLA-4 antitumor immunotherapy (Kim, Francis et al., 2022). F127-grafted gelatin (F127-g-Gelatin) polymer was developed as a lower critical solution temperature polymer to exhibit a gelation at low polymer concentrations (4.0 − 7.0 wt%) at temperatures above 29°C to 31°C. The thermosensitive hydrogel could load GSNO and aCTLA-4 via a simple physical mixing, which allowed to release drugs over sustained times (days) both in vitro and in vivo. As hypothesized, the intratumoral sustained release of GSNO and aCTLA-4 with F127-g-Gelatin hydrogel significantly improved the therapeutic index in the dual B16F10-OVA melanoma model as well as a dual 4T1 breast tumor model, compared with treatment with free GSNO and aCTLA-4 (Kim, Francis et al., 2022).

The effects of exogenous NO on CTLA-4 expression and its potential to potentiate the effects of aCTLA-4 immunotherapy. Exogenous NO induces not only the maturation and activation of DCs but also the elevated expression of CTLA-4 on DCs, macrophages, and MDSCs, which suppresses CD8+ T cell priming and expansion. Cotreatment with aCTLA-4 inhibits CTLA-4-mediated immunosuppression, which results in increased efficiency of DC-mediated CD8+ T cell instruction that is otherwise suppressed by CTLA-4 expressing immune cells. Figure adapted from Kim, Francis et al. (2022).
Synthesizing what can be concluded from the three aforementioned investigations (Sung et al., 2019; Kim et al., 2020; Kim, Francis et al., 2022) (Table 4), NO delivered to the tumor and/or its dLNs promoted the local expansion of macrophages and DC populations, which consist of both effector and regulatory phenotypes (Si et al., 2016; Sung et al., 2019; Kim et al., 2020; Kim et al., 2022). However, MDSCs and Tregs were expanded as well (Kim et al., 2020; Kim et al., 2022), which contributed to fostering an immunosuppressive environment (Nagaraj et al., 2007; Nagaraj et al., 2010; Pan et al., 2010; Molon et al., 2011; Jayaraman et al., 2012; Nagaraj et al., 2013; Markowitz et al., 2017; Stiff et al., 2018; Kim et al., 2020). Accordingly, delivery of high concentrations of NO failed to facilitate effective cancer immunotherapy (Sung et al., 2019; Kim et al., 2020; Kim, Francis et al., 2022). However, lower NO concentrations appeared to exert lower immunosuppressive functions, which allowed the CD8+ T- and M1 macrophage-mediated antitumor immunotherapy in a codelivery with a vaccine (Sung et al., 2019). In particular, aCTLA-4 codelivery with NO led to efficient tumor control by rescuing the immunosuppressive environments fostered by CTLA-4 expressing DCs, macrophages, and MDSCs induced by NO, indicating controlled delivery of NO donors with synergistic agents in combination represents a new potential strategy in NO mediated anticancer immunotherapy (Kim, Francis et al., 2022). Lastly, sustained release of aCTLA-4 and NO through the development of a novel drug-laden micelle-releasing thermosensitive hydrogel not only improved local and systemic therapeutic effects of the drug combination but also achieved dose sparing and obviated the need for repeated administration (Kim, Francis et al., 2022), further demonstrating the utility and potential of sustained delivery systems in NO mediated antitumor therapy.
Immunotherapeutic effects of delivered NO in preclinical in vivo tumor models
VII. Conclusions and Perspective
In summary, exogenous NO directly modulates the immunogenicity of cancer cells and the functions, activation, and differentiation of multiple immune cell subtypes that influence their complex interdependent immune stimulatory or immune-suppressive interactions relevant to antitumor therapy. The application of exogenous sources of NO improves the immunogenicity of cancer cells, expands the populations of activated DCs and M1-polarized macrophages, and helps to differentiate CD4+ T cells into Th1, Th2, Th17, and NO Tregs, while simultaneously manifesting immunosuppressive effects as well that include expansion of regulatory DCs, suppression of intracellular antigen process of DCs and B cells, inhibition of CTL differentiation, proliferation, and migration, and expansion of MDSCs. Three recent studies suggest new ways to exploit these immune-stimulatory functions more selectively; one involves the control of NO dose using a DDS (Sung et al., 2019), and the others explore combination therapy strategies that leverage NO donors used in combination with other immune-modulatory agents (Kim et al., 2020; Kim, Francis et al., 2022).
Due to the concentration-dependent paradoxical functions of NO, there are currently two strategies in antitumor therapy based on NO delivery systems. High concentrations of NO have the potential to induce direct tumor toxicity while simultaneously inducing systemic immune suppression. Accordingly, DDSs capable of delivering high concentrations of NO have been developed to result in direct killing of tumor cells (Lee et al., 2020; Li, Ji et al., 2020). An unintended consequence of such an approach, however, is accelerated tumor progression resulting from immunosuppression (Kim et al., 2020; Kim, Francis et al., 2022) elicited by NO treatment since current NO delivery systems are limited in their capacity in delivering high concentrations of NO only to target sites (Kim et al., 2014; Kim et al., 2017; Park, Im et al., 2019; Kim, Suh et al., 2022). In addition, high systemic dose of NO has a significant risk of arterial pressure reduction due to the vasodilation effects of NO (Carpenter and Schoenfisch, 2012; Kim, Francis et al., 2022). Low NO concentrations on the other hand appear to induce an immune stimulatory environment but do so to extents insufficient to elicit direct antitumor effects as monotherapies (Sung et al., 2019). In this regard, combined NO delivery systems that deliver low concentrations of NO with other conventional agents with antitumor effects have been explored and have demonstrated promising potential.
Another one of the biggest hurdles to the broader utilization of NO in antitumor immunotherapy is the lack of stability and targeting ability of current NO delivery systems. Although a variety of NO donors are physically loaded or chemically conjugated to the DDSs, the resultant NO delivery systems are primarily based on SNP, S-nitrosothiols, N-diazeniumdiolates, and nitrates, which spontaneously release NO under physiologic conditions (Hrabie and Keefer, 2002; Wang et al., 2002; Riccio and Schoenfisch, 2012; Kim et al., 2014; Kim et al., 2017; Yang, Zelikin et al., 2018; Wu et al., 2021; Yang et al., 2021). To surmount this issue, redox-responsive O2-protected N-diazeniumdiolates (Kumar et al., 2010; Park, Im et al., 2019), SIN-1 (Kim, Suh et al., 2022), UV-responsive NO donors (Pramanick et al., 2018), and ultrasound-responsive NO donors (Kang et al., 2019) have been used to develop stimuli-responsive NO delivery systems that do not spontaneously release NO during storage and blood circulation but does so under specific stimuli. In addition, several systems to use cascade reactions have been developed to not only attenuate NO release from NO donors before use but also to facilitate the target-specific NO release (Garcia et al., 2012; Choi et al., 2016). Other alternative strategies are to deliver L-Arg, a substrate of NOSs, to the tumor microenvironment (Kudo and Nagasaki, 2015; Cao et al., 2016; Fan et al., 2017; Jiang et al., 2018; Wan et al., 2018; Wang et al., 2019; Ma et al., 2020; Tao et al., 2022) or to deliver NOS gene (Chen et al., 2002; Cooney et al., 2007; Sharif et al., 2012), which enable the acceleration of endogenous NO production while preventing unintended NO release during storage and blood circulation.
Overall, the understanding of NO’s effects on the immune system has rapidly expanded in recent years, which opens new opportunities to leverage its therapeutic potential. With the continuous efforts in optimizing dose, use in combination with other synergistic agents, and the development of controlled NO donors and delivery systems, NO’s therapeutic potential marches closer to being fully realized.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Kim, Thomas.
Footnotes
- Received September 21, 2021.
- Revision received May 15, 2022.
- Accepted July 5, 2022.
This work was supported by National Institutes of Health National Cancer Institute [Grant R01-CA207619]. The content is solely the responsibility of the authors and does not necessarily represent the official views of NIH.
Abbreviations
- aCTLA-4
- antagonistic monoclonal antibody to CTLA-4
- APC
- antigen presenting cell
- CRT
- calreticulin
- CTL
- cytotoxic T lymphocytes
- CTLA-4
- cytotoxic T-lymphocyte-associated protein-4
- DC
- dendritic cell
- DDS
- drug delivery system
- DETA-NONOate
- diethylenetriamine nitric oxide adduct–diazeniumdiolates, diazen-1-ium-1, 2-diolates
- dLN
- draining lymph node
- DNIC
- dinitrosyl iron complexes
- DOX
- doxorubicin
- Fas
- CD95/APO-1
- FDA
- US Food and Drug Administration
- GSNO
- S-nitrosoglutathione
- GST
- glutathione S-transferase
- HMGB1
- high mobility group protein B1
- ICD
- immunogenic cell death
- IFN
- interferon
- IL
- interleukin
- iNOS
- inducible nitric oxide synthase
- JS-K
- O2 -(2,4-dinitrophenyl) 1-[(4-ethoxycarbonyl)piperazin-1-yl]diazen-1-ium-1,2-diolate
- LPS
- lipopolysaccharide
- MDSC
- myeloid-derived suppressor cell
- MLR
- mixed lymphocyte reaction
- MM
- multiple myeloma
- MSN
- mesoporous silica nanoparticle
- NIR
- near-infrared
- NK
- natural killer cell
- NO
- nitric oxide
- NO donor
- a nitric oxide–releasing small molecule or functional group
- NONOate
- diazeniumdiolate
- NOR-4
- N-(-4-ethyl-2-hydroxyimino-5-nitrohex-3-enyl)pyridine-3-carboxamide
- NOS
- nitric oxide synthases
- NP
- nanoparticle
- NSAID
- nonsteroidal anti-inflammatory drugs
- PD-L1
- programmed cell death ligand 1
- PEG
- poly(ethylene glycol)
- PLGA
- poly(lactic-co-glycolic acid)
- pMHC
- peptide major histocompatibility complex
- PTX
- paclitaxel
- PVR/CD155
- poliovirus receptor
- RBS
- Roussin’s black salt
- ROS
- reactive oxygen species
- RRS
- Roussin’s red salt
- SIN-1
- 3-morphorlinosydnonimine
- SNAP
- S-nitrosopenicillamine
- SNP
- sodium nitroprusside
- TCR
- T cell receptor
- Th
- T helper cell
- TIGIT
- T cell immunoreceptor with Ig and ITIM domains
- Tip-DC
- tumor necrosis factor– and inducible nitric oxide synthase–producing dendritic cells
- TNF
- tumor necrosis factor
- TRAIL
- tumor necrosis factor–related apoptosis-inducing ligand
- Treg
- regulatory T cell.
- Copyright © 2022 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Visual Overview
- Abstract
- I. Introduction
- II. NO-donors
- III. Design of NO Delivery Systems
- IV. Direct Effects of NO on the Immunogenicity of Cancer Cells In Vitro
- V. Direct Effects of NO on Immune Cells In Vitro
- VI. Recent Advances in Antitumor Immunotherapy Leveraging NO-Delivery Systems In Vivo
- VII. Conclusions and Perspective
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters