 Valerio Chiurchiù
Valerio Chiurchiù Alessandro Leuti1,2†
Alessandro Leuti1,2† Mauro Maccarrone
Mauro Maccarrone- 1Department of Medicine, Campus Bio-Medico University of Rome, Rome, Italy
- 2European Center for Brain Research (CERC), Santa Lucia Foundation (IRCCS), Rome, Italy
Inflammation is an immune response that works as a contained fire that is pre-emptively sparked as a defensive process during infections or upon any kind of tissue insult, and that is spontaneously extinguished after elimination or termination of the damage. However, persistent and uncontrolled immune reactions act as a wildfire that promote chronic inflammation, unresolved tissue damage and, eventually, chronic diseases. A wide network of soluble mediators, among which endogenous bioactive lipids, governs all immune processes. They are secreted by basically all cells involved in inflammatory processes and constitute the crucial infrastructure that triggers, coordinates and confines inflammatory mechanisms. However, these molecules are also deeply involved in the detrimental transition from acute to chronic inflammation, be it for persistent or excessive action of pro-inflammatory lipids or for the impairment of the functions carried out by resolving ones. As a matter of fact, bioactive lipids have been linked, to date, to several chronic diseases, including rheumatoid arthritis, atherosclerosis, diabetes, cancer, inflammatory bowel disease, systemic lupus erythematosus, and multiple sclerosis. This review summarizes current knowledge on the involvement of the main classes of endogenous bioactive lipids—namely classical eicosanoids, pro-resolving lipid mediators, lysoglycerophospholipids/sphingolipids, and endocannabinoids—in the cellular and molecular mechanisms that lead to the pathogenesis of chronic disorders.
Introduction
Inflammation represents one of the best known pathophysiological processes and represents a well-conserved mechanism evolved by vertebrates as an adaptive and defensive response to tissue injury and invasion of microorganisms that might attempt to colonize the host (1, 2). Despite the apparent simplicity of its definition, inflammation is instead a rather intricate network of cellular and molecular events, at the core of which, a plethora of pre-formed or newly synthesized mediators is elegantly arranged to obtain specific temporal and spatial responses. Endogenous lipids are arguably the most important mediators not only to be implicated in all phases of inflammation, but also to be involved in the regulation and fine-tuning of its course and cessation. Indeed, lipids are not just the major constituents of cell membranes and very efficient sources of energy, but also as key pathophysiological mediators of several intercellular and intracellular processes. Thus, during the past two decades, they have been termed “bioactive lipids,” due to their pivotal role in immune regulation, inflammation, and maintenance of tissue homeostasis (3, 4). Bioactive lipids, divided into four main families according to their biochemical functions, i.e., classical eicosanoids, specialized pro-resolving mediators (SPMs), lysoglycerophospholipids/sphingolipids and endocannabinoids (eCBs), are generated from ω-6 or ω-3 essential polyunsaturated fatty acids (PUFA) precursors, that are esterified into membrane lipids and act by binding to and activating specific G protein-coupled receptors (GPRs).
Bioactive Lipids and Inflammation
In the event of tissue insults or infections, innate immune cells, such as granulocytes and monocytes/macrophages, are recruited to the damaged site and rapidly generate classical eicosanoids, the class of lipid mediators that is responsible for acute inflammation (or angiophlogosis) characterized by the so-called “cardinal signs” of inflammation: redness, heat, swelling, pain, and loss of function (5). Classical eicosanoids are thus highly pro-inflammatory and ignite the fire during inflammation, with the aim of removing injurious stimuli, a fire that, however, needs to be self-limiting and, eventually, promptly extinguished upon cessation or elimination of the noxious stimulus. During the last process, referred to as “resolution of inflammation” or catabasis (i.e., Dante’s descent into the hell), the very same innate immune cells recruited in the inflammatory milieu, where they produce classical eicosanoids, undergo a temporal lipid mediator class switch and start producing another class of bioactive lipids, the newly discovered SPMs. These lipids actively terminate inflammation and drive the restoration of full tissue homeostasis by activating the signs of resolution: removal, relief, restoration, regeneration, and remission (6, 7). When the fire of inflammation is not properly extinguished, due to impaired resolution, it turns into chronic inflammation (or histophlogosis), resulting in aberrant tissue remodeling and organ dysfunction (8). In this context, the outcome of inflammation depends also on the other two families of bioactive lipids, i.e., lysoglycerophospholipids/sphingolipids and eCBs, which regulate numerous cellular processes that are important for triggering those mechanisms that underlie cell and tissue adaption to inflammatory events (9, 10). Indeed, chronic inflammation represents often the causative agent and the main trigger of the damage associated to many pathologies, such as cancer, autoimmune, metabolic, cardiovascular, and neurodegenerative diseases (11, 12). Thus, it seems that bioactive lipids are largely involved in managing the fire of inflammation, either acting as fire-starters or as fire-fighters, or even as executives of the fire station.
Classical Eicosanoids
These bioactive lipids represent probably the widest and most celebrated family, and include a huge array of molecules that have the ω-6 PUFA arachidonic acid (AA) as their common biosynthetic precursor released from membrane phospholipids by phospholipase A2 (13). AA is then used as a substrate for three different oxygen-incorporating enzymes that together synthesize over 120 heterogeneous and pleiotropic molecules: cyclooxygenases 1 and 2 (COX-1/2) drive the synthesis of prostaglandins (PGs), prostacyclins, and thromboxanes (TXs) (14–16), often referred together as prostanoids; 5-, 12-, and 15-lypooxygenases (5/12/15-LOX) produce leukotrienes (LTs) (17), hydroxyeicosatetraenoids (HETEs) (13), and lipoxins (LX) (6); P450 epoxygenase generates HETEs and epoxyeicosatrienoids (13). Even though all these bioactive lipids are involved in a plethora of physiological and homeostatic processes, including control of vascular tone, platelet aggregation, pain perception as well as ovulation, and embryo implantation (13, 18), they are mostly renowned for their ability to act as fire-starters and initiators of inflammation. Prostanoids, including PGs, such as PGD2, PGE2, PGI2, and PGF2α, represent, to date, a central subject of study among eicosanoids, especially in light of the ability of non-steroidal anti-inflammatory drugs (NSAIDs) to block their synthesis by covalent inhibition of COX-1/2 (19), which in turn results in the forestall of inflammation. The fact that NSAIDs are mostly used to treat acute inflammatory symptoms, such as swelling and pain, while being essentially ineffective on chronic conditions (for which steroidal drugs are preferred as treatment), has led to the idea that prostanoids are far less involved in chronic inflammatory pathologies (13). However, recent studies conducted using knockout mice for each specific GPR of the different classical eicosanoids (e.g., EP1–4, DP1–2, IP, FP, and TP), or specific stimulation by means of selective agonists, unveiled that their role might go well beyond the acute inflammatory response. Indeed, PG signaling—especially the one mediated by PGE2 and PGI2—seems to be involved in the sustained inflammation that causes the transition to chronic inflammation by acting as “cytokine amplifiers” (12, 20). These observations were based on animal and cellular models of chronic inflammatory diseases, such as arthritis (21) and cancer (22), where PGs are known to be involved also in their pathogenesis. In general, PGs induce chronic inflammation through five main mechanisms: (i) enhancement of the pro-inflammatory cytokines release cascade (21); (ii) amplification of innate immunity response to pathogen- and damage-associated molecular patterns (PAMPs and DAMPs) (23); (iii) activation of specific pro-inflammatory subsets of T helper cells, e.g., TH1 and TH17 (24, 25); (iv) recruitment of immune cells associated with chronic inflammation (e.g., macrophages, T and B cells) by synergistically acting with chemokines (12); (v) increase of pro-inflammatory genes induced by cytokines. Consistently, many studies have reported associations between specific PG-related genes (e.g., biosynthesizing enzymes or receptors) and the susceptibility to several chronic diseases, including Crohn’s disease (CD) (26), asthma (27), and multiple sclerosis (MS) (28, 29).
The main role of LTs in acute inflammation is to induce, alongside prostanoids, edema, and neutrophil influx within inflamed tissues (13). However, LTs are also central in perpetuation of inflammatory signals that lead to tissue damage in many chronic diseases. Indeed, LTs and their cysteinyl derivatives have been long known to be intimately connected to the pathogenesis of atherosclerosis, inflammatory bowel disease (IBD), psoriasis, rheumatoid arthritis (RA), as well as bronchial asthma, and MS, acting as chemoattractants for neutrophils, macrophages, eosinophils, and also TH17 lymphocytes (30–32), thus maintaining an ongoing and sustained inflammatory milieu. Of note, discussing the precise role of each LOX-derived eicosanoid in chronic inflammation is not an easy task, mainly due to their vast number (over 70 mediators), their differential action on cellular targets, and their complex and intermingled metabolic destiny. For instance, 5(S)-HpETE is the precursor of LTA4, which in turn is the common precursor of all bioactive leukotrienes (33), and of LX, which instead are anti-inflammatory inasmuch as are involved in the resolution of inflammation, as discussed in the next section.
Specialized Pro-Resolving Lipid Mediators
As mentioned above, at the peak of acute inflammation the very same cells involved in the production of pro-inflammatory lipid mediators undergo a class switch and start producing SPMs from ω-6 AA and even more from ω-3 PUFAs eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and docosapentaenoic acid (DPA), through the stereoselective and concerted action of the same enzymes engaged in classical eicosanoids production: COXs, LOXs, and P450. To date, more than 20 different SPMs have been identified via sophisticated lipidomic approaches in the laboratory of Prof. Serhan, and these can be generally subdivided into six main classes: AA-derived LXs (LXA4 and LXB4); EPA-derived E-series resolvins (RvE1–3); DHA-derived D-series resolvins (RvD1–6); protectins/neuroprotectins (PD1/NPD1 and PDX); and their sulfido-conjugates (PCTRs), maresins (MaR1 and MaR2); and their conjugates (MCTR1–3), as well as the latest class to be identified, namely the DPA-derived 13-series resolvins (RvT1–4) (6, 34, 35) (Figure 1 summarizes the details of their respective biochemical synthesis). The lipid class switch is initiated already in the early phases of inflammation by LXA4 and LXB4, produced by platelets that progressively aggregate at the sites of inflammation (36). Overall, SPMs act as “immunoresolvents,” that is immune-pharmacological agents of resolution, as opposed to immunosuppressive agents, and they induce cessation of further leukocyte infiltration, recruitment and stimulation of nonphlogistic mononuclear cells, promote killing and clearance of pathogens and macrophage-mediated phagocytosis of apoptotic granulocytes (efferocytosis) and cellular debris, inhibit proinflammatory cytokines while inducing the production of anti-inflammatory mediators, shorten the time of resolution and activation of endogenous resolution programs, and promote tissue regeneration and healing (6, 37). Their activity is mediated by five separate GPRs, namely the formyl peptide receptor 2 (FPR2, also known as ALX), GPR32 (or DRV1), chemerin receptor 23 ChemR23 (or ERV), leukotriene B4 receptor 1 (BLT1) and GPR18 (or DRV2), expressed in different cell tissues and with differential affinities for each SPM or other lipid mediators (38). The target receptors of most SPMs are yet to be identified and further studies will be required to characterize the signaling pathways underlying their functions.
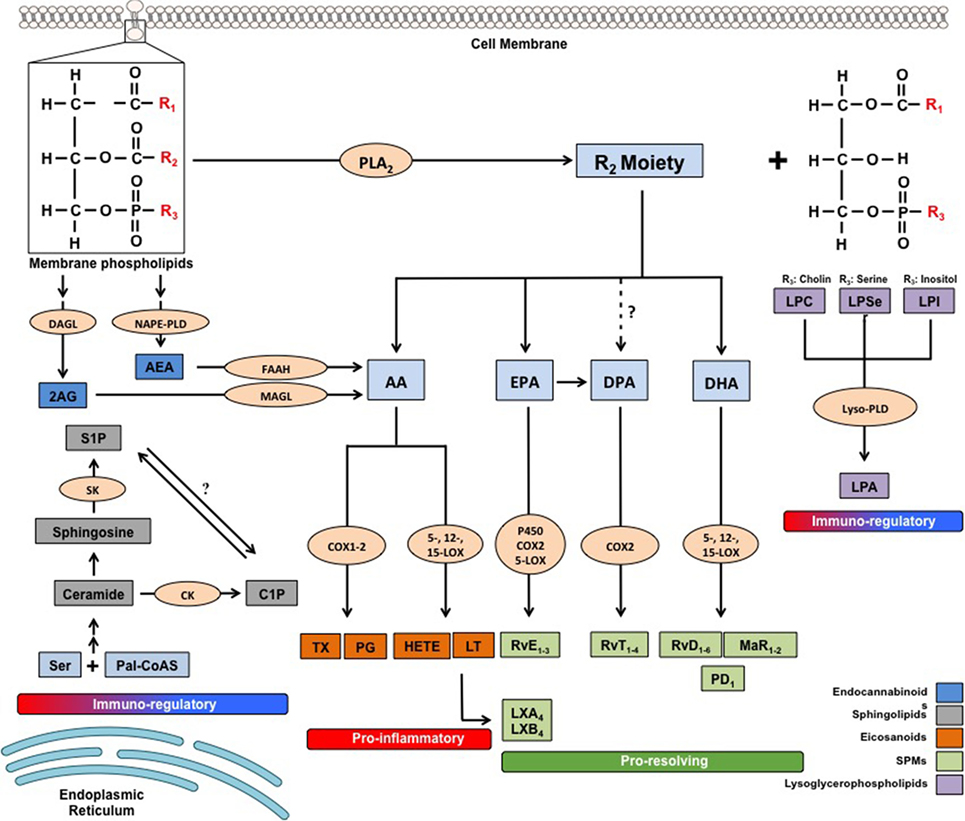
Figure 1. Metabolic pathways of the main families of endogenous bioactive lipids. 2-AG, 2 arachidonoylglycerol; AA, arachidonic acid; AEA, arachidonoylethanolamide; C1P, ceramide-1-phosphate; CK, ceramide kinase; COX, cyclooxygenase; DAGL, diacylglycerol lipase; DHA, docosahexaenoic acid; DPA, docosapentaenoic acid; EPA, eicosapentaenoic acid; FAAH, fatty acid amide hydrolase; HETEs, hydroxyeicosatetraenoic acids; LOX, lipoxygenase; LPA, lysophosphatidic acid; LPC, lysophosphatidilcholine; LPI, lysophosphatidylinositol; LPSer, lysophosphatidylserine; LTs, leukotrienes; LX, lipoxin; Lyso-PLD, lyso-phospholipase D; MAGL, monoacylglycerol lipase; MaR, maresin; NAPE-PLD, N-arachidonoylphosphatidylethanolamide-specific phospholipase D; Pal-CoASH, palmitoyl coenzyme A; PD, protectin; PDX, protectin DX; PGs, prostaglandins; PLA2, phospholipase A2; Rv, resolvin; S1P, sphingosine-1-phosphate; SK, sphingosine kinase; TXs, thromboxanes.
Although most of the insight gathered so far on SPMs concerns their role in modulating acute inflammation innate components, recent investigations have reported their ability to directly modulate adaptive immune cells, such as B and T lymphocytes, which are strongly involved in chronic detrimental inflammation. Indeed, although only RvD1 has been shown to act on B cells by inducing differentiation into plasma cells and promoting IgM and IgG antibody isotype switching (39) while inhibiting IgE production (40), a growing number of studies are now reporting direct or indirect effects of several SPMs on T cells. For instance, LXA4 and LXB4 both inhibited TNF-α secretion from activated human T cells (41), whereby the LXA4-induced effects were dependent on FPR2/ALX, which is expressed on T cells and their subsets (41–43). Additionally, RvE1, RvD1, and PD1 all have been shown to reduce the recruitment of CD4+ and CD8+ T cells (44–47), with the former SPM also limiting CD4-associated production of IFN-γ and IL-4 (45).
Of note, our group recently demonstrated that RvD1, RvD2, and MaR1 are able to hinder the production of pro-inflammatory cytokines in CD4 and CD8 T cells, as well as to inhibit de novo differentiation into TH1 and TH17, while promoting development of Treg cells without exerting any immunosuppressive and cytotoxic effect (43). Moreover, mice genetically unable to produce DHA displayed an increase in TH1/TH17 cells and a decrease in Treg cells (43), implying that SPMs impact on the balance between pathogenic and tolerogenic adaptive immune cells. Taken together, these findings support the view that SPMs may prevent chronicity of inflammation and/or autoimmunity and link resolution to adaptive immune cell responses. Interestingly, recent evidence indicates that pathologic conditions associated with altered SPM metabolism and function can contribute to chronicity and magnitude of persistent inflammatory conditions; as a result current research is centered on investigating the role of SPMs in chronic diseases in several mouse models and humans. Accordingly, decreased production of LX, E-, and D-series resolvins in the airways, as well as disruption of FPR2/ALX signaling have been linked to the pathogenesis of chronic obstructive pulmonary disease (COPD), and their restoration determined beneficial effects (48). Dysfunctional production of D-series resolvins and insufficient resolution has also been observed in mouse models or in human plasma samples of other typical chronic inflammatory and/or autoimmune diseases, such as (i) type-2 diabetes and obesity (49, 50); (ii) RA, in which low levels of RvD3 are associated with delayed resolution in mice and active disease in humans (51) and where RvD1 exerts protective actions on cartilage of murine model of inflammatory arthritis (52); (iii) atherosclerosis, in which evidence for impairment of resolution of vascular inflammation is governed by specific SPMs (53) and reduced RvD1 correlates with atherosclerotic plaque instability (30), and where RvD2, RvE1 and MaR1 have all been reported to have atheroprotective effects (54–57); and (iv) inflammatory bowel disease (58, 59). Accumulating evidence reveals that several neurodegenerative diseases characterized by chronic inflammation, such as MS, Alzheimer’s disease (AD) and amyotrophic lateral sclerosis (ALS), also seem to be associated to failure of activating pro-resolving mechanisms, and interventions with SPMs in vitro or in vivo exert neuroprotective properties. Indeed, a dysfunctional resolution pathway in SPMs and their receptors is present in post-mortem tissues of AD patients (60, 61) and several SPMs promoted neuronal survival and β-amyloid uptake by microglia in vitro (61–64). Additionally, RvD1 strongly inhibited cytokine release from inflammatory macrophages in ALS spinal cord (65), and its daily administration in mouse models of MS decreased disease progression by suppressing autoreactive T cells and by inducing an M2 phenotype of monocytes/macrophages and resident brain microglial cells (66). These findings advocate the stimulation of resolution pathways as a new therapeutic strategy to prevent chronic inflammation. Topical formulation of analogs of resolvin E1 (RX-10001, RX-10008, and RX-10045) or neuroprotectin D1 (RX-20001), resistant to metabolic inactivation, are currently underway in a number of human clinical trials for several chronic conditions, such as dry eye, macular degeneration and diabetic retinopathy, as well as lung, gut and kidney inflammation (67).
Lysophospholipids and Sphingolipids
These bioactive lipids comprise many compounds asymmetrically distributed in plasma membranes with glycerol or sphingosine as respective backbones, and are characterized by a great molecular diversity due to their linkage with other molecules, such as ethanolamine, choline, inositol, serine or and fatty acids (e.g., phosphoinositides, lysoglycerophospholipids, and ceramides). The detailed metabolic steps of these substances are illustrated in Figure 1. Biochemical interconversions between them and also into other classes of bioactive lipids, such as eicosanoids and eCBs, are also possible, thanks to the action of phospholipases, lipid kinases and lipid-phosphate phosphatases (68, 69). The most biologically active lysophospholipids, derived from membrane phospholipids by removal of one or both fatty acids, are lysophosphaditylcholine (LPC) and lysophosphatidilinositol (LPI), and their byproduct lysophosphatidic acid (LPA), which are signaling molecules involved in pivotal aspects of cellular and tissue biology, such as plasma membrane shaping (70), cell growth and death (71), and inflammatory cascades (72). LPC and LPA modulate immune responses mostly by controlling distribution, trafficking and activation of immune cells (72–75), and their sustained activation have been suggested to be linked with several chronic inflammatory diseases, including obesity and diabetes (76, 77), cancer (74), atherosclerosis (78) and RA (79, 80).
On the other hand, the main active sphingolipids, whose peculiar chemical structure has baffled scientists for a long time (hence their name, inspired by the Egyptian Sphynx) (81), are ceramide and their byproducts ceramide 1-phosphate (C1P) and sphingosine 1-phosphate (S1P), shown in Figure 1 (69). Sphingolipids participate in numerous inflammatory processes and are responsible for controlling intracellular trafficking and signaling, cell growth, adhesion, vascularization, survival, and apoptosis (9, 68, 82), even though specific receptors have only been identified for S1P. The role of these three sphingolipids in chronic inflammation has been extensively investigated in the past decade, and have been mostly associated with immune-dependent and vascular-related chronic inflammatory diseases, including diabetes and obesity, COPD, IBD and neuroinflammatory disorders. For instance, an excessive ceramide signaling determines adipose tissue inflammation and insulin resistance, leading to obesity and type-2 diabetes, by inducing overactive immune cells like macrophages and B cells (81–83). Of note, most of the pro-inflammatory activities of ceramide seem to be mediated through its C1P and S1P metabolites. The former enhances both acute and chronic inflammatory responses by promoting phospholipase A2-mediated eicosanoid storm and by inducing cytokine production (84, 85). However, ceramide and C1P have also been shown to negatively regulate some pro-inflammatory cytokines (86, 87), suggesting a more complex role for them in inflammation. Furthermore, C1P has also been reported to impact on insulin resistance-induced type-2 diabetes and metabolic syndrome (82), as well as to induce cell migration in several cellular models of monocytes/macrophages and endothelial cells, as reviewed in Ref. (82), implying that this bioactive lipid might be involved in chronic inflammatory diseases characterized by migration of immune cells to inappropriate sites, such as IBD, atherosclerosis, and MS. Ceramide and its metabolites are also involved in the physiological regulation of endothelial/vascular integrity and function, whereby alterations of these sphingolipids are associated with vascular dysfunctions, and thus with chronic inflammatory states (88, 89).
Sphingosine-1-phosphate is arguably the best-studied molecule of this family of bioactive lipids and its actions are mediated by five identified receptors (S1PR1–5) (90). This lipid is a key mediator for lymphocyte trafficking between lymphoid and non-lymphoid tissues, favoring the egress of effector T and B cells from lymph nodes, thymus, bone marrow, and spleen, and blocking the ability of immature dendritic cells to migrate (91, 92). This function of S1P is particularly important, since T and B cells are the fire-starters of many (if not all) chronic inflammatory conditions and autoimmune diseases, in which modulation of their function is often exploited to develop new therapeutic strategies. Accordingly, the commercially available oral drug Fingolimod was developed as a first-line disease-modifying treatment for MS due to its ability to downregulate S1PR1, and hence to sequester highly pathogenic T cells (i.e., Th1 and Th17 cells) within the lymph nodes, avoiding brain invasion and myelin damage (93, 94). Fingolimod has also been shown to reduce blood–brain barrier dysfunction, a renowned pathogenetic mechanism of MS, by attenuating the production of sphingolipids from reactive astrocytes, including ceramide (95), also because several S1P receptors are significantly upregulated in MS lesions (96, 97). Interestingly, high S1P levels have also been found in patients with IBD and asthma (81, 98) and, accordingly, these conditions were attenuated by genetic deletion of the enzyme responsible for its synthesis in rodent models of disease or by pharmacological modulation of the S1P–S1PR axis (98–100).
Endocannabinoids
Endocannabinoids include a group of bioactive lipids endogenously produced by humans and animals that are able (although with different affinities) to bind to and activate the same receptors as the main psychoactive component of marijuana Δ9-tetrahydrocannabinol, i.e., type-1 and type-2 cannabinoid receptors (CB1 and CB2). Arachidonoylethanolamide (commonly known as anandamide, AEA) and 2-arachidonoylglycerol (2-AG), both identified in the early 1990s, are the two best studied members of the eCB family, which also comprise 2-AG-ether, O-arachidonoylethanolamine, and palmitoylethanolamide (PEA) (10, 101). These molecules are ubiquitously produced by most tissues and immune cells, which are fully capable to metabolize them via a set of specific synthesizing [N-acyl-phosphatidylethanolamine-hydrolyzing phospholipase D (NAPE-PLD) for AEA and its congeners and diacylglycerol lipase (DAGL) for 2-AG] and degrading [fatty acid amide hydrolase FAAH for AEA and monoacylglycerol lipase (MAGL) for 2-AG] enzymes (101). Besides the aforementioned CB1 and CB2 receptors, eCBs also engage other molecular targets that include members of the transient receptor potential (TRP) channels, GPR55, and peroxisome proliferator-activated receptors (PPARs), differentially expressed by body districts also according to their inflammatory state (102, 103). Altogether, eCBs and their enzymes and receptors constitute the so-called “eCB system,” which generally serves as a homeostatic system that controls several physiopathological states ultimately maintaining human health (104). In particular, eCBs are arguably among the most potent immunoregulatory compounds, capable of regulating the functions of several cell subsets of either innate or adaptive immunity (in particular monocytes/macrophages, dendritic cells, granulocytes, and T lymphocytes), with AEA and PEA being mostly anti-inflammatory (105–107) and 2-AG both pro- and anti-inflammatory (10, 108–111). Indeed, due to their role in the overall control of tissue homeostasis, variations in the tone of distinct eCBs within tissues, or in the expression of their metabolic enzymes or receptors have been clearly recognized as central in the pathophysiology of many chronic inflammatory diseases. Indeed, it is now clear that perturbations in all members of the eCB system occur during every chronic inflammatory process, from cancer, metabolic, and gastrointestinal diseases to autoimmune and neuroinflammatory disorders [extensively reviewed in Ref. (112–119)]. This is because every single cell and tissue of our body produces specific eCBs (sometimes also simultaneously) “on demand” and at certain concentrations according to the stimulus and/or the need, in order to autocrinally or paracrinally orchestrate the inflammatory responses of nearby cells through complex interactions between multiple receptors or targets with different but partly overlapping activities (104). Recently, eCBs are also emerging as pro-resolving agents due to the ability of AEA and 2-AG, alongside other congeners (e.g., PEA) to boost resolution programs during neuroinflammation (120, 121), while 2-AG alone can enhance phagocytosis in human macrophages (122). Accordingly, several experimental models of chronic inflammatory diseases have been instrumental, not only to better understand the role of each member of the eCB system in their different pathogenic mechanisms, but mostly to identify in the pharmacological manipulation of either receptors or enzymes (by means of selective activation of specific receptors or inhibition of AEA, 2-AG, or PEA degrading enzymes) a promising therapeutic strategy. In line with this, modulation of the eCB system has been shown to be beneficial by attenuating inflammatory processes that include cytokine release, infiltration of leukocytes at inflamed sites, production of reactive oxygen and nitrogen species, and overall immune cell activation (123–127). These effects were particularly relevant in several neuroinflammatory and neurodegenerative diseases, such as MS and AD (119, 128–131), where chronic inflammation is indeed a hallmark and whereby targeting the eCB system seems to be a promising therapeutic approach in the near future. Interestingly, AEA and 2-AG can also be metabolized by COX-2, LOXs, and P450 into eicosanoid-like PG-ethanolamides and glyceryl esters, hydroxy-anandamides, and hydroxyeicosatetraenoyl-glycerols, respectively (132, 133). The function and biological activity of these lipids is still unclear, but it is plausible that they might play a role in chronic inflammation.
Do Bioactive Lipids Coexist during the Different Stages of Inflammation?
The array of bioactive lipids that lays at the heart of tissue immune homeostasis represents a vastly intertwined network of molecules whose metabolism is rather complex, in that not only they undergo fast biosynthesis, degradation or interconversion, but also they share common metabolic enzymes, the activation or regulation of which is fascinating and still not completely unraveled. Consequently, the full elucidation of their temporal production and their role in the different phases or inflammation (from acute inflammation and its resolution to chronic inflammation) represents conceivably one of the biggest challenges of our time. To date, this has been particularly investigated for eicosanoids and SPMs in terms of their detailed temporal and spatial production and their specific role during the different inflammatory states and this is mainly due to their thorough characterization by means of lipidomics analyses performed locally in inflammatory or self-resolving tissues (i.e., edema). Indeed, both eicosanoids and SPMs are present in all phases of inflammation: the former are massively produced within the first 2–4 h and then show a reduction during the resolution phase, whereas the latter appear already at early phases of inflammation (especially LX and RvTs), usually reach their highest level at the peak of acute inflammation (6–12 h) and some specific molecules (i.e., RvD3) are produced at later stages. During chronic inflammation, as previously described, both families of bioactive lipids are present, with specific molecules being overly or inadequately produced, according to the different inflammatory diseases and tissues. Such temporal and spatial production, although less studied, is beginning to hold true also for the other families of bioactive lipids. Indeed, eCB levels raise rapidly following noxious stimuli and this is generally associated with their role in activating anti-inflammatory and protective mechanisms, although persistent inflammation usually dysregulates the eCB system in a way that their action might even become detrimental (134). On the other hand, other authors have reported diminished levels of eCBs in chronic inflammatory models, even after several days after the induction of inflammation (135). Of note, the temporal production of eCBs can only be inferred from in vitro studies conducted on cell lines activated with different inflammatory stimuli at different times or in tissues of patients or animal models of acute or chronic inflammatory diseases. To date, a fully detailed temporal characterization of each eCB during the different stages of inflammation, namely from an acute model of inflammation to a bona fide model of spontaneous resolution, is still absent and represents a future challenge. This scenario is further complicated by the fact that eCBs exist in dynamic equilibria with different other lipid-derived mediators, including eicosanoids, prostamides, and their recently identified ω-3 congeners (136).
Also sphingolipids are found during different stages of inflammation, and are likely part of the resolution machinery as suggested by the fact that apoptotic cells at the inflammation sites attract pro-resolving macrophages in a S1P-S1PR1-dependent manner (137), while neutrophil apoptosis, which is pivotal in initiating resolution, rely at least in part on the generation of ceramide (138). Furthermore, LPA might represent another brick in the resolution wall, in that it has been recently reported to be rapidly produced during the resolution phase of tissue inflammation and to recruit monocytes via the common pro-resolving receptor ALX/FPR2 (139). Interestingly, treatment of human fibroblasts (key cells involved in tissue healing and regeneration) with TNF-α, a cytokine that is mainly produced during acute or chronic inflammation, results in a significant increase in S1P levels, which rapidly returns to baseline within less than an hour (140), and of COX-2 expression, which can, in turn, temporally generate both eicosanoids, SPMs or even eCBs metabolites.
All these evidences not only account for a coexistence of several families of bioactive lipids during the different stages of inflammation, but also suggest that each inflammatory phase requires the concerted action of such lipid mediators, which are also likely to molecularly interact and engage in physiopathological cross talks.
Concluding Remarks
For a long time, the idea that lipids were mere constituents of cellular membranes and efficient energy sources was indisputable, but over the past two decades, not only it became clear that they actually harbor many functions in the regulation of intercellular and intracellular signaling pathways, but also that they represent bioactive molecules that are able to orchestrate a plethora of biological activities on their own, in order to maintain tissue homeostasis by governing body’s defensive and healing processes like inflammation and its resolution. During these processes, several families of bioactive lipids are temporally and spatially engaged so that the appropriate leukocytes are recruited and the noxious agent or stimulus is eliminated. Accordingly, classical eicosanoids are the fire-starters of the inflammatory processes and engage mainly cells of the innate arm of immunity that execute all possible strategies to quickly eradicate the injury. If the danger ceases or is successfully terminated, the fire of inflammation is elegantly extinguished by SPMs that recruit non-phlogistic innate immune cells and activate resolution pathways, aimed at healing the damaged tissue. On the contrary, if the injurious stimulus is either persistent or not eliminated, perhaps by failure of resolving inflammation, a wildfire of long-lasting inflammatory processes occurs and the flame of inflammation is kept alive mainly by cells of the adaptive arm of immunity, thus leading to many chronic inflammatory diseases. Under these circumstances, tissues activate several adaptation mechanisms that allow cells to cope with the changes induced by the damage, also thanks to other families of bioactive lipids like lysophospholipids, sphingolipids, and eCBs, that ultimately regulate cell growth, differentiation, and destiny with the goal of helping the body to restore homeostatic balance. Most of these bioactive lipids and several elements of their complex metabolism and signaling (i.e., enzymes and receptors) are differentially dysregulated in many chronic inflammatory diseases (Table 1), suggesting that managing the fire within by targeting the endogenous mechanisms involved in the spontaneous fire extinction, or in the modulation of homeostatic processes, rather than simply suppressing inflammation, could be a future and promising therapeutic strategy to be undertaken.
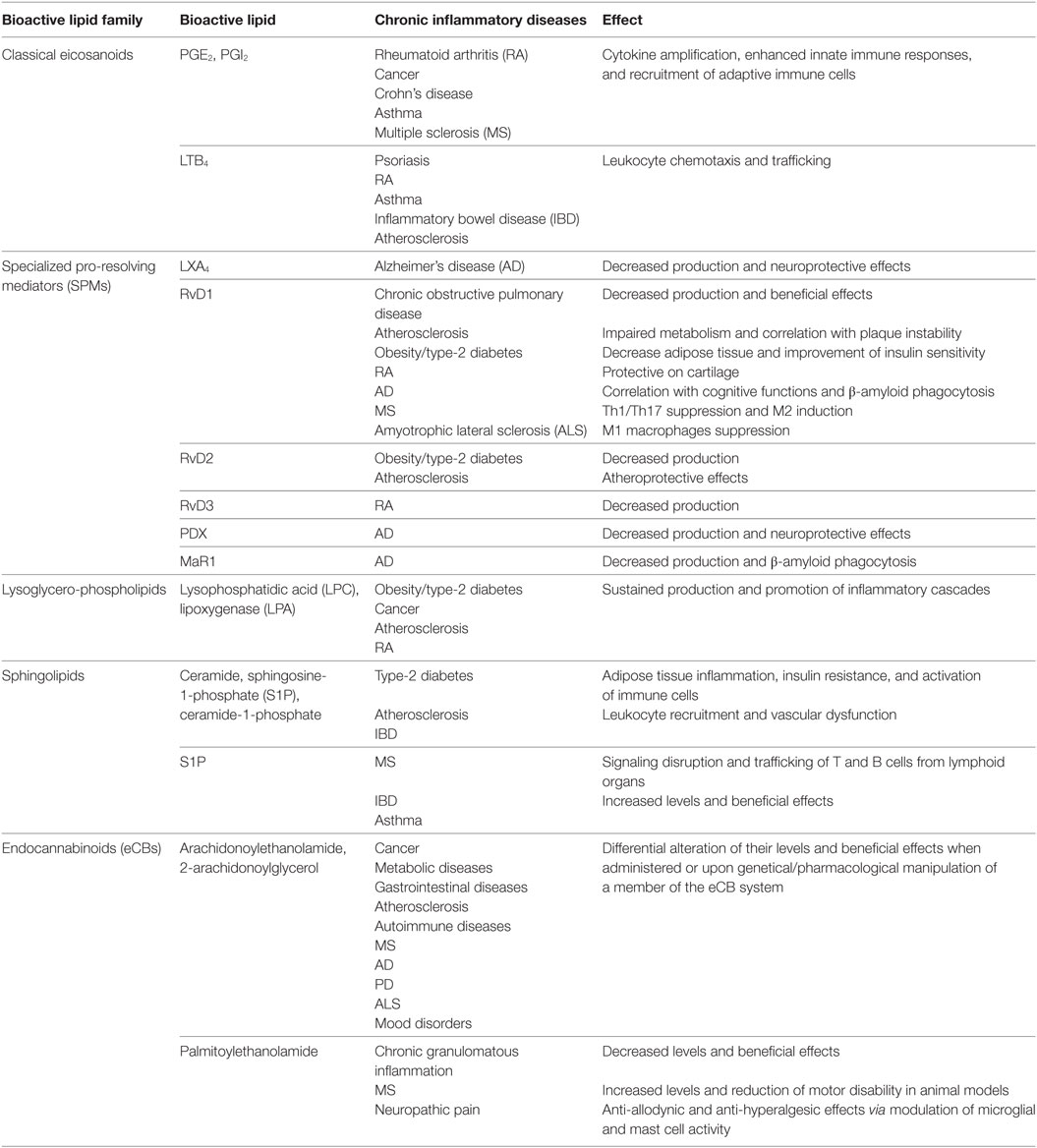
Table 1. Main role of bioactive lipids in chronic inflammatory diseases.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Fondazione Italiana Sclerosi Multipla (competitive FISM grant 2015/R/08 to VC) and by European Union-health EULAC (competitive EULACH16/TO 1032 grant to MM).
References
1. Majno G, Joris I. Cells, Tissues, and Disease. Principles of General Pathology. II ed. Oxford: Oxford University Press (2004).
2. Kumar V, Abbas A, Aster J. Robbins & Cotran Pathologic Basis of Disease. IX ed. Amsterdam: Elsevier (2014).
3. Shimizu T. Lipid mediators in health and disease: enzymes and receptors as therapeutic targets for the regulation of immunity and inflammation. Annu Rev Pharmacol Toxicol (2009) 49:123–50. doi:10.1146/annurev.pharmtox.011008.145616
4. Chiurchiù V, Maccarrone M. Bioactive lipids as modulators of immunity, inflammation and emotions. Curr Opin Pharmacol (2016) 29:54–62. doi:10.1016/j.coph.2016.06.005
6. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature (2014) 510:92–101. doi:10.1038/nature13479
7. Basil MC, Levy BD. Specialized pro-resolving mediators: endogenous regulators of infection and inflammation. Nat Rev Immunol (2016) 16:51–67. doi:10.1038/nri.2015.4
8. Nathan C, Ding A. Nonresolving inflammation. Cell (2010) 140:871–82. doi:10.1016/j.cell.2010.02.029
9. El Alwani M, Wu BX, Obeid LM, Hannun YA. Bioactive sphingolipids in the modulation of the inflammatory response. Pharmacol Ther (2006) 112:171–83. doi:10.1016/j.pharmthera.2006.04.004
10. Chiurchiù V, Battistini L, Maccarrone M. Endocannabinoid signalling in innate and adaptive immunity. Immunology (2015) 144:352–64. doi:10.1111/imm.12441
11. Fullerton JN, Gilroy DW. Resolution of inflammation: a new therapeutic frontier. Nat Rev Drug Discov (2016) 15:551–67. doi:10.1038/nrd.2016.39
12. Aoki T, Narumiya S. Prostaglandins and chronic inflammation. Trends Pharmacol Sci (2012) 33:304–11. doi:10.1016/j.tips.2012.02.004
13. Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol (2015) 15:511–23. doi:10.1038/nri3859
14. Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem (2000) 69:145–82. doi:10.1146/annurev.biochem.69.1.145
15. Stitham J, Midgett C, Martin KA, Hwa J. Prostacyclin: an inflammatory paradox. Front Pharmacol (2011) 11:517–28. doi:10.3389/fphar.2011.00024
16. Kihara Y, Gupta S, Maurya MR, Armando A, Shah I, Quehenberger O, et al. Modeling of eicosanoid fluxes reveals functional coupling between cyclooxygenases and terminal synthases. Biophys J (2014) 106:966–75. doi:10.1016/j.bpj.2014.01.015
17. Mandal AK, Jones PB, Bair AM, Christmas P, Miller D, Ting-ting DY, et al. The nuclear membrane organization of leukotriene synthesis. Proc Natl Acad Sci U S A (2008) 105:20434–9. doi:10.1073/pnas.0808211106
18. Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol (2011) 31:986–1000. doi:10.1161/ATVBAHA.110.207449
19. Vane JR. Biomedicine. Back to an aspirin a day? Science (2002) 296:474–5. doi:10.1126/science.1071702
20. Narumiya S, Furuyashiki T. Fever, inflammation, pain and beyond: prostanoid receptor research during these 25 years. FASEB J (2011) 25:813–8. doi:10.1096/fj.11-0302ufm
21. Honda T, Segi-Nishida E, Miyachi Y, Narumiya S. Prostacyclin-IP signaling and prostaglandin E2-EP2/EP4 signaling both mediate joint inflammation in mouse collagen-induced arthritis. J Exp Med (2006) 203:325–35. doi:10.1084/jem.20051310
22. Sonoshita M, Takaku K, Sasaki N, Sugimoto Y, Ushikubi F, Narumiya S, et al. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc(Delta 716) knockout mice. Nat Med (2001) 7:1048–51. doi:10.1038/nm0901-1048
23. Hirata T, Narumiya S. Prostanoids as regulators of innate and adaptive immunity. Adv Immunol (2012) 116:143–74. doi:10.1016/B978-0-12-394300-2.00005-3
24. Yao C, Sakata D, Esaki Y, Li Y, Matsuoka T, Kuroiwa K, et al. Prostaglandin E2–EP4 signaling promotes immune inflammation through TH1 cell differentiation and TH17 cell expansion. Nat Med (2009) 15:633–40. doi:10.1038/nm.1968
25. Chen Q, Muramoto K, Masaaki N, Ding Y, Yang H, Mackey M, et al. A novel antagonist of the prostaglandin E2 EP4 receptor inhibits Th1 differentiation and Th17 expansion and is orally active in arthritis models. Br J Pharmacol (2010) 160:292–310. doi:10.1111/j.1476-5381.2010.00647
26. Libioulle C, Louis E, Hansoul S, Sandor C, Farnir F, Franchimont D, et al. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genet (2007) 3:e58. doi:10.1371/journal.pgen.0030058
27. Oguma T, Palmer LJ, Birben E, Sonna LA, Asano K, Lilly CM. Role of prostanoid DP receptor variants in susceptibility to asthma. N Engl J Med (2004) 351:1752–63. doi:10.1056/NEJMoa031785
28. Tajouri L, Fernandez F, Griffiths LR. Gene expression studies in multiple sclerosis. Curr Genomics (2007) 8:181–9. doi:10.2174/138920207780833829
29. Sawcer S, Hellenthal G, Pirinen M, Spencer CCA, Patsopoulos NA, Moutsianas L, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature (2011) 476:214–9. doi:10.1038/nature10251
30. Fredman G, Hellmann J, Proto JD, Kuriakose G, Colas RA, Dorweiler B, et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun (2016) 7:12859. doi:10.1038/ncomms12859
31. De Caterina R, Zampolli A. From asthma to atherosclerosis – 5-lipoxygenase, leukotrienes, and inflammation. N Engl J Med (2004) 350:4–7. doi:10.1056/NEJMp038190
32. Lee W, Su Kim H, Lee GR. Leukotrienes induce the migration of Th17 cells. Immunol Cell Biol (2015) 93:472–9. doi:10.1038/icb.2014.104
33. Powell WS, Rokach J. Biosynthesis, biological effects, and receptors of hydroxyeicosatetraenoic acids (HETEs) and oxoeicosatetraenoic acids (oxo-ETEs) derived from arachidonic acid. Biochim Biophys Acta (2015) 1851:340–55. doi:10.1016/j.bbalip.2014.10.008
34. Dalli J, Chiang N, Serhan CN. Elucidation of novel 13-series resolvins that increase with atorvastatin and clear infections. Nat Med (2015) 21:1071–5. doi:10.1038/nm.3911
35. Serhan CN, Chiang N, Dalli J. New pro-resolving n-3 mediators bridge resolution of infectious inflammation to tissue regeneration. Mol Aspects Med (2017). doi:10.1016/j.mam.2017.08.002
36. Chiang N, Arita M, Serhan CN. Anti-inflammatory circuitry: lipoxin, aspirin-triggered lipoxins and their receptor ALX. Prostaglandins Leukot Essent Fatty Acids (2005) 73:163–77. doi:10.1016/j.plefa.2005.05.003
37. Buckley C, Gilroy D, Serhan C. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity (2014) 40:315–27. doi:10.1016/j.immuni.2014.02.009
38. Chiang N, Serhan CN. Structural elucidation and physiologic functions of specialized pro-resolving mediators and their receptors. Mol Aspects Med (2017). doi:10.1016/j.mam.2017.03.005
39. Ramon S, Gao F, Serhan CN, Phipps RP. Specialized proresolving mediators enhance human B cell differentiation to antibody-secreting cells. J Immunol (2012) 189:1036–42. doi:10.4049/jimmunol.1103483
40. Kim N, Ramon S, Thatcher TH, Woeller CF, Sime PJ, Phipps RP. Specialized proresolving mediators (SPMs) inhibit human B-cell IgE production. Eur J Immunol (2016) 46:81–91. doi:10.1002/eji.201545673
41. Ariel A, Chiang N, Arita M, Petasis NA, Serhan CN. Aspirin-triggered lipoxin A4 and B4 analogs block extracellular signal-regulated kinase-dependent TNF-alpha secretion from human T cells. J Immunol (2003) 170:6266–72. doi:10.4049/jimmunol.170.12.6266
42. Spurr L, Nadkarni S, Pederzoli-Ribeil M, Goulding NJ, Perretti M, D’Acquisto F. Comparative analysis of annexin A1-formyl peptide receptor 2/ALX expression in human leukocyte subsets. Int Immunopharmacol (2011) 11:55–66. doi:10.1016/j.intimp.2010.10.006
43. Chiurchiù V, Leuti A, Dalli J, Jacobsson A, Battistini L, Maccarrone M, et al. Proresolving lipid mediators resolvin D1, resolvin D2, and maresin 1 are critical in modulating T cell responses. Sci Transl Med (2016) 8:353ra111. doi:10.1126/scitranslmed.aaf7483
44. Settimio R, Clara DF, Franca F, Francesca S, Michele D. Resolvin D1 reduces the immunoinflammatory response of the rat eye following uveitis. Mediators Inflamm (2012) 2012:318621. doi:10.1155/2012/318621
45. Kim T-H, Kim G-D, Jin Y-H, Park YS, Park C-S. Omega-3 fatty acid-derived mediator, resolvin E1, ameliorates 2,4-dinitrofluorobenzene-induced atopic dermatitis in NC/Nga mice. Int Immunopharmacol (2012) 14:384–91. doi:10.1016/j.intimp.2012.08.005
46. Ariel A, Li P-L, Wang W, Tang W-X, Fredman G, Hong S, et al. The docosatriene protectin D1 is produced by TH2 skewing and promotes human T cell apoptosis via lipid raft clustering. J Biol Chem (2005) 280:43079–86. doi:10.1074/jbc.M509796200
47. Levy BD, Kohli P, Gotlinger K, Haworth O, Hong S, Kazani S, et al. Protectin D1 is generated in asthma and dampens airway inflammation and hyperresponsiveness. J Immunol (2007) 178:496–502. doi:10.4049/jimmunol.178.1.496
48. Hsiao H-M, Thatcher TH, Colas RA, Serhan CN, Phipps RP, Sime PJ. Resolvin D1 reduces emphysema and chronic inflammation. Am J Pathol (2015) 185:3189–201. doi:10.1016/j.ajpath.2015.08.008
49. Hellmann J, Tang Y, Kosuri M, Bhatnagar A, Spite M. Resolvin D1 decreases adipose tissue macrophage accumulation and improves insulin sensitivity in obese-diabetic mice. FASEB J (2011) 25:2399–407. doi:10.1096/fj.10-178657
50. Clària J, Dalli J, Yacoubian S, Gao F, Serhan CN. Resolvin D1 and resolvin D2 govern local inflammatory tone in obese fat. J Immunol (2012) 189:2597–605. doi:10.4049/jimmunol.1201272
51. Perretti M, Norling LV. Actions of SPM in regulating host responses in arthritis. Mol Aspects Med (2017) 58:57–64. doi:10.1016/j.mam.2017.04.005
52. Norling LV, Headland SE, Dalli J, Arnardottir HH, Haworth O, Jones HR, et al. Proresolving and cartilage-protective actions of resolvin D1 in inflammatory arthritis. JCI Insight (2016) 1:e85922. doi:10.1172/jci.insight.85922
53. Merched AJ, Ko K, Gotlinger KH, Serhan CN, Chan L. Atherosclerosis: evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J (2008) 22:3595–606. doi:10.1096/fj.08-112201
54. Ho KJ, Spite M, Owens CD, Lancero H, Kroemer AHK, Pande R, et al. Aspirin-triggered lipoxin and resolvin E1 modulate vascular smooth muscle phenotype and correlate with peripheral atherosclerosis. Am J Pathol (2010) 177:2116–23. doi:10.2353/ajpath.2010.091082
55. Hasturk H, Abdallah R, Kantarci A, Nguyen D, Giordano N, Hamilton J, et al. Resolvin E1 (RvE1) attenuates atherosclerotic plaque formation in diet and inflammation-induced atherogenesis. Arterioscler Thromb Vasc Biol (2015) 35:1123–33. doi:10.1161/ATVBAHA.115.305324
56. Salic K, Morrison MC, Verschuren L, Wielinga PY, Wu L, Kleemann R, et al. Resolvin E1 attenuates atherosclerosis in absence of cholesterol-lowering effects and on top of atorvastatin. Atherosclerosis (2016) 250:158–65. doi:10.1016/j.atherosclerosis.2016.05.001
57. Viola JR, Lemnitzer P, Jansen Y, Csaba G, Winter C, Neideck C, et al. Resolving lipid mediators maresin 1 and resolvin D2 prevent atheroprogression in mice. Circ Res (2016) 119:1030–8. doi:10.1161/CIRCRESAHA.116.309492
58. Bento AF, Claudino RF, Dutra RC, Marcon R, Calixto JB. Omega-3 fatty acid-derived mediators 17(R)-hydroxy docosahexaenoic acid, aspirin-triggered resolvin D1 and resolvin D2 prevent experimental colitis in mice. J Immunol (2011) 187:1957–69. doi:10.4049/jimmunol.1101305
59. Schwanke RC, Marcon R, Bento AF, Calixto JB. EPA- and DHA-derived resolvins’ actions in inflammatory bowel disease. Eur J Pharmacol (2016) 785:156–64. doi:10.1016/j.ejphar.2015.08.050
60. Wang X, Zhu M, Hjorth E, Cortés-Toro V, Eyjolfsdottir H, Graff C, et al. Resolution of inflammation is altered in Alzheimer’s disease. Alzheimers Dement (2015) 11:40.e–50.e. doi:10.1016/j.jalz.2013.12.024
61. Zhu M, Wang X, Hjorth E, Colas RA, Schroeder L, Granholm A-C, et al. Pro-resolving lipid mediators improve neuronal survival and increase Aβ42 phagocytosis. Mol Neurobiol (2016) 53:2733–49. doi:10.1007/s12035-015-9544-0
62. Mizwicki MT, Liu G, Fiala M, Magpantay L, Sayre J, Siani A, et al. 1α,25-Dihydroxyvitamin D3 and resolvin D1 retune the balance between amyloid-β phagocytosis and inflammation in Alzheimer’s disease patients. J Alzheimers Dis (2013) 34:155–70. doi:10.3233/JAD-121735
63. Fiala M, Terrando N, Dalli J. Specialized pro-resolving mediators from omega-3 fatty acids improve amyloid-β phagocytosis and regulate inflammation in patients with minor cognitive impairment. J Alzheimers Dis (2015) 48:293–301. doi:10.3233/JAD-150367
64. Fiala M, Halder RC, Sagong B, Ross O, Sayre J, Porter V, et al. ω-3 supplementation increases amyloid-β phagocytosis and resolvin D1 in patients with minor cognitive impairment. FASEB J (2015) 29:2681–9. doi:10.1096/fj.14-264218
65. Liu G, Fiala M, Mizwicki MT, Sayre J, Magpantay L, Siani A, et al. Neuronal phagocytosis by inflammatory macrophages in ALS spinal cord: inhibition of inflammation by resolvin D1. Am J Neurodegener Dis (2012) 1:60–74.
66. Poisson LM, Suhail H, Singh J, Datta I, Denic A, Labuzek K, et al. Untargeted plasma metabolomics identifies endogenous metabolite with drug-like properties in chronic animal model of multiple sclerosis. J Biol Chem (2015) 290:30697–712. doi:10.1074/jbc.M115.679068
67. Serhan CN. Discovery of specialized pro-resolving mediators marks the dawn of resolution physiology and pharmacology. Mol Aspects Med (2017) 2997(17):30018–30013. doi:10.1016/j.mam.2017.03.001
68. Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol (2008) 9:139–50. doi:10.1038/nrm2329
69. Bartke N, Hannun YA. Bioactive sphingolipids: metabolism and function. J Lipid Res (2009) 50:S91–6. doi:10.1194/jlr.R800080-JLR200
70. Shindou H, Hishikawa D, Harayama T, Eto M, Shimizu T. Generation of membrane diversity by lysophospholipid acyltransferases. J Biochem (2013) 154:21–8. doi:10.1093/jb/mvt048
71. Makide K, Kitamura H, Sato Y, Okutani M, Aoki J. Emerging lysophospholipid mediators, lysophosphatidylserine, lysophosphatidylthreonine, lysophosphatidylethanolamine and lysophosphatidylglycerol. Prostaglandins Other Lipid Mediat (2009) 89:135–9. doi:10.1016/j.prostaglandins.2009.04.009
72. Sevastou I, Kaffe E, Mouratis M-A, Aidinis V. Lysoglycerophospholipids in chronic inflammatory disorders: the PLA2/LPC and ATX/LPA axes. Biochim Biophys Acta (2013) 1831:42–60. doi:10.1016/j.bbalip.2012.07.019
73. Knowlden S, Georas SN. The autotaxin-LPA axis emerges as a novel regulator of lymphocyte homing and inflammation. J Immunol (2014) 192:851–7. doi:10.4049/jimmunol.1302831
74. Piñeiro R, Falasca M. Lysophosphatidylinositol signalling: new wine from an old bottle. Biochim Biophys Acta (2012) 1821:694–705. doi:10.1016/j.bbalip.2012.01.009
75. D’Aquilio F, Procaccini M, Izzi V, Chiurchiu’ V, Giambra V, Carotenuto F, et al. Activatory properties of lysophosphatidic acid on human THP-1 cells. Inflammation (2007) 30:167–77. doi:10.1007/s10753-007-9034-2
76. Heimerl S, Fischer M, Baessler A, Liebisch G, Sigruener A, Wallner S, et al. Alterations of plasma lysophosphatidylcholine species in obesity and weight loss. PLoS One (2014) 9:e111348. doi:10.1371/journal.pone.0111348
77. Moreno-Navarrete JM, Catalan V, Whyte L, Diaz-Arteaga A, Vazquez-Martinez R, Rotellar F, et al. The L-α-lysophosphatidylinositol/GPR55 system and its potential role in human obesity. Diabetes (2012) 61:281–91. doi:10.2337/db11-0649
78. Lee H, Liao JJ, Graeler M, Huang MC, Goetzl EJ. Lysophospholipid regulation of mononuclear phagocytes. Biochim Biophys Acta (2002) 1582:175–7. doi:10.1016/S1388-1981(02)00153-1
79. Fuchs B, Schiller J, Wagner U, Häntzschel H, Arnold K. The phosphatidylcholine/lysophosphatidylcholine ratio in human plasma is an indicator of the severity of rheumatoid arthritis: investigations by 31P NMR and MALDI-TOF MS. Clin Biochem (2005) 38:925–33. doi:10.1016/j.clinbiochem.2005.06.006
80. Nikitopoulou I, Oikonomou N, Karouzakis E, Sevastou I, Nikolaidou-Katsaridou N, Zhao Z, et al. Autotaxin expression from synovial fibroblasts is essential for the pathogenesis of modeled arthritis. J Exp Med (2012) 209:925–33. doi:10.1084/jem.20112012
81. Maceyka M, Spiegel S. Sphingolipid metabolites in inflammatory disease. Nature (2014) 510:58–67. doi:10.1038/nature13475
82. Gomez-Muñoz A, Presa N, Gomez-Larrauri A, Rivera I-G, Trueba M, Ordoñez M. Control of inflammatory responses by ceramide, sphingosine 1-phosphate and ceramide 1-phosphate. Prog Lipid Res (2016) 61:51–62. doi:10.1016/j.plipres.2015.09.002
83. Vandanmagsar B, Youm Y-H, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med (2011) 17:179–88. doi:10.1038/nm.2279
84. Pettus BJ, Bielawska A, Subramanian P, Wijesinghe DS, Maceyka M, Leslie CC, et al. Ceramide 1-phosphate is a direct activator of cytosolic phospholipase A2. J Biol Chem (2004) 279:11320–6. doi:10.1074/jbc.M309262200
85. Simanshu DK, Kamlekar RK, Wijesinghe DS, Zou X, Zhai X, Mishra SK, et al. Non-vesicular trafficking by a ceramide-1-phosphate transfer protein regulates eicosanoids. Nature (2013) 500:463–7. doi:10.1038/nature12332
86. Józefowski S, Czerkies M, Łukasik A, Bielawska A, Bielawski J, Kwiatkowska K, et al. Ceramide and ceramide 1-phosphate are negative regulators of TNF-α production induced by lipopolysaccharide. J Immunol (2010) 185:6960–73. doi:10.4049/jimmunol.0902926
87. Hankins JL, Fox TE, Barth BM, Unrath KA, Kester M. Exogenous ceramide-1-phosphate reduces lipopolysaccharide (LPS)-mediated cytokine expression. J Biol Chem (2011) 286:44357–66. doi:10.1074/jbc.M111.264010
88. Zhang Q-J, Holland WL, Wilson L, Tanner JM, Kearns D, Cahoon JM, et al. Ceramide mediates vascular dysfunction in diet-induced obesity by PP2A-mediated dephosphorylation of the eNOS-Akt complex. Diabetes (2012) 61:1848–59. doi:10.2337/db11-1399
89. Camerer E, Regard JB, Cornelissen I, Srinivasan Y, Duong DN, Palmer D, et al. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Invest (2009) 119:1871–9. doi:10.1172/JCI38575
90. Spiegel S, Milstien S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat Rev Immunol (2011) 11:403–15. doi:10.1038/nri2974
91. Cyster JG, Schwab SR. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol (2012) 30:69–94. doi:10.1146/annurev-immunol-020711-075011
92. Kunkel GT, Maceyka M, Milstien S, Spiegel S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat Rev Drug Discov (2013) 12:688–702. doi:10.1038/nrd4099
93. Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov (2010) 9:883–97. doi:10.1038/nrd3248
94. Chiurchiù V. Novel targets in multiple sclerosis: to oxidative stress and beyond. Curr Top Med Chem (2014) 14:2590–9. doi:10.2174/1568026614666141203143801
95. van Doorn R, Nijland PG, Dekker N, Witte ME, Lopes-Pinheiro MA, van het Hof B, et al. Fingolimod attenuates ceramide-induced blood-brain barrier dysfunction in multiple sclerosis by targeting reactive astrocytes. Acta Neuropathol (2012) 124:397–410. doi:10.1007/s00401-012-1014-4
96. Van Doorn R, Van Horssen J, Verzijl D, Witte M, Ronken E, Van Het Hof B, et al. Sphingosine 1-phosphate receptor 1 and 3 are upregulated in multiple sclerosis lesions. Glia (2010) 58:1465–76. doi:10.1002/glia.21021
97. van Doorn R, Lopes Pinheiro MA, Kooij G, Lakeman K, van het Hof B, van der Pol SMA, et al. Sphingosine 1-phosphate receptor 5 mediates the immune quiescence of the human brain endothelial barrier. J Neuroinflammation (2012) 9:133. doi:10.1186/1742-2094-9-133
98. Snider AJ, Kawamori T, Bradshaw SG, Orr KA, Gilkeson GS, Hannun YA, et al. A role for sphingosine kinase 1 in dextran sulfate sodium-induced colitis. FASEB J (2009) 23:143–52. doi:10.1096/fj.08-118109
99. Proia RL, Hla T. Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. J Clin Invest (2015) 125:1379–87. doi:10.1172/JCI76369
100. Bamias G, Rivera-Nieves J. Targeting S1P receptors, a new mechanism of action for inflammatory bowel disease therapy. Gastroenterology (2016) 151:1025–7. doi:10.1053/j.gastro.2016.09.025
101. Maccarrone M, Guzmán M, Mackie K, Doherty P, Harkany T. Programming of neural cells by (endo)cannabinoids: from physiological rules to emerging therapies. Nat Rev Neurosci (2014) 15:786–801. doi:10.1038/nrn3846
102. Pistis M, Melis M. From surface to nuclear receptors: the endocannabinoid family extends its assets. Curr Med Chem (2010) 17:1450–67. doi:10.2174/092986710790980014
103. Galve-Roperh I, Chiurchiù V, Díaz-Alonso J, Bari M, Guzmán M, Maccarrone M. Cannabinoid receptor signaling in progenitor/stem cell proliferation and differentiation. Prog Lipid Res (2013) 52:633–50. doi:10.1016/j.plipres.2013.05.004
104. Maccarrone M, Bab I, Bíró T, Cabral GA, Dey SK, Di Marzo V, et al. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol Sci (2015) 36:277–96. doi:10.1016/j.tips.2015.02.008
105. Cencioni MT, Chiurchiù V, Catanzaro G, Borsellino G, Bernardi G, Battistini L, et al. Anandamide suppresses proliferation and cytokine release from primary human T-lymphocytes mainly via CB2 receptors. PLoS One (2010) 5:e8688. doi:10.1371/journal.pone.0008688
106. Chiurchiù V, Rapino C, Talamonti E, Leuti A, Lanuti M, Gueniche A, et al. Anandamide suppresses proinflammatory T cell responses in vitro through type-1 cannabinoid receptor-mediated mTOR inhibition in human keratinocytes. J Immunol (2016) 197:3545–53. doi:10.4049/jimmunol.1500546
107. Ribeiro A, Pontis S, Mengatto L, Armirotti A, Chiurchiù V, Capurro V, et al. A potent systemically active N-acylethanolamine acid amidase inhibitor that suppresses inflammation and human macrophage activation. ACS Chem Biol (2015) 10:1838–46. doi:10.1021/acschembio.5b00114
108. Maestroni GJM. The endogenous cannabinoid 2-arachidonoyl glycerol as in vivo chemoattractant for dendritic cells and adjuvant for Th1 response to a soluble protein. FASEB J (2004) 18:1914–6. doi:10.1096/fj.04-2190fje
109. Gasperi V, Evangelista D, Chiurchiù V, Florenzano F, Savini I, Oddi S, et al. 2-Arachidonoylglycerol modulates human endothelial cell/leukocyte interactions by controlling selectin expression through CB1 and CB2 receptors. Int J Biochem Cell Biol (2014) 51:79–88. doi:10.1016/j.biocel.2014.03.028
110. Gallily R, Breuer A, Mechoulam R. 2-Arachidonylglycerol, an endogenous cannabinoid, inhibits tumor necrosis factor-alpha production in murine macrophages, and in mice. Eur J Pharmacol (2000) 406:R5–7. doi:10.1016/S0014-2999(00)00653-1
111. Chang YH, Lee ST, Lin WW. Effects of cannabinoids on LPS-stimulated inflammatory mediator release from macrophages: involvement of eicosanoids. J Cell Biochem (2001) 81:715–23. doi:10.1002/jcb.1103
112. Velasco G, Sánchez C, Guzmán M. Endocannabinoids and cancer. Handb Exp Pharmacol (2015) 231:449–72. doi:10.1007/978-3-319-20825-1_16
113. Mazier W, Saucisse N, Gatta-Cherifi B, Cota D. The endocannabinoid system: pivotal orchestrator of obesity and metabolic disease. Trends Endocrinol Metab (2015) 26:524–37. doi:10.1016/j.tem.2015.07.007
114. Steffens S, Pacher P. The activated endocannabinoid system in atherosclerosis: driving force or protective mechanism? Curr Drug Targets (2015) 16:334–41. doi:10.2174/1389450115666141202113225
115. Cani PD, Plovier H, Van Hul M, Geurts L, Delzenne NM, Druart C, et al. Endocannabinoids – at the crossroads between the gut microbiota and host metabolism. Nat Rev Endocrinol (2016) 12:133–43. doi:10.1038/nrendo.2015.211
116. Alhouayek M, Muccioli GG. The endocannabinoid system in inflammatory bowel diseases: from pathophysiology to therapeutic opportunity. Trends Mol Med (2012) 18:615–25. doi:10.1016/j.molmed.2012.07.009
117. Sido JM, Nagarkatti PS, Nagarkatti M. Role of endocannabinoid activation of peripheral CB1 receptors in the regulation of autoimmune disease. Int Rev Immunol (2015) 34:403–14. doi:10.3109/08830185.2014.921165
118. Maccarrone M. Endocannabinoid signaling and neuroinflammatory diseases. Curr Pharm Des (2008) 14:2252–3. doi:10.2174/138161208785740045
119. Di Marzo V, Stella N, Zimmer A. Endocannabinoid signalling and the deteriorating brain. Nat Rev Neurosci (2015) 16:30–42. doi:10.1038/nrn3876
120. Mecha M, Carrillo-Salinas FJ, Feliú A, Mestre L, Guaza C. Microglia activation states and cannabinoid system: therapeutic implications. Pharmacol Ther (2016) 166:40–55. doi:10.1016/j.pharmthera.2016.06.011
121. Skaper SD, Facci L, Barbierato M, Zusso M, Bruschetta G, Impellizzeri D, et al. Palmitoylethanolamine and neuroinflammation: a novel therapeutic strategy of resolution. Mol Neurobiol (2015) 52:1034–42. doi:10.1007/s12035-015-9253-8
122. Gokoh M, Kishimoto S, Oka S, Sugiura T. 2-Arachidonoylglycerol enhances the phagocytosis of opsonized zymosan by HL-60 cells differentiated into macrophage-like cells. Biol Pharm Bull (2007) 30:1199–205. doi:10.1248/bpb.30.1199
123. Cabral GA, Ferreira GA, Jamerson MJ. Endocannabinoids and the immune system in health and disease. Handb Exp Pharmacol (2015) 231:185–211. doi:10.1007/978-3-319-20825-1_6
124. Lipina C, Hundal HS. Modulation of cellular redox homeostasis by the endocannabinoid system. Open Biol (2016) 6:150276. doi:10.1098/rsob.150276
125. Aizpurua-Olaizola O, Elezgarai I, Rico-Barrio I, Zarandona I, Etxebarria N, Usobiaga A. Targeting the endocannabinoid system: future therapeutic strategies. Drug Discov Today (2017) 22:105–10. doi:10.1016/j.drudis.2016.08.005
126. Chiurchiù V, Cencioni MT, Bisicchia E, De Bardi M, Gasperini C, Borsellino G, et al. Distinct modulation of human myeloid and plasmacytoid dendritic cells by anandamide in multiple sclerosis. Ann Neurol (2013) 73:626–36. doi:10.1002/ana.23875
127. Chiurchiù V, Lanuti M, Catanzaro G, Fezza F, Rapino C, Maccarrone M. Detailed characterization of the endocannabinoid system in human macrophages and foam cells, and anti-inflammatory role of type-2 cannabinoid receptor. Atherosclerosis (2014) 233:55–63. doi:10.1016/j.atherosclerosis.2013.12.042
128. Centonze D, Bari M, Rossi S, Prosperetti C, Furlan R, Fezza F, et al. The endocannabinoid system is dysregulated in multiple sclerosis and in experimental autoimmune encephalomyelitis. Brain (2007) 130:2543–53. doi:10.1093/brain/awm160
129. Centonze D, Finazzi-Agrò A, Bernardi G, Maccarrone M. The endocannabinoid system in targeting inflammatory neurodegenerative diseases. Trends Pharmacol Sci (2007) 28:180–7. doi:10.1016/j.tips.2007.02.004
130. Chiurchiù V, Leuti A, Maccarrone M. Cannabinoid signaling and neuroinflammatory diseases: a melting pot for the regulation of brain immune responses. J Neuroimmune Pharmacol (2015) 10:268–80. doi:10.1007/s11481-015-9584-2
131. Fernández-Ruiz J, Romero J, Ramos JA. Endocannabinoids and neurodegenerative disorders: Parkinson’s disease, Huntington’s chorea, Alzheimer’s disease, and others. Handb Exp Pharmacol (2015) 231:233–59. doi:10.1007/978-3-319-20825-1_8
132. van der Stelt M, van Kuik JA, Bari M, van Zadelhoff G, Leeflang BR, Veldink GA, et al. Oxygenated metabolites of anandamide and 2-arachidonoylglycerol: conformational analysis and interaction with cannabinoid receptors, membrane transporter, and fatty acid amide hydrolase. J Med Chem (2002) 45:3709–20. doi:10.1021/jm020818q
133. Rouzer CA, Marnett LJ. Endocannabinoid oxygenation by cyclooxygenases, lipoxygenases, and cytochromes P450: cross-talk between the eicosanoid and endocannabinoid signaling pathways. Chem Rev (2011) 111:5899–921. doi:10.1021/cr2002799
134. Di Marzo V. Targeting the endocannabinoid system: to enhance or reduce? Nat Rev Drug Discov (2008) 7:438–55. doi:10.1038/nrd2553
135. De Filippis D, D’Amico A, Cipriano M, Petrosino S, Orlando P, Di Marzo V, et al. Levels of endocannabinoids and palmitoylethanolamide and their pharmacological manipulation in chronic granulomatous inflammation in rats. Pharmacol Res (2010) 61:321–8. doi:10.1016/j.phrs.2009.11.005
136. Witkamp R. Fatty acids, endocannabinoids and inflammation. Eur J Pharmacol (2016) 785:96–107. doi:10.1016/j.ejphar.2015.08.051
137. Weichand B, Weis N, Weigert A, Grossmann N, Levkau B, Brüne B. Apoptotic cells enhance sphingosine-1-phosphate receptor 1 dependent macrophage migration. Eur J Immunol (2013) 43:3306–13. doi:10.1002/eji.201343441
138. Scheel-Toellner D, Wang K, Craddock R, Webb PR, McGettrick HM, Assi LK, et al. Reactive oxygen species limit neutrophil life span by activating death receptor signaling. Blood (2004) 104:2557–64. doi:10.1182/blood-2004-01-0191
139. McArthur S, Gobbetti T, Kusters DH, Reutelingsperger CP, Flower RJ, Perretti M. Definition of a novel pathway centered on lysophosphatidic acid to recruit monocytes during the resolution phase of tissue inflammation. J Immunol (2015) 195:1139–51. doi:10.4049/jimmunol.1500733
Keywords: eicosanoids, endocannabinoids, inflammation, resolution, specialized proresolving mediators, sphingolipids
Citation: Chiurchiù V, Leuti A and Maccarrone M (2018) Bioactive Lipids and Chronic Inflammation: Managing the Fire Within. Front. Immunol. 9:38. doi: 10.3389/fimmu.2018.00038
Received: 06 September 2017; Accepted: 05 January 2018;
Published: 29 January 2018
Edited by:
Claudia Monaco, University of Oxford, United KingdomReviewed by:
Yongsheng Li, Third Military Medical University, ChinaHugo Caire Castro-Faria-Neto, Fundação Oswaldo Cruz (Fiocruz), Brazil
Copyright: © 2018 Chiurchiù, Leuti and Maccarrone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valerio Chiurchiù, v.chiurchiu@hsantalucia.it;
Mauro Maccarrone, m.maccarrone@unicampus.it
†These authors have contributed equally to this work.