


Abstract
The objective of the present study was to evaluate the effects of EMD 61753 (asimadoline), a κ-opioid receptor agonist with restricted access to the central nervous system, on postoperative pain in patients who underwent knee surgery and on nociceptive thresholds and inflammation in rats treated with Freund’s complete adjuvant. Patients treated with EMD 61753 (10 mg p.o.) tended to report an increase in pain, as evaluated by a visual analog scale and by the time to the first request for and the total amount of supplemental analgesic medication. The global tolerability of EMD 61753 was assessed as significantly inferior to that of a placebo by the investigator. In rats, the bilateral intraplantar (i.pl.) injection of EMD 61753 (0.1–3.2 mg) resulted in dose-dependent antinociception in both inflamed and noninflamed paws, with a peak at 5 min after injection, as evaluated by the paw pressure method. However, at later time points (1 h–4 days), a significant decrease in the paw pressure threshold was observed, confirming its tendency toward a hyperalgesic action in humans. This was accompanied by an increase in paw volume and paw temperature, with a peak at 6 h after injection. EMD 61753 (1.6 mg)-induced analgesia was blocked by the peripheral opioid receptor antagonist naloxone methiodide (2.5–10 mg/kg s.c.) and by the κ receptor antagonist nor-binaltorphimine (0.1 mg; i.pl.). In contrast, EMD 61753 (1.6 mg)-induced hyperalgesia and increases in paw volume and paw temperature were blocked neither by naloxone methiodide (10–40 mg/kg s.c.) nor by dizocilpine maleate (0.003–0.009 mg i.pl.), aN-methyl-d-aspartic acid receptor antagonist. These data show differentially mediated peripheral actions of EMD 61753: κ-opioid receptor-induced analgesia and nonopioid, non-N-methyl-d-aspartic acid hyperalgesic and proinflammatory effects.
A significant drawback to the acute and chronic use of opiates is a variety of adverse side effects mediated predominantly in the central nervous system (CNS). Although κ-opioid receptor agonists do not cause respiratory depression or constipation and have a reduced potential for abuse compared with μ-opioid receptor agonists (Horwell, 1988), they can produce sedation and dysphoria (Pfeiffer et al., 1986). These CNS side effects can be avoided by the administration of small, systemically inactive doses of κ analgesics directly into injured tissue, where they can produce potent antinociception by activating peripheral opioid receptors (for review, see Barber and Gottschlich, 1992; Stein, 1993, 1995). These effects are particularly prominent under inflammatory conditions and may have a reduced capability to produce tolerance (Stein et al., 1996). A strategy to improve the side-effect profile of opioid analgesics is the restriction of their access to the CNS (Giardina et al. 1995; Barber and Gottschlich, 1997). There is now considerable evidence that peripherally restricted κ-opioid agonists can produce antinociception in different models of inflammatory and neuropathic pain without eliciting unwelcome CNS side effects (for review, see Giardina et al., 1995; Barber and Gottschlich, 1997).
In contrast, κ receptor agonists also can exert nonopioid actions. It was found that dynorphin A(1–17) injected intrathecally produces mechanical hyperalgesia and allodynia in the rat. These effects were not affected by naloxone, but were blocked byN-methyl-d-aspartic acid (NMDA) receptor antagonists (Laughlin et al., 1997). Similar effects also were described for exogenous κ agonists (Kest et al., 1992; Saucier and Kavaliers, 1994). κ receptor ligands also are involved in spinal cord injury, motor impairment, and inflammation through nonopioid mechanisms (for review, see Shukla and Lemaire, 1994).
EMD 61753 (asimadoline) has been described as a selective κ agonist with a restricted ability to cross the blood-brain barrier. This compound contains hydrophilic and hydrophobic groups that determine its peripheral selectivity and high potency at κ-opioid receptors. It has been found to produce dose-dependent, naloxone-reversible antinociception after systemic administration in rodent models of short-lasting inflammation (Barber et al., 1994). Recently, an antiarthritic action of EMD 61753 has been reported in polyarthritic rats (Binder and Walker, 1998). These positive results made EMD 61753 a good candidate for examination in clinical trials. In healthy human volunteers, EMD 61753 was rapidly absorbed into the blood after oral administration and very well tolerated (i.e., the number of adverse events was no different from those reported with a placebo; Barber and Gottschlich, 1997). Here, we investigate the effects of EMD 61753 on postoperative pain in patients who underwent knee surgery. Our unexpected negative results were confirmed by studying the effects of intraplantar (i.pl.) EMD 61753 on pain threshold and inflammation in rats treated with Freund’s complete adjuvant (FCA).
Materials and Methods
Human Studies
Patients.
The study protocol adhered to the ethical guidelines of the International Association for the Study of Pain (Charlton, 1995). Thirty-five patients who underwent diagnostic arthroscopic knee surgery were examined. The criteria for exclusion from the study were evidence of severe cardiovascular, respiratory, metabolic, or neurologic disease and the requirement for postoperative intra-articular drainage. All of the patients were premedicated i.v. with midazolam (0.1 mg/kg) 2 h before surgery. Anesthesia was induced with thiopental (4–5 mg/kg i.v.) and succinylcholine (1 mg/kg), and maintained with oxygen/nitrous oxide and isoflurane, with supplemental doses of vecuronium as needed. No opioids were administered during surgery.
Study Protocol.
Patients (Table1) were examined on the day before surgery. On the day of surgery, they received, in a randomized double-blind manner, either EMD 61753 (n = 17; Merck KGaA, Darmstadt, Germany) or a placebo (n = 18), given p.o. in two separate doses: the first dose (5 mg) was given 30 min before and the second dose (5 mg) was given 1.5 h after surgery. Surgery lasted approximately 1 h. The compounds were applied in color-coded capsules and the codes were broken at the end of the study.
Characteristics of patients undergoing knee surgery
Pain Assessment.
Pain was assessed with a 100-mm visual analog scale (VAS) ranging from no pain (0 mm) to unbearable pain (100 mm), a numerical rating scale (NRS) ranging from 0 to 100, and the McGill Pain Questionnaire (MPQ). The use of these pain measures was explained to each patient before surgery. The scores were taken just after surgery (time 0) and every hour up to 8 h after surgery. Analgesic rescue medication (piritramide i.v.) was available to all patients at all times. The time to the first request of supplemental medication by the patient was measured and the total amount of remedication was calculated up to 8 h after surgery.
Assessment of Vital Signs and Side Effects.
Blood pressure and heart rate were measured on the day before, 30 min before, immediately after, 8 h after, and 1 day after surgery. Side effects such as dizziness, headache, somnolence, paresis, sweating, visual abnormality, diarrhea, nausea, vomiting, and diuresis were recorded. Global tolerability was assessed by the investigator at the end of the observation period with a verbal scale (fair, good, very good, or excellent).
Statistical Analysis.
Data are presented as means ± S.E.M. Differences between the groups were assessed by analysis of covariance (ANCOVA) with respect to the parallel group design (factor medication, covariable baseline at the preoperative examination) for the VAS at each time point (0–8 h). Pairwise comparisons were performed with least-square means of ANCOVA. Differences between the treatment groups regarding the NRS were assessed by an exact test of 2 × k contingency tables, and differences regarding the MPQ were assessed by the Wilcoxon test. Mean differences between measurements at different time points within each treatment group were computed with 95% confidence intervals. If the confidence interval did not include 0, the mean difference was interpreted as statistically significant (paired t test, descriptivep values). The significance level was fixed at α = 5% of two-tailed. All other variables (vital signs and side effects) were analyzed descriptively.
Animal Studies
Animals.
Experiments were carried out in male Wistar rats (250–300 g; Charles River Laboratories, Wilmington, MA) housed individually in cages lined with ground corn cob bedding. Standard laboratory rodent chow and water were available ad libitum. Room temperature was maintained at 22 ± 0.5°C with a relative humidity between 40 and 60%. A 12-h light/dark cycle (7 AM/7 PM) was used. The experiments were performed according to standard ethical guidelines by the International Association for the Study of Pain and by the National Institutes of Health (1985).
Induction and Evaluation of Inflammation.
Rats received an intraplantar (i.pl.) injection of 0.15 ml of FCA (Calbiochem, La Jolla, CA) into the right hind paw under brief halothane anesthesia (Halocarbon Laboratories, North Augusta, SC). The paw volume (PV) was monitored with a plethysmometer (Ugo Basile, Comerio, Italy). The volume of displacement, which is equal to the PV, was indicated on a digital display. The dorsal surface temperature of the paw skin (PT) was measured with a contact thermometer (Cooper Instrument Corporation, Middlefield, CT).
Nociceptive Threshold.
The nociceptive threshold was assessed by the paw pressure test (modified Randall-Selitto test). The animals were gently restrained under paper wadding and incremental pressure was applied via a wedge-shaped, blunt piston onto the dorsal surface of the hind paw by means of an automated gauge (Ugo Basile). The pressure required to elicit paw withdrawal, the paw-pressure threshold (PPT), was determined. A cutoff of 250 g was used. Three consecutive trials, separated by intervals of 10 s, were conducted and the average was determined. The same procedure was performed on the contralateral side; the sequence of sides was alternated between subjects to preclude order effects.
Drugs and Their Administration.
The following substances were used: [3H]diprenorphine (Amersham, Arlington Heights, IL); EMD 61753 (Adolor Corporation, Malvern, PA; synthesized by a published procedure to afford exclusively the active S,S isomer); and naloxone methiodide (NLXM), nor-binaltorphimine (norBNI), and dizocilpine maleate (MK-801 maleate; Research Biochemicals Inc., Natick, MA). NLXM and norBNI were dissolved in sterile water, MK-801 was dissolved in 0.9% saline, and EMD 61753 was dissolved in 5% dimethyl sulfoxide and diluted with sterile water to obtain lower concentrations.
Behavioral Studies.
Experiments were performed at 4 to 6 days after FCA inoculation. EMD 61753 (0.1–3.2 mg/100 μl) was injected i.pl. into both hind paws. A separate group of animals received EMD 61753 s.c. (at the neck) at a dose of 3.2 mg/200 μl (an equivalent of 1.6 mg/paw). PV and PPT were measured before administration (time 0), and at 5 min, 10 min, 30 min, 1 h, 6 h, 24 h, 48 h, or 4 days after administration. PT was measured before and 6 h after injection. Dose-response curves of i.pl. EMD 61753-induced effects and the evaluation of their antagonism were determined at the time of the peak of these effects, i.e., at 5 min for analgesia and at 6 h for increases in PV and PT. Antagonism of hyperalgesic effects of EMD 61753 was tested at 6 and 24 h after injection. For ethical reasons, hyperalgesic effects produced by different doses of EMD 61753 and their antagonism, as well as the antagonism of increases in PV and PT, were evaluated in rats without inflammation. NLXM (2.5–40 mg/kg) was injected s.c. 5 min before i.pl. EMD 61753 (1.6 mg). norBNI was given i.pl. (0.1 mg) concomitantly with EMD 61753 (1.6 mg i.pl.) or s.c. (0.2 mg) immediately before EMD 61753 (1.6 mg i.pl.). MK-801 (0.003–0.009 mg/paw) was injected i.pl. immediately before EMD 61753 (1.6 mg i.pl.). The total volume of i.pl. injections was 100 μl. Control groups were injected with vehicle and tested according to the same schedule as the experimental groups. The number of rats used per group ranged from 6 to 11.
In Vitro Binding Studies
Receptor binding was carried out with Chinese hamster ovary cells that stably express cloned human μ-, δ-, or κ-opioid receptors. Chinese hamster ovary cells were harvested by scraping from the culture flasks, centrifuged at 1000g for 10 min, and then resuspended in assay buffer (Raynor et al., 1994) and centrifuged again. The resulting pellet was resuspended in assay buffer and homogenized with a Polytron homogenizer (Brinkmann, Westbury, NY) for 30 s at a setting of 1. The homogenate was centrifuged at 48,000g for 10 min at 4°C and the pellet was resuspended in 1 mg of protein/ml of assay buffer and stored at −80°C until use.
Binding was performed with [3H]diprenorphine (1 nM; 40,000–45,000 dpm). Membrane proteins (0.05–0.2 mg in 250 μl of assay buffer) were added to mixtures containing EMD 61753 and [3H]diprenorphine in 250 μl of assay buffer in 96-well polystyrene titer plates (Beckman Instruments, Berkeley, CA) and incubated at room temperature for 1 h. The reaction was terminated by vacuum filtration with a Brandel MPXR-96T Harvester through GF/B filters that had been pretreated with a solution of 0.5% polyethylenimine and 0.1% BSA for at least 1 h. The filter-bottom plates were washed four times with 1 ml of ice-cold 50 ml of Tris-HCl at pH 7.8. Thirty microliters of Microscint-20 (Packard Instrument Company, Meriden, CT) was added to each filter, and the radioactivity on the filters was determined by scintillation spectrometry in a Packard TopCount (Packard Instrument Company).
[3H]Diprenorphine had a specific activity of 39 to 45 Ci/mmol. Preliminary experiments were performed to show that no specific binding was lost during the washing of the filters, that binding achieved equilibrium within the incubation time and remained at equilibrium for at least an additional hour, and that binding was linear with regard to protein concentration. Nonspecific binding, determined in the presence of 10 μM unlabeled naloxone, was less than 10% of the total binding.
Statistical Analysis.
Data from behavioral experiments are presented as means ± S.E.M. and are expressed in raw values, as a percentage of the maximum possible effect (%MPE), or as a percentage of the baseline (%BL). The %MPE was calculated according to the formula: (PPTpostinjection − PPTbasal)/(250 − PPTbasal) × 100. Changes in PPT and PV over time were evaluated by the Friedman test and the Dunnett multiple-range test. Changes in PV also were evaluated by two-way repeated measures ANOVA. Two-sample comparisons were made by the Mann-WhitneyU test for independent data and the Wilcoxon test for dependent data. Dose-response curves were analyzed by an ANOVA followed by linear regression. Data from binding studies were fit by nonlinear regression analysis with Prism software (GraphPad Software, Inc., San Diego CA) with the four-parameter equation for one site competition and subsequently calculating Ki from the IC50 with the Cheng-Prusoff equation. Differences were considered significant if p < .05.
Results
Human Studies
All patients were awake and oriented throughout the observation period. There were no significant changes in blood pressure and heart rate between control and EMD 61753-treated patients and no serious side effects were observed (not shown). The NRS and MPQ revealed no statistically significant differences between the two groups (p > .05; 2 × k contingency tables and Wilcoxon test, respectively; not shown). The VAS revealed a significant increase in pain scores at 1.5 h (time 0 on the graph) after the first dose of EMD 61753 compared with controls (p = .03; least-square means, Fig.1A). There were no significant differences in the time to remedication or in the total amount of piritramid consumption (p > .05; ANCOVA; Fig. 1, B and C). The global tolerability of the placebo was judged as significantly better than that of EMD 61753 by the investigator. The investigator recorded the following number of patients choosing the words fair, good, very good, or excellent: placebo, 0, 4, 6, and 7, respectively; EMD 61753, 5, 8, 2, and 2, respectively (p = .011; 2 × k contingency tables).

Effects of oral EMD 61753 (10 mg) on pain threshold in patients who underwent knee surgery, evaluated by the VAS (A), by the time to request for remedication (B), and by the amount of supplemental medication (C). The labels on the x-axis indicate time after surgery. EMD 61753 was given p.o. 30 min before and 90 min after surgery in the amount of 5 mg each (total 10 mg). *p = .03; ANCOVA. Data are expressed as means ± S.E.M.
Animal Studies
Behavioral Studies.
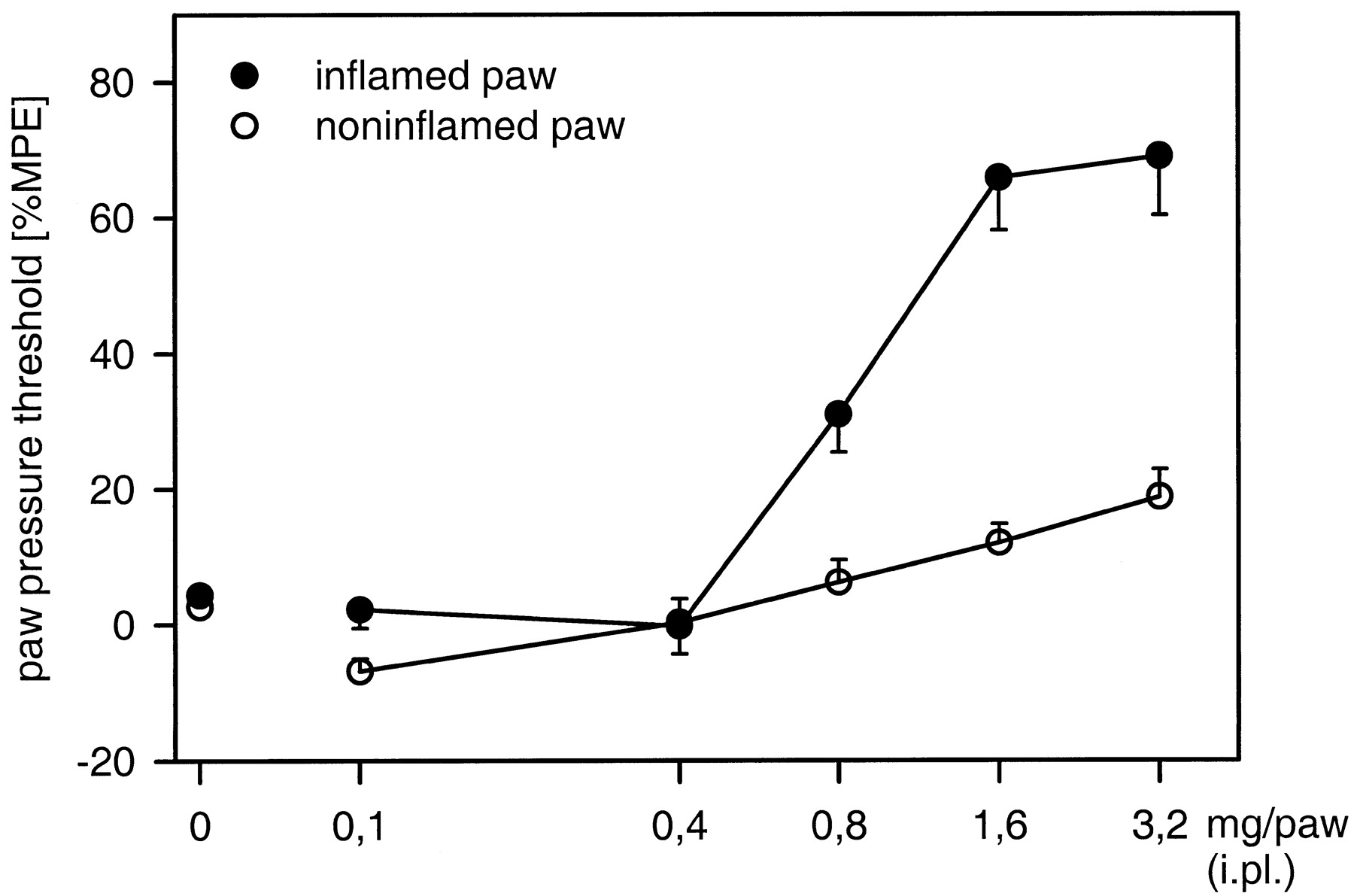
Antinociception. Fig.2, A and B shows a representative time course of PPT alterations after i.pl. EMD 61753 (1.6 mg). An increase in PPT was observed in both inflamed and noninflamed paws at 5 and 10 min, and a return to baseline level was observed at 30 min (p < .05; Dunnett test). The s.c. injection of 3.2 mg (the equivalent dose of 1.6 mg/paw) had no effect on PPT in either inflamed or noninflamed paws (p > .05; Friedman test; not shown). The antinociceptive effects of EMD 61753 (0.1–3.2 mg) were dose-dependent (p < .001; ANOVA, linear regression; Fig. 3).

Effects of i.pl. EMD 61753 (0.1–3.2 mg) on PPT in the rat. PPT in A and B was evaluated in rats with unilateral FCA-induced inflammation and expressed in grams (g). PPT in C is expressed as %MPE and, because of ethical reasons, was evaluated in rats without inflammation. *p < .05; Dunnett test. Data are expressed as means ± S.E.M. m, minutes; h , hours; d, days.

Dose-response relationships of i.pl. EMD 61753 (0.1–3.2 mg)-induced antinociception in rats. Effects are significant in both inflamed and noninflamed paws (p < .001; ANOVA, linear regression). Data are expressed as means ± S.E.M.
EMD 61753 (1.6 mg)-induced antinociception in inflamed paws was dose-dependently blocked by 2.5–10 mg/kg s.c. NLXM ( p< .001; ANOVA, linear regression; Fig.4A). The effect in noninflamed paws also was blocked by 10 mg/kg s.c. NLXM (p = .005; Mann-Whitney U test) but was not dose-dependent (p = .062; ANOVA, linear regression; Fig. 4A). In both inflamed and noninflamed paws, EMD 61753 (1.6 mg)-induced antinociception also was blocked by 0.1 mg of i.pl. norBNI (p < .001 and p = .004, respectively; Mann-Whitney U test) but not by 0.2 mg s.c. of norBNI (p > .05; Mann-Whitney U test; Fig. 4B). Neither NLXM (10 mg/kg s.c.) nor norBNI (0.1 mg i.pl. and 0.2 mg s.c.) alone significantly affected PPT in inflamed or noninflamed paws (p > .05; Mann-Whitney U test; not shown).

Effects of NLXM (2.5–10 mg/kg s.c.; A) and norBNI (0.1 mg/paw i.pl. or 0.2 mg/rat s.c.; B) on i.pl. EMD 61753 (1.6 mg)-induced antinociception in the rat. A, dose-dependent effect appeared in inflamed but not in noninflamed paws, p< .001 and p = .062, respectively; ANOVA, linear regression. B, *p < .001, **p= .004; Mann-Whitney U test. Data are expressed as means ± S.E.M.
Hyperalgesia.
At later time points (6 h–4 days), i.pl. EMD 61753 (1.6 mg) significantly decreased PPT both in inflamed and in noninflamed paws (p < .05; Dunnett test; Fig. 2). As evaluated in noninflamed paws, this hyperalgesic effect was produced by a wide range of doses of EMD 61753 (0.1–3.2 mg; Fig. 2C) and was not dose-dependent. After injection of 0.1 mg, 0.4 mg, or 0.8 mg of EMD 61753, hyperalgesia appeared at 5 min, 10 min, or 1 h, respectively. Two higher doses (1.6 mg and 3.2 mg) decreased PPT at 6 h after injection. The hyperalgesic effect of EMD 61753 appeared to decrease after 6 h (0.1–0.4 mg) or 48 h (0.8–3.2 mg) after injection, but it was still significant up to 4 days after injection (Fig. 2). Pretreatment with NLXM (10–40 mg/kg s.c.) or MK-801 (0.003–0.009 mg i.pl.) did not affect EMD 61753 (1.6 mg)-induced hyperalgesia at 6 h or 24 h after injection (p > .05; Mann-Whitney U test; Table2). Neither NLXM (40 mg/kg s.c.) nor MK-801 (0.009 mg i.pl.) alone significantly affected PPT (p > .05; Mann-Whitney U test; not shown).
Effects of NLXM and MK-801 on i.pl. EMD 61753-induced hyperalgesia and increases in paw volume and temperature
Increases in PV and PT.
Intraplantar EMD 61753 (1.6 mg) increased the PV of both inflamed and noninflamed paws (Fig.5A). In inflamed paws, a statistically significant effect was detected at 1 to 6 h after injection (p < .05; Dunnett test). This effect was not dose-dependent (p > .05; ANOVA, linear regression; Fig. 5B). In noninflamed paws, the increase in PV was stronger, more pronounced, and dose-dependent (p = .003; ANOVA, linear regression; Fig. 5, A and B). It was detectable at 5 min after injection and was significant up to 48 h after injection (p < .05; Dunnett test; Fig. 5A). The EMD 61753-induced increase in PV was significantly different from that found in the control (two-way, repeated-measures ANOVA; inflamed paws,p < .001; noninflamed paws, p = .003).

Time course (A) and dose-response (B) relationships of effects of i.pl. EMD 61753 (0.8–3.2 mg) on PV in the rat. *p < .05; Dunnett test. The effect is dose-dependent in noninflamed but not in inflamed paws,p = .003 and p > .05, respectively; ANOVA, linear regression. Data are expressed as means ± S.E.M. m, minutes; h, hours; d, days.
EMD 61753 (0.8–3.2 mg i.pl.) increased PT of both noninflamed and inflamed paws (Fig. 6). In noninflamed paws, this effect was dose-dependent at 6 h after administration (p = .018; ANOVA, linear regression). The increase in PT of inflamed paws was statistically significant (1.6–3.2 mg;p < .05; Mann-Whitney U test; Fig. 6) but not dose-dependent (p > .05; ANOVA, linear regression).
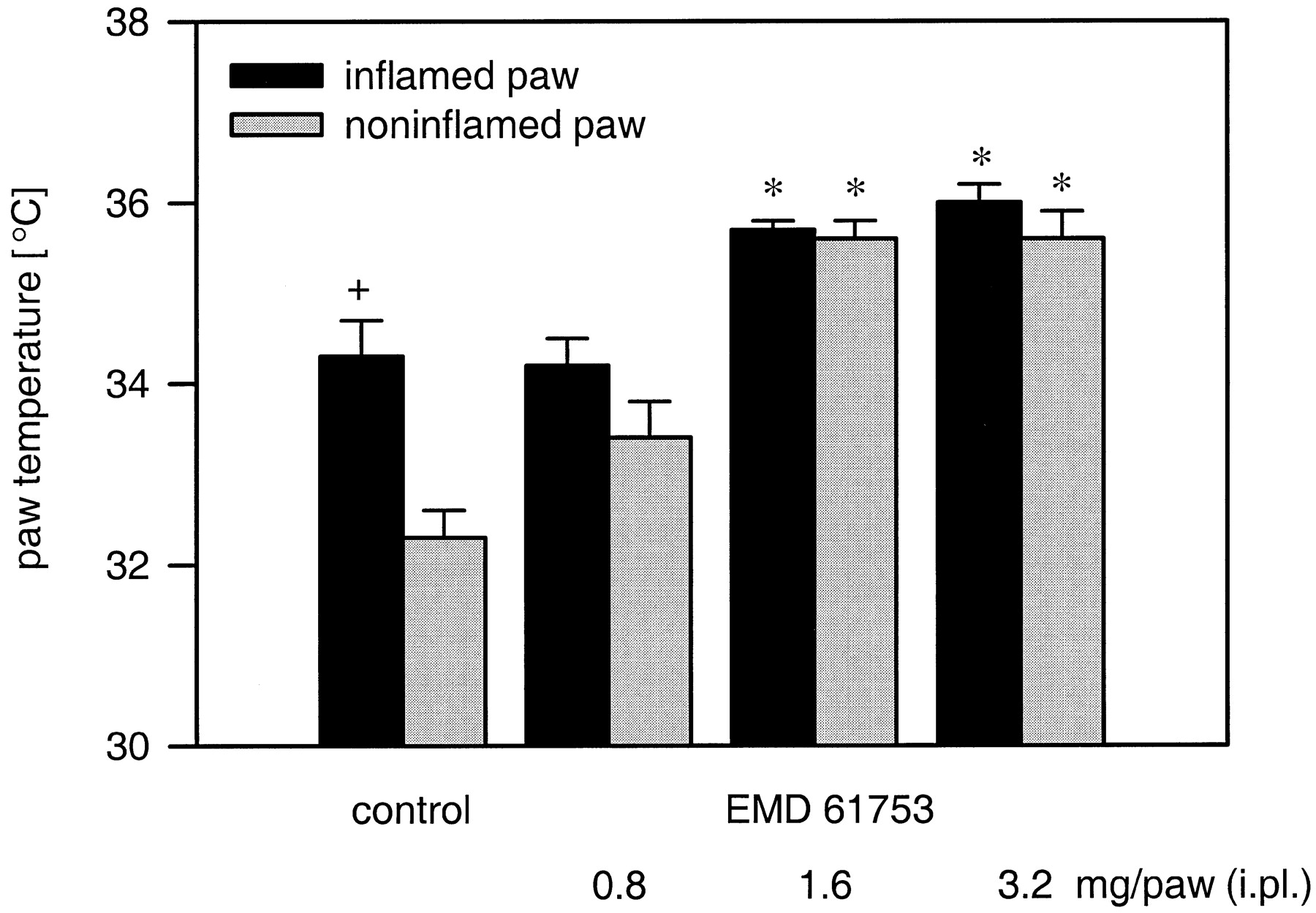
Effects of i.pl. EMD 61753 (0.8–3.2 mg) on paw temperature in the rat. + p < .001; Wilcoxon test, compared with the noninflamed paws of control subjects. *p < .05; Dunnett test, compared with the respective controls. The effect is dose-dependent in noninflamed but not in inflamed paws; p = .018 andp > .05, respectively; ANOVA, linear regression. Data are expressed as means ± S.E.M.
EMD 61753 injected s.c. at a dose of 3.2 mg (an equivalent of 1.6 mg/paw i.pl.) did not affect either PV (p > .05; Friedman test) or PT (p > .05; Mann-WhitneyU test; not shown). Pretreatment with NLXM (10–40 mg/kg s.c.) and MK-801 (0.003 mg i.pl.) did not significantly change EMD 61753 (1.6 mg i.pl.)-induced increases either in PV or PT (p > .05; Mann-Whitney U test; Table 2) in noninflamed paws at 6 h after administration. MK-801 (0.009 mg i.pl.) significantly elevated the EMD 61753 (1.6 mg)-induced increase in PT (p = .026; Mann-Whitney U test). Neither NLXM (40 mg/kg s.c.) nor MK-801 (0.009 mg i.pl.) alone significantly affected PV or PT (p > .05; Mann-WhitneyU test; not shown).
Binding Studies
EMD 61753 binds to κ-opioid receptors with aKi value of 2.4 (2.0–2.9) nM (IC50 ≈ 10 nM). The binding of EMD 61753 to μ- and δ-opioid receptors was much weaker, withKi values higher than 10,000 nM.
Discussion
Our findings indicate that EMD 61753 given p.o. at a dose of 10 mg did not produce clinically satisfactory pain relief in patients who underwent knee surgery. On the contrary, both direct (VAS) and indirect measures (amount of and time to supplemental analgesic medication, global tolerability) suggest a tendency toward hyperalgesic effects. The fact that in rats it was shown that oral EMD 61753 at a dose of 50 mg/kg (i.e., 378 times the dose we used in humans) produced peripherally mediated effects (Barber et al., 1994) also strongly suggests a peripheral action of EMD 61753 in our clinical studies. The above results were unexpected in view of previous preclinical studies showing antinociceptive effects of EMD 61753 (Barber et al., 1994;Binder and Walker, 1998). In agreement with those studies, we found that EMD 61753 injected directly into the rat paw produces dose-dependent antinociception against noxious pressure. This effect was detected shortly after injection (5–30 min) in both inflamed and noninflamed paws, although it was much stronger in the former. This is in accordance with previous studies showing increased efficacy of opioids in reducing pain under pathological conditions (Barber and Gottschlich, 1992; Stein, 1993, 1995; Barber et al., 1994). The antinociceptive effects of EMD 61753 were blocked by NLXM (a peripherally selective opioid antagonist) and by norBNI (a selective κ-opioid antagonist), injected directly into the paw but not injected systemically. Also, EMD 61753 injected systemically in an equivalent dose to that administered i.pl. did not affect nociceptive thresholds. Together, these data indicate that EMD 61753-induced antinociception is mediated by peripheral κ-opioid receptors.
In contrast, in lower doses (0.1–0.4 mg) or at later time points after the injection of higher doses (0.8–3.2 mg), EMD 61753 produced long-lasting (up to 4 days) hyperalgesia. This effect was observed in both paws, although the effect was stronger in the noninflamed paw. This is probably due to the fact that baseline nociceptive thresholds were already decreased in the inflamed paw. Dose-response relationships could not be established because of the highly variable pattern of time courses of effects produced by the different doses. The delay of hyperalgesia produced by the higher doses (0.8–3.2 mg) was apparently caused by the antinociceptive effects at earlier time points after injection (5–30 min; see above). These observations, i.e., both antinociceptive and hyperalgesic effects, are consistent with the dual modulatory mechanisms of opioids proposed by Crain and Shen (1990). However, in our study, EMD 61753-induced hyperalgesia was not affected by NLXM, which implies a nonopioid action. There is now considerable evidence that κ agonists can interact with NMDA receptors. It has been found that intrathecal dynorphin A(1–17), an endogenous ligand at κ-opioid receptors, releases excitatory amino acids in the brain (Faden, 1992) and produces long-lasting mechanical hyperalgesia and tactile, cold, and mechanical allodynia in the rat. The latter effects were not affected by naloxone but were blocked by NMDA receptor antagonists (Laughlin et al., 1997). NMDA antagonists also were found to block antinociception produced by the exogenous κ agonists U-50,488H and U69,593 (Kest et al., 1992; Saucier and Kavaliers, 1994). Furthermore, biochemical and electrophysiological studies suggest direct opioid-NMDA receptor interactions (Faden, 1992; Chen et al., 1995). However, this does not seem to be the case in our studies because EMD 61753-induced hyperalgesia was not influenced by the NMDA receptor antagonist MK-801. Thus, although hyperalgesic effects of EMD 61753 may be caused by the release of pronociceptive neurotransmitters found for κ-opioid receptor agonists (Faden, 1992; Suarez-Roca and Maixner, 1993), in our experiments, they apparently are mediated neither by opioid nor by NMDA receptors.
In addition, EMD 61753, injected i.pl. but not systemically, caused a substantial and dose-dependent increase in the volume and temperature of noninflamed paws. This effect also was observed in inflamed paws, although it was weaker and not dose-dependent, probably due to the preexisting substantial edema and elevated temperature. Similar effects of opioids on PV also were observed in other studies. For example, i.pl. dynorphin A(1–17) and systemic buprenorphine and morphine were shown to increase the volume of inflamed and noninflamed paws (Earl et al., 1994; Hall et al., 1996; Beyer et al., 1997). Opioid-induced edema and hyperthermia are probably due to changes in vascular permeability. There is evidence that opioids, including those acting at κ receptors, injected i.pl. can increase plasma extravasation (Chahl and Chahl, 1986; Haley et al., 1990; Hong and Abbott, 1995), presumably as a result of mast cell degranulation and histamine release (Casale et al., 1984). Also, neurogenic mechanisms cannot be ruled out, e.g., the dynorphin A(1–13)-induced increase in plasma extravasation was partially blocked by capsaicin, a primary afferent neurotoxin (Chahl and Chahl, 1986). Another possibile mechanism is the action of opioids on the immune system. Although there is controversy in the literature, stimulatory effects of opioids on proliferation, chemotaxis, superoxide, and cytokine production by immune cells have been described (Bryant and Holaday, 1993). In our study, the increases in PV and temperature produced by EMD 61753 do not seem to be mediated by opioid receptors because they were not blocked by pretreatment with NLXM. In view of putative opioid-NMDA receptor interactions and the involvement of excitatory amino acids in inflammatory processes (Bong et al., 1996; Yu et al., 1996), we tested this possibility in our study. However, the proinflammatory effects of EMD 61753 were not affected by MK-801 pretreatment.
There are two intriguing observations arising from these data. First, EMD 61753 produces antinociception at the same time points when it significantly increases PV. Some studies suggest that there is no correlation between pain sensitivity and plasma extravasation or edema (Hong and Abbott, 1995) or that plasma extravasation may even protect tissue by diluting locally produced proinflammatory mediators and/or by enhancing their clearance from sites of injury (Coderre et al., 1990). One might speculate that EMD 61753 produces analgesia partially by such a mechanism. Second, hyperalgesic effects produced by lower doses of EMD 61753 (0.1–0.4 mg), which apparently produce little edema, are as strong as those produced by higher doses (1.6–3.2 mg), which elicit substantial increases in PV. This suggests that the magnitude of hyperalgesia is not necessarily related to the degree of inflammation.
The peripheral κ receptor-mediated antinociceptive effects of EMD 61753 observed in our animal experiments are in accordance with previous observations (Barber et al. 1994; Binder and Walker, 1998). In those studies, EMD 61753 also was shown to inhibit neurogenic plasma extravasation (Barber et al., 1994) and to produce antiarthritic actions (Binder and Walker, 1998). This is at variance with the proinflammatory and hyperalgesic effects we observed in our animal and clinical studies. Different binding affinities caused by the different sources of the compounds (Adolor Corporation versus Merck KGaA) apparently do not account for this discrepancy because we found values comparable to those published in the former studies (Barber et al., 1994; Binder and Walker, 1998). However, bell-shaped dose-response curves were observed previously after both oral (Barber et al., 1994) and i.p. (Binder and Walker, 1998) administration of EMD 61753. In the latter study, the effect of the highest dose of EMD 61753 was not affected by NLXM and MR2266, a κ-opioid receptor antagonist. This is in line with the nonopioid hyperalgesic and proinflammatory effects of EMD 61753 observed in the present study and indicates that EMD 61753 has several nonspecific side effects that severely limit its clinical usefulness.
In summary, we have found that EMD 61753 injected directly into a rat paw produces differentially mediated peripheral actions: κ-opioid receptor-mediated antinociception and nonopioid and non-NMDA receptor-mediated proinflammatory and hyperalgesic effects. These results are in line with the lack of analgesic (or even pronociceptive) effects of EMD 61753 in patients who underwent knee surgery. These findings indicate that, similar to the situation in the central nervous system (Laughlin et al., 1997), κ-opioid receptor agonists can exert both beneficial opioid receptor-mediated and adverse nonopioid receptor-mediated effects in the periphery as well.
Acknowledgment
We thank Dr. S. R. Goldberg for his continuous support.
Footnotes
-
Send reprint requests to: Halina Machelska, Klinik für Anaesthesiologie und Operative Intensivmedizin, Klinikum Benjamin Franklin, Freie Universität Berlin, Hindenburgdamm 30, D 12200 Berlin, Germany. E-mail:MACHELSKA{at}zop-admin.ukbf.fu-berlin.de
-
↵1 This work was supported by Merck KGaA, Darmstadt, Germany and by Adolor Corporation, Malvern, PA.
-
↵2 Halina Machelska was on leave from the Department of Molecular Neuropharmacology, Institute of Pharmacology, Polish Academy of Sciences, Krakow, Poland.
- Abbreviations:
- CNS
- central nervous system
- NMDA
- N-methyl-d-aspartic acid
- VAS
- visual analog scale
- NRS
- numerical rating scale
- MPQ
- McGill Pain Questionnaire
- ANCOVA
- analysis of covariance
- i.pl.
- intraplantar
- FCA
- Freund’s complete adjuvant
- PV
- paw volume
- PT
- paw temperature
- PPT
- paw-pressure threshold
- MK-801
- dizocilpine maleate
- NLXM
- naloxone methiodide
- norBNI
- nor-binaltorphimine
- %MPE
- percentage of the maximum possible effect
- %BL
- percentage of the baseline
- Received November 25, 1998.
- Accepted March 2, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}