


Abstract
We investigated the mechanism of the hypotensive effect of Sar-[d-Phe8]des-Arg9-bradykinin (BK) in lipopolysaccharide-treated anesthetized rabbits. The study involved pharmacokinetic and hemodynamic measurements and tests of antagonism with various drugs. The rate of elimination of Sar-[d-Phe8]des-Arg9-BK from the rabbit plasma was slower than that of Lys-BK, a naturally occurring B1 agonist. The amplitude of the hypotensive effect of Sar-[d-Phe8]des-Arg9-BK was not affected by pretreatment with indomethacin, diclofenac, dazmegrel, NG-nitro-l-arginine, glibenclamide, MK-886, BN-50739, atropine or propranolol, but its duration was shortened by indomethacin and diclofenac. Sar-[d-Phe8]des-Arg9-BK-induced hypotension was associated with decreases of total peripheral resistance, cardiac output, carotid, mesenteric and femoral blood flow, transient reductions followed by secondary increases of vascular resistance in the carotid and femoral beds, reductions of central venous pressure, but no change of hematocrit. Animal pretreatment with diclofenac or hexamethonium abolished the secondary increases of carotid bed vascular resistance caused by the B1 agonist. These and other results suggest that peripheral vasodilation leading to a decrease of total peripheral resistance and a decrease of cardiac output may both contribute consecutively to the hypotensive effect of Sar-[d-Phe8]des-Arg9-BK in this animal model. Inappropriate compensatory responses to arterial hypotension, prostaglandin release, and slow rate of elimination of Sar-[d-Phe8]des-Arg9-BK from the rabbit plasma, may all be at the basis of the prolonged duration of the hypotension caused by the B1 agonist.
The existence of a distinct kinin B1 receptor regulated by cytokines has been confirmed recently by the cloning and sequencing of a G protein-coupled receptor from human cells that is selectively stimulated by Lys-des-Arg9-BK and blocked by Lys-[Leu8]des-Arg9-BK (Menke et al., 1994). Constitutive B1- and B2-kinin receptors mediate the cardiovascular effects of kinins in vivo in the dog (Lortie et al., 1992; Nakhostineet al., 1993) and cat (DeWitt et al., 1994), whereas in other species such as the rabbit (Regoli et al., 1981; Drapeau et al., 1991), swine (Siebeck et al., 1989) and rat (Tokumasu et al., 1995), the cardiovascular effects of classical agonists of kinin B1receptors (i.e., des-Arg9-BK and Lys-des-Arg9-BK) are mediated by LPS- or cytokine-inducible kinin B1 receptors, and those of BK itself, by constitutive kinin B2 receptors (Marceau, 1995).
The sensitization of the cardiovascular system of rabbits to B1 agonists by LPS, or by components of the cytokine network (e.g., interleukin-1) (Bouthillier et al., 1987; deBlois et al., 1991), is currently believed to result from an up-regulation of kinin B1 receptors (deBlois et al., 1991; Marceau, 1995). Recent in vitro binding experiments with vascular smooth muscle cells of rabbits strongly support the hypothesis that induction of B1 receptors is responsible for the sensitization of the cardiovascular system of rabbits treated with LPS or cytokines to B1 agonists (Schneck et al., 1994; Galizziet al., 1994; Levesque et al., 1995). Hypotensive responses of LPS-treated rabbits to B1 agonists are inhibited by sequence-related antagonists of B1 receptors (Regoli et al., 1981; Drapeau et al., 1993), but not by HOE 140, a kinin B2 receptor antagonist (Drapeauet al., 1993), thus indicating that kinin B1rather than B2 receptors mediate the hypotensive effect of B1 agonists in LPS-treated rabbits.
The precise mechanism underlying B1 agonist-induced hypotension in LPS-treated rabbits is unknown. B1 agonists are known to relax isolated mesenteric and celiac arteries of rabbits by a PG-mediated process (Churchill and Ward, 1986; deBlois and Marceau, 1987; Ritter et al., 1989). They also increase the production of prostacyclin from isolated rabbit aorta rings and cultured smooth muscle cells derived from the rabbit aorta (Levesqueet al., 1993). However the hypotensive effect of des-Arg9-BK, a prototypical B1 agonist, in LPS-treated rabbits is not reduced by indomethacin treatment (Regoliet al., 1981). On the other hand indomethacin was shown recently to reduce the duration, but not the amplitude of hypotensive episodes elicited by Sar-[d-Phe8]des-Arg9-BK, a metabolically protected B1 agonist, in LPS-treated rabbits (Drapeau et al., 1991).
Des-Arg9-BK, acting through B1 receptors, was shown previously to relax isolated rabbit carotid artery rings (Pruneau and Bélichard, 1993), an effect which was inhibited by endothelium removal and Nω-nitro-l-arginine. This result led the authors to suggest that kinin B1receptors are coupled to the release of endothelium-derived NO. However the participation of NO in the vasorelaxant effect of B1agonists in rabbit isolated vascular tissues may be limited to selected blood vessels, because B1 agonist-induced relaxation of rabbit celiac artery rings is not dependent on an intact endothelium (Ritter et al., 1989). Whether or not NO mediates part of the hypotensive effect of B1 agonists in LPS-treated rabbits is as yet unknown.
The present study investigated the mechanism by which B1agonists elicit hypotension in LPS-treated rabbits, with Sar-[d-Phe8]des-Arg9-BK, a metabolically protected B1 agonist (Drapeau et al., 1991, 1993; Davis and Perkins, 1994a) as prototype, and a variety of experimental approaches (e.g., pharmacokinetic and hemodynamic measurements and inhibitor studies). This study is the first to describe in detail the cardiovascular consequences of a persistent stimulation of B1 receptors for kinins. Its relevance relied on two important points: 1) plasma immunoreactive des-Arg9-BK is increased in animals pretreated with LPS (Raymond et al., 1995); 2) B1 receptors for kinins are up-regulated in LPS-treated animals (see above). Knowledge from such a study might provide a rationale to decide whether the inhibition or stimulation of B1 receptors for kinins is likely to be beneficial to the septic subjects. The results are consistent with the idea that the hypotensive effect of Sar-[d-Phe8]des-Arg9-BK in LPS-treated rabbits involves an early episode of peripheral vasodilation leading to a decrease of TPR, followed by an episode of a decrease of CO.
Methods
Studies were performed with New Zealand White rabbits (1.5–2.0 kg) of either sex. Animals were given free access to standard rabbit chow and tap water, and kept under constant environmental conditions for a few days before use. Experiments were conducted in accordance with the principles and guidelines of the Canadian Council on Animal Care and approved by the local Institutional Committee on Animal Care.
Pharmacokinetic measurements.
The plasma clearance and biological half-life in plasma (T½) of Sar-[d-Phe8]des-Arg9-BK, a metabolically protected agonist of B1 receptors for kinins, and of Lys-des-Arg9-BK, a naturally occurring B1 agonist (Drapeau et al., 1991), were determined and compared with intact (i.e., no pretreatment with LPS) pentobarbital-anesthetized (see below) rabbits. After implantation of a polypropylene catheter (PE-90) into the left carotid artery down to the aorta, animals were given an intraarterial injection of either of the two peptides (3 mg/animal). Arterial blood samples (2 × 1.35 ml) were collected 1 min before (t = 0) and at different times (1, 3, 5, and 10 min) after injection of the B1 agonists in 1.5-ml test tubes containing 1,10-phenanthroline and amastatin (final concentrations, 5 mM and 5 μM, respectively, to prevent the enzymatic degradation of the two B1 agonists), and were centrifuged at 10,000 ×g for 1.5 min. Recovered plasmas (0.75 ml) were supplemented with 4.5 ml of cold (−80°C) ethanol (99% v/v), mixed thoroughly and allowed to rest for 60 min on ice before being centrifuged again at 2000 × g (at 4°C) for 15 min. Supernatants were transferred into polypropylene tubes and evaporated overnight in a Speed-Vac apparatus; the plasma residues were stored at −20°C for 2 to 4 weeks before being analyzed for their content in B1agonists (see above) by HPLC.
The plasma residues were resuspended in 300 μl of trifluoroacetic acid (0.05% v/v), mixed thoroughly, centrifuged at 2500 ×g (4°C) for 40 min, and aliquoted for injection onto the HPLC column. HPLC analysis was performed with a Waters system equipped with a 746 data module and a 486 UV detector set at 214 nm. Separation was achieved with a Vydac 10 μ (3.9 × 300 mm) reverse-phase C18 column with a linear gradient of 5 to 65% acetonitrile/trifluoroacetic acid 0.05% water at 2 ml/min for 20 min. Peptide recoveries from spiked plasma samples were essentially complete in control experiments.
The plasma clearance and T½ values of Sar-[d-Phe8]des-Arg9-BK and Lys-des-Arg9-BK were measured by use of plasma concentration versus time curves obtained from each animal and by equations of first-order drug elimination processes (Rowland and Tozer, 1989). The distribution phase was assumed to be complete at the 1-min time point after injection.
Effect of various inhibitors on the hypotensive effect of Sar-[d-Phe8]des-Arg9-BK.
The experiments were aimed at assessing the potential involvement of secondary mediators in the hypotensive effect of Sar-[d-Phe8]des-Arg9-BK in the LPS-treated rabbit. Animals were pretreated with a sublethal dose of LPS (30 μg/kg) injected into a marginal ear vein 5 h before anesthesia with sodium pentobarbital (30–40 mg/kg i.v., adjusted individually). Lidocaine 2% was applied topically at sites of incision. The trachea was intubated and ventilatory assistance was provided with a Harvard respiratory pump. The body temperature of animals was monitored continuously with a rectal thermoprobe connected to a telethermometer (Yellow Springs Instruments, Yellow Springs, OH), and kept constant at 38–39°C using a heating pad placed underneath the animal. A polyethylene catheter (PE-90) was inserted into the left carotid artery and pushed into the aorta for direct recording of BP with a pressure transducer (Spectramed P23 XL) connected to a Grass polygraph (model 79); a three-way valve connector was attached to the catheter to interrupt the BP recording and to inject drugs intraarterially. Hypotensive responses to Sar-[d-Phe8]des-Arg9-BK (750 ng/kg i.a.) were measured in animals pretreated with various inhibitors (one inhibitor at a time) (see below) and compared with those measured in drug vehicle-pretreated (control) animals. The inhibitors tested were (dose in mg/kg): indomethacin (5), diclofenac (5), NG-nitro-l-arginine (30), dazmegrel (1), glibenclamide (15), MK-886 (3), BN-50739 (10), atropine (5) and propranolol (5). The dose of indomethacin selected was previously shown to acutely suppress the increase of immunoreactive circulating prostanoids (PG, thromboxanes) induced by the anaphylatoxin C5a in the anesthetized rabbit (Lundberg et al., 1987). Diclofenac (also a cyclooxygenase inhibitor) was used at the same dose as indomethacin for the purpose of comparison. The chosen dose of NG-nitro-l-arginine, an inhibitor of NO synthases (Moore et al., 1990), was selected on the basis of its ability to reduce the NO-mediated hypotensive responses to cumulative i.a. infusions of ACH (1, 3, 6 and 12 μg/kg/min) in our animal model. Control animals in these studies had their basal MABP first increased to levels similar to those of NG-nitro-l-arginine-treated animals with phenylephrine (10–50 μg/kg/min i.v.) as vasopressor drug before ACH infusions (Rees et al., 1989, 1990). Dazmegrel, a potent and selective thromboxane synthetase inhibitor, was used at a dose previously shown to reduce serum thromboxane B2 levels by 88% in anesthetized rabbits (Parry et al., 1982). Glibenclamide, an inhibitor of ATP-sensitive K+ channels (Edwards and Weston, 1993), was used at a dose shown in preliminary experiments to reduce by 75% the hypotensive effect of cromakalin (100 μg/kg i.a.), an activator of ATP-sensitive K+ channel (Edwards and Weston, 1993). The selected dose of MK-886 was previously shown to be effective as inhibitor of leukotriene biosynthesis in various animal models (Gillard et al., 1989). BN-50739, an inhibitor of PAF (Yue et al., 1990), was used at a dose found in preliminary experiments to suppress the hypotensive effect of PAF (500 pmol/kg i.a.) in our animal model. Atropine and propranolol were both used at doses shown in preliminary experiments to suppress the hypotensive effect of ACH (3 μg/kg i.a.) and isoproterenol (1 μg/kg i.a.), respectively. Each inhibitor (or its vehicle) was injected intravenously 15 min before injection of Sar-[d-Phe8]des-Arg9-BK.
Hemodynamic effects of Sar-[d-Phe8]des-Arg9-BK.
These experiments were performed to obtain further insight into the mechanism of Sar-[d-Phe8]des-Arg9-BK-induced hypotension in LPS-pretreated rabbits. With use of pentobarbital-anesthetized animals we first examined the effect of bolus injections of Sar-[d-Phe8]des-Arg9-BK on the following hemodynamic parameters: MABP, HR, CBF and CVR. In some of these animals the effect of the B1 agonist on the CVP and HTC was also monitored. BP was measured as described above. HR was derived from the BP signal by use of a Grass tachograph (model 7P 44). CBF was measured with a precalibrated blood flow probe (1.5 mm in internal diameter) (Skalar Medical, Delft, The Netherlands) placed around the right common carotid artery and connected to a Skalar electromagnetic blood flowmeter (model MDL 1401). The flow probe was zeroed in saline at room temperature before placement on the carotid artery and the zero was checked in vivo by transient (∼1–2 sec) clamping of the carotid artery upstream of the carotid flow probe. CBF was recorded on a Harvard recording system (model 52–9545). CVR was calculated by dividing MABP values by CBF values. CVP measurements were made by a PE-90 catheter introduced into the right jugular vein and positioned close to the right atrium of the heart. Pressure transducers (for MABP or CVP) were positioned at the level of the heart. All animals received 400 U of heparin i.v., and a 30-min period was allowed for the stabilization of hemodynamic parameters before injection of Sar-[d-Phe8]des-Arg9-BK (750 ng/kg i.a.). MABP, HR, CBF, CVR, CVP and HTC (via carotid blood samplings) values were measured before (preinjection, control values) (t = −1 min) and different times (0.2, 0.5, 1, 2, 5 or 10 min) after injection of the B1 agonist. Preliminary studies having shown that Sar-[d-Phe8]des-Arg9-BK, despite eliciting relatively prolonged hypotensive episodes in this animal model, decreases CVR values only transiently, we also examined the possibility that compensatory mechanisms involving either the prostanoids and/or the autonomic nervous system contribute to mask the vasodilatory property of the B1 agonist and its effect on CVR values. To test this possibility we measured the hemodynamic effects of Sar-[d-Phe8]des-Arg9-BK in animals pretreated with the cyclooxygenase inhibitor, diclofenac, or with the ganglion-blocking drug, hexamethonium. MABP, HR, CBF and CVR values were measured before (t = −1 min) and at selected times (0.2, 0.5, 1, 2, 5 and 10 min) after injection of Sar-[d-Phe8]des-Arg9-BK (750 ng/kg i.a.). The B1 agonist was injected 15 min after animal pretreatment with diclofenac (5 mg/kg i.v.) or hexamethonium (10 mg/kg i.a.).
Additional experiments were conducted to determine whether the changes of vascular resistance observed in the carotid vascular bed after i.a. injection of Sar-[d-Phe8]des-Arg9-BK were representatives of those occurring in other vascular beds. We first examined the effect of the B1 agonist on the femoral vascular bed with LPS-treated pentobarbital-anesthetized rabbits. Animals were instrumented as described above for MABP and HR measurements, and the blood flow probe (as above) was placed around the left femoral artery. MABP, HR, FBF and FVR were measured before and at different times after injection of the B1 agonist (as above). FVR was obtained by dividing MABP by FBF values. Second, we examined the effect of the B1 agonist on the mesenteric vascular bed. Given the more extensive level of surgery required by such studies, these experiments were done in LPS-pretreated rabbits anesthetized with halothane (1–2% in O2) rather than with barbiturates. Once the anesthesia level was deep enough to prevent the animal responses to noxious stimuli (e.g., cutting of the skin with scissors), the animals were instrumented for BP and HR recording, as well as for i.v. or i.a. injections of drugs (as above). Thereafter, the abdominal cavity was opened longitudinally, and the superior mesenteric artery was located and isolated gently from its surroundings. A precalibrated blood flow probe (1.5 mm in internal diameter) was placed around the superior mesenteric artery and connected to a Skalar electromagnetic blood flowmeter as described above. MBF was recorded on a Harvard recorder (as above). MVR was calculated by dividing MABP values by MBF values. MABP, HR, MBF and MVR values were measured before (t = −1 min) and at selected times (0.2, 0.5, 1, 2, 5 and 10 min) after injection of Sar-[d-Phe8]des-Arg9-BK (750 ng/kg i.a.). In some experiments, Sar-[d-Phe8]des-Arg9-BK was injected in animals in which the basal MVR was artificially raised, to determine if this condition alters the effect of the B1agonist on MVR. The increase of basal MVR was obtained by prior i.v. infusion of animals with the sympathomimetic drug, methoxamine (50–150 μg/min). The B1 agonist was injected 15 min after starting the methoxamine infusion.
Finally, based on the results of regional blood flow and vascular resistance studies, we investigated the effect of Sar-[d-Phe8]des-Arg9-BK on the CO and TPR of LPS-treated rabbits. These experiments were performed in pentobarbital-anesthetized rabbits premedicated 30 min before anesthesia with diazepam (5 mg/kg i.m.). Animals were instrumented for MABP and HR measurements, as well as for i.a. injections of Sar-[d-Phe8]-des-Arg9-BK (750 ng/kg), as described above. All animals were ventilated (open-circuit) with 100% O2 instead of room air, and were given an i.v. infusion of saline (total volume, 25–50 ml) during the surgical period only. The thoracic cavity was opened through a midline longitudinal incision, and the pericardium retracted with small forceps. After careful dissection of the aortic arch, the blood flow probe (4.5 mm in internal diameter) was placed around it and used to measure CO. MABP, HR, CO and TPR were measured before and at different times after injection of the B1 agonist (as above). TPR was derived from the ratio MABP/CO.
Drugs.
LPS, extracted from Escherichia coliserotype O111:B4, was from Difco (Detroit, MI). Lys-des-Arg9-BK and BK were purchased from Peninsula Laboratories (Belmont, CA). Sar-[d-Phe8]des-Arg9-BK was synthetized in our laboratory by solid-phase methodology (Drapeau and Regoli, 1988). Halothane was from M.T.C. Pharmaceuticals, Cambridge, Ontario, and diazepam injectable emulsion from KabiVitrum, Newmarket, Ontario. Methoxamine (Vasoxyl) was from Burroughs Wellcome (Kirkland, Quebec). Indomethacin, sodium diclofenac, NG-nitro-l-arginine, glibenclamide, atropine sulfate, hexamethonium bromide, propranolol hydrochloride, NG-nitro-l-arginine methyl ester, acetylcholine chloride, isoproterenol hydrochloride, phenylephrine hydrochloride andl-α-phosphatidylcholine β-acetyl-γ-O-alkyl (PAF) were all from Sigma Chemical Co. (St. Louis, MO). Dazmegrel was a gift from Pfizer Inc., (Groton, CT). MK-886 (or 3-[1-(4-chlorobenzyl)-3-t-butyl-thio-t-isopropyl-indol-2-yl]-2-2-dimethylpropanoic acid) was provided by Merck Frost (Pointe-Claire, Dorval, Québec). BN-50739 (or 6-(2-chlorophenyl)-9-[3,4-dimethoxyphenylthiomethyl) thiocarbonyl]-1-methyl-7,8,9,10-tetrahydro-4H-pyrido[4′,3′-4,5]-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepine) was a gift from Dr. Pierre Braquet (Institut Henri Beaufour, Le Plessis Robinson, France). Indomethacin and NG-nitro-l-arginine were dissolved with 0.1 M Na2CO3. Dazmegrel was dissolved with 35% (v/v) ethanol in saline. Glibenclamide was dissolved with 100% DMSO. Diclofenac was dissolved with 25% DMSO in 0.1 M Na2CO3. MK-886 was dissolved with 70% (v/v) ethanol in distilled water. BN-50739 was dissolved with 100% DMSO. All other drugs were dissolved with saline. All drug solutions were made fresh daily.
Statistics.
Data are mean ± S.E.M. Statistical analysis was made by the Student’s t test for unpaired data (for results, see fig. 1), or by the Kruskal-Wallis test followed by the Mann-Whitney test (for other results). P ≤ .05 was considered significant.

Time-related decreases of arterial plasma concentrations of Sar-[d-Phe8]des-Arg9-BK (solid bars) and of Lys-des-Arg9-BK (hatched bars) in intact (no LPS pretreatment), pentobarbital-anesthetized rabbits. Each animal was given a single i.a. injection of 3 mg of either one of these peptides. Bars indicate mean values ± S.E.M. (lines at the top of the bars) of three animals. See “Methods” for other details. (Inset) Semilogarithmic plot of the same data (▪, Sar-[d-Phe8]des-Arg9-BK; ○, Lys-des-Arg9-BK; Cp, arterial plasma concentration). *P < .05 and **P < .01, when compared with values of animals given Sar-[d-Phe8]des-Arg9-BK (Student’s t test).
Results
Figure 1 shows the plasma concentrations of Sar-[d-Phe8]des-Arg9-BK and of Lys-des-Arg9-BK in normal (i.e., no pretreatment with LPS), pentobarbital-anesthetized rabbits at various times after an i.a. bolus injection of 3 mg/animal of either one of these peptides. When plotted with a logarithmic ordinate scale, the same data gave straight lines. The correlation coefficients (r) of the regression lines were .99 in both cases. Application of equations of first-order drug disposition processes to the data provided T½ values of 110 ± 10 and 32 ± 6 sec (n = 3 for each peptide) (P < .05) for Sar-[d-Phe8]des-Arg9-BK and Lys-des-Arg9-BK, respectively. Calculated plasma clearance values of both peptides were (in the same order) 54 ± 6 and 162 ± 74 ml/min.
Effect of various inhibitors on the hypotensive effect of Sar-[d-Phe8]des-Arg9-BK.
Hypotensive effects of Sar-[d-Phe8]des-Arg9-BK (750 ng/kg i.a.) were measured in LPS-treated, anesthetized rabbits acutely pretreated with either indomethacin (5 mg/kg), diclofenac (5 mg/kg), NG-nitro-l-arginine (30 mg/kg), dazmegrel (1 mg/kg), glibenclamide (15 mg/kg), MK-886 (3 mg/kg), BN-50739 (10 mg/kg), atropine (5 mg/kg) or propranolol (5 mg/kg), and compared with those measured in drug vehicle-treated (control) animals. None of these inhibitors reduced the amplitudes of hypotensive responses to the B1 agonist. Thus, decreases in MABP elicited by the B1 agonist after pretreatment with indomethacin, diclofenac, NG-nitro-l-arginine, dazmegrel, glibenclamide, MK-886, BN-50739, atropine or propranolol were (in mm Hg): −29 ± 3, −36 ± 4, −33 ± 7, −35 ± 3, −38 ± 9, −47 ± 5, −49 ± 5, −41 ± 4 and −53 ± 9, respectively (from base lines of 78 ± 7, 104 ± 5, 102 ± 11, 91 ± 6, 109 ± 6, 125 ± 6, 99 ± 4, 89 ± 3 and 111 ± 3 mm Hg, respectively;n = 4–7). Decreases in MABP in corresponding drug vehicle-treated (control) animals were (in mm Hg): −30 ± 2, −35 ± 5, −43 ± 8, −46 ± 5, −52 ± 2, −51 ± 11, −47 ± 5, −55 ± 6 and −52 ± 3 (from base lines of 82 ± 3, 89 ± 12, 84 ± 2, 105 ± 4, 109 ± 4, 116 ± 6, 94 ± 8, 98 ± 6 and 97 ± 5 mm Hg; n = 4–7). The times for half-recovery of hypotensive episodes (TR50) caused by Sar-[D-Phe8]des-Arg9-BK were 214 ± 43 sec (n = 7) and 71 ± 31 sec (n = 4) (P < .05 when compared with controls) in animals pretreated with indomethacin or diclofenac, respectively. Corresponding values in drug vehicle-treated (control) animals were 359 ± 33 sec (n = 7) and 624 ± 150 sec (n = 4). None of the other drugs affected the TR50 values of the B1 agonist. Hypotensive responses to ACH (1, 3, 6 and 12 μg/kg/min i.a.) (see “Methods”) in four animals pretreated with the NG-nitro-l-arginine solvent (control) were −14 ± 4, −29 ± 6, −47 ± 6 and −55 ± 4 mmHg, respectively. Corresponding responses in four animals pretreated with NG-nitro-l-arginine (30 mg/kg) were −7 ± 2 (P > .05), −11 ± 3 (P < .05), −18 ± 3 (P < .005) and −26 ± 2 mmHg (P < .001). Basal MABP in these animal groups were similar (control, 110 ± 3 mm Hg; NG-nitro-l-arginine treatment, 104 ± 7 mm Hg) (P > .05).
Hemodynamic effects of Sar-[d-Phe8]des-Arg9-BK.
Figure 2 illustrates the time-related hemodynamic effects of Sar-[d-Phe8]des-Arg9-BK (750 ng/kg i.a.) in LPS-treated, pentobarbital-anesthetized rabbits. Within seconds after injection of the B1 agonist the MABP fell (−40 ± 3 mm Hg) and remained lower than the preinjection value for the next 5 to 10 min. Over the same period the HR decreased gradually to reach its lowest value (−30 ± 7 bpm) 5 min after injection of Sar-[d-Phe8]des-Arg9-BK. Hypotensive episodes caused by the B1 agonist were associated with concomitant reduction of CBF, peak CBF reductions being recorded 1 min after injection of Sar-[d-Phe8]des-Arg9-BK. The CVR fell markedly soon (12 sec) after injection of the B1agonist, returned to the preinjection value at the next time point (30 sec) despite persisting hypotension and later increased significantly above preinjection values. The CVP exhibited a decrease (−1.6 ± 0.6 mm Hg), whereas the HTC remained stable.
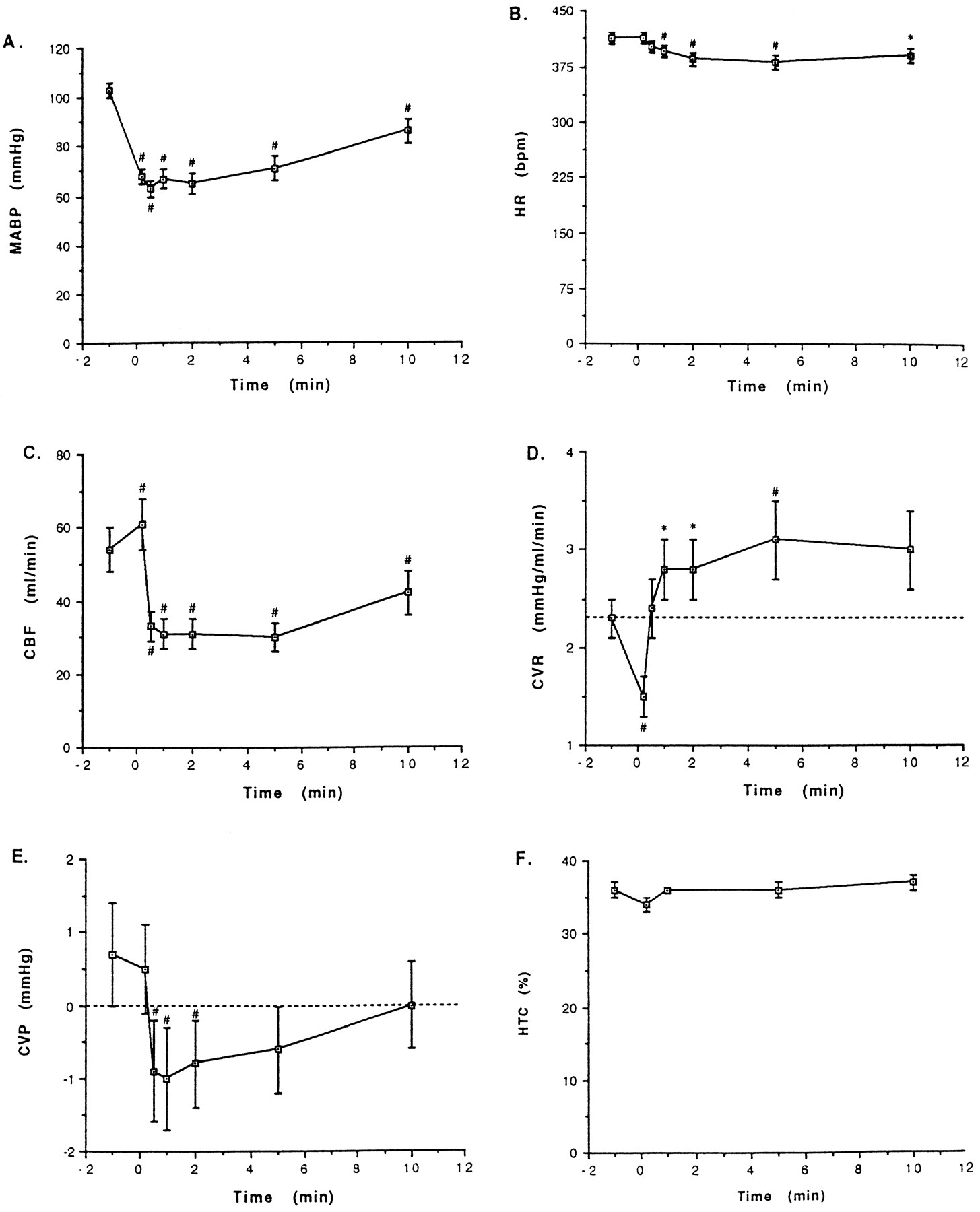
Time-related variations of MABP (A), HR (B), right CBF (C), CVR (D), CVP (E) and HTC (F) in LPS-pretreated, pentobarbital-anesthetized rabbits after bolus i.a. injection of Sar-[d-Phe8]des-Arg9-BK (750 ng/kg). Parameters were measured before (t = −1 min) and at different times (0.2, 0.5, 1, 2, 5 or 10 min) after injection of the B1 receptor agonist. Points indicate mean values and vertical bars the S.E.M. of 21 (MABP, HR, CBF, CVR), 12 (CVP) or 4 (HTC) animals. MABP, HR, CBF, CVR, CVP and HTC are given in absolute values. *P < .05; #P < .01; statistical comparisons were made of the differences (Δ) between pre- and postinjection values rather than of absolute values.
Pretreatment with diclofenac reduced the times for half-recovery of the hypotensive effects of Sar-[d-Phe8]des-Arg9-BK. It was therefore important to examine further the effect of diclofenac on the hemodynamic effects of this peptide. Results are shown in figure3. MABP decreases at the 12-sec time point after injection of the B1 agonist in diclofenac-pretreated animals were similar to corresponding values of control animals. Later on MABP values of diclofenac-pretreated animals were higher than in control animals. The bradycardia and CBF decreases caused by the B1 agonist in control animals were attenuated in diclofenac-pretreated animals. CVR decreases noted at the 12-sec time point after injection of Sar-[d-Phe8]des-Arg9-BK in control animals were of similar amplitude in diclofenac-pretreated animals. However, rebound increases of CVR observed later on in control animals were suppressed by diclofenac.

Time-related changes (Δ) in MABP (A), HR (B), right CBF (C) and CVR (D) elicited by i.a. injection of Sar-[d-Phe8]des-Arg9-BK (750 ng/kg) in LPS-pretreated, pentobarbital-anesthetized rabbits after pretreatment with diclofenac (5 mg/kg i.a.) (○) or hexamethonium (C6) (10 mg/kg i.a.) (▵). Data generated from the latter two groups were compared with those obtained in diclofenac- and C6-free (control) animals (•). Diclofenac and C6 were given by i.a. injection 15 min before the B1 agonist. ΔMABP, ΔHR, ΔCBF and ΔCVR indicate absolute increases or decreases (−) of basal (i.e., preinjection of the B1 agonist) MABP, HR, CBF and CVR, as measured at various times after injection of the B1agonist. Basal MABP, HR, CBF and CVR values in control animals (n = 21) were 103 ± 3 mm Hg, 412 ± 8 bpm, 55 ± 6 ml/min and 2.3 mm Hg/ml/min, respectively. Corresponding values in diclofenac-treated animals (n = 5) were 101 ± 4, 398 ± 21, 57 ± 8, 1.9 ± 0.2, whereas in C6-treated animals (n = 6) these values were 73 ± 8#, 316 ± 7#, 33 ± 4+ and 2.4 ± 0.3. Points indicate mean values and vertical bars show the S.E.M. *P < .05; **P < .01versus corresponding basal (preinjection) hemodynamic values. +P < .05; #P < .01 versuscorresponding values in control animals.
Compensatory mechanisms involving the autonomic nervous system may alter the hemodynamic effects of Sar-[d-Phe8]des-Arg9-BK. To test this hypothesis we assessed the hemodynamic effects of the B1 agonist in animals pretreated with the ganglion blocker hexamethonium (10 mg/kg i.a.). Hexamethonium did not affect the amplitude and time course of hypotensive and bradycardic episodes caused by Sar-[d-Phe8]des-Arg9-BK, despite causing reduction of basal MABP and HR values (fig. 3). However CBF reductions caused by the B1 agonist were lower in hexamethonium-pretreated animals than in control animals. Moreover, CVR decreases caused by Sar-[d-Phe8]des-Arg9-BK were found to persist longer (i.e., there was no rebound increase of CVR) in hexamethonium-pretreated animals compared with control animals. Base-line CBF and CVR values in hexamethonium-pretreated animals were lower than, and similar to, those of control animals, respectively (fig. 3).
In a subgroup of LPS-treated, pentobarbital-anesthetized animals we measured the effect of Sar-[d-Phe8]des-Arg9-BK (750 ng/kg i.a.) on MABP, HR, FBF and FVR. Base-line values of MABP, HR, FBF and FVR were 89 ± 3 mm Hg, 332 ± 5 bpm, 16 ± 2 ml/min and 6.2 ± 0.7 mm Hg/ml/min (n = 9), respectively. ΔMABP values at the 0.2, 0.5, 1, 2, 5 and 10 min time points after injection of the B1 agonist were −25 ± 3, −30 ± 4, −34 ± 3, −33 ± 3, −23 ± 4 and −12 ± 3 mm Hg (P < .01 at all time points). ΔHR values at the same time points were: +3 ± 3, −1 ± 3, −9 ± 5, −17 ± 6, −22 ± 8 and −20 ± 8 bpm (P < .05 at the 1-min time point and on). Corresponding ΔFBF values were 0 ± 0, −1 ± 0, −4 ± 1, −7 ± 1, −8 ± 1 and −6 ± 2 ml/min (P < .01 at the 1-min time point and on). Corresponding ΔFVR values were −1.6 ± 0.3, −2.0 ± 0.3, −1.0 ± 0.4, +1.2 ± 0.9, +4.2 ± 1.1 and +4.0 ± 1.0 mmHg/ml/min (P < .01 at all time points, except 2 min where P > .05).
LPS-pretreated, halothane-anesthetized animals were used to examine the effect of Sar-[d-Phe8]des-Arg9-BK (750 ng/kg i.a.) on MBF and MVR. Results are shown in figure4. Overall changes of MABP and HR elicited by the B1 agonist in halothane-anesthetized animals were similar to those measured in barbiturate-anesthetized animals (compare data of fig. 4 with those of fig. 3). Moreover patterns of MBF and MVR decreases in halothane-anesthetized animals were nearly identical with those of CBF and CVR decreases in barbiturate-anesthetized animals.

Time-related changes (Δ) in MABP (A), HR (B), MBF (C) and MVR (D) elicited by i.a. injection of Sar-[d-Phe8]des-Arg9-BK (750 ng/kg) in LPS-pretreated, halothane-anesthetized rabbits after an i.v. constant infusion of the vasopressor methoxamine (50–150 μg/min) (○) or of saline (control) (•). ΔMABP, ΔHR, ΔMBF and ΔMVR indicate absolute increases or decreases (−) of basal (i.e., preinjection of the B1 agonist) MABP, HR, MBF and MVR, as measured at different times after injection of the B1 agonist. Basal MABP, HR, MBF and MVR values in control animals (n = 8) were 55 ± 6 mm Hg, 325 ± 12 bpm, 54 ± 6 ml/min and 1.2 ± 0.3 mm Hg/ml/min, respectively. Corresponding values in methoxamine-infused animals (n = 5) were 80 ± 5+, 298 ± 12, 25 ± 3+ and 3.5 ± 0.5#. Points indicate mean values and vertical bars the S.E.M.. *P < .05; **P < .01 versuscorresponding basal (preinjection) values. +P < .05; #P < .01 versus corresponding values in control (methoxamine-free) animals.
In a subgroup of halothane-anesthetized rabbits we examined the influence of prior increases of MABP and MVR on hemodynamic effects of Sar-[d-Phe8]des-Arg9-BK (750 ng/kg i.a.) (fig. 4). Prior MABP and MVR increases elicited by infusions of the sympathomimetic drug methoxamine did not alter the hypotensive and bradycardic effects of the B1 agonist. However, MBF and MVR changes in both animal groups differed markedly. In methoxamine-pretreated animals the B1 agonist elicited sustained decreases of MVR, whereas transient decreases were produced in methoxamine-free animals. Furthermore the MBF increased in methoxamine-treated animals, whereas it fell markedly in methoxamine-free animals.
As an attempt to better define the mechanism of the secondary, more persistent phase of the hypotensive episode caused by Sar-[d-Phe8]des-Arg9-BK in our animal model, we also measured the effect of the B1 agonist on CO and TPR. Results are shown in figure 5. Changes of MABP and HR caused by Sar-[d-Phe8]des-Arg9-BK (750 ng/kg i.a.) were similar to those described above. Significant decreases of TPR and CO were observed after injection of the B1 agonist. Decreases of MABP caused by Sar-[d-Phe8]des-Arg9-BK were temporally linked to decreases of TPR up to 2 min postinjection. TPR values were back to base line at the 5-min time point despite persisting hypotension, and above base line at the 10-min time point. Decreases of CO were observed from the 1-min time point and on after injection of the B1 agonist, exhibiting their peak values at the 5-min time point.

Time-related changes (Δ) in MABP (A), HR (B), CO (C) and TPR (D) elicited by i.a. injection of Sar-[d-Phe8]des-Arg9-BK (750 ng/kg) in LPS-pretreated, pentobarbital-anesthetized rabbits. ΔMABP, ΔHR, ΔCO and ΔTPR indicate absolute increases or decreases (−) of basal (i.e., preinjection of the B1agonist) MABP, HR, CO and TPR, as measured at various times after injection of the B1 agonist. Basal MABP, HR, CO and TPR values in these animals (n = 5) were 56 ± 1 mm Hg, 278 ± 7 bpm, 309 ± 15 ml/min and 0.18 ± 0.01 mm Hg/ml/min, respectively. Points indicate mean values and vertical bars the S.E.M.. *P < .05; +P < .01 versuscorresponding basal (preinjection) hemodynamic values.
Discussion
Sar-[d-Phe8]des-Arg9-BK, a Lys-des-Arg9-BK analog that shares the high affinity of this natural sequence, was shown previously to be resistant to enzymatic breakdown by a variety of tissue enzymes from rabbits and to elicit dose-dependent hypotensive effects when injected intraarterially in LPS-treated rabbits (Drapeau et al., 1991). The inhibition of Sar-[d-Phe8]des-Arg9-BK-induced hypotension, in the latter animal model, by sequence-related antagonist of kinin B1 receptors, but not by HOE 140, a kinin B2 receptor, confirmed the participation of kinin B1 receptors in this effect (Drapeau et al., 1993). In addition to being a more potent hypotensive peptide than des-Arg9-BK in LPS-treated animals, Sar-[d-Phe8]des-Arg9-BK produced longer lasting hypotensive episodes than the naturally occurring B1 agonists, des-Arg9-BK and Lys-des-Arg9-BK, in this animal model. The greater metabolic stability of Sar-[d-Phe8]des-Arg9-BK, compared with des-Arg9-BK and Lys-des-Arg9-BK, was responsible for these differences (Drapeau et al., 1991). In the present study we measured and compared the rate of elimination of Sar-[d-Phe8]des-Arg9-BK and of Lys-des-Arg9-BK from the plasma of anesthetized rabbits which were not pretreated with LPS (hereafter called “normal” animals). Our aim was to determine whether longer lasting hypotensive episodes caused by Sar-[d-Phe8]des-Arg9-BK in LPS-treated animals, compared with Lys-des-Arg9-BK, might rely upon differences of their rates of elimination from the plasma. The choice of normal rather than LPS-treated animals was dictated by the fact that high doses (3 mg/animal) of B1 agonists had to be used to raise plasma concentrations to optically measurable levels. Such high doses of B1 agonists were expected to produce cardiovascular collapse in LPS-treated animals but were essentially inert in intact animals (data not shown). The results showed clearly that Sar-[d-Phe8]des-Arg9-BK disappears more slowly from plasma than Lys-des-Arg9-BK. These results suggest that the longer duration of hypotensive effects of Sar-[d-Phe8]des-Arg9-BK in LPS-treated rabbits, compared with Lys-des-Arg9-BK or des-Arg9-BK, may have a metabolic basis. However, this conclusion is contingent upon the existence of identical metabolic pathways in normal and LPS-treated animals.
Endogenous vasodilators such as prostacyclin and NO may participate in the hypotensive effect of Sar-[d-Phe8]des-Arg9-BK in LPS-treated rabbits, for both substances are released from isolated rabbit arteries challenged with B1 agonists (see the introduction). In the present study we showed that indomethacin and diclofenac do not reduce the amplitude of, but quicken the recoveries from, hypotensive episodes caused by Sar-[d-Phe8]des-Arg9-BK. These results add further support to the hypothesis that PG release from blood vessels may contribute to the prolonged hypotension caused by Sar-[d-Phe8]des-Arg9-BK in LPS-treated rabbits (Drapeau et al., 1991). However, the overall effect of PGs in this system is not a vasodilator one, as might have been predicted from isolated vessels systems (see below). As far as the participation of NO in Sar-[d-Phe8]des-Arg9-BK-induced hypotension is concerned, the results are negative (i.e., NO is unlikely to be involved). NG-nitro-l-arginine (NO synthase inhibitor) did not alter the amplitude or duration of Sar-[d-Phe8]des-Arg9-BK-induced hypotension, despite the fact that this arginine derivative at the dose utilized in this study was shown to inhibit the hypotensive effect of ACH, a drug whose blood pressure-lowering effect in rabbits or rats depends at least partially on NO release (Whittle et al., 1989; Rees et al., 1990). Our conclusion that NO does not contribute to the hypotensive action of Sar-[d-Phe8]des-Arg9-BK in our animal model is not in agreement with the results of Pruneau and Belichard (1993) which indicate that the vasorelaxant effect of the B1 agonist des-Arg9-BK in the isolated rabbit carotid artery is mediated by endothelial NO release. However this discrepancy is easily explained if one considers that: 1) the participation of endothelial NO to the vasorelaxant (or contractile) effects of B1 agonists in isolated rabbit arteries is restricted to some blood vessels only (Bouthillier et al., 1987; deBlois and Marceau, 1987; Ritter et al., 1989); and 2) any small contribution by NO to the hypotensive effect of B1 agonists in LPS-treated rabbits is doomed to be masked by presumably more prominent direct and indirect (i.e., PG-mediated) components of hypotensive effects of B1agonists in this animal model.
Hypotensive responses to Sar-[d-Phe8]des-Arg9-BK in LPS-treated rabbits were not inhibited by animal treatment with dazmegrel, a thromboxane synthase inhibitor, MK-886, a leukotriene biosynthesis inhibitor, or by BN-50739, a PAF receptor antagonist. These results suggest that endogenous thromboxanes, leukotrienes and PAF (or PAF receptors), are unlikely to mediate the B1agonist-induced hypotension in LPS-treated rabbits. Glibenclamide, an inhibitor of ATP-sensitive K+ channels, atropine, a nonselective antagonist of muscarinic receptors, and propranolol, a nonselective beta adrenoceptor blocker, did not affect the hypotensive effect of Sar-[d-Phe8]des-Arg9-BK in LPS-treated rabbits. These results indicate that Sar-[d-Phe8]des-Arg9-BK-induced hypotension is not caused by activation of vascular ATP-sensitive K+ channels, by the release of ACH or adrenaline from endogenous stores or by direct activation of vascular muscarinic orbeta adrenergic receptors.
The hypotensive effect of Sar-[d-Phe8]des-Arg9-BK in our animal model was characterized by prolonged decreases of MABP, bradycardia, persistent decreases of CBF, FBF, MBF and CVP, and by large decreases in CVR, FVR and MVR, which subsided within 30 to 0 sec after injection of the B1 agonist and were followed in some cases (CVR, FVR) by rebound increases, despite persisting hypotension. No change of HTC was noted after injection of the B1agonist. These results may be interpreted in the following way. First, the absence of a change in HTC indicates that the hypotensive effect of Sar-[d-Phe8]des-Arg9-BK is not the result of a loss of plasma volume caused by plasma extravasation. Second, the acute decreases in CVR, FVR and MVR occurring within seconds after injection of the B1 agonist, even though transient, suggest that peripheral vasodilation and a decrease of TPR, secondary to the direct effect of the B1 agonist on the blood vessels, may be the primary events responsible for the early decline of MABP. Third, the persistence of the hypotensive episode caused by Sar-[d-Phe8]des-Arg9-BK, despite the early recovery and/or rebound increases of vascular resistance in some vascular beds, suggests that a reduction of CO may contribute to the maintenance of the hypotension caused by the B1 agonist in this animal model. Such interpretation is supported by our observations that initial MABP decreases caused by Sar-[d-Phe8]des-Arg9-BK were associated with decreases of TPR but no significant changes of CO, whereas the late phase of the hypotensive episode caused by the B1 agonist was characterized mainly by a decrease of CO. The reduced CO may be caused by inappropriate compensatory responses (i.e., bradycardia instead of reflex tachycardia, excessive increases of CVR and FVR) to the early acute decrease of MABP. A decrease in cardiac contractility rather than in preload is more likely, at least theoretically, to be responsible for the decrease in CO caused by the B1 agonist, because the CVP (an index of preload) was back to base line at the time the CO was decreased. A detrimental cardiac effect was not expected from a previous study of the Langerdoff preparation based on hearts removed from LPS-treated rabbits where a coronarovasodilator effect mediated by B1receptors for kinins was measured (Regoli et al., 1981). The absence of reflex tachycardia during the hypotensive episode caused by Sar-[d-Phe8]des-Arg9-BK is a relatively surprising event, because increases of HR and CO are the usual compensatory responses to severe arterial hypotension (Rushmer, 1976). This unexpected event may point to a dysfunction of baroreceptor-mediated cardiovascular reflexes in our animal model.
Both the bradycardia and rebound increases of CVR caused by Sar-[d-Phe8]des-Arg9-BK were attenuated in diclofenac-treated animals compared with control. These results suggest that these two processes may be partially PG dependent. Additional mechanisms besides PG release presumably are involved in the HR-decreasing effect of the B1 agonist, because no reflex tachycardia was observed even in diclofenac-treated animals during the hypotensive effect of Sar-[d-Phe8]des-Arg9-BK. The increases of CVR occurring during the hypotensive episode caused by the B1 agonist were converted into prolonged CVR decreases by animal pretreatment with hexamethonium. These results suggest that sympathetic neurons innervating blood vessels may also be involved in such phenomena. The direct and/or indirect (i.e.,via PG) activation of peripheral nociceptors functionally coupled to vascular sympathetic neurons is perhaps one of the most likely mechanisms by which Sar-[d-Phe8]des-Arg9-BK may elicit an increase of CVR (and presumably of MVR and FVR) in our animal model. Evidence that B1 agonists may activate peripheral nociceptors via a PG-dependent mechanism was presented previously (Davis and Perkins, 1994b; Walker et al., 1994).
The hypotensive episode elicited by Sar-[d-Phe8]des-Arg9-BK in LPS-treated, halothane-anesthetized animals was associated with bradycardia, persistent decreases of MBF and transient decreases of MVR. Prior increases of MABP and of MVR with the sympathomimetic vasopressor drug methoxamine produced little or no alteration of the ability of Sar-[d-Phe8]des-Arg9-BK to decrease MABP and HR levels in this animal model. However, the transient decreases of MVR and sustained decreases of MBF noted in methoxamine-free animals were converted into sustained decreases of MVR and sustained increases of MBF in methoxamine-treated animals. These results suggest that important changes of base-line (preinjection) MABP and/or arterial tone may modify both quantitatively and qualitatively the hemodynamic profile of B1 agonists. This conclusion is consistent with the results of DeWitt et al. (1994), which shows that the naturally occurring B1 agonist des-Arg9-BK elicits vasoconstriction under low arterial tone, but vasodilation under high arterial tone, in the pulmonary vascular bed of anesthetized cats.
In summary, i.a. injection of Sar-[d-Phe8]des-Arg9-BK in LPS-treated, anesthetized rabbits causes a prolonged episode of hypotension. Peripheral vasodilation is likely to be the primary event leading to the early decrease of MABP, whereas a reduction of CO may contribute to the maintenance of the hypotension induced by the B1 agonist in this animal model. Inappropriate compensatory responses to arterial hypotension, PG release and the slow rate of elimination of Sar-[d-Phe8]des-Arg9-BK from the rabbit plasma may be at the basis of the prolonged duration of the hypotensive episode elicited by the B1 agonist. These results provide additional circumstantial evidence that endogenous B1 agonists (e.g., des-Arg9-BK, Lys-des-Arg9-BK) may participate in the hemodynamic manifestations of septic shock.
Acknowledgments
We thank Elisabeth Lemay and Gaétane Rioux for typing this manuscript.
Footnotes
-
Send reprint requests to: Francis Rioux, Ph.D., Centre de recherche de l’Hôtel-Dieu de Québec, 11 Côte du Palais, Québec (Québec), Canada G1R 2J6.
-
↵1 Supported in part by the Medical Research Council of Canada (Grant MT-12217) and the Quebec Heart and Stroke Foundation.
-
↵2 Recipient of a Studentship from Fonds pour la Formation de Chercheurs et l’Aide à la Recherche, Québec.
-
↵3 Scholar of the Fonds de la Recherche en Santé du Québec.
- Abbreviations:
- ACH
- acetylcholine
- BK
- bradykinin
- CBF
- carotid blood flow
- CO
- cardiac output
- CVP
- central venous pressure
- CVR
- carotid bed vascular resistance
- DMSO
- dimethyl sulfoxide
- FBF
- femoral blood flow
- FVR
- femoral bed vascular resistance
- HR
- heart rate
- HPLC
- high performance liquid chromatography
- HTC
- hematocrit
- i.a.
- intraarterial
- LPS
- bacterial lipopolysaccharide
- MABP
- mean arterial blood pressure
- MBF
- mesenteric blood flow
- mm Hg
- millimeter of mercury
- MVR
- mesenteric bed vascular resistance
- NO
- nitric oxide
- PAF
- platelet-activating factor
- PG
- prostaglandin
- Sar
- sarcosine
- T½
- biological half-life in plasma
- TPR
- total peripheral resistance
- Received February 6, 1996.
- Accepted September 4, 1996.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}