Article Text

Abstract
Rheumatoid arthritis (RA) is a systemic autoimmune disease characterised by chronic joint inflammation and variable degrees of bone and cartilage erosion. Studies in animal models of arthritis provide insight into elements which can amplify destructive features. The presence of immune complexes in the joint makes arthritis more erosive. Although considerable bone erosion still occurs in the absence of FcγR triggering by immune complexes, through cytokine-induced RANKL and direct osteoclast activation, cartilage erosion is heavily dependent on the FcγR pathway. T cell factors such as IFNγ and IL17 further amplify erosion through upregulation of the damaging FcγRI and stimulation of the influx of granulocytes, respectively. Apart from immune elements, environmental pressure and components of tissue damage contribute through innate pathways. Spontaneous T cell-dependent arthritis in IL1Ra–/– mice is absent under germ-free conditions, and markedly suppressed in TLR4-deficient mice. Moreover, TLR4 blocking with a receptor antagonist suppresses erosive arthritis.
Statistics from Altmetric.com
It is well accepted that activated osteoclasts are the main cell types causing bone erosion. Osteoclast precursors mature under the influence of cytokine-induced RANKL and RANKL is also the main driver of activation of mature RANK-bearing osteoclasts (fig 1). The pivotal cytokines TNF, IL1 and IL17 are all capable of induction of considerable RANKL production, and marked synergy in RANKL upregulation is noted between these cytokines. It is believed that synovial fibroblasts contribute to bone erosion, not by directly attacking the bone, but by RANKL expression and direct cell–cell contact with osteoclasts. Immunostaining in arthritic synovial tissue of mice with collagen arthritis clearly identifies RANKL production by numerous cell types, including macrophages and fibroblasts.

The direct role of cytokines in bone erosion is compatible with the observation of pronounced bone erosion in arthritic joints of TNF transgenic (TNFtg) mice. In contrast, cartilage surface erosion is not a prominent early feature in this animal model and the initial damage is mainly restricted to cartilage proteoglycan loss, which is reversible in the absence of collagen type II damage. In patients with RA bone erosion can be prevented with anti-TNF therapy, identifying TNF as a crucial bone-erosive cytokine in this disease. It is claimed that bone erosion is halted with anti-TNF even under conditions of ongoing joint inflammation. In line with this, treatment with osteoprotegerin (OPG), which is a decoy receptor of RANKL, prevents bone erosion in animal models of arthritis, without an impact on joint inflammation and cartilage erosion. Similar observations have been made with soluble RANK. Recent clinical trials in patients with RA confirm the prevention of bone erosion and lack of effect on joint inflammation.
IMMUNE COMPLEXES AND CARTILAGE EROSION
In contrast to bone erosion, cartilage surface erosion is limited under conditions of cytokine overexpression. Cytokines like TNF, IL1 and IL17 can activate chondrocytes in the articular cartilage and upregulate cartilage-degrading enzymes. However, apart from the pivotal aggrecan-degrading enzyme ADAMTS-51 which needs no further activation, the cytokine-induced metalloproteinases like stromelysin (MMP-3) and collagenase (MMP-13) are in a latent form and still need further activation before collagen breakdown and distinct cartilage erosion occurs. At that stage, the presence of immune complexes in the joint can greatly amplify cartilage erosion in a Fcγ receptor (FcγR)-dependent way. Studies of arthritis models in a range of FcγR knockout mice identified a crucial role of FcgR type I.2–6 When antigen-induced arthritis, which is a mix of T cell and immune complex-driven joint inflammation, is elicited in Fcγ-chain knockout mice, lacking all activating FcγR I, III and IV, inflammation continues, but cartilage erosion is absent. Additional studies in single knockouts demonstrated the crucial inhibitory role of the type IIβ receptor and the cartilage erosive role of type I (table 1).
BONE EROSION
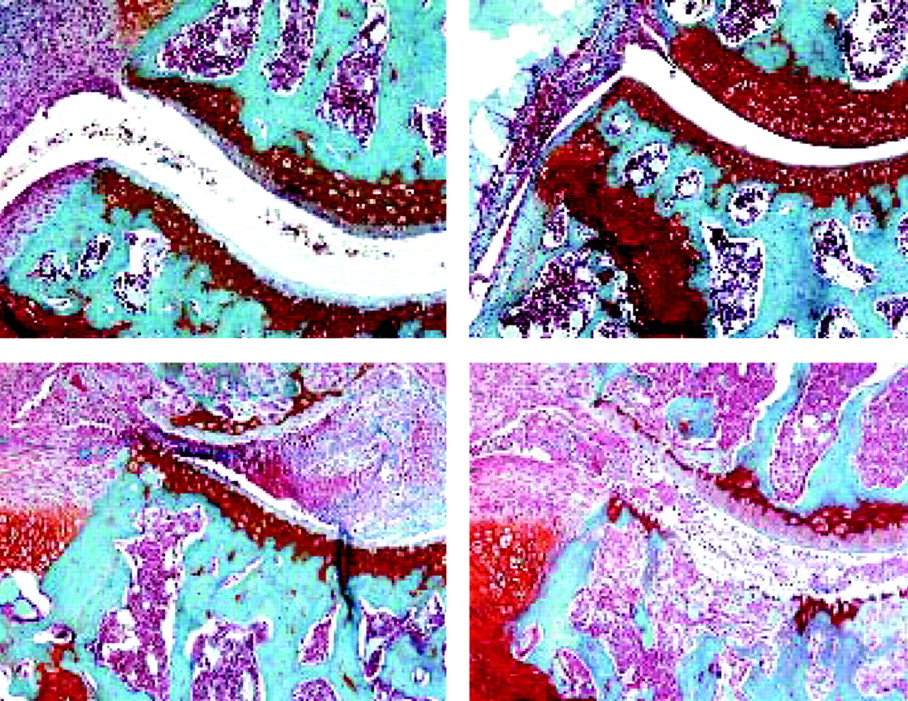
Intriguingly, studies in FcγRI/II/III triple knockout mice confirmed the pivotal role of immune complex (IC)–FcγR interaction in cartilage erosion, but also demonstrated convincingly that FcγR are not directly involved in bone erosion.6 When antigen-induced arthritis is provoked in these triple knockout mice, joint inflammation is markedly enhanced due to a lack of proper clearance of immune complexes/antigen from the joint. Despite tremendous inflammation, cartilage erosion is fully absent, whereas bone erosion is pronounced and roughly follows the degree of inflammation (fig 2). Apparently, there is abundant cytokine generation through sustained Th17 cell activation, with subsequent RANKL production and osteoclast activation. Cathepsin K staining was prominent in osteoclasts at the bone erosion sites. Futher in vitro studies identified downregulation of FcγR expression when bone marrow-derived macrophages mature into osteoclasts, underlining that direct IC–FcγR activation of osteoclasts is unlikely to contribute significantly to bone erosion.

JOINT DAMAGE IN PATIENTS WITH RA
In patients with RA, correlations are found between the erosive character of the disease and the presence of autoantibodies such as rheumatoid factor and anti-CCP (citrullinated protein) antibodies. This includes both cartilage and bone erosion. Arthritogenic potential of anti-CCP antibodies has recently been demonstrated.7 It is argued that immune complexes amplify inflammation and enhance local cytokine levels through FcγR triggering, indirectly enhancing bone erosion. Whether such autoantibodies directly contribute to cartilage damage remains to be seen. Exact localisation of immune complexes, in the synovium or at the cartilage surface, as well as the subclasses of antibodies/Fc determinants involved may influence the erosive phenotype. It is warranted to further improve the detection of joint erosion in upcoming clinical trials. Separate scores of bone and cartilage erosion are often provided nowadays, but joint space narrowing is still an insensitive measure of focal surface erosion of the cartilage. Apart from immune complex-mediated events, synovial fibroblasts contribute to cartilage damage in chronic disease at sites of pannus overgrowth.
AMPLIFYING ROLE OF T CELL-DERIVED IL17
IL17A is the predominant IL17 isoform in arthritis and is further referred to as IL17. This pro-inflammatory and destructive cytokine is produced by T cells. Although initially thought to differentiate from Th1 cells, it is now clear that the IL17-producing Th17 cells are a separate lineage, promoted by IL6, IL1 and IL23.8 9
IL17 can amplify destruction. IL17 alone, when overexpressed in the murine knee joint causes inflammation and bone erosion, but no cartilage destruction. However, when it is overexpressed in an arthritic joint, in combination with an immune complex arthritis such as the passive glucose phosphate isomerase (GPI) model, it greatly amplifies inflammation and destruction, including cartilage erosion.10 Moreover, it overrules the TNF dependency of the GPI-IC arthritis (fig 3). Likewise, its overexpression overrules the IL1 dependency of collagen arthritis. The latter might explain why in patients with RA TNF or IL1-directed therapy is not always successful.
{kind=link}
{kind=link}
{kind=link}
Evidence for its role in arthritis emerged from blocking studies with antibodies or induction of models in IL17 or IL17R deficient mice. The T cell-dependent arthritis in IL1Ra–/– mice is absent in IL17 knockout mice.11 Moreover, treatment with anti-IL17 in established IL1Ra–/– arthritis halted progression (unpublished observation). Late-stage collagen arthritis can still be suppressed with anti-IL17 antibodies, including reduction of erosion. The most impressive therapeutic effect was noted in flares of antigen-induced arthritis, where smouldering arthritis is reactivated with low-dose antigen. IL17 production is prominent in such flares and erosion can be fully blocked with anti-IL17.12 13 IL17 becomes a major therapeutic target at stages where Th17 cells become dominant players. In line with this, in the model of repeated challenge with streptococcal cell wall fragments, arthritis transforms from a macrophage-driven disease to a T cell-dependent, erosive arthritis and that erosive stage is highly reduced in IL17R–/– mice.14
IFNγ/IL17 BALANCE
It is now suggested that Th17 cells are the pathogenic cells inducing erosive arthritis and that Th1 IFNγ producing cells are counterbalancing. This reasoning might explain the well-known enhanced susceptibility for collagen arthritis in non-sensitive mouse strains when crossed with IFNγ deficiency. On the other hand, proteoglycan arthritis is lost in IFNγ–/– mice, arguing that in this model IFNγ is necessary for autoantibody formation and joint pathology.15 Moreover, in passive immune complex arthritis models, co-expression of IFNγ makes arthritis more erosive, through selective upregulation of the Fcγ receptor type I (see above, table 1). IL17 does not upregulate distinct Fcγ receptors but amplifies erosive immune complex arthritis probably by promotion of granulocyte influx and help in enzyme activation (fig 1). So, if IFNγ directly promotes erosive immune complex arthritis its presence cannot be seen as protective. On the other hand, if the generation of Th1 cells prevents Th17 promotion, the relative lack of Th17 cells can be viewed as protective, at least under conditions where T cells are dominant players and IC-driven processes are less involved. Detailed evaluation of the presence and activity of Th17 cells in synovial tissue of various patients with RA needs attention. Recently, the orphan nuclear receptor RORgt has been identified; this directs differentiation and can also be used, in combination with CCR 4 and 6, as a marker for immunostaining.16 17
ENVIRONMENT STIMULI
It has long been suggested that environmental stimuli such as bacteria or viruses contribute to arthritis. More recently, toll-like receptors (TLR) have been identified that recognise pathogen-associated molecular patterns but also damage-associated molecular patterns, including breakdown products of eroded cartilage. In particular, TLR4 is activated by fragments released after tissue damage and subsequent cytokine production including IL1 and TNF might directly contribute to arthritis and further erosion. In addition, TLR4 triggering seems to drive generation of autoimmune Th17 cells. Mice lacking IL1Ra–/–, and therefore displaying uncontrolled IL1 activity, develop a chronic T cell-driven arthritis. However, when these mice are kept under germ-free conditions, arthritis is fully ablated. This suggests that environmental bacterial triggers promote arthritis, either as an antigen, or more likely, as an adjuvant, promoting autoimmune T cells. We investigated the role of TLR4 in this process by crossing IL1Ra–/– mice with TLR4–/– mice. Erosive arthritis was suppressed in the absence of TLR4, and these mice displayed markedly reduced Th17 generation, which seems an obvious underlying principle of the reduced arthritis. In contrast, crossing of IL1Ra–/– with TLR2–/– mice made arthritis more severe. Th17 cells were not changed, but these animals had reduced generation of Treg cells. The arthritis-promoting role of TLR4 was found in IL1Ra–/– mice, and also in collagen arthritis. Treatment with a natural TLR4 antagonist reduced arthritis and erosion, in particular, even when treatment is started in full-blown arthritis.18 It suggests that TLR4 regulates autoimmune responses which might be promoted by environmental triggers as well as cartilage breakdown products, herein creating a vicious circle underlying chronic erosive arthritis. It makes TLR4 an attractive therapeutic target. Of interest, TLR4 co-stimulation with LPS bypassed the IL1 dependency of passive GPI arthritis,19 and abundant presence of natural TLR4 agonists in an arthritic joint might limit efficacy of IL1-directed therapy.
REFERENCES
Footnotes
Competing interests: None declared.
- Abbreviations:
- CCP
- citrullinated protein
- FcγR
- Fcγ receptors
- GPI, glucose phosphate isomerase; IC, immune complex; MMPs
- matrix metalloproteinases
- OPG, osteoprotegerin; RA
- rheumatoid arthritis
- TLR
- toll-like receptor
- TNFtg
- TNF transgenic