




Abstract
Medical intractability, i.e. the absence of any response to anti-epileptic drug (AED) therapy, is an unresolved problem in many patients with epilepsy. Mechanisms of intractability are not well understood, but may include alterations of pharmacological targets and poor penetration of AEDs into the brain because of increased expression of multiple drug-resistance proteins, such as P-glycoprotein (Pgp; ABCB1), capable of active brain extrusion of various drugs, including AEDs. Increased expression of Pgp has been reported in brain tissue of patients with refractory epilepsy, but there is a lack of adequate controls, i.e. brain tissue from patients with drug-responsive epilepsy. In the present study, we used a rat model of temporal lobe epilepsy to examine whether AED responders differ from non-responders in their expression of Pgp in the brain. In this model, spontaneous recurrent seizures develop after status epilepticus induced by prolonged electrical stimulation of the basolateral amygdala. The frequency of these seizures was recorded by continuous video-EEG monitoring before, during and after daily treatment with phenobarbital, which was given at maximum tolerated doses for 2 weeks. Based on their individual response to phenobarbital, rats were grouped into responders (n = 7) and non-responders (n = 4). Pgp expression was studied by immunohistochemistry and showed striking overexpression in non-responders compared with responders in limbic brain regions, including the hippocampus. The Pgp overexpression was confined to brain capillary endothelial cells which form the blood–brain barrier. The present data are the first to demonstrate that rats with drug-resistant spontaneous seizures differ from rats with drug-responsive seizures in their Pgp expression in the brain, thereby substantiating the multidrug transporter hypothesis of intractable epilepsy.
Introduction
Despite advances in anti-epileptic drug (AED) therapy and epilepsy surgery in recent years, intractable epilepsy remains a major clinical problem (Kwan and Brodie, 2000; Arroyo et al., 2002). The consequences of uncontrolled epilepsy can be quite severe and include shortened lifespan, excessive bodily injury, neuropsychological and psychiatric impairment and social disability (Sperling, 2004). An important characteristic of medically intractable (pharmacoresistant) epilepsy is that most patients with refractory epilepsy are resistant to several, if not all, AEDs, even though these drugs act by different mechanisms (Kwan and Brodie, 2000; Sisodiya, 2003). This multidrug resistance argues against epilepsy-induced alterations in specific drug targets as a major cause of pharmacoresistant epilepsy, but rather points to non-specific and possibly adaptive mechanisms, such as decreased drug uptake into the brain by intrinsic or acquired overexpression of multidrug transporters in the blood–brain barrier (BBB) (Sisodiya, 2003). There is now a body of evidence that multidrug transporters such as P-glycoprotein (Pgp; ABCB1) are overexpressed in capillary endothelial cells and astrocytes in epileptogenic brain tissue surgically resected from patients with medically intractable epilepsy (Löscher and Potschka, 2002; Marroni et al., 2003; Sisodiya, 2003). Pgp in the BBB is thought to act as an active defence mechanism, restricting the penetration of lipophilic substances into the brain (Begley, 2004; Löscher and Potschka, 2005). A large variety of compounds, including many lipophilic drugs, are substrates for Pgp (Schinkel and Jonker, 2003). It is thus not surprising that several AEDs, which have been made lipophilic to penetrate into the brain, are substrates for Pgp in the BBB (Löscher and Potschka, 2002; Löscher and Potschka, 2005). Overexpression of such transporters in epileptogenic tissue is thus likely to reduce the amount of drug that reaches the epileptic neurons, which would be a possible explanation for multidrug resistance of epilepsy (Sisodiya, 2003).
However, although the multidrug transporter hypothesis of intractable epilepsy is biologically plausible, proof of principle for this hypothesis is lacking (Sisodiya, 2003). Thus, although high expression of Pgp has been demonstrated in epileptogenic brain tissue of patients with intractable epilepsy, there is a lack of adequate controls, because a direct comparison with Pgp expression in respective tissue from patients responding to AED treatment is not possible. Such tissue is not available as such subjects in general do not undergo surgical treatment for their epilepsy. As a consequence, it is not known whether the increased Pgp expression in patients with drug-resistant epilepsy is really a cause of pharmacoresistance or is just a result of uncontrolled seizures or an epiphenomenon occurring in epileptic brain tissue irrespective of drug responsiveness. An animal model of epilepsy with spontaneous recurrent seizures (SRS) in which it is possible to select subgroups which either respond or not respond to AED treatment would be useful in studying whether non-responders differ from responders in Pgp expression in the BBB and which brain regions are affected in this regard.
In the present study, we examined whether drug-refractoriness of seizures is associated with alterations in Pgp expression in a rat model of temporal lobe epilepsy (TLE). In this model, SRS develop after a latency period following a status epilepticus (SE) which is induced by prolonged electrical stimulation of the basolateral amygdala (Brandt et al., 2003). We have recently shown that daily administration of phenobarbital at maximum tolerable doses in epileptic rats of this model results in two subgroups, i.e. a responder subgroup with control of seizures and a non-responder subgroup without any significant reduction in seizure frequency (Brandt et al., 2004). We suggested that epileptic rats with such AED resistance offer unique approaches to the biological basis of refractoriness, particularly because pathological alterations in such rats can be directly compared with those of rats that respond to AEDs (Brandt et al., 2004). To our knowledge, the present study is the first to examine whether AED responders and non-responders differ in their Pgp expression in the brain, using a rat model of TLE with SRS.
Methods
Animals
Based on our previous experience in different rat strains and genders, female outbred rats of the Sprague–Dawley strain were used for this study because prolonged stimulation of the basolateral nucleus of the amygdala (BLA) results in most of these rats in a generalized convulsive SE which induces development of SRS in >90% of the animals (Brandt et al., 2003). The rats were purchased at a body weight of 200–230 g (Harlan-Winkelmann, Borchen, Germany). Following arrival, the rats were kept under controlled environmental conditions (24–25°C; 50–60% humidity; 12 h light/12 h dark cycle; light on at 6:00 a.m.) with free access to standard laboratory chow (Altromin 1324 standard diet) and tap water. Before being used in the experiments, the rats were allowed to adapt to the new conditions for at least 1 week. All animal experiments were carried out in accordance with the European Communities Council Directive of November 24, 1986 (86/609/EEC) and were formally approved by the animal subjects review board of our institution. All efforts were made to minimize the number of animals used and their suffering.
Electrode implantation
For electrode implantation, rats were anaesthetized with chloral hydrate (360 mg/kg i.p.). A Teflon-isolated bipolar stainless steel electrode was stereotactically implanted into the right anterior BLA as described recently (Brandt et al., 2003) and served as stimulation and recording electrode. A screw, placed above the left parietal cortex, served as the indifferent reference electrode. Additional skull screws and dental acrylic cement anchored the electrode assembly. After surgery, the animals were allowed to recover for a period of at least 2 weeks.
Induction of a self-sustaining status epilepticus
About 2 weeks after electrode implantation, the rats were electrically stimulated via the BLA electrode for induction of a self-sustained SE. The following stimulus parameters were chosen: stimulus duration 25 min; stimulus consisting of 100 ms trains of 1 ms alternating positive and negative square-wave pulses. The trains were given at a frequency of 2/s and the intra-train pulse frequency was 50/s. Peak pulse intensity was 700 μA. For this pulsed-train stimulation, an Accupulser A310C stimulator connected to a Stimulus Isolator A365 (World Precision Instruments, Berlin, Germany) were used. About 90% of rats developed a self-sustained SE with generalized convulsive seizures. After 4 h, SE was interrupted with diazepam (10 mg/kg i.p.) in all rats. If necessary, the application of this dose of diazepam was repeated, but in most rats seizure activity was terminated after the first diazepam injection. By using continuous video and EEG recording for up to 24 h after injection of diazepam, we recently demonstrated that diazepam completely blocked all behavioural seizure activity and the EEG alterations associated with this behaviour and also blocked any reappearance of seizures for 24 h after injection (Brandt et al., 2003).
Starting about 4 weeks later, the rats were monitored by EEG-video recordings for up to 2 months until the first spontaneous seizures were detected, as described recently (Brandt et al., 2003, 2004). Eleven rats with SRS were used for the experiments with phenobarbital.
Treatment with phenobarbital
Phenobarbital was chosen because it is an efficacious AED in rat models of TLE with a sufficiently long half-life to allow maintenance of therapeutic drug levels during prolonged treatment (Löscher and Hönack, 1989; Leite and Cavalheiro, 1995; Brandt et al., 2004). As described in detail recently (Brandt et al., 2004), several preliminary experiments were performed to develop a dosing protocol that would keep plasma drug concentrations within or above the therapeutic range (10–40 µg/ml; Baulac, 2002) for 24 h/day, 7 days/week. Furthermore, we wished to administer phenobarbital at maximum tolerated doses, so that rats were closely observed for adverse effects. Based on these preliminary experiments, a dosing protocol with an i.p. bolus dose of 25 mg/kg in the morning of the first treatment day, followed 10 h later by an administration of 15 mg/kg, and then twice daily 15 mg/kg for the 13 subsequent days, was used in rats with SRS. Before onset of drug treatment, baseline seizure frequency was determined over 2 weeks (predrug control period), then phenobarbital was administered over 2 weeks, followed by a postdrug control period of 2 weeks. Blood was sampled 10 h after the first drug injection and 12 h after the last drug injection for drug analysis in plasma by high-performance liquid chromatography with ultraviolet detection (Potschka et al., 2002). In each of the 6 weeks of the experiment, seizures were continuously (24 h/day, 7 days/week) monitored by video-EEG recording, as described in detail recently (Brandt et al., 2004). A schematic illustration of the drug trial is shown in Fig. 1.
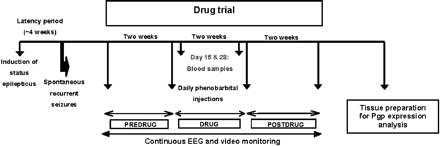
Schematic illustration of the experimental protocol used to select drug-resistant and drug-responsive rats with spontaneous recurrent seizures for determination of P-glycoprotein (Pgp) in brain tissue.
Tissue preparation and immunohistochemistry
Two months after termination of the drug experiment, rats were decapitated and brains were immediately embedded in Tissue Freezing Medium® (Jung, Nussloch, Germany), rapidly removed and snap-frozen in liquid nitrogen. Frozen brains were stored at −80°C at least overnight. Frozen transverse sections of forebrain and midbrain were cut at 14 µm using a cryostat (HM 560 M; Microm, Walldorf, Germany) and mounted onto TESPA (3-aminopropyl-triethoxysilane)-coated glass slides. The sections were postfixed in acetone and stored at −80°C until immunohistological staining. The following section levels of both hemispheres were chosen for immunohistochemistry: −2.3, −3.8 and −5.8 mm relative to bregma (Paxinos and Watson, 1998).
Brain sections of responders and non-responders (see Results) were processed simultaneously in the same reaction trays to obtain comparable staining intensity. Immunohistochemical analysis of Pgp expression was performed using a labelled streptavidin–biotin–peroxidase method with a monoclonal mouse antibody (C219; 1:100; Calbiochem, Bad Soden, Germany) directed against Pgp, according to the protocol described by us in detail recently (Volk et al., 2004, 2005).
In each brain region, the area labelled for Pgp (labelled surface area) and its optical density (OD) were analysed with a computer-assisted image analysis system, as described in detail recently (Volk et al., 2004). The hardware consisted of an Axioskop microscope with a Plan-Neofluar lens (Zeiss, Germany), a single chip charge-coupled device (CCD) colour camera (Axiocam; Zeiss, Göttingen, Hallbergmoos, Germany), and a Pentium III-based computer equipped with an image capture interface card (V7-Mirage; Spea, USA). For analysis of brain sections, 400× magnification was used. The captured images were 1300×1030 pixels in dimensions and were processed by means of the image analysis software KS400 (Windows Release 3.0; Carl Zeiss Vision, Germany), as described by Volk et al. (2004).
Pgp expression was investigated in ipsilateral (i.e. the hemisphere with the stimulation electrode) and contralateral sections of the cerebral cortex (at three section levels per rat; Table 1), piriform cortex (two section levels), hippocampus (two or three section levels) and substantia nigra pars reticulata (one section level). In each region, fields of 43 434 µm2 were randomly chosen for analysis of Pgp expression, using three to 10 fields per section level. The area of positive reactions relative to the total area (%) of the fields was measured per region, side and animal. Background correction of the OD measurements was performed as described recently (Volk et al., 2004). In all experiments, image analysis was done in a blinded fashion, i.e. the experimenter was not aware whether sections were from responders or non-responders.
Quantitative evaluation of immunostaining for P-glycoprotein (Pgp) in responders and non-responders in terms of relative labelled area measured by computer-assisted image analysis
| Brain region | Bregma | Area (%) labelled for Pgp | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Responders | Non-responders | |||||||||
| Contralateral | Ipsilateral | Contralateral | Ipsilateral | |||||||
| Cortex | −2.3 | 10.30 ± 1.56 | 26.24 ± 6.52 | 15.53 ± 2.92 | 22.49 ± 5.77 | |||||
| −3.8 | 16.89 ± 3.93 | 20.51 ± 4.37 | 19.37 ± 4.04 | 25.21 ± 1.93 | ||||||
| −5.8 | 21.31 ± 6.94 | 22.87 ± 4.35 | 7.86 ± 0.56 | 25.21 ± 1.93 | ||||||
| Piriform cortex | −2.3 | 3.23 ± 0.91 | 2.04 ± 0.12 | 9.66 ± 1.89* | 8.89 ± 1.58* | |||||
| −3.8 | 3.47 ± 0.66 | 3.67 ± 1.21 | 7.80 ± 1.37* | 8.49 ± 1.78* | ||||||
| Hippocampal formation | ||||||||||
| Dentate gyrus | ||||||||||
| Granule cell layer | −2.3 | 4.89 ± 1.50 | 2.62 ± 0.16 | 5.11 ± 0.16 | 4.53 ± 0.32* | |||||
| −3.8 | 2.57 ± 0.47 | 3.71 ± 0.62 | 6.10 ± 1.42* | 4.53 ± 0.50 | ||||||
| Hilus | −2.3 | 7.01 ± 2.86 | 4.40 ± 1.02 | 7.12 ± 0.42 | 6.24 ± 1.41 | |||||
| −3.8 | 7.01 ± 2.86 | 4.40 ± 1.02 | 7.12 ± 0.42 | 6.24 ± 1.41 | ||||||
| CA1 | −2.3 | 5.41 ± 1.68 | 3.57 ± 0.97 | 6.09 ± 0.39 | 7.53 ± 1.16* | |||||
| −3.8 | 3.84 ± 0.77 | 3.73 ± 0.87 | 7.79 ± 2.22* | 10.11 ± 2.69* | ||||||
| −5.8 | 5.95 ± 2.00 | 10.90 ± 4.85 | 9.58 ± 2.38 | 19.07 ± 8.48 | ||||||
| CA3 | −2.3 | 5.42 ± 0.92 | 9.08 ± 2.67 | 8.53 ± 2.15 | 11.63 ± 4.22 | |||||
| −3.8 | 10.06 ± 1.19 | 9.08 ± 2.67 | 5.41 ± 1.10 | 11.63 ± 4.22 | ||||||
| −5.8 | 9.27 ± 3.50 | 7.43 ± 2.80 | 9.83 ± 3.81 | 8.15 ± 1.64 | ||||||
| Substantia nigra | −5.8 | 17.37 ± 6.40 | 10.00 ± 3.99 | 23.88 ± 3.91 | 17.89 ± 8.43 | |||||
| Brain region | Bregma | Area (%) labelled for Pgp | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Responders | Non-responders | |||||||||
| Contralateral | Ipsilateral | Contralateral | Ipsilateral | |||||||
| Cortex | −2.3 | 10.30 ± 1.56 | 26.24 ± 6.52 | 15.53 ± 2.92 | 22.49 ± 5.77 | |||||
| −3.8 | 16.89 ± 3.93 | 20.51 ± 4.37 | 19.37 ± 4.04 | 25.21 ± 1.93 | ||||||
| −5.8 | 21.31 ± 6.94 | 22.87 ± 4.35 | 7.86 ± 0.56 | 25.21 ± 1.93 | ||||||
| Piriform cortex | −2.3 | 3.23 ± 0.91 | 2.04 ± 0.12 | 9.66 ± 1.89* | 8.89 ± 1.58* | |||||
| −3.8 | 3.47 ± 0.66 | 3.67 ± 1.21 | 7.80 ± 1.37* | 8.49 ± 1.78* | ||||||
| Hippocampal formation | ||||||||||
| Dentate gyrus | ||||||||||
| Granule cell layer | −2.3 | 4.89 ± 1.50 | 2.62 ± 0.16 | 5.11 ± 0.16 | 4.53 ± 0.32* | |||||
| −3.8 | 2.57 ± 0.47 | 3.71 ± 0.62 | 6.10 ± 1.42* | 4.53 ± 0.50 | ||||||
| Hilus | −2.3 | 7.01 ± 2.86 | 4.40 ± 1.02 | 7.12 ± 0.42 | 6.24 ± 1.41 | |||||
| −3.8 | 7.01 ± 2.86 | 4.40 ± 1.02 | 7.12 ± 0.42 | 6.24 ± 1.41 | ||||||
| CA1 | −2.3 | 5.41 ± 1.68 | 3.57 ± 0.97 | 6.09 ± 0.39 | 7.53 ± 1.16* | |||||
| −3.8 | 3.84 ± 0.77 | 3.73 ± 0.87 | 7.79 ± 2.22* | 10.11 ± 2.69* | ||||||
| −5.8 | 5.95 ± 2.00 | 10.90 ± 4.85 | 9.58 ± 2.38 | 19.07 ± 8.48 | ||||||
| CA3 | −2.3 | 5.42 ± 0.92 | 9.08 ± 2.67 | 8.53 ± 2.15 | 11.63 ± 4.22 | |||||
| −3.8 | 10.06 ± 1.19 | 9.08 ± 2.67 | 5.41 ± 1.10 | 11.63 ± 4.22 | ||||||
| −5.8 | 9.27 ± 3.50 | 7.43 ± 2.80 | 9.83 ± 3.81 | 8.15 ± 1.64 | ||||||
| Substantia nigra | −5.8 | 17.37 ± 6.40 | 10.00 ± 3.99 | 23.88 ± 3.91 | 17.89 ± 8.43 | |||||
In sections of the brain regions shown in the table, the area of positive Pgp-immunostaining relative to the total area of the section was determined per region, hemisphere and rat, using 3–10 fields per section in each hemisphere. The average values of each rat were used for calculation of group values. Data are mean ± SE for seven responders and four non-responders. For each region, the anterior–posterior coordinate (in mm from bregma) of the section(s) is indicated (Paxinos and Watson, 1998). ‘Ipsilateral’ refers to the hemisphere with the stimulation/recording electrode. Significant differences between non-responders and responders (P < 0.05) are indicated by an asterisk.
Quantitative evaluation of immunostaining for P-glycoprotein (Pgp) in responders and non-responders in terms of relative labelled area measured by computer-assisted image analysis
| Brain region | Bregma | Area (%) labelled for Pgp | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Responders | Non-responders | |||||||||
| Contralateral | Ipsilateral | Contralateral | Ipsilateral | |||||||
| Cortex | −2.3 | 10.30 ± 1.56 | 26.24 ± 6.52 | 15.53 ± 2.92 | 22.49 ± 5.77 | |||||
| −3.8 | 16.89 ± 3.93 | 20.51 ± 4.37 | 19.37 ± 4.04 | 25.21 ± 1.93 | ||||||
| −5.8 | 21.31 ± 6.94 | 22.87 ± 4.35 | 7.86 ± 0.56 | 25.21 ± 1.93 | ||||||
| Piriform cortex | −2.3 | 3.23 ± 0.91 | 2.04 ± 0.12 | 9.66 ± 1.89* | 8.89 ± 1.58* | |||||
| −3.8 | 3.47 ± 0.66 | 3.67 ± 1.21 | 7.80 ± 1.37* | 8.49 ± 1.78* | ||||||
| Hippocampal formation | ||||||||||
| Dentate gyrus | ||||||||||
| Granule cell layer | −2.3 | 4.89 ± 1.50 | 2.62 ± 0.16 | 5.11 ± 0.16 | 4.53 ± 0.32* | |||||
| −3.8 | 2.57 ± 0.47 | 3.71 ± 0.62 | 6.10 ± 1.42* | 4.53 ± 0.50 | ||||||
| Hilus | −2.3 | 7.01 ± 2.86 | 4.40 ± 1.02 | 7.12 ± 0.42 | 6.24 ± 1.41 | |||||
| −3.8 | 7.01 ± 2.86 | 4.40 ± 1.02 | 7.12 ± 0.42 | 6.24 ± 1.41 | ||||||
| CA1 | −2.3 | 5.41 ± 1.68 | 3.57 ± 0.97 | 6.09 ± 0.39 | 7.53 ± 1.16* | |||||
| −3.8 | 3.84 ± 0.77 | 3.73 ± 0.87 | 7.79 ± 2.22* | 10.11 ± 2.69* | ||||||
| −5.8 | 5.95 ± 2.00 | 10.90 ± 4.85 | 9.58 ± 2.38 | 19.07 ± 8.48 | ||||||
| CA3 | −2.3 | 5.42 ± 0.92 | 9.08 ± 2.67 | 8.53 ± 2.15 | 11.63 ± 4.22 | |||||
| −3.8 | 10.06 ± 1.19 | 9.08 ± 2.67 | 5.41 ± 1.10 | 11.63 ± 4.22 | ||||||
| −5.8 | 9.27 ± 3.50 | 7.43 ± 2.80 | 9.83 ± 3.81 | 8.15 ± 1.64 | ||||||
| Substantia nigra | −5.8 | 17.37 ± 6.40 | 10.00 ± 3.99 | 23.88 ± 3.91 | 17.89 ± 8.43 | |||||
| Brain region | Bregma | Area (%) labelled for Pgp | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Responders | Non-responders | |||||||||
| Contralateral | Ipsilateral | Contralateral | Ipsilateral | |||||||
| Cortex | −2.3 | 10.30 ± 1.56 | 26.24 ± 6.52 | 15.53 ± 2.92 | 22.49 ± 5.77 | |||||
| −3.8 | 16.89 ± 3.93 | 20.51 ± 4.37 | 19.37 ± 4.04 | 25.21 ± 1.93 | ||||||
| −5.8 | 21.31 ± 6.94 | 22.87 ± 4.35 | 7.86 ± 0.56 | 25.21 ± 1.93 | ||||||
| Piriform cortex | −2.3 | 3.23 ± 0.91 | 2.04 ± 0.12 | 9.66 ± 1.89* | 8.89 ± 1.58* | |||||
| −3.8 | 3.47 ± 0.66 | 3.67 ± 1.21 | 7.80 ± 1.37* | 8.49 ± 1.78* | ||||||
| Hippocampal formation | ||||||||||
| Dentate gyrus | ||||||||||
| Granule cell layer | −2.3 | 4.89 ± 1.50 | 2.62 ± 0.16 | 5.11 ± 0.16 | 4.53 ± 0.32* | |||||
| −3.8 | 2.57 ± 0.47 | 3.71 ± 0.62 | 6.10 ± 1.42* | 4.53 ± 0.50 | ||||||
| Hilus | −2.3 | 7.01 ± 2.86 | 4.40 ± 1.02 | 7.12 ± 0.42 | 6.24 ± 1.41 | |||||
| −3.8 | 7.01 ± 2.86 | 4.40 ± 1.02 | 7.12 ± 0.42 | 6.24 ± 1.41 | ||||||
| CA1 | −2.3 | 5.41 ± 1.68 | 3.57 ± 0.97 | 6.09 ± 0.39 | 7.53 ± 1.16* | |||||
| −3.8 | 3.84 ± 0.77 | 3.73 ± 0.87 | 7.79 ± 2.22* | 10.11 ± 2.69* | ||||||
| −5.8 | 5.95 ± 2.00 | 10.90 ± 4.85 | 9.58 ± 2.38 | 19.07 ± 8.48 | ||||||
| CA3 | −2.3 | 5.42 ± 0.92 | 9.08 ± 2.67 | 8.53 ± 2.15 | 11.63 ± 4.22 | |||||
| −3.8 | 10.06 ± 1.19 | 9.08 ± 2.67 | 5.41 ± 1.10 | 11.63 ± 4.22 | ||||||
| −5.8 | 9.27 ± 3.50 | 7.43 ± 2.80 | 9.83 ± 3.81 | 8.15 ± 1.64 | ||||||
| Substantia nigra | −5.8 | 17.37 ± 6.40 | 10.00 ± 3.99 | 23.88 ± 3.91 | 17.89 ± 8.43 | |||||
In sections of the brain regions shown in the table, the area of positive Pgp-immunostaining relative to the total area of the section was determined per region, hemisphere and rat, using 3–10 fields per section in each hemisphere. The average values of each rat were used for calculation of group values. Data are mean ± SE for seven responders and four non-responders. For each region, the anterior–posterior coordinate (in mm from bregma) of the section(s) is indicated (Paxinos and Watson, 1998). ‘Ipsilateral’ refers to the hemisphere with the stimulation/recording electrode. Significant differences between non-responders and responders (P < 0.05) are indicated by an asterisk.
For quantification of vascular density, the brain-type glucose transporter (GLUT-1) was immunostained with a polyclonal rabbit antibody against GLUT-1 (1 : 1000; Chemicon, Hofheim, Germany) as described recently (Volk et al., 2004). In the brain, GLUT-1 is selectively localized in brain capillary endothelial cells (Pardridge et al., 1990; Farrell and Pardridge, 1991; Dobrogowska and Vorbrodt, 1999). The immunohistochemical procedure was performed using a labelled streptavidin–biotin–peroxidase method as described in detail recently (Volk et al., 2004a). Analysis of GLUT-1-labelled surface area and of its OD in diaminobentidine-stained sections was performed as described above for Pgp. All analyses were done in a blinded fashion. Data from GLUT-1 staining were also used to calculate the relative amount of Pgp-expressing capillaries to total capillaries in brain sections. In this respect it has to be noted that adjacent sections had to be used for this comparison, because we did not perform experiments with double-staining of Pgp and GLUT-1 in the same sections.
Statistics
All values were expressed as mean ± SEM and were statistically analysed with Student's t-test for non-paired (differences between groups) comparisons. For correlation analysis between seizure frequency and Pgp or GLUT-1 expression, the Pearson correlation coefficient was calculated. P < 0.05 was considered significant.
Results
Selection of responders and non-responders
Detailed results of selection with phenobarbital have been described recently (Brandt et al., 2004), so that responders and non-responders will be characterized only briefly here. All rats received phenobarbital at maximum tolerated doses, as indicated by the marked sedation which was observed in all rats during treatment. Analysis of plasma drug concentrations showed that drug concentrations within the therapeutic range (10–40 µg/ml) were maintained in all rats throughout the period of treatment. In six of 11 rats with SRS, complete control of seizures was achieved and another rat exhibited a >90% reduction in seizure frequency. These seven rats were considered responders (Fig. 2). Three animals (DS107, DS110, DS118) of the remaining four rats showed no anticonvulsant response but an increase in seizure frequency during drug treatment (Fig. 2). The fourth rat (DS113) showed only moderate (<50%) reduction in seizure frequency. These four rats were therefore considered non-responders (Fig. 2).
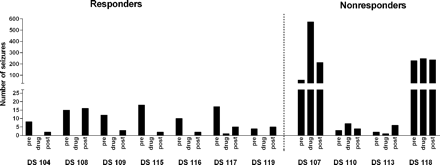
Number of spontaneous seizures in the predrug, drug and postdrug periods in phenobarbital responders and non-responders. Numbers of seizures occurring per 2-week period are shown for each individual rat. The daily variability of seizure frequency in the responders and non-responders has been illustrated elsewhere (Brandt et al., 2004). In responders (n = 7), only one rat (DS117) exhibited one seizure during the treatment period (on day 1), whereas all other responders were seizure-free during treatment. In contrast, such suppression of seizures was not observed in non-responders during the treatment period.
Plasma drug concentrations did not differ significantly between responders and non-responders. Average plasma concentrations determined 10 h after the bolus administration of phenobarbital (25 mg/kg) were 25.1 ± 0.8 in responders versus 24.4 ± 1.2 µg/ml in non-responders. Twelve hours after the last phenobarbital injection of the treatment period, average plasma levels of 28.7 ± 3.8 µg/ml were determined in responders versus 25.6 ± 5.7 µg/ml in non-responders.
Two of the non-responders (DS107 and DS118) exhibited an extremely high seizure frequency during the control and treatment periods of the experiment, while the other two non-responders did not differ from responders in seizure frequency during the control periods (Fig. 2). The type of SRS was the same in all responders and non-responders, i.e. generalized convulsive seizures, resembling stage 4 or 5 seizures of the Racine scale (Racine, 1972). Furthermore, the severity of the initial, electrically induced SE was not different between responders and non-responders, indicating that the same severity and duration of SE produces two subgroups of epileptic rats, AED responders and non-responders.
Pgp expression in the brain of responders and non-responders
As previously reported for rat brain sections stained by the immunohistochemical protocol used in the present study (Volk et al., 2004, 2005), immunolocalization of the Pgp antibody C 219 was observed exclusively in microvessel endothelial cells (Fig. 3). In the piriform cortex and hippocampal formation, markedly more capillary endothelial cells were stained for Pgp in non-responders (Fig. 3B, D) than in responders (Fig. 3A, C). This was seen in all non-responders, irrespective of the individual seizure frequency of the rats.
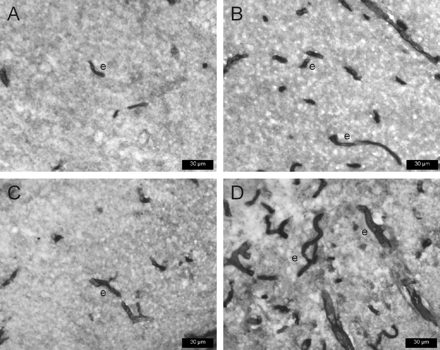
P-glycoprotein (Pgp) expression in representative transverse sections of rat brain, using 3,3′-diaminobenzidine as chromogen and the monoclonal antibody C219 for Pgp immunostaining. Prominent immunoreactivity is evident in endothelial cells of capillaries (e). A and B are sections from the dentate gyrus; C and D are sections from the piriform cortex. Sections are from the ipsilateral hemisphere 3.8 mm posterior to bregma (Paxinos and Watson, 1998). The sections shown in A and C are from phenobarbital responders (DS104 and DS115, respectively); sections in B and D are from a non-responder (DS118), illustrating the striking increase in the number of capillaries expressing Pgp in non-responders.
For quantification of the increase of Pgp expression in non-responders, computer-assisted image analysis was used. Because under the conditions of the present experiments Pgp was exclusively detected in capillary endothelial cells, the Pgp-stained area determined by computer-assisted image analysis in each region was related to endothelial Pgp (Fig. 4, Table 1). As shown in Fig. 4, the Pgp-labelled area, as assessed by analysis of labelled surface area, was significantly higher in the ipsilateral piriform cortex, dentate gyrus and CA1 sector of the hippocampus of non-responders compared with the responder group. The most marked, >4-fold increase in Pgp-labelled area was determined for the piriform cortex (at −2.3), whereas most other increases in limbic brain regions were about 2-fold on average (Fig. 4). A significant increase in Pgp-labelled area of similar magnitude was also observed in limbic brain regions of the contralateral hemisphere (Table 1). In contrast, no significant differences in Pgp-labelled area between responders and non-responders were observed in the cerebral cortex and substantia nigra (Table 1), indicating that the difference was restricted to limbic brain regions.
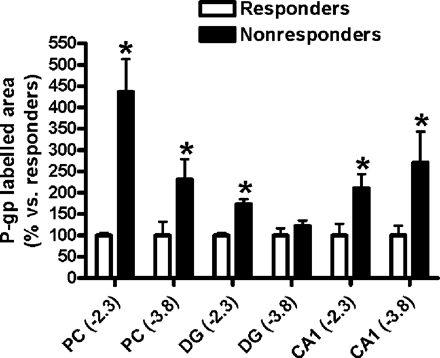
Relative increases in P-glycoprotein (Pgp) expression in limbic brain regions of phenobarbital non-responders compared with responders (which are set to 100%). Data are from analysis of the Pgp-labelled area as assessed by measurement of labelled surface area (see Methods). For absolute values see Table 1. Data are shown as mean ± SEM of seven responders and four non-responders. Significant differences between groups are indicated by an asterisk (P < 0.05). All regions are from the ipsilateral hemisphere. Numbers in brackets indicate the section level relative to bregma. PC = piriform cortex; DG = granule cell layer of the dentate gyrus.
For quantification of the intensity of Pgp staining, the OD of staining was determined by computer-assisted image analysis. As shown in Table 2, the only regional difference between non-responders and responders in the ipsilateral hemisphere was a moderate but statistically significant increase in Pgp staining intensity in the CA3 sector of the hippocampus. In the contralateral hemisphere, significantly enhanced staining intensity was also determined in the granule cell layer and hilus of the dentate gyrus and the CA1 sector of the hippocampus (Table 2).
Quantitative evaluation of immunostaining for P-glycoprotein (Pgp) in responders and non-responders in terms of staining intensity as measured by computer-assisted analysis of optical density
| Brain regions | Bregma | Optical density measurement | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Responders | Non-responders | |||||||||
| Contralateral | Ipsilateral | Contralateral | Ipsilateral | |||||||
| Cortex | −2.3 | 0.35 ± 0.02 | 0.28 ± 0.02 | 0.35 ± 0.03 | 0.31 ± 0.03 | |||||
| −3.8 | 0.30 ± 0.03 | 0.28 ± 0.03 | 0.30 ± 0.02 | 0.29 ± 0.02 | ||||||
| −5.8 | 0.41 ± 0.02 | 0.29 ± 0.02 | 0.30 ± 0.03 | 0.28 ± 0.03 | ||||||
| Piriform cortex | −2.3 | 0.48 ± 0.04 | 0.48 ± 0.01 | 0.51 ± 0.02 | 0.49 ± 0.01 | |||||
| −3.8 | 0.45 ± 0.03 | 0.45 ± 0.04 | 0.49 ± 0.02 | 0.50 ± 0.02 | ||||||
| Hippocampal formation | ||||||||||
| Dentate gyrus | ||||||||||
| Granule cell layer | −2.3 | 0.43 ± 0.02 | 0.46 ± 0.03 | 0.50 ± 0.01* | 0.50 ± 0.01 | |||||
| −3.8 | 0.43 ± 0.01 | 0.43 ± 0.02 | 0.49 ± 0.03* | 0.49 ± 0.02 | ||||||
| Hilus | −2.3 | 0.31 ± 0.02 | 0.41 ± 0.05 | 0.50 ± 0.02* | 0.46 ± 0.03 | |||||
| −3.8 | 0.31 ± 0.02 | 0.41 ± 0.05 | 0.50 ± 0.02* | 0.46 ± 0.03 | ||||||
| CA1 | −2.3 | 0.40 ± 0.04 | 0.41 ± 0.01 | 0.48 ± 0.03 | 0.45 ± 0.03 | |||||
| −3.8 | 0.39 ± 0.02 | 0.42 ± 0.03 | 0.50 ± 0.03* | 0.43 ± 0.03 | ||||||
| −5.8 | 0.36 ± 0.03 | 0.35 ± 0.03 | 0.45 ± 0.04 | 0.39 ± 0.04 | ||||||
| CA3 | −2.3 | 0.38 ± 0.01 | 0.30 ± 0.02 | 0.44 ± 0.04* | 0.38 ± 0.03* | |||||
| −3.8 | 0.38 ± 0.02 | 0.30 ± 0.02 | 0.36 ± 0.01 | 0.38 ± 0.03* | ||||||
| −5.8 | 0.51 ± 0.04 | 0.37 ± 0.04 | 0.37 ± 0.04 | 0.48 ± 0.02* | ||||||
| Substantia nigra | −5.8 | 0.33 ± 0.04 | 0.36 ± 0.03 | 0.34 ± 0.04 | 0.37 ± 0.02 | |||||
| Brain regions | Bregma | Optical density measurement | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Responders | Non-responders | |||||||||
| Contralateral | Ipsilateral | Contralateral | Ipsilateral | |||||||
| Cortex | −2.3 | 0.35 ± 0.02 | 0.28 ± 0.02 | 0.35 ± 0.03 | 0.31 ± 0.03 | |||||
| −3.8 | 0.30 ± 0.03 | 0.28 ± 0.03 | 0.30 ± 0.02 | 0.29 ± 0.02 | ||||||
| −5.8 | 0.41 ± 0.02 | 0.29 ± 0.02 | 0.30 ± 0.03 | 0.28 ± 0.03 | ||||||
| Piriform cortex | −2.3 | 0.48 ± 0.04 | 0.48 ± 0.01 | 0.51 ± 0.02 | 0.49 ± 0.01 | |||||
| −3.8 | 0.45 ± 0.03 | 0.45 ± 0.04 | 0.49 ± 0.02 | 0.50 ± 0.02 | ||||||
| Hippocampal formation | ||||||||||
| Dentate gyrus | ||||||||||
| Granule cell layer | −2.3 | 0.43 ± 0.02 | 0.46 ± 0.03 | 0.50 ± 0.01* | 0.50 ± 0.01 | |||||
| −3.8 | 0.43 ± 0.01 | 0.43 ± 0.02 | 0.49 ± 0.03* | 0.49 ± 0.02 | ||||||
| Hilus | −2.3 | 0.31 ± 0.02 | 0.41 ± 0.05 | 0.50 ± 0.02* | 0.46 ± 0.03 | |||||
| −3.8 | 0.31 ± 0.02 | 0.41 ± 0.05 | 0.50 ± 0.02* | 0.46 ± 0.03 | ||||||
| CA1 | −2.3 | 0.40 ± 0.04 | 0.41 ± 0.01 | 0.48 ± 0.03 | 0.45 ± 0.03 | |||||
| −3.8 | 0.39 ± 0.02 | 0.42 ± 0.03 | 0.50 ± 0.03* | 0.43 ± 0.03 | ||||||
| −5.8 | 0.36 ± 0.03 | 0.35 ± 0.03 | 0.45 ± 0.04 | 0.39 ± 0.04 | ||||||
| CA3 | −2.3 | 0.38 ± 0.01 | 0.30 ± 0.02 | 0.44 ± 0.04* | 0.38 ± 0.03* | |||||
| −3.8 | 0.38 ± 0.02 | 0.30 ± 0.02 | 0.36 ± 0.01 | 0.38 ± 0.03* | ||||||
| −5.8 | 0.51 ± 0.04 | 0.37 ± 0.04 | 0.37 ± 0.04 | 0.48 ± 0.02* | ||||||
| Substantia nigra | −5.8 | 0.33 ± 0.04 | 0.36 ± 0.03 | 0.34 ± 0.04 | 0.37 ± 0.02 | |||||
In sections of the brain regions shown in the table, the intensity of positive Pgp-immunostaining was determined per region, hemisphere and rat, using 3–10 fields per section in each hemisphere. The average values of each rat were used for calculation of group values. Data are mean ± SE for seven responders and four non-responders. For each region, the anterior–posterior coordinate (in mm from bregma) of the section(s) is indicated (Paxinos and Watson, 1998). ‘Ipsilateral’ refers to the hemisphere with the stimulation/recording electrode. Significant differences between non-responders and responders (P < 0.05) are indicated by an asterisk.
Quantitative evaluation of immunostaining for P-glycoprotein (Pgp) in responders and non-responders in terms of staining intensity as measured by computer-assisted analysis of optical density
| Brain regions | Bregma | Optical density measurement | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Responders | Non-responders | |||||||||
| Contralateral | Ipsilateral | Contralateral | Ipsilateral | |||||||
| Cortex | −2.3 | 0.35 ± 0.02 | 0.28 ± 0.02 | 0.35 ± 0.03 | 0.31 ± 0.03 | |||||
| −3.8 | 0.30 ± 0.03 | 0.28 ± 0.03 | 0.30 ± 0.02 | 0.29 ± 0.02 | ||||||
| −5.8 | 0.41 ± 0.02 | 0.29 ± 0.02 | 0.30 ± 0.03 | 0.28 ± 0.03 | ||||||
| Piriform cortex | −2.3 | 0.48 ± 0.04 | 0.48 ± 0.01 | 0.51 ± 0.02 | 0.49 ± 0.01 | |||||
| −3.8 | 0.45 ± 0.03 | 0.45 ± 0.04 | 0.49 ± 0.02 | 0.50 ± 0.02 | ||||||
| Hippocampal formation | ||||||||||
| Dentate gyrus | ||||||||||
| Granule cell layer | −2.3 | 0.43 ± 0.02 | 0.46 ± 0.03 | 0.50 ± 0.01* | 0.50 ± 0.01 | |||||
| −3.8 | 0.43 ± 0.01 | 0.43 ± 0.02 | 0.49 ± 0.03* | 0.49 ± 0.02 | ||||||
| Hilus | −2.3 | 0.31 ± 0.02 | 0.41 ± 0.05 | 0.50 ± 0.02* | 0.46 ± 0.03 | |||||
| −3.8 | 0.31 ± 0.02 | 0.41 ± 0.05 | 0.50 ± 0.02* | 0.46 ± 0.03 | ||||||
| CA1 | −2.3 | 0.40 ± 0.04 | 0.41 ± 0.01 | 0.48 ± 0.03 | 0.45 ± 0.03 | |||||
| −3.8 | 0.39 ± 0.02 | 0.42 ± 0.03 | 0.50 ± 0.03* | 0.43 ± 0.03 | ||||||
| −5.8 | 0.36 ± 0.03 | 0.35 ± 0.03 | 0.45 ± 0.04 | 0.39 ± 0.04 | ||||||
| CA3 | −2.3 | 0.38 ± 0.01 | 0.30 ± 0.02 | 0.44 ± 0.04* | 0.38 ± 0.03* | |||||
| −3.8 | 0.38 ± 0.02 | 0.30 ± 0.02 | 0.36 ± 0.01 | 0.38 ± 0.03* | ||||||
| −5.8 | 0.51 ± 0.04 | 0.37 ± 0.04 | 0.37 ± 0.04 | 0.48 ± 0.02* | ||||||
| Substantia nigra | −5.8 | 0.33 ± 0.04 | 0.36 ± 0.03 | 0.34 ± 0.04 | 0.37 ± 0.02 | |||||
| Brain regions | Bregma | Optical density measurement | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Responders | Non-responders | |||||||||
| Contralateral | Ipsilateral | Contralateral | Ipsilateral | |||||||
| Cortex | −2.3 | 0.35 ± 0.02 | 0.28 ± 0.02 | 0.35 ± 0.03 | 0.31 ± 0.03 | |||||
| −3.8 | 0.30 ± 0.03 | 0.28 ± 0.03 | 0.30 ± 0.02 | 0.29 ± 0.02 | ||||||
| −5.8 | 0.41 ± 0.02 | 0.29 ± 0.02 | 0.30 ± 0.03 | 0.28 ± 0.03 | ||||||
| Piriform cortex | −2.3 | 0.48 ± 0.04 | 0.48 ± 0.01 | 0.51 ± 0.02 | 0.49 ± 0.01 | |||||
| −3.8 | 0.45 ± 0.03 | 0.45 ± 0.04 | 0.49 ± 0.02 | 0.50 ± 0.02 | ||||||
| Hippocampal formation | ||||||||||
| Dentate gyrus | ||||||||||
| Granule cell layer | −2.3 | 0.43 ± 0.02 | 0.46 ± 0.03 | 0.50 ± 0.01* | 0.50 ± 0.01 | |||||
| −3.8 | 0.43 ± 0.01 | 0.43 ± 0.02 | 0.49 ± 0.03* | 0.49 ± 0.02 | ||||||
| Hilus | −2.3 | 0.31 ± 0.02 | 0.41 ± 0.05 | 0.50 ± 0.02* | 0.46 ± 0.03 | |||||
| −3.8 | 0.31 ± 0.02 | 0.41 ± 0.05 | 0.50 ± 0.02* | 0.46 ± 0.03 | ||||||
| CA1 | −2.3 | 0.40 ± 0.04 | 0.41 ± 0.01 | 0.48 ± 0.03 | 0.45 ± 0.03 | |||||
| −3.8 | 0.39 ± 0.02 | 0.42 ± 0.03 | 0.50 ± 0.03* | 0.43 ± 0.03 | ||||||
| −5.8 | 0.36 ± 0.03 | 0.35 ± 0.03 | 0.45 ± 0.04 | 0.39 ± 0.04 | ||||||
| CA3 | −2.3 | 0.38 ± 0.01 | 0.30 ± 0.02 | 0.44 ± 0.04* | 0.38 ± 0.03* | |||||
| −3.8 | 0.38 ± 0.02 | 0.30 ± 0.02 | 0.36 ± 0.01 | 0.38 ± 0.03* | ||||||
| −5.8 | 0.51 ± 0.04 | 0.37 ± 0.04 | 0.37 ± 0.04 | 0.48 ± 0.02* | ||||||
| Substantia nigra | −5.8 | 0.33 ± 0.04 | 0.36 ± 0.03 | 0.34 ± 0.04 | 0.37 ± 0.02 | |||||
In sections of the brain regions shown in the table, the intensity of positive Pgp-immunostaining was determined per region, hemisphere and rat, using 3–10 fields per section in each hemisphere. The average values of each rat were used for calculation of group values. Data are mean ± SE for seven responders and four non-responders. For each region, the anterior–posterior coordinate (in mm from bregma) of the section(s) is indicated (Paxinos and Watson, 1998). ‘Ipsilateral’ refers to the hemisphere with the stimulation/recording electrode. Significant differences between non-responders and responders (P < 0.05) are indicated by an asterisk.
As described above, two of the four non-responders had a very high seizure frequency (Fig. 2). We therefore performed a correlation analysis over all 11 rats to determine whether the increased Pgp expression in non-responders was related to high seizure frequency. No significant correlation between seizure frequency (as measured during control periods) and Pgp expression was obtained. Furthermore, two of the four non-responders which did not differ from responders in seizure frequency (DS110 and DS113 in Fig. 2) exhibited an increase in Pgp expression similar to that of the two other non-responders, arguing against the possibility that the enhanced Pgp expression in non-responders was related to high seizure frequency.
GLUT-1 expression in the brain of responders and non-responders
In order to evaluate whether the increased capillary expression of Pgp in limbic areas in drug-resistant animals is a result of reactive capillary proliferation, brain sections from regions with increased Pgp expression were labelled with the brain capillary endothelial cell marker GLUT-1. Visual inspection and image analysis of the GLUT-1 labelled sections did not reveal any significant differences in the density of GLUT-1 labelled capillaries between non-responders and responders in such brain sections (not illustrated). These data indicate that the differences in Pgp expression between non-responders and responders were not a result of variation in reactive capillary proliferation after seizures. This was also substantiated by calculating the relative amount of Pgp-expressing capillaries to total (GLUT-1-expressing) capillaries in the respective regions. For instance, in the piriform cortex, the region with the highest Pgp expression in non-responders (Fig. 3, Table 1), the relative amount of Pgp-expressing capillaries to total capillaries was 85.3 ± 22.7% in non-responders compared with 41.7 ± 15.7% in responders, demonstrating that a considerably higher percentage of capillaries expressed Pgp in non-responders than in responders. With respect to the possible effect of SRS on vessel density in limbic brain regions such as the piriform cortex, there was no significant correlation between individual frequency of SRS and vessel density.
Discussion
The present study closes an important gap in the multidrug transporter hypothesis of medically intractable epilepsy (Sisodiya, 2003) by demonstrating that AED non-responders exhibit strikingly higher Pgp expression in brain capillary endothelial cells than AED responders. This higher expression was not secondary to high seizure frequency but occurred in all non-responders, independent of their individual seizure frequency. Furthermore, not all brain regions examined were involved in overexpression of Pgp but the increased expression in non-responders exclusively occurred in the hippocampus, dentate gyrus and piriform cortex, i.e. limbic brain regions thought to be part of an epileptic network underlying TLE (Gale, 1988; Löscher and Ebert, 1996; Lowenstein, 1996; Sloviter, 1999; Nair et al., 2004). In these brain regions of non-responders, remarkably more brain capillary endothelial cells expressed Pgp compared with responders. Because brain capillary endothelial cells form the BBB (Pardridge, 1999), enhanced expression of the multidrug efflux transporter Pgp in these cells would restrict the brain penetration of all drugs which are recognized as substrates by this transporter (Begley, 2004). Phenobarbital is a substrate for Pgp (Schuetz et al., 1996; Potschka et al., 2002), so that the enhanced Pgp expression in phenobarbital non-responders is a plausible explanation for the inability of phenobarbital to suppress seizures in these rats.
Unfortunately, the experimental protocol used for the present study (Fig. 1) did not allow the measurement of phenobarbital levels in the brains of non-responders versus responders to unequivocally show that the lack of response to phenobarbital is due to reduced drug levels in the brain of non-responders. As described in the Methods section, the rats were killed 2 months after termination of the drug experiment. This was done to exclude any effect of drug treatment on expression of Pgp. Furthermore, it took us about 2 months to analyse the EEGs and videos for SRS which were continuously monitored over the 6 weeks of the drug trial in the 11 epileptic rats of this study. Only then did we know that we had succeeded in selecting drug responders and non-responders, and killed the rats to examine our hypothesis that non-responders differ in their Pgp expression from responders. Now that we know that non-responders exhibit strikingly higher Pgp expression than responders in several limbic brain regions, including the hippocampus, we are planning to perform a prospective drug trial in the same model in which we will evaluate different AEDs and subsequently determine AED levels in these brain regions of responders and non-responders after selection. However, even without this important piece of information, it is very likely that the strikingly enhanced expression of Pgp in non-responders results in decreased levels of phenobarbital (and other AEDs that are Pgp substrates) in respective limbic brain regions. Thus, Rizzi et al. (2002) have shown that overexpression of Pgp mRNA following kainate-induced seizures is associated with a significant 30% decrease in the brain/plasma ratio of phenytoin in the hippocampus. A decrease in phenytoin concentrations of similar magnitude was also determined in the hippocampus of amygdala-kindled rats, another model of TLE (Potschka and Löscher, 2002). Like phenobarbital, phenytoin is a substrate for Pgp, explaining reduced brain drug levels associated with Pgp overexpression (Rizzi et al., 2002).
The present finding that Pgp overexpression in phenobarbital non-responders was restricted to limbic brain regions explains why adverse effects of phenobarbital, i.e. the marked sedation associated with the maximum tolerated doses used in the present study, did not differ between non-responders and responders. Similarly, resected histologically normal brain tissue adjacent to epileptogenic tissue from patients with refractory epilepsy showed lower immunoreactivity for Pgp compared with epileptogenic tissue, indicating that Pgp overexpression in patients is restricted to epileptogenic tissue (Sisodiya et al., 2002). The mechanisms underlying this locally restricted increase in Pgp expression in the brain are only poorly understood, but paroxysmal activity and the structural and functional alterations occurring in epileptogenic brain regions are certainly involved (Sisodiya et al., 2002; Aronica et al., 2003). The present evidence suggests that overexpression of Pgp in the brain of patients with epilepsy may be either constitutive or induced (acquired), depending on the specific type of epilepsy and its underlying neuropathology (Sisodiya et al., 2002; Sisodiya, 2003). In animal models, it has been shown that seizures induce transient and regionally selective overexpression of Pgp (Zhang et al., 1999; Rizzi et al., 2002; Seegers et al., 2002; Volk et al., 2004). As discussed above, this induced Pgp overexpression is associated with reduced penetration of AEDs into the affected brain regions (Potschka and Löscher, 2002; Rizzi et al., 2002). Constitutive overexpression of Pgp could be a result of polymorphisms in the MDR1 gene, which encodes Pgp (Sisodiya, 2003). Hitherto more than 50 single-nucleotide polymorphisms (SNPs) and insertion/deletion polymorphisms in the ABCB1 gene have been reported, and mutations at positions 2677 and 3435 were associated with alteration of Pgp expression and/or function (Brinkmann and Eichelbaum, 2001; Eichelbaum et al., 2004). In a recent study in 315 patients with epilepsy, classified as drug-resistant in 200 and drug-responsive in 115, patients with drug-resistant epilepsy were more likely to have the CC genotype of the MDR1 C3435T polymorphism, which is associated with increased expression of Pgp (Siddiqui et al., 2003). To our knowledge, it is not known whether such functionally relevant genetic polymorphisms also occur in the two genes (mdr1a, mdr1b) that encode Pgp in the brain of rodents (Demeule et al., 2002). The present finding that Pgp expression was not correlated with seizure frequency in rats with SRS would argue in favour of genetic polymorphisms in these genes, for instance in the promoter region that regulates gene expression, to explain the overexpression of Pgp in drug-resistant rats. The observed differences between non-responders and responders could also be the consequence of translational regulation or of aberrant transport of the protein. For further evaluation, we have started to search for polymorphisms in the gene (mdr1a) that encodes Pgp in brain capillary endothelial cells in rats. Sequencing analysis of the mdr1a gene in different rat strains yielded three SNPs in the exon–intron boundaries of exon 8 which were associated with differences in pharmacosensitivity in the kindling model of TLE (Baars et al., 2004). Whether these SNPs also affect AED response in the present TLE model remains to be determined.
The concept that constitutive overexpression of Pgp in the BBB may be involved in drug resistance in epilepsy has been substantiated by a recent study by our group in another animal model of refractory TLE, phenytoin-resistant kindled rats (Potschka et al., 2004). In this study, Pgp expression was determined in subgroups of amygdala-kindled rats which were sensitive or resistant to the anticonvulsant effect of phenytoin. Immunohistochemical analysis of Pgp expression was performed 2 days after the last kindled seizure, so that all rats had exhibited the same number of seizures with comparable severity and duration (Potschka et al., 2004). In the ipsilateral amygdala of phenytoin-resistant rats, the area labelled for Pgp was more than twice as large than that in phenytoin-sensitive rats, whereas such a difference was not seen in the adjacent piriform cortex (Potschka et al., 2004). Phenytoin-resistant kindled rats exhibit resistance against a variety of AEDs (Löscher, 2002), which could be explained by overexpression of Pgp in these animals. We do not know yet whether the phenobarbital-resistant rats used in the present study are also resistant to other AEDs, but in view of the markedly enhanced expression of Pgp in the BBB this appears very likely.
Various western immunoblotting and immunohistochemical studies, using confocal and electron microscopy in different species, including humans, have demonstrated that Pgp in the brain is predominantly localized in the apical (luminal) membrane of capillary endothelial cells, where it contributes critically to BBB function (Schinkel, 1999; Lee et al., 2001; Abbott et al., 2002; Begley, 2004). In order to achieve optimal immunohistochemical detection of Pgp in the brain capillary endothelial cells that form the BBB, we used a fixation and staining protocol (acetone-fixed cryostat sections of snap-frozen tissue) previously demonstrated to allow the best possible detection and quantification of Pgp in these cells (Volk et al., 2005). In contrast to the expression of Pgp in brain microvessel endothelial cells, this multidrug transporter is normally not expressed by astrocytes, but astrocytic expression may occur in epileptogenic tissue (Marroni et al., 2003). For optimal immunohistochemical detection of Pgp in astrocytes, other fixation protocols, e.g. by using paraformaldehyde, have to be used; however, these markedly reduce Pgp staining in capillaries (Volk et al., 2005). Thus, with the protocol used in the present study we cannot exclude the possibility that non-responders also exhibited enhanced Pgp expression in the parenchymal and/or perivascular astrocytes that contribute to BBB function (Marroni et al., 2003). We have recently demonstrated that fixation and staining variables have a striking effect on the immunolocalization of Pgp in the brain and detection of its increased expression in epileptogenic tissue (Volk et al., 2005). It has been reported previously that immunohistological assessment of Pgp in brain capillaries can be optimized by fixation in acetone (Thiebaut et al., 1989), and that epitope integrity of Pgp is better maintained in frozen sections than in formalin-fixed, paraffin-embedded tissues (Toth et al., 1994).
Recently, electroconvulsive seizures have been shown to induce endothelial cell proliferation in the hippocampus of rats (Hellsten et al., 2004). Potential variation in reactive capillary proliferation after electrical induction of seizures or SRS could form a bias in studies analysing Pgp expression in AED responders and non-responders. We therefore used GLUT-1 immunostaining to compare brain capillary density in responders and non-responders. No significant difference in GLUT-1 staining was observed, strongly indicating that the increased Pgp expression in non-responders was not secondary to variation in enhanced formation of new blood vessels. This was substantiated by calculating the relative amount of Pgp-expressing capillaries to total capillaries in limbic brain regions such as the piriform cortex, showing that the percentage of Pgp-expressing capillaries was markedly increased in non-responders. These data do not exclude the possibility that seizures resulted in the formation of new blood vessels in the present experiments but they indicate that variation in such reactive capillary formation was not involved in the marked difference in Pgp expression between responders and non-responders.
In conclusion, the present data further substantiate the multidrug transporter hypothesis of drug-resistant epilepsy. Overexpression of multidrug transporters such as Pgp at the level of the BBB is likely to impair the penetration of various AEDs into the brain, leading to decreased drug concentration at their brain targets and thereby contributing to multidrug resistance in epilepsy. Inhibitors of Pgp that are currently being evaluated in clinical trials in multidrug resistant cancer patients (Bates et al., 2002; Leonard et al., 2002; Kemper et al., 2004) may prove useful to prevent or overcome multidrug resistance in epilepsy, particularly because the regionally specific increase in Pgp raises the possibility of improving the therapeutic efficacy of AEDs without simultaneously increasing their toxicity (Rizzi et al., 2002; Summers et al., 2004; Löscher and Potschka, 2005). Another potentially promising approach to enhancing the penetration of AEDs in the presence of increased Pgp expression in the BBB is to bypass efflux transporters at the BBB without direct inhibition of these transporters (Fricker and Miller, 2004; Löscher and Potschka, 2005). One strategy in this respect uses antibody-coupled immunoliposomes to transport Pgp substrates through the BBB, avoiding direct interaction with Pgp or other transporters (Fricker and Miller, 2004). However, although overexpression of multidrug transporters such as Pgp is a reasonable hypothesis to explain multidrug resistance in epilepsy, further studies are needed to establish this concept. Furthermore, multidrug resistance is likely to be a multifactorial process, and different mechanisms, including overexpression of multidrug transporters and alterations in drug targets, are not necessarily mutually exclusive but may occur in the same tissue (Remy et al., 2003; Sisodiya, 2003; Löscher and Schmidt, 2004). As shown in the present study, animal models of TLE with SRS not adequately responding to clinically established AEDs are useful in investigating the basis of drug resistance in epilepsy and may help to identify new treatment approaches.
Present address: The Queen Mother Hospital for Animals, The Royal Veterinary College, Hatfield, UK
We thank Dr Heidrun Potschka for helpful discussions and advice during preparation of the manuscript and Christiane Bartling and Michael Weissing for skilful technical assistance. The determination of GLUT-1 immunoreactivity by Dr Claudia Brandt and Dr Heidrun Potschka is gratefully acknowledged. The study was supported by a grant (Lo 274/9) from the Deutsche Forschungsgemeinschaft (Bonn, Germany), the Studienstiftung des deutschen Volkes (Bonn, Germany) and a grant (1 R21 NS049592–01) from the National Institutes of Health (Bethesda, MD, USA).
References
Abbott NJ, Khan EU, Rollinson CMS, Reichel A, Janigro D, Dombrowski SM, et al., Drug resistance in epilepsy: the role of the blood-brain barrier. In: Ling V, editor. Mechanisms of drug resistance in epilepsy. Lessons from oncology. Chichester: Wiley;
Aronica E, Gorter JA, Jansen GH, van Veelen CW, van Rijen PC, Leenstra S, et al. Expression and cellular distribution of multidrug transporter proteins in two major causes of medically intractable epilepsy: focal cortical dysplasia and glioneuronal tumors.
Arroyo S, Brodie MJ, Avanzini G, Baumgartner C, Chiron C, Dulac O, et al. Is refractory epilepsy preventable?
Baars C, Potschka H, Volk HA, Becker A, Leeb T, Löscher W. mdr1a-Polymorphisms and their impact on pharmacoresistance in the amygdala-kindling model of epilepsy [abstract]. Washinton (DC): Society for Neuroscience,
Bates SF, Chen C, Robey R, Kang M, Figg WD, Fojo T. Reversal of multidrug resistance: lessons from clinical oncology.
Baulac M. Phenobarbital and other barbiturates: clinical efficacy and use in epilepsy. In: Levy RH, Mattson RH, Meldrum BS, Perucca E, editors. Antiepileptic drugs. 5th edn. Philadelphia: Lippincott Williams & Wilkins;
Brandt C, Glien M, Potschka H, Volk H, Löscher W. Epileptogenesis and neuropathology after different types of status epilepticus induced by prolonged electrical stimulation of the basolateral amygdala in rats.
Brandt C, Volk HA, Löscher W. Striking differences in individual anticonvulsant response to phenobarbital in rats with spontaneous seizures after status epilepticus.
Brinkmann U, Eichelbaum M. Polymorphisms in the ABC drug transporter gene MDR1.
Demeule M, Regina A, Jodoin J, Laplante A, Dagenais C, Berthelel F, et al. Drug transport to the brain: key roles for the efflux pump P-glycoprotein in the blood-brain barrier.
Dobrogowska DH, Vorbrodt AW. Quantitative immunocytochemical study of blood-brain barrier glucose transporter (GLUT-1) in four regions of mouse brain.
Eichelbaum M, Fromm MF, Schwab M. Clinical aspects of the MDR1 (ABCB1) gene polymorphism.
Farrell CL, Pardridge WM. Blood-brain barrier glucose transporter is asymmetrically distributed on brain capillary endothelial luminal and abluminal membranes: an electron microscopic immunogold study.
Fricker G, Miller DS. Modulation of drug transporters at the blood-brain barrier.
Gale K. Progression and generalization of seizure discharge: anatomical and neurochemical substrates.
Hellsten J, Wennstrom M, Bengzon J, Mohapel P, Tingstrom A. Electroconvulsive seizures induce endothelial cell proliferation in adult rat hippocampus.
Kemper EM, Boogerd W, Thuis I, Beijnen JH, van Tellingen O. Modulation of the blood-brain barrier in oncology: therapeutic opportunities for the treatment of brain tumours?
Lee G, Dallas S, Hong M, Bendayan R. Drug transporters in the central nervous system: brain barriers and brain parenchyma considerations.
Leite JP, Cavalheiro EA. Effects of conventional antiepileptic drugs in a model of spontaneous recurrent seizures in rats.
Leonard GD, Polgar O, Bates SE. ABC transporters and inhibitors: new targets, new agents.
Lowenstein DH. Recent advances related to basic mechanisms of epileptogenesis.
Löscher W. Animal models of drug-resistant epilepsy.
Löscher W, Ebert U. Basic mechanisms of seizure propagation: Targets for rational drug design and rational polypharmacy.
Löscher W, Hönack D. Comparison of the anticonvulsant efficacy of primidone and phenobarbital during chronic treatment of amygdala-kindled rats.
Löscher W, Potschka H. Role of multidrug transporters in pharmacoresistance to antiepileptic drugs.
Löscher W, Potschka H. Blood-brain barrier active efflux transporters: ATP-binding cassette (ABC) gene family.
Löscher W, Schmidt D. New horizons in the development of antiepileptic drugs: the search for new targets.
Marroni M, Marchi N, Cucullo L, Abbott NJ, Signorelli K, Janigro D. Vascular and parenchymal mechanisms in multiple drug resistance: a lesson from human epilepsy.
Nair DR, Mohamed A, Burgess R, Luders H. A critical review of the different conceptual hypotheses framing human focal epilepsy.
Pardridge WM, Boado RJ, Farrell CR. Brain-type glucose transporter (GLUT-1) is selectively localized to the blood-brain barrier. Studies with quantitative western blotting and in situ hybridization.
Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 4th edition.
Potschka H, Löscher W. A comparison of extracellular levels of phenytoin in amygdala and hippocampus of kindled and non-kindled rats.
Potschka H, Fedrowitz M, Löscher W. P-Glycoprotein-mediated efflux of phenobarbital, lamotrigine, and felbamate at the blood-brain barrier: evidence from microdialysis experiments in rats.
Potschka H, Volk HA, Löscher W. Pharmacoresistance and expression of multidrug transporter P-glycoprotein in kindled rats.
Racine RJ. Modification of seizure activity by electrical stimulation: II. Motor seizure.
Remy S, Gabriel S, Urban BW, Dietrich D, Lohmann TN, Elger CE, et al. A novel mechanism underlying drug resistance in chronic epilepsy.
Rizzi M, Caccia S, Guiso G, Richichi C, Gorter JA, Aronica E, et al. Limbic seizures induce P-glycoprotein in rodent brain: functional implications for pharmacoresistance.
Schinkel AH. P-Glycoprotein, a gatekeeper in the blood-brain barrier.
Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview.
Schuetz EG, Beck WT, Schuetz JD. Modulators and substrates of P-glycoprotein and cytochrome P4503A coordinately up-regulate these proteins in human colon carcinoma cells.
Seegers U, Potschka H, Löscher W. Transient increase of P-glycoprotein expression in endothelium and parenchyma of limbic brain regions in the kainate model of temporal lobe epilepsy.
Siddiqui A, Kerb R, Weale ME, Brinkmann U, Smith A, Goldstein DE, et al. Association of multidrug resistance in epilepsy with a polymorphism in the drug-transporter gene ABCB1.
Sisodiya SM, Lin WR, Harding BN, Squier MV, Thom M. Drug resistance in epilepsy: expression of drug resistance proteins in common causes of refractory epilepsy.
Sloviter RS. Status epilepticus-induced neuronal injury and network reorganization.
Summers MA, Moore JL, McAuley JW. Use of verapamil as a potential P-glycoprotein inhibitor in a patient with refractory epilepsy.
Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC. Immunohistochemical localization in normal tissues of different epitopes in the multidrug transport protein P170: evidence for localization in brain capillaries and crossreactivity of one antibody with a muscle protein.
Toth K, Vaughan MM, Slocum HK, Arredondo MA, Takita H, Baker RM et al. New immunohistochemical ‘sandwich’ staining method for mdr1 P-glycoprotein detection with JSB-1 monoclonal antibody in formalin-fixed, paraffin-embedded human tissues.
Volk HA, Potschka H, Löscher W. Increased expression of the multidrug transporter P-glycoprotein in limbic brain regions after amygdala-kindled seizures in rats.
Volk H, Potschka H, Löscher W. Immunohistochemical localization of P-glycoprotein in rat brain and detection of its increased expression by seizures are sensitive to fixation and staining variables. J Histochem Cytochem. In press


{kind=link}
{kind=link}
{kind=link}
{kind=link}