
Abstract
Dendritic cell (DC)-based immunotherapy is a promising strategy for the elimination of minimal residual disease in patients with acute myeloid leukemia (AML). Particularly, patients with a high risk of relapse who are not eligible for hematopoietic stem cell transplantation could benefit from such a therapeutic approach. Here, we review our extensive studies on the development of a protocol for the generation of DCs with improved immunogenicity and optimized for the use in cell-based immunotherapy. This new generation DC vaccine combines the production of DCs in only 3 days with Toll-like receptor-signaling-induced cell maturation. These mature DCs are then loaded with RNA encoding the leukemia-associated antigens Wilm’s tumor protein 1 and preferentially expressed antigen in melanoma in order to stimulate an AML-specific T-cell-based immune response. In vitro as well as in vivo studies demonstrated the enhanced capacity of these improved DCs for the induction of tumor-specific immune responses. Finally, a proof-of-concept Phase I/II clinical trial is discussed for post-remission AML patients with high risk for disease relapse.
Similar content being viewed by others


Introduction
In adult acute myeloid leukemia (AML), intensive induction chemotherapy induces complete cytomorphological remission in up to 80 % of patients. However, most patients experience subsequent recurrence of disease. Relapse rates are substantially higher in patients over the age of 60 and vary according to cytogenetic and molecular risk groups [1]. Post-remission therapy is critical for the elimination of minimal residual disease (MRD) and is a prerequisite for achieving cure. Allogeneic hematopoietic stem cell transplantation (HSCT) was shown to provide the most potent immunological antileukemic effect, with the lowest rate of relapse and a relevant benefit for overall survival (OS) in certain age groups [2]. However, this approach is restricted to only a subset of patients due to patient-associated comorbidities, donor availability, and age. Targeted immunotherapies may provide suitable alternate therapeutic approaches to eliminate MRD in patients not eligible for HSCT [3].
Recent advances in the field of tumor immunology have resulted in the identification of a number of leukemia-associated antigens (LAAs): specific cytotoxic T-lymphocyte (CTL) responses were detected against the Wilm’s tumor protein 1 (WT1), proteinase 3, preferentially expressed antigen in melanoma (PRAME), human telomerase reverse transcriptase (hTERT), and Fms-like tyrosine kinase 3 internal tandem duplication (FLT3-ITD), to name only a few. For several of these LAAs, clinical peptide vaccination trials were initiated. So far, clinical Phase I/II trials have demonstrated immunological and molecular responses that translate into clinical efficacy in up to half of the vaccinated AML patients [4]. A major challenge in peptide vaccination, however, is the conversion of T-cell tolerance into specific CD8+ CTL and T helper 1 (Th1) type CD4+ T-cell responses directed against self-restricted LAAs. It is known that tumor immunity against AML is conferred by both these cell types and by natural killer (NK) cells.
Dendritic cells (DCs) are “nature’s adjuvants” for eliciting cellular and humoral immunity, and many animal experiments have shown that the injection of tumor antigen-loaded DCs reliably induced tumor-specific CTL responses, tumor resistance, and in some cases regression of metastases. Therefore, DCs have the potential to be more successful than peptide vaccination in eliciting immunological responses needed for the eradication of MRD. RNA loading of DCs is an attractive technique to overcome the need for patient selection due to MHC restriction and allows development of multiplex immune responses mediated by both CD4+ and CD8+ T cells using all MHC molecules of the patient. In a recently updated Phase I/II trial, the clinical efficacy of an autologous, mRNA-electroporated DC vaccination was clearly shown in 17 patients with AML [5, 6].
With increasing knowledge of how to optimize protocols for DC maturation, there is potential for the improvement of clinical benefits by DC vaccination. We developed a three-day manufacturing protocol using a cytokine cocktail containing a synthetic Toll-like receptor (TLR)7/8 agonist for the generation of monocyte-derived mature DCs with improved immunogenicity.
In this review, we discuss our approach for the development of DC vaccines, addressing the questions of DC maturation, route of vaccination as well as dose and schedule of application for post-remission therapy of patients with AML. The setup of the clinical Phase I/II trial is introduced, which is currently recruiting patients.
A three-day protocol for the rapid production of DCs
Most protocols for the generation of clinical-grade monocyte-derived DCs require approximately seven days (7d) of cell culture [7]. Thereby, these protocols are time- and labor-intensive, especially when performed in a clean room facility. In order to develop more feasible strategies for clinical translation, generation of mature DCs within 2–5 days was addressed by several groups in recent years. These fast DCs displayed mature phenotypes and had the capacity to stimulate antigen-specific T-cell responses [8, 9]. Faster generation of clinical-grade mature DCs reduces manufacturing costs, and some evidence suggests that rapid DC differentiation may better reflect the situation in vivo [10].
Our procedures allow us to differentiate and mature DCs within only 3 days (3d) of culture. Hereby, monocytes are stimulated with GM-CSF and IL-4 on the day of isolation and are further activated on day two for maturation over 24 h.
These young DCs were comparable to cells generated with a standard 7d protocol with respect to their mature surface phenotype as well as their capacities for antigen uptake and presentation and antigen-specific T-cell stimulation. Higher yields of viable DCs could be obtained using the 3d protocol. Additionally, improvements in some of the characteristics of young DCs could be observed. The relative expression of the co-stimulatory molecules CD80 and CD86 compared to inhibitory molecules, such as CD274, was higher in 3d-DCs, indicating that shortened culture is beneficial for supporting a positive stimulatory phenotype. Furthermore, 3d-DCs showed a somewhat better migratory capacity than 7d-DCs in vitro, accompanied by high expression of the chemokine receptor CCR7 [11]. These results were observed in numerous experiments using DCs generated with our 3d protocol compared to more standard 7–9d procedures, supporting the hypothesis that fast DCs will be superior in priming naïve T cells to specific antigens. This protocol could be easily transferred into a GMP cell facility and was therefore selected for further development of a DC-based vaccine study for AML patients.
A TLR7/8 agonist-containing maturation cocktail for the generation of Th1-polarizing DCs
The ability of DCs to activate effective antitumor immune responses is highly dependent on their capacity to secret inflammatory cytokines that induce strong effector functions in T cells. IL-12p70 secretion by DCs plays a critical role in this process by polarizing CD4+ cells into the direction of Th1 responses [12]. CD4+ Th1 cells, in turn, support the activation of CD8+ CTLs, which have the capacity to kill tumor cells in an antigen-specific manner. In addition, IL-12p70 has an important role in the activation of NK cells, which also display cytolytic activity against tumor cells. Thus, IL-12p70 secreting DCs can orchestrate both innate and adaptive immune cells to build effective immune responses against tumors.
Various maturation stimuli have been studied for their capacity to induce IL-12p70 secretion by DCs. Activation of DCs via TLRs, which normally signal via pathogen-derived products, has been extensively analyzed. The signaling pathways activated by TLR7/8 and TLR3 stimulations were found to synergize in induction of IL-12p70 secretion [13, 14]. Recent evidence also suggests that stimulation of DCs via the TLR7/8 signaling pathway can overcome IL-12p70 production defects in patient-derived DCs [15].
We established a maturation cocktail containing the synthetic TLR7/8 agonist R848 and the TLR3 ligand poly(I:C), in combination with TNFα, IL1β, IFNγ, and PGE2 [16]. Monocyte-derived 3d- and 7d-DCs were matured with this new cocktail or with a gold standard cocktail consisting of TNFα, IL1β, IL-6, and PGE2 [7], which has been used extensively for the generation of DCs tested in the clinic. TLR agonist-activated 3d-DCs had a mature phenotype and displayed a positive stimulatory profile, as exhibited by increased expression of co-stimulatory molecules compared to co-inhibitory markers. Additionally, only TLR ligand-stimulated DCs could secrete high amounts of IL-12p70 upon CD40–CD40-ligand interactions. Secretion of IL-10 by TLR-activated DCs was low. This is important since IL-10 can counteract the polarizing effects of IL-12p70 [17].
The TLR-activated 3d-DCs displayed several other advantageous features in vitro. They showed an improved capacity for Th1 polarization of both CD4+ and CD8+ T cells as well as improved capacity for antigen-specific activation of autologous T cells. A strong impact on functional activation of NK cells was also demonstrated. All these effects were significantly weaker with DCs generated with the standard cocktail [16, 18, 19].
Since earlier studies demonstrated a synergism of TLR7/8 and TLR3 activation on IL-12p70 secretion by 7d-DCs [13, 14], we were interested to determine whether these results would also be found with 3d-DCs. A side-by-side comparison of 3d-DCs matured with cocktails containing either R848 plus poly(I:C) or R848 alone revealed no significant differences between the two different cell preparations, according to phenotype and function. Therefore, we elected to remove poly(I:C) from our maturation cocktail. This protocol was then assessed for the generation of mature 3d-DCs from monocytes of AML patients in remission and shown to result in pronounced innate and adaptive antileukemic immune responses in vitro [20].
In vitro-transcribed mRNA for efficient antigen loading of DCs
The specificity of an adaptive immune response is determined by the identity of the antigenic peptides presented to the CD8+ or CD4+ T cells in association with MHC class-I or class-II molecules, respectively (i.e., pMHC ligands). Tumor cells may escape immune recognition through down-regulation of single tumor-associated antigens (TAAs) [21]. Therefore, activation of T cells recognizing multiple pMHC ligands from multiple TAAs is important to guard against tumor escape variants.
To date, numerous strategies for antigen loading of DCs have been evaluated. Pulsing DCs with synthetic TAA-derived peptides has been analyzed extensively in recent years. In this case, the activated T-cell response is not only limited to the selected epitope but also restricted to a specific HLA molecule, and progressive growth of antigen-loss variants may occur [22]. Although loading of DCs with peptides derived from multiple TAAs may prevent immune escape, this approach is still limited to patients with particular MHC allotypes where knowledge of suitable peptides is available. To allow activation of T-cell responses directed against multiple TAAs, independent of the patient`s genetic background, use of tumor lysates or amplified tumor-derived mRNA has been supposed to be more advantageous [23]. DCs loaded with these sources of antigens should be able to present a patient`s own tumor set of known and unknown TAAs. However, these sources need to be prepared individually from each patient, and availability of tumor material might be a limiting factor.
Instead, use of in vitro-transcribed messenger RNA (ivt-mRNA) encoding defined antigens offers an attractive alternative for loading of DCs with message of either single or multiple TAAs. Furthermore, sufficient amounts of RNA can be produced independently of the need for access to patient tumor material. We analyzed the capacity of DCs loaded with (1) total tumor-derived RNA; (2) amplified tumor-derived RNA, prepared from a tumor cell line; or (3) ivt-mRNA encoding a single antigen (single-species RNA), known to be present in the total tumor-derived RNA preparation, for the reactivation of antigen-specific effector-memory CTLs. Here, DCs loaded with single-species RNA showed a clear advantage in CTL activation [24]. By loading DCs with single-species RNA, high amounts of RNA molecules could be introduced into DCs, and TAA-derived epitopes could be processed and presented via MHC molecules in sufficient numbers for efficient T-cell recognition. In tumor lysates or native or amplified total tumor-derived mRNA, adequate amounts of some immunogenic antigens or antigen-encoding messenger RNA may not be sufficient for efficient antigen presentation at the DC surface, leading to inadequate activation of antigen-specific T cells [24]. Therefore, single-species ivt-mRNA as the selected form for antigen loading of DCs offers an attractive strategy to provide mature DCs with sufficient amounts of different peptides of individual or multiple TAAs.
Further, we established methods to efficiently transfect 3d and 7d mature DCs with TAA-encoding RNA by means of electroporation without changing their phenotype [11, 24]. It was clearly demonstrated that RNA-loaded DCs were able to efficiently process and present peptides derived from numerous transfected antigens, allowing activation of antigen-specific memory CTL clones as well as activation and expansion of naïve antigen-specific T cells [24–26].
Since the development of immune responses to multiple TAAs is advantageous, loading of DCs with pools of different ivt-mRNA species would seem warranted. We compared the stimulatory capacities of DCs transfected with either single-species ivt-mRNA or with pools of three or six RNA species, respectively. Cells transfected with a pool of three different RNAs showed an approximate 80 % capacity for CTL activation per antigen compared to single antigen-loaded DCs. Transfection with a pool of six different RNA species further reduced this capacity to 40 % [25]. Therefore, we elected to only introduce single antigens into separate aliquots of mature DCs. As a consequence of these results, in our clinical vaccination protocol, aliquots from one DC batch production are individually loaded with a selected antigen and then DCs with different antigens are administered simultaneously to the patient to allow stimulation of T-cell responses specific for multiple TAAs.
A humanized mouse model for the preclinical evaluation of DC vaccines
For preclinical evaluation of our DC-based vaccines, it was important to analyze the induction of specific T-cell responses in an appropriate in vivo model. Such an animal model needs to bear a functional human immune system to ensure that therapeutically administered human DCs have a network of human immune cells in order to activate T cells and NK cells. The immunodeficient mouse strains NOD/SCID Il2rg−/− and BALB/c-Rag2−/−Il2rg−/− are currently considered to be state-of-the art models for such experiments [27, 28]. These mice can be engrafted with functional human immune cells by transplanting them with human hematopoietic stem cells (HSCs) or peripheral blood mononuclear cells (PBMCs) [29, 30].
We chose the NOD/SCID Il2rg‐/‐ (NSG) mouse model to analyze our vaccine strategy and to assess the functionality of our new generation DCs. Mice were engrafted with human PBMC and vaccinated twice with autologous human DCs on days 14 and 21 after reconstitution. Approximately one week later, the human lymphocytes were analyzed for their immunological properties. By choosing this short-time protocol, we avoided the xeno reactivity that normally occurs in longer engraftment and vaccination protocols. We compared the stimulatory capacity of standard 7d-DCs and 3d-DCs. Both DC types were matured with a standard maturation cocktail lacking a TLR ligand. Additionally, 3d-DCs were included that were matured with our TLR7/8 and TLR3 ligand-containing cocktail. MART-1 was chosen as the antigen, and DCs were transfected with ivt-mRNA encoding this antigen. Splenic-derived T cells isolated from mice vaccinated with the 3d-DCs matured with the TLR agonist-containing maturation cocktail showed the best MART-1-specific immune responses, as assessed by antigen-specific killing capacity and IFNγ secretion. In comparison, T cells isolated from mice vaccinated with standard 7d-DCs showed no or only low antigen-specific immune responses [31].
This NSG mouse model allowed a direct comparison of different DC vaccine cells, thereby the superiority of TLR-stimulated 3d-DCs in the activation of antigen-specific T-cell stimulation based on extensive in vitro experiments could be confirmed in vivo.
TLR-activated 3d-DCs for immunotherapy of AML: a proof-of-concept clinical trial
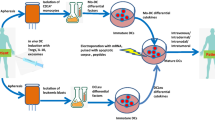
As demonstrated in the work above, our 3d, TLR-activated DCs have optimized characteristics for the induction of potent CD4+ and CD8+ Th1-polarized T-cell responses as well as NK cell responses when compared to DC vaccines used in previous clinical trials of DC vaccination in AML patients, which have already shown some promising results [5]. Therefore, we were encouraged to design a clinical trial to test this type of DCs in post-remission AML patients. We developed a GMP-compliant process for the production of DCs within 3 days of culture from autologous monocytes, including maturation using our TLR7/8 agonist-containing cocktail. Subsequent antigen loading is performed by electroporation of the mature cells with TAA-encoding ivt-mRNA (Fig. 1). In a head-to-head comparison, we were able to show that DCs differentiated from monocytes taken from AML patients in complete remission were comparable to DCs of healthy donors with respect to phenotype and function [20]. A proof-of-concept Phase I/II clinical trial was initiated to evaluate our new generation mature DC vaccines loaded with mRNA for WT1, PRAME, and hCMVpp65 (see below for choice of antigens) as post-remission therapy of AML patients with non-favorable prognosis (NCT01734304), and we are currently recruiting patients. As the primary end point of our trial is feasibility and safety, we adapted our DC dose and vaccination schedule to prior protocols (see below) [32, 33]. This enables us to deduce observed differences in clinical and immunological responses to the properties of the DCs rather than to DC dose or vaccination schedule. Patients are vaccinated with 5 × 106 DCs transfected with one of the three antigens, accumulating to a total of 15 × 106 DCs at each vaccination time point. The first 4 vaccinations are given weekly, followed by up to 6 vaccinations at monthly intervals. Secondary end points of our trial are immune responses and disease control with particular focus on MRD conversion as detected by real-time quantitative polymerase chain reaction (RQ-PCR) and multiparameter flow cytometry (MFC) (Fig. 2). Importantly, innate and adaptive immune responses against “self” (WT1 and PRAME) and “viral” (hCMVpp65) antigens will be closely analyzed using at least two different immune monitoring methods according to the recommendations of the MIATA association (http://miataproject.org/).

Manufacturing of new generation DCs. Monocytes are differentiated to immature DCs by GM-CSF and IL-4 within 48 h. Addition of a novel TLR7/8 agonist-containing maturation cocktail for another 24 h leads to the development of mature DCs that are characterized by a positive stimulatory profile and high production of bioactive IL-12p70. After electroporation with antigen-encoding RNA, these DCs are capable of polarizing Th1 responses and stimulating antigen-specific CTLs in addition to NK cell activation

Protocol for the proof-of-concept clinical trial with new generation DCs in AML. Patients with AML and non-favorable risk profile in complete remission after intensive induction therapy are able to enroll. Standard exclusion criteria apply, and patients have to be ineligible for allogeneic SCT. Patients are vaccinated intradermally with one batch of 5 × 106 DCs for each of the three antigens (WT1, PRAME, and hCMVpp65) up to 10 times within 26 weeks. The primary end point of the trial is feasibility and toxicity; secondary end points are immune responses and disease control, with particular focus on MRD conversion
The study antigens WT1, PRAME, and hCMVpp65
In recent years, the identification of TAAs has provided many potential targets for directed immunotherapy. However, the choice of a suitable TAA for antigen-specific immunotherapy needs careful consideration in order to achieve efficient immune responses and to avoid unwanted on-target as well as off-target toxicity. In a systematic approach to the selection of the most suitable TAA, a priority-ranked list of target antigens was developed [34]. The evaluation of 75 representative cancer antigens according to predefined and preweighted criteria revealed that none had all the characteristics required to be an “ideal” cancer antigen. For our DC vaccine, we chose the LAAs WT1 and PRAME because they fulfill most of these criteria (Table 1). Importantly, these antigens were shown to be safe in prior peptide or DC vaccination studies [35–37]. Furthermore, potentially curative immunotherapy needs to target leukemic stem cells (LSCs); thus, these target antigens were selected since they are overexpressed in LSCs compared to normal hematopoietic stem cells (HSCs) [38, 39].
Based on these characteristics and its proven immunogenicity, WT1 was placed at the top of the prioritized list of potential target antigens for cancer immunotherapy [34]. Despite the fact that WT1 is a self-antigen, T cells specific for WT1 are detected in healthy donors and AML patients, and the immunogenicity of WT1 in an autologous setting is exemplified by the natural development of humoral [40] and cellular [41] immunity against WT1 in patients with WT1-expressing malignancies, including AML. Furthermore, naturally activated WT1-specific T cells were reported to contribute to the graft versus leukemia effect in patients after allogeneic HSCT [42]. In clinical trials, in vivo immunological responses to WT1 peptide-based vaccines were measured in as many as 88 % of AML patients. Clinically, decreased numbers of leukemic cells and decreased levels of WT1 expression, disease stabilization, and complete hematological remission were observed in these trials. In some cases, clinical responses were at least associated, if not clearly correlated, with the observed immunological effects [36, 43, 44].
PRAME seems to inhibit cell differentiation, growth arrest, and apoptosis by acting as a repressor of retinoic acid receptor signaling. For this reason, it is thought that PRAME may contribute to oncogenesis in general, and that it may be an important contributory factor in AML disease progression in particular [45]. PRAME has been detected in bone marrow (BM) and peripheral blood (PB) of 30–64 % of leukemia patients [46, 47]. In one study, a strong correlation between PRAME expression and unique leukemia markers was found [48], and in another one, blast counts in marrow samples were correlated with PRAME expression [47]. Furthermore, the development of PRAME-specific T cells in vivo and in vitro was reported in several studies, underscoring the processing and immunogenicity of different PRAME-derived epitopes [49, 50].
Human cytomegalovirus (hCMV) is common in all parts of the world, with infection rates between 60 % in developed countries and up to 100 % in developing countries. The hCMV 65-kDa phosphoprotein (pp65) has been identified as a major, immunogenic and immunodominant target antigen for hCMV-specific, MHC class-I-restricted CTLs. Due to its high immunogenicity as a foreign antigen, pp65 is a promising target for immunotherapy of immunocompromized patients, such as HSCT recipients at risk of developing an hCMV infection [51].
It has been well known for the past several decades that simultaneous immunization against two different antigens may result in different outcomes of the immune response to each antigen. Classically, antigen competition has been seen by the fact that a good immune response occurs against one antigen while the immune response specific for the second antigen is suppressed. More recently, the molecular and cellular mechanisms that underlie antigen competition have been elucidated. At the molecular level, antigenic peptides from one antigen might be able to out-compete peptides from a second antigen for presentation at the surface of the DC. As a consequence, the DC will display a superior capacity to prime T cells to the dominant epitope(s) of the former antigen while being deficient in their capacity to prime T cells to poorly competing epitopes derived from the second antigen.
At the cellular level, predominance in particular pMHC complexes at the surface of the DC will allow it to attract and interact with greater numbers of T cells specific for the first antigen over time. Even if the DC expresses small amounts of pMHC from the second antigen, specific T cells for the second antigen may be unable to adequately recognize and interact with the DC due to overcrowding at the surface of the DC with T cells responding to the first antigen [52].
We completely avoid the molecular and cellular antigen competition in our DC vaccines despite the use of three different antigens, anyone of which might carry immunodominant epitope(s). This is accomplished by separating the DCs into three aliquots prior to antigen loading. Each aliquot of DCs is then electroporated separately with ivt-mRNA encoding only one of the three different antigens. This means that each of the three subfractions of DCs only needs to process and present peptides from one antigen, resulting in separate populations of DCs for each antigen.
Dosing and administration of the DC vaccine
A literature review of clinical studies of DC-based therapeutics administered intradermally to adult cancer patients revealed that doses of up to 1 × 108 DCs per injection can be applied multiple times without the danger of patients developing high-grade toxicities, including autoimmune reactions. In general, DC vaccinations have been observed to be safe irrespective of the tumor type, DC maturation stage, antigen identity, and antigen form used to load the DCs. On average, patients received a total of 3 or 4 vaccinations at two-week intervals. Immunological responses were observed in all of these trials and were associated or even correlated with clinical responses on several occasions. Dose-limiting toxicities were not reported in any of the studies in which the dose was escalated in the 1–100 × 106 DC range [53–55]. In fact, few differences were observed among the different doses.
Scarce adverse events have been reported in the few trials with maximum DC doses ranging from 50 to 100 × 106 cells. Furthermore, toxicities higher than grade 2 (according to the National Cancer Institute’s Common Toxicity Criteria) were not observed [54, 56]. In one melanoma trial, 10 × 106 DCs loaded with peptides and tumor lysates were administered up to 60 times per patient at weekly or biweekly intervals. While most subjects experienced mild and transient pain at the vaccine injection sites, in several patients, a grade 3 autosensitization dermatitis-like eruption and transient eosinophilia was observed, but none of these adverse events were considered to be severe [57]. In consideration of all the published data on DC dose and schedule, our choice to use 15 × 106 DCs per injection, ranging from 4 to 10 applications, according to availability and feasibility is judged to be acceptable for a first-in-man study.
MRD diagnostics in AML
Monitoring of MRD in patients with AML has become an important diagnostic tool. Improved MRD techniques and new sensitive markers are used for the assessment of response to therapy and to monitor the individual course of the disease in each patient. Reflecting initial chemosensitivity, MRD levels constitute a prognostic marker that combines various biological properties of an individual AML. Available methods to determine the levels of MRD from BM samples are RQ-PCR (“molecular MRD”) and MFC (“flow MRD”). RQ-PCR offers very high sensitivity (up to 1 cell in 10.000 to 1 cell in 100.000). The high predictive value of NPM1 mutation MRD has been demonstrated in several retrospective studies. An increase over 200 NPM1 copies per 104 copies of the housekeeping gene ABL1 was associated with disease recurrence after a median time of 3 months [58]. WT1 is a ubiquitously expressed target antigen available for molecular MRD assessment. Measurable WT1 levels have been shown to correlate with the clinical course of disease in over 85 % of the cases [59, 60]. Therefore, WT1 is a suitable surrogate marker to monitor the success of a therapeutic approach. However, WT1 detection is less suitable to trigger an intervention, because an increase in WT1 levels is associated with a rather rapid relapse after a median of only 38 days (range 8–180 days), if monitored in >4 week intervals [61].
The use of MFC for the detection of MRD is applicable in the majority of patients, offering a sensitivity of up to 1 cell in 10.000 cells, depending on the number of events acquired. Initial studies revealed high prognostic value of flow MRD assessment for relapse risk, relapse-free survival (RFS), and OS after induction and consolidation therapy [62, 63]. The prognostic impact of flow MRD at these time points was subsequently confirmed in several other studies [64, 65]. In conclusion, molecular MRD allows the prediction of relapse over the course of the disease, with variable sensitivity (NPM1 > WT1) and different time intervals. Flow MRD at early time points of disease appears to be most suitable for improved risk stratification and identification of patients at high risk of relapse [66]. Combining the strategies of molecular and flow MRD enables risk-adapted therapy to be made while using a surrogate marker for clinical efficacy that is critical for AML monitoring in Phase I/II trials during post-remission therapy.
Concluding remarks
Within the last decade, we have covered the long distance from bench to bedside in the development of a GMP-compatible protocol for a new generation of DCs with optimized characteristics for cancer immunotherapy. We established a time- and labor-efficient protocol for DC generation within only 3 days. In combination with a novel maturation cocktail, including a synthetic TLR7/8 ligand, this protocol yields DCs with several highly advantageous properties for application in cancer immunotherapy when compared to other more conventional monocyte-derived DCs. Specifically, these DCs display a predominant positive stimulatory profile and high secretion of IL-12p70 upon interaction with T cells. Both of these signals are responsible for the superior capacity of the DCs to polarize CD4+ T cells toward a Th1-dominant phenotype and to activate both CTLs and NK cells. Antigen loading via electroporation with ivt-mRNA encoding LAAs was established and shown to be efficient for the induction of antigen-specific T-cell immune responses. This was demonstrated not only in vitro, but also in a humanized mouse model. Based on this extensive preclinical data, we developed a GMP-compliant process to produce these new generation DCs from leukapheresis products and have initiated a clinical Phase I/II trial to test their application in AML patients.
Acute myeloid leukemia was chosen as the first clinical entity for the assessment of these new generation DCs for several reasons: After intensive induction therapy, patients have low disease burdens, rendering post-remission the ideal time point for immunotherapy. Leukemic blasts in the BM and PB can be more easily accessed by the immune system than stroma-rich solid tumors. MRD measurements provide an excellent means to determine the efficacy of our treatment approach. Finally, the identification of molecular characteristics and immunogenic epitopes in AML is advanced. We chose WT1 and PRAME as antigens for the stimulation of T cells, accompanied by the very immunogenic viral antigen hCMVpp65, both as an adjuvant and as an internal immune monitoring control. According to the protocol of our clinical trial that is actively recruiting study participants, AML patients with non-favorable prognosis are vaccinated 4–10 times with 5 × 106 DCs for each of these antigens. We believe that this DC-based vaccine has the potential to significantly improve the clinical outcome of AML patients with a non-favorable genetic risk profile. Furthermore, by selecting appropriate TAAs, our platform technology for DC generation allows us to develop vaccine formulations specific for any type of malignancy.
Parts of this work have been presented and published in abstract books of the following scientific meetings
Lichtenegger FS, Beck B, Bigalke I, Geiger C, Hiddemann W, Henschler R, Kvalheim G, Schendel, DJ, Subklewe M (2014) Dendritic cell vaccination for postremission therapy in AML. 1st Immunotherapy of Cancer Conference (ITOC1), Munich, Germany, poster, published in Journal for Immunotherapy of Cancer, 2014; 2(Suppl 2):P29.
Geiger C, Beck B, Bigalke I, Lichtenegger FS, Heemskerk M, Saboe-Larssen S, Kvalheim G, Schendel DJ, Subklewe M (2013) A DC-based therapeutic vaccine for AML patients: preclinical evaluation of a new generation of DCs expressing the leukemia-associated antigens WT1 and PRAME. Cellular Therapy, Erlangen, Germany, abstract book, poster C2.
Lichtenegger FS, Beck B, Geiger C, Munker D, Schlueter M, Draenert R, Schnorfeil FM, Kvalheim G, Hiddemann W, Schendel, DJ, Subklewe M (2013) Improving Efficacy of Dendritic Cell Vaccination in AML: Optimization of the DC Generation Protocol and Maximization of T Cell Responses by Immune Checkpoint Blockade. ASH Annual Meeting, New Orleans, LA, USA, poster, published in Blood, 2013; 122:3492.
Subklewe M, Lichtenegger FS, Beck B, Geiger C, Bigalke I, Kvalheim G, Hiddemann W, Schendel DJ (2013) Dendritic Cell Vaccination for Post-remission Therapy in AML. Acute Leukemias XIV, Munich, Gemany, talk, published in Annals of Hematology, 2013; 92(1S):S58.
Geiger C, Beck B, Bigalke I, Lichtenegger FS, Heemskerk M, Saboe-Larssen S, Schendel DJ, Subklewe M (2012) Development of a DC-based vaccine for AML patients: Characterization of GMP-grade TLR-agonist matured 3-day DCs expressing the leukemia-associated antigens WT1 and PRAME. CIMT 10th Annual Meeting, Mainz, Germany, abstract book, poster 171:225.
Lichtenegger FS, Mueller K, Hiddemann W, Schendel DJ, Subklewe M (2011) Dendritic Cells Matured with a TLR7/8 Agonist Induce T Helper 1 Cell Polarization, Activate NK Cells and Are Thus Highly Suitable for Application in Cancer Immunotherapy. ASH Annual Meeting, San Diego, CA, USA, talk, published in Blood, 2011; 118:168.
Geiger C, Beck B, Dörfel D, Lichtenegger FS, Tippmer S, Bigalke I, Spranger S, Eichenlaub E, Schendel DJ, Subklewe M (2010) mRNA-loaded three-day mature dendritic cells for immunotherapy in acute myeloid leukemia patients in a Phase I clinical trial. DC2010: Forum on Vaccine Science, Lugano, Switzerland, abstract book, poster P05-038.
Abbreviations
- AML:
-
Acute myeloid leukemia
- BM:
-
Bone marrow
- CTL:
-
Cytotoxic T lymphocytes
- DC:
-
Dendritic cells
- GMP:
-
Good manufacturing practice
- hCMV:
-
Human cytomegalovirus
- HSC:
-
Hematopoietic stem cell
- HSCT:
-
Hematopoietic stem cell transplantation
- Ivt:
-
In vitro-transcribed
- LAA:
-
Leukemia-associated antigen
- LSC:
-
Leukemic stem cell
- MFC:
-
Multiparameter flow cytometry
- MRD:
-
Minimal residual disease
- NK:
-
Natural killer
- OS:
-
Overall survival
- PB:
-
Peripheral blood
- PBMC:
-
Peripheral blood mononuclear cells
- PRAME:
-
Preferentially expressed antigen in melanoma
- RFS:
-
Relapse-free survival
- RQ-PCR:
-
Real-time quantitative polymerase chain reaction
- TAA:
-
Tumor-associated antigen
- Th1:
-
T helper 1
- TLR:
-
Toll-like receptor
- WT1:
-
Wilm’s tumor protein 1
References
Ferrara F, Schiffer CA (2013) Acute myeloid leukaemia in adults. Lancet 381(9865):484–495. doi:10.1016/S0140-6736(12)61727-9
Stelljes M, Krug U, Beelen DW, Braess J, Sauerland MC, Heinecke A, Ligges S, Sauer T, Tschanter P, Thoennissen GB, Berning B, Kolb HJ, Reichle A, Holler E, Schwerdtfeger R, Arnold R, Scheid C, Muller-Tidow C, Woermann BJ, Hiddemann W, Berdel WE, Buchner T (2014) Allogeneic transplantation versus chemotherapy as postremission therapy for acute myeloid leukemia: a prospective matched pairs analysis. J Clin Oncol 32(4):288–296. doi:10.1200/JCO.2013.50.5768
Lichtenegger FS, Schnorfeil FM, Hiddemann W, Subklewe M (2013) Current strategies in immunotherapy for acute myeloid leukemia. Immunotherapy 5(1):63–78. doi:10.2217/imt.12.145
Schmitt M, Casalegno-Garduno R, Xu X, Schmitt A (2009) Peptide vaccines for patients with acute myeloid leukemia. Expert Rev Vaccines 8(10):1415–1425. doi:10.1586/erv.09.90
Van Tendeloo VF, Van de Velde A, Van Driessche A, Cools N, Anguille S, Ladell K, Gostick E, Vermeulen K, Pieters K, Nijs G, Stein B, Smits EL, Schroyens WA, Gadisseur AP, Vrelust I, Jorens PG, Goossens H, de Vries IJ, Price DA, Oji Y, Oka Y, Sugiyama H, Berneman ZN (2010) Induction of complete and molecular remissions in acute myeloid leukemia by Wilms’ tumor 1 antigen-targeted dendritic cell vaccination. Proc Natl Acad Sci USA 107(31):13824–13829. doi:10.1073/pnas.1008051107
Berneman ZN VdVA, Anguille S, Cools N, Van Driessche A, Nijs G, Stein B et al (2012) WT1-targeted dendritic cell vaccination as a postremission treatment to prevent or delay relapse in acute myeloid leukemia. J Clin Oncol 30 (suppl; abstr 2506)
Jonuleit H, Kuhn U, Muller G, Steinbrink K, Paragnik L, Schmitt E, Knop J, Enk AH (1997) Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol 27(12):3135–3142. doi:10.1002/eji.1830271209
Dauer M, Obermaier B, Herten J, Haerle C, Pohl K, Rothenfusser S, Schnurr M, Endres S, Eigler A (2003) Mature dendritic cells derived from human monocytes within 48 hours: a novel strategy for dendritic cell differentiation from blood precursors. J Immunol 170(8):4069–4076
Kvistborg P, Boegh M, Pedersen AW, Claesson MH, Zocca MB (2009) Fast generation of dendritic cells. Cell Immunol 260(1):56–62. doi:10.1016/j.cellimm.2009.09.003
Randolph GJ, Beaulieu S, Lebecque S, Steinman RM, Muller WA (1998) Differentiation of monocytes into dendritic cells in a model of transendothelial trafficking. Science 282(5388):480–483
Burdek M, Spranger S, Wilde S, Frankenberger B, Schendel DJ, Geiger C (2010) Three-day dendritic cells for vaccine development: antigen uptake, processing and presentation. J Transl Med 8:90. doi:10.1186/1479-5876-8-90
Mescher MF, Curtsinger JM, Agarwal P, Casey KA, Gerner M, Hammerbeck CD, Popescu F, Xiao Z (2006) Signals required for programming effector and memory development by CD8+ T cells. Immunol Rev 211:81–92. doi:10.1111/j.0105-2896.2006.00382.x
Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A (2005) Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol 6(8):769–776. doi:10.1038/ni1223
Gautier G, Humbert M, Deauvieau F, Scuiller M, Hiscott J, Bates EE, Trinchieri G, Caux C, Garrone P (2005) A type I interferon autocrine-paracrine loop is involved in Toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J Exp Med 201(9):1435–1446. doi:10.1084/jem.20041964
Carreno BM, Becker-Hapak M, Huang A, Chan M, Alyasiry A, Lie WR, Aft RL, Cornelius LA, Trinkaus KM, Linette GP (2013) IL-12p70-producing patient DC vaccine elicits Tc1-polarized immunity. J Clin Invest 123(8):3383–3394. doi:10.1172/JCI68395
Zobywalski A, Javorovic M, Frankenberger B, Pohla H, Kremmer E, Bigalke I, Schendel DJ (2007) Generation of clinical grade dendritic cells with capacity to produce biologically active IL-12p70. J Transl Med 5:18. doi:10.1186/1479-5876-5-18
Trinchieri G (2003) Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol 3(2):133–146. doi:10.1038/nri1001
Spranger S, Javorovic M, Burdek M, Wilde S, Mosetter B, Tippmer S, Bigalke I, Geiger C, Schendel DJ, Frankenberger B (2010) Generation of Th1-polarizing dendritic cells using the TLR7/8 agonist CL075. J Immunol 185(1):738–747. doi:10.4049/jimmunol.1000060
Lichtenegger FS, Mueller K, Otte B, Beck B, Hiddemann W, Schendel DJ, Subklewe M (2012) CD86 and IL-12p70 are key players for T helper 1 polarization and natural killer cell activation by Toll-like receptor-induced dendritic cells. PLoS ONE 7(9):e44266. doi:10.1371/journal.pone.0044266
Beck B, Dorfel D, Lichtenegger FS, Geiger C, Lindner L, Merk M, Schendel DJ, Subklewe M (2011) Effects of TLR agonists on maturation and function of 3-day dendritic cells from AML patients in complete remission. J Transl Med 9:151. doi:10.1186/1479-5876-9-151
Jager E, Jager D, Knuth A (2002) Clinical cancer vaccine trials. Curr Opin Immunol 14(2):178–182
Butterfield LH (2013) Dendritic cells in cancer immunotherapy clinical trials: are we making progress? Front Immunol 4:454. doi:10.3389/fimmu.2013.00454
Gilboa E, Vieweg J (2004) Cancer immunotherapy with mRNA-transfected dendritic cells. Immunol Rev 199:251–263. doi:10.1111/j.0105-2896.2004.00139.x
Javorovic M, Pohla H, Frankenberger B, Wolfel T, Schendel DJ (2005) RNA transfer by electroporation into mature dendritic cells leading to reactivation of effector-memory cytotoxic T lymphocytes: a quantitative analysis. Mol Ther 12(4):734–743. doi:10.1016/j.ymthe.2005.03.034
Javorovic M, Wilde S, Zobywalski A, Noessner E, Lennerz V, Wolfel T, Schendel DJ (2008) Inhibitory effect of RNA pool complexity on stimulatory capacity of RNA-pulsed dendritic cells. J Immunother 31(1):52–62. doi:10.1097/CJI.0b013e31815a1202
Wilde S, Sommermeyer D, Frankenberger B, Schiemann M, Milosevic S, Spranger S, Pohla H, Uckert W, Busch DH, Schendel DJ (2009) Dendritic cells pulsed with RNA encoding allogeneic MHC and antigen induce T cells with superior antitumor activity and higher TCR functional avidity. Blood 114(10):2131–2139. doi:10.1182/blood-2009-03-209387
Shultz LD, Ishikawa F, Greiner DL (2007) Humanized mice in translational biomedical research. Nat Rev Immunol 7(2):118–130. doi:10.1038/nri2017
Zhou Q, Facciponte J, Jin M, Shen Q, Lin Q (2014) Humanized NOD-SCID IL2rg(-/-) mice as a preclinical model for cancer research and its potential use for individualized cancer therapies. Cancer Lett 344(1):13–19. doi:10.1016/j.canlet.2013.10.015
Harui A, Kiertscher SM, Roth MD (2011) Reconstitution of huPBL-NSG mice with donor-matched dendritic cells enables antigen-specific T-cell activation. J Neuroimmune Pharmacol 6(1):148–157. doi:10.1007/s11481-010-9223-x
Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, Ueyama Y, Koyanagi Y, Sugamura K, Tsuji K, Heike T, Nakahata T (2002) NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood 100(9):3175–3182. doi:10.1182/blood-2001-12-0207
Spranger S, Frankenberger B, Schendel DJ (2012) NOD/scid IL-2Rg(null) mice: a preclinical model system to evaluate human dendritic cell-based vaccine strategies in vivo. J Transl Med 10:30. doi:10.1186/1479-5876-10-30
Mu LJ, Kyte JA, Kvalheim G, Aamdal S, Dueland S, Hauser M, Hammerstad H, Waehre H, Raabe N, Gaudernack G (2005) Immunotherapy with allotumour mRNA-transfected dendritic cells in androgen-resistant prostate cancer patients. Br J Cancer 93(7):749–756. doi:10.1038/sj.bjc.6602761
Vik-Mo EO, Nyakas M, Mikkelsen BV, Moe MC, Due-Tonnesen P, Suso EM, Saeboe-Larssen S, Sandberg C, Brinchmann JE, Helseth E, Rasmussen AM, Lote K, Aamdal S, Gaudernack G, Kvalheim G, Langmoen IA (2013) Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol Immunother 62(9):1499–1509. doi:10.1007/s00262-013-1453-3
Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, Mellman I, Prindiville SA, Viner JL, Weiner LM, Matrisian LM (2009) The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res 15(17):5323–5337. doi:10.1158/1078-0432.CCR-09-0737
Maslak PG, Dao T, Krug LM, Chanel S, Korontsvit T, Zakhaleva V, Zhang R, Wolchok JD, Yuan J, Pinilla-Ibarz J, Berman E, Weiss M, Jurcic J, Frattini MG, Scheinberg DA (2010) Vaccination with synthetic analog peptides derived from WT1 oncoprotein induces T-cell responses in patients with complete remission from acute myeloid leukemia. Blood 116(2):171–179. doi:10.1182/blood-2009-10-250993
Keilholz U, Letsch A, Busse A, Asemissen AM, Bauer S, Blau IW, Hofmann WK, Uharek L, Thiel E, Scheibenbogen C (2009) A clinical and immunologic phase 2 trial of Wilms tumor gene product 1 (WT1) peptide vaccination in patients with AML and MDS. Blood 113(26):6541–6548. doi:10.1182/blood-2009-02-202598
Li L, Giannopoulos K, Reinhardt P, Tabarkiewicz J, Schmitt A, Greiner J, Rolinski J, Hus I, Dmoszynska A, Wiesneth M, Schmitt M (2006) Immunotherapy for patients with acute myeloid leukemia using autologous dendritic cells generated from leukemic blasts. Int J Oncol 28(4):855–861
Saito Y, Kitamura H, Hijikata A, Tomizawa-Murasawa M, Tanaka S, Takagi S, Uchida N, Suzuki N, Sone A, Najima Y, Ozawa H, Wake A, Taniguchi S, Shultz LD, Ohara O, Ishikawa F (2010) Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci Transl Med 2 (17):17ra19. doi:10.1126/scitranslmed.3000349
Gerber JM, Qin L, Kowalski J, Smith BD, Griffin CA, Vala MS, Collector MI, Perkins B, Zahurak M, Matsui W, Gocke CD, Sharkis SJ, Levitsky HI, Jones RJ (2011) Characterization of chronic myeloid leukemia stem cells. Am J Hematol 86(1):31–37. doi:10.1002/ajh.21915
Elisseeva OA, Oka Y, Tsuboi A, Ogata K, Wu F, Kim EH, Soma T, Tamaki H, Kawakami M, Oji Y, Hosen N, Kubota T, Nakagawa M, Yamagami T, Hiraoka A, Tsukaguchi M, Udaka K, Ogawa H, Kishimoto T, Nomura T, Sugiyama H (2002) Humoral immune responses against Wilms tumor gene WT1 product in patients with hematopoietic malignancies. Blood 99(9):3272–3279
Scheibenbogen C, Letsch A, Thiel E, Schmittel A, Mailaender V, Baerwolf S, Nagorsen D, Keilholz U (2002) CD8 T-cell responses to Wilms tumor gene product WT1 and proteinase 3 in patients with acute myeloid leukemia. Blood 100(6):2132–2137. doi:10.1182/blood-2002-01-0163
Rezvani K, Yong AS, Savani BN, Mielke S, Keyvanfar K, Gostick E, Price DA, Douek DC, Barrett AJ (2007) Graft-versus-leukemia effects associated with detectable Wilms tumor-1 specific T lymphocytes after allogeneic stem-cell transplantation for acute lymphoblastic leukemia. Blood 110(6):1924–1932. doi:10.1182/blood-2007-03-076844
Rezvani K, Yong AS, Mielke S, Savani BN, Musse L, Superata J, Jafarpour B, Boss C, Barrett AJ (2008) Leukemia-associated antigen-specific T-cell responses following combined PR1 and WT1 peptide vaccination in patients with myeloid malignancies. Blood 111(1):236–242. doi:10.1182/blood-2007-08-108241
Mailander V, Scheibenbogen C, Thiel E, Letsch A, Blau IW, Keilholz U (2004) Complete remission in a patient with recurrent acute myeloid leukemia induced by vaccination with WT1 peptide in the absence of hematological or renal toxicity. Leukemia 18(1):165–166. doi:10.1038/sj.leu.2403186
Wadelin F, Fulton J, McEwan PA, Spriggs KA, Emsley J, Heery DM (2010) Leucine-rich repeat protein PRAME: expression, potential functions and clinical implications for leukaemia. Mol Cancer 9:226. doi:10.1186/1476-4598-9-226
Greiner J, Ringhoffer M, Taniguchi M, Li L, Schmitt A, Shiku H, Dohner H, Schmitt M (2004) mRNA expression of leukemia-associated antigens in patients with acute myeloid leukemia for the development of specific immunotherapies. Int J Cancer 108(5):704–711. doi:10.1002/ijc.11623
Tajeddine N, Millard I, Gailly P, Gala JL (2006) Real-time RT-PCR quantification of PRAME gene expression for monitoring minimal residual disease in acute myeloblastic leukaemia. Clin Chem Lab Med 44(5):548–555. doi:10.1515/CCLM.2006.106
Rezvani K, Yong AS, Tawab A, Jafarpour B, Eniafe R, Mielke S, Savani BN, Keyvanfar K, Li Y, Kurlander R, Barrett AJ (2009) Ex vivo characterization of polyclonal memory CD8+ T-cell responses to PRAME-specific peptides in patients with acute lymphoblastic leukemia and acute and chronic myeloid leukemia. Blood 113(10):2245–2255. doi:10.1182/blood-2008-03-144071
Griffioen M, Kessler JH, Borghi M, van Soest RA, van der Minne CE, Nouta J, van der Burg SH, Medema JP, Schrier PI, Falkenburg JH, Osanto S, Melief CJ (2006) Detection and functional analysis of CD8+ T cells specific for PRAME: a target for T-cell therapy. Clin Cancer Res 12(10):3130–3136. doi:10.1158/1078-0432.CCR-05-2578
Quintarelli C, Dotti G, Hasan ST, De Angelis B, Hoyos V, Errichiello S, Mims M, Luciano L, Shafer J, Leen AM, Heslop HE, Rooney CM, Pane F, Brenner MK, Savoldo B (2011) High-avidity cytotoxic T lymphocytes specific for a new PRAME-derived peptide can target leukemic and leukemic-precursor cells. Blood 117(12):3353–3362. doi:10.1182/blood-2010-08-300376
Grigoleit GU, Kapp M, Hebart H, Fick K, Beck R, Jahn G, Einsele H (2007) Dendritic cell vaccination in allogeneic stem cell recipients: induction of human cytomegalovirus (HCMV)-specific cytotoxic T lymphocyte responses even in patients receiving a transplant from an HCMV-seronegative donor. J Infect Dis 196(5):699–704. doi:10.1086/520538
Kedl RM, Kappler JW, Marrack P (2003) Epitope dominance, competition and T cell affinity maturation. Curr Opin Immunol 15(1):120–127
Van Driessche A, Van de Velde AL, Nijs G, Braeckman T, Stein B, De Vries JM, Berneman ZN, Van Tendeloo VF (2009) Clinical-grade manufacturing of autologous mature mRNA-electroporated dendritic cells and safety testing in acute myeloid leukemia patients in a phase I dose-escalation clinical trial. Cytotherapy 11(5):653–668. doi:10.1080/14653240902960411
Chang AE, Redman BG, Whitfield JR, Nickoloff BJ, Braun TM, Lee PP, Geiger JD, Mule JJ (2002) A phase I trial of tumor lysate-pulsed dendritic cells in the treatment of advanced cancer. Clin Cancer Res 8(4):1021–1032
Butterfield LH, Ribas A, Dissette VB, Lee Y, Yang JQ, De la Rocha P, Duran SD, Hernandez J, Seja E, Potter DM, McBride WH, Finn R, Glaspy JA, Economou JS (2006) A phase I/II trial testing immunization of hepatocellular carcinoma patients with dendritic cells pulsed with four alpha-fetoprotein peptides. Clin Cancer Res 12(9):2817–2825. doi:10.1158/1078-0432.CCR-05-2856
Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Herndon JE 2nd, Lally-Goss D, McGehee-Norman S, Paolino A, Reardon DA, Friedman AH, Friedman HS, Bigner DD (2009) An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol Cancer Ther 8(10):2773–2779. doi:10.1158/1535-7163.MCT-09-0124
Nakai N, Asai J, Ueda E, Takenaka H, Katoh N, Kishimoto S (2006) Vaccination of Japanese patients with advanced melanoma with peptide, tumor lysate or both peptide and tumor lysate-pulsed mature, monocyte-derived dendritic cells. J Dermatol 33(7):462–472. doi:10.1111/j.1346-8138.2006.00110.x
Kronke J, Schlenk RF, Jensen KO, Tschurtz F, Corbacioglu A, Gaidzik VI, Paschka P, Onken S, Eiwen K, Habdank M, Spath D, Lubbert M, Wattad M, Kindler T, Salih HR, Held G, Nachbaur D, von Lilienfeld-Toal M, Germing U, Haase D, Mergenthaler HG, Krauter J, Ganser A, Gohring G, Schlegelberger B, Dohner H, Dohner K (2011) Monitoring of minimal residual disease in NPM1-mutated acute myeloid leukemia: a study from the German-Austrian acute myeloid leukemia study group. J Clin Oncol 29(19):2709–2716. doi:10.1200/JCO.2011.35.0371
Cilloni D, Renneville A, Hermitte F, Hills RK, Daly S, Jovanovic JV, Gottardi E, Fava M, Schnittger S, Weiss T, Izzo B, Nomdedeu J, van der Heijden A, van der Reijden BA, Jansen JH, van der Velden VH, Ommen H, Preudhomme C, Saglio G, Grimwade D (2009) Real-time quantitative polymerase chain reaction detection of minimal residual disease by standardized WT1 assay to enhance risk stratification in acute myeloid leukemia: a European LeukemiaNet study. J Clin Oncol 27(31):5195–5201. doi:10.1200/JCO.2009.22.4865
Ommen HB, Nyvold CG, Braendstrup K, Andersen BL, Ommen IB, Hasle H, Hokland P, Ostergaard M (2008) Relapse prediction in acute myeloid leukaemia patients in complete remission using WT1 as a molecular marker: development of a mathematical model to predict time from molecular to clinical relapse and define optimal sampling intervals. Br J Haematol 141(6):782–791. doi:10.1111/j.1365-2141.2008.07132.x
Weisser M, Kern W, Rauhut S, Schoch C, Hiddemann W, Haferlach T, Schnittger S (2005) Prognostic impact of RT-PCR-based quantification of WT1 gene expression during MRD monitoring of acute myeloid leukemia. Leukemia 19(8):1416–1423. doi:10.1038/sj.leu.2403809
Buccisano F, Maurillo L, Spagnoli A, Del Principe MI, Fraboni D, Panetta P, Ottone T, Consalvo MI, Lavorgna S, Bulian P, Ammatuna E, Angelini DF, Diamantini A, Campagna S, Ottaviani L, Sarlo C, Gattei V, Del Poeta G, Arcese W, Amadori S, Lo Coco F, Venditti A (2010) Cytogenetic and molecular diagnostic characterization combined to postconsolidation minimal residual disease assessment by flow cytometry improves risk stratification in adult acute myeloid leukemia. Blood 116(13):2295–2303. doi:10.1182/blood-2009-12-258178
Al-Mawali A, Gillis D, Lewis I (2009) The role of multiparameter flow cytometry for detection of minimal residual disease in acute myeloid leukemia. Am J Clin Pathol 131(1):16–26. doi:10.1309/AJCP5TSD3DZXFLCX
Terwijn M, van Putten WL, Kelder A, van der Velden VH, Brooimans RA, Pabst T, Maertens J, Boeckx N, de Greef GE, Valk PJ, Preijers FW, Huijgens PC, Drager AM, Schanz U, Jongen-Lavrecic M, Biemond BJ, Passweg JR, van Gelder M, Wijermans P, Graux C, Bargetzi M, Legdeur MC, Kuball J, de Weerdt O, Chalandon Y, Hess U, Verdonck LF, Gratama JW, Oussoren YJ, Scholten WJ, Slomp J, Snel AN, Vekemans MC, Lowenberg B, Ossenkoppele GJ, Schuurhuis GJ (2013) High prognostic impact of flow cytometric minimal residual disease detection in acute myeloid leukemia: data from the HOVON/SAKK AML 42A study. J Clin Oncol 31(31):3889–3897. doi:10.1200/JCO.2012.45.9628
Freeman SD, Virgo P, Couzens S, Grimwade D, Russell N, Hills RK, Burnett AK (2013) Prognostic relevance of treatment response measured by flow cytometric residual disease detection in older patients with acute myeloid leukemia. J Clin Oncol 31(32):4123–4131. doi:10.1200/JCO.2013.49.1753
Kohnke T, Sauter D, Ringel K, Hoster E, Laubender RP, Hubmann M, Bohlander SK, Kakadia PM, Schneider S, Dufour A, Sauerland MC, Berdel WE, Buchner T, Wormann B, Braess J, Hiddemann W, Spiekermann K, Subklewe M (2014) Early assessment of minimal residual disease in aml by flow cytometry during aplasia identifies patients at increased risk of relapse. Leukemia. doi:10.1038/leu.2014.186
Acknowledgments
We thank Frauke Schnorfeil for help with Figs. 1 and 2. This work was supported in part by funds from BayImmuNet, the Bavarian Immunotherapy Network (www.bayimmunet.de), and the Helmholtz Alliance for Immunotherapy of Cancer.
Conflict of interest
Dolores J. Schendel is Managing Director of Trianta Immunotherapies GmbH and Chief Scientific Officer of Medigene AG. Christiane Geiger is Associate Director of DC Vaccine Development of Trianta Immunotherapies GmbH. All other authors declare that they have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper is a Focussed Research Review based on a presentation given at the Thirteenth International Conference on Progress in Vaccination against Cancer (PIVAC 13), held in Amsterdam, the Netherlands, 2nd – 4th October 2013. It is part of a CII series of Focussed Research Reviews and meeting report.
Rights and permissions
About this article

Cite this article
Subklewe, M., Geiger, C., Lichtenegger, F.S. et al. New generation dendritic cell vaccine for immunotherapy of acute myeloid leukemia. Cancer Immunol Immunother 63, 1093–1103 (2014). https://doi.org/10.1007/s00262-014-1600-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-014-1600-5