
Abstract
The bile salt, sodium deoxycholate (NaDOC), is a natural detergent that promotes digestion of fats. At high physiologic levels, NaDOC activates many stress-response pathways and induces apoptosis in various cell types. NaDOC induces DNA damage and activates poly(ADP-ribose) polymerase (PARP), an enzyme that utilizes NAD+ as a substrate to repair DNA. NaDOC also induces oxidative stress, endoplasmic reticulum (ER) stress and contributes to protein malfolding. The NAD+ precursors, nicotinic acid (NA) and nicotinamide (NAM) were found to protect cells against NaDOC-induced apoptosis. NA and NAM also decreased constitutive levels of both activated NF-κB and GRP78, two proteins that respond to oxidative stress. However, the mechanism by which NA and NAM protects cells against apoptosis does not involve a reduction in constitutive levels of oxidative stress. NA or NAM treatment increased the protein levels of glyceraldehyde-3-phosphate dehydrogense (GAPDH), a multi-functional enzyme, in the nucleus and cytoplasm, respectively. NAM did not activate the promoter/response elements of 13 stress response genes nor reduce intracellular non-protein thiols, suggesting that it is non-toxic to cells. NAM thus has promise as a dietary supplement to help prevent disorders involving excessive apoptosis.
Similar content being viewed by others


Introduction
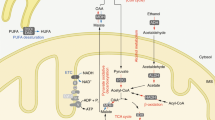
Bile salts are natural detergents produced in the liver from cholesterol1 and function to emulsify fats as an aid to digestion. However, high concentrations of certain bile salts are cytotoxic and induce apoptosis in hepatocytes and/or colonic epithelial cells. Apoptosis-inducing bile salts include deoxycholate (DOC),2,3,4,5,6 glycodeoxycholate,7,8 chenodeoxycholate,2 glycochenodeoxycholate9,10 and taurochenodeoxycholate.11 High concentrations of bile salts in the liver are found in association with cholestatic liver disease, in which a deficiency in the secretion of bile salts12 or an inborn error in bile acid metabolism13 results in the accumulation of hydrophobic bile acids within hepatocytes. High concentrations of bile salts in the colon usually result from a typical Western-style high fat/low fiber diet, and is associated with increased risk for colon cancer.1,14,15 The possible mechanisms by which the cytotoxic bile salts induce apoptosis include an increase in oxidative stress,16,17,18,19,20,21,22,23,24,25,26 an increase in DNA damage18,26,27,28,29,30,31,32,33,34,35 and the release of Ca2+ from the endoplasmic reticulum.36 Recent work from our laboratory indicated that DOC activates many stress-response proteins.27,30,37 These are NF-κB, a redox-sensitive transcription factor,38 poly(ADP-ribose)polymerase (PARP), a DNA repair enzyme,39,40 GADD153, a 153-kD growth arrest and DNA damage responsive protein involved in cell regulation,41 HSP70, a major heat shock-related chaperone protein42 and GRP78, a chaperone protein43 that functions within the ER.44 We have also shown, through the use of pharmacologic inhibitors, that the activation of NF-κB and PARP is protective against apoptosis.27
Protective PARP activity against DOC-induced apoptosis may be limited by substrate availability. As part of its DNA repair function, PARP consumes nicotinamide adenine dinucleotide (NAD+) to make ADP-ribose polymers that covalently attach to nuclear proteins45,46 and add negative charge to areas of DNA damage.40 This drains the NAD+ pool, which may compromise the further repair of DNA.47 A major decrease in NAD+ levels also has serious consequences for mitochondrial function. Since NAD+ is an electron carrier in mitochondrial respiration and a source of reducing equivalents, it contributes to the maintenance of the redox state of mitochondria, the formation of the mitochondrial membrane potential and the generation of ATP. ATP is also consumed in the synthesis of NAD+ from NAM in an endogenous biochemical pathway to replenish the NAD+ consumed by PARP activity. A major drain on NAD+ pools would, therefore, eventually compromise energy production, resulting in the death of the cell. The addition of the NAD+ precursors, nicotinic acid (NA) and nicotinamide (NAM) can however, effectively increase intracellular levels of NAD+,48,49,50 thereby protecting cells against excessive DNA damage, ATP depletion and cell death.
We now report that both NA and NAM dramatically protect cells against DOC-induced apoptosis. Potential mechanisms by which NA and NAM are protective, in addition to increasing substrate availability for PARP activity, were also explored in this study. We found that NA and NAM induce a low sub-lethal level of oxidative stress, down-regulate NF-κB, increase IκB-γ, decrease GRP78 and increase GAPDH levels in Jurkat cells.
Since increased oxidative damage, DNA damage and apoptosis are associated with numerous disorders,51,52,53,54 such as AIDS,55,56,57 inflammatory diseases,53 dermatologic-diseases,58 cardiovascular disease,59 neurodegenerative diseases,60 aging61,62,63 and cancer,64,65,66,67 NAD+ precursors may prove useful as chemopreventive68 and therapeutic agents.69,70 An assessment of relative cellular toxicities of NA and NAM (using 13 different promoter/response element CAT reporter constructs in the present study) indicates that NAM may be the preferred chemopreventive agent.
Results
Sodium deoxycholate induces classic apoptosis in Jurkat cells
Since transmission electron microscopy (TEM) is the gold standard for the identification of apoptotic cells,71,72,73,74 we first determined if DOC induced classic apoptosis in Jurkat cells which were derived from a T-cell lymphoma. Jurkat cells were either untreated or treated with 0.5 mM NaDOC for 2 h and then fixed for brightfield and TEM. Untreated cells showed normal morphology by light microscopy and TEM (Figure 1A,C). NaDOC-treated cells showed chromatin condensation, nuclear fragmentation and cellular shrinkage by light microscopy, and margination of chromatin with crescent formation and increased electron density by TEM, the classic morphologic features of apoptosis (Figure 1B,D). Jurkat cells (Figure 1D) exhibit less apoptotic body formation compared with HCT-116 cells (Figure 2), since the former have a higher nuclear/cytoplasmic ratio. The electron micrographs of HCT-116 cells depicted in Figure 2 show the early stages of nuclear margination with nucleolar segregation (Figure 2A) and the later stages of extensive apoptotic body formation (Figure 2B).

Light (A,B) and electron micrographs (C,D) comparing untreated Jurkat cells (A,C) with those treated with 0.5 mM NaDOC for 2 h (B,D). (A) The chromatin is evenly dispersed throughout the nucleus. (B) Several apoptotic cells are present and show chromatin condensation and nuclear fragmentation. (C) The chromatin is evenly dispersed and shows no margination or crescent formation. (D) Three apoptotic cells are shown. An increase in chromatin condensation is seen in all three cells. The cell at the upper right shows crescent formation and nucleolar segregation. The electron density of the cytoplasm of the cell at the bottom is markedly increased compared to untreated cells in (C). [Modified Giemsa stain, 100× oil immersion lens (A,B); uranyl acetate, lead citrate (C,D)]

Electron micrographs of HCT-116 cells reacted with 0.5 mM NaDOC for 2 h. (A) Two apoptotic cells are present in this field. The cell indicated by the arrow exhibits some margination of the chromatin, nucleolar segretation and an increase in electron density in both the nucleus and cytoplasm compared with the normal-appearing cell at the left. (B) This apoptotic cell is at a more advanced stage of apoptosis, evidenced by the more extensive chromatin condensation, a marked increase in electron density and apoptotic body formation (uranyl acetate, lead citrate)
Nicotinic acid and nicotinamide protect against NaDOC-induced apoptosis
Dose-response curves (NA/NAM concentration vs apoptosis/necrosis) were first carried out over a 24 h period to determine the maximum concentration of the NAD+ precursors that did not induce cell death in the absence of NaDOC. A non-lethal concentration of 10 mM NA or NAM was chosen to pretreat Jurkat cells prior to NaDOC treatment. Cells were incubated for 24 h in 10 mM NA or NAM followed by the addition of 0.5 mM NaDOC to the culture media. NA- and NAM-pretreated cells that did not receive any NaDOC, as well as cells with no pretreatment, served as controls. Aliquots of cells were removed after 1, 2 or 4 h of incubation and the cells were assessed for viability by dye exclusion and for apoptosis by morphological analysis. Apoptosis after NaDOC treatment was substantially reduced by pretreatment with either NA or NAM (Figure 3). Similar results were obtained in two additional experiments, with NA consistently being more protective than NAM.

Effect of preincubation with NAD+ precursors on NaDOC-induced apoptosis
Nicotinic acid and nicotinamide do not affect cellular proliferation
We have found that Jurkat cells in the very early phase of logarithmic growth are considerably more resistant to DOC-induced apoptosis than cells in the very late phase of logarithmic growth (unpublished results). Thus, we determined whether pretreatment with NA and NAM affected cellular proliferation in order to rule out a growth phase effect as a possible mechanism of the observed apoptosis resistance. Cells in early logarithmic growth phase were incubated in the presence or absence of 10 mM NA or 10 mM NAM for 4 days. At 24 h, the period of preincubation used in the previous apoptosis experiments, there was little difference between growth of untreated vs treated cells (Figure 4). After 4 days of treatment, only a slight decrease in growth of treated cells was observed (Figure 4). Similar results were obtained in two further experiments. These growth curves do not rule out small, transient cell cycle perturbations, but convincingly indicate that apoptosis resistance observed with NAD+ precursors is not caused by growth inhibition.

Growth of Jurkat cells in unsupplemented medium, or media supplemented with 10 mM NA or 10 mM NAM for 4 days
Nicotinic acid and nicotinamide reduce the constitutive levels of activated NF-κB
Jurkat, HT-29, and HCT-116 cells were grown in media alone, or in the presence of 10 mM NA or 10 mM NAM for 24 h. The cells were then fixed and stained with a monoclonal antibody against activated NF-κB, followed by immunofluorescent detection and confocal microscopy. It was found that NA and NAM treatment reduced the intracellular levels of activated NF-κB relative to untreated control cells (Figure 5). Evaluation of fluorescent intensity values, using semi-quantitative digital image analysis, revealed that NA and NAM significantly reduced the level of activated NF-κB in the nuclei of Jurkat cells, with NAM causing the greatest reduction (44%) (Table 1). NAM and NA caused a small but significant reduction in the constitutive levels of activated NF-κB in the nuclei of HT-29 and HCT-116 cells (Table 1).

Confocal images of Jurkat cells stained for activated NF-κB using a monoclonal antibody that detects an epitope on the p65 protein that includes the NLS sequence. Cells grown in media supplemented with 10 mM NAM (A) or 10 mM NA (B) show lower levels of activated NF-κB than cells grown in unsupplemented medium (C). The red color indicates activated NF-κB detected by biotin/streptavidin (Cy5) labeling. The green color indicates DNA stained with YOYO-1 after RNAase treatment
Nicotinic acid and nicotinamide reduce the protein levels of NF-κB
There are at least three explanations by which activated NF-κB can be reduced by NA and NAM pretreatment: a decrease in protein levels of NF-κB proteins, an increase in protein levels of IκB proteins (inhibitors of NF-κB activation) or a decrease in constitutive levels of oxidative stress, since NF-κB is a redox-sensitive transcription factor.38 We used polyclonal antibodies against p50 and p65, the two proteins that make up the classic NF-κB heterodimer, and examined the immunostained Jurkat cells using confocal microscopy. Control cells express p50 and p65 proteins in both the nucleus and cytoplasm (Figure 6). Treatment of cells with 10 mM NA or 10 mM NAM for 24 h resulted in a decrease in both the p50 and p65 proteins; this decrease was evident in both the nucleus and cytoplasm (Figure 6). In addition, since a decrease in NF-κB activation is controlled by the amount of IκB inhibitory proteins, we evaluated NA and NAM modulation of the protein levels of IκB-α, IκB-β and IκB-γ, three major members of the IκB family of NF-κB inhibitors.75,76 We found that the protein levels of IκB-γ were increased over that of control cells after pretreatment with NA but not with NAM (Figure 6). These findings indicate that one way in which both NA and NAM can decrease activated NF-κB is through a down regulation of NF-κB(p50) and NF-κB(p65) protein levels. NA treatment also resulted in an up-regulation of IκB-γ protein levels.

Confocal images of NF-κB/IκB expression in untreated (control) Jurkat cells compared with those that had been incubated with 10 mM NA or 10 mM NAM for 24 h. The immuno controls represent cells that had been reacted with the secondary antibody and the Cy5-streptavidin fluorochrome in the absence of the primary antibodies
Nicotinic acid and nicotinamide induce a sub-lethal increase in oxidative stress
It is possible that a decrease in the expression of NF-κB could be caused by a general decrease in constitutive levels of oxidative stress. Therefore, we reacted Jurkat cells for 1 h with an oxidative stress-reactive fluorochrome, carboxy-H2-DCFDA [5-(and)-6-carboxy-2′,7′-dichlorodihydro-fluorescein diacetate] after 24 h of pre-incubation with 10 mM NA or 10 mM NAM. The cells were fixed and then examined using confocal microscopy. We found that both NA and NAM increased the level of oxidative stress over that of control Jurkat cells (Figure 7). Since this concentration of both NA and NAM did not induce apoptosis (Figure 3) or cause a significant decrease in cellular proliferation (Figure 4), the induced oxidative stress was well tolerated and sub-lethal in nature. In conclusion, a reduction in constitutive levels of oxidative stress was not the reason for the decrease in constitutive levels of NF-κB and GRP78 caused by NA and NAM.

Confocal images of Jurkat cells treated with NA and NAM for 24 h, followed by incubation with the oxidative stress-sensitive fluorochrome, Carboxy-H2-DCFDA. The fluorescence of the control cells represent background autofluorescence
Nicotinic acid and nicotinamide reduce the constitutive levels of GRP78
To determine if NA and NAM also reduce constitutive levels of another major stress-response protein induced by NaDOC, we examined the effect of these NAD+ precursors on GRP78 protein levels. Jurkat cells were grown in media alone or in media supplemented with 10 mM NA or 10 mM NAM for 24 h. Cells were then fixed and stained for GRP78 using a polyclonal antibody, followed by immunofluorescent detection and confocal microscopy. Semi-quantitative digital image analysis indicated that NA reduced nuclear protein levels of GRP78 by 43% and cytoplasmic protein levels by 20%; NAM caused a small but significant reduction of cytoplasmic, but not nuclear, protein levels of GRP78 (Table 2).
NA and NAM increase protein levels of glyceraldehyde-3-phosphate dehydrogenase (GAPDH)
We have previously reported77 that NA and NAM increased the mRNA levels of GAPDH, a multifunctional enzyme involved in energy production, DNA repair, DNA replication, apoptosis, cytoskeletal control of endocytosis and control of gene expression (e.g. through RNA chaperone activity).78,79,80,81,82,83 Therefore, we tested the hypothesis that one mechanism by which NA and NAM could protect against apoptosis is through elevation of GAPDH protein levels. We used a monoclonal antibody to GAPDH, followed by fluorescent detection and confocal microscopy. Jurkat cells were incubated in medium alone, or in the presence of 10 mM NAM or 10 mM NA for 24 h. NA increased the levels of GAPDH predominantly in the nucleus, whereas NAM increased GAPDH levels predominantly in the cytoplasm (Figure 8).

Confocal images of Jurkat cells stained for GAPDH. Cells grown in medium supplemented with 10 mM NAM (B) show increased levels of GAPDH compared to cells grown in unsupplemented medium (C), while cells grown in the presence of 10 mM NA (A) have increased levels of GAPDH in the nuclei (C). Jurkat cells stained without the primary antibody present (background control) are shown in panel (D)
Nicotinamide is non-toxic to cells
Two different methods that measure intracellular stresses were used to compare the relative toxicities of NA and NAM, the first involving activation of promoter/response element CAT reporter constructs, and the second involving measurement of cellular non-protein thiol levels.
Gene promoter/response element activity of stress-response genes
To assess genotoxic, oxidative and ER stress, HepG2 cells, transfected with 13 different promoter/response element CAT reporter constructs, were treated with 10 mM NAM or 10 mM NA for 24 h. Gene activation was measured using a CAT ELISA assay. NAM failed to activate promoters or response elements of any of the 13 genes associated with cellular stresses. NA produced only marginal but statistically significant induction of grp78 promoter activation levels (1.1-fold induction) and hsp70 promoter activation levels (1.1-fold induction). Previous positive control tests of these stress-related promoter/response element constructs with mitomycin C, H2O2, camptothecin, and methyl methanesulfonate showed substantial positive responses.84 Thus, NA and NAM at 10 mM cause little, if any, genotoxic, oxidative or ER stress to HepG2 cells. [Note:- the slight promoter activation of grp78 by NA contrast, somewhat, with the reduced protein expression (Table 2). Since there are multiple points of regulation between promoter activation and the final level of protein expression, it is probable that events downstream of transcriptional activation are critical in controlling the observed levels of grp78 protein expression].
Nicotinamide does not reduce intracellular non-protein thiol levels
Non-protein thiol levels were measured in Jurkat cells by staining with mercury orange and detecting fluorescence intensity using flow cytometry. Cells treated with 10 mM NAM had no apparent effect on the intensity of staining with mercury orange (Figure 9). On the other hand, cells treated with 10 mM NA had reduced mercury orange fluorescent intensity levels, comparable to those achieved after treatment of cells with 5 mM DL-buthionine-S,R-sulfoximine (BSO), but not to the low levels obtained when cells were treated with 5 mM diethylmaleate (DEM). BSO specifically decreases the synthesis of glutathione (GSH) by inhibiting γ-glutamylcysteine synthase,85 whereas DEM is a general non-protein thiol-depleting agent.86

FACS analysis of mercury orange fluorescence of Jurkat cells with the peak of fluorescence indicated for each treatment: mercury orange alone (HgO control), BSO in the presence of HgO (BSO), DEM in the presence of HgO (DEM), NA in the presence of HgO (NA), NAM in the presence of HgO (NAM), untreated (NEG control)
A reduction in glutathione levels does not protect cells against NaDOC-induced apoptosis
Our flow cytometry data indicate that a possible mechanism by which NA has a somewhat greater protective effect on NaDOC-induced apoptosis compared with NAM is that decreased GSH levels produced by NA could have an anti-apoptotic effect as shown in certain other experimental situations.87 Jurkat cells were pretreated with 5 mM BSO for 24 h to deplete cellular GSH levels and then challenged with 0.5 mM NaDOC. We found that BSO treatment dramatically sensitized cells to NaDOC-induced apoptosis (Figure 10). Therefore, the marked reduction in NaDOC-induced apoptosis after pretreatment with NA is not caused by decreased GSH levels.

Effect of preincubation with 5 mM BSO on NaDOC-induced apoptosis
Discussion
We have found that the NAD+ precursors, NA and NAM, protect against apoptosis induced by the natural detergent, NaDOC. This hydrophobic bile acid induces several cellular stresses, including DNA damage, oxidative stress, ER stress and protein malfolding.37 Several of these stresses presumably act upstream of the classic pro-apoptotic gene products, such as bax and caspase-3 activation. Therefore, these NAD+ precursors may be involved in increasing viability through multiple mechanisms directed at the different stresses. NA and NAM are precursors for NAD+ and NADH, and probably aid in DNA repair. An increase in ability to repair DNA is suggested by the role of NAD+ as a substrate for PARP, a DNA repair enzyme, and the induction of GAPDH, which also functions in DNA repair through its uracil glycosylase activity. We have previously reported27 that PARP is protective against apoptosis, since 3-aminobenzamide, a strong PARP inhibitor,88 sensitized cells to NaDOC-induced apoptosis. In the present study, we found that both NAD+ precursors were effective at preventing apoptosis, with NA consistently showing a greater protective effect. This could be explained by the fact that NAM is a weak PARP inhibitor, whereas NA has no effect on PARP activity.88 A protective effect of NAM against apoptosis was also demonstrated in the in vivo studies of Klaidman et al.89 and Mukherjee et al.,90 who showed that NAM treatment prevented neuronal apoptosis produced by tertiary-butylhydroperoxide,89 an organic peroxide, and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine,90 a neurotoxin.
A major finding of the present study was that both NA and NAM decreased NF-κB, as measured by a decrease in the activated form of NF-κB and a decrease in protein expression of the NF-κB(p50) and NF-κB(p65) proteins. These findings corroborate the findings of Pero et al.91 who showed that NAM reduced the levels of TNF-α whose promoter is under the control of the transcription factor, NF-κB. They also showed that MAC (metoclopramide), a model N-substituted benzamide, decreased NF-κB activity using a plasmid comprised of a promoter containing 3 NF-κB sites and a CAT reporter gene.91 We chose to first understand the mechanism(s) by which NA and NAM reduced the levels of NF-κB. Two plausible mechanisms to reduce the level of NF-κB proteins and/or the activated form of NF-κB (both of which were measured in this study) is to reduce constitutive levels of oxidative stress and/or to increase the levels of one or more NF-κB inhibitory proteins. The fact that NAM was more effective than NA in reducing the levels of activated NF-κB, in the present study, may result from the free-radical scavenging ability of NAM90,92 and its ability to inhibit xanthine oxidase activity.93 Therefore, to test the oxidative stress hypothesis, we treated cells with carboxy-H2-DCFDA, a fluorochrome that fluoresces in the presence of increased reactive oxygen species (ROS).94 Pretreatment of Jurkat cells with either NA or NAM resulted in an increase in oxidative stress. Thus, a reduction in oxidative stress does not appear to be a major mechanism responsible for the decrease in constitutive levels of activated NF-κB. To test the hypothesis that these NAD+ precursors decrease activated NF-κB through a general decrease in NF-κB protein levels or an increase in IκB inhibitor protein levels, we used polyclonal antibodies against two major members of the NF-κB family of proteins, p50 and p65, and three major members of the IκB family of inhibitory proteins, IκB-α, IκB-β and IκB-γ. We found that both NA and NAM decreased protein levels of p50 and p65. NA, but not NAM, increased the protein levels of IκB-γ over that of control, whereas both NA and NAM reduced the protein levels of IκB-α and IκB-β compared to those of the control. Therefore, at least two mechanisms by which these NAD+ precursors decrease activated NF-κB are through a general decrease in the levels of NF-κB proteins and an increase in the inhibitory protein, IκB-γ. The mechanism by which IκB-γ remains elevated in the presence of an increase in ROS is not known.
It is somewhat paradoxical that NA and NAM both dramatically protect cells against apoptosis, although the protein levels and the activated form of NF-κB, an anti-apoptotic protein,95,96,97,98 is decreased in the nucleus and cytoplasm after NA and NAM treatments. We have previously reported27 on the anti-apoptotic nature of NF-κB using pyrrolidine dithiocarbamate (PDTC), a potent inhibitor of NF-κB activation.99 Pretreatment of cells with PDTC sensitized cells to NaDOC-induced apoptosis in Jurkat cells, and decreased the levels of activated NF-κB in both Jurkat and HCT-116 cells.27 Since we measured oxidative stress levels and protein levels of NF-κB and IκB proteins after 24 h of pre-treatment with NA or NAM, it is possible that the increase in oxidative stress might have occurred early on resulting in a subsequent increase in expression of antioxidant defense genes. The increase in antioxidant defense genes may then have relieved oxidative stress, causing a reduction in the level of NF-κB proteins by 24 h. Therefore, a mild, sub-lethal increase in oxidative stress at an earlier time may induce a response sufficient to protect cells against a greater oxidant challenge, such as that caused by hydrophobic cytotoxic bile acids, at a later time.
It is probable that NA and NAM can modulate gene expression and that this modulation occurs through diverse mechanisms. We previously reported that these NAD+ precursors increased the mRNA level of GAPDH,77 a multifunctional enzyme involved in energy production, DNA repair, DNA replication, apoptosis, cytoskeletal control of endocytosis and control of gene expression (e.g. through RNA chaperone activity).78,79,80,81,82,83,100 In the present study, we determined whether NA and NAM also modulated gene expression of GAPDH at the protein level. We found that NA increased the level of GAPDH in the nucleus, whereas NAM increased the levels of GAPDH in the cytoplasm. This subcellular distribution of GAPDH may have consequences for protection against apoptosis.82,83,101,102 GAPDH has been previously shown to translocate to the nucleus,83,101,102 where it could participate in DNA repair, DNA replication or activate gene transcription.79 GAPDH can also participate in protecting against oxidative stress and increasing energy production by catalyzing an increase in NADH levels.77
We also found that NA and NAM reduced the constitutive levels of GRP78, a chaperone protein that responds to ER stress.44,103 Our results are consistent with the results obtained by Chatterjee et al.,104 who showed that treatment of cells with 6-aminonicotinamide, which pharmacologically produces niacin deficiency,105,106 induces GRP78 activity. NA and NAM may reduce ER stress by protecting against protein malfolding in the ER and/or prevention of Ca2+ pool depletion within the ER, although there is no evidence from the literature for either of these possibilities.
Since many disorders (see Introduction) are associated with oxidative stress, DNA damage, and an increase in apoptosis,107,108 we hypothesize that NA and NAM may be protective as dietary supplements for such conditions by increasing protective defenses against these deleterious effects. Although NA was more protective against apoptosis than NAM, NAM appeared to be the least toxic to cells in our in vitro experiments. This was evidenced by the absence of activation of promoters or response elements for 13 stress response genes and a lack of reduction in non-protein thiols by NAM. NA treatment, on the other hand, caused marginal induction of GRP78 and HSP70 promoters and reduced non-protein thiols to some extent. The major non-protein thiol in mammalian cells is glutathione, a cysteine-containing tripeptide that catalyzes reactions for the detoxification of xenobiotic compounds and regulates cellular redox balance.109 GSH has been reported to protect cells against induced apoptosis.110,111,112 Although NA reduced GSH levels in Jurkat cells, it markedly protected against apoptosis. A reduction in GSH levels has also been shown to protect cells against apoptosis,87 and could have been a possible mechanism by which NA was so protective against apoptosis in the present study. However, we tested this hypothesis by depleting cells of GSH with BSO and then reacting these cells with the apoptosis-inducing agent, NaDOC. The depletion of GSH sensitized cells to NaDOC-induced apoptosis; therefore, a reduction in GSH levels is not a mechanism by which NA protects cells against apoptosis.
Our in vitro toxicity studies are consistent with the in vivo clinical trials which used NAM for the prevention or delay of Type 1 (insulin-dependent) diabetes mellitus in 173 children over a 7 year period with no observable liver toxicity.113 In addition, if NAM is to be considered as a dietary supplement for the prevention of certain diseases, it is noteworthy that it induces a considerably greater increase in the tissue concentration of NAD+ than does NA. This increase in tissue NAD+ concentrations after NAM treatment most probably results from the much longer half-life of NAM in liver and blood compared with NA.114
In conclusion, we have shown that NAM reduces apoptosis, decreases constitutive levels of activated NF-κB and GRP78, increases intracellular levels of GAPDH and is non-toxic to cells in millimolar concentrations. We suggest that NAM may be useful as a dietary supplement to reduce oxidative damage, enhance DNA repair and to alleviate cellular stresses leading to apoptosis. NA may also prove beneficial as a chemopreventive agent against neurodegenerative diseases that are associated with increased neuronal apoptosis since it is more protective against apoptosis than NAM.
Materials and Methods
Chemicals
Sodium deoxycholate (NaDOC) was obtained from ICN Biochemicals (Cleveland, Ohio), and was stored as a 50 mM stock solution in water at −20°C. NAM and NA were obtained from Sigma (St. Louis, MO, USA). NAM and NA solutions were made fresh for each use. Chemicals used in the flow cytometry experiments, mercury orange [1(4-chloromercuryphenyl-azo-2-naphthol)], diethylmaleate, and DL-buthionine-S,R-sulfoximine were obtained from Sigma, and solutions were made fresh for each use. Mercury orange was dissolved in acetone, while BSO and DEM were dissolved in water. Carboxy-H2-DCFDA was obtained from Molecular Probes, Inc. (Eugene, OR, USA).
Cell culture
The Jurkat cell line [human T-cell lymphoma, American Type Culture Collection (ATCC), Bethesda, MD; ATCC #TIB 152] was maintained in RPMI 1640 medium (Irvine Scientific, Santa Ana, CA, USA) supplemented with 10% fetal calf serum (Omega Scientific, Inc., Tarzana, CA, USA), 100 U/ml penicillin, 100 μg/ml streptomycin, and 3.44 mg/ml L-glutamine (Gibco BRL Life Technologies, Grand Island, NY, USA). Two colon adenocarcinoma cell lines, HCT-116 (ATCC #CCL 247) and HT-29 (ATCC #HTB 38), were grown in DMEM medium supplemented with 10% fetal calf serum, penicillin/streptomycin/glutamine (same concentrations as for Jurkat cells), and 1% MEM non-essential amino acids (Sigma, St. Louis, MO, USA). A transformed human hepatoma cell line, HepG2 (ATCC #HB 8065), was grown in MEM medium containing 10% fetal calf serum, 50 μg/ml streptomycin, 50 U/ml penicillin, 100 μM MEM non-essential amino acids, 2 mM L-glutamine, and 1 mM sodium pyruvate (Gibco). Experiments were performed between passages 3 and 15, and at concentrations of 4–5×105 cells/ml. This cellular density placed the cells approximately in the middle of the growth curve. This was an important variable to control in our apoptosis experiments, since cells in very early (1–2×105 cells/ml) or very late (8–9×105 cells/ml) logarithmic phase of cell growth are unusually resistant or sensitive, respectively, to NaDOC-induced apoptosis (unpublished data).
Determination of membrane integrity
Trypan Blue dye exclusion was used as a measure of membrane integrity. One hundred cells were counted using a hemocytometer and the percentage of cells that excluded trypan blue was determined.
Quantification of apoptosis using brightfield microscopy
At various times after NaDOC treatment, cytospins were prepared using a Shandon Cytospin 3 cytocentrifuge. Cells were then fixed with 100% methanol for 1 min, and after drying, stained overnight with a modified Giemsa stain in methanol (Sigma Diagnostics, St. Louis, MO, USA) [1 : 20 dilution of 0.4% w/v (pH 6.9)]. Under a 100×-oil immersion objective employing brightfield microscopy, 200 cells were counted from each slide and scored for normal, apoptotic, and mitotic cells. Apoptotic cells were identified by the presence of condensed chromatin, fragmented nuclei, cell shrinkage, cytoplasmic vacuolization, and formation of apoptotic bodies, as previously described.115,116
Preparation of cells for confocal microscopy
Suspension cells (Jurkat) were spun onto 0.17 mm thick coverslips using the Shandon Cytospin 3 cytocentrifuge, while the adherent cells, HCT-116 and HT-29, were grown directly on coverslips. The coverslips were fixed in 4% methanol-free formaldehyde for 20 min at room temperature, and then further permeabilized at −20° in 100% methanol for 6 min, air-dried, and stored at −20°C until immunostained.
Immunofluorescent procedures
NF-κB
A monoclonal antibody (Boehringer Mannheim, Indianapolis, IN, USA) was used that recognizes an epitope of the p65 subunit of NF-κB that includes the nuclear localization signal (NLS) sequence.117Positive staining with this antibody therefore measures presence of activated NF-κB. The antibody was used at a dilution of 1 : 100 in 1% BSA (bovine serum albumin)/PBS. An assessment of the expression of two proteins present in the classic NF-κB dimer, NF-κB(p50) and NF-κB(p65), was made using polyclonal antibodies obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Treatment with a biotinylated goat anti-mouse secondary antibody (Vector Laboratories, Inc, Burlingame, CA, USA) followed by Cy5-conjugated streptavidin (Vector Laboratories) was used as the detection system for the primary monoclonal antibody; biotinylated goat anti-rabbit secondary antibody (Vector Laboratories, Inc, Burlingame, CA, USA) followed by Cy5-conjugated streptavidin (Vector Laboratories) was used as the detection system for the polyclonal antibodies. Nuclei were identified using YOYO-1 staining after RNase digestion, as previously described.27
IκB
Polyclonal antibodies to IκB-α, IκB-β and IκB-γ were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Biotinylated goat anti-rabbit secondary antibody (Vector Laboratories, Inc, Burlingame, CA, USA) followed by Cy5-conjugated streptavidin (Vector Laboratories) was used as the detection system for the polyclonal antibodies. Nuclei were identified using YOYO-1 staining after RNase digestion, as previously described.27
GRP78
An affinity-purified goat polyclonal antibody that recognizes an amino acid sequence at the amino terminus of the 78 kDa glucose-regulated protein (GRP78) precursor of human origin was obtained from Santa Cruz Biotechnology, Inc (Santa Cruz, CA, USA). The antibody was used at a dilution of 1 : 50. A biotinylated goat anti-rabbit secondary antibody (Vector Laboratories) followed by Cy5-conjugated streptavidin (Vector Laboratories) was used as the detection system. Nuclei were identified using YOYO-1 staining after RNase digestion.
GAPDH
A monoclonal antibody that recognizes GAPDH from human erythrocytes, skeletal and heart muscle was obtained from Biogenesis Ltd (England, UK). The antibody was used at a dilution of 1 : 50. A biotinylated goat anti-mouse secondary antibody (Vector Laboratories) followed by Cy5-conjugated streptavidin (Vector Laboratories) was used as the detection system.
Semi-quantitative digital image analysis
A LEICA LSM10 laser scanning confocal microscope was used to obtain confocal images. To compare relative fluorescent intensity values from confocal images, laser scans were carried out by keeping the laser power and voltage on the photomultiplier (PMT) at a constant setting, and performing the same number of line averages as previously described.27,118 Scanned and processed images were saved to a ZIP diskette and imported to the hard drive of a PC. Fluorescent intensity levels in the nuclei and cytoplasm of the pixelated images were assessed using Image Pro-Plus (Media Cybernetics, Silver Spring, MD, USA), an automated image analysis software package, as previously described.27,118
Statistical analysis of immunofluorescence intensity levels
The mean and standard error of the gray level intensity was computed for each cell line under some of the different experimental conditions used. Statistical comparison of the mean gray level between NA or NAM treated cells versus untreated control cells was performed using a one-way analysis of variance. An alternative method used the number of pixels in each nucleus to perform a weighted analysis. The results of the weighted analyses were statistically equivalent to the results of the unweighted analyses. The unweighted analyses are, therefore, presented in this study for simplicity. When images were visually distinct in certain experimental situations, no image analysis was performed.
CAT ELISA assays
J Schneider and S Beard at Xenometrix, Inc. (Boulder, CO, USA) performed these assays. Briefly, HepG2 cells were transfected with the pSP-CAT plasmid by electroporation using a gene pulser (Bio-Rad Laboratories, Richmond, CA, USA), as previously described.84 After treatment of HepG2 cells with NA or NAM, the cells were lysed and the lysate transferred to 8-well microtiter strips coated with anti-CAT antibodies. The CAT ELISA was performed according to the protocol described in the Xenometrix CAT-Tox assay manual. Microtiter plates were read at an absorbance of 405 nm in a microtiter plate reader (Bio-Tek Instruments, Inc). Xenometrix software was used to collect and analyze data.84
Confirmation of apoptotic cells by transmission electron microscopy (TEM)
Jurkat and HCT-116 cells were treated with 0.5 mM NaDOC for 2 h, then fixed overnight in 3% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2). The cells were pelleted, post-fixed in 2% osmium tetroxide, dehydrated in a graded series of ethanols, and embedded in epoxy resin. Electron microscopy is the gold standard for the identification of apoptotic cells.6,71,74,119 Ultrastructural features characteristic of apoptosis include condensation and margination of chromatin, fragmentation of the nucleus into double membrane-bound bodies, an increase in electron density, cytoplasmic vacuolization, normal appearing mitochondria, and apoptotic body formation.
Mercury orange fluorescence and flow cytometry
Jurkat cells were pretreated with 10 mM NA or 10 mM NAM for 24 h before harvest. As controls, Jurkat cells were pretreated for 4 h with 5 mM diethylmaleate (DEM), 18 h with 5 mM DL-buthionine-S,R-sulfoximine (BSO), or were not pretreated. DEM induces non-protein thiol depletion through oxidation of sulfhydryl groups,86 while specific depletion of GSH occurs with BSO, an inhibitor of γ-glutamylcysteine synthase.85 After pretreatments, cells were centrifuged for 5 min at 1200 r.p.m. The cell pellets were resuspended in 100 μM mercury orange (MO) for 5 min on ice, then centrifuged for another 5 min at 1200 r.p.m.20 The pellets were resuspended in ice-cold PBS supplemented with 1% FCS.
Mercury orange fluorescence was detected using a FACStarPLUS (Becton Dickinson Immunocytometry Systems, San Jose, CA, USA) flow cytometer, utilizing a Coherent 90-5 (Palo Alto, CA, USA) water cooled argon laser tuned to 488 nm at 100 milliwatts. Fluorescence emission was captured through a 575/26 band-pass filter. A minimum of 10 000 events were collected, and debris and fragments were excluded from analysis based on forward scatter and side scatter. Data were acquired and analyzed using Lysis II (BD, San Jose, CA, USA) software. Results are presented as single parameter histograms with four-decade log scale.
Oxidative stress measurements
Carboxy-H2-DCFDA [5-(and-6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate] was obtained from Molecular Probes, Inc. (Eugene, OR, USA) and used at a final concentration of 5 μM. Jurkat cells were pretreated with 10 mM NA or 10 mM NAM for 24 h, and then incubated for 1 h in Carboxy-H2-DCFDA. Cytospins were prepared and pelleted cells were fixed in 4% formaldehyde (methanol-free)(Ted Pella, Inc., Redding, CA, USA) for 20 min, washed with PBS and mounted using DAKO mounting media (DAKO Corp., Carpinteria, CA, USA). Images were scanned on a LEICA confocal microscope using the 488 laser line. Images were compared by leaving the laser power and the voltage on the PMT at the same settings.
Abbreviations
- BSO:
-
DL-buthionine-S, R-sulfoximine
- carboxy-H2-DCFDA [5-(and)-6-carboxy-2′:
-
7′-dichlorodihydro-fluorescein diacetate]
- DEM:
-
diethylmaleate
- GAPDH:
-
glyceraldehyde-3-phosphate dehydrogenase
- NA:
-
nicotinic acid
- NAD:
-
nicotinamide adenine dinucleotide
- NaDOC:
-
sodium deoxycholate
- NAM:
-
nicotinamide
- PARP:
-
poly(ADP-ribose) polymerase
- ROS:
-
reactive oxygen species
- TEM:
-
transmission electron microscopy
References
Reddy BS . (1981) Dietary fat and its relationship to large bowel cancer. Cancer Res. 41: 3700–3705
Martinez JD, Stratagoules ED, LaRue JM, Powell AA, Gause PR, Craven MT, Payne CM, Powell MB, Gerner EW and Earnest DL . (1998) Different bile acids exhibit distinct biological effects: The tumor promoter deoxycholic acid induces apoptosis and the chemopreventive agent ursodeoxycholic acid inhibits cell proliferation. Nutrition Cancer 31: 111–118
Marchetti MC, Migliorati G, Moraca R, Riccardi C, Nicoletti FR, Mastrandrea V and Morozzi G . (1997) Possible mechanisms involved in apoptosis of colon tumor cell lines induced by deoxycholic acid, short-chain fatty acids, and their mixtures. Nutrition Cancer 28: 74–80
Samaha H, Bernstein C, Payne CM and Asher E . (1995) Evaluation of cell death in EBV-transformed lymphocytes using agarose gel electrophoresis, light microscopy and electron microscopy. I. Induction of classic apoptosis by the bile salt, sodium deoxycholate. Leuk. Lymph. 19: 95–105
Hague A, Elder DJE, Hicks DJ and Paraskeva C . (1995) Apoptosis in colorectal tumour cells: Induction by the short chain fatty acids butyrate, propionate and acetate and by the bile salt deoxycholate. Int. J. Cancer 60: 400–406
Payne CM, Bernstein H, Bernstein C and Garewal H . (1995) Role of apoptosis in biology and pathology: Resistance to apoptosis in colon carcinogenesis. Ultrastruct. Pathol. 19: 221–248
Kwo P, Patel T, Bronk SF and Gores GJ . (1995) Nuclear serine protease activity contributes to bile acid-induced apoptosis in hepatocytes. Am. J. Physiol. 268: G613–G621
Patel T, Bronk SF and Gores GJ . (1994) Increase of intracellular magnesium promote glycodeoxycholate-induced apoptosis in rat hepatocytes. J. Clin. Invest. 94: 2183–2192
Patel T and Gores GJ . (1997) Inhibition of bile salt-induced hepatocyte apoptosis by the antioxidant lazaroid U83836E. Toxicol. Appl. Pharmacol. 142: 116–122
Patel T, Arora A and Gores GJ . (1995) A fluorometric assay for quantitating DNA strand breaks during apoptosis. Anal. Biochem. 229: 229–235
Chieco P, Romagnoli E, Aicardi G, Suozzi A, Forti GC and Roda A . (1997) Apoptosis induced in rat hepatocytes by in vivo exposure to taurochenodeoxycholate. Histochem. J. 29: 875–883
Greim H, Trulzsch D, Czygan P, Rudick J, Hutterer F, Schaffner F and Popper H . (1972) Mechanism of cholestasis. 6. Bile acids in human livers with or without biliary obstruction. Gastroenterology 63: 846–850
Setchell KDR, Schwartz M, O'Connell NC, Lund EG, Davis DL, Lathe R, Thompson HR, Tyson RW, Sokol RJ and Russell DW . (1998) Identification of a new inborn error in bile acid synthesis: mutation of the oxysterol 7α-hydroxylase gene causes severe neonatal liver disease. J. Clin. Invest. 102: 1690–1703
Hill MJ . (1986) Bile acids and colorectal cancer in humans. In: Dietary Fiber– Basic and Clinical Aspects. Vahouny GV and Kritchevsky D, eds (New York City: Plenum) pp. 497–513
Cohen BI and Mosbach EH . (1986) The role of bile acids in colon cancer. In: Dietary Fiber. Basic and Clinical Aspects, Vahouny GV and Kritchevsky D, eds (New York: Plenum Press) pp. 487–496
Shivaram KN, Winklhofer-Roob BM, Straka MS, Devereaux MW, Everson G, Mierau GW and Sokol RJ . (1998) The effect of idebenone, a coenzyme Q analogue, on hydrophobic bile acid toxicity to isolated rat hepatocytes and hepatic mitochondria. Free Radical Biol. Med. 25: 480–492
Rodrigues CMP, Fan G, Ma X, Kren BT and Steer CJ . (1998) A novel role for ursodeoxycholic acid in inhibiting apoptosis by modulating mitochondrial membrane perturbation. J. Clin. Invest. 101: 2790–2799
Booth LA, Gilmore IT and Bilton RF . (1997) Secondary bile acid induced DNA damage in HT29 cells: are free radicals involved? Free Radical Res. 26: 135–144
Bomzon A, Holt S and Moore K . (1997) Bile acids, oxidative stress, and renal function in biliary obstruction. Semin. Nephrol. 17: 549–562
Sokol RJ, Devereaux M, Khandwala R and O'Brien K . (1993) Evidence for involvement of oxygen free radicals in bile acid toxicity to isolated rat hepatocytes. Hepatology 17: 869–881
Sokol RJ, Winklhofer-Roob BM, Devereaux MW and McKim Jr JM . (1995) Generation of hydroperoxides in isolated rat hepatocytes and hepatic mitochondria exposed to hydrophobic bile acids. Gastroenterology 109: 1249–1256
Craven PA, Pfanstiel J and DeRubertis FR . (1987) Role of activation of protein kinaseC in the stimulation of colonic epithelial proliferation and reactive oxygen formation by bile acids. J. Clin. Invest. 79: 532–541
Craven PA, Pfanstiel J, Saito R and DeRubertis FR . (1987) Actions of sulfasalazine and 5-aminosalicylic acid as reactive oxygen scavengers in the suppression of bile acid-induced increases in colonic epithelial cell loss and proliferative activity. Gastroenterology 92: 1998–2008
Craven PA, Pfanstiel J and DeRubertis FR . (1986) Role of reactive oxygen in bile salt stimulation of colonic epithelial proliferation. J. Clin. Invest. 77: 850–859
DeRubertis FR, Craven PA and Saito R . (1984) Bile salt stimulation of colonic epithelial proliferation. Evidence for involvement of lipoxygenase products. J. Clin. Invest. 74: 1614–1624
Venturi M, Hambly RJ, Glinghammer B, Rafter JJ and Rowland IR . (1997) Genotoxic activity in human faecal water and the role of bile acids: a study using the alkaline comet assay. Carcinogenesis 18: 2353–2359
Payne CM, Crowley C, Washo-Stultz D, Briehl M, Bernstein H, Bernstein C, Beard S, Holubec H and Warneke J . (1998) The stress-response proteins poly(ADP-ribose) polymerase and NF-κB protect against bile salt-induced apoptosis. Cell Death Differ. 5: 623–636
Masamune K, Kunitomo K, Sasaki K, Yagi K, Komi N and Tashiro S . (1997) Bile-induced DNA strand breaks and biochemical analysis of bile acids in an experimental model of anomalous arrangement of the pancreaticobiliary ducts. J. Med. Invest. 44: 47–51
Pool-Zobel BL and Leucht U . (1997) Induction of DNA damage by risk factors of colon cancer in human colon cells derived from biopsies. Mutation Res. 375: 105–115
Zheng Z-Y, Bernstein H, Bernstein C, Payne CM, Martinez JD and Gerner EW . (1996) Bile acid activation of the gadd153 promoter and of p53-independent apoptosis: Relevance to colon cancer. Cell Death Diff. 3: 407–414
Zheng Z-Y and Bernstein C . (1992) Bile salt/acid induction of DNA damage in bacterial cells: effect of taurine conjugation. Nutrition Cancer 18: 157–164
Kandell RL and Bernstein C . (1991) Bile salt/acid induction of DNA damage in bacterial and mammalian cells: Implications for colon cancer. Nutrition Cancer 16: 227–238
Watabe J and Bernstein H . (1985) The mutagenicity of bile acids using a fluctuation test. Mutation Res. 158: 45–51
Kulkarni MS and Yielding KL . (1985) DNA damage and repair in epithelial (mucous) cells and crypt cells from isolated colon. Chem.-Biol. Interact. 52: 311–318
Kulkarni MS, Cox BA and Yielding KL . (1982) Requirements for induction of DNA strand breaks by lithocholic acid. Cancer Res. 42: 2792–2795
Combettes L, Dumont M, Berthon B, Erlinger S and Claret M . (1988) Release of calcium from the endoplasmic reticulum by bile acids in rat liver cells. J. Biol. Chem. 263: 2299–2302
Bernstein H, Payne CM, Bernstein C, Schneider J, Beard SE and Crowley CL . (1999) Activation of the promoters of genes associated with DNA damage, oxidative stress, ER stress and protein malfolding by the bile salt, deoxycholate. Toxicol. Lett. 108: 37–46
Schreck R, Rieber P and Baeuerle PA . (1991) Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappaB transcription factor and HIV-1. EMBO J. 10: 2247–2258
Wintersberger U and Wintersberger E . (1985) Poly ADP-ribosylation–A cellular emergency reaction? FEBS Lett. 188: 189–191
Shall S . (1984) ADP-ribose in DNA repair: a new component of DNA excision repair. Adv. Rad. Biol. 11: 1–69
Luethy JD and Holbrook NJ . (1992) Activation of the gadd153 promoter by genotoxic agents: a rapid and specific response to DNA damage. Cancer Res. 52: 5–10
Fink AL . (1999) Chaperone-mediated protein folding. Physiol. Rev. 79: 425–449
Nigam SK, Goldberg AL, Ho S, Rohde MF, Bush KT and Sherman MY . (1994) A set of endoplasmic reticulum proteins possessing properties of molecular chaperones includes Ca2+-binding proteins and members of the thioredoxin superfamily. J. Biol. Chem. 269: 1744–1749
Liu H, Bowes III RC, van de Water B, Sillence C, Nagelkerke JF and Stevens JL . (1997) Endoplasmic reticulum chaperones GRP78 and calreticulin prevent oxidative stress, Ca2+ disturbances, and cell death in renal epithelial cells. J. Biol. Chem. 272: 21751–21759
De Murcia G and de Murcia JM . (1994) Poly(ADP-ribose)polymerase: A molecular nick-sensor. TIBS 19: 172–176
De Murcia G, Menissier-de Murcia J and Schreiber V . (1991) Poly(ADP-ribose) polymerase: molecular biological aspects. BioEssays 13: 455–462
Wielckens K, Schmidt A, George E, Bredehorst R and Hilz H . (1982) DNA fragmentation and NAD depletion. Their relationship to the turnover of endogenous mono(ADP-ribosyl) and poly(ADP-ribosyl) proteins. J. Biol. Chem. 257: 12872–12877
Grant RS and Kapoor V . (1998) Murine glial cells regenerate NAD, after peroxide-induced depletion, using either nicotinic acid, nicotinamide, or quinolinic acid as substrates. J. Neurochem. 70: 1759–1763
Weitberg AB . (1989) Effect of nicotinic acid supplementation in vivo on oxygen radical-induced genetic damage in human lymphocytes. Mutat. Res. 216: 197–201
Jackson TM, Rawling JM, Roebuck BD and Kirkland JB . (1995) Large supplements of nicotinic acid and nicotinamide increase tissue NAD+ and poly(ADP-ribose) levels but do not affect diethylnitrosamine-induced altered hepatic foci in Fischer-344 rats. J. Nutr. 125: 1455–1461
Giacosa A and Filiberti R . (1996) Free radicals, oxidative damage and degenerative diseases. Eur. J. Cancer Prevent. 5: 307–312
Stohs SJ . (1995) The role of free radicals in toxicity and disease. J. Basic Clin. Physiol. Pharmacol. 6: 205–228
Davies KJA . (1995) Oxidative stress: the paradox of aerobic life. Biochemical Society Symposia 61: 1–31
Cross CE, Halliwell B, Borish ET, Pryor WA, Ames BN, Saul RL, McCord JM and Harman D . (1987) Oxygen radicals and human disease. Ann. Intern. Med. 107: 526–545
Peterhans E . (1997) Oxidants and antioxidants in viral diseases: disease mechanisms and metabolic regulation. J. Nutr. 127: 962S–965S
Schwartz KB . (1996) Oxidative stress during viral infection: A review. Free Radical Biol. Med. 21: 641–649
Pace GW and Leaf CD . (1995) The role of oxidative stress in HIV disease. Free Rad. Biol. Med. 19: 523–528
Boh EE . (1996) Role of reactive oxygen species in dermatologic diseases. Clin. Dermatol. 14: 343–352
Griendling KK and Alexander RW . (1997) Oxidative stress and cardiovascular disease. Circulation 96: 3264–3265
Joseph JA, Denisova N, Fisher D, Bickford P, Prior R and Cao G . (1998) Age-related neurodegeneration and oxidative stress: putative nutritional intervention. Neurol. Clin. 16: 747–755
Warner HR, Hodes RJ and Pocinki K . (1997) What does cell death have to do with aging? J. Amer. Geriatr. Soc. 45: 1140–1146
Cutler RG . (1992) Genetic stability and oxidative stress: common mechanisms in aging and cancer. EXS 62: 31–46
Bernstein C and Bernstein H . (1991) Aging, Sex, and DNA Repair New York City: Academic Press, Inc. pp. 1–382
Dreher D and Junod AF . (1996) Role of oxygen free radicals in cancer development. Eur. J. Cancer 32A: 30–38
Cerutti PA and Trump BF . (1991) Inflammation and oxidative stress in carcinogenesis. Cancer Cells 3: 1–7
Breimer LH . (1990) Molecular mechanisms of oxygen radical carcinogenesis and mutagenesis: The role of DNA base damage. Mol. Carcinogen. 3: 188–197
Cerutti PA . (1985) Pro-oxidant states and tumor promotion. Science 227: 375–381
Gensler HL . (1997) Prevention of photoimmunosuppression and photocarcinogenesis by topical nicotinamide. Nutrition Cancer 29: 157–162
Reddy S, Bibby NJ, Wu D, Swinney C, Barrow G and Elliott RB . (1995) A combined casein-free-nicotinamide diet prevents diabetes in the NOD mouse with minimum insulitis. Diabetes Res. Clin. Pract. 29: 83–92
Yamada K, Nonaka K, Hanafusa T, Miyazaki A, Toyoshima H and Tarui S . (1982) Preventive and therapeutic effects of large-dose nicotinamide injections on diabetes associated with insulitis. An observation in non-obese diabetic (NOD) mice. Diabetes 31: 749–753
Payne CM, Bernstein C and Bernstein H . (1995a) Apoptosis overview emphasizing the role of oxidative stress, DNA damage and signal-transduction pathways. Leuk. Lymph. 19: 43–93
Searle J, Kerr JFR and Bishop CJ . (1982) Necrosis and Apoptosis: distinct modes of cell death with fundamentally different significance. Pathol. Annual 17: 229–259
Wyllie AH, Kerr JFR and Currie AR . (1980) Cell death: The significance of apoptosis. Int. Rev. Cytol. 68: 251–306
Kerr JFR, Wyllie AH and Currie AR . (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26: 239–257
Verma IM, Stevenson JK, Schwartz EM, Van Antwerp D and Miyamoto S . (1995) Rel/NF-kB/IkB family: Intimate tales of association and dissociation. Genes Dev. 9: 2723–2735
Beauparlant P and Hiscott J . (1996) Biological and biochemical inhibitors of the NF-kB/rel proteins and cytokine synthesis. Cytokine Growth Factor Rev. 7: 175–190
Yan Q, Briehl M, Crowley C, Payne CM, Bernstein H and Bernstein C . (1999) The NAD+ precursors, nicotinic acid and nicotinamide, upregulate mRNA levels of glyceraldehyde dehydrogenase and glucose-6-phosphate dehydrogenase in Jurkat cells. Biochem. Biophys. Res. Comm. 255: 133–136
Meyer-Siegler K, Mauro DJ, Seal G, Wurzer J, deRiel JK and Sirover MA . (1991) A human nuclear uracil DNA glycosylase is the 37-kDa subunit of glyceraldehyde-3-phospate dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 88: 8460–8464
Sirover MA . (1996) Emerging new functions of the glycolytic protein, glyceraldehyde-3-phosphate dehydrogenase, in mammalian cells. Life Sci. 58: 2271–2277
Ito Y, Pagano PJ, Tornheim K, Brecher P and Cohen RA . (1996) Oxidative stress increases glyceraldehyde-3-phosphate dehydrogenase mRNA levels in isolated rabbit aorta. Am. J. Physiol. 270: H81–H87
Sirover MA . (1997) Role of the glycolytic protein, glyceraldehyde-3-phosphate dehydrogenase, in normal cell function and in cell pathology. J. Cell. Biochem. 66: 133–140
Nakazawa M, Uehara T and Nomura Y . (1997) Koningic acid (a potent glyceraldehyde-3-phosphate dehydrogenase inhibitor)-induced fragmentation and condensation of DNA in NG108-15 cells. J. Neurochem. 68: 2493–2499
Saunders PA, Chen R-W and Chuang D-M . (1999) Nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase isoforms during neuronal apoptosis. J. Neurochem. 72: 925–932
Todd MD, Lee MJ, Williams JL, Nalezny JM, Gee P, Benjamin MB and Farr SB . (1995) The CAT-Tox (L) assay: a sensitive and specific measure of stress-induced transcription in transformed human liver cells. Fund. Appl. Toxicol. 28: 118–128
Griffith OW and Meister A . (1979) Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine). J. Biol. Chem. 254: 7558–7560
Plummer JL, Smith BR, Sies H and Bend JR . (1981) Chemical depletion of glutathione in vivo. Methods Enzymol. 77: 50–59
Boggs SE, McCormick TS and Lapetina EG . (1998) Glutathione levels determine apoptosis in macrophages. Biochem. Biophys. Res. Comm. 247: 229–233
Purnell MR and Whish WJD . (1980) Novel inhibitors of poly(ADP-ribose)synthetase. Biochem. J. 185: 775–777
Klaidman LK, Mukherjee SK, Hutchin TP and Adams JD . (1996) Nicotinamide as a precursor for NAD+ prevents apoptosis in the mouse brain induced by tertiary-butylhydroperoxide. Neurosci. Lett. 206: 5–8
Mukherjee SK, Klaidman LK, Yasharel R and Adams Jr JD . (1997) Increased brain NAD prevents neuronal apoptosis in vivo. Eur. J. Pharmacol. 330: 27–34
Pero RW, Axelsson B, Siemann D, Chaplin D and Dougherty G . (1999) Newly discovered anti-inflammatory properties of the benzamides and nicotinamides. Mol. Cell. Biochem. 193: 119–125
LeDoux SP, Hall CR, Forbes PM, Patton NJ and Wilson GL . (1988) Mechanisms of nicotinamide and thymidine protection from alloxan and streptozocin toxicity. Diabetes 37: 1015–1019
Di Stefano A, Pizzichini M and Marinello E . (1979) Nicotinamide and liver xanthine oxidase. Adv. Exp. Med. Biol. 122B: 189–196
Karbowski M, Kurono C, Wozniak M, Ostrowski M, Teranishi M, Nishizawa Y, Usukura J, Soji T and Wakabayashi T . (1999) Free radical-induced megamitochondria formation and apoptosis. Free Radical Biol. Med. 26: 396–409
Beg AA and Baltimore D . (1996) An essential role for NF-κB in preventing TNF-α-induced cell death. Science 274: 782–784
Baeuerle PA and Baltimore D . (1996) NF-κB: Ten years later. Cell 87: 13–20
Bellas RE, FitzGerald MJ, Fausto N and Sonenshein GE . (1997) Inhibition of NF-κB activity induces apoptosis in murine hepatocytes. Am. J. Pathol. 151: 891–896
Mayo MW, Wang C-Y, Cogswell PC, Rogers-Graham KS, Lowe SW, Der CJ and Baldwin Jr AS . (1997) Requirement of NF-κB activation to suppress p53-independent apoptosis induced by oncogenic ras. Science 278: 1812–1815
Schreck R, Meier B, Mannel DN, Droge W and Baeuerle PA . (1992) Dithiocarbamates as potent inhibitors of nuclear factor κB activation in intact cells. J. Exp. Med. 175: 1181–1194
Morgenegg G, Winkler GC, Hubscher U, Heizmann CW, Mous J and Kuenzle CC . (1986) Glyceraldehyde-3-phosphate dehydrogenase is a nonhistone protein and a possible activator of transcription in neurons. J. Neurochem. 47: 54–62
Saunders PA, Chalecka-Franaszek E and Chuang D-M . (1997) Subcellular distribution of glyceraldehyde-3-phosphate dehydrogenase in cerebellar granule cells undergoing cytosine arabinoside-induced apoptosis. J. Neurochem. 69: 1820–1828
Sawa A, Khan AA, Hester LD and Snyder SH . (1997) Glyceraldehyde-3-phosphate dehydrogenase: nuclear translocation participates in neuronal and nonneuronal cell death. Proc. Natl. Acad. Sci. U.S.A. 94: 11669–11674
Liu H, Miller E, van de Water B and Stevens JL . (1998) Endoplasmic reticulum stress proteins block oxidant-induced Ca2+ increases and cell death. J. Biol. Chem. 273: 12858–12862
Chatterjee S, Cheng M-F, Berger RB, Berger SJ and Berger NA . (1995) Effect of inhibitors of poly(ADP-ribose) polymerase on the induction of GRP78 and subsequent development of resistance to etoposide. Cancer Res. 55: 868–873
Hothersall JS, Gordge M and Noronha-Dutra AA . (1998) Inhibition of NADPH supply by 6-aminonicotinamide: effect on glutathione, nitric oxide and superoxide in J774 cells. FEBS Lett. 434: 97–100
Dietrich LS, Friedland IM and Kaplan LA . (1958) Pyridine nucleotide metabolism: mechanism of action of the niacin antagonist, 6-aminonicotinamide. J. Biol. Chem. 233: 964–968
Adams JD, Mukherjee SK, Klaidman LK, Chang ML and Yasharel R . (1996) Apoptosis and oxidative stress in the aging brain. Ann. N.Y. Acad. Sci. 786: 135–151
Thompson CB . (1995) Apoptosis in the pathogenesis and treatment of disease. Science 267: 1456–1462
Bray TM and Taylor CG . (1993) Tissue glutathione, nutrition, and oxidative stress. Can. J. Physiol. Pharmacol. 71: 746–751
Shan X, Jones DP, Hashmi M and Anders MW . (1993) Selective depletion of mitochondrial glutathione concentrations by (R,S)-3-hydroxy-4-pentenoate potentiates oxidative cell death. Chem. Res. Toxicol. 6: 75–81
Mirkovic N, Voehringer DW, Story MD, McConkey DJ, McDonnell TJ and Meyn RE . (1997) Resistance to radiation-induced apoptosis in bcl-2-expressing cells is reversed by depleting cellular thiols. Oncogene 15: 1461–1470
Hall AG . (1999) Review. The role of glutathione in the regulation of apoptosis. Eur. J. Clin. Invest. 29: 238–245
Elliott RB, Pilcher CC, Fergusson DM and Stewart AW . (1996) A population based strategy to prevent insulin-dependent diabetes using nicotinamide. J. Pediatr. Endocrinol. Metab. 9: 501–509
Petrack B, Greengard P and Kalinsky H . (1996) On the relative efficacy of nicotinamide and nicotinic acid as precursors of nicotinamide adenine dinucleotide. J. Biol. Chem. 241: 2367–2372
Payne CM, Glasser L, Tischler ME, Wyckoff D, Cromey D, Fiederlein R and Bohnert O . (1994) Programmed cell death of the normal human neutrophil: An in vitro model of senescence. Microsc. Res. Tech. 28: 327–344
Payne CM, Bjore Jr CG and Schultz DA . (1992) Change in the frequency of apoptosis after low- and high-dose X-irradiation of human lymphocytes. J. Leuk. Biol. 52: 433–440
Kaltschmidt C, Kaltschmidt B, Henkel T, Stockinger H and Baeuerle PA . (1995) Selective recognition of the activated form of transcription factor NF-κB by a monoclonal antibody. Biol. Chem. Hoppe-Seyler 376: 9–16
Baker A, Payne CM, Briehl MM and Powis G . (1997) Thioredoxin, a gene found over-expressed in human cancer, inhibits apoptosis in vitro and in vivo. Cancer Res. 57: 5162–5167
Hockenbery D . (1995) Defining apoptosis. Am. J. Pathol. 146: 16–19
O'Connor JE, Kimler BF, Morgan MC and Tempas KJ . (1988) A flow cytometric assay for intracellular nonprotein thiols using mercury orange. Cytometry 9: 529–532
Acknowledgements
This work was supported in part by NIEHS grant #ES06694 (Experimental Pathology Core support: Southwest Environmental Health Sciences Center small grant award), Arizona Disease Control Research Commission grant #10016, NIH Institutional Core Grant #CA23074, NIH PPG #CA72008, Innovite, Inc. (Tigard, Oregon) and Biomedical Diagnostics & Research, Inc. (Tucson, Arizona).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by RA Lockshin
Rights and permissions
About this article
Cite this article
Crowley, C., Payne, C., Bernstein, H. et al. The NAD+ precursors, nicotinic acid and nicotinamide protect cells against apoptosis induced by a multiple stress inducer, deoxycholate. Cell Death Differ 7, 314–326 (2000). https://doi.org/10.1038/sj.cdd.4400658
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4400658
Keywords
This article is cited by
-
Combined nature and human selections reshaped peach fruit metabolome
Genome Biology (2022)
-
Rapamycin rescues mitochondrial dysfunction in cells carrying the m.8344A > G mutation in the mitochondrial tRNALys
Molecular Medicine (2022)
-
NAD(H)-loaded nanoparticles for efficient sepsis therapy via modulating immune and vascular homeostasis
Nature Nanotechnology (2022)
-
Involvement of non-protein thiols, mitochondrial dysfunction, reactive oxygen species and p53 in honey-induced apoptosis
Investigational New Drugs (2010)
-
Micronutrients and their supplementation in chronic cardiac failure. An update beyond theoretical perspectives
Heart Failure Reviews (2006)
