


Abstract
Phosphatidylinositol 3-kinases (PI3Ks) are key elements in the signaling cascades that lie downstream of many cellular receptors. In particular, PI3K δ and γ isoforms contribute to inflammatory cell recruitment and subsequent activation. For this reason, in a series of preclinical studies, we tested the potential of a recently developed small-molecule inhibitor of these two isoforms, TG100-115 [3-[2,4-diamino-6-(3-hydroxyphenyl)pteridin-7-yl]phenol], as a form of anti-inflammatory therapy for respiratory diseases such as asthma and chronic obstructive pulmonary disease (COPD). To determine pharmacokinetic profiles, aerosolized formulations of the drug were delivered to mice by a nose-only inhalation route, yielding high pulmonary TG100-115 levels with minimal systemic exposure. Safety assessments were favorable, with no clinical or histological changes noted after 21 days of daily dosing. In a murine asthma model, aerosolized TG100-115 markedly reduced the pulmonary eosinophilia and the concomitant interleukin-13 and mucin accumulation characteristic of this disease. As a functional benefit, interventional dosing schedules of this inhibitor also reduced airway hyper-responsiveness. To model the pulmonary neutrophilia characteristic of COPD, mice were exposed to either intranasal lipopolysaccharide or inhaled smoke. Aerosolized TG100-115 again inhibited these inflammatory patterns, most notably in the smoke model, where interventional therapy overcame the steroid-resistant nature of the pulmonary inflammation. In conclusion, aerosolized TG100-115 displays pharmacokinetic, safety, and biological activity profiles favorable for further development as a therapy for both asthma and COPD. Furthermore, these studies support the hypothesis that PI3K δ and γ are suitable molecular targets for these diseases.
Phosphoinositide 3-kinases (PI3Ks) phosphorylate inositol lipids within cell membranes as an early step in the signaling cascades initiated by many ligand-receptor interactions (Vanhaesebroeck et al., 2001). The PI3K family can be divided into different classes based on subunit arrangement and substrate utility. For example, class IA isoforms, PI3Kα/β/δ, interact with tyrosine kinases such as growth factor receptor tyrosine kinases, and the class IB member PI3Kγ interacts with G-protein-coupled receptors. PI3Kα and β are broadly expressed across many tissues and control fundamental processes such as cellular proliferation; genetic deletion of either isoform is embryonically lethal (Vanhaesebroeck et al., 2005). In contrast, PI3Kδ and γ have a more restricted cellular distribution and a more focused role of mediating inflammatory responses; genetically deleted mice are not only viable but display reduced inflammatory responses (Hirsch et al., 2000; Yum et al., 2001; Hannigan et al., 2002; Laffargue et al., 2002; Ali et al., 2004).
Based on these profiles, one can appreciate that PI3Kδ and γ represent promising molecular targets in the development of novel anti-inflammatory agents. A relatively small number of PI3Kδ- and/or γ-specific inhibitors have been tested in preclinical disease models (Sadhu et al., 2003; Barber et al., 2005; Camps et al., 2005; Doukas et al., 2006; Lee et al., 2006). We reported that a dual PI3Kγ/δ inhibitor named TG100-115 can diminish reperfusion injury to ischemic tissue after acute myocardial infarction (Doukas et al., 2006). Although TG100-115 was extended into the clinic for this indication, we also recognized its potential for use in other inflammatory-based settings because the combination of δ and γ isoform inhibition presents the opportunity to intervene across a broad range of cellular responses, including those induced by both receptor tyrosine kinases and G-protein-coupled receptors. For example, these two isoforms play nonredundant roles in the recruitment and activation of immune cells during inflammatory responses (Rommel et al., 2007); therefore, as an anti-inflammatory, a dual PI3Kγ/δ inhibitor may have extended benefits over isoform monoselective compounds. This is of particular relevance because there is building interest regarding the therapeutic potential of PI3K inhibitors in respiratory diseases, such as asthma and chronic obstructive pulmonary disease (COPD) (Finan and Thomas, 2004; Adcock et al., 2006b; Ito et al., 2007; Krymskaya, 2007).
Asthma and COPD are driven by distinct immunologic processes (Barnes, 2008). Asthma is an allergic hypersensitivity involving a Th2-type immune response mounted by CD4+ T cells, with mast cells and eosinophils also playing important roles and neutrophils contributing in chronic severe disease. COPD initiates as an abnormal inflammatory response involving CD8+ T cells releasing Th1-type cytokines, with contributions from macrophages and neutrophils (Doherty, 2004; Welte and Groneberg, 2006; Baraldo et al., 2007). Clinical pictures also differ for the two diseases. Asthma presents as a nonprogressive airflow limitation that in most cases can be controlled with bronchodilators and corticosteroids. However, for many patients, symptom control is inadequate, and for the steroid-nonresponsive patient, new anti-inflammatories would be particularly welcome (Adcock and Ito, 2004; Barnes, 2004). COPD, in contrast, displays progressive airflow limitation, making longer term disease of increasingly serious impact. The disease is generally steroid nonresponsive; thus, anti-inflammatories that can prove efficacious in this setting are a clear need (Bailey and Tashkin, 2007).
As a means of assessing the potential of PI3K inhibitors as respiratory disease therapies, we conducted a series of preclinical studies focusing on aerosolized TG100-115. Aerosolization allowed for delivery of this compound to test animals by an inhalation route, the goal being to model a delivery system that could readily transfer to a clinical setting. In addition to favorable pharmacokinetic and safety profiles, we documented readily demonstrable anti-inflammatory activity in murine models of asthma and pulmonary neutrophilia (including smoke-induced neutrophilia, a steroid-resistant model of COPD) and functional improvements in the former.
Materials and Methods
Aerosolized TG100-115. TG100-115 was generated as previously published (Doukas et al., 2006; Palanki et al., 2007). To aerosolize TG100-115, it was first milled using a magicLAB apparatus (IKEA, Wilmington, NC) and then suspended in sterile water containing tyloxapol (as a dispersing agent) at a 4:1 w/v ratio of TG100-115/tyloxapol. TG100-115 suspensions (or water/tyloxapol vehicle) were aerosolized using a nebulizer (LC Plus nebulizer and Proneb Ultra II compressor; Pari Respiratory Equipment, Midlothian, VA) and delivered to mice as an airstream at an output flow rate of 9 l/min using a nose-only directed flow inhalation exposure system [CH Technologies (USA) Inc., Westwood, NJ]. Exposure times were held constant (30 min), whereas suspension strengths were varied to achieve the desired TG100-115 dosage.
To monitor exposure levels, flow from one of the 24 ports of the inhalation exposure system was collected onto a Pallflex 47-mm filter (Pall Corporation, East Hills, NY). Filters were extracted in acetonitrile/water [50:50 (v/v)] containing 0.05% trifluoroacetic acid and analyzed via the transferred method on an Agilent 1100 HPLC/UV (Agilent Technologies, Santa Clara, CA) with an 1100 variable wavelength detector at a wavelength of 215 nm. In brief, mobile phase (MP) A was water with 0.05% trifluoroacetic acid, and MP B was acetonitrile with 0.05% trifluoroacetic acid. Initial conditions of 72:28 MP A/MP B at 1.0 ml/min were ramped linearly for 15 min to 48:52, at which time flow was restored to initial conditions and allowed to equilibrate for 5 min until the next injection. Chromatography was conducted on a Supelco Discovery HS F5 column (Sigma-Aldrich, St. Louis, MO). To measure particle size [mass median aerodynamic diameter (MMAD)], samples were collected directly from the breathing zone of the inhalation exposure system and measured using a Mercer-style impactor (In-Tox Products, Moriarity, NM) operated at 2 1/min. Impactor substrates were chemically extracted and assayed via the transferred HPLC/UV method as described previously. All studies presented used aerosolized suspensions with an MMAD of 1 to 2 μM.
Pharmacokinetic and Safety Studies. All animal studies followed the Guide for Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996). BALB/c mice (18-25 g) were exposed to aerosolized TG100-115 by a nose-only inhalation route as described above. Exposure was performed once daily for up to 21 days, depending on the study design. For pharmacokinetic determinations, plasma was first sampled from tail veins, and bronchoalveolar lung fluid (BALF) was then collected (by lavaging lungs in situ with 3 × 1-ml volumes of PBS containing 0.1% bovine serum albumin); finally, lungs were harvested. Lungs were homogenized in radioimmunoprecipitation assay buffer using a FastPrep homogenizer (Thermo Fisher Scientific, Waltham, MA) followed by extraction in acetonitrile (containing internal standards), and then supernatants were dried and reconstituted into dimethyl sulfoxide/water (8:2). Plasma and BALF samples were extracted in a 2-fold excess of acetonitrile (containing internal standards) followed by centrifugation to obtain supernatants. Processed samples were then quantitated by LC/MS/MS against external calibration standards prepared in naive mouse tissues. The LC/MS/MS system consisted of a Sciex API3000 triple quadrupolar mass spectrometer (Applied Biosystems/MDS Sciex, Foster City, CA), an Agilent 1100 HPLC system, and a CTC autosampler (LEAP Technologies, Carrboro, NC). LC separations were performed on a Zorbax SB reverse phase HPLC column (Agilent Technologies); mass spectrometric detection was achieved using electrospray ionization operating in positive ionization mode. Pharmacokinetic parameters were estimated using WinNonlin software (Pharsight, Mountain View, CA). For safety assessments, mice were assessed for changes in clinical signs (respiration rate, piloerection, body posture, activity level) 30 min and 4 h after TG100-115 dosing, and at 24 h after 21 days of daily dosing, selected organs (heart, lungs, liver, kidneys, stomach, intestines) were processed for histopathologic assessment by a board-certified doctor of veterinary medicine.
Asthma Model. Ovalbumin (OVA) was prepared by mixing 5 mg with 500 mg of alum in a 100-ml volume of PBS. BALB/c mice (6-8 weeks) were injected with the OVA/alum mix (0.2 ml i.p.) on days 1 and 6. On days 13 and 14, mice were then challenged with aerosolized OVA by whole-body exposure to a 0.5% solution (in PBS but without alum) using a nebulizer for delivery. Control groups included mice injected with PBS alone rather than OVA/alum or OVA-sensitized animals challenged with PBS alone. Pharmacological treatments included nose-only exposure to aerosolized TG100-115 (or water/tyloxapol vehicle as a control) or systemic exposure to dexamethasone (3 mg/kg i.p. q.d. on days 8-14). Endpoints were determined on day 16.

Pharmacokinetics of aerosolized TG100-115. TG100-115 was delivered as aerosolized microsuspensions to mice via a nose-only inhalation route as a single 30-min exposure daily for 1, 5, or 21 days; TG100-115 formulations used were 0.004, 0.04, or 0.4% strength. On the desired study day, 30 min after dosing, lung (A), BALF (B), and plasma (C) samples were collected from each animal and processed for LC/MS/MS determination of TG100-115 levels. Data are shown as means ± S.E.M. (n = 5). For plasma samples, TG100-115 was detectable in four of five animals in the 0.4% TG100-115 group but in two out five animals at most in the lower dose groups.
For cell count and cytokine level measures, BALF was harvested as described previously. After centrifugation, supernatants were processed for cytokine enzyme-linked immunosorbent assays using commercial kits (R&D Systems, Minneapolis, MN), and cell pellets raised in 1 ml of RPMI medium were deposited onto glass slides using a Cytospin cytocentrifuge (Thermo Fisher Scientific). Slides were then processed as differential stains using the Diff-Quick system (IMEB, San Marcos, CA). For histological assessments, lungs were fixed in situ with formalin, then processed as either hematoxylin/eosin-stained or nuclear fast red/Alcian Blue-stained paraffin sections. For functional endpoint determinations, airway resistance was measured in anesthetized mice (paralyzed with pancuronium) using a flexiVent device according to the manufacturer's instructions (SCIREQ, Montreal, QC, Canada), with nebulized methacholine as the bronchoconstrictor. Arterial oxygenation levels were monitored using a pulse oximeter (Starr Life Sciences, Oakmont, PA) to ensure proper ventilation.
Pulmonary Neutrophilia Models. Two models of pulmonary neutrophilia were used. In the first, LPS (Escherichia coli 055:B5 source; Sigma-Aldrich) was delivered by syringe to each naris of BALB/c mice (6-8 weeks) as 5 ng in a 25-μl volume of PBS; control animals received PBS alone. After 30 min, mice were exposed to aerosolized TG100-115 or vehicle (as described previously) or dexamethasone (0.3 mg/kg i.p. q.d.), and 3 h later (i.e., 4 h after LPS exposure) BALF was harvested and analyzed for cell counts and cytokine enzyme-linked immunosorbent assays as described previously.
In a second model, BALB/c mice were whole-body exposed once per day for 3 days to the smoke from five commercial-grade cigarettes, delivered over a 50-min period in a stream of air using a small animal ventilator (Harvard Apparatus Inc., Holliston, MA) set to a rate of 30 strokes/min. Control mice were exposed to air only. Treatments consisted of aerosolized water/tyloxapol vehicle (delivered as described previously each day of smoke exposure), dexamethasone (0.3 mg/kg i.p. q.d. for 3 days), or aerosolized TG100-115 (delivered on some or all days of smoke exposure depending on the specific treatment group). Twenty-four hours after the last smoke exposure (day 4), BALF was harvested and analyzed for cell counts as described previously.
Statistics. Two-group comparisons were made using unpaired Student's t tests and multiple group analyses using one-way analyses of variance with post hoc Dunnett's tests (SigmaStat software; SPSS Inc., Chicago, IL). If Dunnett's tests showed statistical differences for more than one group compared with controls, a second analysis of variance for intergroup differences was performed using the Student-Newman-Keuls method as the post hoc test. Statistical significance was defined as P < 0.05.
Results
Pharmacokinetics and Safety of Aerosolized TG100-115. Microsuspensions of TG100-115 were aerosolized using a nebulizer and delivered to mice via a nose-only inhalation route; total dose was controlled by varying formulation strength while holding exposure time constant. To determine basic pharmacokinetic profiles, mice were exposed to aerosols for a single 30-min period; then, 30 min post-treatment, lung tissues, plasma, and BALF were analyzed for TG100-115 content. As shown in Fig. 1, it was possible to achieve relatively high TG100-115 levels in lungs while at the same time limiting systemic exposure. In particular, formulation strengths of 0.004, 0.04, and 0.4% (which delivered 1, 10, and 100 μg/kg, respectively) yielded a broad range of lung exposures while still falling within a pharmacologically relevant range. Lung tissues contained an average of 74 nM TG100-115 after a single exposure to the 0.004% formulation and 664 and 5242 nM after the 0.04 and 0.4% formulations, respectively. For BALF, these values were 25, 386, and 2428 nM, respectively, or, in other words, 34 to 58% that found in the corresponding lung tissue. In contrast, the highest plasma levels reached (which, as expected, occurred in the highest dose group) averaged only 20 nM, corresponding to <0.004% of the level present in the lung tissues and <0.009% of that in the BALF. As for the two lower dose groups, all plasma levels fell below the lower level of quantitation (5 nM), except for one mouse (of five) in the middle dose group.
Exposing mice to TG100-115 in this same manner for 5 or 21 days revealed a 2.5- to 4-fold increase in lung tissue levels between days 1 and 5 but then only an additional 1- to 1.5-fold accumulation at day 21 (Fig. 1). This pattern indicates that steady state levels were achieved relatively quickly and agrees with other studies in which exposure was followed over a 24-h period to calculate a tissue half-life [t½] of approximately 24 h (data not shown). BALF levels followed a slightly different pattern, with accumulation at both days 5 and 21 increasing by 1- to 2-fold. As for plasma, repeated dosing did not meaningfully drive TG100-115 exposure, and the drug remained detectable beyond random animals only in the highest dose group; the mean plasma concentration for the 0.4% formulation group at day 21 was 27 nM, in the same general range as that seen after a single dose exposure.
To correlate these exposure levels with safety, animals were also monitored for changes in clinical signs. No changes in clinical signs were observed whether mice were exposed to aerosolized vehicle or aerosolized TG100-115 (0.004, 0.04, or 0.4% formulations) for 21 days. A sampling of relevant organs (lungs, heart, liver, kidneys, and digestive tract) showed no histological changes after treatment compared with organs taken from naive mice.
Aerosolized TG100-115 Reduces Inflammation Associated with Asthma Development in a Murine Model. Having established a means of achieving meaningful pulmonary TG100-115 exposure in mice without appreciable systemic exposure or observable safety signals, we next tested whether this might translate into therapeutic benefits using animal models of respiratory diseases, beginning with a standard murine model of asthma. OVA-immunized mice were challenged with aerosolized OVA for 2 consecutive days and then analyzed for pulmonary inflammation 48 h later. As shown in Table 1, OVA-sensitized mice challenged with only saline contain relatively few eosinophils in their lungs. Challenge with OVA induced a readily detectable eosinophil accumulation, and aerosolized TG100-115 markedly reduced this eosinophilia when delivered as a preventative therapeutic 1 day before and on each day of OVA challenge. A 79% reduction in eosinophil numbers was achieved when dosing with a 0.4% formulation (P < 0.001), statistically equivalent to the effect (88% reduction) seen after systemic dexamethasone delivery. In addition to the reduction in eosinophilia, BALF levels of the classic Th2 cytokine IL-13 were also reduced after TG100-115 treatment; however, for this measure, dexamethasone treatment did achieve a statistically greater effect than aerosolized TG100-115.
Pulmonary inflammation in a murine asthma model: eosinophil and IL-13 accumulation in BALF
Mice sensitized to OVA by systemic exposure (days 1 and 6) were challenged with either aerosolized saline or OVA (days 13 and 14), the latter inducing an asthmatic response. TG100-115 was administered as an aerosolized 0.4% formulation (days 12-14) and dexamethasone as a systemic treatment (days 8-14). BALF collected on day 16 was analyzed to determine eosinophil counts (represented as percentage of total cells) and IL-13 levels. Data shown as means ± S.E.M. (n = 8-9). For both endpoints, the vehicle treatment/OVA challenge group differed from all others by P < 0.001. For the IL-13 measure only, TG100-115 and dexamethasone groups differ by P < 0.01.
Pulmonary inflammation was also assessed by histology. Building from the data set presented in Table 1, the TG100-115 dose was stepped down to 10 μg/kg by using a 0.04% aerosolized formulation, and a clear therapeutic benefit was again seen, with markedly reduced perivascular and peribronchial leukocyte accumulation similar to that observed with systemic steroid treatment (Fig. 2). Bronchiolar mucin accumulation, an additional hallmark most commonly associated with the chronic asthmatic response, was also reduced in a similar pattern.
Aerosolized TG100-115 Improves Functional Responses in Asthmatic Mice. One functional consequence of the inflammatory process that underlies asthma is a hyperresponsiveness to bronchoconstrictors. Therefore, we examined whether aerosolized TG100-115 also influenced this endpoint. As shown in Table 2, aerosolized TG100-115 delivered across the range of 1 to 100 μg/kg (0.004-0.4% formulation strengths) reduced airway hyper-responsiveness (AHR) in asthmatic mice by approximately 50% (P < 0.05); response levels were statistically equivalent across these doses, indicating that maximal efficacy had been reached with the 0.004% formulation. Additional studies confirmed this because response to the drug did not drop off until formulations below 0.001% (data not shown). Although a trend toward a greater efficacy response was seen in animals systemically dosed with dexamethasone, this increase was not statistically significant.
AHR responses in a murine asthma model
Mice were treated as described in Table 1, except that airway resistance was measured as the study endpoint. All animals were sensitized to OVA and challenged with nebulized methacholine (50 mg/ml) as a bronchoconstrictor but differed as to whether they had been challenged with aerosolized OVA (to induce asthma) or saline (nonasthmatics) and as to the therapeutic intervention administered (aerosolized water/tyloxapol, aerosolized TG100-115 at various formulation strengths, or systemic steroid). Data are shown as airway resistance (centimeters of H2O per second per milliliter, means ± S.E.M.) and percentage reduction (where reducing resistance to that of the vehicle-treated nonasthmatic control group would equal −100%). Study 1, n = 9 to 11, the vehicle-treated asthmatic group differs from all others (P < 0.04), which do not differ from one another. Study 2, n = 8 to 9, the vehicle-treated asthmatic group differs from all others (P < 0.05), which do not differ from one another.
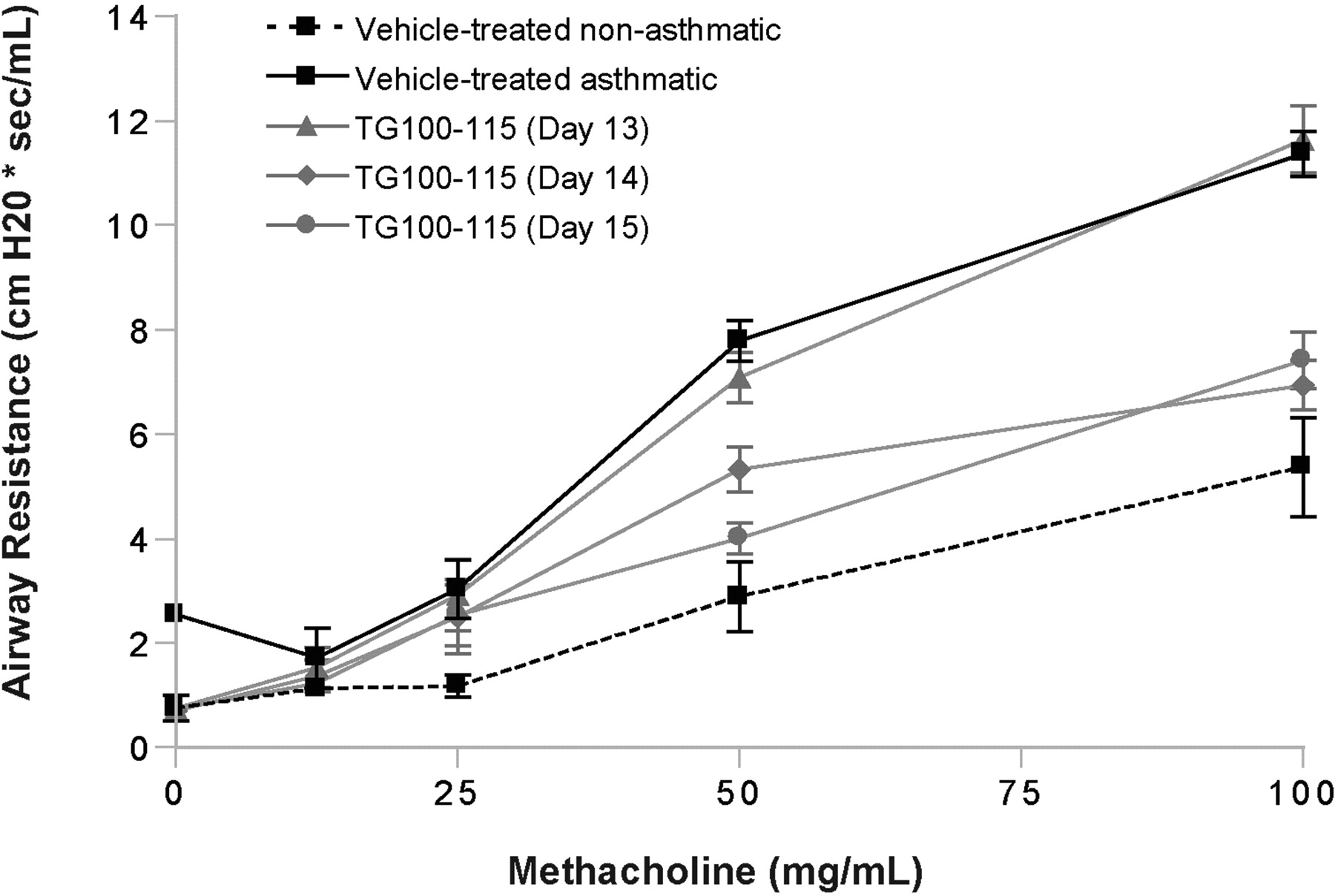
The asthma model studies presented to this point employed a preventative dosing schedule, in which TG100-115 was delivered before asthma induction (by aerosolized OVA challenge). As a higher hurdle, aerosolized TG100-115 was next delivered as an interventional therapy. Asthma was induced in mice as described previously by OVA immunization on days 1 and 6 followed by OVA challenge on days 13 and 14, but now aerosolized TG100-115 was delivered on single days only, at days 13, 14, or 15; AHR was then determined on day 16. Aerosolized TG100-115 (0.1% formulation) showed an equivalent therapeutic efficacy when delivered at either day 14 or 15; the bronchoconstriction response of asthmatic mice was normalized toward that of nonasthmatic animals across a range of methacholine doses (Fig. 3). Consistent with these results, inflammatory cell influx to the lungs also decreased with TG100-115 delivery on day 14 or 15 (data not shown). In contrast, delivery as a single dose on day 13 had no therapeutic effect.
To confirm that TG100-115 reduction of AHR was not simply because of a blockade of methacholine signaling through muscarinic receptors, the influence of aerosolized TG100-115 on methacholine-induced bronchoconstriction was examined in nonasthmatic mice. No changes in bronchoconstriction were seen (data not shown).
Aerosolized TG100-115 Reduces LPS- and Smoke-Induced Pulmonary Inflammation. Having established efficacy in asthma models, we next examined whether TG100-115 could reduce the inflammatory patterns characteristic of COPD, specifically pulmonary neutrophilia. In our first model, LPS was instilled in the nares of BALB/c mice, which rapidly induces an inflammatory response dominated initially by neutrophils. Aerosolized TG100-115 reduced neutrophil accumulation across a range of doses (Table 3). In one study, a 100 μg/kg dose (0.4% formulation) reduced neutrophil accumulation by 42% (P = 0.001), whereas systemic dexamethasone achieved a 70% reduction (P < 0.001). In a second study, both 0.4 and 0.04% aerosolized formulations of TG100-115 reduced neutrophilia by ∼50% (54 and 46%, respectively, P ≤ 0.007), whereas the greatest activity was seen with a 0.004% formulation (95% reduction, P < 0.001). In addition to neutrophil accumulation, aerosolized TG100-115 also reduced accumulation of the classic Th1 cytokine TNFα, with similar dose-response relationships.
LPS-induced pulmonary neutrophilia
LPS was instilled into the nares of BALB/c mice, followed 30 min later by treatment with aerosolized vehicle (water/tyloxapol), aerosolized TG100-115, or systemic steroid. Four hours after LPS administration, BALF was collected and analyzed for neutrophil counts (presented as percentage of total cells present) and TNFα levels. Data are presented as means ± S.E.M. Study 1, n = 4, all groups differ from one another by P < 0.01 for both measures. Study 2, n = 5 to 6, all groups differ from one another by P < 0.01 for both measures except for the 0.04 and 0.4% TG100-115 groups and the 0.004% TG100-115 and non-LPS control groups.

Pulmonary inflammation in a murine asthma model: leukocyte and mucin accumulation. Mice were treated as described in Table 1, except that lungs were processed for histological examination as the study endpoint. Representative photomicrographs are shown, taken from OVA-sensitized and challenged animals treated with aerosolized water/tyloxapol vehicle (A and D), aerosolized 0.04% TG100-115 (B and E), or systemic dexamethasone (C and F). Paraffin sections were stained with either hematoxylin/eosin (A-C; original magnifications, 100×) to highlight infiltrating leukocytes or nuclear fast red/Alcian Blue (D-F; original magnifications, 200×) to reveal mucin as blue-stained material within the airway.
In a second model system, pulmonary neutrophilia was induced in mice by cigarette smoke exposure. As with the LPS model, an inflammatory response dominated by neutrophils is produced, but unlike the former model, smoke exposure offers a closer mimic of human COPD in that not only is inflammation induced by the relevant trigger but also that the neutrophilia is nonresponsive to steroids. Building from the studies presented to date, we selected a single TG100-115 dose to test (30-min exposure to a 0.004% formulation to deliver 1 μg/kg) but altered the timing of aerosol delivery to ask whether interventional therapy was possible (Fig. 4). Therefore, mice were exposed to smoke on days 1 to 3 and to aerosolized TG100-115 on days 1 to 3, 2 to 3, or 3 only, and then BALF analyzed on day 4. We observed a marked reduction in neutrophil accumulation, statistically equivalent across all TG100-115 treatment groups (81-92% reduction, P < 0.001). As expected, systemic dexamethasone treatment had no effect.
Discussion
In this report, we present our initial exploration into the development of aerosolized PI3K inhibitors for the treatment of respiratory diseases. Selecting TG100-115 as a lead candidate, based on previous success with this compound as an anti-inflammatory in preclinical models (Doukas et al., 2006), we first validated a method of pulmonary delivery using aerosolized microsuspensions. Delivery was both efficient and controlled in that linear dosing could be achieved over a several log range and in that high pulmonary delivery was achieved without meaningful systemic exposure. In subsequent studies in murine models of asthma and acute stages of COPD, aerosolized TG100-115 demonstrated not only markedly inhibited anti-inflammatory activity but also, in the case of the asthma model, improved functional outcome for the test animals.

AHR responses in a murine asthma model: interventional delivery of aerosolized TG100-115. Mice were treated as described in Table 2, except that aerosolized vehicle (water/tyloxapol) and TG100-115 (0.1% formulation) were delivered as single-day treatments on days 13, 14, or 15. In addition, for AHR measurements on day 16, a range of methacholine doses were administered to determine response curves. Data are presented as means ± S.E.M. (n = 8-9). The TG100-115/day 14 and TG100-115/day 15 groups differ from both vehicle-treated groups and the TG100-115/day 13 group at the 50 and 100 mg/ml methacholine doses (P < 0.01); however, they do not differ from one another.
Our goal in exploring aerosolized formulations was to achieve, if possible, high pulmonary drug levels without concurrent systemic exposure. Kinase inhibitors can affect multiple cellular processes in multiple organs; therefore, a logical approach to maximizing therapeutic index is to restrict exposure as much as possible to the target organ. As a first step, we confirmed that our delivery methods achieved the desired exposure pattern. At the highest dose examined (100 μg/kg), over 2 logs more TG100-115 was deposited in the lung, compared with the plasma compartment. At lower doses, plasma levels fell below the level of quantitation for most animals. Repeat dosing drove pulmonary levels of TG100-115 higher but did not meaningfully alter plasma levels. We hypothesize that this distribution pattern in part reflects physical properties of TG100-115, which is not orally bioavailable (unpublished data). Such properties would not only prevent systemic exposure from any drug that gained access to the oropharyngeal and esophageal regions but might also limit transport of the drug across the pulmonary epithelial and endothelial barriers. Finally, daily delivery of TG100-115 for 21 days did not produce clinical signs of toxicity (decreased body weight, etc.) or histological changes in normal mice, even at doses 2 logs higher than that subsequently seen as sufficient for anti-inflammatory efficacy, supporting our hope that localized delivery would help maximize therapeutic index. Although dual genetic deletion of PI3Kδ and γ in mice produces a phenotype marked by T cell lymphopenia and multiorgan inflammation, this is probably the result of faulty thymic selection (Ji et al., 2007). Therefore, it is not surprising that we did not observe similar pathologies in adults exposed to TG100-115 because thymic deletion of autoreactive T cells would have been completed in these animals.
A second goal for our drug delivery approach was to best model clinical use by employing a delivery device compatible with clinical practice. Previous reports had demonstrated efficacy of PI3K inhibitors in murine asthma models; however, these had employed the more preclinical approach of intratracheal instillation (Tigani et al., 2001; Kwak et al., 2003; Lee et al., 2006). Inflammatory cells are distributed throughout the lung, so delivery of an anti-inflammatory will probably be best served by aerosolized microsuspensions with MMAD < 2 to 3 μM based on the general understanding that this size range is appropriate for reaching the distal airways (Labiris and Dolovich, 2003). Therefore, microsuspensions of TG100-115 were designed with an MMAD of 1 to 2 μM (achieved by controlling the first step of their production, a milling process). In contrast, bronchodilators are often better served by targeting larger airways with larger sized particles. These and other points, such as the use of metered dose inhalers and dry powder inhalers beyond the scope of our initial pharmacologic assessments, do warrant future explorations and, therefore, are planned as topics for future assessment.
Having achieved favorable delivery and exposure patterns, we first explored the potential efficacy of TG100-115 in asthma models, based on previous work demonstrating the role of PI3K (Tigani et al., 2001; Kwak et al., 2003) and particularly the δ isoform (Lee et al., 2006; Nashed et al., 2007) in this disease. Aerosolized TG100-115 not only reduced asthma's characteristic inflammatory pattern, limiting the accumulation of eosinophils and IL-13 in BALF and of mucin in the airway, but also improved lung function, reducing the AHR, which compromises pulmonary function in this disease. Of particular note for potential human applications, the full benefit of AHR reduction was achieved with interventional dosing. A single delivery of TG100-115 reduced AHR up to 48 after treatment, whereas 72 h later, aerosolized TG100-115 no longer affected AHR. Although PI3K inhibitors (or any anti-inflammatory) are unlikely to serve as fast-acting rescue therapies on par with bronchodilators, these data do suggest a potential for relatively short on/off times that at the least could offer an alternative to corticosteroids and chromones in current use. In addition, although we have focused on the anti-inflammatory aspects of TG100-115, PI3K has also been proposed as a respiratory disease target based on its role in mediating smooth muscle and endothelial cell proliferation and by extension airway remodeling (Goncharova et al., 2002; Krymskaya et al., 2005). Although we do not believe that such activity played major roles in our relatively acute models, it does represent another area worthy of future research.
In addition to asthma, we also wished to model the inflammatory pattern that underlies COPD. Unlike asthma's Th2-driven inflammation, COPD is a Th1 response dominated by neutrophil accumulation (Barnes, 2008). Aerosolized TG100-115 inhibited pulmonary neutrophilia induced by both intranasal LPS and smoke exposure. Signs of an inverse dose response were observed in the LPS-induced inflammatory model, where LPS is delivered intranasally, but not in the OVA or smoke-induced models, where the inflammatory triggers are delivered by inhalation (following the same route as TG100-115). We suggest that differences in the deposition patterns of LPS, OVA, smoke particulates, and TG100-115 may underlie these apparent differences in dose-response curves.

Smoke-induced pulmonary neutrophilia: interventional delivery of aerosolized TG100-115. Mice were exposed to cigarette smoke (or air only as a control) daily for 3 days, and then on day 4, neutrophils in BALF were counted. Treatments consisted of: 1) aerosolized TG100-115 (0.004% formulation) delivered daily on days 1 to 3, daily on days 2 to 3, or once on day 3; 2) vehicle (water/tyloxapol) delivered daily on days 1 to 3; or 3) dexamethasone (steroid) delivered daily on days 1 to 3 at 0.3 mg/kg i.p. Data are shown as means ± S.E.M. (n = 6). The vehicle-treated smoke-exposed group differs from all others, with the exception of the dexamethasone-treated, smoke-exposed groups by P < 0.001. In addition, the TG100-115 groups do not differ from one another or from the vehicle-treated air-exposed group.
To our knowledge, these data are the first demonstration that PI3K inhibitors can influence smoke-induced pulmonary inflammation. The smoke model was of particular interest in that like human COPD, it is steroid resistant. Steroid resistance is the result of several alterations within the cell, including dysregulation of the glucocorticoid receptor, attenuated histone deacetylase activity, and increased proinflammatory gene transcription (Barnes et al., 2004; Matthews et al., 2004; Adcock et al., 2006a). Our data demonstrating that aerosolized TG100-115 provides efficacy as an interventional therapy in our smoke-induced neutrophilia model, therefore, suggest the potential use of TG100-115 as a means of over-coming this therapeutic challenge, not only for COPD patients but also for steroid-resistant severe asthmatics (Adcock and Ito, 2004; Adcock et al., 2008).
Acute lung injuries such as acute respiratory distress syndrome represent additional pulmonary indications where PI3K inhibitors are likely to offer therapeutic benefits. These conditions are driven not only by an influx of inflammatory cells but also by a loss of endothelial and epithelial barrier function (Matthay et al., 2003). Because TG100-115 reduces not only cellular inflammation but also vascular permeability (Doukas et al., 2006), we explored its therapeutic potential in studies using rat models of acute respiratory distress syndrome (induced by acid injury and/or hyperventilation). Although beyond the scope of the current report, we can say that this compound markedly reduced the development of pulmonary edema in such studies (data not shown).
TG100-115 targets both PI3Kδ and γ, a pattern that we (Doukas et al., 2006) and others (Rommel et al., 2007) have suggested as particularly appropriate for providing broad-based anti-inflammatory activity. Inhibition of both a Th2-driven allergic hypersensitivity and Th1-driven inflammatory responses, as described in this article, provides an example of such broad activity. With regard to asthma, the activity of TG100-115 against PI3Kδ may be of primary importance. For example, the early phase response in asthma is largely driven by mast cell degranulation (Bloemen et al., 2007), and although both PI3Kδ and γ contribute to mast cell activation (Laffargue et al., 2002; Ali et al., 2008), the former seems to predominate (Ali et al., 2008). Subsequent events during the late phase response, such as IgE hyperproduction, may also depend primarily on PI3Kδ (Jou et al., 2002; Okkenhaug et al., 2002; Al-Alwan et al., 2007). In contrast, LPS- and smoke-induced pulmonary inflammations are probably driven by chemokine-induced neutrophil recruitment, and PI3Kγ is the isoform primarily responsible for this response (Hannigan et al., 2002; Thomas et al., 2005). It should be acknowledged, however, that in achieving the high intrapulmonary TG100-115 levels we reported, inhibition of other PI3K isoforms (e.g., α and β) may also have occurred (Doukas et al., 2006), thereby making the task of assigning biological outcomes to inhibition of specific isoforms difficult. Regardless, although our studies cannot define the relative contributions of PI3K isoforms, they do support the value of a dual specificity inhibitor such as TG100-115 in treating distinct respiratory diseases such as asthma and COPD.
Footnotes
-
doi:10.1124/jpet.108.144311.
-
ABBREVIATIONS: PI3K, phosphatidylinositol 3-kinase; TG100-115, 3-[2,4-diamino-6-(3-hydroxyphenyl)pteridin-7-yl]phenol; COPD, chronic obstructive pulmonary disease; Th, T helper; HPLC, high-performance liquid chromatography; MP, mobile phase; MMAD, mass median aerodynamic diameter; BALF, bronchoalveolar lung fluid; LC, liquid chromatography; MS/MS, tandem mass spectrometry; OVA, ovalbumin; LPS, lipopolysaccharide; IL, interleukin; AHR, airway hyper-responsiveness; TNF, tumor necrosis factor.
- Received August 4, 2008.
- Accepted November 21, 2008.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}