


Abstract
The interaction of an agonist-bound G-protein-coupled receptor (GPCR) with its cognate G-protein initiates a sequence of experimentally quantifiable changes in both the GPCR and G-protein. These include the release of GDP from Gα, the formation of a ternary complex between the nucleotide-free G-protein and the GPCR, which has a high affinity for agonist, followed by the binding of GTP to Gα, the dissociation of the GPCR/G-protein complex, and the hydrolysis of GTP. The efficacy of an agonist is a measure of its ability to activate this cascade. It has been proposed that efficacy reflects the ability of the agonist to stabilize the active state of the GPCR. We examined a series of β2-adrenoceptor (β2AR) agonists (weak partial agonists to full agonists) for their efficacy at promoting two different steps of the G-protein activation/deactivation cycle: stabilizing the ternary complex (high-affinity, GTP-sensitive agonist binding), and steady-state GTPase activity. We obtained results for the wild-type β2AR and a constitutively active mutant of the β2AR (β2ARCAM) using fusion proteins between the GPCRs and Gsα to facilitate GPCR/G-protein interactions. There was no correlation between efficacy of ligands in activating GTPase and their ability to stabilize the ternary complex at β2ARCAM. Our results suggest that the GPCR state that optimally promotes the GDP release and GTP binding is different from the GPCR state that stabilizes the ternary complex. By strongly stabilizing the ternary complex, certain partial agonists may reduce the rate of G-protein turnover relative to a full agonist.
Many hormones and neurotransmitters exert their effects through GPCRs that activate heterotrimeric G-proteins and, thereby, change the activity of effector systems (Gilman, 1987; Kobilka, 1992). Binding of an agonist to a GPCR induces distinct conformational changes in the receptor protein that enable GPCR to promote GDP release from Gα (Wess, 1997; Gether and Kobilka, 1998). The agonist-occupied GPCR forms a high-affinity ternary complex with guanine nucleotide-free Gα (Kent et al., 1980;Kobilka, 1992; Seifert et al., 1998a,b). Upon binding of GTP or the hydrolysis-resistant GTP analog GTPγS to Gα, the high-affinity ternary complex is disrupted, and both Gα and Gβγ can regulate the activity of effector systems (Kent et al., 1980; Gilman, 1987; Kobilka, 1992; Seifert et al., 1998b). G-protein deactivation is accomplished by the GTPase activity of Gα (Gilman, 1987).
Several models have been developed to conceptualize the as yet incompletely understood mechanisms of GPCR activation (for a historical perspective, see Kenakin, 1996a; Colquhoun, 1998). The ternary complex model of GPCR activation states that there is a correlation between the efficacy of agonists at stabilizing the ternary complex and at promoting multiple G-protein activation/deactivation cycles (Kent et al., 1980). The ternary complex model has been elaborated into the extended ternary complex model to explain the finding that GPCRs can activate G-proteins even in the absence of agonist and that certain receptor ligands, namely, inverse agonists, suppress G-protein activation by agonist-free GPCRs (Costa and Herz, 1989; Lefkowitz et al., 1993; Gether and Kobilka, 1998). The agonist-independent GPCR activity is referred to as “constitutive activity”. The extended ternary complex model assumes that agonists stabilize GPCR in the active (R*) state, while inverse agonists stabilize the inactive (R) state. It has been proposed that for constitutively active mutant (CAM) GPCRs, the equilibrium between R and R* is shifted toward R* as a result of diminished structural constraints that control spontaneous R to R* transition. Experimentally, this results in increased agonist affinity and potency and increased efficacy of partial agonists at CAM-GPCRs compared with wild-type GPCRs (Samama et al., 1993). In addition, the relative inhibitory effects of inverse agonists at CAM-GPCRs are larger than at wild-type GPCRs (Samama et al., 1994;Gether and Kobilka, 1998).
Of interest, an increasing number of experimental results cannot readily be explained by the extended ternary complex model. As an example, certain β2AR ligands behave like “protean drugs”, i.e., they act as agonist in one setting, but as inverse agonist in another (Chidiac et al., 1994). Additionally, the efficacy and potency of β2AR agonists is strongly dependent on the specific purine nucleotide present for G-protein activation and the specific G-protein to which the β2AR couples (Seifert et al., 1999a;Wenzel-Seifert and Seifert, 2000). Moreover, the agonist-free and agonist-occupied β2AR differ from each other with respect to their ability to activate different cellular effectors (Zhou et al., 1999). These and other experimental findings (Keith et al., 1996; Blake et al., 1997; Krumins and Barber, 1997; Zuscik et al., 1998; Thomas et al., 2000) could be reconciled by a multistate model of GPCR activation in which ligands stabilize unique and ligand-specific GPCR conformations, which enable GPCR to modulate its cognate G-protein(s) in a ligand-specific manner (Kenakin, 1996a,b; Tucek, 1997; Gether and Kobilka, 1998).
The aim of this study was to examine the hypothesis that the functional properties of partial agonists are due to their ability to stabilize distinct conformational states in GPCRs. Therefore, we characterized functional differences between full agonists and partial agonists using fusion proteins between the wild-type β2AR and Gsα(β2ARGsα) and β2ARCAM (Samama et al., 1993; Gether et al., 1997) and Gsα(β2ARCAMGsα) expressed in Sf9 insect cells. We and others have shown that fusion proteins are a very sensitive experimental system for studying receptor/G-protein interactions (Seifert et al., 1999b; Milligan, 2000). Because of the defined 1:1 stoichiometry of GPCR to Gα, fusion proteins eliminate bias in the analysis of ligand potencies and efficacies caused by varying ratios of receptor to G-protein (Hoyer and Boddeke, 1993; Kenakin, 1997). In addition, fusion proteins allow precise determination of agonist and inverse agonist efficacy in an expression level-independent manner by measurement of steady-state GTP hydrolysis and ternary complex formation (Seifert et al., 1999b; Milligan, 2000). The results of our present study suggest that agonists have different efficacies with respect to their ability to promote different steps of the G-protein activation/deactivation cycle and provide evidence for the existence of multiple ligand-specific conformational GPCR states. Finally, our results suggest that one possible mechanism for partial agonism is strong stabilization of the ternary complex, thereby reducing G-protein turnover.
Experimental Procedures
Materials.
The DNA encoding β2ARCAM was kindly donated by Dr. R. J. Lefkowitz (Duke University, Durham, NC) (Samama et al., 1993). Sources of other materials have been described elsewhere (Seifert et al., 1998a,b).
Construction of β2ARCAMGsα Fusion Protein DNAs.
The fusion proteins used in our present study contained the long splice variant of Gsα. The construction of β2ARGsα DNA was described recently (Seifert et al., 1998a,b). For construction of β2ARCAMGsαDNA, the KpnI/EcoRV-fragment of the β2AR was excised from pGEM-3Z-β2ARGsα and replaced by the corresponding KpnI/EcoRV-fragment of β2ARCAM DNA. TheKpnI/EcoRV fragment of β2ARCAM DNA differs from the corresponding β2AR DNA fragment by mutations that encode for four discrete amino acid substitutions in the third intracellular loop of the receptor (Samama et al., 1993). The β2ARCAMGsαDNA was cloned into the baculovirus expression vector pVL 1392.
Cell Culture and Membrane Preparation.
Recombinant baculoviruses were generated in Sf9 cells using the BaculoGOLD transfection kit (Pharmingen, San Diego, CA). After initial transfection, recombinant baculoviruses were isolated by plaque purification. High-titer virus stocks were generated by three sequential amplifications of plaque-purified virus colonies. Culture of Sf9 cells and membrane preparation were performed as described recently (Seifert et al., 1998a,b).
[3H]DHA Binding.
Membranes were thawed and sedimented by a 15-min centrifugation at 4°C and 15,000gto remove residual endogenous guanine nucleotides as far as possible and resuspended in binding buffer (12.5 mM MgCl2, 1 mM EDTA, and 75 mM Tris/HCl, pH 7.4). Expression levels of fusion proteins were determined by incubating Sf9 membranes (15–25 μg of protein per tube) in the presence of [3H]DHA at a concentration of 10 nM. Nonspecific binding was determined in the presence of [3H]DHA (10 nM) plus 10 μM (±)-alprenolol. The total volume of the binding reaction was 500 μl. Incubations were performed for 90 min at 25°C and shaking at 250 rpm. Competition binding experiments were carried out with Sf9 membranes expressing β2ARGsα or β2ARCAMGsα(15–40 μg of protein per tube), 1 nM [3H]DHA and agonists at various concentrations with or without GTPγS (10 μM). Ligand-competition studies were performed in triplicates. Bound [3H]DHA was separated from free [3H]DHA by filtration through GF/C filters using a 48-well harvester (model M-48R; Brandel, Gaithersburg, MD), followed by three washes with 2 ml of binding buffer (4°C). Filter-bound radioactivity was determined by liquid scintillation counting using Cytoscint cocktail from ICN (Irvine, CA). The experimental conditions chosen ensured that not more than 10% of the total amount of [3H]DHA added to binding tubes was bound to filters.
Steady-State GTPase Activity.
Membranes were thawed and sedimented by a 15-min centrifugation at 4°C and 15,000gto remove residual endogenous guanine nucleotides as far as possible and resuspended in 10 mM Tris/HCl, pH 7.4. Assay tubes contained Sf9 membranes expressing β2ARGsα or β2ARCAMGsα(10 μg of protein per tube), 1.0 mM MgCl2, 0.1 mM EDTA, 0.1 mM ATP, 1 mM adenylyl imidodiphosphate, 100 nM GTP, 5 mM creatine phosphate, 40 μg of creatine kinase, 0.2% (w/v) bovine serum albumin in 50 mM Tris/HCl, pH 7.4, and β2AR ligands at various concentrations. Reaction mixtures (80 μl) were incubated for 3 min at 25°C before the addition of 20 μl of [γ-32P]GTP (0.2–0.5 μCi/tube). Reactions were conducted for 20 min at 25°C. Reactions were terminated by the addition of 900 μl of a slurry consisting of 5% (w/v) activated charcoal and 50 mM NaH2PO4, pH 2.0. Charcoal absorbs nucleotides but not Pi. Charcoal-quenched reaction mixtures were centrifuged for 15 min at room temperature at 15,000g. Seven hundred microliters of the supernatant fluid of reaction mixtures was carefully removed to avoid any aspiration of charcoal, and 32Pi was determined by liquid scintillation counting. Enzyme activities were corrected for spontaneous degradation of [γ-32P]GTP. Spontaneous [γ-32P]GTP degradation was determined in tubes containing all of the above-described components plus a very high concentration of unlabeled GTP (1 mM) that, by competition with [γ-32P]GTP, prevents [γ-32P]GTP hydrolysis by enzymatic activities present in Sf9 membranes. Spontaneous [γ-32P]GTP degradation was <1% of the total amount of radioactivity added. The experimental conditions chosen ensured that not more than 10% of the total amount of [γ-32P]GTP added was converted to32Pi.
Miscellaneous.
Protein was determined using the Bio-Rad DC protein assay kit (Bio-Rad, Hercules, CA). Immunoblotting studies were performed as described (Seifert et al., 1998a,b). Ligand competition curves and concentration-response curves were analyzed by nonlinear regression, using the Prism program (GraphPad, San Diego, CA).
Results
Expression of β2ARGsα and β2ARCAMGsα in Sf9 Membranes.
Similar to nonfused β2AR and β2ARCAM (Gether et al., 1997), the maximum expression level of β2ARCAMGsαin Sf9 cells was ∼2.5-fold lower than the expression level of β2ARGsα(β2ARGsα, 7.2 ± 2.1 pmol/mg; β2ARCAMGsα, 3.2 ± 1.2 pmol/mg). However, the different expression levels of the fusion proteins were not of relevance for our studies since the efficacies of ligands at stabilizing the ternary complex and at promoting GTP hydrolysis are expression level-independent (Seifert et al., 1999; Milligan, 2000). Immunoblotting studies with the M1 antibody recognizing the N-terminal FLAG epitope of β2AR and β2ARCAM (Gether et al., 1995, 1997) and anti-Gsα Ig confirmed that β2ARGsα and β2ARCAMGsαexpressed in Sf9 membranes were structurally intact (data not shown).
Agonist and Inverse Agonist Regulation of Steady-State GTPase Activity in Sf9 Membranes Expressing β2ARGsα and β2ARCAMGsα.
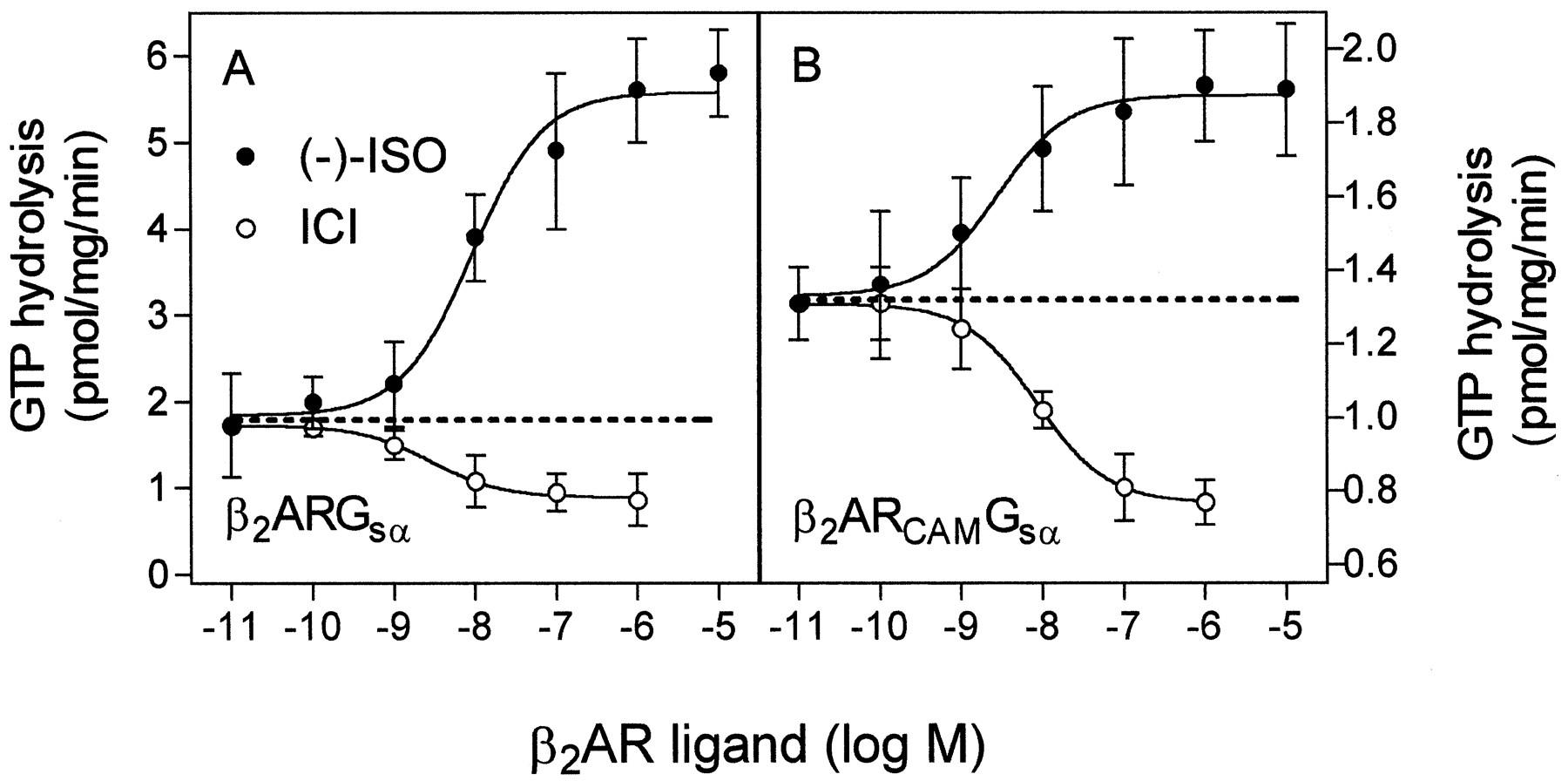
An important hallmark of constitutive GPCR activation is the increased relative inhibitory effect of inverse agonist at CAM-GPCR compared with wild-type GPCR (Lefkowitz et al., 1993; Gether and Kobilka, 1998). In addition, the agonist-affinity of CAM-GPCRs is increased relative to wild type-GPCRs, while the inverse agonist-affinity is decreased. The full agonist (−)-ISO was more potent at activating GTP hydrolysis at β2ARCAMGsα(EC50 = 2.4 ± 1.1 nM) than at β2ARGsα, (EC50 = 13 ± 3 nM), while the inverse agonist ICI was less potent at β2ARCAMGsα(IC50 = 8.5 ± 2.5 nM) than at β2ARGsα(IC50 = 2.8 ± 1.0 nM) (Fig.1). For β2ARGsα, 17% of the ligand-regulated GTPase activity (the difference between maximum (−)-ISO-stimulated and minimum ICI-inhibited GTP hydrolysis) was attributable to the inverse agonist, while for β2ARCAMGsα, 48% of the ligand-regulated GTPase activity was attributable to ICI.

Effects of (−)-ISO and ICI on GTPase activity in Sf9 membranes expressing β2ARGsα and β2ARCAMGsα. GTPase activity in membranes expressing β2ARGsα (6.5 pmol/mg) (A) or β2ARCAMGsα (2.5 pmol/mg) (B) was determined as described under Experimental Procedures. Reaction mixtures contained (−)-ISO or ICI at the concentrations indicated on the abscissa. The dashed lines are extrapolations of basal GTPase activities to illustrate the relative contributions of (−)-ISO and ICI at the ligand-regulated enzyme activity. To reliably quantitate the inhibitory effect of ICI on GTPase in membranes expressing β2ARGsα, this construct was expressed at a higher level than β2ARCAMGsα. Therefore, the absolute GTPase activities in A are higher than in B. To facilitate comparison of the two constructs, the scales of the ordinates in A and B are different. Data shown are the means ± S.D. of three independent experiments performed in duplicate.

Competition by (−)-ISO, SAL, and DOB of [3H]DHA binding in Sf9 membranes expressing β2ARGsα or β2ARCAMGsα: effect of GTPγS. [3H]DHA binding in Sf9 membranes was performed as described under Experimental Procedures. Reaction mixtures contained Sf9 membranes (15–40 μg of protein per tube) expressing β2ARGsα (3.3–7.5 pmol/mg) (A–C) or β2ARCAMGsα (2.5–4.9 pmol/mg) (D–F), 1 nM [3H]DHA, and ligands at the concentrations indicated on the abscissa. Reaction mixtures additionally contained distilled water (control) or GTPγS (10 μM). Data shown are the means ± S.D. of four to seven independent experiments performed in triplicate.
Ligand Binding Properties of β2ARGsαand β2ARCAMGsα.
Sf9 membranes expressing β2ARGsα and β2ARCAMGsαbound the β2AR antagonist [3H]DHA in a monophasic and saturable manner and with similar Kd values (β2ARGsα, 0.36 ± 0.03 nM; β2ARCAMGsα, 0.24 ± 0.05 nM). We also studied the agonist and inverse agonist binding properties of β2ARGsα and β2ARCAMGsα. The ligand competition curves are shown in Figs.2-4, and Table 1 summarizes the nonlinear regression analysis of the binding data. In membranes expressing β2ARGsα, strong agonists [(−)-ISO, (+)-ISO, SAL, and DOB] efficiently stabilized the ternary complex as is shown by the GTPγS-sensitive high-affinity agonist binding. With these ligands, ∼40% of the β2ARs displayed high agonist affinity. For the partial agonist EPH, distinct high-affinity binding sites at β2ARGsα could not be discriminated, but GTPγS could still shift the EPH competition curve ∼5-fold to the right. The binding of another partial agonist, DCI, to β2ARGsα, was virtually GTPγS insensitive. GTPγS did not shift the ICI-competition curve in membranes expressing β2ARGsα.
Ligand binding properties of β2ARGsα and β2ARCAMGsα expressed in Sf9 membranes: effect of GTPγS
The ligand binding properties of β2ARCAMGsαdiffered considerably from the ligand binding properties of β2ARGsα. The high-affinity Ki values of (−)-ISO, (+)-ISO, SAL, and DOB at β2ARCAMGsαwere about 4 to 10 times lower than at β2ARGsα. While there was no large change in the fraction of β2ARCAM displaying high agonist affinity when liganded to (−)-ISO and SAL compared with β2ARGsα, the fraction of receptors displaying high agonist-affinity upon binding of DOB and (+)-ISO in β2ARCAMGsαwas substantially higher than in β2ARGsα. In contrast to β2ARGsα, where distinct high-affinity binding sites for EPH and DCI could not be distinguished, highly effective formation of GTPγS-sensitive ternary complexes with EPH and DCI was observed at β2ARCAMGsα. In agreement with data obtained for nonfused β2AR and β2ARCAM (Samama et al., 1994), the affinity of ICI for β2ARCAMGsαwas about 2-fold lower than for β2ARGsα. GTPγS increased the affinity of ICI for β2ARCAMGsαby ∼2.5-fold.
Efficacies of Partial Agonists on Steady-State GTPase Activity in Sf9 Membranes Expressing β2ARGsα and β2ARCAMGsα.
The precise determination of partial agonist efficacy constitutes a major problem since efficacy is influenced by numerous variables such as the expression level of GPCR and the availability of G-protein and effector system (Hoyer and Boddeke 1993; Kenakin 1996a, 1997). The fusion protein approach offers a unique possibility to assess agonist efficacy in an expression level-independent manner by measuring steady-state GTPase activity (Seifert et al., 1999; Milligan, 2000). At β2ARGsα, ligands activated GTP hydrolysis in the order of efficacy (−)-ISO ∼ SAL ≥ (+)-ISO > DOB > EPH > DCI (Figs. 5 and6; Table2). In accordance with data obtained for effector system activation by nonfused β2AR and β2ARCAM (Samama et al., 1993), the potencies of agonists for GTPase activation at β2ARCAMGsαwere higher than at β2ARGsα. There was a small but significant increase in the efficacy of the partial agonists DOB, EPH, and DCI at activating GTPase in membranes expressing β2ARCAMGsαcompared with membranes expressing β2ARGsα. For SAL and (+)-ISO, the increase in efficacy at β2ARCAMGsαversus β2ARGsα was not significant.

Concentration-response curves for the stimulatory effects of (−)-ISO, SAL, DOB, EPH, and DCI on steady-state GTPase activity in membranes expressing β2ARGsαand β2ARCAMGsα. GTPase activity in membranes expressing β2ARGsα (3.3–7.5 pmol/mg) (A) or β2ARCAMGsα(2.5–4.9 pmol/mg) (B) was determined as described underExperimental Procedures. Reaction mixtures contained ligands at the concentrations indicated on the abscissa. For each construct, the stimulatory effect of (−)-ISO (10 μM) on GTP hydrolysis was set 100%, and all data points were referred to this stimulation. Data shown are the means ± S.D. of three to six independent experiments performed in duplicate.

Concentration-response curves for the stimulatory effects of (−)-ISO and (+)-ISO on steady-state GTPase activity in membranes expressing β2ARGsα and β2ARCAMGsα. GTPase activity in membranes expressing β2ARGsα (3.3–7.5 pmol/mg) (A) or β2ARCAMGsα(2.5–4.9 pmol/mg) (B) was determined as described underExperimental Procedures. Reaction mixtures contained ligands at the concentrations indicated on the abscissa. For each construct, the stimulatory effect of (−)-ISO (10 μM) on GTP hydrolysis was set 100%, and all data points were referred to this stimulation. Data shown are the means ± S.D. of three to six independent experiments performed in duplicate.
Efficacies and potencies of β2AR ligands at the GTPase of β2ARGsα and β2ARCAMGsα
Discussion
Sf9 Cell Membranes Expressing GPCR-Gα Fusion Proteins as Model System for the Analysis of Ligand-Receptor Interactions.
In the present study, we expressed a wild-type GPCR and a CAM-GPCR fused to Gα in Sf9 cells to study ligand-receptor interactions. Compared with conventional coexpression systems, the fusion protein approach offers several advantages. Specifically, the defined 1:1 stoichiometry of GPCR and Gα eliminates bias in the analysis of ligand potencies and efficacies because of varying GPCR/Gα ratio (Seifert et al., 199b; Milligan, 2000). In addition, the physical proximity of GPCR and Gα ensures efficient interaction of the proteins. Fusion proteins allow for the sensitive analysis of ligand potencies and efficacies in an expression level-independent manner directly at the G-protein level by measuring steady-state GTP hydrolysis. This is a very important point because nonfused β2ARCAM expresses at considerably lower levels than β2AR (Samama et al., 1993; Gether et al., 1997), resulting in different GPCR/G-protein ratios. Finally, for GPCRs mediating adenylyl cyclase activation such as the β2AR, the number of effector molecules limits signal output (Alousi et al., 1991), introducing additional bias into the analysis of ligand potencies and efficacies.
GPCR-Gα fusion proteins are not naturally occurring so that caution must be exerted when extrapolating conclusions obtained with fused proteins to the in vivo situation. Perhaps most evident, in native membranes there is a large excess of Gs molecules relative to β2AR molecules (Ransnäs and Insel, 1988). However, the mere excess of G-protein relative to GPCR does not automatically imply that one GPCR molecule activates multiple Gα molecules. Rather, recent data from various GPCR/G-protein combinations, including the β2AR/Gs pair indicate that even in coexpression systems with a high G-protein to GPCR ratio, GPCRs activate G-proteins only linearly (Seifert et al., 1998a;Wenzel-Seifert et al., 1999; Wenzel-Seifert and Seifert, 2000). Thus, GPCR-Gα fusion proteins may mimic the close association of GPCRs and G-proteins in vivo (Seifert et al., 1999b).
Another issue that needs to be considered is the GTP concentration in our experiments. Ternary complex formation in membranes was studied in washed membranes, i.e., in the virtual absence of GTP. GTPase studies were conducted with a substrate concentration of 100 nM (underExperimental Procedures). It could be argued that such studies are not of relevance for the in vivo situation because the bulk intracellular GTP concentration is in the high micromolar range (Otero, 1990; Jinnah et al., 1993). In addition, adenylyl cyclase assays in membranes have been routinely performed with GTP concentrations in the high micromolar range (Samama et al., 1993; Chidiac et al., 1994;Gether et al., 1995). However, the concentration of GTP available to G-proteins in vivo has not been determined. First, the access of GTP to G-proteins is restricted (Otero, 1990; Wieland and Jakobs, 1992;Klinker et al., 1996). Second, there is a remarkable discrepancy between the high intracellular GTP concentration and the lowKm of G-proteins for GTP (∼0.1 μM) (Seifert et al., 1998a; Wenzel-Seifert et al., 1999), and in vivo, most enzymes work at substrate concentrations around theKm value. These data suggest that even in vivo G-proteins may operate under nonsaturating GTP concentrations, allowing for at least some ternary complex formation and efficient regulation of G-protein turnover by modulating the GTP concentration in the G-protein vicinity.
Can the extended ternary complex model explain the functional properties of β2ARGsα and β2ARCAMGsα?
Several of our results are in agreement with the extended ternary complex model, which proposes that GPCRs exist in an equilibrium between an inactive R state and an active R* state (Lefkowitz et al., 1993; Gether and Kobilka 1998). The model proposes that in CAM-GPCRs, R to R* isomerization occurs more readily than in wild-type GPCRs, but there is no fundamental difference in the R and R* states of CAM-GPCRs. Thus, the model predicts that the relative inhibitory effects of inverse agonists at CAM-GPCRs are higher than at wild type-GPCRs. The increased relative inhibitory effect of ICI at β2ARCAMGsα compared with β2ARGsα is in agreement with this model (Fig. 1). The extended ternary complex model also predicts that the inverse agonist affinity and potency at CAM-GPCR is lower than at wild-type GPCR (Samama et al., 1994). Again, our findings with fusion proteins are in agreement with the model (Figs. 1 and 3). Another postulate of the extended ternary complex model is that guanine nucleotides increase inverse agonist affinity of GPCR, presumably by uncoupling of GPCR from G-protein and, thereby, stabilizing the R state (Barker et al., 1994; Leeb-Lundberg et al., 1994). In agreement with this postulate, we found an increase in inverse agonist-affinity of β2ARCAMGsα by GTPγS (Fig. 3; Table 1).

Competition by EPH, DCI, and ICI of [3H]DHA binding in Sf9 membranes expressing β2ARGsα or β2ARCAMGsα: effect of GTPγS. [3H]DHA binding in Sf9 membranes was performed as described under Experimental Procedures. Reaction mixtures contained Sf9 membranes (15–40 μg of protein per tube) expressing β2ARGsα (3.3–7.5 pmol/mg) (A–C) or β2ARCAMGsα (2.5–4.9 pmol/mg) (D–F), 1 nM [3H]DHA, and ligands at the concentrations indicated on the abscissa. Reaction mixtures additionally contained distilled water (control) or GTPγS (10 μM). Data shown are the means ± S.D. of four to seven independent experiments performed in triplicate.
An additional prediction of the extended ternary complex model is that CAM-GPCRs possess a higher agonist affinity and potency than wild-type GPCRs (Lefkowitz et al., 1993; Samama et al., 1993). Our data with β2ARGsα and β2ARCAMGsαshow that this property of GPCRs is preserved in fusion proteins (Tables 1 and 2). Moreover, the extended ternary complex model predicts that the efficacies of partial agonists are higher at CAM-GPCR than at wild-type GPCR (Samama et al., 1993). This was also the case for β2ARGsα and β2ARCAMGsα, but the differences were small (Table 2). The explanation for these only small differences in agonist efficacies between the two fusion proteins presumably is that we studied coupling of the β2AR and β2ARCAM to the long splice variant of Gsα and that this G-protein confers to the wild-type β2AR properties of high constitutive activity in terms of partial agonist efficacy (Seifert et al., 1998b). It is likely that the differences in partial agonist efficacies between β2AR and β2ARCAM would have been more evident if coupling of these GPCRs to the short splice variant of Gsα had been analyzed. However, we did not construct a fusion protein of β2ARCAM and the short splice variant of Gsα because a fusion protein of the β2AR and this G-protein expresses less well than a fusion protein of the β2AR and the long splice variant of Gsα (Seifert et al., 1998a,b). Given the fact that β2ARCAM expresses much less well than β2AR (see Results;Gether et al., 1997), we predicted that the expression levels of a fusion protein of β2ARCAMand the short splice variant of Gsα would be too low for sensitive quantitative analysis of ternary complex formation and particularly GTPase activation.
Certain effects of agonists on the functional properties of β2ARGsα and β2ARCAMGsαare less readily explained by the extended ternary complex model. Specifically, the extended ternary complex model also predicts that there is a correlation between the efficacy of agonists at stabilizing the ternary complex and their efficacy at promoting multiple G-protein activation/deactivation cycles. This concept is based on the observed highly significant correlation between the efficacy of agonists at stabilizing the ternary complex and the efficacy of agonists at promoting effector system activation (Kent et al., 1980). We studied the outcome of multiple G-protein activation/deactivation cycles more directly by assessing steady-state GTPase activity. In agreement with the results of a previous study (Kent et al., 1980), we observed significant correlations between the efficacy of agonists at stabilizing the ternary complex (as determined by the percentage of high-affinity agonist-binding sites and the ratioKlGTPγS/Kh) and the efficacy of agonists at activating GTPase for β2ARGsα (Fig.7, A and C). However, no such correlation was observed for β2ARCAMGsα(Fig. 7, B and D). Most strikingly, at β2ARCAMGSα, EPH, DCI, and particularly DOB exhibited a higher percentage of high-affinity binding sites than (−)-ISO. In contrast, all of these ligands had lower efficacies than (−)-ISO at stimulating GTPase. In addition, (−)-ISO and (+)-ISO, which differ from each other only in the chirality of the β-carbon hydroxyl group of the ethylamine side chain of the phenyl ring, differed considerably from each other in their ability to stabilize the ternary complex at β2ARCAMGsα(Fig. 4). However, the efficacies of (−)-ISO and (+)-ISO at activating GTPase in membranes expressing β2ARCAMGsαwere similar (Fig. 6; Table 2).

Relation between the efficacy of agonists at stabilizing the ternary complex and the ligand efficacy at activating GTPase in Sf9 membranes expressing β2ARGsαand β2ARCAMGsα. The efficacies of ligands at stabilizing the ternary complex in membranes expressing β2ARGsα and β2ARCAMGsα (Figs. 2-4; Table1) were plotted against the respective efficacies of these ligands at activating GTPase (Figs. 5 and 6; Table 2). Data were analyzed by linear regression analysis. In A and B, ternary complex formation is expressed as the percentage of receptors displaying high agonist affinity (Rh, %). A,r2 = 0.71; p = 0.035. B, r2 = 0.08; p, = 0.586 (slope not significantly different from zero). In C and D, ternary complex formation is expressed as the ratioKlGTPγS/Kh. For EPH and DCI, distinct Kh values at β2ARGsα could not be determined. Therefore, we used the values listed under Kl in Table1 to calculate the ratio. C, r2 = 0.86;p = 0.007. D, r2 = 0.04; p = 0.716 (slope not significantly different from zero). The dotted lines indicate the 95% confidence interval of the regression lines.

Competition by (−)-ISO and (+)-ISO of [3H]DHA binding in Sf9 membranes expressing β2ARGsα or β2ARCAMGsα: effect of GTPγS. [3H]DHA binding in Sf9 membranes was performed underExperimental Procedures. Reaction mixtures contained Sf9 membranes (15–40 μg of protein per tube) expressing β2ARGsα (3.3–7.5 pmol/mg) (A and B) or β2ARCAMGsα (2.5–4.9 pmol/mg) (C and D), 1 nM [3H]DHA, and ligands at the concentrations indicated on the abscissa. Reaction mixtures additionally contained distilled water (control) or GTPγS (10 μM). Data shown are the means ± S.D. of four to seven independent experiments performed in triplicate.
Dissociation in the efficacy of agonists at stabilizing the ternary complex and promoting multiple cycles of G-protein activation/deactivation were observed previously. First, when the β2AR couples to either the long or the short splice variant of Gsα, SAL and DOB are similarly efficient at stabilizing the ternary complex. However, with respect to GTPase activation, the two ligands are more effective at the β2AR coupled to the long splice variant of Gsα compared with the β2AR coupled to the short splice variant of Gsα (Seifert et al., 1998b). Second, at the β2AR and β2ARCAM expressed in Chinese hamster ovary cells, DOB is more effective than SAL at stabilizing the ternary complex, but not at activating adenylyl cyclase (Samama et al., 1993). Third, at certain mutants of the β2AR (Hausdorff et al., 1990), histamine H2 receptor (Smit et al., 1996), prostaglandin EP3 receptor (Irie et al., 1994), 5-hydroxytryptamine2A receptor (Roth et al., 1997), and M4 muscarinic acetylcholine receptor (Van Koppen et al., 1994) the ability of agonist to promote multiple G-protein activation/deactivation cycles is severely reduced compared with their wild-type counterparts, but the ability of mutant GPCRs to form ternary complexes is normal. Nonsignaling ternary complexes were also reported for the α1B-adrenergic receptor (Chen et al., 2000), cannabinoid CB1 receptor (Bouaboula et al., 1997), and the MOR-1 opioid receptor (Brown and Pasternak, 1998). Based on these observations encompassing multiple GPCRs coupled to G-proteins from different families it can be concluded that the GPCR conformation that optimally promotes multiple G-protein activation/deactivation cycles is distinct from the GPCR conformation that promotes high-affinity interactions between GPCR and G-protein. These observations suggest that one possible mechanism of partial agonism is stabilization of the ternary complex relative to GTP binding and G-protein dissociation. A highly stable ternary complex would be less likely to bind GTP, thereby reducing G-protein turnover.
Mechanisms of Action of Full and Partial Agonists.
Of interest, the ratio of EC50 for GTPase activation to Kh for agonists at β2ARCAMGsαshows a significant correlation with the efficacy of agonists at activating GTPase (Fig. 8). The data obtained for β2ARGsαshow a similar trend as for β2ARCAMGsαbut do not reach significance. It is surprising that the ratio of EC50 to Kh is significantly higher for full agonists than partial agonists. This suggests that occupancy of a GPCR by a full agonist is less likely to lead to a complete G-protein activation/deactivation cycle than is GPCR occupancy by a partial agonist. A possible mechanism for this hypothesis is illustrated in Fig. 9. This model proposes that the ternary complex for partial agonists is more stable, relative to the other GPCR and G-protein states (PR, G, GGDP, GGTP), than is the ternary complex for full agonists, relative to the other GPCR and G-protein states (FR, G, GGDP, GGTP). Thus, rate constantsk1 andk2 for full and partial agonists may be similar. However, if the rate constantsk−1,k−2, andk3 are all smaller for partial agonists, compared with these constants for full agonists, the relative amount of the ternary complex (partial agonists) will be greater and the number of total G-protein hydrolysis cycles will be smaller, compared with full agonists. The model shown in Fig. 9 also proposes that the ternary complex for full agonists is more likely to dissociate without binding GTP, thus k−1 andk−2 are greater for full agonists than for partial agonists. This could explain the larger EC50 to Kh ratio for full agonists. A larger receptor occupancy would be required to compensate for larger k−1 andk−2 rates for full agonists as compared to partial agonists.

Relation between the ratio EC50/Kh and efficacy of agonists at activating GTPase in Sf9 membranes expressing β2ARGsα and β2ARCAMGsα. The ratios of the EC50 for GTPase activation (Figs. 5 and 6; Table 2) andKh values (Figs. 2 and 3; Table 1) for various agonists at β2ARGsα and β2ARCAMGsα were plotted against the respective efficacies of these ligands at activating GTPase (Figs.5 and 6; Table 2). Data were analyzed by linear regression analysis. A,r2 = 0.653; p = 0.192. B, r2 = 0.82;p = 0.014. The dotted lines indicate the 95% confidence interval of the regression line.
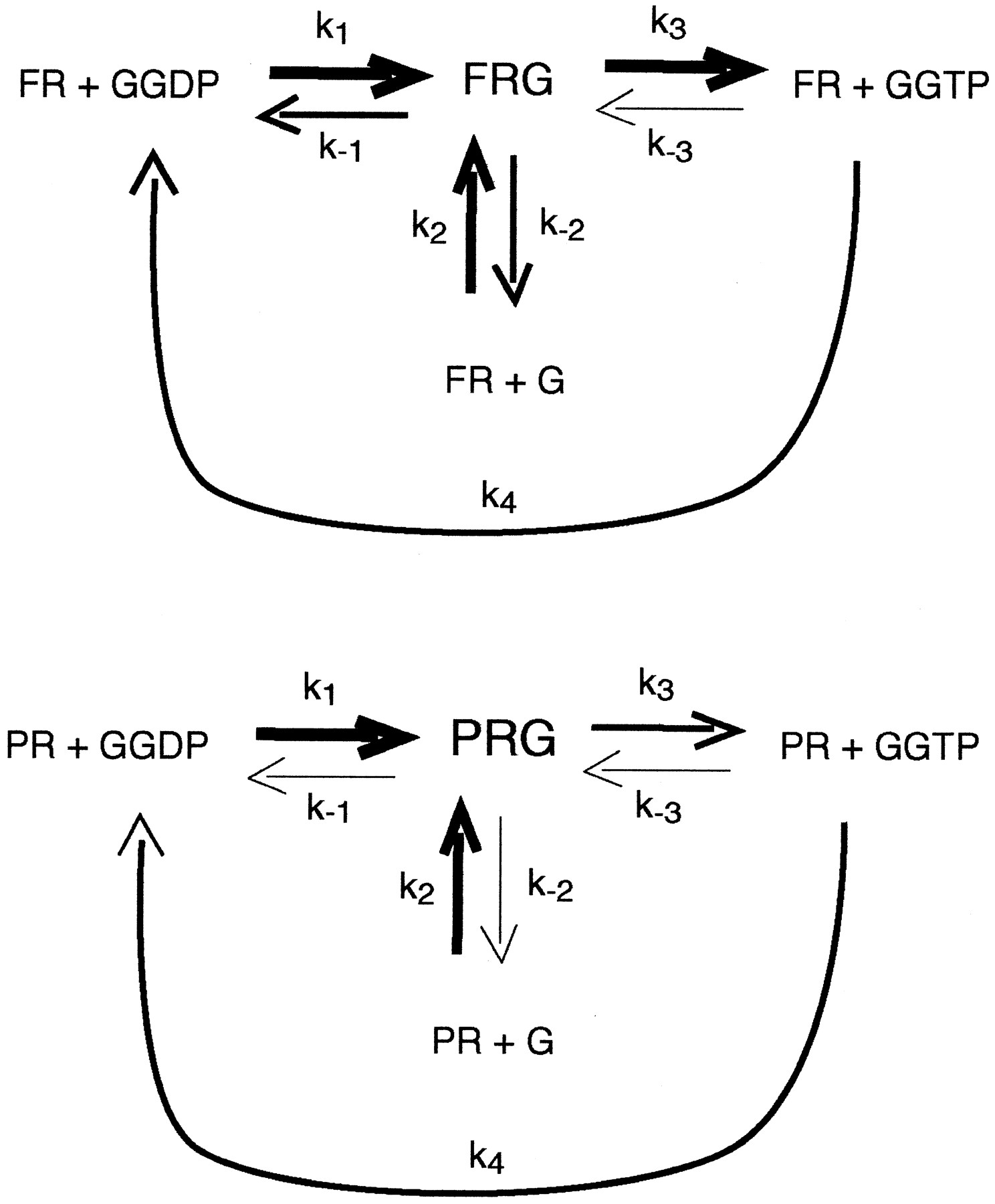
Model of the different effects of full and partial agonists on the G-protein activation/deactivation cycle. The arrows symbolize the relative rate constants for interconversion of the various GPCR and G-protein states. G, guanine nucleotide-free G-protein; GGDP, GDP-liganded G-protein; GGTP, GTP-liganded G-protein; F, full agonist; P, partial agonist; R, G-protein-coupled receptor.
Conclusions.
Our present data and an increasing body of data obtained with wild-type GPCRs (Chidiac et al., 1994; Chiu et al., 1996;Keith et al., 1996; Blake et al., 1997; Bouaboula et al., 1997) and constitutively active GPCR mutants (Zuscik et al., 1998; Thomas et al., 2000) and theoretical considerations (Kenakin 1996a,b; Tucek 1997) suggest that ligands stabilize distinct GPCR conformations that differ from each other in their ability to interact with G-proteins. Thus, the conformations stabilized by (−)-ISO, (+)-ISO, and SAL in both β2AR and β2ARCAM are functionally distinguishable from those induced by agonists with lower efficacies, i.e., DOB, EPH, and DCI. Additionally, our results suggest that some GPCR ligands act as partial agonists because the GPCR conformation that they stabilize promotes a more stable ternary complex than the conformation stabilized by full agonists. By stabilizing the guanine nucleotide-free ternary complex these partial agonists reduce G-protein turnover.
Acknowledgments
We thank Dr. Terry Kenakin (Glaxo-Wellcome, Research Triangle Park, NC) for stimulating discussions and Maria Bakk for help with the cell culture.
Footnotes
-
Send reprint requests to: Brian Kobilka, M.D., Howard Hughes Medical Institute, B-157, Beckman Center, Stanford University Medical School, Stanford, CA 94305-5428. E-mail:kobilka{at}cmgm.stanford.edu
-
↵1 Present address: Department of Pharmacology and Toxicology, The University of Kansas, 5064 Malott Hall, Lawrence, KS 66045.
-
↵2 Present address: Department of Cellular Physiology, Institute of Medical Physiology 12.5, The Panum Institute, University of Copenhagen, Blegdamsvej 3, DK-2200 Copenhagen N, Denmark.
-
R.S. and K.W.-S. were the recipients of a research fellowship of the Deutsche Forschungsgemeinschaft.
- Abbreviations:
- GPCR
- G-protein-coupled receptor
- GTPγS
- guanosine 5′-O-(3-thiotriphosphate)
- CAM
- constitutively active mutant
- β2AR
- β2-adrenoceptor
- β2ARGsα
- fusion protein consisting of the β2-adrenoceptor and Gsα
- β2ARCAM
- constitutively active mutant of the β2-adrenoceptor
- β2ARCAMGsα
- fusion protein consisting of the constitutively active mutant of the β2-adrenoceptor and Gsα
- DHA
- dihydroalprenolol
- ISO
- isoproterenol
- SAL
- salbutamol
- DOB
- dobutamine
- EPH
- (−)-ephedrine
- DCI
- dichloroisoproterenol
- ICI
- ICI 118,55 ([erythro-dl-1(7-methylindan-4-yloxy)-3-isopropylaminobutan-2-ol])
- Received December 7, 2000.
- Accepted March 8, 2001.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}