
Abstract
Adenosine is a potent extracellular messenger that is produced in high concentrations under metabolically unfavourable conditions. Tissue hypoxia, consequent to a compromised cellular energy status, is followed by the enhanced breakdown of ATP leading to the release of adenosine. Through the interaction with A2 and A3 membrane receptors, adenosine is devoted to the restoration of tissue homeostasis, acting as a retaliatory metabolite. Several aspects of the immune response have to be taken into consideration and even though in general it is very important to dampen inflammation, in some circumstances, such as the case of cancer, it is also necessary to increase the activity of immune cells against pathogens. Therefore, adenosine receptors that are defined as ‘sensors–of metabolic changes in the local tissue environment may be very important targets for modulation of immune responses and drugs devoted to regulating the adenosinergic system are promising in different clinical situations.
Similar content being viewed by others


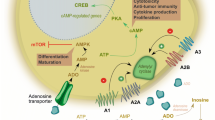
Introduction
Adenosine is an endogenous purine nucleoside that is constitutively present at low levels outside the cells but might dramatically increase its concentrations following metabolic stress conditions like those induced by hypoxia or ischaemia. After its release adenosine induces its biological effects through the interaction with four cell surface receptors classified by molecular, biochemical and pharmacological data into four subtypes: A1, A2A, A2B and A3 [1]. Each of these receptors, with the exception of the A1 subtype, are expressed on human and mouse T lymphocytes and it appears that their activation represents a potent endogenous immunosuppressive pathway that regulates the excessive immune response against potent external insults. The interest in the immunomodulatory effects of adenosine arose after the discovery that hereditary deficiency of the enzyme adenosine deaminase (ADA) was associated with severe combined immune deficiency disease (SCID) [2]. ADA converts adenosine to inosine and deoxyadenosine to deoxyinosine and its activity is greater in T cells in comparison with B cells or erythrocytes. ADA deficiency is one of the most severe immunodeficiencies due to the sensitivity of T cells to the accumulation of the ADA substrates adenosine and 2–deoxyadenosine. SCID is a disease characterized by severe lymphocytopenia, affecting both B and T cells, and a marked susceptibility to infection. The accumulation of adenosine and the activation of adenosine receptors in T cells may lead to the depletion of lymphocytes and impairment of their function [3, 4]. Moreover, ADA deficiency in mice causes impairment of intrathymic T cell development and enhanced thymocyte apoptosis supporting the hypothesis that adenosine is responsible for depletion of T cells occurring in ADA SCID [5]. Indeed, the studies aimed at elucidating the mechanisms by which the absence of ADA leads to immunodeficiency first suggested the presence of adenosine receptors on lymphocytes to suppress or dampen the immune response [6]. Therefore, to understand the pathophysiological implications of adenosine-triggered effects in T cells we intend to review the main actions attributed to adenosine by receptor subtype activation.
Adenosine metabolism and adenosine receptor activation under physiological and pathological conditions
Adenosine has several physiological effects through the interaction with four known adenosine receptors, A1 and A3 that through the interaction with Gi/0 inhibit adenylyl cyclase activity, and A2A and A2B that increase cAMP production (a strong immunosuppressive agent) via Gs. In addition, they can modulate the activity of phospholipase C, D, A2, cGMP, K+ and Ca2+ channels and mitogen-activated protein kinases (MAPKs) regulating a variety of cellular effects [1]. It has been suggested that adenosine receptors act as ‘sensors–and that extracellular adenosine acts as a ‘reporter–of metabolic changes in the local tissue environment [7]. Adenosine concentrations are normally regulated through its catabolism by ADA and through phosphorylation by adenosine kinase. In ADA deficiency the capacity of adenosine kinase is saturated and adenosine levels increase from 100–00 nM to 10 μM [2], which could excessively stepwise activate all adenosine receptors. Moreover, an increase of intracellular adenosine inhibits the hydrolysis of S-adenosylhomocysteine that is a potent inhibitor of transmethylation reactions. The lack of ADA leads also to the accumulation of deoxyadenosine, a product of DNA degradation, that in contrast to adenosine does not play a regulatory role in physiological conditions but determines an increase in deoxyATP that in turn inhibits DNA synthesis and activates apoptosis suggesting the possibility that deoxyadenosine itself may also have pathological effects [2]. Inflammatory tissue conditions are often associated with a low oxygen tension, or hypoxia, that is the cause of an associated production of adenosine in this environment. Therefore, the prevalent activity of 5’nucleotidase over activity of adenosine kinase [8, 9] occurring in hypoxic conditions is responsible for a significant adenosine accumulation in sites of tissue injury [10]. 5′Nucleotidase represents the major enzyme responsible for the extracellular production of adenosine from AMP and has a critical role in the functional activation of T cells. Deficiency of 5′nucleotidase activity has been associated with a number of immunodeficiency diseases, such as lymphoproliferative disorders and systemic lupus erythematosus. Abnormal levels of these enzymes may be associated with an autoimmune pathology [11].
Therefore, under normal conditions adenosine, which is continuously produced intracellularly and extracellularly and maintained at low intracellular levels (about 100 nM) through the metabolic activity exerted by adenosine kinase and adenosine deaminase, interacts with the high-affinity A1 and A2A receptor subtypes. In ADA deficiency or in hypoxic, ischaemic or inflamed conditions, the intracellular production of adenosine is increased at very high micromolar concentrations and transported across cell membranes by specific agents finally leading to the stepwise activation of all adenosine receptors, including the low-affinity A2B and A3 subtypes. Adenosine has been classified as a ‘retaliatory metabolite–as a consequence of its ability to mediate an autoregulatory loop, the function of which is to protect organs from injury following the initiating stressful stimuli [12, 13]. There are different mechanisms by which adenosine receptor activation protects organs. First, it decreases the energy demand of the tissue, second it can augment oxygen and nutrient supply through vasodilation, and finally, it regulates the exuberant immune response to harmful external agents. As basal adenosine levels have only a marginal influence on the immune response, ischaemic and inflammatory conditions represent scenarios in which adenosine levels become high enough to have significant immunomodulatory and specifically immunosuppressive effects [14].
Adenosine inhibits lymphocyte activation through A2 receptor activation
A body of literature concerning in vitro and in vivo studies clearly shows the beneficial role of adenosine as an immune modulator. Activation of T lymphocytes starts from the recognition of antigen by T cell antigen receptor and CD4 or CD8 coreceptors that initiates a cascade of signalling events resulting in cytokine secretion, cellular cytotoxicity and T cell proliferation. The immunosuppressive effects of adenosine on cytotoxic T lymphocyte (CTL) actions may be explained by activation of A2A receptors followed by sustained increases in cAMP that, in turn, antagonize T cell receptor (TCR)-triggered signalling. In particular, adenosine modulates TCR-mediated interleukin (IL)-2 production, expression of CD25 and CD69, granule exocytosis, Fas ligand up-regulation and cell proliferation through the interaction with A2A receptors [15, 16]. The predominant expression of A2A receptors, which has been established in functional assays using selective agonists and antagonists of A2A receptors, was confirmed by Northern blot studies [15, 16]. The role of A2A adenosine receptors in the regulation of the immune response has been investigated by determining the expression levels of this receptor in different subsets of functional lymphocytes [17]. The levels of expression of A2A receptors have been found to be much higher among T than B cells. T cell subsets are distinguished by the expression of TCR coreceptor molecules CD8+ and CD4+ involved in recognition of class I and class II major histocompatibility complex, respectively. More CD4+ than CD8+ T cells express A2A adenosine receptors. CD8+ T cells are mostly cytotoxic effector cells, whereas CD4+ cells have been implicated in T-helper cell activities. Studies of T-helper cell subsets (TH1 and TH2) reveal that lymphokine-producing cells are much more likely to express A2A receptors than are cells that do not produce lymphokines. A possible explanation was that inhibitory A2A receptors are induced selectively in cells that produce cytokines, as a means of limiting cytokine release [17]. In agreement with these results, it has been reported that activation of CD4+ lymphocytes induced A2A receptor mRNA expression and resulted in a dose-dependent inhibition of TCR-mediated interferon gamma (IFN-γ) production, which is the most important effector molecule synthesized by Th1 cells [18]. Recently, the effect of adenosine on IL-2 receptor-associated signalling pathways was explored in T lymphocytes by Zhang and co-workers [19]. IL-2 is an immunomodulatory molecule crucial for the proliferation of activated T cells, and its receptor consists of three subunits indicated as alpha, beta and gamma. Signalling through this receptor for the transduction of a proliferative signal includes Jak1, Jak3 and STAT5. Jak1 and Jak3 activation leads to phosphorylation of tyrosine residues on IL-2 receptors. Primed by these events the IL-2 complex generates two major proliferative signals. One of these signals is mediated by the adaptor protein Shc and activates p42/p44 MAPK and phosphatidylinositol 3-kinase (PI3K) signalling pathways. The second proliferative signal transduced by the IL-2 receptor complex involves the transcription factor STAT5 that after phosphorylation translocates to the nucleus to control gene expression. Adenosine, by activating A2A and A2B receptors, inhibits IL-2-dependent proliferation through inhibition of STAT5 phosphorylation. Because T cells play a major role in antitumour immunity and considering that adenosine is highly increased on the microenvironment of solid carcinomas, the adenosine-induced defect in IL-2-stimulated proliferative signalling would be expected to result in a defective T lymphocyte response in cancer patients and generally in impaired antitumour immunity (Fig. 1). Impairment of proliferation has been recently investigated also in anergic B lymphocytes [20]. In B cells adenosine inhibits activation of NF-kappa B in response to B cell antigen receptor (BCR) triggering (Fig. 1). Stimuli known to enhance 3–5–cyclic adenosine monophosphate (cAMP) are capable of selectively suppressing the activation both of NF-kB downstream of the BCR and Toll-like receptor 4 in splenic B lymphocytes. This suppression is reached by blocking phosphorylation and subsequent degradation of the inhibitor of NF-kappa B. This suggests that adenosine-mediated signals represent an important step in the molecular decision process controlling inflammation versus anergic immune responses. In this way adenosine might affect B cell dysfunction in ADA deficiency and may explain defective B cell proliferation and activation found in ADA-deficient mice [21]. Convincing evidence of a link between adenosine receptors and NF-kB activation comes also from in vivo studies performed in A2A adenosine receptor knockout (KO) mice. Lipopolysaccharide (LPS), given to these animals, induced a greater inflammatory response and a higher binding of NF-kB to nuclear DNA and consequently a higher cytokine gene transcription in macrophages [10].

Role of adenosinereceptors in the regulationof immune responses
In vivo evidence of the immunosuppressive effects played by adenosine through A2A receptors
Although it has been demonstrated that adenosine in vitro acted as an immunosuppressing agent it was not clear, until genetically modified mice were available, whether its effects were present also under pathophysiological conditions in vivo. There are many G protein-coupled receptors that increase cAMP levels showing immunosuppressive pharmacological effects and that have been considered as potential anti-inflammatory agents, e.g. catecholamines, prostaglandins, dopamine and histamine. However, although pharmacological evidence underlines their importance as potential immunosuppressive molecules, there is no real proof whether their recruitment does occur during physiological control of inflammation in vivo. Such evidence has been obtained for adenosine A2A receptors. In wild-type mice, activation of A2A receptors blocked tissue damage induced by inflammation demonstrating its role in counteracting inflammation [22, 23]. More recently, the availability of A2A receptor-deficient mice in models of acute liver inflammation and sepsis provided the first conclusive in vivo evidence of A2A receptors playing a critical role in the suppression of acute inflammation. Evaluation of T cell-, macrophage- and cytokine-dependent tissue injury in A2A adenosine receptor-deficient mice revealed a dramatic increase of local tissue damage and an increase of proinflammatory cytokines such as tumour necrosis factor α (TNF-α), IFN-γ and IL-12 in these mice, but not in wild-type animals [24, 25]. Moreover, low doses of inflammatory stimuli, which were not damaging in wild-type animals, were detrimental in terms of liver status and cytokine levels in knockout mice. These data furnished the first indisputable demonstration of an in vivo role played by A2A adenosine receptor in the regulation of inflammation and underlined a non-redundant role in the down-regulation of inflammation in vivo. Importantly, A2A receptors seem to be essential as demonstrated by the failure of all other anti-inflammatory mechanisms, e.g. β-adrenergic and prostaglandin receptors, to overcome the lack of A2A receptors in preventing severe tissue injury. Experiments performed in thymocytes and T cells from heterozygous A2A receptor mice showed that there was a correlation between reduction of A2A receptors and reduction of cAMP response of the cells after activation with adenosine and suggesting the lack of A2A spare receptors in T cells and a lack of compensation by the A2B receptors [26]. Also the level of apoptosis of thymocytes from these animals was less pronounced following A2A activation in comparison to that obtained in wild-type mice. The expression of other adenosine subtypes like A1, A2B or A3 did not compensate for the lack of A2A in lymphoid organ of deficient mice as suggested by real-time reverse transcription polymerase chain reaction (RT-PCR) experiments indicating again that in acute models of inflammation, the A2A receptors play a crucial and non-redundant role in the protection of tissue from damage derived by excessive inflammation [27]. It has been also suggested that in non-exacerbated inflammatory conditions different anti-inflammatory stimuli, such as other adenosine receptor subtypes and other anti-inflammatory mediators, may act in concert to reduce the extent of damage. Therefore, based on the evidences reported above, the treatment of a series of diseases such as sepsis, wound healing and rheumatoid arthritis, might improve following A2A receptor stimulation. Indeed, methotrexate (MTX), which is used for treatment of rheumatoid arthritis, may mediate its effects through the release of endogenous adenosine [28]. Even though its mechanism of action remains unclear, it has been shown that MTX caused a dose-dependent suppression of T cell activation and adhesion molecule expression, and this was not due to lymphocyte apoptosis. The suppression of intercellular adhesion molecule (ICAM)-1 was adenosine and folate dependent, suggesting that the suppression of T cell activation and T cell adhesion molecule expression, rather than apoptosis, mediated in part by adenosine or polyglutamated MTX or both, are important mechanisms in the anti-inflammatory action of MTX [29]. Besides methotrexate, there is evidence that other therapeutic agents, such as sulfasalazine and FK-506, could exert their anti-inflammatory effects by promoting adenosine release [30, 31].
Adenosine inhibits killer T cell activation via A3 and A2A stimulation: implications in cancer
The ability of immune cells to recognize and eliminate tumour cells is fundamental for successful host defence against cancer. It has been suggested that adenosine, the concentration of which increases within hypoxic regions of solid tumours, may interfere with the recognition of tumour cells by cytolytic effector cells of the immune system [32, 33]. Adoptive immunotherapy with lymphokine-activated killer (LAK) cells has shown some promise in the treatment of certain cancers that are unresponsive to conventional treatment approaches. However, colon adenocarcinomas tend to respond poorly to LAK therapy, possibly as a result of tumour-induced immunosuppression. In 1994 Hoskin and co-workers demonstrated that colon adenocarcinoma cells inhibited anti-CD3-activated killer cell induction through the production of a tumour-associated soluble factor that was distinct from transforming growth factor beta or prostaglandins [34]. In the same period these authors indicated adenosine as a possible inhibitor of killer T cell activation in the microenvironment of solid tumours [35] and showed that 2-chloroadenosine (2CA), a stable analogue of adenosine, reduced MHC-unrestricted killing of P815 tumour target cells by anti-CD3-activated killer (AK) lymphocytes. 2CA exerted this effect by interfering with the recognition/adhesion phase of cytolysis. Treatment with dipyridamole to block cellular uptake of 2CA increased the inhibition of cytolysis, suggesting the involvement of a cell surface receptor. However, neither DPCPX nor DMPX, the A1 and A2 receptor antagonists, respectively, were able to reduce the inhibitory effect of 2CA on AK lymphocyte function. Similarly, the nonselective A1 and A2 receptor blockers, theophylline and 8-phenyltheophylline, had no effect on 2CA-mediated inhibition of AK cell activity. These data clearly demonstrated that 2CA inhibited the cytolytic activity of AK lymphocytes by interacting with a novel non-A1/A2 cell surface receptor [36]. Then, MacKenzie and co-workers [37] evaluated the adhesion of murine spleen-derived anti-CD3-activated killer (AK) lymphocytes to syngeneic MCA-38 colon adenocarcinoma cells. Adenosine, in the presence of the adenosine deaminase inhibitor coformycin, reduced adhesion by up to 60%. The inhibitory effect of adenosine was exerted on AK cells and not on the MCA-38 targets and was mediated by cell surface receptors as adenosine-induced inhibition of adhesion was not abrogated following treatment with dipyridamole, a blocker of adenosine uptake. The agonist potency profile indicated that the A3 receptor subtype might be responsible for the inhibition of adhesion. The authors suggested that this mechanism of immunosuppression, secondary to tissue hypoxia, may be important in the resistance of colorectal and other solid cancers to immunotherapy. In addition, the same authors demonstrated that adenosine exerts a strong inhibitory effect on the induction of mouse cytotoxic T cells [38]. Diminished tumouricidal activity correlated with reduced expression of mRNAs coding for granzyme B, perforin, Fas ligand and TNF-related apoptosis-inducing ligand (TRAIL) (Fig. 1). IL-2 and IFN-γ synthesis by AK-T cells was also inhibited by adenosine. Also in this case the inhibitory effect of adenosine on AK-T cell proliferation was blocked by an A3 receptor antagonist, suggesting that adenosine acts through A3 receptors to prevent AK-T cell induction. Tumour-associated adenosine may act through the same mechanism to impair the development of tumour-reactive T cells in cancer patients. Therefore, the suppression of T killer cell function suggests that adenosine may act as a local immunosuppressant within the microenvironment of solid tumours. The same authors reported that adenosine partially inhibits the interaction of T lymphocytes with tumour cells by blocking the function of integrin α4β7, which is the major cell adhesion molecule involved in the adhesion of T cells to syngeneic MCA-38 adenocarcinoma cells [39]. Importantly, involvement of α4β7 has also been postulated in leukocyte localization in inflammatory disorders such as asthma, intestinal inflammation and rheumatic disorders and may be regulated by adenosine [40–42]. As adenosine is known also for its anti-inflammatory effects in addition to its immunosuppressive actions, it is possible to hypothesize that adenosine-mediated inhibition of adhesion through α4β7 found in T cells may be important in a variety of other inflammatory diseases. Tumour microenvironment can suppress the function of tumour-infiltrating T cells. Therefore, the effect of adenosine has been investigated on the expression of costimulatory molecules by T cells in resting and activated conditions. One of the most important costimulatory molecules present on the T cells–surface are CD2 and CD28 acting in concert to achieve optimal costimulation of T lymphocytes during interaction with antigen-presenting cells. It has been demonstrated that adenosine interfered with activation-induced expression of the costimulatory molecules CD2 and CD28 and that their up-regulation was IL-2 dependent. This effect could not be attributed to cyclic AMP (cAMP) accumulation resulting from the stimulation of adenylyl cyclase-coupled adenosine receptors, even though cAMP at concentrations much higher than those generated following adenosine stimulation was inhibitory for both CD2 and CD28 expression. Therefore, it has been proposed that adenosine interferes with IL-2-dependent T cell expression of costimulatory molecules via a mechanism that does not involve the accumulation of intracellular cAMP and through a cell surface receptor that is coupled to signalling pathways different from those involving adenylyl cyclase activity, possibly the A3 subtype [43]. However, the relevance of adenosine-mediated pathways in tumour-infiltrating T cells, considering the expression on T cells of adenosine deaminase in association with CD26 (dipeptidyl aminopeptidase), remains to be elucidated [44]. CD26 is a multifunctional type II cell surface glycoprotein widely expressed on T, B and natural killer (NK) cells. One of its functions is that of an adenosine deaminase binding site. Binding of ADA to CD26 is capable of reducing the local concentration of adenosine and has an enzymatic role in protecting T cells from an adenosine-mediated inhibition of proliferation [44, 45]. Indeed, it has been reported that adenosine deaminase activity is significantly lower in the peripheral lymphocytes of cancer patients, suggesting increased susceptibility of T cells to adenosine-mediated inhibition within the tumour microenvironment [46]. Moreover, cytotoxic T cells show very low levels of adenosine deaminase activity, leaving us to hypothesize that cytotoxic cells would be quite sensitive to the immune inhibitory effect of adenosine accumulated within tumours. In this regard, it is relevant to underline that the presence of the A3 adenosine receptor has been demonstrated in Jurkat cells, a human leukaemic cell line, where its level is very high, and in human lymphocytes in which an up-regulation occurs after T cell activation [47, 48]. In addition to the A3 receptors, antigen activation has been shown to alter the expression of other adenosine subtypes. For example it has been reported that A2B receptors are also up-regulated by phytohemagglutinin (PHA) and/or anti-CD3 in both CD4+ and CD8+ cells [49], and as for A2A subtypes there are contrasting reports suggesting both that the activation process increases their expression predominantly in CD8+ and/or CD4+ T cells [17, 18]. These data suggest that adenosine may stepwise recruit different adenosine receptors following antigen activation. In human lymphocytes, e.g. A2A and A2B receptors are coexpressed, leading to synergy in cAMP production [50]; in addition, adenosine may mediate different signals if each of these receptors is linked to different effector molecules, as is the case of the A3 subtype. In contrast to the immunosuppressive role of adenosine in the environment of solid tumours, recent findings from in vivo studies of Fishmans–group report a stimulatory effect of A3 agonists in the synthesis of IL-12 that is dependent from inhibition of cAMP levels and protein kinase A expression. This cytokine is a potent stimulatory agent of NK cells and is a cytotoxic factor that exerts a potent antitumour effect in vivo. It induces IFN-γ production by activated T and NK cells and augments cytotoxic activity of these cells via perforin, Fas and TRAIL-dependent mechanisms. Therefore, it enhances NK cell activity and probably NK cell-mediated destruction of tumour cells [51]. This antitumour effect played in immune cells is in line with other findings of the same group demonstrating a direct inhibitory action of A3 receptor activation on tumour cell growth [52].
However, in general, it has to be remarked that contrasting results obtained from pharmacological studies may be explained by the limitations due to the poor selectivity of agonists and antagonists for adenosine receptors. Recent data obtained from studies using adenosine receptor KO mice examined the capability of adenosine and its analogues to inhibit the ability of lymphokine-activated killer cells (LAK) to kill tumour cells. This work demonstrated that adenosine and adenosine A2A ligands suppress the cytotoxicity of LAK cells in parallel with their ability to increase cAMP levels. These effects were produced by interfering with both perforin-mediated and Fas ligand-mediated killing pathways. Studies with LAK cells generated from A1 and A3 adenosine receptor KO mice indicated the lack of any involvement of these adenosine subtypes in the inhibitory effect exerted by adenosine, whereas LAK cells obtained from A2 adenosine receptor KO mice were resistant to the inhibitory effect of this nucleoside. Only very high concentrations of the nonselective agonists NECA or CADO produced mild inhibition of LAK cytotoxicity that were possibly induced through A2B activation, suggesting a predominant role of A2A subtype in inhibition of LAK cell toxicity [53]. Therefore, adenosine has been indicated as an important intra-tumour factor that inhibits the effector function of NK and T cells and protects tumours from immune destruction. Being that these effects are mediated by A2A receptors, the authors suggest the introduction of A2A antagonists to increase the efficacy of immunotherapy.
Conclusion and perspectives
The evidences summarized in this review indicate that adenosine through the interaction with A2 and A3 receptors plays a crucial role in the regulation of immune cells. The most important therapeutic implication arising from the data summarized above is that A2A receptors, proposed as ‘natural–brakes of inflammation, appear to represent a promising pharmacological target to treat a wide variety of diseases characterized by a strong immunoinflammatory component. On the other hand, it may be advantageous in some circumstances to enhance certain aspects of inflammation in order to eliminate the causative agent, as in the case of cancer. In fact, it has to be remarked that tumour defence mechanisms are akin to inflammatory processes. Solid tumours, due to their abnormal vasculature, are often hypoxic and show increased levels of adenosine that may be an important mediator of tumour-associated immunosuppression. It is likely that killer T cells that may be recruited against cancer cells fail to act in an effective manner due to the high level of adenosine in the tumour microenvironment. Because several of these immunosuppressive functions have been attributed to the stimulation of A3 and A2A receptors, expressed on the surface of T cells, adenosine antagonists of these subtypes may be potentially useful in the immunotherapy of cancer.
Abbreviations
- IL:
-
interleukin
- IFN-γ:
-
interferon gamma
- TNF-α:
-
tumour necrosis factor α
- LPS:
-
lipopolysaccharide
- PHA:
-
phytohemagglutinin
- NK cells:
-
natural killer cells
References
Fredholm BB, Ijzerman AP, Jacobson KA, Klotz KN, Linden J (2001) International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 53:527–52
Hershfield MS (2005) New insights into adenosine-receptor-mediated immunosuppression and the role of adenosine in causing the immunodeficiency associated with adenosine deaminase deficiency. Eur J Immunol 35:25–0
Buckley RH (2004) Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol 22:625–55
Hershfield MS (2004) Combined immune deficiencies due to purine enzyme defects. In: Sthiem ER, Ochs HD, Winkelstein JA (eds) Immunologic disorders in infants and children, 5th edn. Saunders, Philadelphia, pp 480–04
Apasov SG, Blackburn MR, Kellems RE, Smith PT, Sitkovsky MV (2001) Adenosine deaminase deficiency increases thymic apoptosis and causes defective T cell receptor signalling. J Clin Invest 108:131–41
Wolberg G, Zimmerman TP, Hiemstra K, Winston M, Chu LC (1975) Adenosine inhibition of lymphocyte-mediated cytolysis: possible role of cyclic adenosine monophosphate. Science 87:957–59
Sitkovsky M, Ohta A (2005) The danger sensors that stop the immune response: the A2 adenosine receptors? Trends Immunol 26:299–04
Decking UKM (1997) Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ Res 81:154–64
Ledoux S (2003) Hypoxia enhances ecto-5–nucleotidase activity and cell surface expression in endothelial cells. Circ Res 92:848–55
Lukashev D, Ohta A, Apasov S, Chen JF, Sitkovsky M (2004) Cutting edge: physiologic attenuation of proinflammatory transcription by the Gs protein-coupled A2A adenosine receptor in vivo. J Immunol 173:21–4
Vivekanandhan S, Soundararajan CC, Tripathi M, Maheshwari MC (2005) Adenosine deaminase and 5′nucleotidase activities in peripheral blood T cells of multiple sclerosis patients. Neurochem Res 30:453–56
Newby AC (1984) Adenosine and the concept of retaliatory metabolites. Trends Biochem Sci 9:42–4
Hasko G, Cronstein BN (2004) Adenosine: an endogenous regulator of innate immunity. Trends Immunol 25:33–9
Gessi S, Varani K, Merighi S, Ongini E, Borea PA (1999) A2A adenosine receptors in human peripheral blood cells. Br J Pharmacol 129:2–1
Huang S, Apasov S, Koshiba M, Sitkovsky M (1997) Role of A2A extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood 90(4):1600–610
Koshiba M, Kojima H, Huang S, Apasov S, Sitkovsky M (1997) Memory of extracellular adenosine A2A purinergic receptor-mediated signaling in murine T cells. J Biol Chem 272:25881–5889
Koshiba M, Rosin D, Nobuhide H, Linden J, Sitkovsky V (1999) Patterns of A2A extracellular adenosine receptor expression in different functional subsets of human peripheral T cells. Flow cytometry studies with anti-A2A receptor monoclonal antibodies. Mol Pharmacol 55:614–24
Lappas MC, Rieger JM, Linden J (2005) A2A adenosine receptor induction inhibits IFN-gamma production in murine CD4+ T cells. J Immunol 174:1073–080
Zhang H, Conrad DM, Butler JJ, Zhao C, Blay J, Hoskin DW (2004) Adenosine acts through A2 receptors to inhibit IL-2-induced tyrosine phosphorylation of STAT5 in T lymphocytes: role of cyclic adenosine 3–5–monophosphate and phosphatases. J Immunol 173(2):932–44
Minguet S, Huber M, Rosenkranz L, Schamel WWA, Reth M, Brummer T (2005) Adenosine and cAMP are potent inhibitors of the NF-kB pathway downstream of immunoreceptors. Eur J Immunol 35:31–1
Aldrich MB, Chen W, Blackburn MR, Martinez-Valdez H, Datta SK, Kellems RE (2003) Impaired germinal center maturation in adenosine deaminase deficiency. J Immunol 171:5562–570
Cronstein BN (1994) Adenosine, an endogenous anti-inflammatory agent. J Appl Physiol 76(1):5–3
Hasko G, Szabo C, Nemeth ZH, Kvetan V, Pastores SM, Vizi ES (1996) Adenosine receptor agonists differentially regulate IL-10, TNF-alpha, and nitric oxide production in RAW 264.7 macrophages and in endotoxemic mice. J Immunol 157:4634–640
Ohta A, Sitkovsky M (2001) Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 414:916–20
Sitkovsky MV (2003) Use of the A2A adenosine receptor as a physiological immunosuppressor and to engineer inflammation in vivo. Biochem Pharmacol 65:493–01
Armstrong JM, Chen JF, Schwarzschild MA, Apasov S, Smith PT, Caldwell C, Chen P, Figler H, Sullivan G, Fink S, Linden J, Sitkovsky MV (2001) Gene dose effect reveals no Gs protein-coupled A2A adenosine receptor reserve in murine T-lymphocytes: studies of cells from A2A-receptor-gene-deficient mice. Biochem J 354:123–30
Lukashev DE, Smith PT, Caldwell CC, Ohta A, Apasov SG, Sitkovsky M (2003) Analysis of A2A receptor-deficient mice reveals no significant compensatory increases in the expression of A2B, A1, and A3 adenosine receptors in lymphoid organs. Biochem Pharmacol 65(12):2081–090
Chan ES, Cronstein BN (2002) Molecular action of methotrexate in inflammatory disease. Arthritis Res 4:266–73
Johnston A, Gudjonsson JE, Sigmundsdottir H, Ludviksson BR, Valdimarsson H (2005) The anti-inflammatory action of methotrexate is not mediated by lymphocyte apoptosis, but by the suppression of activation and adhesion molecules. Clin Immunol 114(2):154–63
Gadangi P, Longaker M, Naime D et al (1996) The anti-inflammatory mechanism of sulfasalazine is related to adenosine release at inflamed sites. J Immunol 156:1937–941
Hwang KK, Hall CS, Spielman WS et al (2001) FK506 promotes adenosine release from endothelial cells via inhibition of adenosine kinase. Eur J Pharmacol 425:85–3
Blay J, White TD, Hoskin DW (1997) The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res 57:2602–605
Merighi S, Mirandola P, Varani K, Gessi S, Leung E, Baraldi PG, Tabrizi MA, Borea PA (2003) A glance at adenosine receptors: novel target for antitumor therapy. Pharmacol Ther 100:31–8
Hoskin DW, Reynolds T, Blay J (1994) Colon adenocarcinoma cells inhibit anti-CD3-activated killer cell induction. Cancer Immunol Immunother 38(3):201–07
Hoskin DW, Reynolds T, Blay J (1994) Adenosine as a possible inhibitor of killer T-cell activation in the microenvironment of solid tumours. Int J Cancer 59(6):854–55
Hoskin DW, Reynolds T, Blay J (1994) 2-Chloroadenosine inhibits the MHC-unrestricted cytolytic activity of anti-CD3-activated killer cells: evidence for the involvement of a non- A1/A2 cell-surface adenosine receptor. Cell Immunol 159(1):85–3
MacKenzie WM, Hoskin DW, Blay J (1994) Adenosine inhibits the adhesion of anti-CD3-activated killer lymphocytes to adenocarcinoma cells through an A3 receptor. Cancer Res 54:3521–526
Hoskin DW, Buttler JJ, Drapeau D, Haeryfar SM, Blay J (2002) Adenosine acts through an A3 receptor to prevent the induction of murine anti-CD3-activated killer T cells. Int J Cancer 99:386–95
MacKenzie WM, Hoskin DW, Blay J (2002) Adenosine suppresses α4β7 integrin-mediated adhesion of T lymphocytes to colon adenocarcinoma cells. Exp Cell Res 276:90–00
Walsh GM, Symon FA, Lazarovits AI, Wardlaw AJ (1996) Integrin α4β7 mediates human eosinophils interaction with MadCAM1, VCAM1 and fibronectin. Immunology 89:112–19
Michetti M, Kelly CP, Kraehenbuhul JP, Bouzourene H, Michetti P (2000) Gastric mucosal alpha(4)beta(7)-integrin-positive CD4 T lymphocytes and immune protection against helicobacter infection in mice. Gastroenterology 119:109–18
Elewaut D, De Keyser F, Van Den Bosch F, Lazarovits AI, De Vos M, Cuvelier C, Verbruggen G, Mielants H, Veys EM (1998) Enrichment of T cells carrying β7 integrins in inflamed synovial tissue from patients with early spondyloarthropathy, compared to rheumatoid arthritis. J Rheumatol 25:1932–937
Butler JJ, Mader JS, Watson CL, Zhang H, Blay J, Hoskin DW (2003) Adenosine inhibits activation-induced T cell expression of CD2 and CD28 co-stimulatory molecules: role of interleukin-2 and cyclic AMP signaling pathways. J Cell Biochem 89:975–91
Dong RP, Kameoka J, Hegen M, Tanaka T, Xu Y, Schlossman SF, Morimoto C (1996) Characterization of adenosine deaminase binding to human CD26 on T cells and its biologic role in immune response. J Immunol 156:1349–355
Gorrell MD, Gysbers V, Mccaughan GW (2001) CD26: a multifunctional integral membrane and secreted protein of activated lymphocytes. Scand J Immunol 54:249–64
Dasmahapatra KS, Hill HZ, Dasmahapatra A, Suarez S (1986) Evaluation of adenosine deaminase activity in patients with head and neck cancer. J Surg Res 40:368–73
Gessi S, Varani K, Merighi S, Morelli A, Ferrari D, Leung E, Baraldi PG, Spalluto G, Borea PA (2001) Pharmacological and biochemical characterization of A3 adenosine receptors in Jurkat T cells. Br J Pharmacol 134:116–26
Gessi S, Varani K, Merighi S, Cattabriga E, Avitabile A, Gavioli R, Fortini C, Klotz KN, Leung E, MacLennan S, Borea PA (2004) Expression of A3 adenosine receptors in human lymphocytes: up-regulation in T cell activation. Mol Pharmacol 65:711–19
Mirabet M, Herrera C, Cordero OJ, Mallol J, Lluis C, Franco R (1999) Expression of A2B adenosine receptors in human lymphocytes: their role in T cell activation. J Cell Sci 112:491–02
Gessi S, Varani K, Merighi S, Cattabriga E, Pancaldi C, Szabadkai Y, Rizzuto R, Klotz KN, Leung E, MacLennan S, Borea PA (2005) Expression, pharmacological profile, and functional coupling of A2B receptors in a recombinant system and in peripheral blood cells by using a novel selective antagonist radioligand, [3H]-MRE 2029-F20. Mol Pharmacol 67:2137–147
Harish A, Hohana G, Fishman P, Arnon O, Bar-Yehuda S (2003) A3 adenosine receptor agonist potentiates natural killer cell activity. Int J Oncol 23(4):1245–249
Fishman P, Bar-Yehuda S, Barer F, Madi L, Multani AS, Pathak S (2001) The A3 adenosine receptor as a new target for cancer therapy and chemoprotection. Exp Cell Res 269:230–36
Raskovalova T, Huang X, Sitkovsky M, Zacharia LC, Jackson EK, Gorelik E (2005) Gs protein-coupled adenosine receptor signaling and lytic function of activated NK cells. J Immunol 175:4383–391
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Gessi, S., Varani, K., Merighi, S. et al. Adenosine and lymphocyte regulation. Purinergic Signalling 3, 109–116 (2007). https://doi.org/10.1007/s11302-006-9042-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-006-9042-y