Article Text

Abstract
OBJECTIVE To evaluate the correlation between changes in platelet monoamine oxidase type B (MAO-B) activity and plasma β-phenylethylamine (PEA) concentrations in patients with Parkinson's disease and controls.
METHODS Platelet MAO-B activity and plasma PEA were measured with gas chromatography-mass spectrometry (GC-MS) in patients with Parkinson's disease treated with levodopa (12 men and 12 women) or selegiline (three men and three women), and physically healthy subjects as a control group (10 men and 10 women).
RESULTS Platelet MAO-B activity was significantly higher in the Parkinson's disease group (mean 542 (SD 318) pmol/107 platelets/30 min) than in the control group (mean 349 (SD 307) pmol/107 platelets/30 min) (p<0.05). By contrast, the plasma PEA concentrations in patients with Parkinson's disease were significantly lower than in the control group (mean 532 (SD 243) pg/ml; 931 (SD 560) pg/ml) (p<0.01). The plasma PEA concentrations in patients with Parkinson's disease treated with selegiline were prominently higher than in patients with no selegiline treatment (p<0.001). There was a significantly negative correlation between platelet MAO-B activity and plasma PEA concentrations in patients (n=24, r=−0.466, p<0.001).
CONCLUSIONS The increase in platelet MAO-B activity and decrease in plasma PEA concentrations in patients with Parkinson's disease may be involved in the pathophysiological processes of the disease, and these changes are reversed by treatment with selegiline.
- monoamine oxidase B
- β-phenylethylamine
- Parkinson's disease
Statistics from Altmetric.com
The pathophysiological basis of Parkinson's disease is nigrostriatal neuronal degeneration, characterised by a deficiency of the nigrostriatal dopaminergic system. The physiological role of monoamine oxidase (MAO) is in the regulation of the concentrations of biogenic amines in the brain by metabolising neurotransmitters or neuromodulators. Monoamine oxidase type B (MAO-B) catabolises phenylethylamine and is inhibited by selegiline. Activity of MAO in human blood thrombocytes, which is almost exclusively of the B type, is thought to reflect brain MAO-B activity,1 and this enzyme has been studied widely in several neuropsychiatric disorders. Although there are conflicting results in patients with Parkinson's disease, platelet MAO-B concentrations are still considered to play a part in the pathophysiology of the disease.2 It has been determined that MAO-B mediates the toxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) by catalysing the formation of the neurotoxic 1-methyl-4-phenylpyridine (MPP+) which produces parkinsonism in intravenous drug misusers. Thus, the oxidative stress compounds that may cause Parkinson's disease would be synthesised by MAO-B in primates.3
β-Phenylethylamine (PEA) is an endogenous trace amine, synthesised by decarboxylation of phenylalanine in dopaminergic neurons of the nigrostriatal system, and may act as a neuromodulator of catecholamine neurotransmission in the brain.4 We first showed that the concentrations of PEA in the CSF in patients with Parkinson's disease are markedly decreased compared with those in controls, and reported the novel role of PEA in patients with Parkinson's disease.5 Moreover, PEA is a typical substrate for MAO-B.6 However, the relation between platelet MAO-B activity and plasma PEA concentrations in patients with Parkinson's disease is not yet understood in detail. To evaluate the alteration in platelet MAO-B activity and plasma PEA concentrations in patients with Parkinson's disease and controls, we established a method for the determination of platelet MAO-B activity by gas chromatography-mass spectrometry (GC-MS).
Patients and methods
SUBJECTS
All except two patients with Parkinson's disease, including patients treated with selegiline, had been taking antiparkinsonian drugs, including levodopa-benserazide and bromocriptine or pergolide. All subjects were non-smokers.
The Parkinson's disease group was made up of 24 patients treated with levodopa (12 men, mean age 64.9 (SD 4), range 56–71 years; 12 women, mean 63.6 (SD 10), range 42–76 years). Two patients who had not been treated with levodopa-benserazid were also included in this group. The mean amount of levodopa given was 240 (SD 80) mg/day, ranging from 100–400 mg/day. The mean duration of Parkinson's disease was 7.7 (SD 4.5) with a range of 1–17 years. The severity of Parkinson's disease was rated according to Hoehn and Yahr stage7 (Hoehn and Yahr stage I, n = 3; stage II, n= 5; stage III, n=3; stage IV, n=11; stage V, n=2).
Six patients were treated with selegiline (three men and three women, mean age 55 (SD 6) years; Hoehn and Yahr stage I, n=2; stage III, n=2; stage IV, n=2). The mean amount of levodopa given was 200 (SD 50) mg/day. Selegiline was given at a rate of 3.75 mg/day for each patient for 3–6 weeks, and they were neurologically evaluated before and after selegiline administration.
The control group was composed of 20 physically healthy subjects with no concurrent disease and claiming to be drug free (10 men, mean age 60.2 (SD 8.5) range 48–73 years; 10 women, mean age 61.3 (SD 6.6), range 49−72 years).
With informed consent, 7 ml fasting venous blood was drawn into two plastic tubes containing an anticoagulant agent in the morning between 8 00 and 9 00 am and after bed rest for 30 minutes. They were not allowed to eat cheese or chocolate or drink wine for a few days, and no drugs were taken in the morning. Platelet rich plasma for MAO-B measurements was obtained by gentle centrifugation at 200g for 10 minutes, and the number of platelets in the platelet rich plasma was determined. Another sample was centrifuged at 3000 g for 10 minutes for the PEA measurement. Plasma and plasma rich plasma were stored separately at −80°C until assay.
DETERMINATION OF MAO-B ACTIVITY
Platelet MAO-B activity was determined by GC-MS (GC-17 A and QP-5000 Mass Spectrometer, Shimadzu, Kyoto, Japan). Sample preparation and the incubation of samples were performed according to the method of Husseini et al 8 with a slight modification. Briefly, the platelet pellet was resuspended with saline to obtain a concentration of 107 platelets/ml and then sonicated for 10 seconds. After preincubation of 50 μl suspension with 80 μl 100 mM KH2PO4 at 37°C for 5 minutes, the suspension was incubated with 20 μM PEA and 0.15 units aldehyde dehydrogenase at 37°C for 30 minutes. One sample was incubated at 0°C. To another was added 0.24 mM pargyline, an MAO-B inhibitor, as a blank. A capillary column (0.23 mm internal diameter, 30 m long, J and W Scientific Co, Folsom, CA, USA) coated with DB-5 was used. The mass numbers used for the quantitative analysis were m/z 268, corresponding to phenylacetic acid (PAA), and m/z 282, corresponding top-methylphenyl acetic acid (mPAA). A peak area measurement was used to estimate the ion current. The calibration curve was highly correlated in standard samples (r=0.947) in the range of 10–500 ng/ml. There was no production of PAA in the samples incubated at 0°C or with added pargyline. Enzyme activity was calculated from the production of PAA and expressed in pmol product /107platelets/30 min.
The determination of PEA was performed by GC-MS, as described previously, and expressed in pg/ml.9
STATISTICS
Data are summarised as the mean (SD). For statistical analysis, we used analysis of variance (ANOVA) followed by the Fisher's PLSD test or the Student's t test (two tailed) for comparing group means. The relation between platelet MAO-B activity and PEA concentration was analyzed with Pearson's correlation. A p value<0.05 was considered significant.
Results
The platelet MAO-B activity was significantly higher in patients with Parkinson's disease (n=24, mean 542 (SD 318) pmol/107platelets/30 min) than in the control group (n=20, mean 349 (SD 307) pmol/107 platelets/30 min) (p<0.05). The MAO-B activity in the patients treated with selegiline was the lowest among the three groups (n=6, mean 240 (SD 244) pmol/107 platelets/30 min; p<0.05 when compared with that of patients without selegiline, fig 1). By contrast, the plasma PEA concentrations in patients with Parkinson's disease were significantly lower than those in the control group (n=24, mean 532 (SD 243) pg/ml for Parkinson's disease; n=20, mean 931 (SD 560) pg/ml for control) (p<0.01). The plasma PEA concentrations in the two patients not treated with levodopa-benserazid were 375 pg/ml and 235 pg/ml.
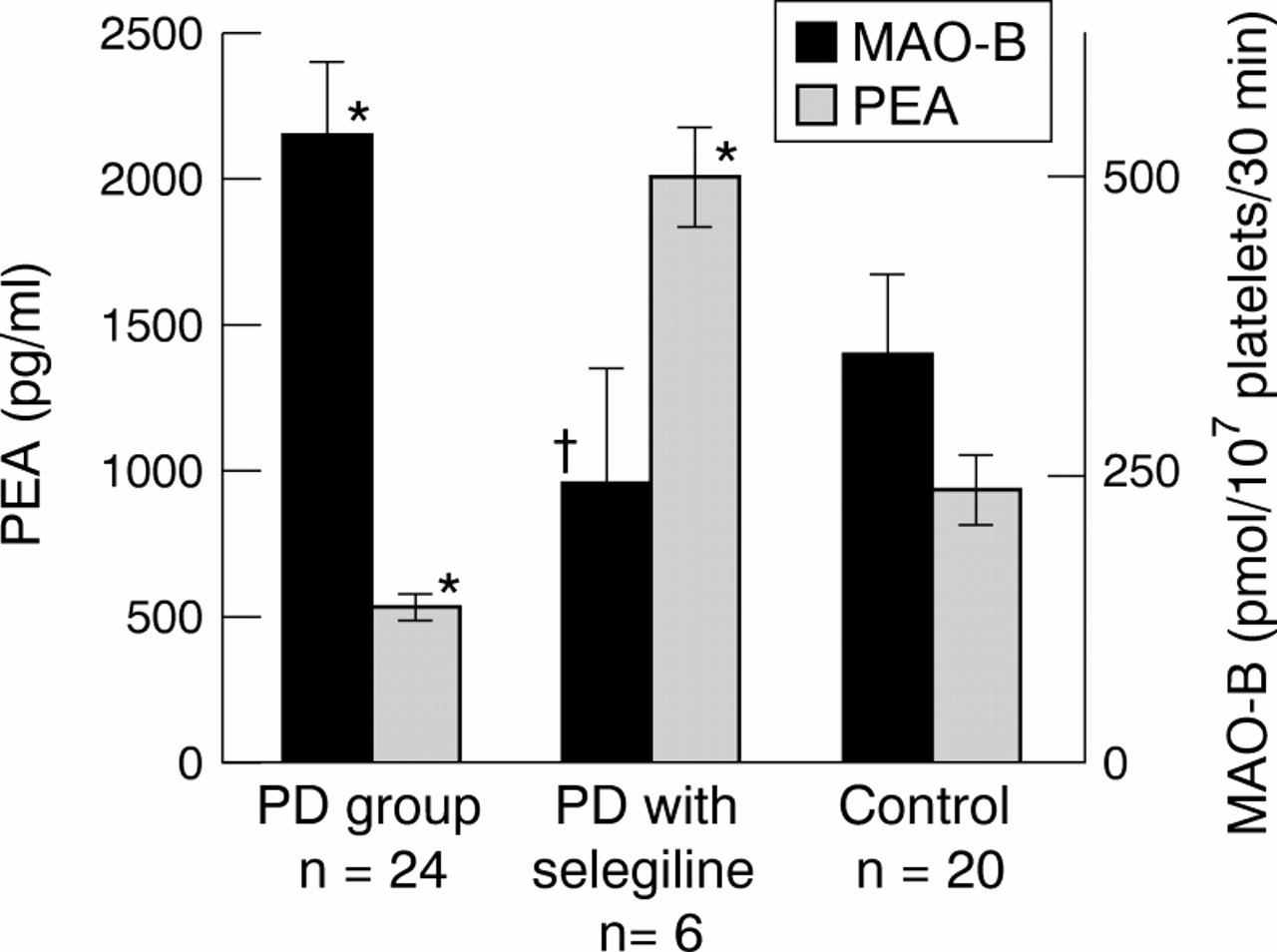
Results are given as mean (SD). Platelet MAO-B activity is expressed in pmol/107 platelets/30 min; plasma PEA concentration is expressed in pg/ml. *Significant differences compared with the control group; †significant difference compared with Parkinson's disease group.
The six patients treated with selegiline showed significantly higher concentrations of plasma PEA than patients treated without selegiline (mean 2010 (SD 425) pg/ml, p<0.001, fig 1). The three patients showed a rapid improvement in their neurological symptoms, although their Hoehn and Yahr stage was unchanged.
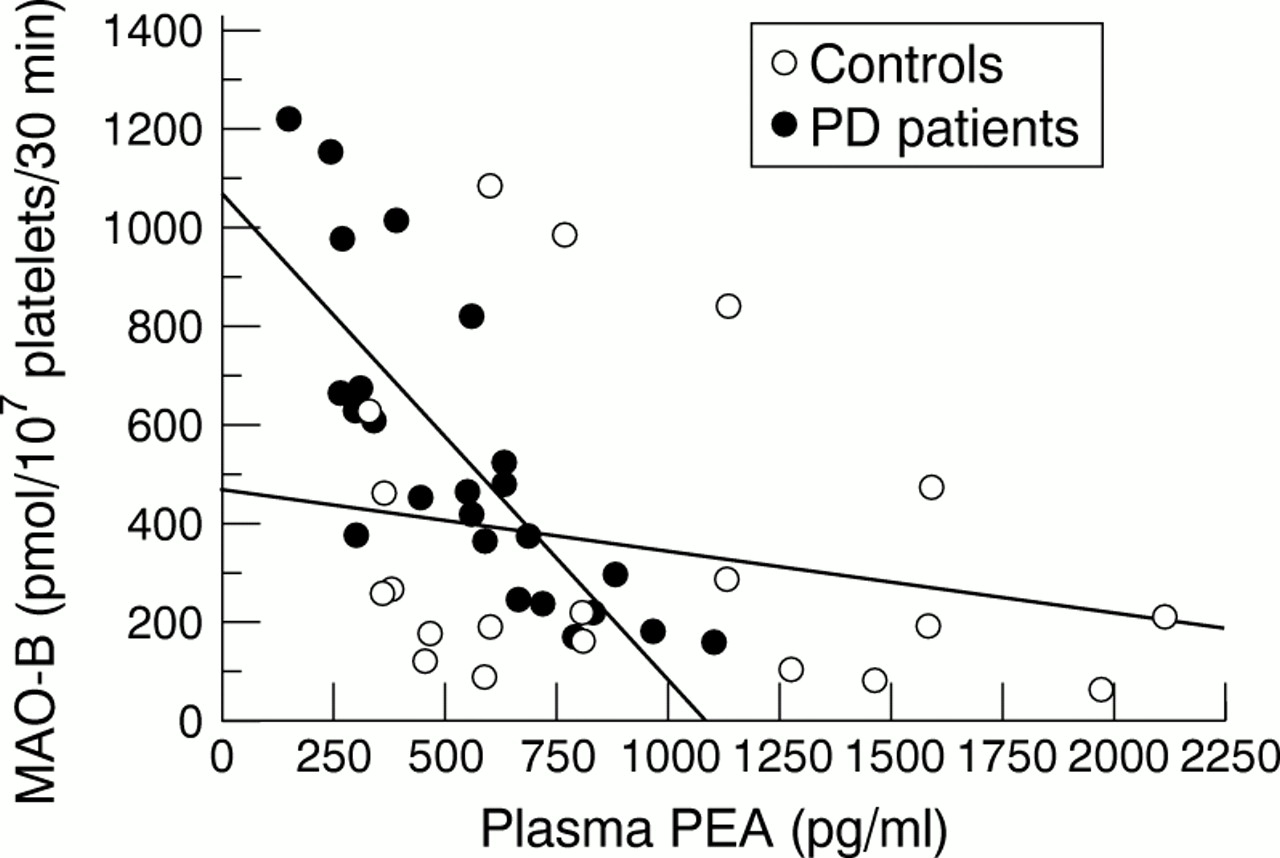
There was a significant negative correlation between platelet MAO-B activity and plasma PEA concentrations in the patients and the control group (n=44, r=−0.466, p<0.001 for all subjects; n=24, r=−0.74, p<0.0001 for patients, fig 2). No significant correlation was found between PEA or MAO-B and the duration of Parkinson's disease, the severity of disease, or the amount of levodopa.

{kind=link}
{kind=link}
Plasma PEA is negatively correlated with platelet MAO-B activity (n=44, r=−0.466, p<0.001), especially in patients with Parkinson's disease (n=24, r=−0.74, p<0.0001).
Discussion
The platelet MAO-B activity in our patients was higher than in healthy controls. The activity of this enzyme in patients with Parkinson's disease has been variously reported to be reduced,10 not different,11 12 and significantly increased.13 14 The conflicting results might be explained by the use of different substrates. Activity of MAO-B has been reported to be higher in untreated patients with Parkinson's disease than in controls when using PEA as a substrate, which is compatible with the present result, but it decreases significantly when using dopamine as a substrate.13 In addition, it is also possible that treatment with exogenous levodopa may cause an increase in MAO-B activity.
Our present results show a significantly reduced concentration of plasma PEA in patients with Parkinson's disease. We have previously reported reduced CSF PEA concentrations in Parkinson's disease and in Rett syndrome—which may reflect an impairment of the dopamine system—compared with concentrations in age matched controls.15 It remains unclear whether the reduced concentrations of PEA are due to the degeneration of dopaminergic neurons or the effect of the levodopa treatment. However, the two patients who did not take levodopa in our study also presented reduced concentrations of PEA.
Based on our present data, there is a significant negative correlation between MAO-B activity and the concentration of PEA, as previously reported in normal subjects.11 It seems that a change in MAO-B activity could directly affect the plasma PEA concentrations. It has been reported that 10 mg/day selegiline can cause a more than 80% inhibition of MAO-B activity in achieving a functional effect.16 The amount of selegiline used in the present study was small, as the patients were examined just at the time of the initial dose of selegiline. However, the low dose of selegiline did markedly increase the plasma PEA concentrations, with some clinical improvement. It has been reported that endogenous concentrations of PEA antagonise the quinpirole induced reduction of evoked dopamine release from rat striatum in vitro,17 and cause turning behaviour in hemistriatal lesioned rats. These data and our results suggest that PEA may be important as a neuromodulator to improve symptoms of Parkinson's disease, and that it may provide another way to explore the role of selegiline.18 Because of the few patients in this preliminary study, further studies are needed to elucidate this link. Thus, the measurement of plasma PEA as well as platelet MAO-B activity might provide some information on pathophysiological processes of Parkinson's disease and the action mechanism of selegiline.
Acknowledgments
We thank Miss M Fukamachi for assistance in the collection of plasma and platelet rich plasma samples.