


Abstract
Endomorphin-1 and endomorphin-2 are tetrapeptides of the brain whose binding profiles and analgesic activities indicate that they are endogenous ligands at μ opioid receptors. To analyze the classes of G transducer proteins activated by these opioids in the production of supraspinal antinociception, the expression of α subunits of the Gi protein class, Gi1, Gi2, Gi3, Go1, Go2, and Gz, and those of the Gq protein family, Gq and G11, was reduced by administration of antisense oligodeoxynucleotides (ODNs) complementary to sequences in their respective mRNAs. The ODN treatments promoted differences in the analgesic effects displayed by morphine, [d-Ala2,N-MePhe4,Gly-ol5]enkephalin (DAMGO), and the novel opioids endomorphin-1 and endomorphin-2. The impairment of Gi1α and Gi3α function led to a weaker analgesic response to the endomorphins and to the α2-adrenoceptor agonist clonidine, whereas the effects of morphine and DAMGO were not affected. An antisense probe targeting Gi2α blocked the antinociceptive effects of endomorphin-2, morphine, DAMGO, and clonidine but was without effect on the activity of endomorphin-1. Mice receiving the ODN to Gzα subunits showed impaired response to all agonists. The knockdown of either Go1α, Go2α, Gqα, or G11α had little or no influence on the antinociception induced by any of the opioids in the study. Thus, agonists exhibit differences in activating the variety of GTP-binding proteins regulated by μ opioid receptors.
Receptors signaling through GTP-binding proteins (G proteins) vary in the amino acid sequences that interact with the α subunits of these transducer proteins (i.e., the receptor loop that links the fifth and sixth transmembrane regions and the C-terminal tail; Strosberg, 1991). It has been shown that a single type of receptor regulates several classes of G proteins [e.g., kyotorphin (tyrosine-arginine) receptors; (Ueda et al., 1989), muscarinic acetylcholine receptors (Offermanns et al., 1994), somatostatin receptors (Law et al., 1994), and dopamine receptors (Lui et al., 1994)]. This has been verified in in vitro systems and cultured cell lines by using specific antibodies and antisense oligodeoxynucleotides (ODNs) to reduce the function and/or expression of different G protein subunits (Eason et al., 1992;Garzón et al., 1997a,b). The identification of the G protein subtypes involved in the in vivo effects of neuroactive substances has also been accomplished using similar experimental methods. Single intracerebroventricular (i.c.v.) injection into mice of IgGs directed to Gα subunits and subchronic administration of antisense ODNs to specifically “knockdown” a particular class of Gα subunits have both demonstrated the diversity of G proteins involved in the supraspinal antinociception mediated by opioid and nonopioid receptors (Sánchez-Blázquez et al., 1993,1995, 1996; Sánchez-Blázquez and Garzón, 1993, 1998;Raffa et al., 1994; Rossi et al., 1995; Standifer et al., 1996;Garzón et al., 1999).
Recently, two peptides designated as endomorphins because they possess morphine-like properties were isolated from bovine brain extracts and proposed as natural ligands of the μ opioid receptor (Zadina et al., 1997). Studies in mice have shown that i.c.v. administration of endomorphin-1 induces a long-lasting antinociceptive effect that can be blocked by pretreatment with the μ-selective antagonist β-funaltrexamine. Moreover, in brain regions that are involved in nociception, endomorphin-1 stimulates the binding of [35S]guanosine-5′-O-(3-thio)triphosphate to G proteins through the activation of μ opioid receptors (Sim et al., 1998). However, the G protein subtypes that mediate the antinociceptive effects of both endomorphin-1 and endomorphin-2 have yet to be identified.
The present study was designed to explore the participation of different G proteins of the Gi and Gqfamilies in supraspinal antinociception promoted by endomorphins. Therefore, the expression of the α subunits of the Gi1, Gi2, Gi3, Go1, Go2, Gz, Gq, and G11 transducer proteins was reduced by the administration of ODNs complementary to sequences of their respective mRNAs. The results were compared with those obtained with other analgesic compounds, such as the μ opioid receptor ligands morphine and [d-Ala2,N-MePhe4,Gly-ol5]enkephalin (DAMGO) and the α2-adrenoceptor agonist clonidine. The participation of multiple classes of G proteins in the antinociception induced by endomorphin-1 and endomorphin-2 was revealed. The pattern of G protein activation exhibited by the endogenous peptides in the production of supraspinal analgesia differs from those of morphine or DAMGO. Furthermore, differences were observed between the endomorphins because impairment of Gi2α function reduced the antinociception of endomorphin-2 but had no effect on the activity of endomorphin-1. This further suggests that after binding to μ opioid receptors, agonists can promote activation of different G proteins.
Materials and Methods
Animals and Evaluation of Analgesia.
Albino male mice CD-1 (Charles River, Barcelona, Spain) weighing 22 to 25 g were used throughout. Animals were kept at 22°C, and a 12-h light/dark cycle (8:00 AM/8:00 PM) was established. Food and water were provided ad libitum. Mice were housed and used strictly in accordance with the guidelines of the European Community regarding the care and use of laboratory animals. To reduce the possibility of interference from spinal events, all substances were injected i.c.v. into the right lateral ventricle, as described previously (Sánchez-Blázquez et al., 1995;Sánchez-Blázquez and Garzón, 1998). The warm water (52°C) tail-flick test was used to measure the antinociceptive effects. Latencies were determined before treatment (basal latency) and after the administration of the substance under study. Baseline latencies ranged from 1.3 to 2.2 s and were not affected by ODN administration. A cut-off time of 10 s was allotted to minimize the risk of tissue damage. Antinociception was expressed as a percentage of the maximum analgesic effect (MAE) according to the following equation: MAE (%) = 100 × (test latency − baseline latency)/(cut-off time − baseline latency). Opioid agonists were injected i.c.v., and antinociception was determined at its peak (i.e., 30 min after morphine or clonidine, 15 min after DAMGO, and 10 min after endomorphin-1 and endomorphin-2). All compounds were dissolved in distilled water, and solutions were made up immediately before use. Statistical significance was determined by ANOVA followed by the Student-Newman-Keuls test. The level of significance was set atP < .05.
Synthesis of ODNs.
Synthetic end-capped phosphorothioate antisense ODNs were prepared by solid phase phosphoramidite chemistry using a CODER 300 DNA synthesizer (DuPont, Wilmington, DE) at the 1-μmol scale. The introduction of phosphorothioate linkages was achieved by tetraethylthiuram disulfide sulfurization. Crude ODNs were purified by conventional reverse-phase chromatography through a 5-μm C18 column (Spherisorb ODS-2, 150 × 4.6 mm) using 0.1 M triethylammonium acetate (pH 7.0) and acetonitrile as the mobile phase. The eluted ODNs were then desecated (Speed Vac Plus; Savant, Farmingdale, NY) and stored at −20°C until use. Sequences were as follows: ODN-Gi1α, 5′-G∗C∗TGTCCTTCCACAGTCTCTTTATGACGCCG∗G∗C-3′, corresponding to nucleotides 588 to 621 of the rat Gi1α gene sequence; ODN-Gi2α, 5′-A∗T∗GGTCAGCCCAGAGCCTCCGGATGACGCCC∗G∗A-3′, corresponding to nucleotides 523 to 556 of the murine Gi2α gene sequence; ODN-Gi3α, 5′-G∗C∗CATCTCGCCATAAACGTTTAATCACGCCT∗G∗C-3′, corresponding to nucleotides 554 to 587 of the rat Gi3α gene sequence; and ODN-Gzα, 5′-C∗G∗TGATCTCACCCTTGCTCTCTGCCGGGCCA∗G∗T-3′, corresponding to nucleotides 330 to 363 of the rat Gzα gene sequence (see Sánchez-Blázquez et al., 1995). The antisense Gi1α, Gi3α, and Gzα ODNs directed to rat sequences form RNA hybrids in NG108x15 cells of murine origin (McKenzie and Milligan, 1990) and have been shown to have effect on the murine target proteins (Sánchez-Blázquez et al., 1995). ODN-Go1α, 5′-A∗G∗GC AGCTGCATCTTCATAGGTG∗T∗T-3′, a 25-base ODN, corresponds to nucleotides 882 to 906 of the murine Go1α gene sequence; ODN-Go2α, 5′-G∗A∗GCCACAGCTTCTGTGAAGGCA∗C∗T-3′, corresponds to nucleotides 882 to 906 of the murine Go2α sequence; ODN-Gqα, 5′-C∗G∗GCTACACGGTCCAAGTC∗A∗T-3′, corresponds to nucleotides 484 to 504 of the murine Gqα gene sequence; and ODN-G11α, 5′-C∗T∗GTGGCGATGCGGTCCAC∗G∗T-3′, corresponds to nucleotides 487 to 507 of the murine G11α sequence (see Sánchez-Blázquez and Garzón, 1998). These sequences displayed no homology to other relevant cloned proteins (GenBank database). A random ODN (ODN-RD) with the sequence 5′-C∗C∗CTTATTTACTACTTTC∗G∗C-3′ served as a control. The reducing activity of all these ODNs to Gα subunits on the target proteins in mice has been demonstrated (Sánchez-Blázquez et al., 1995; Sánchez-Blázquez and Garzón, 1998).
Administration of ODNs.
ODN solutions were made up in the appropriate volume of sterile water immediately before use. Animals received either the vehicle (control), the ODN-RD, or the antisense ODN injected i.c.v. into the right lateral ventricle. Subsequent administrations were performed on the same side. Each ODN treatment was performed on a distinct group of 15 to 20 mice using the following schedule: on days 1 and 2 with 1 nmol, days 3 and 4 with 2 nmol, and day 5 with 3 nmol. On day 6, the opioid agonists were injected i.c.v., and their antinociceptive activity was evaluated by the warm water tail-flick test. An interval of 24 h was selected between ODN administrations to minimize the neurotoxic damage. This schedule of administration did not alter the normal behavior of the mice.
Chemicals.
Endomorphin-1 and endomorphin-2 were obtained from Tocris Cookson (Bristol, UK). Morphine sulfate was obtained from Merck (Darmstadt, Germany). Clonidine hydrochloride was purchased from Sigma-Aldrich Quı́mica (Madrid, Spain). Naloxonazine and ICI 174,864 (N,N-diallyl-Tyr-Aib-Aib-Phe-Leu) were obtained from Research Biochemicals, Inc. (Natick, MA). DAMGO and Cys2,Tyr3,Orn5,Pen7-amide (CTOP) were purchased from Peninsula Laboratories (San Carlos, CA).
Results
Bioactivity of Endomorphin-1 and Endomorphin-2 as Analgesics in Warm Water Tail-Flick Test.
Endomorphins induced a dose-dependent antinociception after i.c.v. injection to mice. The peak of the effect was obtained 10 min after their administration. The antinociception induced by these peptides (6 to 20 nmol/mouse) exhibited a steady plateau at 42 ± 4% (n = 25) of the MAE (Fig.1). In this test, morphine and DAMGO are able to produce MAEs (see e.g., Garzón and Sánchez-Blázquez, 1995). The ED50 values (nmol/mouse; 95% confidence limits) and apparent maximum antinociceptive effects for these opioids were: endomorphin-1, 0.25 (0.16–0.7) and 40% MAE; endomorphin-2, 1.26 (0.90–1.76) and 40% MAE; morphine, 4.2 (3.0–5.9) and 100% MAE, DAMGO, 0.051 (0.037–0.068) and 100% MAE.

Effect of the opioid antagonists CTOP and ICI 174,864 on the supraspinal antinociceptive effect induced by endomorphins in the warm water tail-flick test. Dose-response curves for antinociception of endomorphin-1 and endomorphin-2 were constructed for mice in absence and presence of 0.6 nmol/mouse of antagonists at δ (ICI 174,864) or μ (CTOP) opioid receptors. All compounds were injected i.c.v. in volumes of 4 μl. Antinociception is expressed as a percentage of the MAE. Values are the mean ± S.E. from groups of 10 to 15 mice. The peak of 10 min for the analgesic effect of endomorphins was determined in pilot experiments. *Significantly different from the control group receiving saline instead the antagonist (ANOVA, Student-Newman-Keuls test, P < .05).
The antinociception induced by endomorphin-1 and endomorphin-2 was reversed by the selective antagonist at μ opioid receptors, CTOP (0.6 nmol/mouse, i.c.v.), but not by the administration of 0.6 nmol/mouse of the selective δ-antagonist ICI 174,864 (Fig. 1), thus indicating that the antinociceptive action of both endomorphins is mediated through μ opioid receptors.
Effect of In Vivo i.c.v. Administration of ODNs to Gα Subunits on Supraspinal Analgesia Induced by Various Analgesic Compounds.
In an effort to discard the possibility of toxicity within the central nervous system, particularly that associated with the presence of phosphorothioates, only end capped ODNs and the minimal doses of these needed to observe an effect were used. The possibility of nonspecific actions was therefore minimized. No signs of neurotoxic damage were observed in thionine-stained consecutive brain slices (not shown). The analgesic substances produced comparable effects in mice that received i.c.v. the vehicle, the ODN-RD, or in noninjected (naive) animals. Thus, the responsiveness of the mice was not altered by the experimental procedure alone.
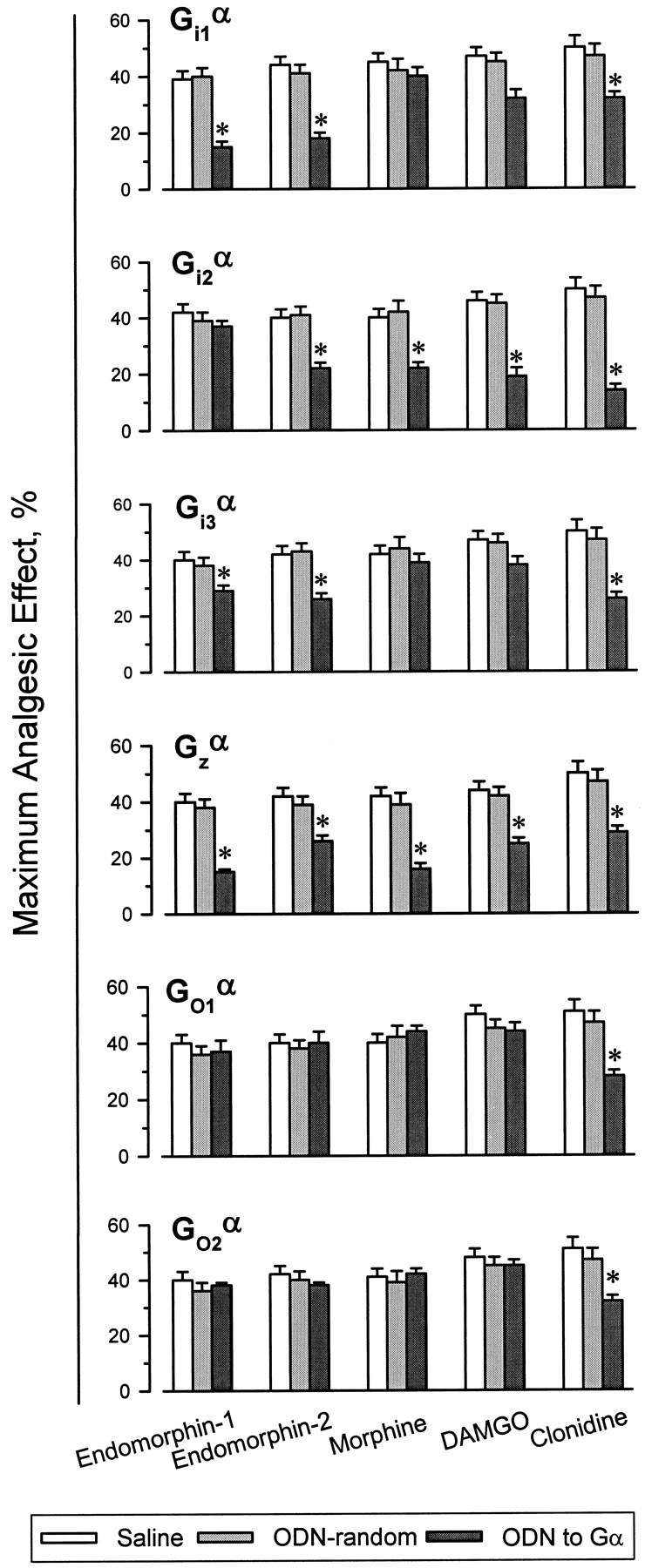
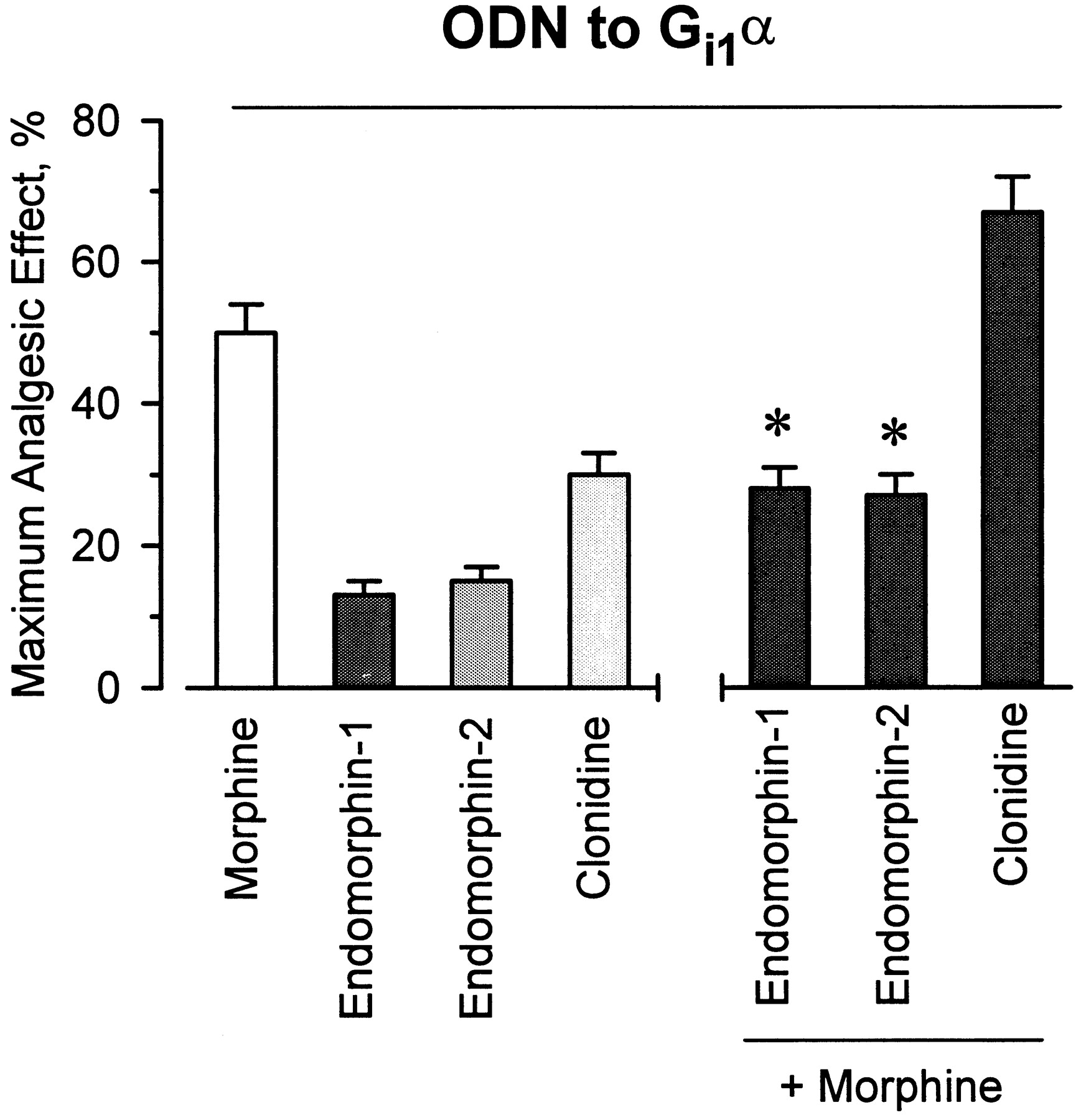
In a previous paper it was shown that in vivo administration of ODNs directed against Gi2α and Gzα subunits, but not those against Gi1α, blocked morphine- and DAMGO-evoked analgesia (Sánchez-Blázquez et al., 1995). The current study confirmed the ability of those ODNs to prevent morphine analgesia and also examined other analgesics: the novel opioid peptides endomorphin-1 and −2, and the α2-adrenergic ligand clonidine. Although the impairment of Gi1α function did not change the response of the mice to morphine and DAMGO, it did lead to a decrease in the effect of both endomorphins and that of the α2-adrenoceptor agonist, clonidine (Figs.2 and 3). Similar results were observed after i.c.v. injections of the antisense probe to Gi3α subunits. The antisense ODN to Gi2α produced distinct effects: endomorphin-1-induced antinociception was unchanged, whereas the activities of endomorphin-2, morphine, DAMGO, and clonidine were diminished. The administration of an ODN to the pertussis toxin-insensitive Gzα subunits was followed by a significant decrease in the antinociception evoked by all the agonists under study (Fig. 3). The profile of endomorphin-1 to activate Gi1- and Gi2 proteins is similar to that described for the morphine metabolite, morphine-6β-glucuronide (Rossi et al., 1995). In mice where the expression of Gi1α subunits was reduced, endomorphin-1 and endomorphin-2 antagonized the capacity of morphine to produce antinociception (Fig. 4). This result suggests that these agonists all share the μ opioid receptor for producing antinociception. In the Gi1α-knockdown mice, coadministration of clonidine and morphine potentiated the effect of either agonist (Fig.4). This is expected for agonists acting on different receptors, i.e., the α2-adrenoceptors and μ opioid receptors.

Effect of subchronic i.c.v. administration of ODNs to Gi1α and Gi2α subunits on the antinociception induced by endomorphin-1. Dose-response curves for antinociception of endomorphin-1 were constructed for control (ODN-RD) mice and for animals receiving increasing amounts of the antisense ODNs on a once-daily schedule for 5 consecutive days (see Materials and Methods). Antinociception is expressed as a percentage of the MAE. Values are the mean ± S.E. from groups of 10 to 15 mice. *Significantly different from the control group receiving saline instead the antagonist (ANOVA, Student-Newman-Keuls test,P < .05).

Effect of subchronic i.c.v. administration of ODNs to Giα, Goα, and Gzα subunits on supraspinal antinociception induced by endomorphins, morphine, DAMGO, and clonidine. Mice were injected with increasing amounts of ODNs on a once-daily schedule for 5 consecutive days (see Materials and Methods). On day 6, the antinociceptive activity of opioids at the supraspinal level was evaluated by the tail-flick test. For each opioid and treatment (saline, ODN-RD, and ODNs), a different group of animals was used. Latencies were measured 30 min after administration of clonidine (150 nmol/mouse) or morphine (3 nmol/mouse), 15 min after DAMGO (0.07 nmol/mouse), and 10 min after the endomorphins (6 nmol/mouse). Antinociception is expressed as a percentage of the MAE. Values are the mean ± S.E.M. from groups of 10 to 15 mice each. *Significantly different from the control group receiving saline or the ODN-RD instead of the ODN to the corresponding Gα subunit (ANOVA, Student-Newman-Keuls test, P< .05).

Agonist-antagonist activities of endomorphins in mice after pretreatment with ODN-Gi1α. The ODN was given as described in Materials and Methods. To analyze the interaction between agonists and morphine, the alkaloid was coinjected with clonidine or given 20 min before the endomorphins. Analgesia was measured 10 min later. Control groups received the random ODN instead of the ODN to the Gi1α. Values are the mean ± S.E. from groups of 10 to 15 mice each. *Significantly difference with respect to the effect of morphine of the control group. Other details as in the legend of Fig. 3.
The supraspinal antinociceptive effects of clonidine were highly reduced by the administration of the ODNs directed to both Go1α or Go2α subunits, whereas neither endomorphins nor morphine or DAMGO antinociception was influenced by these treatments (Fig. 3). Finally, the antisense ODNs to Gqα and G11α subunit-mRNAs produced dissimilar effects; DAMGO-induced antinociception was reduced whereas the activity of endomorphins, as well as that exhibited by morphine, was unaltered (Fig. 5).

Effect of antisense ODNs to Gqα and G11α subunits on the supraspinal analgesia induced by μ opioid agonists. Mice received repeated i.c.v. injections of ODNs to Gqα or G11α subunits on a once-daily schedule (see Materials and Methods). Latencies were measured 30 min after morphine, 15 min after DAMGO, and 10 min after endomorphins. Details as in the legend of Fig. 3.
Discussion
There is increasing evidence that endomorphins from bovine brain extracts are natural ligands of the μ opioid receptor. Studies in animal models have revealed that i.c.v. administration of endomorphin-1 to mice induces a long-lasting antinociceptive effect that can be blocked by pretreatment with the μ-selective antagonist β-funaltrexamine (Zadina et al., 1997). [3H]Endomorphin shows no detectable binding in brain membranes from mice lacking the μ receptor gene (Borsodi et al., 1998). Endomorphin-2 displays no analgesic effect in such animals (Loh et al., 1998). This confirms the activity of these endogenous tetrapeptides at μ opioid receptors. In brain regions involved in nociception, the incorporation of [35S]guanosine-5′-O-(3-thio)triphosphate to G proteins evoked by endomorphin-1 is weaker than that promoted by DAMGO (Sim et al., 1998). Thus, in contrast to the remarkable affinity displayed by endomorphin-1 in vitro (Zadina et al., 1997), this peptide seems to behave as a partial agonist at the receptors bound by DAMGO. This might account for the inefficiency of this compound to produce the levels of supraspinal analgesia observed for other μ receptor-binding opioids [i.e., morphine and DAMGO (present work)].
The administration of antisense ODNs to Gα subunit mRNAs is used to selectively impair the function of a single class of mouse G regulatory proteins. After five consecutive days of repeated i.c.v. injections, decreases of 20 to 60% on these Gα-like immunoreactivities are observed in neural structures of mouse brain (Sánchez-Blázquez et al., 1995; Sánchez-Blázquez and Garzón, 1998). Similar reductions in the expression of Gα subunits in rodent CNS have also been reported by other groups using chronic delivery of the ODNs (Standifer et al., 1996) or single high-dose treatments (Shen et al., 1998). Mismatched ODNs, or a ODN-RD, did not significantly change Gα immunoreactivity compared with that of naive mice. These treatments showed no cross effect on other Gα subunits or on the immunoreactivity associated with nonrelated proteins (Sánchez-Blázquez et al., 1995;Sánchez-Blázquez and Garzón, 1998).
Although in the promotion of supraspinal antinociception the agonist at α2-adrenoceptors, clonidine, showed activity with most of the G proteins evaluated (Sánchez-Blázquez et al., 1996; present work), the opioids morphine, DAMGO, and the endomorphins after binding μ receptors (Matthes et al., 1996, 1998;Sora et al., 1997; Loh et al., 1998) showed distinct patterns of regulating these classes of G proteins (Fig.6). The pertussis toxin-insensitive Gz protein was activated by all the opioids. Gi1 and Gi3 were regulated by both endomorphins but not by morphine or DAMGO. Gi2 was regulated by all except endomorphin-1. The Go1 and Go2 proteins were activated by clonidine but by neither of the μ-binding opioids. DAMGO was the only μ-binding opioid agonist to show some activity with Gq proteins (Garzón et al., 1995; present work). The implication of a phosphoinositide second messenger pathway in the antinociceptive effects of μ receptor-binding agonists has been suggested (Raffa et al., 1992). Thus, besides the involvement of the cAMP signaling pathway, the activation of phospholipase C appears to be implicated in both μ and δ receptor-mediated analgesic effects (Raffa et al., 1992; Sánchez-Blázquez and Garzón, 1998; present work).

Assignment of G proteins to different agonists at μ opioid receptors in the production of supraspinal analgesia. Solid lines denote significant reductions in analgesic efficacy in mice undergoing treatment with ODNs to these G proteins. G proteins without arrows are those not regulated by the agonist in the production of this effect.
The G protein activation promoted by the μ-binding agonists shows differences from that described for agonists at δ opioid receptors. The Gi2, Gi3, Go2, and G11 proteins are activated by both subtypes of δ opioid receptors in the production of supraspinal antinociception (Sánchez-Blázquez et al., 1995;Sánchez-Blázquez and Garzón, 1998). Furthermore, δ-mediated supraspinal analgesia is reduced by antisense ODNs complementary to mRNA sequences of Go1α and Gqα subunits. Go1 proteins seem to be selectively activated by δ1 receptors, whereas δ2 receptors show preference for the pertussis toxin-insensitive Gq proteins in this effect (Sánchez-Blázquez and Garzón, 1998). Thus, agonists at μ and δ opioid receptors exert their analgesic effects via the activation of both pertussis toxin-sensitive and -insensitive G proteins. The present study shows that a variety of G transducer proteins, Gi1, Gi2, Gi3, and Gz, are also involved in the supraspinal analgesic effects of the novel opioid peptides endomorphin-1 and endomorphin-2.
There is ample literature describing pleiotropic agonist responses at a single receptor. Differences in the activation profiles of agonists can then be explained on the basis of heterogeneous transduction and efficacy in the activation of all, or the most efficiently coupled, G proteins (Kenakin and Morgan, 1988). However, in certain circumstances, agonists acting at the same receptor show reversal of potency. This has already been described in the production of antinociception for opioid agonists at μ or δ opioid receptors. The study of the supraspinal antinociceptive effect of opioids mediated by μ opioid receptors has shown that the impairment of a single class of G proteins brings about decreases in the efficacy of some, but not all, agonists. Certain ligands even exhibit antagonist properties (Sánchez-Blázquez and Garzón, 1988, 1998;Garzón et al., 1994). After reducing the availability of Gi2 proteins in mice, [d-Ala2,d-Leu5]enkephalin antagonized the analgesic effect promoted by morphine. Conversely, the reduction in functional Gz proteins brought about the antagonism of [d-Ala2,d-Leu5]enkephalin-evoked antinociception by morphine (Garzón et al., 1994). Antagonism was also described in δ opioid receptor-mediated activation of G proteins (Garzón et al., 1997a). After impairing the synthesis of Go1α subunits, [d-Pen2,d-Pen5]enkephalin exhibited an antagonistic activity on the antinociception produced by [d-Ala2]deltorphin II (Sánchez-Blázquez and Garzón, 1998). The present study reports the antagonism of endomorphin-1 and endomorphin-2, but not of clonidine, on morphine-evoked analgesia in mice undergoing Gi1α knockdown.
Pharmacological studies have revealed that opioid agonists of peptide and nonpeptide classes interact with μ opioid receptors in a different manner (Ward et al., 1986; Sánchez-Blázquez and Garzón, 1988; Garzón and Sánchez-Blázquez, 1991). Studies with site-directed mutagenesis have indicated differences in the binding profiles of agonists and antagonists (Surratt et al., 1994; Wang et al., 1995). Moreover, small nonpeptide ligands with agonist properties, such as sufentanyl or morphine, bind to regions of the μ opioid receptor that are partially distinct from those bound by peptide agonists such as DAMGO (Fukuda et al., 1995;Wang et al., 1995; Xue et al., 1995). These differences have also been described for the binding of the selective ligands at δ opioid receptors (Befort et al., 1996) and κ opioid receptors (Meng et al., 1995). Differences in the interaction of agonists with receptors also reside in their capacity to bind with greater affinity when the receptor is coupled to a particular type of G protein (Garzón et al., 1998). Patterns of G protein-dependent agonist-receptor interactions might also account for differences of cAMP-dependent protein kinase phosphorylation of μ opioid receptors (Chakrabarti et al., 1998). Such results suggest that efficacy of agonists depends on the classes of G proteins activated by the liganded receptor. This has been determined in certain expression systems: theDrosophila octopamine-tyramine receptor in Chinese hamster ovary cells (Robb et al., 1994), the pituitary adenylyl cyclase-activating polypeptide receptor transfected into LLCPK1 cells (Spengler et al., 1993), and δ opioid receptor-binding opioids in membranes from mouse periaqueductal gray matter (Garzón et al., 1997a). Considering the capacity of receptors to discriminate between G proteins and the agonist-dependent binding domains of the receptor, some agonists might promote one receptor/G protein complex, whereas others favor the association of the receptor with a different G protein (Garzón et al., 1994, 1998).
Thus, the different patterns of G protein activation observed for the agonists at μ opioid receptors in the present work might account for the low efficacy exhibited by the endomorphins in the production of μ opioid receptor-mediated supraspinal antinociception.
Footnotes
-
Send reprint requests to: Dr. Pilar Sánchez-Blázquez, Instituto Cajal, CSIC, Avenida Doctor Arce 37, E-28002, Madrid, Spain. E-mail: jgarzon{at}cajal.csic.es
-
↵1 This work was supported by Comisión Interministerial de Ciencia y Tecnologı́a Grant CICYT SAF98-0057, Comunidad Autónoma de Madrid (CAM) Grant 08.8/0011/1998, and Fondo de Investigaciones Sanitarias (FIS) Grant FIS97/0506. M.R.-D. is supported by CAM. I.D. is supported by FIS. A preliminary report of this work was presented at the 29th International Narcotic Research Conference, Gasmisch-Partenkirchen, July 1998.
- Abbreviations:
- G protein
- GTP-binding protein
- i.c.v.
- intracerebroventricular
- ODN
- oligodeoxynucleotide
- MAE
- maximum analgesic effect
- RD
- random sequence
- DAMGO
- [d-Ala2,N-MePhe4,Gly-ol5]enkephalin
- ICI 174,864
- N,N-diallyl-Tyr-Aib-Aib-Phe-Leu
- CTOP
- Cys2,Tyr3,Orn5,Pen7-amide (somatostatin analog)
- Received February 23, 1999.
- Accepted May 28, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}