


Abstract
Two orphan nuclear receptors, constitutive active (or androstane) receptor (CAR) and pregnane X receptor (PXR), are among the most important mediators of ligand-activated transcriptional induction of liver microsomal cytochrome P450 drug-metabolizing enzymes. CAR and PXR belong to the same NR1I receptor subfamily and show high sequence homology to each other. The vitamin D receptor (VDR) also belongs to the NR1I subfamily and has the second highest homology to CAR in the ligand binding domain. A 3D model of the ligand binding domain of human CAR (hCAR) was constructed based on the available X-ray structures of human PXR (hPXR) and VDR (hVDR). The model shows that the size of the ligand binding cavities of hCAR and hPXR are similar, but larger than that of hVDR. hPXR's capability of binding to extremely large ligands, such as rifampicin, implies that its binding cavity may be able to expand further through the flexibility of a surface loop. In contrast, hCAR does not have this loop so that its cavity cannot expand, suggesting that hCAR would not bind to the largest hPXR ligands. Docking calculations of selected ligands to hCAR, based on the structural model, are consistent with previously reported receptor binding data. The results from this study indicate that structural modeling will be a useful tool for understanding ligand binding to hCAR and for design of drugs free of hCAR-mediated enzyme induction.
Constitutive active (or androstane) receptor (CAR1), a member of the orphan nuclear receptor family, is a major regulator of hepatic microsomal cytochrome P450 2B1/2 in rats (Muangmoonchai et al., 2001), CYP2B10 in mice (Honkakoski and Negishi, 1997; Kawamoto et al., 1999) and CYP2B6 in humans (CYP2B) (Sueyoshi et al., 1999). A group of structurally unrelated compounds, exemplified by phenobarbital (PB), have been shown to be very potent inducers of the CYP2B family. In response to exposure to inducers, CAR translocates into the nucleus, forms a heterodimer with the 9-cis retinoic acid receptor (RXR), and activates the 51-base pair PB responsive enhancer module that is located upstream of the CYP2B gene (Honkakoski and Negishi, 1997, 1998; Honkakoski et al., 1998a,b). Homologous CAR-mediated enhancer sequences have been found in many other PB-regulated genes, such as CYP3A and human bilirubin UDP-glucuronosyltransferase UGT1A1 (Smirlis et al., 2001). In addition, CAR has been shown to regulate as many as twenty hepatic genes following exposure to PB (Ueda et al., 2002). Therefore, CAR may have a diverse role in regulating enzymes involved in drug metabolism and other pharmacological processes. It is not known if PB induces CYP2B by serving as an agonist ligand for CAR since no direct binding of PB to CAR has been observed (Sueyoshi and Negishi, 2001). However, several compounds, such as TCPOBOP, clotrimazole, and 5β-pregnane-3,20-dione, act directly as conventional agonist ligands to increase CAR transactivation (Moore et al., 2000; Tzameli et al., 2000).
Both CAR and PXR belong to the same NR1I orphan nuclear receptor gene subfamily (Nuclear Receptors Nomenclature Committee, 1999) and have high sequence homology. VDR belongs to this NR1I subfamily too. The homology between CAR and VDR is relatively high but only moderate for peroxisome proliferator activator receptor (PPAR) and RXR. CAR and PXR share some common xenobiotic and steroid ligands (Moore et al., 2000). These two nuclear receptor signaling pathways have been reported to have cross talk, which is probably due to ligand-sharing and sharing of the DNA responsive elements (Xie et al., 2000; Smirlis et al., 2001).
The X-ray crystal structure of the ligand binding domain of human CAR (hCAR) has not been determined. However, X-ray structures of several other nuclear receptors have been solved (Moras and Gronemeyer, 1998;Weatherman et al., 1999; Bourguet et al., 2000a; Steinmetz et al., 2001), including a recent structure of the ligand binding domain of human PXR (hPXR) (Watkins et al., 2001, 2002). Because of the fairly high sequence identity among hCAR, hPXR and human VDR (hVDR) in the ligand binding domain, we have built a reliable homology model of hCAR using hPXR and hVDR as the templates. Selected ligands were docked into this model to demonstrate potential binding interactions with hCAR.
Materials and Methods
Alignment.
The sequence of the ligand binding domain of hCAR (SWISS-PROT; NRI3_HUMAN) was searched against the nonredundant and PDB sequence databases of GenBank at the NCBI using the BLAST program to obtain its homologs (Altschul et al., 1997). For each hCAR homolog that has a known X-ray structure, the database SCOP was used to select a unique Protein Data Bank (PDB) structure file (Murzin et al., 1995). The sequences of the ligand binding domain of these nuclear receptors were extracted directly from their PDB files. A multiple sequence alignment was obtained using the program Clustal W (Thompson et al., 1994). The pair-wise sequence identities were calculated for hCAR and its homologs using the vector-NTI Suite (InforMax, Inc., Bethesda, MD). Structure comparison and alignment of these nuclear receptors were carried out using INSIGHT II (Accelrys, Princeton, NJ). In addition, a superposition of the X-ray structure of the PXR ligand binding domain with its structural neighbors in the PDB provided by the combinatorial extension database (Shindyalov and Bourne, 1998) was used.
Model Building.
Homology models of the hCAR ligand binding domain were constructed using MODELLER (Accelrys) (Sali and Blundell, 1993; Sanchez and Sali, 1997, 1998). From the alignment, spatial restraints, including distance restraints and torsion angle restraints were derived and used in the 3D-model construction of the protein. All models were further optimized with the internal optimizer of MODELLER. Before docking the ligands into the binding site of the selected final model, several residue side chains around the binding cavity were manually adjusted so that their side-chain torsion angles were similar to those of the templates. These side chains were further minimized using the Tripos forcefield, Amber charges, and distance-dependent dielectric.
The volume of the ligand binding cavity of a protein was calculated using GRASP (Nicholls et al., 1991). First, water molecules were added as a 5Å layer to the protein structure to close off the binding cavity from the surface by using INSIGHT II. A molecular surface was then constructed using the default atomic radii implemented in GRASP. Cavity surfaces were then built, and the cavity volumes were calculated. The cavity corresponding to the ligand binding pocket was identified by visual inspection.
Docking.
The program GOLD (version 1.2; CCDC, Cambridge, UK) (Jones et al., 1997) was used to dock the ligands to the hCAR ligand binding site. The ligand 3D structures were constructed using SYBYL version 6.7 accessed via CORCORD (Tripos Inc., St. Louis, MO; Pearlman, 2000). The active site radius was set to be 15Å. During the docking calculations, all residues of the protein were fixed while ligands were treated as flexible. Twenty genetic algorithm runs were performed for each ligand. No constraint was required. The 20 binding modes obtained for each ligand were grouped into several unique binding modes using distance clustering and a 2Å cutoff.
Results
Selection of Templates.
The hCAR sequence was searched against the nonredundant sequence database of GenBank at NCBI using the BLAST program. The sequences of hPXR and hVDR (Rochel et al., 2000) were most homologous to hCAR. A BLAST search against the PDB showed that several additional nuclear receptors with known X-ray structures are homologous to hCAR (Table1). The sequence identities with hCAR are 43% for hPXR, 34% for hVDR, and <30% for other nuclear receptors.
Pair-wise sequence comparison of human CAR ligand binding domain with other nuclear receptors
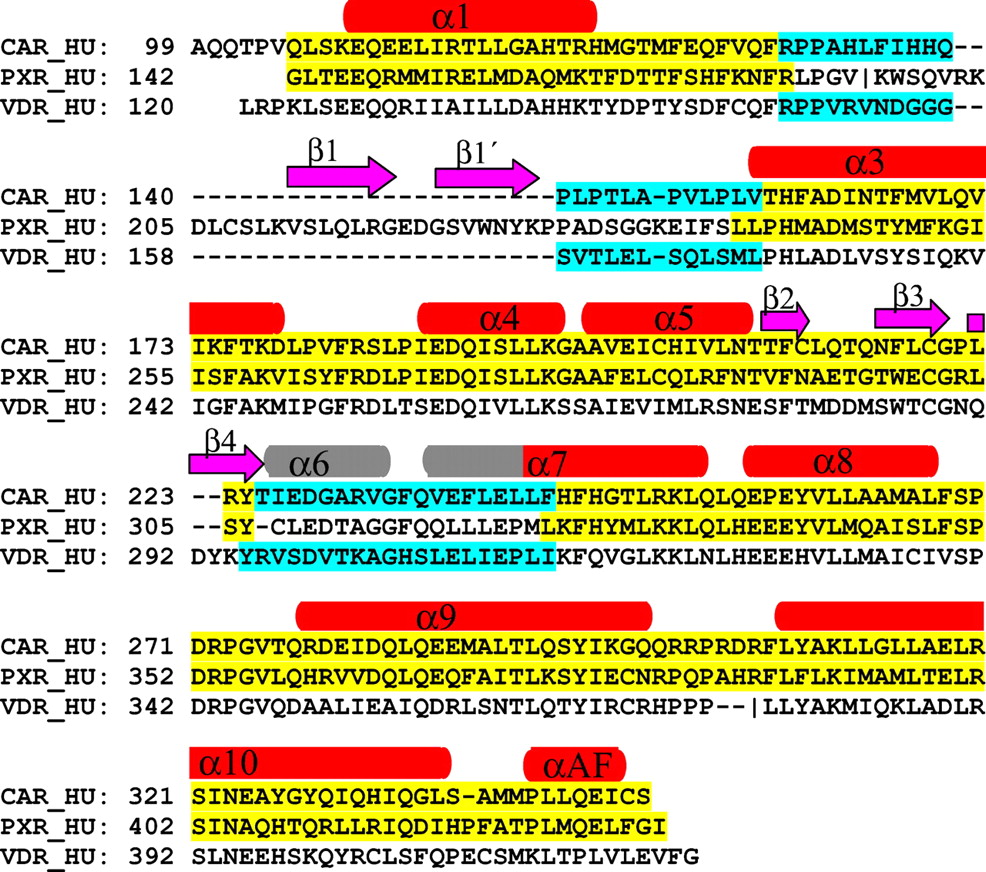
Structural comparisons among known nuclear receptors ligand binding domains have shown that their overall folds are similar. The canonical fold comprises 10 α helices and 3 β strands. Among the structures listed in Table 1, hPXR differs the most from the canonical structure. It has two extra β strands (β1 and β 1′), with β1 occupying the space where α6 is located in the canonical fold. The hPXR sequence corresponding to α6 forms a surface-exposed loop [residues 309–321 (Fig. 1)]. An engineered hVDR construct has a canonical nuclear receptor structure (Rochel et al., 2000). An “insertion domain” corresponding to the β1 and β 1′ sequence was engineered out before crystallization and structure determination. Therefore, even though the original full-length sequence of hVDR is similar to hPXR, the engineered hVDR has no β1 and β 1′ strands but an intact α6 helix. The variable region between α1 and α3 is also unique in hPXR.

Alignment of hPXR, hVDR, and hCAR used in the model building.
The secondary structures are labeled on the top. In red and gray are α helices and in magenta are β strands. The assignment of the secondary structures was taken from the hPXR X-ray structure except the α6 helix, where it was defined according to the hVDR X-ray structure and is colored in gray. Regions taken from hPXR are in yellow and regions taken from hVDR in cyan.
The overall high sequence identity between hCAR and hPXR in the ligand binding domain indicates that hPXR would be a good template for a hCAR homology model. However, multiple sequence alignment shows that hCAR should have the canonical fold rather than matching the unique β1 and β 1′, unfolded α6 loop and α1 and α3 loop of hPXR. In these regions of high sequence homology between hCAR and the engineered hVDR, the hVDR structure was used as the template. For the remainder, the hPXR structure was the template.
The alignment used in the model building was obtained from a Clustal W multiple sequence alignment of hCAR with hPXR and hVDR. The structural information was then manually input into the alignment, and several adjustments were made. The final alignment of hCAR with hPXR and hVDR is shown in Fig. 1. Regions from each protein used as the template are highlighted.
Ligand Binding Domain.
The alignment and template selection shown in Fig. 1 were used to generate five models of hCAR using the MODELLER software package. Each of these models was optimized using the “high” option. The one with optimal MODELLER scores was selected as the final model. This model was further validated with Profile-3D (INSIGHT II). It exhibits a φ,ψ density of 90% in the most favorable region of the Ramachandran plot. Since the sequence identity between the templates, hPXR and hVDR, and the model of hCAR is far above the 30% confidence limit of MODELLER, it should be highly reliable.
As shown in Fig. 2, the hCAR model has the canonical nuclear receptor fold. The root mean square deviation between the hCAR model and hPXR over 203 pairs of superimposed Cα positions is 0.25Å, and the root mean square deviation between the hCAR model and hVDR over 43 pairs of superimposed Cα positions is 0.65Å. Similar to other nuclear receptors with the common canonical fold, hCAR comprises 10 helices and 3 β strands. The ligand binding cavity is located in the bottom half of the protein (shown in the circled region in Fig. 2).
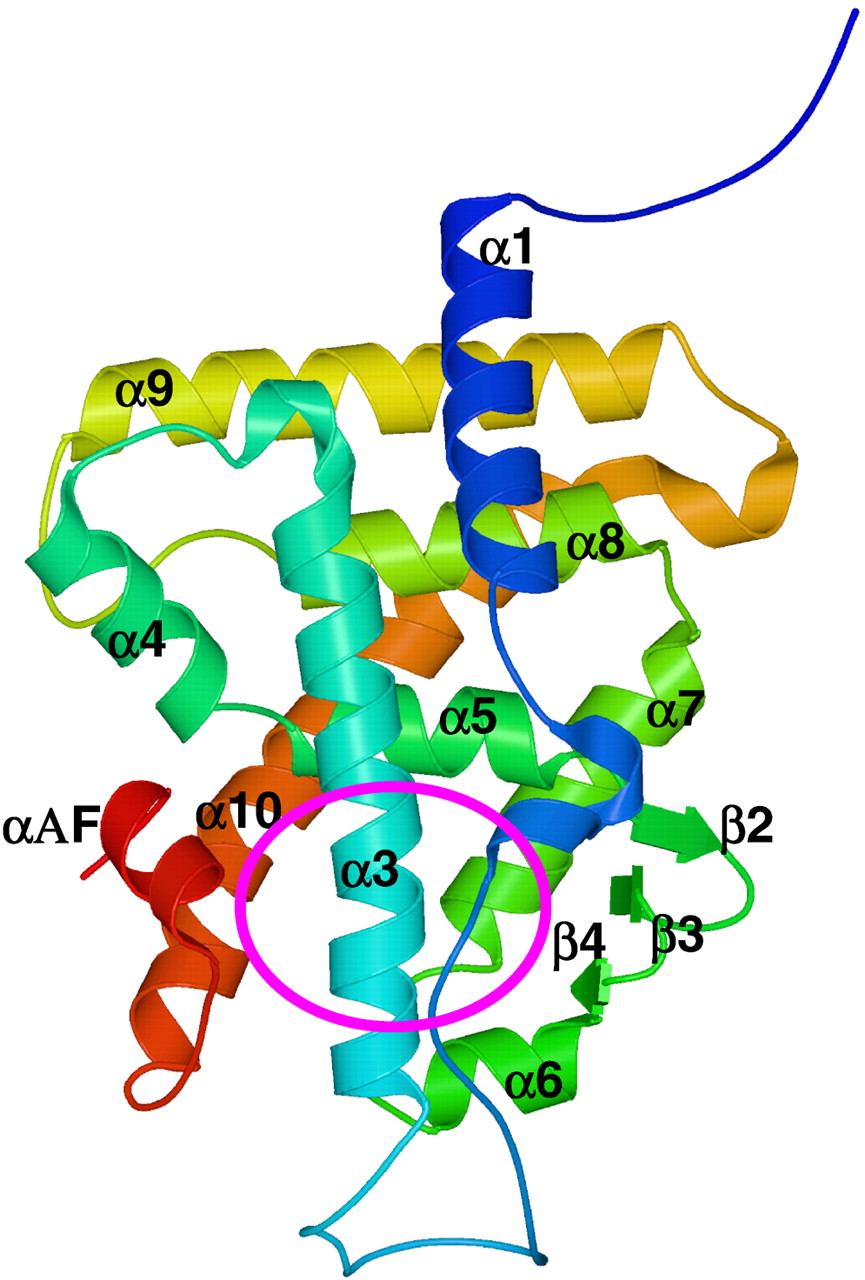
The structural model of hCAR-LBD.
Residues 99 to 348 are shown using the amino-acids rainbow color scheme where the N-terminus is in blue and the C-terminus in red. The secondary structural elements are numbered according to the hPXR X-ray structure. The location of the ligand binding site is indicated by the circled area. LBD, ligand-binding domain.
Ligand Binding Cavity.
Table 2 lists the residues lining the ligand binding cavity of hCAR. These residues are located on α3 (using the secondary structure index in the PXR X-ray structure) in the front of the cavity, on α5 at the back top, on β3 and β4 on the right, on α7 at the back bottom, on α10 and αF on the left, and on α6 (using the secondary structure index in the VDR X-ray structure) at the bottom (Fig. 2). The number of residues lining the cavity is 34 in hCAR versus 28 in hPXR. Similar to hPXR, most of these residues are hydrophobic. Among these 34 residues, 10 are conserved between hPXR and hCAR, almost one third of the total. Three charged or partially charged residues were observed in the cavity, His203, His246, and Asp228. His203 and His246 are adjacent to each other in space, possibly forming a hydrogen bond between their imidazole nitrogens. The side chain of Asp228 points to the surface with its backbone atoms in the cavity. Three polar residues, Asn165, Thr225, and Asn323, seen on α3, β4, and α10, spread far apart from each other in the cavity. Moreover, only the backbone atoms of Thr225 are able to interact with ligands whereas its side-chain atoms are on the protein surface. Therefore, similar to hPXR, the inner surface of the ligand binding cavity of hCAR is relatively uncharged and hydrophobic, which suggests that its ligands should be mostly hydrophobic.
Comparison of the ligand binding sites of hPXR/hCAR2-a
The size of the ligand binding cavity of hCAR is similar to hPXR although slightly smaller. Calculated by using the program GRASP with default parameters, the volumes of the cavities of hCAR, hPXR, and hVDR are 1170Å3, 1220Å3, and 910Å3, respectively. Figure3 shows the cavity surfaces of these proteins. The cavities of all three proteins are bordered by an aromatic residue on the right-hand side (Trp299 in hPXR, F217 in hCAR, Trp286 in hVDR). The central cavity of hPXR can be enlarged by opening a constriction into a second cavity bounded by a flexible surface loop (residues 309–321). The resulting new cavity would have a volume of 1600Å3. A docking calculation indicated this enlarged cavity would allow hPXR to bind extremely large ligands, such as rifampicin (results not shown). In contrast, hCAR does not have this loop. Its sequence segment corresponding to this loop is folded as α6. Therefore, hCAR would not be able to bind to very large ligands like rifampicin.
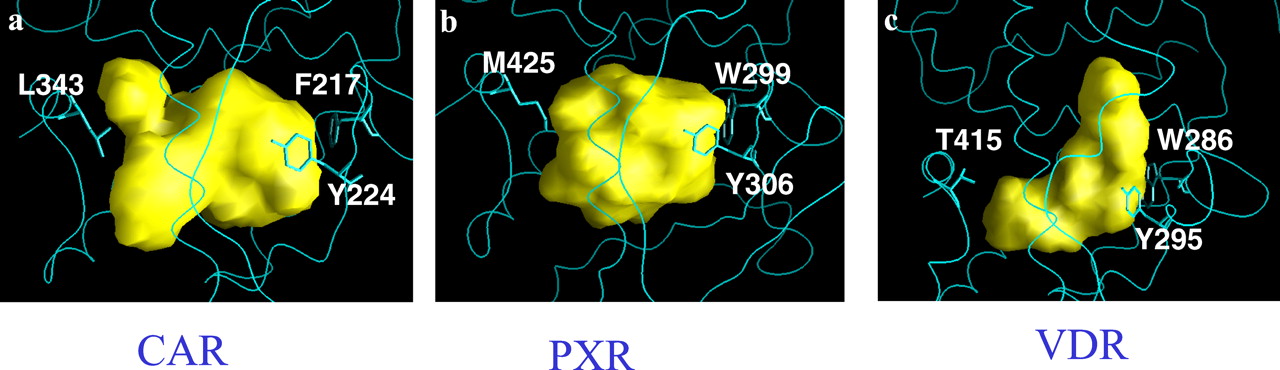
The ligand binding cavity surfaces generated with GRASP.
The protein structures around the cavity are shown in blue. Side chains of three residues are also shown as stick representations. a, hCAR; b, hPXR; c, hVDR.
Docking Ligands to hCAR.
Four ligands, clotrimazole, 5α-androstan-17α-ol, 5β-pregnanedione, and TCPOBOP, were docked into the ligand binding cavity of the hCAR model. All of the ligands bind well in the pocket, and three of them exhibit multiple binding modes. The best binding modes of each ligand given by the program GOLD are shown in Fig.4 except protonated clotrimazole, where the binding mode shown in Fig. 4aII has a GOLD score very close to that of the best binding mode.

Docking of clotrimazole, 5β-pregnanedione, 5α-androstan-17α-ol, and TCPOBOP to hCAR.
Colors, active site residue carbon atoms in cyan, ligand carbon atoms in light pink, all nitrogen atoms in blue, all oxygen atoms in red, sulfur in yellow, chlorine in light green. 4aI, nonprotonated clotrimazole; 4aII, protonated clotrimazole, a H-bond is shown between the ligand and residue His246; 4b, 5β-pregnanedione; 4c, 5α-androstan-17α-ol; 4d, TCPOBOP.
Clotrimazole.
Clotrimazole occupies only part of the cavity (i.e., the region covered by residues Phe161, Ile164, Asn165, Cys202, His203, Leu206, Phe217, Cys219, Tyr224, Leu239, and Phe243). None of the residues on α10 and αΑF contacts the ligand. Although the molecule displays seven potential binding modes in the cavity, its center of mass shifts very little. Large changes occur in the orientations of the four rings of the molecule. Besides strong hydrophobic interactions between hCAR and clotrimazole, we also observed significant interactions between the aromatic groups in hCAR and clotrimazole. In the binding mode shown in Fig. 4aI, the aromatic interactions between the chlorophenyl ring of the compound and Phe217 phenyl side chain (edge-to-face), between the imidazole ring of the compound with Tyr224 hydroxylphenyl side chain (face-to-face), and between the two other phenyl rings of the compound with Phe161 phenyl side chain (edge-to-face) are clearly seen. The 3-nitrogen of the imidazole ring of the compound has a distance of 2.9Å to the Tyr224 hydroxyl oxygen atom. Although the geometry is not perfect for the formation of a hydrogen bond, the optimal distance suggests potential electrostatic interactions.
The 3-nitrogen of the imidazole ring of clotrimazole can be partially protonated at neutral pH. To investigate the effect of protonation of the 3-nitrogen on hCAR binding, the protonated state of this ligand was also docked into the protein. The general location of the ligand in the cavity remains the same, but the orientation of the four rings changes significantly. We observed a hydrogen bond (2.6Å distance and 180 degree angle) between the 3-nitrogen of the imidazole ring of the compound and one of the nitrogen atoms from the His246 imidazole side chain. In the binding mode shown in Fig. 4aII, a few aromatic interactions between the ligand and the protein can be seen. For example, the edge-to-face interaction between the chlorophenyl ring of the compound with the Phe243 phenyl ring at the top and the Phe161 phenyl ring from the bottom.
It has been shown that clotrimazole is a potent ∼100 nM inhibitor of hCAR (Moore et al., 2000; Tzameli et al., 2000). Our modeling results suggest that the strong binding of clotrimazole to hCAR arises primarily from hydrophobic interactions and aromatic interactions with smaller contributions from electrostatic interactions.
5β-Pregnanedione.
This molecule shows six distinct binding modes. The center of the molecule can occupy various locations in the cavity, encompassing the entire volume of the cavity (Table 2). In one of the binding modes of 5β-pregnanedione (Fig. 4b), no electrostatic interaction is observed between the two ligand C=O groups and the protein. The steroid nucleus of this ligand is in van der Waals contact with residues Phe161, His203, Leu206, Phe217, Tyr224, I226, Val232, Phe234, Phe238, Leu239, Leu242, Phe243, and Tyr326. The majority of these residues are hydrophobic, indicating that the strong binding of the molecule to hCAR would be due to hydrophobic interactions.
5α-Androstan-17α-ol.
Like clotrimazole, this molecule only binds to one region of the cavity and a single binding mode was observed (Fig. 4c). The C17-hydroxyl oxygen is situated between the side chains of His203 and Asn165, displaying some degree of electrostatic interactions among the hydroxyl oxygen and the imidazole nitrogen of His203 and the amide nitrogen of Asn165. The C18 and C19 methyl groups of the ligand are in van der Waals contact with the side chains of Ile164, Met168, Leu206, and Tyr224. Additional hydrophobic interactions between the steroid core and hCAR were also observed.
TCPOBOP.
Six distinct binding modes were found for this molecule. The center of the molecule was seen at different locations in the cavity, encompassing the entire binding pocket. Figure 4d shows the most favorable binding mode from the program GOLD. The ligand is found to interact with residues Phe161, Ile164, Asn165, Cys202, His203, Leu206, Phe217, Cys219, Tyr224, Leu239, and Phe243. In this binding mode, interactions between the ligand and hCAR are primarily hydrophobic.
Noncompetitive Binding of TCPOBOP to Clotrimazole Bound hCAR.
Experimental data have shown that both 5α-androstan-17-ol and 5β-pregnanedione compete with clotrimazole for binding to hCAR, whereas TCPOBOP does not compete with clotrimazole. After analyzing all of the binding modes of 5α-androstan-17-ol and 5β-pregnanedione, it appears that each binding mode of these two ligands has some overlap with the binding region of clotrimazole. Therefore, binding of clotrimazole to hCAR may prevent these two ligands from binding to hCAR. On the other hand, in one of its binding modes, TCPOBOP is found to locate at a totally different part of the pocket from clotrimazole. To further confirm this finding, we carried out a calculation in which we took the bound clotrimazole as part of the protein and docked TCPOBOP into the complex. Two different binding modes were found for TCPOBOP. Figure 5 shows the best binding mode found for this dual-ligand complex. In this dual-ligand binding mode, the ligands are surrounded by residues Phe161, Ile164, Asn165, Met168, Val199, Cys202, His203, Leu206, Phe217, Try224, Phe234, Phe238, Leu242, His246, and Tyr326. One of the pyridine rings of the ligand stacks on the side chain of Phe234 (edge-to-face).

A stereoview of noncompetitive binding of TCPOBOP to clotrimazole bound hCAR.
Colors, active site reside carbon atoms in cyan, clotrimazole carbon atoms in gray, TCPOBOP carbon atoms in light pink, all nitrogen atoms in blue, oxygen atoms in red, sulfur in yellow, chlorine in light green. Labels for residues V199 and C202 were removed for clarity.
Similar docking experiments on both 5α-androstan-17-ol and 5β-pregnanedione showed that these two ligands cannot bind to hCAR in the presence of clotrimazole.
Discussion
According to Baker and Sali (2001), medium-high accuracy models may be obtained when more than 30% sequence identity exists between the templates and the modeling target. hCAR is most closely related to hPXR, sharing 43% sequence identity in the ligand binding domain. Both of receptors belong to the same NR1I subfamily. A common fold would be expected. However, hPXR contains a large insertion around the region of the ligand binding cavity and several structural changes that deviate from the canonical nuclear receptor fold. The next most closely related protein is hVDR, from the same NR1I subfamily, sharing 34% sequence identity in the ligand binding domain, which is high enough to surpass the statistical limit for having a similar structure. In this study, we combine the structural features of hPXR and hVDR to create a hybrid model for hCAR. This model displays a canonical fold that is shared by many other nuclear receptors.
hPXR has been shown to bind to a set of structurally diverse ligands, including small molecules like SR12813 (Watkins et al., 2001) to the extremely large ligand rifampicin. The size of the ligand binding cavity is almost the same in hCAR and hPXR. However, hPXR has a flexible surface loop adjacent to the ligand binding site. It is likely that a constriction can open to enlarge the binding cavity by including the volume enclosed by the surface loop. Rifampicin cannot fit into the cavity in the current hPXR X-ray structure. Structural changes have to occur to accommodate this large ligand in hPXR. In contrast to hPXR, hCAR lacks structural flexibility around the binding cavity. Therefore, it is likely that large molecules, such as rifampicin, will not be able to bind.
The ligand binding cavities of hCAR and hPXR share many similar features. Both are largely hydrophobic and are lined by a similar number of amino acid residues. The binding cavity volumes of hPXR and hCAR are similar and are substantially larger than those of many other nuclear receptors. We believe that this large binding cavity may be responsible for their capability to bind many structurally diverse hydrophobic compounds (Moore et al., 2000). In addition, both hPXR and hCAR share similar xenobiotic and steroid ligands. This type of substrate promiscuity has not been observed in the steroid, retinoid, and thyroid receptors, which are highly specific for their cognate hormones. The large binding cavities of hCAR and hPXR permit multiple binding modes as demonstrated in this study and for SR12813 bound hPXR (Watkins et al., 2001).
A diverse range of compounds have been identified to be ligands of hCAR by competitive ligand binding and reporter gene assays (Moore et al., 2000). These ligands include clotrimazole, 5α-androstan-17-ol and 5β-pregnanedione. Clotrimazole and 5β–pregnanedione are agonists for hCAR, whereas 5α-androstan-17-ol is an antagonist. Although TCPOBOP has not been demonstrated to be a ligand of hCAR, it can induce CYP2B in human tissues (Smith et al., 1993). Docking of these ligands to the modeled hCAR ligand binding site showed that all of them fit well inside the cavity. The interactions between the ligands and the protein are largely hydrophobic. We found that TCPOBOP and clotrimazole could occupy the cavity simultaneously with each compound occupying a different part of the cavity. On the other hand, the binding sites of 5α-androstan-17-ol and 5β-pregnanedione overlap with that of clotrimazole in the docking model. These results are consistent with the findings that TCPOBOP does not compete with clotrimazole in the direct binding assay, whereas 5β–pregnanedione and 5α-androstan-17-ol do. To confirm this structural model, we are in the progress of determining the 3D structure of hCAR by X-ray crystallography and by NMR.
Conformational changes in the ligand binding domain have been observed among the apo, agonist-bound, antagonist-bound, and partial agonist-bound forms in many nuclear receptor X-ray structures (Bourguet et al., 2000a; Steinmetz et al., 2001). The two templates used here to build the hCAR model are both in the ligand-bound form and similar to the agonist-bound conformation, although the reported structures of the apo and ligand bound forms of the hPXR are almost identical (Watkins et al., 2001). In modeling of hCAR we have made a reasonable and practical assumption that there is no conformational change upon ligand binding.
This study has highlighted similarities and differences in CAR and PXR ligand binding. Continued modeling and structural studies will facilitate development of drugs lacking side effects due to enzyme induction.
Acknowledgments
We thank Drs. Diana Montgomery and Anthony Y. H. Lu for critically reading the manuscript.
Footnotes
- Abbreviations used are::
- CAR
- constitutive active (or androstane) receptor
- PB
- phenobarbital
- RXR
- 9-cisretinoic acid receptor
- TCPOBOP
- 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene
- PXR
- pregnane X receptor
- VDR
- vitamin D receptor
- PPAR
- peroxisome proliferator activator receptor
- hCAR
- human CAR
- hVDR
- human VDR
- PDB
- Protein Data Bank
- Received April 19, 2002.
- Accepted June 3, 2002.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}