


Abstract
The mechanisms underlying the neuroprotective effects of the group II metabotropic glutamate receptor (mGluR) agonist LY379268 were investigated in a gerbil model of global ischemia. LY379268 (10 mg/kg i.p.) 30 or 60 min after 5-min bilateral carotid artery occlusion (BCAO) attenuated the ischemia-induced hyperactivity and provided protection in the CA1 hippocampal cells. This neuroprotective effect was maintained (P < .001) when histological analysis was performed 14 and 28 days after BCAO. Furthermore, 24- or 48-h pretreatment with LY379268, 10 mg/kg i.p., before 5-min BCAO markedly reduced (P < .001 andP < .05, respectively) the damage to CA1 hippocampal neurons. This result is consistent with the induction of neuroprotective factors or a very long brain half-life. To study the possible induction of neuroprotective factors as contributing to this action of LY379268, brains were examined for expression of neurotrophic factors. Results indicated that LY379268 (10 mg/kg i.p.) failed to alter the expression of transforming growth factor-β, brain-derived neurotrophic factor, nerve growth factor, and basic fibroblast growth factor in the hippocampal regions of brains taken from gerbils sacrificed at 6, 24, 72, and 120 h postinjection. The new group II mGlu antagonist, LY341495, administered 1 h before 5-min BCAO, attenuated the neuroprotective effect of LY379268 administered 24 h before 5-min BCAO. Complementary pharmacokinetic studies showed that a significant receptor-active concentration persisted in the brain 24 h after LY379268 10 mg/kg i.p. We conclude that group II mGluR occupancy, rather than induction of neuroprotective factors, explains the long-lasting neuroprotective effect of LY379268 in the gerbil model of global ischemia.
Glutamate is the major excitatory neurotransmitter in the mammalian central nervous system, activating ionotropic glutamate (iGlu) and metabotropic glutamate (mGlu) receptors. It also is thought to play a central role in acute neurodegeneration after cerebral ischemia (Meldrum and Garthwaite, 1990). However, for many years, investigators have focused mainly on the roles of iGlu receptors and, in particular, ofN-methyl-d-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-isoxazole-4-propionic acid (AMPA), and kainate receptors in ischemia (McCulloch, 1991; Gill and Lodge, 1997). Because ionotropic receptors play such a critical role in fast synaptic transmission, it has proved difficult to block these receptors without side effects, and to date no such compounds have therapeutic use in human stroke. Recent advances have provided selective ligands for metabotropic glutamate receptor (mGluR) subtypes (Pin et al., 1999;Schoepp et al., 1999), allowing further investigation of the proposed role of mGluRs in aspects of neurodegeneration (Schoepp and Conn, 1993;Buisson et al., 1996; Nicoletti et al., 1996).
To date, eight subtypes of the G protein-coupled mGluRs have been cloned and classified into three groups according to their second-messenger association, sequence homology, and agonist selectivity (Pin and Duvoisin, 1995; Pin et al., 1999). Group II (mGlu2 and -3) and group III (mGlu4, -6, -7, and -8) mGluRs are negatively coupled to adenyl cyclase and thought to act as presynaptic autoreceptors, regulating glutamate transmission (Shigemoto et al., 1995). Recent evidence indicates that mGlu3 receptors are also expressed by astrocytes and glia (Petralia et al., 1996). Activation of group II mGluRs is reported to protect neurons against excitotoxic degeneration by the inhibition of glutamate release (Buisson and Choi, 1995; Buisson et al., 1996). In support of this idea, the selective group II mGluR agonist LY354740 (Schoepp et al., 1997) reduces veratradine-evoked striatal amino acid release (Battaglia et al., 1997) and field excitatory postsynaptic potentials in rat hippocampal slices (Kilbride et al., 1998).
Although reduction of glutamate release is an attractive hypothesis, there is a body of evidence in vitro that suggests this may not always account for the neuroprotective activity of group II mGluR agonists. For example, Bruno et al. (1997) suggested that astrocytes, after exposure to group II mGluR agonists, produce a heat-sensitive factor in the culture medium, which itself has neuroprotective properties in cortical cells. More recently, this group reported that transforming growth factors-β1 (TGF-β1) and -β2 were released from astrocytes exposed to group II mGluR agonists and that antibodies that neutralized the actions of TGF-β1 or -β2 prevented the neuroprotective effects of DCG-IV and 4C3HPG in cultured cortical neurons (Bruno et al., 1998).
Group II mGlu agonists are also reported to be neuroprotective in vivo (Chiamulera et al., 1996; Miyamoto et al., 1997), but these earlier compounds were not very selective. A more selective group II agonist, LY354740, has been shown to provide some neuroprotection in gerbil global ischemia (Bond et al., 1998) but none in rat focal ischemia (Lam et al., 1998). Recently, a more potent and highly selective group II agonist, LY379268 [(1R,4R,5S,6R)-2-oxa-4-aminobicyclo[3.1.0]hexane-4,6-dicarboxylate], was discovered that has EC50 values of 2.69 ± 0.26 and 4.48 ± 0.04 nM at human mGluR2 and mGlu3 receptors, respectively (Monn et al., 1999). LY379268 provided neuroprotection against NMDA-mediated cell death in vitro (Kingston et al., 1999) and almost complete protection against CA1 hippocampal damage after global ischemia in gerbils but failed to show neuroprotection against focal ischemia in the rat (Bond et al., 1999b). Therefore, the mechanism of neuroprotection in the gerbil model remains to be elucidated.
In this study, we extended our original findings by investigating neuroprotection in terms of both histological and functional behavioral improvement in gerbils subjected to 5-min bilateral carotid artery occlusion (BCAO). We have undertaken detailed time course studies to evaluate the effects of LY379268 both during the induction of ischemic brain injury and at later time points after occlusion. To understand the mechanism of action of LY379268, we investigated the hypothesis that group II mGlu agonists may induce neurotrophic factors with neuroprotective properties, by examining its effects on the expression of TGF-β1, TGF-β2, brain-derived neurotrophic factor (BDNF), nerve growth factor (NGF), and basic fibroblast growth factor (bFGF) in brain tissue using immunocytochemistry. We have also investigated the interaction of LY379268 with the induction of “ischemic tolerance” in the gerbil (Kirino et al., 1991). Finally, we examined whether the protection afforded by 24-h pretreatment with LY379268 is blocked by the new selective group II antagonist, LY341495 (Kingston et al., 1998) and made measurements of brain levels of LY379268 at various time points after drug administration.
Materials and Methods
Animals and Surgery.
Male Mongolian gerbils (Bantin and Kingman, Hull, UK) at least 3 months old and weighing in excess of 60 g were maintained in standard lighting conditions with food and water available ad libitum. The animals were anesthetized with a 5% halothane/oxygen mixture and maintained using 2% halothane delivered with oxygen at 1 l/min via a face mask throughout the operation. Through a midline cervical incision, both common carotid arteries were exposed, freed from surrounding connective tissue, and clamped for 5 min, except for preconditioning experiments where clamps were applied for 2 min. At the end of the occlusion period, blood flow was reestablished. In sham-operated animals, the arteries were exposed but not occluded. The wound was then sutured, and the animals were allowed to recover. Throughout surgery, body temperature was maintained at 37°C using a K-TEMP temperature controller/heating pad (International Market Supply, Cheshire, UK). After surgery, the animals were placed in a four-compartment Thermacage (Beta Medical and Scientific, UK) that maintained the environmental temperature at 28°C, and rectal temperatures were measured for a 6-h period after occlusion.
General Histology.
Five days (or, in some experiments, 6, 24, 72, or 120 h or 14 or 28 days) after surgery, the animals were perfused transcardially with 30 ml of 0.9% saline followed by 100 ml of 10% buffered Formalin solution. The brains were removed and placed in 10% Formalin for 3 days, processed, and embedded in paraffin wax. Coronal sections (6 μm) were taken 1.5, 1.7, and 1.9 mm caudal to bregma using a microtome (Leitz 1400 sledge microtome; Leica, Milton-Keynes, UK). The slices were stained with H&E, and the neuronal density in the CA1 subfield of the hippocampus was measured using a microscope with grid lines (0.05 × 0.05 mm) as described previously (O'Neill et al., 1998). The neuronal density was expressed as number of viable cells per millimeter of CA1 hippocampus. Statistical analysis of histological data was assessed using a two-tailed unpaired Student's t test, with Pvalues of <.05 being considered statistically significant.
Locomotor Activity.
At 24 and 120 h postocclusion, locomotor activity was measured in clear Perspex boxes (30 × 30 × 30 cm) with a metal base with a 2-cm covering of fine sawdust. Each of the 16 cages had five, equally spaced horizontal photocell beams 5.0 cm above the cage floor. Each beam break was recorded as a photocell count. All the boxes were individually connected to a Compaq PC, and the photocell interruptions were recorded as the number of counts using software provided by Greenacre and Misac Instruments (Ware, Hertfordshire, UK). All experiments were videotaped for subsequent analysis. The animals were placed in individual boxes for 60 min, and photocell interruptions were recorded every minute.
Immunocytochemical Methods.
Immunohistochemical staining was performed on paraffin-embedded coronal sections (6 μm) of hippocampus. Hippocampal sections were deparaffinized in xylene and rehydrated in ethanol and distilled water. Sections were incubated in 0.3% hydrogen peroxide (H2O2) for 30 min to block endogenous peroxidase activity, rinsed, incubated in pepsin/HCl for 30 min, rinsed in water, and then rinsed in PBS. Normal goat serum (1.5% in PBS) was used as a preblock for 20 min, after which the sections were incubated with the various primary antibodies: TGF-β1 and -β2 (Santa Cruz Biotechnology, Santa Cruz, CA) 1:100 for 2 h; bFGF (Biogenex, San Ramon, CA) ready-to-use antibody for 2 h; and NGF at 1:2000 and BDNF at 1:500 (Chemicon International, Temecula, CA), both for 24 h at room temperature in a humidified immunostaining chamber. Slides that were exposed to the bFGF antibody underwent a high temperature antigen retrieval pretreatment with Citra solution (Biogenex) immediately before the blocking step in 0.3% H2O2. Briefly, sections were rinsed in water, placed in Citra solution, microwaved on high until solution boiled, and then set to boil for 10 s every 30 s for a period of 15 min. Slides were allowed to cool for 30 min before continuing the procedure.
After the incubation with primary antibodies, sections were rinsed in PBS (3 × 5 min) and incubated in biotinylated secondary antibody (goat anti-rabbit Vector ABC Elite kit; 1:200 dilution in PBS with 1.5% normal goat serum; Vector Laboratories, Peterborough, UK) for 30 min at room temperature, after which they were again washed in PBS (3 × 5 min). Subsequently, sections were incubated in avidin-conjugated horseradish peroxidase (Vector ABC Elite kit, diluted in PBS) for 30 min at room temperature and washed in PBS (3 × 5 min). Finally, sections were incubated with nickel-enhanced 3,3′-diaminobenzidine chromagen (Vector kit) until color developed and then washed in tap water, dehydrated, coverslipped using DPX mountant, and examined under a light microscope. A detailed examination of the immunostaining product observed in the CA1 area of the hippocampus was performed, and digitized images were obtained using an Optimus 5.2 image analysis system (Datacell, Finchampstead, England). The number of immunoreactive cells in the CA1 area was indicated by the use of the scoring system in which − indicates no staining; +, little staining; ++, moderate staining; +++, intense staining; and ++++, most intense staining.
Bioavailability Studies.
Gerbils were dosed with LY379268 at 10 mg/kg i.p. and housed in incubator cages at 28°C for up to 8 h. At 15 and 30 min and 1, 2, 4, 6, 8, and 24 h after drug administration, animals were sacrificed and brain levels of LY379268 were determined by HPLC/tandem mass spectrometry.
Ischemic Tolerance.
Gerbils were subjected to a 2-min “preconditioning” ischemia and 48 h later were subjected to a 5-min “test” ischemia. Animals subjected to a 2-min ischemia before the 5-min ischemia are protected with ischemic tolerance. The effects of LY379268 (10 mg/kg i.p.) 30 min after the initial 2-min ischemia were assessed.
Results
Neuroprotective Effects of LY379268: Locomotor Activity.
Locomotor activity was assessed 24 and 120 h postocclusion. Results indicated that 5-min BCAO gerbils exhibited a large increase in locomotor activity 24 h postocclusion (over a 60-min period) in comparison with sham-operated animals (Fig.1A). LY379268 produced a marked attenuation (P < .001) in the ischemia-induced hyperactivity when administered 30 or 60 min after occlusion (Fig. 1, A and B).

The effects of LY379268 on ischemia-induced hyperactivity 24 h postocclusion. Locomotor activity was measured for a 60-min period and is illustrated as the number of counts (mean ± S.E.; n = 8 animals/group) in 10-min bins at 10, 20, 30, 40, 50, and 60 min (A) or total locomotor activity counts for a 60-min period (B). Sham-operated animals habituated rapidly to the apparatus and maintained low locomotor activity counts. In contrast, 5 min of BCAO caused a large increase in locomotor activity counts that was sustained for the 60-min period. LY379268 (10 mg/kg i.p.) commenced 30 or 60 min after the BCAO attenuated the ischemia-induced hyperactivity (Student's t test). ***P < .001 versus sham control.+++P < .001 versus BCAO control.
When tested at 120 h postocclusion, 5-min BCAO gerbils also exhibited an increase in locomotor activity (over a 60-min period) in comparison with sham-operated animals (Fig.2A). This hyperactivity was much smaller than that observed at 24 h (Fig. 2B). LY379268 attenuated (P < .05) the ischemia-induced hyperactivity when administered 30 or 60 min after occlusion (Fig. 2, A and B).

The effects of LY379268 on ischemia-induced hyperactivity 120 h postocclusion. Locomotor activity was measured for a 60-min period and is illustrated as the number of counts (mean ± S.E.; n = 8 animals/group) in 10-min bins at 10, 20, 30, 40, 50, and 60 min (A) or total locomotor activity counts for a 60-min period (B). Sham-operated animals habituated rapidly to the apparatus and maintained low locomotor activity counts. In contrast, 5 min of BCAO caused a large increase in locomotor activity counts that was sustained for the 60-min period. LY379268 (10 mg/kg i.p.) commenced 30 or 60 min after the BCAO attenuated the ischemia-induced hyperactivity (Student's t test). *P < .05, **P < .01, ***P < .001 versus sham control.+P < .05 versus BCAO control.
Neuroprotective Effects of LY379268: Histological Assessment.
At the end of behavioral testing, the brains were removed for histological analysis. Results indicated that there was a severe loss of CA1 hippocampal cells 120 h postocclusion. LY379268 protected against this ischemia-induced cell death when administered 30 or 60 min postocclusion. The protection was greatest when administration was initiated 30 min after occlusion (85%, P < .001), but significant protection was also observed when the compound was administered 60 min postocclusion (47%, P < .05) as shown in Fig. 3.

mGluR selectivity profile of LY379268 and the effects of LY379268 (10 mg/kg i.p.) administered 30 or 60 min after occlusion on the neuronal density at three stereotaxic levels in the CA1 region of the hippocampus 5 days after surgery. Histological results are expressed as mean ± S.E. viable cells per millimeter of CA1 hippocampal region (n = 8 animals/group). The 5-min BCAO produced severe damage to the CA1 region (P < .001), whereas LY379268 produced protection when administration was initiated 30 min after (P < .001) or 60 min after (P < .05) occlusion (Student's ttest). ***P < .001 versus sham control.+P < .05,+++P < .001 versus 5-min BCAO control.
In separate studies, we examined the effects of LY379268 (10 mg/kg i.p., 30 min postocclusion), on histological outcome 6, 24, 72, and 120 h and 14 and 28 days post-BCAO (5 min). Results indicated that there was no cell damage in any of the treatment groups at 6 and 24 h postocclusion (Fig. 4). However, we did observe a large loss in CA1 cells in 5-min BCAO animals at 72 and 120 h and 14 and 28 days postocclusion (P < .001). This form of “delayed neuronal death” is well established in this gerbil model of transient global cerebral ischemia (see Kirino, 1982). This 30-min postocclusion treatment with LY379268 provided significant protection (P < .001) against this ischemia-induced cell death at all of the time points (Fig. 4). The results at 14 and 28 days indicate that LY379268 is not delaying the onset of cell death but rather is producing a sustained neuroprotection against BCAO-induced damage to CA1 cells.

The effects of LY379268 (10 mg/kg i.p.) administered 30 min after occlusion on the neuronal density in the CA1 region of the hippocampus (1.7 mm caudal to bregma) at 6 h and 1, 3, 5, 14, and 28 days after surgery. Histological results are expressed as mean ± S.E. viable cells per millimeter of CA1 hippocampal region (n = 8 animals/group). The 5-min BCAO produced severe damage to the CA1 region (P < .001), whereas LY379268 produced protection when administration was initiated 30 min after occlusion (P < .001) (Student'st test). ***P < .001 versus sham control. +++P < .001 versus 5-min BCAO control.
Effect of Pretreatment with LY379268.
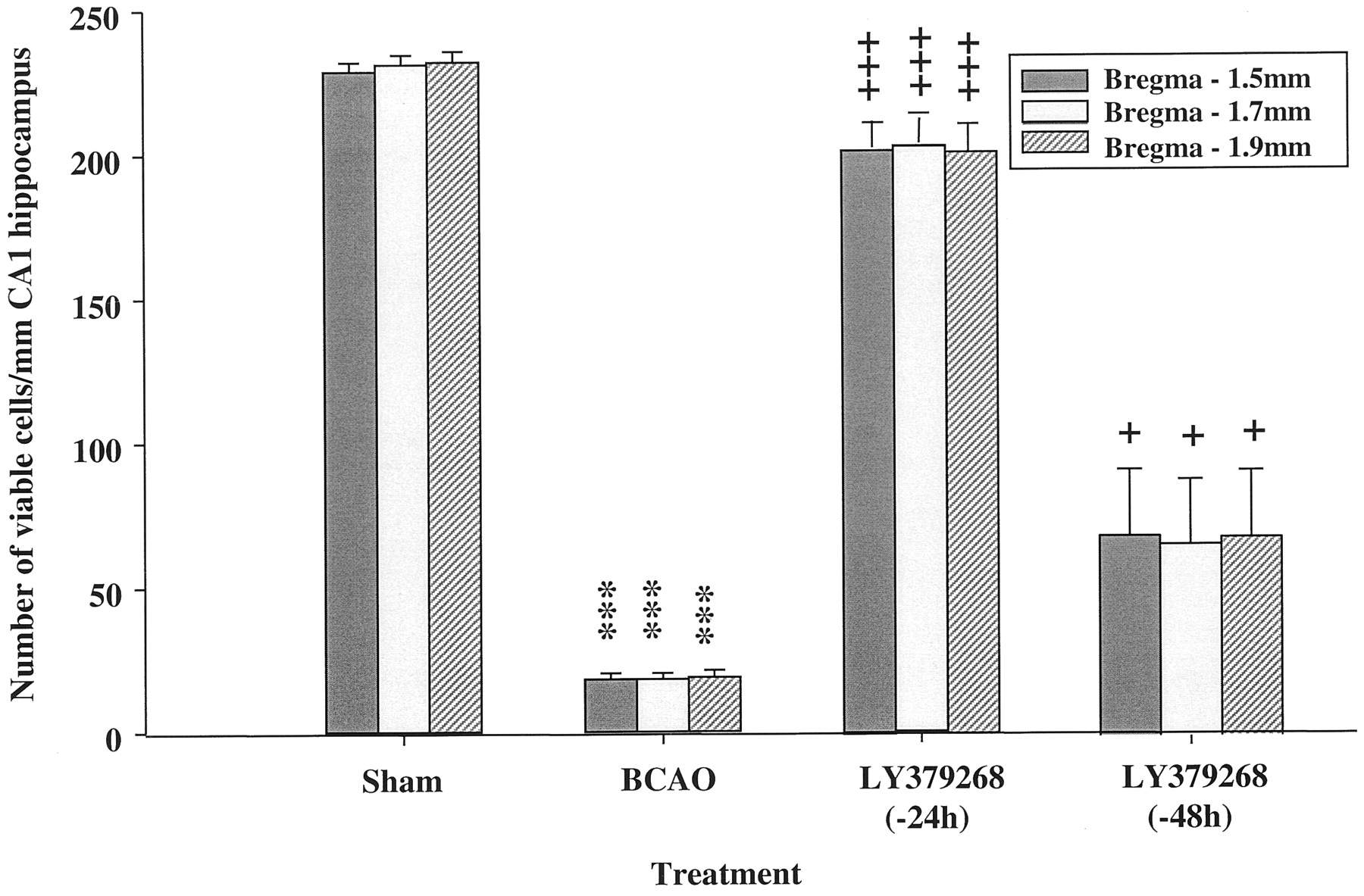
To investigate the potential of LY379268 to induce expression of some neurotrophic factors, LY379268 (10 mg/kg i.p.) was administered 24 or 48 h before BCAO to allow time for new protein synthesis to occur. The results clearly show that LY379268 at 10 mg/kg i.p. produced 93% neuroprotection (P < .001) when dosed at 24 h and retained significant neuroprotective effects (29%; P< .05) when dosed 48 h before 5-min BCAO (Fig.5).

The effects of LY379268 (10 mg/kg i.p.) administered 24 or 48 h before occlusion on the neuronal density at three stereotaxic levels in the CA1 region of the hippocampus 5 days after surgery. Histological results are expressed as mean ± S.E. viable cells per millimeter of CA1 hippocampal region (n = 8 animals/group). The 5-min BCAO produced severe damage to the CA1 region (P < .001), whereas LY379268 produced protection when administration was initiated 24 h before (P < .001) or 48 h before (P < .05) occlusion (Student's ttest). ***P < .001 versus sham control.+P < .05,+++P < .001 versus 5-min BCAO control.
Neuroprotective Effect of LY379268 on Induction of Ischemic Tolerance.
This result raised the possibility that activation of group II mGluRs may underlie the mechanism of ischemic tolerance (Kirino et al., 1991) (i.e., glutamate released by the “preconditioning ischemia” could activate group II receptors and up-regulate some neurotrophic factor or factors). To investigate this, we coadministered LY379268 with a 2-min preconditioning BCAO, hypothesizing that if the above mechanism were true, they would act additively. In fact, LY379268 (10 mg/kg i.p.) produced a marked reduction (P < .001) in the induced tolerance to BCAO tested 48 h later (Fig. 6).

Effect of LY379268 on induced ischemic tolerance in the gerbil hippocampus. From left to right, mean ± S.E. (n = 8) of the number of viable CA1 pyramidal cells after 1) sham occlusion, 2) a preconditioning 2-min ischemia alone, 3) a test 5-min ischemia alone, 4) a 2-min ischemia followed 2 days later by a 5-min ischemia (tolerance), and 5) drug pretreatment before the 2-min preconditioning ischemia, followed 2 days later by a 5-min ischemia (no drugs). LY379268 was dosed at 10 mg/kg i.p. and produced a significant reduction (P < .01) in the induced tolerance. **P < .01, ***P < .001.
Effects of LY379268 on Induction of Neuroprotective Growth Factors.
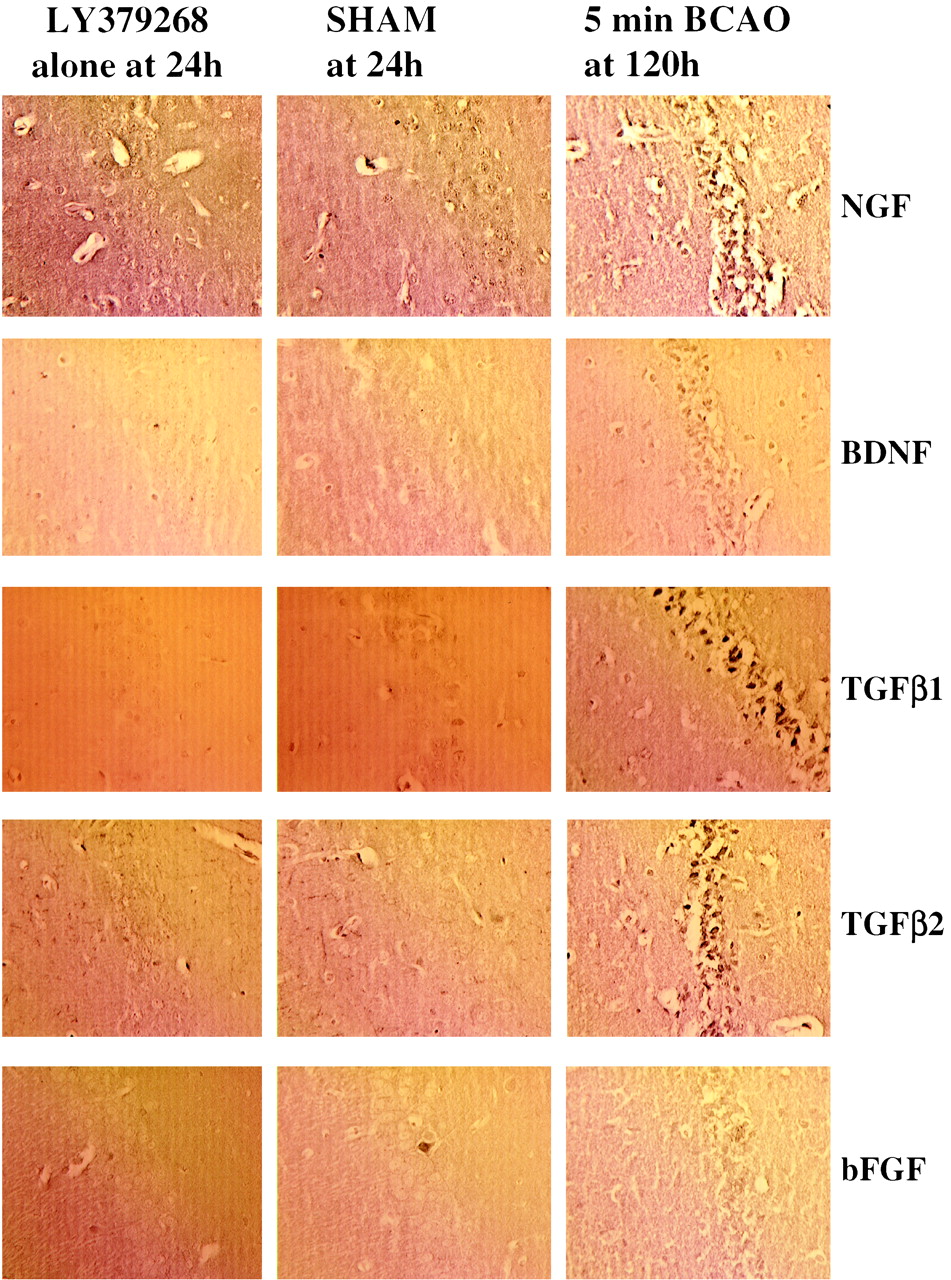
To investigate directly whether there was any up-regulation of the candidate factors bFGF, BDNF, NGF, TGF-β1, and TGF-β2, we examined the effects of 10 mg/kg LY379268 i.p. on their expression at 6, 24, 72, and 120 h in nonischemic gerbils. Results show that LY379268 alone did not stimulate the expression of any of these neurotrophic factors (Table 1). Representative photomicrographs demonstrating immunostaining for bFGF, BDNF, NGF, TGF-β1, and TGF-β2 in LY379268-treated and saline control-treated gerbils are shown in Fig.7. In additional experiments, we also demonstrated that chronic administration (20 mg/kg i.p. for 4 days) failed to alter the expression of neurotrophic factors in rat brain (data not shown).
A summary of immunohistochemical staining observed for antibodies to BDNF, NGF, bFGF, TGF-β1, and TGF-β2 in tissue from saline and LY379268 (10 mg/kg i.p.)-treated gerbils at 6, 24, 72, and 120 h postinjection
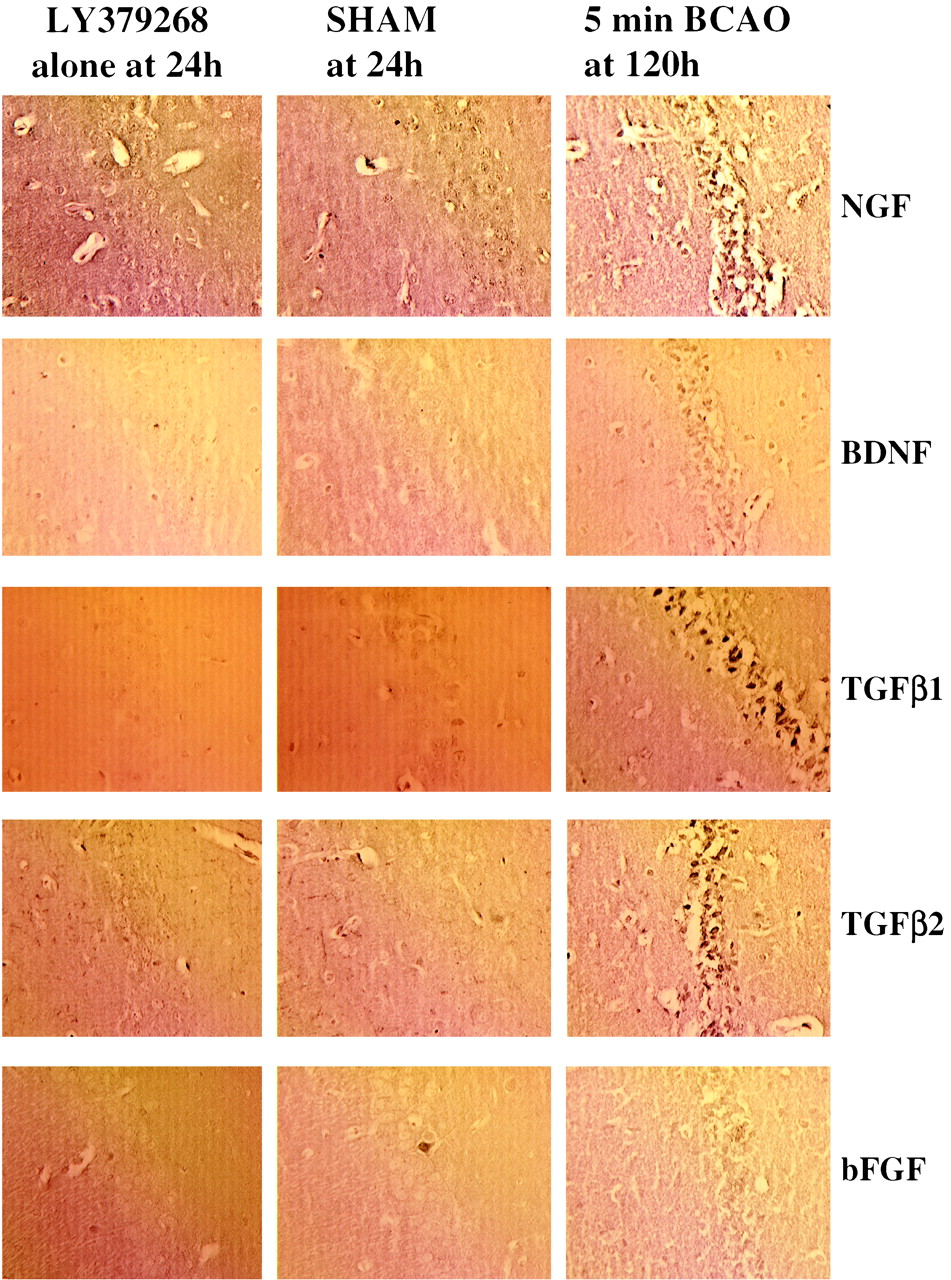
Effects of saline or LY379268 (10 mg/kg i.p.) on TGF-β1, TGF-β2, BDNF, NGF, and bFGF immunostaining in the hippocampus 24 h after injection. The 5-min ischemic animal results at 120 h are included as a positive control.
Duration of Action of LY379268.
These results suggest the possibility that LY379268 remains in the brain for more than 24 h at levels sufficient to activate group II receptors. To investigate this possibility, we tested whether the new group II mGlu antagonist LY341495 (Kingston et al., 1998) would prevent the protective effects of the prior administration of LY379268. Dosing 1 h before 3-min BCAO, LY341495 (5 mg/kg i.p.) alone did not cause a reduction or exacerbation of the neuronal damage induced by a mild 3-min ischemia (Fig. 8). Importantly, however, LY341495 at 5 mg/kg i.p. administered 24 h after LY379268 10 mg/kg i.p. and 60 min before 5-min BCAO reduced the level of neuroprotection from 97 to 20% (P < .001; Fig.9).This ability of the competitive antagonist LY341495 to reverse the effects of the agonist under these conditions indicated that mGlu2/3 receptors were still occupied by the agonist 24 h after LY379268 dosing.

Structure and selectivity of the mGluR antagonist LY341495 and the effects of LY341495 (5 mg/kg i.p.) administered 1 h before 3-min BCAO on neuronal density at three stereotaxic levels in the CA1 region of the hippocampus 5 days after surgery. Histological results are expressed as mean ± S.E. viable cells per millimeter of CA1 hippocampal region (n = 8 animals/group). The 3-min BCAO produced damage to the CA1 region (P< .01). LY341495 did not provide any potentiation (or protection) against this damage (Student's t test).

The effects of LY379268 (10 mg/kg i.p.) administered 24 h before occlusion alone or when dosed in combination with LY341495 (5 mg/kg i.p., 1 h before 5-min BCAO) on the neuronal density at three stereotaxic levels in the CA1 region of the hippocampus 5 days after surgery. Histological results are expressed as mean ± S.E. viable cells per millimeter of CA1 hippocampal region (n = 8 animals/group). The 5-min BCAO produced severe damage to the CA1 region (P < .001). LY379268 produced almost complete protection when administration was initiated 24 h before occlusion (P < .001). This protection provided with LY379268 was prevented by LY341495 (P < .001) administered 1 h before occlusion (Student's t test). ***P < .001.
Pharmacokinetic studies also demonstrated that at 24 h after a 10 mg/kg i.p. dose of LY379268, a significant receptor-active concentration of the compound was present in brain tissue (Fig.10).

Line graph showing the brain concentrations of LY379268 after a single dose of 10 mg/kg i.p. Ordinate, concentration of LY379268 (ng/g of brain tissue); abscissa, time (h).
Discussion
mGluRs are increasingly being considered as targets for the therapeutic intervention into neurodegenerative disorders, because their activation affects intracellular events that contribute to both the induction and progression of neuronal damage (Schoepp and Conn, 1993; Buisson et al., 1996; Nicoletti et al., 1996; Bruno et al., 1998). The lack of selective agents has made it difficult to clarify the exact contribution of the various mGluRs to neurodegenerative diseases. However, recently, several new ligands for mGluRs have been synthesized that have allowed the role of mGluRs in ischemia to be studied in detail (Pin et al., 1999; Schoepp et al., 1999). It has recently been demonstrated that the newer group II mGluR agonists LY354740 and LY379268 are systemically active (Bond et al., 1998,1999b). Here, we have provided further insight into possible mechanisms whereby the selective group II mGluR agonist LY379268 has neuroprotective actions in a model of global ischemia in vivo.
Acute Neuroprotection.
In this study, we extended our previous finding that LY379268 is neuroprotective by demonstrating that LY379268 provides not only histological protection but also functional improvement when dosed after ischemia. A large increase in locomotor activity was observed in control animals after BCAO, as noted previously by several groups (e.g., Mileson and Schwartz, 1991), and it has been suggested that this hyperactivity correlates with loss of hippocampal neurons (Anderson et al., 1997). In agreement with this, both the hyperactivity and the cell loss were considerably attenuated by LY379268, administered 30 or 60 min after BCAO (Fig. 1-3). We also demonstrated that the good neuroprotection of CA1 neurons previously observed with LY379268 (Bond et al., 1999b) is long-lasting. This maintained neuroprotection is in contrast to the reported short-lasting effects of ionotropic antagonists (e.g., NBQX; Nurse and Corbett, 1996). Because in our model of global ischemia cell death occurs by 3 days and LY379268 maintains protection up to 28 days (Fig. 4), it appears that LY379268 is blocking, rather than merely delaying, the process of cell death. The slow death of cells in our model is in accordance with the delayed neuronal death that has been described in this model (Kirino, 1982). Interestingly, the level of protection observed with LY379268 in this model is much greater than that we previously observed using the same protocol with calcium channel blockers (NCC 09-0026, CNS1237; O'Neill et al., 1997), NMDA antagonists (MK-801, ACEA1021, GV150526A; Hicks et al., 1999), and AMPA antagonists (GYKI52466, LY300164; Lodge et al., 1996) and similar to what we have seen with the iGluR5 antagonist (LY377770; O'Neill et al., 1998).
Other studies have shown that group II mGluR agonists are neuroprotective in vitro and in vivo. In mouse cortical neurons, DCG-IV, 4C3HPG, and L-CCG-1 were shown to protect against NMDA-induced excitotoxicity (Bruno et al., 1995), and the in vivo administration of DCG-IV directly into the cerebral ventricles can block kainate-induced degeneration in rats (Shinozaki, 1994). However, these compounds are not selective for group II mGluRs and have additional pharmacology that could account for their neuroprotective effects. LY379268, being more selective, confirms the involvement of group II mGluRs. Interestingly, neither LY354740 nor LY379268 is protective in focal ischemia (Lam et al., 1998; Behrens et al., 1999; Bond et al., 1999b). The exact reason for this is not clear. One possible explanation may be that the mechanisms of cell death are different in these models. Other recent in vitro studies have reported that both LY354740 and LY379268 are more potent against apoptotic indices of neuronal injury in vitro at concentrations that are more commensurate with their actions at group II mGluRs than those required to reduce lactate dehydrogenase release, which occurs as a result of both apoptotic as well as necrotic cell death (Kingston et al., 1999). In another recent study, LY354740 failed to attenuate acute (24 h) excitotoxicity of NMDA or oxygen-glucose deprivation in vitro or of intrastriatal NMDA injections in vivo (Behrens et al., 1999). Taken together, these results indicate that selective group II mGlu agonists appear to have little or no effect in conditions where there is rapid development of neuronal cell loss such as acute excitotoxicity and focal ischemia, but they appear to be most effective in circumstances of slowly developing neuronal cell death (i.e., global ischemia). In this study, we have demonstrated further convincing evidence for the group II mGluR agonist LY379268 being neuroprotective in global ischemia using various dosing protocols.
Mechanism of Action of LY379268.
The neuroprotective activity of group II mGluR agonists can be ascribed to their presynaptic actions at mGlu2 receptors and the prevention of glutamate release (Pin and Duvoisin, 1995). However, this mechanism of action does not account for neuroprotection seen with this class of compound against apoptosis induced by oxygen-glucose deprivation and β-amyloid peptide (Copani et al., 1995), nor for the demonstration that DCG-IV can protect cultured neurons even when applied after a toxic pulse of NMDA, when large amounts of endogenous glutamate have already accumulated in the extracellular medium (Nicoletti et al., 1996). Furthermore, protein synthesis inhibitors can block the neuroprotective actions of group II mGluR agonists in vitro against NMDA-induced toxicity in cortical neurons (Bruno et al., 1997; Kingston et al., 1999). A recent report demonstrated that transient exposure of astrocytes to DCG-IV or 4C3HPG led to an increase in TGF-β and that neutralizing antibodies to TGF-β1 or -β2 prevented the neuroprotection afforded by DCG-IV and 4C3HPG in mixed cortical cultures. This then suggested that activation of mGlu3 receptors in astrocytes led to the production of TGF-β, which protects cells (Bruno et al., 1998).
In this study, we sought to investigate this latter possibility. First, we demonstrated that LY379268 was neuroprotective when dosed 24 h (P < .001) and 48 h (P < .01) before a 5-min period of BCAO (Fig. 5), a result consistent with up-regulation of a neurotrophic factor, such as TGF-β (Bruno et al., 1998). However, LY379268 did not induce expression of the specific neuroprotective factors TGF-β1, TGF-β2, bFGF, BDNF, and NGF.
The prolonged activity of LY379268 after 24- and 48-h pretreatment raises the possibility that it shares a common mechanism with that of ischemic tolerance (Kirino et al., 1991), whereby glutamate released by a preconditioning ischemia may activate group II mGluRs and induce a neuroprotective factor. We argued that if LY379268 induced a neuroprotective factor, it should act additively with the preconditioning ischemia. Our results indicate that LY379268 almost blocked the degree of tolerance induced by a preconditioning ischemia. It has previously been demonstrated that NMDA antagonists (Kato et al., 1992; Bond et al., 1999a), but not AMPA antagonists, attenuated the induction of tolerance in this model (Bond et al., 1999a). These results suggest that LY379268 is acting via mGlu2 presynaptic autoreceptors to reduce glutamate release, and hence NMDA receptor activation, to interfere with the development of tolerance.
These results leave open the possibility that even at 24 and 48 h, sufficient LY379268 remains in the brain to reduce the degree of damage after BCAO in the gerbil. We investigated this in two ways. First, we took advantage of the newly available group II mGlu antagonist LY341495 (Kingston et al., 1998), which given alone was without effect on the outcome of a submaximal global ischemia (Fig. 8). Given 24 h after 10 mg/kg LY379268 and 1 h before a 5-min BCAO, 5 mg/kg LY341495 virtually prevented the neuroprotective effect of LY379268. Second, we assayed the levels of LY379268 in the brain at time points up to 24 h postadministration (Fig. 10). Although there was a rapid decline in central nervous system levels between 0.5 and 8 h, they plateaued at a level above that required for group II mGluR activation.
Taken together, these results support a mechanism that is directly dependent on receptor activation by LY379268 rather than through a mechanism that involves a long-lasting induction of other factors that would be farther downstream of receptor activation and not affected by the actions of an mGlu antagonist such as LY341495.
In conclusion, we have shown that LY379268 provides marked long-lasting neuroprotection against ischemia-induced damage in the gerbil. In this model of global ischemia, the mechanism of action of LY379268 does not appear to be via induction of neurotrophic factors such as NGF, BDNF, or TGF but most likely occurs via a mechanism that is closely coupled with receptor activation. Such a mechanism may involve inhibition of glutamate release and/or activation of biochemical pathways that inhibit programmed cell death. However, these findings do not rule out the possibility that other mechanisms may also contribute to the quite remarkable neuroprotection seen with LY379268 and encourage further investigation of the use of group II mGlu agonists in chronic rather than acute neurodegenerative disorders.
Footnotes
-
Send reprint requests to: Dr. Michael J. O'Neill, Eli Lilly & Co. Ltd., Lilly Research Centre, Erl Wood Manor, Windlesham, Surrey GU20 6PH, UK. E-mail: ONEILL_MICHAEL_J{at}Lilly.com
- Abbreviations:
- iGlu
- ionotropic glutamate
- mGlu
- metabotropic glutamate
- mGluR
- mGlu receptor
- BCAO
- bilateral carotid artery occlusion
- TGF-β1
- transforming growth factor-β1
- TGF-β2
- transforming growth factor-β2
- BDNF
- brain-derived neurotrophic factor
- NGF
- nerve growth factor
- bFGF
- basic fibroblast growth factor
- NMDA
- N-methyl-d-aspartate
- AMPA
- α-amino-3-hydroxy-5-methyl-isoxazole-4-propionic acid
- DCG-IV
- (2S,1′R,2′R,3′R)-2-(2,3-dicarboxycyclopropyl)glycine
- 4C3HPG
- (S)-4-carboxy-3-hydroxyphenylglycine
- L-CCG-1
- (2S,1S,2S)-2(carboxycyclopropyl) glycine
- LY354740
- (+)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid
- LY379268
- (1R,4R,5S,6R)-2-oxa-4-aminobicyclo[3.1.0.]hexane-4,6-dicarboxylate
- LY341495
- 2S-2-amino-2-(1S,2S-2-caroxycyclopropy-1-yl)-30(xanth-9-yl)propionic acid
- NBQX
- 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline
- MK-801
- (5R,10S)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d] cyclohepten-5,10-imine
- ACEA1021
- 5-nitro-6,7-dichloro-2,3-quinoxalinedione
- GV150526A
- [(E)-3[(phenylcarbamoyl) ethenyl]-4,6-dichloroindole-2-carboxylic acid
- GYKI52466
- 1-(aminophenyl)-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine
- LY300164
- (+)-3-N-acetyl-1(aminophenyl)-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine
- Received February 17, 2000.
- Accepted April 27, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}