
Abstract
Aims/hypothesis
In obesity, beta cells activate compensatory mechanisms to adapt to the higher insulin demand. Interleukin-1 receptor antagonist (IL-1Ra) prevents obesity-induced hyperglycaemia and is a potent target for the treatment of diabetes, but the mechanisms of its secretion and regulation in obesity are unknown. In the present study, we hypothesise the regulation of IL-1Ra secretion by purinergic P2X7 receptors in islets.
Methods
Production and regulation of P2X7 were studied in pancreatic sections from lean and obese diabetic patients, non-diabetic controls and in isolated islets. IL-1Ra, IL-1β and insulin secretion, glucose tolerance and beta cell mass were studied in P2x7 (also known as P2Rx7)-knockout mice.
Results
P2X7 levels were elevated in beta cells of obese patients, but downregulated in patients with type 2 diabetes mellitus. Elevated glucose and non-esterified fatty acids rapidly activated P2X7 and IL-1Ra secretion in human islets, and this was inhibited by P2X7 blockade. In line with our results in vitro, P2x7-knockout mice had a lower capacity to secrete IL-1Ra. They exhibited severe and rapid hyperglycaemia, glucose intolerance and impaired beta cell function in response to a high-fat/high-sucrose diet, were unable to compensate by increasing their beta cell mass in response to the diet and showed increased beta cell apoptosis.
Conclusions/interpretation
Our study shows a tight correlation of P2X7 activation, IL-1Ra secretion and regulation of beta cell mass and function. The increase in P2X7 production is one mechanism that may explain how beta cells compensate by adapting to the higher insulin demand. Disturbances within that system may result in the progression of diabetes.
Similar content being viewed by others


Introduction
An alarming increase in obesity and type 2 diabetes mellitus has been recorded among the global population. The rise in type 2 diabetes is tightly connected with a dramatic increase in obesity, a major risk factor for the development of type 2 diabetes [1]. Most obese people have become insulin resistant but compensate for the enhanced insulin demand by increasing insulin secretion [2]. In order to explore how beta cells increase mass under physiological conditions, it is important to stimulate beta cell expansion in vivo and in vitro. The pathology of type 2 diabetes comprises initial hyperinsulinaemia with, thereafter, a continuous decline in beta cell function in association with a progressive decrease in beta cell mass due to an increasing rate of beta cell apoptosis [3]. A previously described mechanism for glucose-induced beta cell apoptosis involves the production and secretion of interleukin-1β (IL-1β) by the beta cell itself [4], which is blocked by an anti-inflammatory cytokine and naturally occurring antagonist of interleukin-1α (IL-1α) and β [5–7], the interleukin-1 receptor antagonist (IL-1Ra) [8]. Similarly to IL-1β, IL-1Ra binds to type 1 and 2 IL-1 receptors (IL-1R1, IL-1R2), but lacks a second binding domain. IL-1Ra does not recruit the IL-1 receptor accessory protein, the second component of the receptor complex, and blocks IL-1R1 signalling. Endogenous production and secretion of IL-1Ra has been shown to limit inflammation and tissue damage.
Exogenous IL-1Ra protects against IL-1β-induced beta cell damage [9, 10], counteracts low-dose-streptozotocin-induced diabetes [11] and autoimmune diabetes [12]. Results from a clinical study show that IL-1Ra improves glycaemic control and beta cell function in patients with type 2 diabetes [13].
The secretory mechanisms and those regulating changes of IL-1Ra are unknown. An increasing body of evidence suggests that IL-1β, as well as intracellular IL-1Ra, is secreted by a leaderless pathway via activation of the P2X7 receptor (P2X7; [14]). P2X7, a member of the purinergic receptor family, is a G-protein-coupled ionotropic receptor for extracellular ATP that forms a pore in response to ligand stimulation, regulating cell permeability, cytokine release and apoptosis [15]. The cytokine IL-1β, which lacks a leader signal sequence, does not follow the classic endoplasmic-reticulum-to-Golgi pathway of secretion. Its secretion is governed by P2X7-mediated pathways [16, 17]. This raises a question about the mechanisms of its release; it has been demonstrated that it is secreted upon activation of P2X7 receptors, leading to shedding of microvesicles that contain the cytokine [18].
The pore-forming ATP receptor P2X7 is also responsible for IL-1Ra release in endothelial cells and macrophages [14]. Consequently, we hypothesised that P2X7 could be involved in IL-1Ra release in beta cells. The work presented here evaluates an important role for P2X7 in beta cells, not only for cytokine release but also for insulin release and apoptosis. We have found upregulation of P2X7 in islets from obese individuals, while receptors were almost undetectable in type 2 diabetes. We then investigated whether P2X7 is involved in the compensatory mechanisms of the beta cells in response to an increased insulin demand in insulin resistance.
Methods
Animals
P2x7 (also known as P2rx7)-knockout mice (B6.129P2-P2rx7 tm1Gab/J; P2X7-KO) backcrossed to C57BL/6 mice for ten generations and C57BL/6 J wild-type (WT) mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA) at 4 weeks of age. Beginning at 5 weeks of age and continuing for 16 weeks, knockout as well as wild-type animals were fed a normal diet (ND; P2X7-KO-ND and WT-ND, respectively; normal diet was Harlan Teklad Rodent Diet 8604, containing 12.2, 57.6 and 30.2% energy from fat, carbohydrate and protein, respectively) or a high-fat/high-sucrose diet (HFD; P2X7-KO-HFD and WT-HFD; high-fat/high-sucrose diet was Surwit diet, Research Diets, New Brunswick, NJ, USA containing 58, 26 and 16% energy from fat, carbohydrate and protein, respectively [19]). Further details are described in the Electronic supplementary material (ESM).
Intraperitoneal glucose and insulin tolerance tests
At the beginning of the experiment and after 4, 8, 12 and 16 weeks of diet, all animals underwent in vivo studies. For intraperitoneal glucose tolerance tests (IPGTTs), glucose at a dose of 1 g/kg body weight and for intraperitoneal insulin tolerance tests (IPITTs), recombinant human insulin (Novolin; Novo Nordisk, Bagsværd, Denmark) at a dose of 0.5 U/kg was injected intraperitoneally as described before [9].
Islet isolation and culture
After 12 and 16 weeks of diet and treatment, mice were killed, blood was taken by cardiac puncture and serum stored at −80°C until further analysis. Thereafter, islets from all groups were isolated as described previously [20]. Human islets were isolated from pancreases of six healthy organ donors as described previously [21]. Mouse islets were cultured in RPMI-1640 medium (11.1 mmol/l glucose), and human islets in CMRL-1066 medium (5.5 mmol/l glucose), as described previously [22].
Functional analysis of P2X7 in human islets
Freshly isolated human islets were grown on dishes coated with extracellular matrix until a monolayer formed. Cells were loaded with 10 μmol/l YO-PRO-1 (0.375 kDa; Molecular Probes/Invitrogen, Carlsbad, CA, USA) in PBS after a protocol by Narcisse et al. [15]. Further details are described in the ESM.
Glucose-stimulated insulin secretion
Islets used to perform glucose-stimulated insulin secretion (GSIS) experiments were kept in culture medium for 24 h and GSIS was performed as described previously [9]. Islets were extracted with 0.18 mol/l HCl in 70% (vol./vol.) ethanol for determination of insulin content. Islet insulin was determined using mouse or human insulin ELISA (ALPCO, Salem, NH, USA).
IL-1Ra and IL-1β analysis
Serum obtained from retro-orbital bleedings and cell culture supernatant fractions from mouse isolated islets were evaluated using mouse IL-1Ra or IL-1β Quantikine ELISA (R&D Systems, Minneapolis, MN, USA) (see ESM).
Beta cell mass and histochemical analyses
After 12 weeks of diet and treatment, pancreases were weighed and fixed overnight in 4% (vol./vol.) paraformaldehyde at 4°C under continuous shaking followed by paraffin embedding, orienting pancreases such that sections were cut along the head–tail axis. Beta cell mass and apoptosis were analysed as described previously [8] (P2X7 staining is described in the ESM). Human pancreatic tissue was obtained at autopsy from non-diabetic individuals and from people with type 2 diabetes (all with documented fasting plasma glucose >8.048 mmol/l). ESM Table 1 summarises age, BMI, sex, glucose levels and diagnosis.
RNA extraction and PCR analysis
The total RNA of isolated islets was extracted after overnight culture as described previously [8]. The primers were: P2X7 5′AGATCGTGGAGAATGGAGTG3/5′TCTCGTGGTGTAGTTGTGG3′; tubulin 5′TCCACGTTGGCCAGGCTGGTGTCCAG3′/5′CTGTGATGAGCTGCTCAGGGTGG3′.
Western blot analysis
Islet lysates were prepared from 100 islets per treatment condition as described previously [23]. PVDF filters (Amersham Biosciences/GE Healthcare, Uppsala, Sweden) were incubated with rabbit anti-P2X7 (Almone Labs, Jerusalem, Israel), rabbit-anti-IL-1Ra (R&D Systems) and anti-actin (#4967; Cell Signaling Technology, Beverly, MA, USA), followed by incubation with horseradish-peroxidase-linked anti-rabbit IgG.
Statistical analysis
Samples were evaluated in a randomised manner by two investigators (R. Glas and K. Maedler) who were blinded to the treatment conditions. Data are presented as means ± SE and were analysed by paired Student’s t test or by analysis of variance with a Bonferroni correction for multiple group comparisons.
Results
P2X7 is produced in beta cells, upregulated in obesity and decreased in diabetes
To investigate the role of P2X7 in islets, we analysed its production in isolated human islets. The presence of P2X7 transcripts is shown by mRNA analysis (Fig. 1a) and western blotting (Fig. 1b). The majority of the beta cells stained positive for P2X7 (Fig. 1c). We also found the presence of P2X7 in alpha cells (not shown). To analyse regulation of P2X7 in vivo, we compared staining and production of P2X7 in beta cells in pancreatic sections obtained at autopsy from both obese and lean non-diabetic individuals and patients with type 2 diabetes. Double immunostaining for P2X7 and insulin (Fig. 1d) confirmed the presence of P2X7 localised in the beta cells in human pancreatic sections from lean control patients (first row), which was highly upregulated in obese non-diabetic patients (second row). In contrast, in both lean (third row) and obese (fourth row) diabetic groups, P2X7 was barely detectable; no islet P2X7 receptors were found in any of the diabetic pancreases.

P2X7 is produced in beta cells, upregulated in obesity and decreased in diabetes. a Agarose gel of the PCR products P2X7 and tubulin obtained using cDNA from isolated human islets that was pretested for insulin production; mRNA served as a negative control (−). RT, reverse transcriptase. b Western blot to detect P2X7 in human islets. c Immunofluorescence to detect P2X7 and insulin in human pancreatic islets cultured for 24 h at 5.5 mmol/l glucose. P2X7 is shown in red and insulin, a marker for beta cells, in green. Co-localisation (‘Merge’) is visible as yellow. The results are representative of at least three independent experiments using islets from three donors. d Paraffin-embedded human pancreatic sections from a lean non-diabetic control (first row), an obese control (second row), a lean patient with type 2 diabetes (third row) and an obese patient with type 2 diabetes (fourth row) were analysed for the presence of P2X7 (shown in red) and insulin (green). Co-localisation is visible in yellow. The results represent five independent cases from each group
P2X7 is upregulated by short-term exposure to elevated glucose and NEFA in isolated human islets
The functional production of P2X7 in beta cells was verified with the nucleic acid stain YO-PRO-1 (0.375 kDa, Molecular Probes). This is based on the pore-forming character of P2X7; its activation leads to the formation of a pore that is passable to molecules up to 0.9 kDa [24]. YO-PRO-1 has been widely used to visualise P2X7 activity [15], although YO-PRO-1 can also access the beta cell in the presence of membrane blebbing during apoptosis and is also used for the detection of apoptotic cells [25]. Freshly isolated human islets plated on dishes coated with extracellular matrix were loaded with 10 μmol/l YO-PRO-1 10 min before receptor activation by addition of 100 μmol/l benzoyl ATP (BzATP). Within 5 min of exposure, YO-PRO-1 was taken up by 45 ± 6.01% of the islet cells; this was inhibited by pre-incubation with the specific P2X7 inhibitor KN-62, which has been demonstrated to block YO-PRO-1 uptake initiated by prolonged P2X7 activation previously [26]. The ATP-degrading enzyme apyrase (a less specific inhibitor of P2X7) had similar effects on YO-PRO-1 uptake (Fig. 2a). These data show production of functional P2X7 in islets. In contrast, <5% of the untreated cells showed YO-PRO-1 uptake.

P2X7 is upregulated by short-term exposure to elevated glucose and NEFA in human isolated islets. a–d Freshly isolated human islets were grown on dishes coated with extracellular matrix until monolayer formation (72 h). Uptake of the nucleic stain YO-PRO-1 was allowed by inducing P2X7 activation via stimulation with a 100 μmol/l BzATP (black triangles), b 33.3 mmol/l glucose (black triangles), c 11.1 mmol/l glucose (black triangles) and d 0.5 mmol/l palmitate at 5.5 mmol/l glucose (black triangles), respectively, or inhibited with 30 min pre-incubation with the specific inhibitor of P2X7, KN-62 (crosses), or apyrase (white squares). As the control, human islets were incubated in media containing 5.5 mmol/l glucose (black squares). Quantification was accomplished by verifying YO-PRO-1 emission at 488 nm and counting fluorescence nuclei per visual field (four fields per condition) every 5 min for 1 h; n = 3 experiments from three different donors. Data are expressed as percentage of YO-PRO-positive islet cells. e P2X7 mRNA levels and f, g P2X7 protein levels from human islets in response to incubation with 11.1 mmol/l or 33.3 mmol/l glucose or palmitate (NEFA) for 10 min. The results are presented after normalising to tubulin mRNA levels and as relative numbers to the corresponding unstimulated control (5.5 mmol/l glucose). Actin was used as loading control. h IL-1Ra and i insulin secretion from human islets exposed for 1 h to elevated glucose concentrations or palmitic acid (NEFA) at 5.5 mmol/l glucose with (black bars) or without (white bars) 30 min pre-treatment with KN-62. IL-1Ra was analysed by the LINCOplex technology and insulin by ELISA from the same cell culture supernatant fractions, which were collected from three independent experiments from three donors from three dishes for each experiment. Data are shown as means of insulin divided by corresponding insulin content ± SE and normalised to control conditions at 5.5 mmol/l glucose (i only). e–i Values on x-axis are glucose (mmol/l). j The rise in IL-1Ra was confirmed by a time-dependent experiment using isolated human islets exposed to 11.1 and 33.3 mmol/l glucose and 0.5 mmol/l palmitate (NEFA) for 30 min to 24 h. Western blot analysis was performed for IL-1Ra and β-actin as loading control on the same blot. Representative blots from three experiments from three donors are shown. *p < 0.05 vs untreated control at 5.5 mmol/l glucose; **p < 0.05 vs islets treated with BzATP, 11.1 or 33.3 mmol/l glucose or NEFA, respectively
As we have observed changes in P2X7 levels in obesity and diabetes, we tested whether P2X7 is activated by a diabetic milieu in culture. Islets were exposed to increased glucose concentrations (5.5–33.3 mmol/l, Fig. 2b,c) or 0.5 mmol/l palmitic acid (Fig. 2d) for 10 min after YO-PRO-1 loading. Elevated glucose concentration activated P2X7, as did palmitic acid; we observed a nine-fold activation by 33.3 mmol/l glucose, six-fold activation by 11.1 mmol/l glucose and a nine-fold activation by palmitic acid within 5 to 10 min of exposure (p < 0.001). Peak levels slightly decreased after 15 min exposure, but remained elevated compared with control conditions throughout the 1 h incubation period, similar to the finding with BzATP stimulation. In all assays, co-culture with the P2X7 inhibitors KN-62 (Fig. 2a–d) and apyrase (Fig. 2a, b, d), reduced the P2X7 activation induced by BzATP, elevated glucose or palmitic acid.
As BzATP evokes P2X7 effects very briefly, even within 1 to 30 s [27], we wanted to see if there is a change in P2X7 level in response to the activation. Elevated glucose concentrations increased P2X7 mRNA levels (Fig. 2e: 1.7-fold increase with 11.1 mmol/l glucose, 2.5-fold increase with 33.3 mmol/l glucose and 1.8-fold increase with palmitic acid, compared with control [untreated, with 5.5 mmol/l glucose], p < 0.05) after 10 min of exposure. This was confirmed by western blot analysis (Fig. 2f, g). Changes in mRNA level and protein production were detected within this short time, whereas values returned to basal after 24 h of exposure (not shown). Previous reports show that IL-1β induces increases in P2X7 at 6 h, with levels decreasing after 48 h of treatment [15]. One explanation for the increase in P2X7 within this narrow timeframe would be a rapid de novo synthesis of P2X7 mRNA and P2X7 protein, given weight by its relatively small size (595 amino acids/68 kDa).
Recently, we have shown that IL-1Ra can protect from impaired beta cell function and death in vivo and in vitro [4, 8]. The mechanisms of its secretion from the beta cell are unknown. We tested whether activation of P2X7 leads to IL-1Ra secretion. Long-term glucose exposure had no effect on IL-1Ra secretion (data not shown and [28, 29]). But short-term (30 min) exposure of islets to elevated glucose, palmitic acid or BzATP induced IL-1Ra secretion, which was inhibited by KN-62 (Fig. 2h). Analysis of insulin secretion in the same supernatant fractions showed similar regulation of insulin secretion. Elevated glucose levels (11.1 and 33.3 mmol/l), 0.5 mmol/l palmitic acid and 100 μmol/l BzATP induced insulin secretion, which was prevented by co-culture with KN-62 (Fig. 2i).
Regulation of IL-1Ra production by glucose and palmitic acid was confirmed by western blot analysis. Short-term exposure of human islets to elevated glucose and palmitic acid (30 min–8 h) induced IL-1Ra secretion, whereas no differences were observed at 24 h treatment. These data indicate a parallel induction of IL-1Ra and P2X7 in pancreatic islets.
P2X7-deficient mice have impaired glucose tolerance, beta cell mass and function and fail to adapt IL-1Ra secretion to a high-fat diet
The changes in P2X7 activation in human islets in vitro were compared with glucose tolerance, beta cell survival and IL-1Ra secretion in the P2X7-KO mice. Male C57Bl/6 WT and P2X7-KO mice were fed an ND or an HFD for up to 16 weeks. Mice fed the HFD gained more weight than the ND control group; this was not influenced by the P2X7-deficiency (Fig. 3a). Before and throughout the treatment period, we measured the fasting and fed blood glucose level every 4 weeks. Average fasted glucose levels before the beginning of the study in 5-week-old mice were 3.88 ± 0.33 mmol/l in the wild-type mice and 5.27 ± 0.44 mmol/l in the P2X7-KO mice (p < 0.05).

P2X7-deficient mice have impaired glucose tolerance and beta cell mass and function and do not adapt IL-1Ra secretion during a high-fat diet. a Changes in body weight in response to the HFD were measured every week during the study and results are shown from WT-ND (black squares), WT-HFD (black triangles), P2X7-KO-ND (white squares) and P2X7-KO-HFD (white triangles) mice from three different studies (12 mice per group). b Blood glucose following i.p. injection of glucose at 1 g/kg body weight are shown from WT-ND (black squares), WT-HFD (black triangles), P2X7-KO-ND (white squares) and P2X7-KO-HFD (white triangles) mice after 12 weeks of treatment. c Serum insulin levels following i.p. injection of glucose at 1 g/kg body weight are shown from mice after 12 weeks of treatment; open bars show levels at 0 min, solid bars show levels at 30 min. The stimulatory index (d) denotes the ratio of stimulated and basal insulin levels in WT (black bars) and P2X7-KO (white bars) mice. e Blood glucose concentrations in mice undergoing an insulin tolerance test after 12 weeks of treatment. Insulin was injected i.p. at a concentration of 0.5 U/kg body weight; WT-ND (black squares), WT-HFD (black triangles), P2X7-KO-ND (white squares) and P2X7-KO-HFD (white triangles). f Immunostaining for insulin in green (magnification ×125) shows disrupted islets in the P2X7-KO mice, which was confirmed by analysis of beta cell mass (g) in tissue sections spanning the width of the whole pancreas from (1) WT-ND and (2) WT-HFD (black bars), (3) P2X7-KO-ND and (4) P2X7-KO-HFD (white bars) mice after 12 and 16 weeks of diet (n = 4 mice for each group). Beta cell mass per pancreas was estimated as the product of the relative cross-sectional area of beta cells (determined by quantification of the cross-sectional area occupied by beta cells divided by the cross-sectional area of total tissue) and the weight of the pancreas. h Analysis of beta cell apoptosis by triple staining for TUNEL, insulin and DAPI (a fluorescent stain that binds strongly to DNA) was performed on the same pancreatic sections. Results are expressed as percentage of TUNEL-positive beta cells ± SE normalised to control ND islets. The mean number of beta cells scored was 6,239 ± 1,035 for each treatment condition from six individual mice per treatment (solid bars represent WT, open bars P2X7-KO mice). i Serum levels of IL-1Ra were measured after 4, 8, 12 and 16 weeks of diet (black bars represent WT, white bars P2X7-KO mice). a p < 0.05 between mice on ND and HFD; b p < 0.05 between P2X7-KO and WT mice; c p < 0.05 GSIS: 30 min vs 0 min; d p < 0.05 beta cell mass: 16 weeks vs 12 weeks
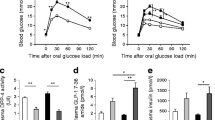
After 12 weeks of the diet, high-fat feeding increased glucose levels and impaired glucose tolerance. The highest glucose levels were found in the P2X7-KO mice fed an HFD, with differences significant at all time points during an IPGTT compared with all other groups (WT-ND, WT-HFD, P2X7-KO-ND; Fig. 3b, p < 0.01). Glucose tolerance further deteriorated after 16 weeks of HFD diet (not shown).
Figure 3c shows serum insulin levels during an IPGTT after 12 weeks of diet and treatment. As previously described [30], high-fat feeding resulted in hyperglycaemia (Fig. 3b) as well as hyperinsulinaemia (Fig. 3c, p < 0.001) compared with control diet mice. Compared with mice consuming an ND, fasted insulin levels were 3.4-fold higher in HFD-fed WT mice and 5.7-fold higher in the P2X7-KO mice (p < 0.01). The HFD-fed WT mice had a much lower capacity to increase their glucose-stimulated insulin release (Fig. 3d, p < 0.01), whereas the P2X7-KO mice on the HFD completely failed to further increase their insulin levels in response to a glucose challenge (Fig. 3d, p < 0.001). P2X7-KO-ND mice had a 1.3-fold lower stimulatory index than the WT control mice (p < 0.05). These data indicate that P2X7-KO mice have an impaired beta cell function, which deteriorates under the challenge of an HFD treatment.
To determine insulin sensitivity, we performed insulin tolerance tests by measuring glucose concentrations after intraperitoneal insulin injection of 0.5 U/kg body weight. Glucose levels from all mice were significantly higher at all time points during the IPITT compared with control wild-type mice on an ND (unchanged in P2X7-KO-ND at 120 min, Fig. 3e). To investigate whether the impaired glucose tolerance and insulin secretion was due to a defect in beta cell survival together with beta cell function, we analysed beta cell mass from the four groups after 12 and 16 weeks of treatment. As reported previously [8], after 12 weeks of diet, islet beta cell mass was increased by high-fat feeding in the WT mice compared with those on a normal diet (1.6-fold increase, p < 0.01; Fig. 3f,g). In contrast, P2X7-KO mice had no such compensatory capacity in response to the HFD. There was no significant change in beta cell mass in the P2X7-KO mice fed the HFD compared with the P2X7-KO-ND mice. Moreover, beta cell mass was significantly decreased compared with the wild-type mice, independent of the diet (1.5-fold with ND and 1.5-fold with HFD, p < 0.0.01, Fig. 3f, g). This was confirmed after 16 weeks of diet. We have previously reported the tendency to beta cell apoptosis in mice after 12 weeks of HFD despite an increased beta cell mass at that time point [8]. Here, we analysed beta cell mass after 12 and 16 weeks of diet. In contrast to 12 weeks, there was no increase in beta cell mass in wild-type mice fed an HFD compared with ND-fed animals after 16 weeks. To investigate whether the changes in beta cell mass correlate with the increased rate of apoptosis, we analysed TUNEL-positive beta cells in pancreatic sections. After 16 weeks of diet, we observed a clear increase in beta cell apoptosis in the control HFD group. This was further increased in the P2X7-KO-HFD mice (1.5-fold raise in apoptosis in the WT-HFD and 11.2-fold in the P2X7-KO-HFD compared with ND controls, p < 0.01, Fig. 3h). As P2X7 regulates IL-1Ra secretion, we tested whether IL-1Ra levels are altered in P2X7-KO mice. The serum concentration of IL-1Ra was measured in WT and P2X7-KO mice during the 16 weeks of HFD feeding. In WT-ND mice, IL-1Ra levels were unchanged during the experiment. IL-1Ra levels were significantly lower in P2X7-KO-ND mice at 8, 12 and 16 weeks. Interestingly, after 4 weeks of diet, IL-1Ra increased 2.2-fold in the WT-HFD mice; these high levels were maintained at 8 weeks (1.9-fold compared with WT) and time-dependently decreased again after 12 and 16 weeks of diet (1.5-fold and 2.4-fold decrease from 8 weeks, respectively). After 16 weeks, IL-1Ra levels with HFD were lower than with the ND (2.6-fold decrease), which correlates with the changes in beta cell mass. P2X7-KO mice, which had beta cells unable to compensate via increasing beta cell mass, had no adaptive increase in IL-1Ra levels (Fig. 3i). We have also measured IL-1β serum levels together with IL-1Ra in the mouse studies. Unfortunately, the levels were very low and mostly under the lower level of the ELISA kit. Positive IL-1β readings were found for some but not all samples from the HFD-fed control mice at 12 and 16 weeks of diet, but none were found in the ND or P2X7-KO mice, making statistical data analysis not possible.
Isolated islets from P2X7-deficient mice display a defect in insulin, IL-1Ra and IL-1β secretion
As we observed impaired glucose tolerance and survival in the P2X7-deficient mice in vivo, we investigated whether impaired beta cell insulin secretion could explain this effect. Islets were isolated from all four treatment groups and insulin secretion into the culture medium was measured. Acute glucose-stimulated insulin secretion was abolished in islets derived from wild-type mice fed a high-fat diet and was further impaired in P2X7-KO-HFD mice (Fig. 4a). The stimulatory index (the ratio of stimulated to basal insulin secretion) was decreased 1.2-fold in the P2X7-KO-ND mice, 2.4-fold in the WT-HFD mice and 3.4-fold in the P2X7-KO-HFD mice compared with WT-ND control mice (Fig. 4b). These data confirm the impaired beta cell function in P2X7-KO mice from our in vivo studies on the level of isolated islets. We then incubated the islets for 8 h at 33.3 mmol/l glucose or 0.5 mmol/l palmitate. As expected, glucose and palmitate increased insulin secretion in the wild-type islets. In contrast, islets from P2X7-KO mice did not increase insulin secretion in response to the stimulation (Fig. 4c). These data confirm the lack of stimulation of insulin secretion in the P2X7-KO mice. Under the same culture conditions, 72 h incubation of islets with 0.5 mmol/l palmitate increased IL-1Ra as well as IL-1β secretion from the wild-type but not the P2X7-KO islets (Fig. 4d).

Isolated islets from P2X7-deficient mice display a defect in insulin secretion. a, b For GSIS experiments, islets from all four treatment groups (WT-ND, WT-HFD, P2X7-KO-ND and P2X7-KO-HFD) were isolated after 12 weeks of diet and pre-cultured for 24 h on dishes coated with extracellular matrix. a Glucose-stimulated insulin secretion of islets. Basal (white bars) and stimulated (black bars) insulin secretion during successive 1 h incubations at 2.8 mmol/l (basal) and 16.7 mmol/l (stimulated) glucose divided by the corresponding insulin content and expressed as means ± SE, normalised to control conditions at 11.1 mmol/l glucose. b The stimulatory index denotes the ratio between stimulated and basal values of insulin secretion. The assay was performed in triplicate with samples from four individual mice (black bars represent WT, white bars P2X7-KO mice). c Chronic insulin secretion over 8 h from islets isolated from ND-fed WT (black bars) and P2X7-KO (white bars) mice exposed to 11.1 or 33.3 mmol/l glucose, or 11.1 mmol/l glucose and 0.5 mmol/l palmitate (NEFA). Values on x-axis shown glucose (mmol/l). Glucose and palmitate stimulated insulin secretion in the wild-type but not in the P2X7-deficient islets. d IL-1Ra and IL-1β secretion from the same islets after 72 h of treatment. Values on x-axis shown glucose (mmol/l). a p < 0.05 GSIS: stimulated vs basal glucose; b p < 0.05 between ND- and HFD-treated mice; c p < 0.05 between P2X7-KO and WT mice; d p < 0.05 treated (33.3 mmol/l glucose or NEFA) vs untreated control
Discussion
The present study shows that P2X7 is produced in human islets in the beta cell and is regulated during the progression of type 2 diabetes. In human obesity, P2X7 is produced in large amounts, but is downregulated in pancreases obtained from patients with type 2 diabetes, in lean as well as in obese individuals. These data suggest that P2X7 plays an important role in beta cell compensation. This is confirmed in our subsequent studies using P2X7-KO mice. These mice show impaired glucose tolerance and glucose-stimulated insulin secretion together with an increased number of apoptotic beta cells, which leads to a decrease in beta cell mass. When P2X7-KO mice were challenged with a diet enriched with fat and sucrose, they became severely glucose intolerant and were completely unable to upregulate insulin secretion in response to glucose during an IPGTT in vivo or a GSIS assay in vitro. Although they had increased basal insulin levels, which indicate an attempt to adapt for the higher insulin demand, they were unable to compensate by increasing beta cell mass during the diet. Beta cell mass adaptation is an important mechanism to maintain normoglycaemia in obese insulin-resistant individuals, in whom beta cell mass is almost doubled compared with lean individuals [31]. Similar increases in beta cell mass in diet-induced obesity were measured in mice using a diet enriched with fat in a previous [32] or the present study. Interestingly, the high-fat/high-sucrose ‘Surwit diet’ induces an early reduction in beta cell proliferation at a time point when beta cell mass is still increased [8], indicating a development towards fewer cells. This tendency is substantiated in the present study, where we show a decrease in beta cell mass with time together with an induction of beta cell apoptosis after 16 weeks of diet. In comparison, P2X7-KO mice show a higher apoptosis rate under the ND as well as under the HFD. The increase in basal insulin levels in response to the diet together with a defect in the compensation of beta cell mass indicates that, despite the lower beta cell mass, high amounts of insulin can be secreted. However, the functional failure is apparent in the abolished response to glucose stimulation. A complete loss of P2X7 was also observed in the NOD mouse with manifest diabetes [33].
Little is known about the purinergic receptor family in the beta cell and the effects on insulin release are controversial. A low concentration of ATP stimulates insulin secretion via the metabotropic purinergic receptor subtype P2Y rather than via P2X receptors (the ionotropic selective cation channel receptors) [34]. P2X7 was found in rat alpha but not beta cells [33], its production in human islets has not been studied before. Here, we show clearly the production and regulation of P2X7 in the beta cell.
Fibroblasts from donors with type 2 diabetes have increased P2X7-mediated responses compared with non-diabetic donors, but levels of P2X7 were unchanged [24]. In contrast, we report here a decrease in the level of P2X7 in beta cells in type 2 diabetes. Consistent with the results from fibroblasts, P2X7 activation was enhanced in isolated islets by a diabetic milieu (high glucose and palmitic acid), an effect prevented by P2X7 inhibition. High glucose induces caspase 3 activation in beta cells as well as in fibroblasts. It is possible that beta cell P2X7 receptors are activated and produced in high numbers at a time when IL-1Ra is highly secreted (e.g. in obesity) and when diabetes develops, levels are lowered in specific cells.
Our in vitro studies on isolated islets show that P2X7 is directly involved in insulin secretion. Activation of P2X7 by BzATP (which specifically targets P2X7 receptors [27, 35, 36]) or acute stimulation of insulin secretion by glucose or palmitic acid for 1 h resulted in insulin release that was partially inhibited by two P2X7 inhibitors.
Although secretory pathways of insulin and IL-1Ra are distinct, one possible common step could be the calcium exchange; activation of P2X7 increases intracellular calcium levels by calcium influx from the extracellular space. This leads to activation of the leaderless IL-1Ra-releasing pathway through pore formation. Increased intracellular calcium levels would also be a trigger for exocytosis of insulin-containing secretory granules.
High glucose can also activate P2X7 in fibroblasts [37]. In parallel, BzATP, glucose and palmitic acid induced short-term secretion of IL-1Ra, which was also inhibited by KN-62. Apyrase, a less specific P2X7 inhibitor that affects the ligands of all P2X7 receptors, showed similarities to the specific P2X7 inhibitor KN-62 in blocking the effect of BzATP on islets, suggesting that P2X7 is mainly activated by glucose and palmitic acid in islets.
Intracellular IL-1Ra has no signal peptide. A possible mechanism for release of intracellular IL-1Ra from the cells to bind to IL-1R1 is through vesicle shedding, which occurs through activation of the P2X7 by ATP [38]. Our data indicate that IL-1Ra secretion is an ATP-dependent process that requires P2X7 activation. This may be how intracellular IL-1Ra, as well as IL-1β, is released from beta cells. Similar results are obtained from macrophages and endothelial cells, demonstrating that intracellular IL-1Ra and IL-1β are secreted by an extracellular ATP-dependent mechanism involving P2X7 activation [14]. We hypothesised that the balance of IL-1β and IL-1Ra is crucial for the onset of diabetes. P2X7 receptors have a dual role in this system: they mediate the release of both pro-inflammatory IL-1β and anti-inflammatory IL-1Ra. From in vitro experiments we know that an excess of 10- to 1,000-fold IL-1Ra is needed to inhibit the IL-1β effects, dependent on the exposure time [4, 9]. Βeta cell dysfunction and increased apoptosis may occur rapidly when IL-1Ra levels are decreased.
Corresponding with the levels of P2X7, increased IL-1Ra serum levels correlate with obesity and insulin resistance [39–42], whereas IL-1Ra is decreased in type 2 diabetes [43]. Recent results from the Whitehall II study show that IL-1Ra levels are increased before the onset of type 2 diabetes [44], which is consistent with our findings in mice fed with an HFD. The increase in IL-1Ra could be an attempt by the body to counteract the deleterious effects of IL-1β and to preserve beta cell survival, insulin secretion and insulin sensitivity. In contrast, it has been speculated that increased IL-1Ra could have an additional metabolic effect that leads to insulin resistance. This is not supported by our results, as daily IL-1Ra injections over 12 weeks did not impair insulin sensitivity in C57Bl/6 mice in a previous study [8], and increased IL-1Ra levels did not lead to changes in insulin sensitivity in the present study. As shown by the bell-shaped curve in Fig. 3i, decreased IL-1Ra levels occurred together with the increase in beta cell apoptosis and the development of diabetes in our study. It will now be important to determine whether long-standing type 2 diabetes leads to decreased IL-1Ra levels in humans in a follow-up study.
Abbreviations
- BzATP:
-
Benzoyl ATP
- GSIS:
-
Glucose-stimulated insulin secretion
- HFD:
-
High-fat/high-sucrose diet
- IL-1:
-
Interleukin-1
- IL-1Ra:
-
Interleukin-1 receptor antagonist
- IPGTT:
-
Intraperitoneal glucose tolerance test
- IPITT:
-
Intraperitoneal insulin tolerance test
- ND:
-
Normal diet
- P2X7-KO:
-
P2x7 knockout
- P2X7 :
-
P2X7 receptor
- WT:
-
Wild-type
References
Zimmet P, Alberti KG, Shaw J (2001) Global and societal implications of the diabetes epidemic. Nature 414:782–787
Martin BC, Warram JH, Krolewski AS, Bergman RN, Soeldner JS, Kahn CR (1992) Role of glucose and insulin resistance in development of type 2 diabetes mellitus: results of a 25-year follow-up study. Lancet 340:925–929
Donath MY, Halban PA (2004) Decreased beta-cell mass in diabetes: significance, mechanisms and therapeutic implications. Diabetologia 47:581–589
Maedler K, Sergeev P, Ris F et al (2002) Glucose-induced beta-cell production of interleukin-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 110:851–860
Dinarello CA (2000) The role of the interleukin-1-receptor antagonist in blocking inflammation mediated by interleukin-1. N Engl J Med 343:732–734
Seckinger P, Lowenthal JW, Williamson K, Dayer JM, MacDonald HR (1987) A urine inhibitor of interleukin 1 activity that blocks ligand binding. J Immunol 139:1546–1549
Seckinger P, Williamson K, Balavoine JF et al (1987) A urine inhibitor of interleukin 1 activity affects both interleukin 1 alpha and 1 beta but not tumor necrosis factor alpha. J Immunol 139:1541–1545
Sauter NS, Schulthess FT, Galasso R, Castellani LW, Maedler K (2008) The antiinflammatory cytokine interleukin-1 receptor antagonist protects from high-fat diet-induced hyperglycemia. Endocrinology 149:2208–2218
Eizirik DL, Tracey DE, Bendtzen K, Sandler S (1991) An interleukin-1 receptor antagonist protein protects insulin-producing beta cells against suppressive effects of interleukin-1 beta. Diabetologia 34:445–448
Giannoukakis N, Rudert WA, Ghivizzani SC et al (1999) Adenoviral gene transfer of the interleukin-1 receptor antagonist protein to human islets prevents IL-1beta-induced beta-cell impairment and activation of islet cell apoptosis in vitro. Diabetes 48:1730–1736
Sandberg JO, Andersson A, Eizirik DL, Sandler S (1994) Interleukin-1 receptor antagonist prevents low dose streptozotocin induced diabetes in mice. Biochem Biophys Res Commun 202:543–548
Nicoletti F, Di Marco R, Barcellini W et al (1994) Protection from experimental autoimmune diabetes in the non-obese diabetic mouse with soluble interleukin-1 receptor. Eur J Immunol 24:1843–1847
Larsen CM, Faulenbach M, Vaag A et al (2007) Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 356:1517–1526
Wilson HL, Francis SE, Dower SK, Crossman DC (2004) Secretion of intracellular IL-1 receptor antagonist (type 1) is dependent on P2X7 receptor activation. J Immunol 173:1202–1208
Narcisse L, Scemes E, Zhao Y, Lee SC, Brosnan CF (2005) The cytokine IL-1beta transiently enhances P2X7 receptor expression and function in human astrocytes. Glia 49:245–258
Piccini A, Carta S, Tassi S, Lasiglie D, Fossati G, Rubartelli A (2008) ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc Natl Acad Sci U S A 105:8067–8072
Ferrari D, Pizzirani C, Adinolfi E et al (2006) The P2X7 receptor: a key player in IL-1 processing and release. J Immunol 176:3877–3883
Bianco F, Pravettoni E, Colombo A et al (2005) Astrocyte-derived ATP induces vesicle shedding and IL-1 beta release from microglia. J Immunol 174:7268–7277
Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN (1988) Diet-induced type II diabetes in C57BL/6J mice. Diabetes 37:1163–1167
Maedler K, Spinas GA, Dyntar D, Moritz W, Kaiser N, Donath MY (2001) Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 50:69–76
Oberholzer J, Triponez F, Mage R et al (2000) Human islet transplantation: lessons from 13 autologous and 13 allogeneic transplantations. Transplantation 69:1115–1123
Schulthess FT, Paroni F, Sauter NS et al (2009) CXCL10 impairs beta cell function and viability in diabetes through TLR4 signaling. Cell Metab 9:125–139
Maedler K, Schumann DM, Sauter N et al (2006) Low concentration of interleukin-1beta induces FLICE-inhibitory protein-mediated beta-cell proliferation in human pancreatic islets. Diabetes 55:2713–2722
Solini A, Chiozzi P, Morelli A et al (2004) Enhanced P2X7 activity in human fibroblasts from diabetic patients: a possible pathogenetic mechanism for vascular damage in diabetes. Arterioscler Thromb Vasc Biol 24:1240–1245
Boffa DJ, Waka J, Thomas D et al (2005) Measurement of apoptosis of intact human islets by confocal optical sectioning and stereologic analysis of YO-PRO®-1-stained islets. Transplantation 79:842–845
Wiley JS, Gargett CE, Zhang W, Snook MB, Jamieson GP (1998) Partial agonists and antagonists reveal a second permeability state of human lymphocyte P2Z/P2X7 channel. Am J Physiol 275:C1224–1231
Surprenant A, Rassendren F, Kawashima E, North RA, Buell G (1996) The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 272:735–738
Maedler K, Sergeev P, Ehses JA et al (2004) Leptin modulates beta cell expression of IL-1 receptor antagonist and release of IL-1beta in human islets. Proc Natl Acad Sci U S A 101:8138–8143
Welsh N, Cnop M, Kharroubi I et al (2005) Is there a role for locally produced interleukin-1 in the deleterious effects of high glucose or the type 2 diabetes milieu to human pancreatic islets? Diabetes 54:3238–3244
Surwit RS, Feinglos MN, Rodin J et al (1995) Differential effects of fat and sucrose on the development of obesity and diabetes in C57BL/6 J and A/J mice. Metabolism 44:645–651
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC (2003) Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52:102–110
Hull RL, Kodama K, Utzschneider KM, Carr DB, Prigeon RL, Kahn SE (2005) Dietary-fat-induced obesity in mice results in beta cell hyperplasia but not increased insulin release: evidence for specificity of impaired beta cell adaptation. Diabetologia 48:1350–1358
Coutinho-Silva R, Parsons M, Robson T, Lincoln J, Burnstock G (2003) P2X and P2Y purinoceptor expression in pancreas from streptozotocin-diabetic rats. Mol Cell Endocrinol 204:141–154
Verspohl EJ, Johannwille B, Waheed A, Neye H (2002) Effect of purinergic agonists and antagonists on insulin secretion from INS-1 cells (insulinoma cell line) and rat pancreatic islets. Can J Physiol Pharmacol 80:562–568
Armstrong S, Pereverzev A, Dixon SJ, Sims SM (2009) Activation of P2X7 receptors causes isoform-specific translocation of protein kinase C in osteoclasts. J Cell Sci 122:136–144
Rampe D, Wang L, Ringheim GE (2004) P2X7 receptor modulation of beta-amyloid- and LPS-induced cytokine secretion from human macrophages and microglia. J Neuroimmunol 147:56–61
Solini A, Chiozzi P, Falzoni S, Morelli A, Fellin R, Di Virgilio F (2000) High glucose modulates P2X7 receptor-mediated function in human primary fibroblasts. Diabetologia 43:1248–1256
MacKenzie A, Wilson HL, Kiss-Toth E, Dower SK, North RA, Surprenant A (2001) Rapid secretion of interleukin-1beta by microvesicle shedding. Immunity 15:825–835
Abbatecola AM, Ferrucci L, Grella R et al (2004) Diverse effect of inflammatory markers on insulin resistance and insulin-resistance syndrome in the elderly. J Am Geriatr Soc 52:399–404
Meier CA, Bobbioni E, Gabay C, Assimacopoulos-Jeannet F, Golay A, Dayer JM (2002) IL-1 receptor antagonist serum levels are increased in human obesity: a possible link to the resistance to leptin? J Clin Endocrinol Metab 87:1184–1188
Ruotsalainen E, Salmenniemi U, Vauhkonen I et al (2006) Changes in inflammatory cytokines are related to impaired glucose tolerance in offspring of type 2 diabetic subjects. Diabetes Care 29:2714–2720
Salmenniemi U, Ruotsalainen E, Pihlajamaki J et al (2004) Multiple abnormalities in glucose and energy metabolism and coordinated changes in levels of adiponectin, cytokines, and adhesion molecules in subjects with metabolic syndrome. Circulation 110:3842–3848
Marculescu R, Endler G, Schillinger M et al (2002) Interleukin-1 receptor antagonist genotype is associated with coronary atherosclerosis in patients with type 2 diabetes. Diabetes 51:3582–3585
Herder C, Brunner EJ, Rathmann W et al (2009) Elevated levels of the anti-inflammatory interleukin-1 receptor antagonist (IL-1Ra) precede the onset of type 2 diabetes (Whitehall II Study). Diabetes Care 32:421–423
Acknowledgements
This work was supported by the American Diabetes Association (Junior Faculty Grant 706JF41), the German Research Foundation (DFG, Emmy Noether Program, MA 4172/1-1) and the European Foundation for the Study of Diabetes (EFSD)/Merck Sharp & Dohme. R. Glas is a recipient of the German Research Foundation Fellowship award (Schr 1166/1-1), N. S. Sauter and F. T. Schulthess are recipients of the Swiss National Foundation Fellowship award. Human islets were provided through the Islet Cell Resource Consortium, administered by the Administrative Bioinformatics Coordinating Center (ABCC) and supported by the National Center for Research Resources (NCRR), the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and the Juvenile Diabetes Research Foundation (JDRF). We thank J. Bergemann (Uni Bremen), H. Gerber, M. Kennedy and L. N. Le (UCLA) for excellent technical assistance, C. Lyon (UCLA) for help with the luminex assays and P. U. Heitz, P. Komminoth, P. Saremaslani (University Hospital Zurich) and the National Disease Research Interchange for providing human pancreatic sections.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article is available at http://dx.doi.org/10.1007/s00125-015-3681-x.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 75.5 kb)
ESM Table 1
Clinical data from the autopsy study, results from which are shown in Fig. 1d (PDF 22.3 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Glas, R., Sauter, N.S., Schulthess, F.T. et al. Purinergic P2X7 receptors regulate secretion of interleukin-1 receptor antagonist and beta cell function and survival. Diabetologia 52, 1579–1588 (2009). https://doi.org/10.1007/s00125-009-1349-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-009-1349-0