

Abstract
Axonal regeneration is minimal after CNS injuries in adult mammals and medical treatments to recover neurological deficits caused by axon disconnection are extremely limited. The failure of axonal elongation is principally attributed to the nonpermissive environment and reduced intrinsic growth capacity. In this report, we studied the role of glycogen synthase kinase-3 (GSK-3) inactivation on neurite and axon growth from adult neurons via combined in vitro and in vivo approaches. We found that the major CNS inhibiting substrates including chondroitin sulfate proteoglycans could inactivate protein kinase B (Akt) and activate GSK-3β signals in neurons. GSK-3 inactivation with pharmacologic inhibitors enhances neurite outgrowth of dorsal root ganglion neurons derived from adult mice or cerebellar granule neurons from postnatal rodents cultured on CNS inhibitors. Application of GSK-3 inhibitors stimulates axon formation and elongation of mature neurons whether in presence or absence of inhibitory substrates. Systemic application of the GSK-3 inhibitor lithium to spinal cord-lesioned rats suppresses the activity of this kinase around lesion. Treatments with GSK-3 inhibitors including a clinical dose of lithium to rats with thoracic spinal cord transection or contusion injuries induce significant descending corticospinal and serotonergic axon sprouting in caudal spinal cord and promote locomotor functional recovery. Our studies suggest that GSK-3 signal is an important therapeutic target for promoting functional recovery of adult CNS injuries and that administration of GSK-3 inhibitors may facilitate the development of an effective treatment to white matter injuries including spinal cord trauma given the wide use of lithium in humans.
Introduction
The reduced intrinsic growth capacity of adult CNS neurons and the poor environment for axon extension greatly contribute to the failure of CNS axonal regeneration (David and Aguayo, 1981; Chen et al., 1995; Goldberg et al., 2002). The robust growth capability of CNS neurons to elongate axons is markedly reduced during neuronal maturation. Chondroitin sulfate proteoglycans generated by astroglial scar around lesions strongly suppress axonal extension via unknown receptor(s) (Bradbury et al., 2002; Silver and Miller, 2004). Myelin components including myelin-associated glycoprotein (MAG), oligodendrocyte-myelin glycoprotein, Nogo, and netrin1 potently inhibit CNS axonal regeneration (Chen et al., 2000; GrandPré et al., 2000; Domeniconi et al., 2002; Wang et al., 2002; Löw et al., 2008). Lack of neurotrophic factors may partly attribute to the regenerative failure in adult CNS (Jones et al., 2001). Moreover, axon guidance molecules semaphorin 3A, 5A, and ephrin-3B/Eph-A4 may restrict axonal elongation in injured CNS (Goldberg et al., 2004; Goldshmit et al., 2004; Benson et al., 2005). Thus, manipulating multiple factors for axon growth failure may facilitate the development of more effective therapies for CNS axonal injuries (Mueller et al., 2005; Zhou and Snider, 2006).
Glycogen synthase kinase-3α (GSK-3α) and -3β, especially GSK-3β, are critical in regulating multiple biological processes of neurons. Highly expressed in neurons during neurite remodeling, GSK-3β is critical for establishing neuronal polarity and turning mature neurites into axons in CNS neurons (Jiang et al., 2005; Yoshimura et al., 2005). Several attractive extracellular cues promote axonal elongation via inactivation of GSK-3β and subsequent regulation of growth binding proteins (Woodgett, 2001; Del Río et al., 2004). Nerve growth factor (NGF) suppresses GSK-3β via phosphorylation at serine-9 (Zhou et al., 2004). Wnt-7a enhances axon branching by inactivating GSK-3β through Frizzled/dishevelled receptors independently of serine-9 phosphorylation (Lucas et al., 1998; Arévalo and Chao, 2005). The reduced GSK-3β activity enhances microtubule assembly and axon elongation. In contrast, inhibitory molecules semaphorin 3A and 4D repel axon growth via inhibiting serine-9 phosphorylation and activating GSK-3β (Eickholt et al., 2002; Ito et al., 2006). Thus, regulating GSK-3 activity may overcome axonal growth suppression by altering the responses of local growth axons to various inhibitory factors. Furthermore, GSK-3 inactivation may mimic the growth-promoting effects of neurotrophic factors via upregulating growth-related genes and promoting the assembly of cytoskeletal proteins in axons (Bomze et al., 2001; Zhou and Snider, 2006).
Given the critical function of GSK-3 in regulating axon genesis and elongation, modulation of GSK-3 activity may be an important strategy for developing successful therapies for CNS axonal injuries. In this report, we focus on the role of GSK-3β inactivation on neurite and axonal growth in adult neurons via combined in vitro and in vivo approaches. We demonstrate that pharmacological inhibition of GSK-3β can overcome growth suppression of CNS inhibitory substrates, stimulate significant neurite elongation and axonal growth in vitro and promote axonal regeneration and functional recovery in vivo after traumatic CNS axonal injuries.
Materials and Methods
Neuronal cultures and Akt and GSK-3β assays.
PC12 cells were grown on collagen-coated dishes in MEM Ham's nutrient medium F-12 supplemented with 5% fetal bovine serum and 10% horse serum, 100 μg/ml penicillin, 100 μg/ml streptomycin, and 2 mm glutamine. These cells were differentiated for 24 h with NGF (50 ng/ml) in the same medium. DRGs were dissected out from C57BL/6 mice aged 7–10 weeks. After incubation with collagenase and 0.25% trypsin/EDTA and washing with culture medium, the dissociated DRG neurons were plated onto plastic coverslips in a 24-well plate and grown in culture medium (F-12 plus 10% fetal bovine serum, 1% glutamine, 100 μg/ml penicillin, and 100 μg/ml streptomycin supplied by 100 ng/ml NGF) for 24 h at 37°C (Fu et al., 2007). The primary cerebellar granular neuron (CGN) cultures were prepared from cerebelli of postnatal day 7 (P7)–P9 d C57BL/6 mice. After trypsin digestion, the cells were dissociated and grown in culture medium (DMEM/F-12 containing 10% fetal bovine serum, 2 mm glutamine, 100 μg/ml penicillin, 100 μg/ml streptomycin, 25 mm KCl, and 25 mm glucose) for 24 h at 37°C. After being coated with 100 μg/ml poly-l-lysine, the cell culture dishes or coverslips were incubated with 10 μg/ml laminin for 2 h at 37°C before cell plating.
For Akt and GSK-3β signal assays, PC12 and CGN cultures were treated with soluble inhibitory substrates. In brief, after 24 h growth in NGF medium, PC12 cells or CGNs were incubated with chondroitin sulfate proteoglycans (CSPGs; 1.5 μg/ml) or MAG (570 ng/ml) for 2, 5, 10, or 30 min. After washing with ice-cold PBS, cells in each dish (60 mm) were prepared in 400 μl of cold lysis buffer supplemented with protease inhibitors (1 mm phenylmethylsulfonyl fluoride, 2 mm orthovanadate, 10 μg/ml leupeptin, and 10 μg/ml aprotinin) and clarified by centrifugation at 15,000 × g for 6 min at 4°C. After total protein quantification in lysates with Bio-Rad DC protein assay reagents, samples containing same amount of protein were prepared for Western blot analysis using antibodies against phosphorylated Akt ser473 (Akt-p), GSK-3β Ser9 (GSK-p), and total Akt or GSK-3β (Cell Signaling Technology). After stimulation of extracellular factors, phosphorylation of Akt by phosphoinositide 3 kinase activates Akt, which subsequently inactivates GSK-3β via phosphorylation. Thus, the levels of phosphorylated Akt and GSK-3β represent active Akt and inactive GSK-3β, respectively. Proteins were transferred to nitrocellulose membrane and bands were visualized with enhanced chemiluminescence reagents (Amersham). For blot densitometry, the images of Akt-p and GSK-p bands were captured with a Bio-Rad Gel Doc XR documentation system and band density was determined using Quantity One software (Fu et al., 2007). The arbitrary units for band density represent the signal density per unit area calculated by the software. For each inhibitory substrate and cell type, 4–6 separate experiments were performed and the values of band density measured from cells collected at different time points were compared with those treated with vehicle (PBS).
For Akt and GSK-3β activity assays in lesioned spinal cord, a moderate contusion injury was performed in adult female rats at T8 level. One hour after injury, a vehicle or lithium (3 mEq/kg/d) was administered with subcutaneous injections. Twenty-four hours after injury, rats were perfused through the heart with cold PBS for 5 min. Two tissue blocks of fresh spinal cord were collected on dry ice and stored at −80°C, with one block 3 mm rostral to and caudal to lesion center (6 mm long containing the lesion area) and the other block 4–10 mm rostral to lesion center. Then, each of the spinal cord blocks was prepared in 1 ml of lysis buffer with protease inhibitors for signal protein analyses with Western blot as described above. To visualize Akt-p and GSK-p signals in individual cells, we directly stained the tissue sections of fixed rat spinal cord harvested 24 h after a contusion injury at T8 (5 mm rostral and caudal to the lesion, 40 μm thick). The parasagittal tissue sections containing the lesioned area were incubated with anti-Akt-p or GSK-p antibodies. To localize the signals to definite cells, cell-type-specific antibodies (NeuN neurons, neurofilament axon cylinders, GFAP astrocytes, and adenomatous polyposis coli oligodendrocytes) were applied in some sections. The levels of Akt-p and GSK-p were determined by measuring the intensity of staining signals with Photoshop and NIH Image software under the same conditions. The Akt-p and GSK-p signals in the white matter areas were calculated from five to seven representative images in each group.
Neurite outgrowth and axonal formation and elongation in neuronal cultures.
The neurite outgrowth from neurons was performed in DRG and CGN cultures 24 h after growth. In some cases, coverslips were coated with CNS myelin (50 μg/ml) purified from bovine brain white matter or CSPGs (1.5 μg/ml; Millipore). NGF was applied to neuronal cultures in myelin inhibition experiments but not in CSPG experiments because NGF has been shown to overcome the inhibitory effect of CSPGs in conditioning lesioned neurons via integrin-linked kinase (ILK) signaling pathway (Zhou et al., 2006). The dissociated neurons were treated with vehicle, lithium chloride (3 mm), or 3-[(3-chloro-4-hydroxyphenyl)amino]-4-(2-nitrophenyl)-1H-pyrrole-2,5-dione [SB415286 (SB); 7.5 μm, a selective GSK-3 inhibitor]. For neurite outgrowth, neuronal cells were fixed 24 h after plating and stained with rhodamine phalloidin (Invitrogen). Images of each culture were captured with a Nikon image-collecting system and neurite outgrowth was quantified with Photoshop and NIH Image software (GrandPré et al., 2002; Fu et al., 2007). For outgrowth quantification, the total neurite length for each neuron was determined from 40–150 representative DRG neurons or from 120–180 CGNs in each experiment. The mean values were calculated from 6–10 separate experiments.
We assessed axonal formation and elongation in DRG neurons derived from 7- to 10-week-old mice 4 d after cell plating. Briefly, mouse DRG neurons were dissected out and cultured on coverslips as described above. For myelin inhibition experiments, coverslips were coated with purified CNS myelin (20 μg/ml) after poly-l-lysine (50 μg/ml) coating. For drug treatments, vehicle, lithium chloride (3 mm), or SB (7.5 μm) were applied to cultures after cell plating. After 4 d growth, cells were fixed with 2% formaldehyde in 10% sucrose and PBS. After blocking with 10% goat serum, primary antibody against neurofilament and secondary fluorescent antibody were applied to visualize neuronal bodies and axons. The general structures of all the cultured cells were visualized via double staining F-actin with rhodamine phalloidin. The ratio of axon-forming neurons was determined from each culture under fluorescence microscopy by counting the number of neurons with identifiable axons and total neurons (250–550 neurons/cover) in several random areas of a coverslip away from the border area. The total length of axons in each axon-forming neuron was traced manually and measured with Photoshop and NIH Image software. The axonal branches along an individual axon were counted under high magnification (40×). For axonal length and branching analyses, 30–50 axon-forming neurons were used from each coverslip. The mean values were reported from five to six separate experiments for the axonal assay experiments.
Unilateral transection lesions of spinal cord and axonal tracing in mice.
Adult female C57BL/6 mice (8–10 weeks of age; Jackson Laboratories) were deeply anesthetized with intramuscular ketamine (100 mg/kg) and intraperitoneal xylazine (15 mg/kg). A complete laminectomy was performed, and the dorsal part of spinal cord was fully exposed at levels T6 and T7 (Li and Strittmatter, 2003). The entire left half and part of the spinal cord on the right side (200 μm from midline) was transected at T7 with a 30-gauge needle with an aid of a stereotaxic apparatus (see Fig. 5A). The lesioned area of the spinal cord was ensured by passing the needle several times across the transected spinal cord, including all of the corticospinal tracts (CSTs) on the left spinal cord (dorsal, lateral, dorsolateral, and ventral CSTs). In addition to CST fibers on the left side, this model transected part of the spinal cord on the right side, including dorsal and ventral CSTs, although the CST tracts on the right side were not traced and measured in this study. Immediately after spinal cord injury (SCI), an Alzet osmotic minipump (model 2004; Alza) was implanted and positioned to deliver reagents to subcutaneous space. The pump was filled with saline or GSK-3 inhibitor lithium chloride (3 mEq/kg/d) in 0.9% saline. The vehicle or lithium was delivered continuously at a rate of 0.3 μl/hr for 28 d. The muscle layers over the laminectomies were sutured, and the skin on the back was closed with surgical staples. To trace CSTs, a burr hole overlying cerebral cortex on the right side was made into the skull 2 weeks after the injury. Biotin dextran amine [BDA; molecular weight (MW), 10,000; 10% in PBS; Invitrogen] was applied at four injection sites at a depth of 0.7 mm from the cortical surface.
Spinal cord lesions and axonal tracing in rats.
Two rat SCI models were used for evaluating axonal growth and recovery after GSK-3 inhibitor treatments. Female Sprague Dawley rats (weight 180–220 g) were deeply anesthetized with ketamine (70 mg/kg) and xylazine (8 mg/kg). For dorsal transection, laminectomy was conducted and spinal cord was exposed at spinal levels of T6–T7. The dorsal part of the cord was cut with a pair of microscissors to sever the dorsal parts of corticospinal tracts. The depth of lesion (∼1.9 mm) was assured by passing the sharp part of a No.11 scalpel blade across the dorsal spinal cord. This model typically transected most of the spinal cord from dorsal entry including dorsal, dorsolateral and lateral CST fibers, but spared some of the ventral CST axons (see Fig. 6B) (Li et al., 2004; Fu et al., 2007). Any rats with incomplete transections in dorsal, dorsolateral, or lateral CST axons were excluded from the study verified by staining BDA-labeled CST fibers from transverse sections caudal to the lesion. A total of 22 rats underwent the dorsal transection injury procedure, 12 receiving vehicle, and 10 receiving lithium. Two rats died in saline group during the first 6 d after SCI because of poor general health. Of the 20 surviving rats, four rats were excluded from the anatomical and behavioral analyses because of the incomplete lesions documented by one to four uninjured dorsal or dorsolateral CST fibers in transverse sections of the caudal spinal cord (three in the control group and one in the lithium group). Thus, seven vehicle and nine lithium-treated animals completed the study and were used for axonal quantification and behavioral analysis in the dorsal transection injury model.
For contusion SCI, the laminectomy was performed at T7–T8 and a moderate lesion was made at T8 with the New York University Impactor. A total of 29 rats were used for the contusion injury procedure, 10 in each of the vehicle and lithium groups and 9 in SB415286 group. One rat in the lithium group was killed 2 d after SCI because of a mild spinal cord injury [Basso, Beattie, and Bresnahan (BBB) locomotor score, 10 on day 2 after SCI]. Thus, 10 animals in vehicle group and 9 in each of lithium and SB groups were used for anatomical and behavioral analysis in the contusion injury model.
The contusion-injured rats received subcutaneous injections of vehicle (saline), lithium (3 mEq/kg/d), or SB (1 mg/kg/d) with syringes (two times per day). This dose of SB has been shown to be protective to injured spinal cord after systemic application in rodents (Cuzzocrea et al., 2006). Treatments were initiated 1 h after spinal cord contusion and continued for 3 weeks. Six weeks after the lesion, burr holes were made on each side of the skull overlying sensorimotor cortex of the lower limbs. Tracer biotin dextran amine (MW, 10,000; 10% in PBS, 3.5 μl/cortex) was applied at five injection sites at a depth of 1.5 mm from cortical surface on each side. Two weeks after BDA injections (8 weeks after SCI), the animals were killed by perfusion with cold PBS, followed by 4% paraformaldehyde. The spinal cord was dissected and postfixed overnight in the same fixative. For in vivo Akt and GSK-3 signal assays, some rats with a contusion injury were perfused with cold PBS (for lysates), or cold PBS plus 4% paraformaldehyde (for sections) 24 h after SCI.
Histology.
The mice with unilateral SCI were perfused transcardially 4 weeks after injury with PBS, followed by 4% paraformaldehyde. After postfixation in the same fixative, the spinal cord 4 mm rostral and 4 mm caudal to the lesion site (8 mm long) was embedded in a glutaraldehyde-polymerized albumin matrix and cut parasagittally on a vibratome (30 μm thick). Transverse sections (40 μm) were collected from the spinal cord 5–7 mm rostral and 5–7 mm caudal to the injury site. To detect the BDA-labeled axons around lesion, the free-floating sections were preincubated with 0.5% BSA/TBS for 1 h and then processed with avidin-HRP (Vector Laboratories), followed by a nickel-enhanced diaminobenzidine reaction (GrandPré et al., 2002). To visualize the lesion area, the longitudinal sections were double-stained with antibodies directed against GFAP (Sigma). The sections were mounted, dehydrated, and covered with mounting medium.
The rat spinal cord tissue 10 mm rostral to and 10 mm caudal to the lesion site (20 mm long, containing lesion area) was cut parasagittally on a vibratome. Transverse sections were collected from the spinal cord 11–15 mm rostral to and 11–15 mm caudal to the injury site. After 1 h preincubation with 0.5% bovine serum albumin and 1% Triton in Tris-buffered saline, sections were incubated with avidin-biotin-peroxidase complex in Tris-buffered saline with 0.1% bovine serum albumin, 0.5% Triton for 3–4 d at room temperature. The BDA tracer was visualized by nickel-enhanced diaminobenzidine HRP reaction. To visualize serotonergic fibers and lesion area, all of the sagittal sections and some transverse sections were double stained with antibodies against 5-HT and astrocytic marker GFAP. The microscopic slices were pasted on glass slides and analyzed with a Nikon microscope. For CST quantification in the sagittal sections containing the lesion site, the BDA-labeled fibers outside of ventral CST area were counted at various distances caudal to the lesion center from all of the sagittal sections in each rat if the length of an individual fiber or the longest axis of a sprouting complex (a group of disorganized axonal sprouts) was >100 μm. For CST fiber quantification from transverse sections, the averaged number of BDA-labeled axons was obtained from three random sections at 11–15 mm caudal to the lesion site in each animal. To determine serotonergic fibers at the spinal cord 11–15 mm caudal to the lesion site, the individual fibers stained by 5-HT antibody in the ventral and dorsal half of spinal cord were traced and the mean density of traced 5-HT fibers was calculated from two random transverse sections in each rat using NIH software.
Behavioral tests.
BBB locomotor scale, grid walking, and footprint analysis were used to evaluate functional recovery in SCI rats. For grid walking, the rats were trained to walk on a wire grid (90 cm long, 25 mm between grids) before surgery. Six weeks or eight after injury, grid walk was videotaped and the number of instances in which the hindpaw dropped below the grid plane was counted. For footprint analysis, the walking patterns of hindpaws were recorded with ink during a continuous locomotion across a 90 cm runway at 6 (dorsal transection) or 8 (contusion) weeks after SCI. The stride length on each side and stride width between two sides were calculated (Metz et al., 2000a; Li et al., 2004; Fu et al., 2007). In this study, statistical analyses were performed using Student's t test. The differences with p < 0.05 were considered significant between the control and drug groups.
Results
Axonal growth inhibitors transiently inactivate Akt and activate GSK-3β in neuronal cultures
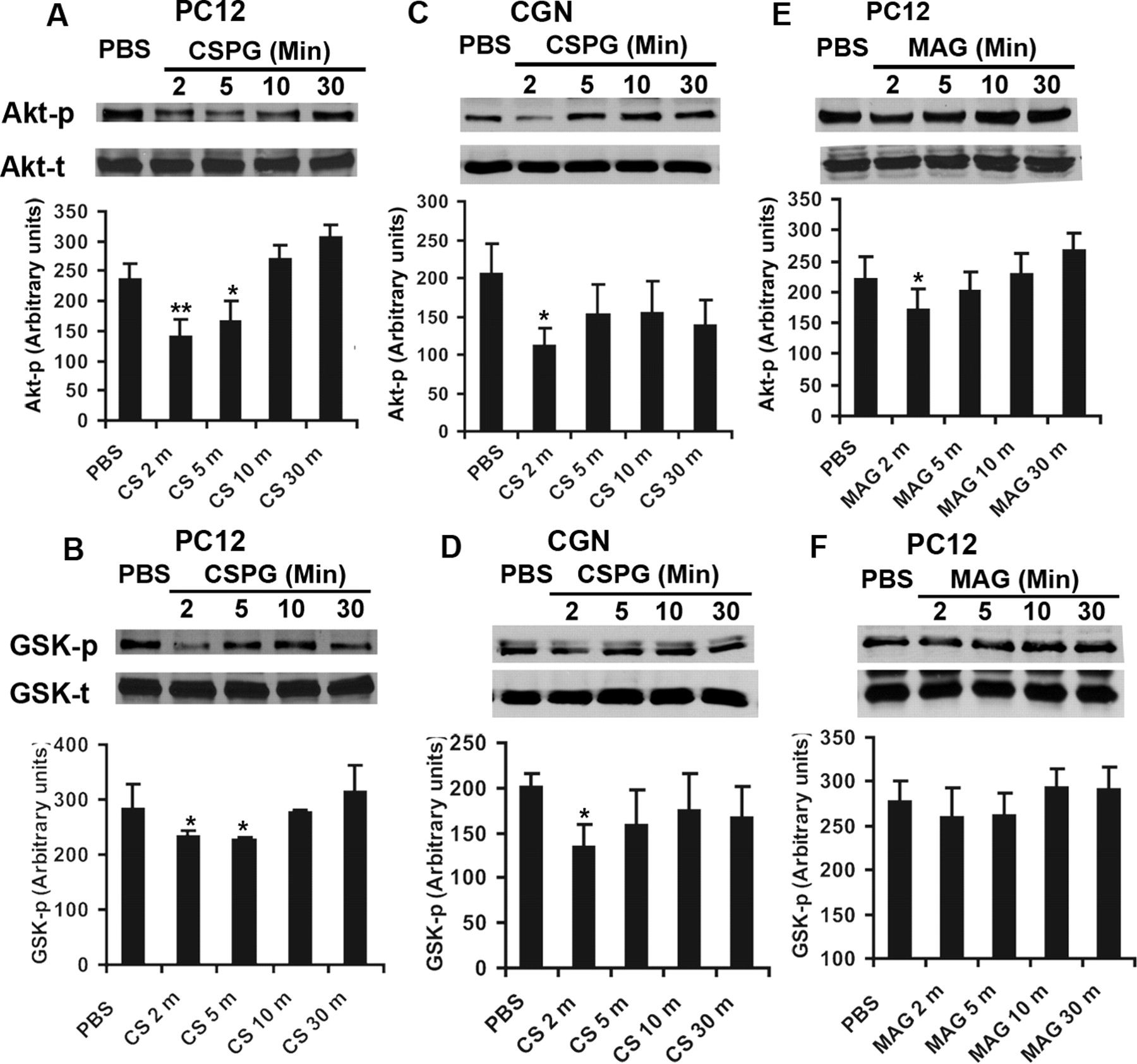
CSPGs from astroglial scars and several proteins in CNS myelin partially mediate axonal growth inhibition via intracellular ILK and/or RhoA signals (McGee and Strittmatter, 2003; Monnier et al., 2003; Jain et al., 2004; Fu et al., 2007). Considering the potential role of ILK and RhoA in regulating Akt and GSK-3β activities in neurons (Eickholt et al., 2002; Sayas et al., 2002; Zhou et al., 2006; Guo et al., 2007), we tested whether CNS inhibitory molecules alter the activities of GSK-3β and its upstream modulator Akt. Measurement of phosphorylated Akt and GSK-3β has been widely used to determine their activities (Sayas et al., 2002; Zhou et al., 2004, 2006; Chadborn et al., 2006). Phosphorylated Akt at residue Ser473 (Akt-p) in its C-terminal hydrophobic motif is necessary for full activation of Akt. The cellular levels of Akt-p at Ser473 correlate with the activity of this kinase. GSK-3β, however, is constitutively active in neurons under resting conditions and is inactivated by extracellular signals through phosphorylation of an N-terminal serine residue Ser-9. The levels of phosphorylated GSK-3β at Ser9 (GSK-p) negatively correlate with GSK-3β activity in neurons (Zhou et al., 2004). Thus, we examined the levels of Akt-p and GSK-p in the lysates of cultured PC12 cells exposed to axon growth inhibitors. After 24 h differentiation with NGF, PC12 cells were treated with CSPGs (1.5 μg/ml) or with myelin inhibitor MAG for 2, 5, 10, and 30 min. The levels of Akt-p and GSK-p in lysates were detected via Western blot. Application of CSPGs at 1.5 μg/ml induced significant reduction of phosphorylated Akt and GSK-3β at 2–5 min after exposure (Fig. 1A,B). To confirm this finding in CNS neurons, we determined the levels of these phosphorylated proteins in primary cultured CGNs, a type of central neuron derived from postnatal 7–9 d mice. We detected the similar reductions of Akt-p and GSK-p levels after CSPG incubations (Fig. 1C,D). Myelin protein MAG has been shown to suppress axonal growth by binding to neuronal membrane Nogo receptors or membrane surface gangliosides (Domeniconi et al., 2002; Liu et al., 2002; Vyas et al., 2002). Similarly, we applied MAG to cultured PC12 cells and found a degree of Akt-p reduction 2 min after exposure although it did not significantly alter GSK-p levels (Fig. 1E,F). Our results suggest that the major CNS axonal inhibitors, particularly CSPGs, suppress Akt but activate GSK-3β signals in neurons. These findings support the potential role of Akt/GSK-3β signaling pathways in regulating growth cone collapse of neurons after exposure to CNS axon growth inhibitors.

Axon growth inhibitors transiently alter the activities of Akt and GSK-3β signals in cultured neuronal cells. A, B, Western blot indicates the levels of phosphorylated Akt (Akt-p, active form) and GSK-3β (GSK-p, inactive form) in the lysates of PC12 cells 24 h after differentiation with NGF (50 ng/ml). Application of inhibitor CSPGs (CS, 1.5 μg/ml) significantly reduced the levels of Akt-p (A) and GSK-p (B) 2–5 min after exposures, although the total protein levels (Akt-t, GSK-t) are similar in these lysates. The graphs at bottom indicate the level of Akt-p and GSK-p in PC12 lysates quantified from several separate experiments. C, D, Western blot indicates the levels of Akt-p and GSK-p in the lysates of CGNs 24 h after cultures derived from P7–P9 mice. Similarly, CSPGs (1.5 μg/ml) significantly attenuated the levels of Akt-p (C) and GSK-p (D) a few minutes after exposures. E, F, The levels of Akt-p and GSK-p in the lysates of PC12 cells 24 h after differentiation with NGF (50 ng/ml) were determined from PC12 cell lysates after application of myelin inhibitor MAG (570 ng/ml). MAG moderately reduced the levels of Akt-p (E), although it did not significantly affect levels of GSK-p after exposure. The bar graphs indicate means ± SEM from four to six separate experiments. The differences indicated are compared with the PBS-treated controls without CSPGs or MAG (*p < 0.05, **p < 0.01, Student's t test). CS, CSPG.
GSK-3β inactivation promotes neurite outgrowth from cultured neurons on inhibitory substrates
Neurite growth assay is one of the most frequently used in vitro models for studying axonal growth in neurons (Fournier et al., 2001; GrandPré et al., 2002; Fu et al., 2007). We next examined the effect of GSK-3β inactivation on neurite outgrowth from cultured DRG neurons exposed to CNS axon inhibitors. As expected, purified CNS myelin and CSPGs remarkably attenuated the number and length of neurites in dissociated DRG neurons derived from adult mice 24 h after growth. Treatments with lithium chloride (3 mm) or SB415286 (7.5 μm), two well known inhibitors for GSK-3β, significantly increased the neurite length of DRG neurons when cultured on these inhibitory substrates (Fig. 2A–C). To explore the role of GSK-3β activation in limiting axon elongation in central neurons, we assessed the neurite growth from cultured cerebellar granular neurons derived from P7–P9 mice, a model widely used for studying axon growth in vitro (Wang et al., 2002; Hata et al., 2006). Consistently, CGNs treated with lithium or SB415286 dramatically overcame the growth suppression of CNS myelin and CSPGs 24 h after cell plating (Fig. 2D–F). Lithium is a direct and indirect noncompetitive inhibitor of GSK-3β (Stambolic et al., 1996; Ryves and Harwood, 2001). SB415286 is a potent and competitive specific inhibitor of GSK-3β (Coghlan et al., 2000). Although both inhibitors inactivate GSK-3β via different mechanisms, they induced a similar degree of neurite extension on axon inhibitors, suggesting that inhibiting GSK-3β rather than other enzymes mediates the growth-enhancing activity detected here. Our findings support that GSK-3β activation contributes to the neurite growth suppression of CNS inhibitory components and that GSK-3β inactivation considerably overcomes the growth repression of these CNS inhibitors. Our results are consistent with the reports that GSK-3β activation causes growth cone collapse and axon formation suppression in neuronal cultures (Zhou et al., 2004; Jiang et al., 2005; Chadborn et al., 2006). The detected beneficial effects of GSK-3 inhibitors might be attributable to the suppression to GSK-3β plus GSK-3α given the high sequence identities in their kinase domains (>98%) and the abundant distribution of both isoforms in axons (Woodgett, 1990; Kim et al., 2006).

GSK-3 inhibitors stimulate neurite outgrowth from cultured DRG neurons or CGNs on CNS myelin or CSPG substrates. A, The representative examples of dissociated DRG neurons from 7–10 week mice were treated with lithium (Li; 3 mm), SB (7.5 μm), or vehicles (Veh) starting after cell plating on PBS, myelin (Mye; 50 μg/ml), or CSPG (1.5 μg/ml) substrates. Cells were fixed and stained with rhodamine phalloidin 24 h after growth. Lithium and SB415286 stimulated neurite growth on myelin or CSPGs. B, C, The neurite outgrowth per DRG neuron was manually traced and quantified 24 h after cell plating on CNS myelin (B) or CSPG (C) substrates using NIH and Photoshop software. The bar graphs indicate means ± SEM from five to nine separate experiments (30–80 representative neurons were analyzed from 7 images in each experiment). The differences indicated are compared with the vehicle-treated controls. Treatments with Li or SB significantly increased neurite length of DRG neurons on CNS myelin and CSPGs 24 h after cell plating. D, The representative examples of dissociated CGNs derived from P7–P9 mice were treated with lithium (3 mm), SB415286 (7.5 μm), or vehicles starting after cell plating on PBS, myelin (50 μg/ml), or CSPG (1.5 μg/ml) substrates. The cells were fixed and stained with rhodamine phalloidin 24 h after growth. E, F, The neurite outgrowth per neuron was quantified 24 h after cell plating on CNS myelin (E) or CSPGs (F). The numbers in bar graphs indicate means ± SEM from six to eight separate experiments (100–150 representative neurons were analyzed from 7 images in each experiment). The differences indicated are compared with the vehicle-treated controls (*p < 0.05, **p < 0.01, Student's t test). Treatments with Li or SB significantly increased neurite length of CGNs cultured on CNS myelin and CSPGs. Scale bars, 50 μm.
GSK-3β inactivation enhances axon formation and elongation in DRG cultures derived from adult mice
During development, a neuron establishes neuronal polarity usually by forming a single axon and multiple dendrites, which are important for sending and receiving signals, respectively. Inactivation of GSK-3β has been shown to stimulate neuronal polarization by converting the dendrites of developing neurons into axons. Thus, we wondered whether inhibition of GSK-3β would promote axon formation and extension in mature neurons derived from adult rodents. Neuronal cultures derived from embryonic rodents have been frequently used to study axonal formation and growth (Zhou et al., 2004; Jiang et al., 2005). However, analysis of in vitro axogenesis and axon growth in mature neurons derived from adult mammals is more relevant to the repair of CNS axonal injuries in patients. We cultured dissociated DRGs from adult mice (7–9 weeks), which normally send ascending central axons into spinal cord in addition to peripheral nerves into other tissues. Surprisingly, a portion of DRG neurons formed one to three axons (1.6 axons/neuron on average) with several branches (approximately seven branches per axon) 4 d after culture. Given that DRGs, the only adult neurons that can be readily cultured to study axon growth, are very sensitive to various CNS axon-growth inhibitors and physiologically possess both central and peripheral axons, this culture system could become a reasonable model for studying CNS axonal regeneration in vitro. Indeed, adult DRG cultures have been used to study axonal growth in response to CNS inhibitors (Zhou et al., 2006), although the central axons could not be distinguished from peripheral branches in vitro. Thus, we tested axonal formation and elongation after pharmacological inactivation of GSK-3β using this model. In the vehicle-treated controls, ∼40% of DRG neurons derived from adult mice formed axonal structures after 4-day growth on laminin-coated coverslips in presence of NGF (50 ng/ml) (Fig. 3A,B). However, treatment with GSK-3β inhibitor lithium (3 mm) or SB415286 (7.5 μm) significantly increased the number of axon-forming neurons to 59% (p < 0.01). Moreover, GSK-3β inhibitors significantly increased the length of axons in each axon-forming neuron although they only slightly increased the number of branches in each axon (Fig. 3C,D). Our results suggest that GSK-3β inhibition mimics the neurotrophic effects of positive factors or enhance the growth capability of adult neurons by promoting axon formation and extension.

GSK-3β inhibitors promote axonal formation and enhance axonal length in DRG neuronal cultures derived from adult mice. A, The representative examples of DRG neurons with axons cultured from 8 wk C57BL/6 mice 4 d after cell plating. The neurons were treated with vehicle (Veh; saline), lithium (Li; 3 mm), or SB (7.5 μm) in absence (top row) or presence (bottom row) of purified CNS myelin (20 μg/ml). After fixation, the cells were stained with axonal marker neurofilament (arrows). B–D, Bar graphs indicate the percentage of axon-forming neurons (B), the length of axons in each neuron with axons (C), and branching numbers in each axon (D) of DRG cultures without exposure to CNS myelin axon inhibitors. E–G, Bar graphs indicate the percentage of axon-forming neurons (E), the length of axons in each axon-forming neuron (F), and branching numbers of each axon (G) in DRG cultures in presence of CNS myelin. The means ± SEM are reported from five to seven separate experiments. Multiple images (7–8) were collected and quantified from an individual experiment in each group (*p < 0.05, **p < 0.01 compared with vehicle-treated cultures, Student's t test). Treatments with lithium (3 mm) or SB (7.5 μm) significantly enhanced the number of axon-forming neurons and the length of axons in axon-bearing neurons on CNS myelin (E, F). H, Synaptic marker synaptophysin was visualized via immunostaining in DRG neurons derived from postnatal 2 d mice 5 d after cell plating. GSK-3 inhibitors enhanced the expression of synaptic structural protein in neuronal bodies (arrows) as well as in axonal cylinders (arrowheads). Scale bars, 50 μm.
We next determined whether GSK-3β suppression could stimulate axonal formation and elongation in mature neurons in presence of CNS growth inhibitors. Toward this end, we characterized the growth of adult DRG neurons cultured on coverslips coated with purified CNS myelin. CNS myelin remarkably restricted axon initiation by reducing the number of axon-forming neurons and axon extension by decreasing axon length 4 d after growth. However, treatments with lithium (3 mm) or SB (7.5 μm) promoted axon initiation and elongation on CNS myelin by significantly recovering the number of axon-forming neurons and the length of axons in the axon-bearing neurons (Fig. 3E,F). GSK-3β inhibitors did not dramatically alter the number of axons per neuron (data not shown) or the number of branches in each axon (Fig. 3G). These results demonstrate that GSK-3β activation negatively regulates axonal genesis and elongation in adult neurons in presence or absence of nonpermissive molecules, suggesting that GSK-3β repression is effective in promoting axon growth in mature neurons.
Reestablishing synaptic connections is essential for functional recovery during axonal regeneration. Clustering of synaptophysin, a marker for presynaptic differentiation, can occur in the absence of postsynaptic targets during axonal remodeling (Fletcher et al., 1991). We examined expression of the presynaptic structural protein synaptophysin in DRG neurons derived from adult or postnatal day 2 mice. In controls treated with vehicle, only a small number of neurons exhibited faint staining for synaptophysin in somatic region after 5–6 d in vitro (Fig. 3H). In contrast, a number of neurons treated with GSK-3 inhibitors displayed higher levels of staining signal for synaptophysin along axons as well as in neuronal bodies. This finding suggests that GSK-3β inhibition not only stimulates axonal formation and extension, but also promotes presynaptic structure formation. Consistently, GSK-3 inhibition with lithium or stimulation with WNT-7a, an axonal guidance cue that suppresses GSK-3, has been shown to enhance the expression of presynaptic protein synapsin I during axon remodeling in immature cerebellar neuronal cultures (Lucas and Salinas, 1997; Hall et al., 2000).
Systemic application of lithium represses GSK-3β activity in the lesioned rat spinal cord
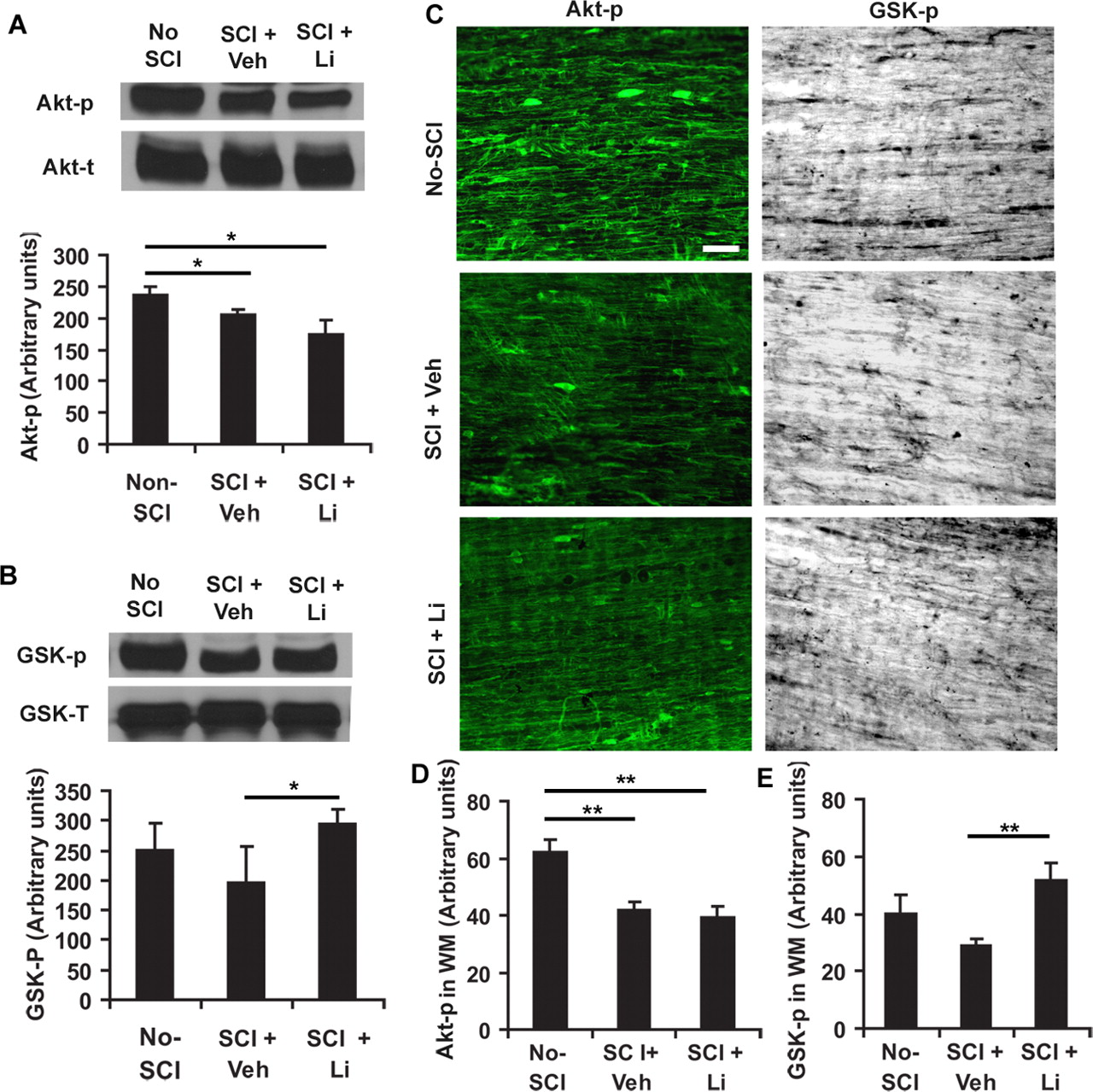
To determine endogenous alterations of Akt and GSK-3β activity after a CNS traumatic injury, we examined the levels of phosphorylated Akt Ser-473 and GSK-3β Ser-9 in fresh spinal cord lysates or in fixed tissue sections 24 h after a contusion SCI at T8 in rats. Compared with levels in normal spinal cord at T7–9, the Akt-p levels in the injured spinal cord (3 mm rostral and caudal to the lesion) were modestly decreased 24 h after lesion in vehicle- or lithium-treated rats (Fig. 4A). The GSK-p levels in lesioned spinal cord were similar between normal and SCI rats treated with vehicle, but the SCI rats treated with systemic lithium (3 mEq/kg/d) exhibited higher GSK-p levels at the same spinal cord levels than vehicle-treated SCI controls 24 h after SCI (Fig. 4B). Further characterization of Akt-p and GSK-p levels in the lysates of spinal cord 4–10 mm rostral to the lesion center demonstrated a similarly altered pattern of Akt and GSK-3β phosphorylation 24 h after injury (data not shown).

Traumatic injury attenuated Akt activity and treatment with lithium (Li) suppressed GSK-3β activity by enhancing GSK-p level in the lesioned rat spinal cord. A, B, Akt-p and GSK-p were analyzed from the tissue lysates of fresh spinal cord 3 mm rostral to and 3 mm caudal to a lesion 24 h after injury at T8 (or the same level of spinal cord in no-SCI controls) via Western blot with antibody specific to phosphorylated Akt at Ser473 (active) and GSK-3β at Ser-9 (inactive). Total Akt and GSK-3β from the same tissue samples were detected via immunoblotting (bottom bands). Bar graph below bands indicates the densitometric analysis of Akt-p and GSK-p levels in the lesioned spinal cord (means ± SEM; n = 4 from 4 rats in each group). C, The levels of Akt-p and GSK-p were examined from the fixed sagittal sections of spinal cord white matter (2–4 mm from lesion) at midthoracic level from three groups of rats 24 h after SCI. Both Akt-p and GSK-p were visualized via immunostaining against these proteins. The signal for Akt-p was attenuated in spinal cord around lesion compared with noninjury controls at the same spinal cord level. Lithium treatment enhanced GSK-p signal. Scale bar, 50 μm. D, E, Akt-p and GSK-p signals were quantified from white matter of spinal cord sections 2–4 mm rostral to and caudal to lesion (means ± SEM; n = 5–7 from 5–7 images in each group; *p < 0.05, **p < 0.01 compared with non-SCI for Akt-p or with SCI controls for GSK-p, Student's t test).
To visualize alterations of Akt and GSK-3β activity in projecting axons of spinal cord, we examined the cellular distributions of Akt-p and GSK-p in white matter areas via immunohistochemistry 24 h after SCI in rats. Immunoreactive signals for Akt-p and GSK-p were detected in glial cells as well as axonal cylinders (Fig. 4C). Consistently, we found reduced levels of Akt-p 24 h after contusion SCI (Fig. 4D). The injury did not significantly alter GSK-p levels compared with normal controls, but treatment with lithium considerably increased GSK-p signal compared with vehicle-treated SCI controls (Fig. 4E). Thus, traumatic injury modestly attenuates Akt activity in the lesioned spinal cord 1 d after injury, but treatment with systemic lithium significantly suppresses GSK-3β activity by enhancing the levels of GSK-p (inactive form of GSK-3β) around the lesion, including along white matter tracts.
Systemic lithium stimulates corticospinal tract fiber sprouting rostral to lesion in mice with unilateral transection injury
Given the inhibiting effect of GSK-3β on axonal extension in vitro, GSK-3β inactivation via pharmacologic approach may enhance axonal growth after a CNS injury in vivo. We tested this hypothesis using three SCI models in rodents in this study. Previously, we demonstrated that blockade of myelin inhibitory protein Nogo with its antagonists or deletion of Nogo in knock-out mice stimulates significant CST fiber sprouting rostral to a transection injury in the mouse spinal cord (Kim et al., 2003; Li and Strittmatter, 2003; S. Li et al., 2005). GSK-3 inhibition may have a similar effect on CST sprouting rostral to a SCI given that GSK-3 antagonists overcome growth restriction of CNS axon inhibitors (Figs. 2, 3). Thus, we first systemically delivered lithium, a well-known GSK-3β inhibitor clinically used as a mood stabilizer, to mice with a unilateral transection injury at T7 (Fig. 5A) via osmotic pumps for 4 weeks (3 mEq/kg/d). The integrity of CST fibers on the lesioned half of spinal cord (left site) was traced by injecting BDA into the right motor cortex. In saline-treated controls, a few BDA-labeled collateral sprouts projected into gray matter in transverse sections 5–7 mm rostral to the lesion. In contrast, a higher density of BDA-labeled fibers outside of dorsal CST were present in lithium-treated samples at the same level (Fig. 5B–E). Tracing of individual CST axonal sprouts outside of predominant CST indicates a greater number of CST sprouts in the gray matter area in lithium-treated mice (6524 ± 1056 μm per transverse section vs 4415 ± 295 μm in vehicle-treated controls; p < 0.05, Student's t test). Longitudinal sections including the lesion site display the projection of some CST sprouts into the lesion center (data not shown) compared with termination of CST axons without obvious sprouting in SCI controls (Kim et al., 2003; Li and Strittmatter, 2003). However, most CST sprouts failed to extend into the caudal spinal cord because transverse sections of lumbar spinal cord (∼6 mm caudal to lesion) exhibit only a few BDA-labeled fibers in lithium-treated mice (data not shown) compared with no BDA-labeled axons in the controls. The failure of CST sprouts to project into the caudal spinal cord is probably attributable to growth barriers from scar tissue around the lesion and from midline structures, such as the anterior commissure, and expression of repulsive ephrin-B3 at midline spinal cord (Kullander et al., 2001; Benson et al., 2005). Our result suggests that lithium treatment stimulates CST growth rostral to a transection, although these sprouts mostly failed to cross the growth barriers in this model system.

Systemic application of lithium stimulates corticospinal fiber sprouting rostral to a unilateral transection in mouse spinal cord. A, Schematic of transverse spinal cord illustrates the transected area of cord in mice. The small arrow indicates the dorsal CST area labeled with BDA. B–E, BDA-labeled dorsal CST axons are visualized in transverse sections 5–7 mm rostral to SCI in vehicle- (B, C) or lithium (Li)-treated (D, E) animals. A few BDA-labeled sprouts (arrowheads) project into the gray matter in controls (dorsal is up). Treatment with lithium for 4 weeks exhibits a higher density of CST sprouts extending laterally into gray matter 4 weeks after SCI. Scale bars: A, 200 μm; (in B) B, D, 50 μm; (in B) C, E, 25 μm.
GSK-3β inhibitors stimulate axonal growth of CST fibers in rats with a dorsal transection or a contusion SCI
To further characterize the role of GSK-3β activation in limiting axonal growth along descending tracts in vivo, we examined GSK-3β inhibitor lithium on the integrity of CST fibers in rats after a dorsal transection. This model is widely used for axonal regeneration and functional recovery studies (Huang et al., 1999; GrandPré et al., 2002; Rosenzweig and McDonald, 2004; Fu et al., 2007) because it interrupts a group of defined pathways and provides the tissue bridging for regenerating axons in the ventral spinal cord. After a lesion in the controls (GrandPré et al., 2002; Li et al., 2004; Fu et al., 2007), the dorsal CST fibers are disrupted and only partial ventral CST axons are detectable in the caudal spinal cord by BDA labeling. No obvious CST sprouts are seen in either rostral or caudal spinal cord (Fig. 6A,C). In contrast, treatment with lithium induced numerous CST fiber sprouts rostral to the lesion and many BDA-labeled fibers were observed in the caudal spinal cord outside of ventral CST area (Fig. 6D–G), particularly in the gray matter areas. Counts of CST fibers ≥100 μm in sprout length from sagittal sections demonstrate a greater number of CST axons in the caudal spinal cord in lithium-treated rats (81.1 ± 19.6 axons per rat at 1–8 mm caudal to SCI) than in saline-treated controls (6.2 ± 2.5 axons per rat at the same spinal cord level; p < 0.01, Student's t test). Examination of transverse spinal cord sections at the more caudal level (11–15 mm caudal to the lesion) revealed more BDA-labeled CST axons in ventral and dorsal spinal cord in the rats treated with lithium (Fig. 6H,I).

Systemic application of GSK-3 inhibitors promotes CST axonal growth around lesion in rats with dorsal transection or contusion injuries. A, C, Sagittal sections in SCI controls show no regenerative CST growth in rostral and caudal spinal cord 6 weeks after a dorsal transection injury at T7. B, Schematic drawing illustrates transverse spinal cord and transected area (shaded) in rat dorsal transection model. D, Similar section from a lithium (Li)-treated rat indicates a number of CST fiber sprouts rostral and caudal to the transection lesion, particularly in the gray matter (GM) areas (the dorsal is up in all of these sections). E, F, Higher-magnification images from D demonstrate the meandering course of CST sprouting fibers (arrowheads) around lesion. G, Quantification of CST fibers ≥100 μm in fiber length or in longest axis of sprouting complex outside of ventral CST is illustrated at various distances from longitudinal sections caudal to the transection lesion (mean ± SEM; n = 7, 9 rats in the control and lithium groups; *p < 0.05, Student's t test). H, Transverse sections of spinal cord 11–15 mm caudal to the transection lesion from vehicle-treated rats indicated a few spared ventral CST axons (arrowhead), but few BDA-labeled CST fibers were found in the dorsal part of spinal cord. Transverse sections of spinal cord at same level from lithium-treated rats displayed tracer-labeled CST axons in both ventral and dorsal part of spinal cord (arrowheads). I, Counting of BDA-labeled CST fibers from transverse sections 11–15 mm caudal to the transection lesion indicates more BDA-labeled fibers in the lithium group (mean + SEM; n = 7, 9 rats in the control and lithium groups, *p < 0.05, Student's t test). J, A parasagittal section containing the injury site (arrow) from a lithium-treated rat demonstrates the lesion area after a contusion injury. A large cavity is visualized in the lesion epicenter of the spinal cord. K, L, Bar graphs indicate quantification of CST fibers ≥100 μm in sprouting length from longitudinal sections 1–10 mm caudal to a moderate contusion lesion 8 weeks after injury in the vehicle- lithium-, and SB-treated rats. Treatments with lithium or SB significantly increase the CST fiber number caudal to the contusion. WM, White matter. M, Counting of BDA-labeled CST fibers from transverse sections 11–15 mm caudal to the contusion lesion displays more BDA-labeled fibers in the lithium or SB group 8 weeks after SCI (mean ± SEM; n = 10, 9 and 9 rats with contusion SCI in three groups, respectively; *p < 0.05, **p < 0.01, Student's t test). Scale bars: (in A) A, D, J, 500 μm; B, 200 μm; (in C) C, E, F, 100 μm; H, 20 μm.
Although dorsal transection is frequently used for studying in vivo axonal regeneration, this model is different from spinal cord injuries in most SCI patients. Thus, we further evaluated the effect of GSK-3 suppression on axonal growth after a moderate rat contusion injury at T8. This model produces a lesion mimicking the complex lesions of many SCI patients (Metz et al., 2000b), including remarkable central gray matter damage and variable white matter injuries. Considering the possible effects of lithium on enzymes other than GSK-3, we also systemically applied SB415286, a selective GSK-3 inhibitor, to the rats with contusion SCI. To provide more valuable information for treatment of SCI patients, we delivered these GSK-3 inhibitors via systemic bolus injections with syringes for 3 weeks after SCI instead of the continuous infusion with implanted Alzet pumps for 4 weeks. Eight weeks after SCI, parasagittal sections across the lesion in three groups displayed a large cavity in the lesion epicenter surrounded by numerous small cavitations and scar tissue (Fig. 6J) (Fu et al., 2007). The lesion damaged almost all of the gray matter and most of the white matter in the spinal cord. Rostral to the lesion, the linear projecting CST axons terminated at a few hundred micrometers to several millimeters above the lesion in the vehicle-treated rats. In contrast, equivalent sections from lithium- or SB-treated animals display some of BDA-labeled CST fibers extending into the lesion area. Caudal to the lesion, very few BDA-labeled CST fibers were observed in SCI controls, suggesting that this model disconnected almost all of the CST axons around the lesion. In contrast, a number of BDA-labeled fibers are detectable in the lithium- and SB-treated animals (Fig. 6K,L). Counts of BDA-labeled fibers ≥100 μm in sprout length from all of the sagittal sections per rat 1–10 mm caudal to the lesion indicate significant increase of CST fibers in the inhibitor-treated rats than in the SCI controls. Evaluation of BDA-labeled axons from transverse spinal cord sections 11–15 mm caudal to the lesion also revealed more CST fibers in the rats treated with lithium or SB (Fig. 6M). These increased caudal CST fibers in GSK-3 inhibitor-treated rats might derive from the sprouting of spared CST axons or the regenerating CST fibers from lesioned axons after GSK-3 inactivation.
GSK-3 inhibitors promote serotonergic axonal growth in injured rat spinal cord
A few descending axonal pathways contribute to the extent of behavioral recovery after SCI, including descending raphespinal fiber tracts (Kim et al., 2004; Li et al., 2004). Thus, we characterized serotonergic fibers from transverse sections in the SCI rats treated with vehicle or GSK-3 inhibitors via immunostaining with an antibody against 5-HT. Rostral to the lesion, a high density of 5-HT fibers is detected in the transverse sections of spinal cord, particularly in ventral horns and central and intermediolateral areas (Fig. 7A,B). Six weeks after the dorsal transection injury, the density of 5-HT-positive fibers was dramatically reduced in the caudal spinal cord compared with the sections rostral to the lesion (Fig. 7C,D). However, in samples from injured rats treated with lithium, the density of 5-HT fibers in the caudal spinal cord was significantly increased compared with the SCI controls (Fig. 7E–G), although the density of 5-HT fibers 11–15 mm rostral to the lesion was almost identical between two groups (42.5 + 4.5 vs 42.4 + 2.5 mm/section; n = 7 and 9 rats in the control and lithium groups, respectively).
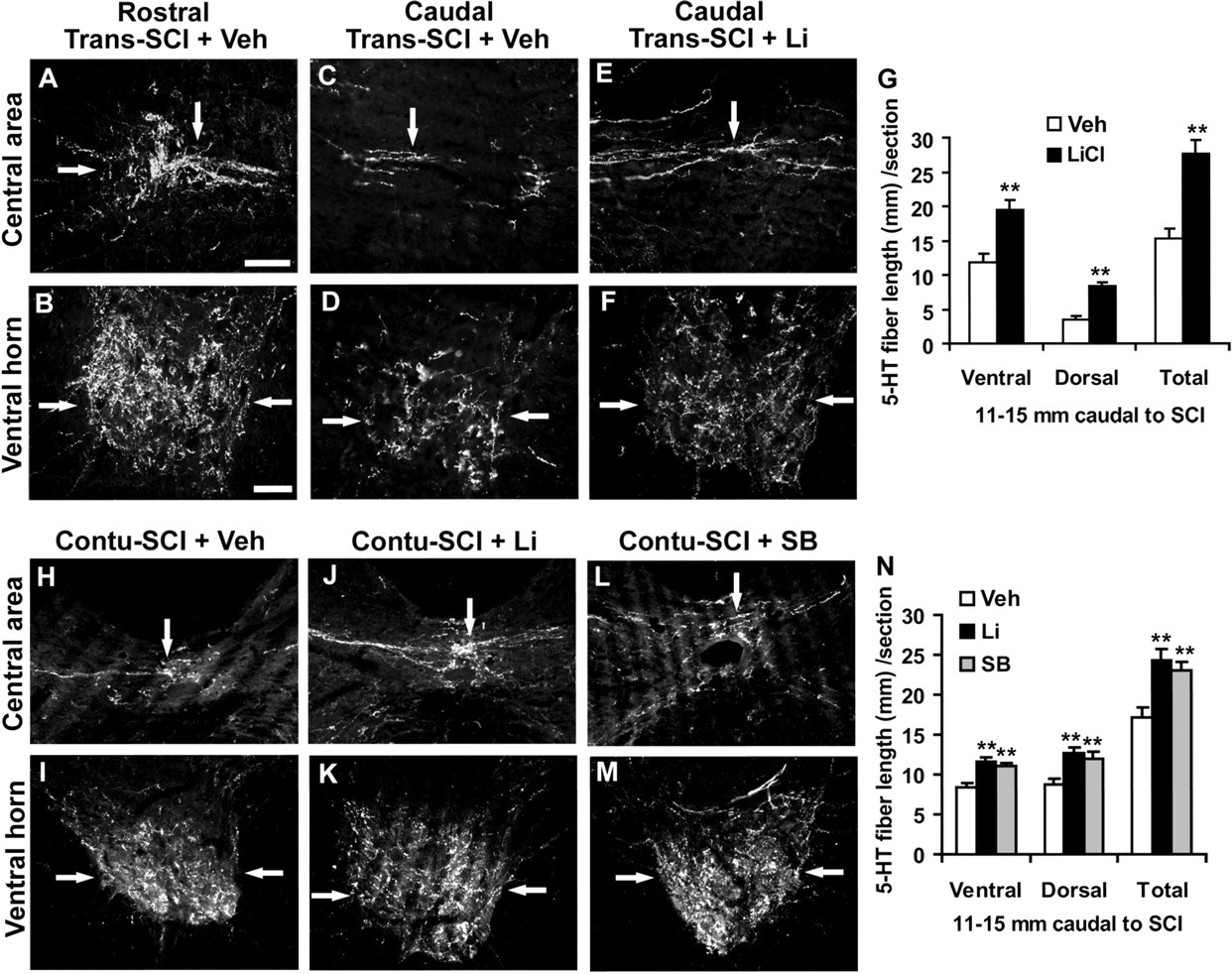
Systemic GSK-3 inhibitors enhance serotonergic fibers in the spinal cord 11–15 mm caudal to a dorsal transection or a contusion injury in rats. A, B, Transverse sections of central and ventral spinal cord 11–15 mm rostral to SCI reveal the high density of serotonin fibers (arrows) in SCI-control rats. C–F, Transverse sections of spinal cord 11–15 mm caudal to the lesion displayed the reduced 5-HT fibers (arrows) 6 weeks after the transection injury at T7 (Trans-SCI), but treatment with lithium increased serotonergic fibers (arrows) in both central and ventral part of spinal cord (E, F) compared with those from vehicle-treated rats (C, D). G, Serotonin fiber length was measured from gray and white matter in dorsocentral areas and from gray matter in ventral horn of the spinal cord 11–15 mm caudal to transection lesion. The means ± SEM from 7 and 9 rats in control and lithium groups are reported. H–M, Transverse sections of spinal cord 11–15 mm caudal to the lesion displayed the reduced 5-HT fibers (arrows) 8 weeks after a contusion injury at T8 (Contu-SCI). Treatments with lithium or SB increased serotonergic fibers (arrows) in both central and ventral part of the spinal cord (J–M) compared with those from vehicle-treated rats (H, I). Scale bars: A (for A, C, E, H, J, L), 50 μm; B (for B, D, F, I, K, M), 50 μm. N, Serotonin fiber length was measured in dorsocentral and ventral areas of spinal cord 11–15 mm caudal to the contusion lesion. The means ± SEM from 10, 9 and 9 rats with contusion SCI in control, lithium, and SB groups are reported. **p < 0.01, Student's t test.
Moreover, serotonergic fibers were examined in rostral and caudal spinal cord in the rats with contusion SCI. Similar to dorsal transection, the contusion injury damaged most of serotonergic fibers at the site of injury. However, treatment with systemic lithium or SB dramatically increased the number of 5-HT axons in the spinal cord 11–15 mm caudal to SCI (Fig. 7H–N), although the lesion areas visualized by GFAP staining were generally similar in vehicle- or inhibitor-treated rats (data not shown). Thus, treatments with systemic GSK-3 inhibitors promote the regrowth of both corticospinal and raphespinal axons after a dorsal transection or contusion SCI. The increased 5-HT fibers in the caudal spinal cord might be the result of collateral sprouting in the caudal spinal cord from spared fibers or long distance axonal growth from severed axons.
Treatment with GSK-3 inhibitors enhances locomotor recovery in SCI rats
The anatomical analyses demonstrate increased axonal growth of descending fiber tracts after treatments with systematic GSK-3 inhibitors. Next, we evaluated whether treatment with GSK-3 inhibitors would promote functional recovery several weeks after SCI in these rats using several behavioral tasks. The standardized rat locomotor BBB scores were used to assess the outcome (Basso et al., 1995; GrandPré et al., 2002; Fu et al., 2007). The rats usually show no observable movement or slight-extensive movement of one joint (BBB score, 0–2) 2 d after a dorsal transection injury (Fig. 8A). A few weeks after dorsal transection injury, the control rats exhibit partial recovery, probably because of the reorganization of segmental circuitry including propriospinal reconnections (Courtine et al., 2008) and the short-range sprouting from spared fibers. The recovery reaches a stable level by 3–4 weeks after SCI. Generally, the control rats have consistent weight-supported plantar steps, frequent or consistent forelimb–hindlimb (FL–HL) coordination, but rotate the predominant paw position when making the initial contact with a testing surface. In contrast, the locomotor BBB score in the rats treated with lithium initiated 1 h after injury continues to increase 4–6 weeks after SCI and the majority of them have consistent FL–HL coordination plus parallel paw position at initial contact with the testing surface (Fig. 8A).

Treatments with GSK-3 inhibitors improve the behavioral recovery after a dorsal transection or a moderate contusion injury in rats. A, Graph indicates the locomotor BBB score in the vehicle or lithium-treated rats after a dorsal transection injury (n = 7, 9 rats in the control and lithium groups; **p < 0.01, Student's t test). B, The examples of representative footprints from control (top) and lithium (Li, bottom) animals after a dorsal transection injury. The double-end arrows indicate the stride distances measured from the footprints. C, Footprint analysis revealed a greater stride length and a shorter stride width in lithium-treated animals than in controls. The grid-walk and footprint tests were performed 6 weeks after the dorsal transection injury. D, E, Graphs indicate BBB score (D) and grid-walking errors (E) in the contusion-injured rats treated with vehicle, lithium, or SB. F, The examples of representative footprints from control (top), lithium (middle), and SB (bottom) animals after a contusion injury. The double-ended arrows illustrate the stride width measured from the footprints. G, Footprint analysis revealed a shorter stride width in lithium- or SB-treated animals than in the controls. The grid-walk and footprint tests were performed at 8 weeks after the contusion. Means ± SEM are reported. The differences indicated are compared with the saline-treated group (*p < 0.05, **p < 0.01, Student's t test).
To further characterize the locomotion recovery in SCI rats, two more tests were performed in these rats. In the second test, rats walked on a metal grid runway (25 mm between grids) and the excursions of hindlimbs below the grid plane were counted from videotaping under a slow speed by an observer blind to experimental condition (Metz et al., 2000a). Uninjured rats made no errors on this test. Six weeks after injury, control rats made numerous errors by misplacing their hindpaws and falling into grid holes, but the rats treated with lithium made significantly fewer errors (53 vs 79%; p < 0.01, Student's t test) by correctly placing hindpaws on the grid. Also, analysis of hindpaw footprints 6 weeks after lesion revealed the decreased stride length and increased stance width in the saline-treated rats, but treatment with lithium significantly recovered the stride length and width (Fig. 8B,C).
The locomotor recovery of rats with moderate contusion injury was also monitored during 8 week survival. Two days after contusion SCI, all of the rats exhibited no detectable movement of the legs (BBB score, 0) or slight extensive movement of one or two joints (BBB scores, 1–3) (Fig. 8D). Several weeks after contusion, the majority of control rats had consistent plantar steps and consistent FL–HL coordination, but frequent toe clearance and rotated paw position at lift-off. However, most rats treated with GSK-3 inhibitors exhibited consistent toe clearance and parallel paw position at lift off, in addition to consistent plantar steps and consistent FL–HL coordination. The rats treated with systemic GSK-3 inhibitors had shorter stride length 8 week post-SCI (Fig. 8F,G). Also, the GSK-3 inhibitor-treated rats performed better on the grid-walk task by making fewer errors, although this measure did not reach statistical significance (p = 0.1 and 0.2 in lithium and SB groups, respectively) (Fig. 8E). Thus, these data indicate that treatments with systemic GSK-3 inhibitors significantly improve the behavioral recovery in the adult rodents with either dorsal transection or contusion SCI.
Discussion
CNS inhibitory substrates regulate Akt and GSK-3β activities in neurons
Some attractive extracellular molecules, such as NGF, have been shown to promote axon growth via transiently elevating Akt signal and subsequently suppressing GSK-3 activity (Zhou et al., 2004). In contrast, a few repulsive molecules, such as Sema 3A and 4D, can suppress axon extension by transiently activating GSK-3 at the leading edge of neuronal growth cones (Eickholt et al., 2002; Ito et al., 2006). In this study, we found that axon growth inhibitors CSPGs, the major chemical barrier for axon elongation after a CNS injury, and MAG, a myelin inhibitor, could repress Akt activity in neurons evidenced by a reduction of phosphorylated Akt. Moreover, CSPGs significantly altered the activity of GSK-3β, a major substrate of Akt. After a CNS injury, exposure to high levels of axon inhibitors, including CSPGs and CNS myelin proteins, directly interact with the growth cones of injured axons. As a result, it is likely that the transient changes of Akt and GSK-3 triggered by CNS myelin proteins and CSPGs are sufficient to activate the downstream cascades and cause growth cone collapse, which leads to axon growth inhibition. Thus, our results suggest that altered GSK-3β activity contributes to the growth suppression induced by CNS axon growth inhibitors.
There are several possibilities for the linkage between CNS inhibitors and Akt/GSK-3 signals in neurons. First, RhoA, an important transducer for CSPG and myelin inhibitors (McGee and Strittmatter, 2003), is able to regulate GSK-3β activity in neurons (Eickholt et al., 2002; Sayas et al., 2002) and to activate PTEN (phosphatase/tensin homolog on chromosome 10), a upstream positive regulator of GSK-3β (Shi et al., 2003; Jiang et al., 2005; Z. Li et al., 2005). Thus, RhoA activation after CNS inhibitor exposures might couple the axon inhibitors with GSK-3β activation in neurons. Second, CSPGs could induce neurite growth failure by repressing integrin and ILK, which can negatively regulate GSK-3β activity in neurons (Zhou et al., 2006; Guo et al., 2007; Oinuma et al., 2007). Also, it is unclear whether unknown CSPG receptors could regulate GSK-3β activity in neurons via alternative mechanisms.
GSK-3 inactivation remarkably overcomes the growth suppression of CNS inhibitors
Analyses of neurite growth of immature and mature neurons from chickens or rodents have been frequently used to study in vitro axonal regeneration shortly after cell cultures. Taking advantage of this model, we found that pharmacological inhibition of GSK-3 considerably increased the neurite outgrowth from cultured mouse DRG neurons or CGNs when incubated on CSPG and CNS myelin substrates. Our finding supports the role of GSK-3 activation in restricting axon growth of neurons on CNS inhibitors. Similarly, GSK-3β antagonists have been shown to prevent the growth cone collapse induced by Sema 3A (Eickholt et al., 2002; Chadborn et al., 2006). During maturation, neurons usually establish polarity by forming one or two axons and multiple dendrites to ensure unidirectional signal conduction and GSK-3β exhibits a polarized distribution during neuronal polarity formation, with higher GSK-3β activity in axons than in dendrites (Jiang et al., 2005; Yoshimura et al., 2005). Considering the distinct mechanisms for regulating the formation of axons and dendrites, we further characterized axogenesis and growth in cultured DRG neurons derived from adult mice after GSK-3 inhibition and a longer period of culture (4 d). Although DRGs are a type of peripheral neurons, they normally form both peripheral and CNS axons and are very sensitive to various CNS growth inhibitors, including CSPG and CNS myelin proteins. Thus, analysis of axon generation and extension of adult DRGs may provide valuable information on CNS axon regeneration of mature neurons. Consistently, both lithium and SB dramatically enhance the number of axon-forming neurons and the length of axons in each axon-forming neuron, supporting the important role of GSK-3 activation in restricting axon genesis and outgrowth of mature neurons after exposure to a nonpermissive environment. Moreover, repressing GSK-3 activity appears to enhance the inherent growth ability of mature neurons because GSK-3 inhibitors exhibit similar axon growth-promoting effects in cultures without inhibitory substrates. The increased growth capacity after GSK-3 inhibition is probably related to enhanced transcription of growth-related genes in neurons (Zhou and Snider, 2006). Unlike developing or conditioning lesioned neurons (Jiang et al., 2005), the mature neurons did not exhibit remarkable changes in numbers of axons and branching after GSK-3 inhibition. The cellular mechanisms for GSK-3β regulating axon morphogenesis involve altered phosphorylation status of several microtubule-binding proteins and the assembly of microtubules (Shi et al., 2004; Zhou et al., 2004; Yoshimura et al., 2005; Zhou and Snider, 2006). Together, GSK-3 suppression appears to be an effective approach to stimulate axon regeneration of mature neurons.
Our results, together with others (Eickholt et al., 2002; Jiang et al., 2005; Yoshimura et al., 2005), support the increased axon growth of neurons after GSK-3 suppression. However, some studies indicate reduced axon elongation of neurons after GSK-3 inhibition caused by repression of microtubule-associated protein-1B phosphorylation in growth cones (Lucas et al., 1998; Owen and Gordon-Weeks, 2003). Recently, a group further characterized axogenesis in embryonic neurons with different extents of GSK-3 inhibition (Kim et al., 2006). Strong inhibition of GSK-3 activity with high concentrations of inhibitor or a short hairpin RNA targeting GSK-3α and GSK-3β dramatically reduced axon growth in immature and adult DRG neurons. In contrast, incomplete GSK-3 inactivation with low concentrations of inhibitor increased axon length. Indeed, we also observed the reduced neurite outgrowth of neurons at high concentrations of GSK-3 inhibitors, particularly with potent inhibitor SB415286 (data not shown). Thus, it seems that GSK-3 activation contributes to axon growth repression, but a degree of GSK-3 activity is necessary for efficient axon extension because of the dual roles of GSK-3 in activating and inactivating different substrates during microtubule assembly (Kim et al., 2006).
The basis for axonal growth and improved functional recovery in the injured CNS after GSK-3 inhibitor application
Using different spinal cord injury models, we consistently demonstrate that systemic application of inhibitors remarkably enhances the axonal growth of descending corticospinal and raphespinal tracts as well as the locomotor functional recovery in adult rodents. The GSK-3 inhibition by lithium and SB is most likely to be the basis for the increased axonal growth and improved behavioral recovery of rodents in this study. First, GSK-3 suppression with various inhibitors or GSK-3 knockdown with siRNA has been shown to stimulate axogenesis and extension of embryonic neurons (Jiang et al., 2005; Yoshimura et al., 2005) or adult neurons (Fig. 3). Second, after systemic lithium treatment, we demonstrate the reduced levels of GSK-3β activity in the lesioned rat spinal cord. Third, SB415286 is a potent and specific inhibitor for GSK-3 and does not have obvious inhibitory effects on other protein kinases (Coghlan et al., 2000). Both lithium and SB induce a similar degree of axon growth under in vitro and in vivo conditions, although they repress GSK-3 via different mechanisms. Also, we applied the moderate doses of GSK-3 inhibitors, which were unlikely to block other kinases.
After SCI, the recovery extent of locomotor function depends on reorganization of segmental circuitry and restoration of supraspinal input. In control SCI rats treated with vehicle, the segmental mechanisms probably play a major role in locomotor recovery because of lack of obvious axonal regeneration. However, in rats treated with GSK-3 inhibitors, the restoration of supraspinal input to caudal spinal cord might participate in functional improvement, in addition to the enhanced plasticity at segmental levels (Courtine et al., 2008). Treatment with GSK-3 inhibitors induces the axonal growth in spinal cord below the lesion and significantly increases the fiber number of descending corticospinal and raphespinal tracts. Both CST and 5-HT axonal tracts play a role in locomotor recovery of rodents after SCI (Weidner et al., 2001; Li et al., 2004). Actually, the total number of regenerated CST fibers should be much greater than that reported here because anterograde tracing with BDA only labels a fraction of total CST fibers (Brösamle and Schwab, 1997). Also, GSK-3 inhibition might promote the formation of presynaptic structures (Fig. 3) and possibly neuronal connections. We cannot, however, exclude the possibility that GSK-3 inhibition benefits recovery via protecting injured tissues and/or stimulating proliferation and differentiation of endogenous stem cells (Cuzzocrea et al., 2006; Su et al., 2007; Fornai et al., 2008).
Therapeutic potential of GSK-3 inhibitors in patients with CNS axonal injuries
Regulating GSK-3 activity via pharmacological intervention has become an important target for treating a few neurological disorders including bipolar disorder, Alzheimer's disease, Parkinson's disease, ischemic brain injury, and amyotrophic lateral sclerosis (Meijer et al., 2004; Fornai et al., 2008). Particularly, application of lithium appears to protect neuronal cell death via upregulating cell survival molecules and repressing various proapoptotic factors. In this study, we report that GSK-3 inhibitors, including lithium, could significantly stimulate axonal growth and promote behavioral recovery after the CNS injuries. We recently reported that pain reliever ibuprofen improved axon growth and functional recovery in SCI rodents when applied at a dose of 60 mg/kg/d, which is a higher dose than that used clinically as a cyclooxygenase inhibitor (Fu et al., 2007). However, we delivered lithium to SCI rodents with a clinically relevant dose (3 mEq/kg/d) and identified a similar beneficial effect to SCI recovery. Because moderate GSK-3 inhibition appears to be appropriate for stimulating axon elongation and the dosage of lithium is an important determining factor for the degree of axon growth and functional recovery, we might achieve a greater extent of recovery by identifying the optimal dose of GSK-3 inhibitors for axon growth in vivo. Together, this study suggests that GSK-3 signal is an important therapeutic target for promoting functional recovery in adult mammals with CNS injuries, supporting the potential therapeutic use of these GSK-3 inhibitors in patients with white matter injuries.
Footnotes
-
This work was supported in part by a research grant from Paralyzed Veterans of America to S.L. We thank Ya Ren, Qiao Fu, and Qin Li for technical assistance, and Drs. Craig M. Powell and Jeffrey Elliott for proofreading this manuscript.
- Correspondence should be addressed to Shuxin Li, Department of Neurology, University of Texas Southwestern Medical Center at Dallas, 5323 Harry Hines Boulevard, Dallas, TX 75390-8813. shuxin.li{at}utsouthwestern.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}