

Abstract
Functional plasticity of synaptic networks in the dentate gyrus has been implicated in the development of posttraumatic epilepsy and in cognitive dysfunction after traumatic brain injury, but little is known about potentially pathogenic changes in inhibitory circuits. We examined synaptic inhibition of dentate granule cells and excitability of surviving GABAergic hilar interneurons 8–13 weeks after cortical contusion brain injury in transgenic mice that express enhanced green fluorescent protein in a subpopulation of inhibitory neurons. Whole-cell voltage-clamp recordings in granule cells revealed a reduction in spontaneous and miniature IPSC frequency after head injury; no concurrent change in paired-pulse ratio was found in granule cells after paired electrical stimulation of the hilus. Despite reduced inhibitory input to granule cells, action potential and EPSC frequencies were increased in hilar GABA neurons from slices ipsilateral to the injury versus those from control or contralateral slices. Furthermore, increased excitatory synaptic activity was detected in hilar GABA neurons ipsilateral to the injury after glutamate photostimulation of either the granule cell or CA3 pyramidal cell layers. Together, these findings suggest that excitatory drive to surviving hilar GABA neurons is enhanced by convergent input from both pyramidal and granule cells, but synaptic inhibition of granule cells is not fully restored after injury. This rewiring of circuitry regulating hilar inhibitory neurons may reflect an important compensatory mechanism, but it may also contribute to network destabilization by increasing the relative impact of surviving individual interneurons in controlling granule cell excitability in the posttraumatic dentate gyrus.
Introduction
Reactive plasticity of granule cell axons is a defining feature of the injured dentate gyrus, and it is associated with the emergence of new recurrent excitatory connections among granule cells after traumatic brain injury (TBI) (Hunt et al., 2009, 2010) and in temporal lobe epilepsy (Dudek and Spitz, 1997). Enhanced excitatory innervation of subsets of hilar GABAergic interneurons after neuronal injury has also been proposed (Chang and Lowenstein, 2003; Sloviter et al., 2003). This possibility is supported by increased mossy fiber length in the hilus of epileptic rodents (Buckmaster and Dudek, 1997). However, little is known about how excitatory inputs to dentate inhibitory neurons are reorganized after injury.
The input–output activity of granule cells is tightly regulated by hilar interneurons, many of which are lost after experimental TBI (Lowenstein et al., 1992; Santhakumar et al., 2000; Grady et al., 2003; Hall et al., 2005; Kharatishvili et al., 2006) and in human posttraumatic epilepsy (Swartz et al., 2006). One week after TBI, the frequency of miniature IPSCs (mIPSCs) is reduced in granule cells (Toth et al., 1997; Witgen et al., 2005) and evoked extracellular burst discharges are increased in the granule cell layer in vivo (Lowenstein et al., 1992). However, whether there are long-term deficits in the inhibitory control of granule cells after TBI remains unresolved. Levels of GABA immunobinding and GAD67 expression increase in the hippocampus after TBI (Reeves et al., 1997; Kobori and Dash, 2006). Some studies have suggested enhanced inhibition of granule cells weeks after TBI (Reeves et al., 1997; Santhakumar et al., 2001; Hunt et al., 2009), whereas others found decreased inhibitory synaptic input to granule cells (Mtchedlishvili et al., 2010).
Hilar somatostatin-immunoreactive interneurons represent one subtype of local-circuit GABAergic interneuron in the dentate gyrus (Esclapez and Houser, 1995; Freund and Buzsáki, 1996). These neurons provide important feedback inhibition to distal dendrites of granule cells (Bakst et al., 1986; Leranth et al., 1990; Katona et al., 1999), but they are vulnerable to cell death after a variety of brain insults including ischemic injury (Johansen et al., 1987), repeated seizures (Sloviter, 1987; de Lanerolle et al., 1989; Mathern et al., 1995; Buckmaster and Dudek, 1997; Kobayashi and Buckmaster, 2003; Sun et al., 2007), and TBI (Lowenstein et al., 1992). Their dendrites, which concentrate in the hilus, receive excitatory input primarily from granule cells (Leranth et al., 1990; Katona et al., 1999). Moreover, hilar somatostatin-immunoreactive GABA neurons that survive pilocarpine-induced seizures have larger somata, increased dendritic branching, and receive increased excitatory input (Zhang et al., 2009; Halabisky et al., 2010) relative to controls. Although these features suggest enhanced excitatory innervation of these neurons after seizures, the source of new afferent inputs is unknown.
We used mice which express enhanced green fluorescent protein (eGFP) in a subpopulation of somatostatin interneurons (Oliva et al., 2000) to test whether synaptic inhibition of granule cells was compromised, and whether the pattern and degree of excitatory input to surviving hilar eGFP-GABA neurons was altered several weeks after experimental brain injury.
Materials and Methods
Animals.
Seven- to 8-week-old adult mice that express eGFP in a subset of inhibitory interneurons [i.e., GIN mice; FVB-Tg(GadGFP)4570Swn/J; The Jackson Laboratory) (Oliva et al., 2000)] weighing 25–32 g were housed under a normal 12 h light/dark cycle. We used 28 mice subjected to TBI (26 males and 2 females) and 17 uninjured controls (all males). No significant gender differences were observed. Water and food were available ad libitum. Mice were bred in house, and all procedures were first approved by the University of Kentucky Animal Care and Use Committee and adhered to National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. All experiments were performed on mice 8–13 weeks after injury.
Head injury.
Mice were subjected to a severe unilateral cortical contusion injury by controlled cortical impact (CCI) as previously described (Scheff et al., 1997; Hunt et al., 2009, 2010). Briefly, mice were anesthetized by 2% isoflurane inhalation and placed in a stereotaxic frame. The skull was exposed by midline incision, and a 4 mm craniotomy was made lateral to the sagittal suture and centered between bregma and lambda. The skull cap was removed without damage to the exposed underlying dura. The contusion device consisted of a computer-controlled, pneumatically driven impactor fitted with a beveled stainless-steel tip 3 mm in diameter (Precision Systems and Instrumentation). Brain injury was delivered using this device to compress the cortex to a depth of 1.0 mm at a velocity of 3.5 m/s and 500 ms duration. The hemostatic agent Surgicel (Johnson & Johnson) was placed over the dura after injury, the incision was sutured, and the animal was allowed to recover for 8–13 weeks. A qualitative postoperative health assessment was performed daily for 7 d after TBI and periodically thereafter. Deficits in contralateral hindlimb function were apparent in brain-injured mice for several days. A significant percentage of identically treated mice have previously been shown to exhibit spontaneous seizures (Hunt et al., 2009, 2010), but mice in the present study were not monitored for seizure activity. All brain-injured mice survived and remained otherwise healthy until the day of experimentation.
Slice preparation.
Mice were deeply anesthetized by isoflurane inhalation and decapitated. The brain was removed and immersed in ice-cold (2–4°C) oxygenated artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 3 KCl, 1.3 CaCl2, 26 NaHCO3, 1.3 MgCl2, 1.4 NaH2PO4, 11 glucose, and equilibrated with 95% O2–5% CO2, pH 7.2–7.4, 290–305 mOsm/kg. Brains were blocked, glued to a sectioning stage, and 400-μm-thick transverse slices were cut in ice-cold, oxygenated ACSF using a vibrating microtome (Vibratome Series 1000; Technical Products International). Slices were cut in the coronal plane to ensure consistency of transverse slices from the dorsal one-third of the hippocampus. The hippocampus was isolated from all surrounding tissue, making sure to completely remove the entorhinal cortex. Slices were transferred to a storage chamber containing oxygenated ACSF at 34–36°C and their order maintained so that the location relative to the injury within each hippocampus was known.
Electrophysiology.
After an equilibration period of at least 1 h, slices were transferred to a recording chamber on an upright, fixed-stage microscope equipped with infrared, differential interference contrast and epifluorescence optics (Olympus BX50WI), where they were continuously perfused with warmed (32–34°C) ACSF. Recordings were performed on granule cells and hilar eGFP-labeled neurons, which were identified by epifluorescent illumination. Patch pipettes were pulled from borosilicate glass (1.65 mm outer diameter and 0.45 mm wall thickness; King Precision Glass) with a P-87 puller (Sutter Instrument). The intracellular solution contained the following (in mm): 140 Cs+ gluconate (or 140 K+ gluconate for voltage recordings), 1 NaCl, 5 EGTA, 10 HEPES, 1 MgCl2, 1 CaCl2, 3 KOH, 2 ATP, and 0.2% biocytin, pH 7.15–7.3. The activity-dependent sodium channel blocker QX-314 bromide [N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide] (5 mm) was added to the intracellular solution for electrical stimulation recordings. Open tip resistance was 2–5 MΩ. Recordings were obtained with an Axopatch 200B or Multiclamp 700B amplifier (Molecular Devices), low-pass filtered at 2–5 kHz, digitized at 88 kHz (Neuro-corder; Cygnus Technology), and recorded to pClamp 10.2 (Clampfit; Molecular Devices). Cell attached recordings of spontaneous action potentials were recorded in voltage-clamp mode at −60 mV. For whole-cell patch-clamp recordings, cells were first voltage clamped for ∼5 min at −60 or −70 mV (i.e., near resting membrane potential) to allow equilibration of intracellular and recording pipette contents. Spontaneous EPSCs (sEPSCs), miniature EPSCs (mEPSCs), and evoked EPSCs (eEPSCs) were examined at a holding potential of −60 mV, and inhibitory currents (sIPSCs, mIPSCs, and eIPSCs) were examined at a holding potential of 0 mV. Series resistance was typically <16 MΩ (12.7 ± 3.7 MΩ) and was monitored throughout the recordings. Data were only used for analysis if the series resistance remained <20 MΩ and changed by ≤20% during the recordings. Added to the ACSF for specific experiments were the following: tetrodotoxin (TTX) (2 μm; Sigma-Aldrich), bicuculline methiodide (BMI) (30 μm; Sigma-Aldrich), and kynurenic acid (1 mm; Sigma-Aldrich).
Electrical stimulation.
A concentric bipolar stimulating electrode made of platinum–iridium wire (125 μm diameter; FHC) was used to apply paired stimuli to the hilus. The stimulus duration was 80 μs, and stimulus intensity was adjusted to produce an eIPSC at 50% of maximum amplitude. Pairs of stimuli were delivered to the hilus at pairing intervals of 30, 60, 90, 120, and 200 ms. Eight to 10 consecutive responses were obtained at 0.1 Hz for each pairing frequency. Amplitudes of eEPSCs were measured from the baseline at which the stimulus was applied to the IPSC peak. Responses at each pairing frequency were averaged, and the ratio of the amplitude of the second eIPSC to the first (eIPSC2/eIPSC1) was calculated to establish the paired-pulse ratio (PPR).
Glutamate photostimulation.
Slices were perfused with γ-(carboxy-2-nitrobenzyl)ester, trifluoroacetic acid salt (i.e., CNB-caged glutamate) (250 μm; Invitrogen) added to recirculating ACSF. Brief pulses of fluorescent light (30 ms exposure; UV filter; Chroma Technology) were directed into the slice through the 40× objective (Bhaskaran and Smith, 2010; Hunt et al., 2010). The objective was initially positioned to “uncage” glutamate directly over the recorded cell. This consistently resulted in a large inward current (n = 19). No difference in direct current amplitude (p = 0.5) or duration (p = 0.3) was detected between groups. The effective radius of stimulation (<50 μm) was established by manually moving the focal point of the stimulus until a direct inward current after stimulation was no longer observed. This was repeated in three directions to establish the effective uncaging area. The objective was then moved away from the recorded cell, and photostimulation was applied focally to sites along the entire extent of the granule cell layer and in the CA3 region. A series of five stimuli were applied per stimulation site at 0.1 Hz.
Histology.
After the conclusion of the recording experiment, slices were placed in 0.37% sodium sulfide solution in 0.1 m NaHPO4 for 20 min, followed by 4% paraformaldehyde in 0.15 m phosphate buffer overnight to fix the slices. Slices were then rinsed three times with PBS (0.01 m), pH 7.4, and placed in a 30% sucrose solution in PBS overnight or until they sank for cryoprotection. Slices were sectioned at 20 μm on a cryostat, rinsed, mounted on charged slides (Superfrost Plus; Thermo Fisher Scientific), dried overnight, and treated according to previous protocols using Timm stain to reveal mossy fibers and Nissl counterstained with cresyl violet to visualize cell bodies (Tauck and Nadler, 1985; Shibley and Smith, 2002; Hunt et al., 2009, 2010). To semiquantitatively assess mossy fiber sprouting after CCI, sections from hemispheres ipsilateral and contralateral to the injury were examined by an investigator who was blind to the electrophysiological outcomes. Slices were assigned Timm scores from 0, little to no sprouting, to 3, robust mossy fiber sprouting (Tauck and Nadler, 1985; Patrylo and Dudek, 1998; Shibley and Smith, 2002; Hunt et al., 2009, 2010). Regions of the dentate gyrus with Timm scores >1 were considered to have an abnormal degree of mossy fiber sprouting (Patrylo and Dudek, 1998; Shibley and Smith, 2002; Hunt et al., 2009, 2010). Before cryoprotection, slices in which a recording from an eGFP-GABA neuron was obtained were examined for biocytin labeling. Slices were immersed in avidin conjugated to Texas Red (1:400; Vector Laboratories) in PBS containing 0.5% Triton X-100 and incubated for 3–4 h at room temperature to identify biocytin-filled neurons and confirm the location and eGFP content of the recorded cell. Slices were then rinsed three times with PBS, mounted on slides, covered with Vectashield containing DAPI (4′,6-diamidino-2-phenylindole) (Vector Laboratories), and coverslipped. Images were taken with an Olympus BX40 microscope and captured with a Spot RT camera (Diagnostic Instruments). Slices were then washed in PBS, placed in 30% sucrose solution, and processed for Timm and Nissl stains.
Data analysis and statistics.
Data analysis was performed using pClamp 10.2 (Clampfit; Molecular Devices), MiniAnalysis 6.0 (Synaptosoft), Instat (GraphPad Software), and IBM SPSS Statistics 18 (SPSS) programs. A 2 min sample recording per cell was used for measuring synaptic event frequency, amplitude, and kinetics. Events characterized by a typical fast rising phase and exponential decay phase were manually detected using MiniAnalysis. Multiple and complex peaks were detected by exponential extrapolation of decay using this program. Only currents with amplitudes greater than three times the root mean square noise level were included for analysis. Event frequency, mean amplitude, and kinetics were averaged across neurons (i.e., n = neurons) and groups were compared by one-way ANOVA followed by Tukey's post hoc test or two-tailed t test. Paired-pulse responses were analyzed by two-way (treatment group by pairing interval) repeated-measures (pairing interval) ANOVA. For photostimulation experiments, EPSC frequency was analyzed every 100 ms for 500 ms before and after each stimulation trial and averaged for each stimulation site (i.e., n = stimulation sites). A response at a given location was considered to be positive (i.e., a local synaptic connection was evoked) if the number of EPSCs in at least one of the first two 100 ms segments after stimulation was greater than the mean number of events per 100 ms before stimulation + 3SD and a response was observed in at least three of five trials time-locked to the stimulus, demonstrating that the response was repeatable (Hunt et al., 2010). Positive responses that were time-locked to the stimulus, exhibiting relatively short and constant latency to the first evoked EPSC on repetitive stimulation, were interpreted to be attributable to direct connections between the stimulated and recorded cell. Direct photostimulation responses (i.e., because of postsynaptic activation of the recorded neuron) could be easily distinguished from synaptic responses based on a lack of onset latency and slow rise time, and they were revealed by addition of 2 μm TTX, which blocked evoked synaptic events but not direct currents (Callaway and Katz, 1993; Dantzker and Callaway, 2000; Waurin and Dudek, 2001). Stimulation sites in which the selection criteria were not met were considered to have a negative response. Responses evoked by glutamate photostimulation were analyzed within a time window between 10 and 200 ms after photostimulation. We chose this relatively short time window, because all directly evoked action potentials in principal neurons occurred within 10–200 ms after photostimulation, with most occurring in the first 100 ms (Hunt et al., 2010). EPSCs that occurred within 10 ms of photostimulation were considered to be either spontaneous events or attributable to the direct postsynaptic activation of the recorded neurons and were disregarded, because spike onset in principal neurons occurred ≥10 ms after direct photostimulation. The number of eEPSCs for each stimulation site was calculated by subtracting the number of EPSCs in the first 190 ms before stimulation from the number of EPSCs between 10 and 200 ms after stimulation. To analyze differences in evoked activity after glutamate stimulation, data from control and contralateral slices, which acted as an internal control, were combined and compared with responses ipsilateral to the injury by two-tailed Mann–Whitney U or Fisher's exact test. Data are expressed as mean ± SD, and significance was set at p < 0.05.
Results
Mossy fiber sprouting
Mossy fiber sprouting into the inner molecular layer is a reliable marker of synaptic reorganization associated with posttraumatic epilepsy (Kharatishvili et al., 2006; Swartz et al., 2006; Hunt et al., 2009, 2010). Mossy fiber sprouting was present in sections of the dentate gyrus near the injury site (n = 23 of 28 animals) (Fig. 1), but never in the contralateral hemisphere (n = 0 of 28 animals) (Fig. 1) or in controls (n = 0 of 10 animals). These findings verified the presence of axon sprouting in most animals, suggestive of network reorganization common to the injured dentate gyrus.

Mossy fiber sprouting in GIN mice after severe traumatic brain injury. A, Timm- and Nissl-stained section from a slice contralateral to the injury. B, Timm- and Nissl-stained section from a slice ipsilateral to the injury. The arrows indicate mossy fiber sprouting into the inner molecular layer. m, Molecular layer; g, granule cell layer; h, hilus.
Reduced synaptic inhibition of dentate granule cells after TBI
There are fewer hilar GABA neurons after TBI (Lowenstein et al., 1992; Toth et al., 1997; Santhakumar et al., 2000), but it is unclear whether there are long-term changes in synaptic inhibition of granule cells after severe TBI. To test whether overall inhibitory synaptic input to dentate granule cells was altered after CCI injury, whole-cell voltage-clamp recordings of sIPSCs were obtained from granule cells in controls (n = 10 cells from 5 animals) and in slices contralateral (n = 12 cells in 11 animals) and ipsilateral (n = 20 cells in 11 animals) to the injury. Recordings were made from cells in the apex or outer blade regions of the granule cell layer in slices at or adjacent to the injury site. Spontaneous IPSCs were recorded at a holding potential of 0 mV. At this potential, application of 30 μm BMI completely blocked sIPSCs (n = 5), indicating that these currents were mediated by GABAA receptors. Representative recordings for each group are shown in Figure 2A–C. The mean sIPSC frequency was significantly lower in granule cells ipsilateral to the injury versus other groups, as detected by one-way ANOVA with Tukey's post hoc test (control, 3.31 ± 0.5 Hz; contralateral, 3.38 ± 1.07 Hz; ipsilateral, 1.55 ± 0.47 Hz; F(2,39) = 34.5; p < 0.001) (Fig. 2D).

Reduced sIPSC frequency in granule cells in slices from the ipsilateral dentate gyrus. A–C, Example whole-cell patch-clamp recording from granule cells in a control slice (A), a slice contralateral to the injury (B), and a slice ipsilateral to the injury (C). The line below each trace indicates region of the trace expanded below (arrows). D, Average sIPSC frequency for cells in each treatment group. Significance is indicated by an asterisk (*). E, Average sIPSC frequency for cells in ipsilateral slices in the presence and absence of mossy fiber sprouting (MFS). F, Average sIPSC amplitude for cells in each treatment group. Error bars indicate SD.
To determine whether sIPSCs were related to the presence of mossy fiber sprouting, event frequency from slices ipsilateral to the injury after CCI was assessed based on post hoc identification of the presence (Timm scores >1; n = 13) or absence (Timm scores ≤1; n = 7) of mossy fiber sprouting in the inner molecular layer. We did not detect a significant difference in sIPSC frequency between slices with and without mossy fiber sprouting (p = 0.84, t test) (Fig. 2E). A difference in sIPSC amplitudes was also not detected between groups (control, 17.06 ± 3.8 pA; contralateral, 18.04 ± 3.16 pA; ipsilateral, 20.3 ± 6.11 pA; F(2,39) = 1.7; p = 0.2) (Fig. 2F). However, one-way ANOVA found a significant difference in mean sIPSC 10–90% rise time (control, 1.5 ± 0.4 ms; contralateral, 1.9 ± 0.5 ms; ipsilateral, 2.5 ± 0.8 ms; F(2,39) = 7.4; p < 0.01) and decay time constants (control, 13.2 ± 4.8 ms; contralateral, 13.8 ± 4.1 ms; ipsilateral, 18.6 ± 5.6 ms; F(2,39) = 5.8; p < 0.01). Although event kinetics in controls and contralateral slices were comparable with previously reported values for granule cells from control animals (Kobayashi and Buckmaster, 2003; Shao and Dudek, 2005), post hoc evaluations revealed slower sIPSC kinetics in granule cells ipsilateral to the injury.
The reduced sIPSC frequency in granule cells after TBI could be attributable to fewer inhibitory synapses onto granule cells, altered probability of vesicle release, or reduced activity of presynaptic inhibitory interneurons. If there were fewer inhibitory synapses onto granule cells, the frequency of mIPSCs should also be reduced. mIPSC characteristics were measured in the presence of 2 μm TTX in some of the same granule cells used to measure sIPSCs. Representative recordings for each group are shown in Figure 3A–C. Similar to spontaneous events, mIPSC frequency was lower in granule cells ipsilateral to the injury (control, 2.56 ± 0.85 Hz; contralateral, 2.7 ± 1.08 Hz; ipsilateral, 1.19 ± 0.42 Hz; F(2,31) = 14.3; p < 0.001) (Fig. 3D). A significant difference in mIPSC frequency was not found between slices from injured animals with mossy fiber sprouting (n = 9) versus those without sprouting (n = 6) (p = 0.93; t test) (see Fig. 3E). Additionally, a difference in mIPSC amplitude was not detected between groups by one-way ANOVA (control, 12.05 ± 2.35 pA; contralateral, 14.17 ± 2.2 pA; ipsilateral, 14.14 ± 3.19 pA; F(2,31) = 2.0; p = 0.16) (Fig. 3F), implying a presynaptic change in synaptic inhibition of granule cells after head injury. Unlike spontaneous events, a significant difference was not detected in mean event 10–90% rise time (control, 1.7 ± 0.4 ms; contralateral, 1.6 ± 0.3 ms; ipsilateral, 1.9 ± 0.7 ms; F(2,39) = 0.85; p < 0.85) or decay time constant (control, 13.2 ± 3.7 ms; contralateral, 11.2 ± 2.4 ms; ipsilateral, 14.9 ± 5.9 ms; F(2,39) = 2.0; p = 0.15).

Reduced mIPSCs in granule cells of slices from the ipsilateral dentate gyrus. A–C, Example whole-cell patch-clamp recording from granule cells in a control slice (A), a slice contralateral to the injury (B), and a slice ipsilateral to the injury (C). The line below each trace indicates area of the trace expanded below (arrows). D, Average mIPSC frequency for cells in each treatment group. Significance is indicated by an asterisk (*). E, Average mIPSC frequency for cells in ipsilateral slices in the presence and absence of MFS. F, Average mIPSC amplitude for cells in each treatment group. Error bars indicate SD.
Hilar-evoked PPRs are unchanged
Reduced mIPSC frequency could be attributable to dysfunction of GABAergic synaptic efficacy to granule cells or altered probability of vesicle release (Hirsch et al., 1999). Paired-pulse stimulation has previously been used to examine the strength of synaptic inhibition and as a measure of release probability in an epilepsy model (Kobayashi and Buckmaster, 2003; Halabisky et al., 2010). Paired stimuli were applied to the hilus at pairing intervals of 30, 60, 90, 120, and 200 ms (50% maximum threshold, 80 μs, 0.1 Hz) in the presence of 1 mm kynurenic acid. Paired-pulse depression of the eIPSC amplitude was observed at all intervals (Fig. 4), and application of 2 μm TTX (n = 3) or 30 μm BMI (n = 3) completely blocked hilar-evoked IPSCs. Two-way repeated-measures ANOVA detected a significant effect of pairing interval (F(4,116) = 3.56; p < 0.01), but there was no effect of treatment group (F(2,29) = 0.48; p = 0.63) or the pairing interval by treatment group interaction (F(8,116) = 0.71; p = 0.68). These findings suggest there is no significant change in IPSC PPRs evoked from the hilus after injury.

No change in PPRs of hilar-evoked GABAA receptor-mediated IPSCs in granule cells after brain injury. A–C, Superimposed pairs of eIPSCs at pairing intervals of 30, 60, 90, 120, and 200 ms (average of 8–10 responses for each trial) in granule cells from a control slice (A), a slice contralateral to the injury (B), and a slice ipsilateral to the injury (C). Note that application of 2 μm TTX blocked hilar-evoked IPSCs. D, Average paired-pulse responses across cells for each treatment group showed no differences between groups. Error bars indicate SD.
Increased excitatory input to hilar eGFP-GABA interneurons
In granule cells, sIPSCs result from the release of GABA at a mixture of perisomatic and dendritic synapses, whereas mIPSCs primarily result from the action potential-independent release of GABA restricted to the somatic/perisomatic region (Soltesz et al., 1995). Therefore, an increase in slow-rising sIPSCs, but not mIPSCs, might be attributable to an increase in excitability of hilar interneurons that project to granule cell dendrites after TBI. To determine whether excitability of GABAergic hilar interneurons was altered after TBI, eGFP-labeled hilar neurons were first recorded in the cell-attached configuration to examine spontaneous action potential firing without intracellular disruption of the cell (Cossart et al., 2001; Jones and Baraban, 2007). A significant difference in the mean unit firing frequency between treatment groups was detected by one-way ANOVA (control, 2.4 ± 1.4 Hz; contralateral, 3.9 ± 1.8 Hz; ipsilateral, 9.2 ± 6.1 Hz; F(2,28) = 9.08; p < 0.001) (Fig. 5). Post hoc comparisons revealed that interneurons from ipsilateral slices had a significantly higher action potential frequency versus other groups.

Increased spontaneous action potential firing in hilar eGFP-GABA interneurons ipsilateral to the injury. A–C, Cell-attached recordings of activity in a hilar eGFP-GABA interneuron in a control slice (A), a slice contralateral to the injury (B), and a slice ipsilateral to the injury (C). D, Average action potential frequency for cells in each treatment group. Significance is indicated by an asterisk (*). Error bars indicate SD.
Excitatory synaptic input to hilar eGFP-GABA neurons was evaluated by whole-cell voltage-clamp recordings of sEPSCs at a holding potential of −60 mV in controls (n = 21 cells from 13 animals) and in slices contralateral (n = 13 cells in 11 animals) and ipsilateral (n = 21 cells in 15 animals) to the injury. Representative recordings for each group are shown in Figure 6A–C. A difference in the mean event frequency was detected by one-way ANOVA (control, 4.5 ± 1.8 Hz; contralateral, 5.3 ± 1.9 Hz; ipsilateral, 20.9 ± 12.9 Hz; F(2,52) = 25.5; p < 0.001) (Fig. 6D). Post hoc comparisons revealed increased sEPSC frequency in hilar interneurons ipsilateral to the injury versus other groups. This fourfold increase in event frequency is comparable with the increase observed in granule cells after TBI (Hunt et al., 2010). We did not find a significant difference in sEPSC frequency between slices from injured animals with mossy fiber sprouting (19.0 ± 11.4 Hz; n = 10) versus those without sprouting (23.5 ± 13.6 Hz; n = 11) (p = 0.44; t test). The mean amplitude of sEPSCs for each treatment group was as follows: control, −19.4 ± 6.7 pA; contralateral, −18.8 ± 4.7 pA; and ipsilateral, −20.9 ± 7.6 pA. A significant difference between groups was not detected (F(2,52) = 0.43; p = 0.65) (Fig. 6E). Kinetics of sEPSCs were comparable with reported values in cortical eGFP-somatostatin interneurons in these same transgenic mice (Halabisky et al., 2006, 2010). One-way ANOVA did not detect a difference between groups in mean 10–90% rise times (control, 1.6 ± 0.4 ms; contralateral, 1.7 ± 0.5 ms; ipsilateral, 1.7 ± 0.4 ms; F(2,52) = 1.0; p = 0.4) or decay time constant (control, 5.3 ± 2.0 ms; contralateral, 6.6 ± 2.2 ms; ipsilateral, 6.8 ± 0.4 ms; F(2,52) = 2.4; p = 0.11).

Increased sEPSC frequency in hilar eGFP-GABA interneurons in slices from the ipsilateral dentate gyrus. A–C, Example whole-cell patch-clamp recordings from eGFP-GABA neurons from a control slice (A), a slice contralateral to the injury (B), and a slice ipsilateral to the injury (C). The line above each trace indicates area of the trace expanded below (arrows). D, Average sEPSC frequency for cells in each treatment group. Significance is indicated by an asterisk (*). E, Average sEPSC amplitude for cells in each treatment group. The inset shows example fluorescent illumination of a biocytin-filled eGFP neuron: eGFP-labeled neuron visualized in a live brain slice before recording (a); post hoc examination shows eGFP labeling (b); biocytin labeling of the same neuron (identified with an avidin Texas Red label) (c); and merged image shows coexpression of eGFP and biocytin labeling (d). Error bars indicate SD.
Miniature EPSCs were recorded in the presence of 2 μm TTX in some of the same cells used to examine spontaneous activity (controls, n = 9; contralateral, n = 8; ipsilateral, n = 11). Representative recordings for each group are shown in Figure 7A–C. Similar to spontaneous events, mEPSC frequency was significantly increased in hilar interneurons ipsilateral to the injury (control, 2.7 ± 1.3 Hz; contralateral, 3.1 ± 0.9 Hz; ipsilateral, 10.1 ± 6.5 Hz; F(2,52) = 9.72; p < 0.001) (Fig. 7D). We did not detect a significant difference in mEPSC frequency between slices from injured animals with mossy fiber sprouting (13.4 ± 8.5 Hz; n = 5) versus those without sprouting (7.3 ± 2.5 Hz; n = 6) (p = 0.13; t test). The ratio of the frequency of mEPSCs to sEPSCs (mEPSC/sEPSC) was calculated to examine the relative contribution of network-driven inputs. The mEPSC/sEPSC ratio was significantly reduced in eGFP-GABA neurons ipsilateral to the injury (control, 0.71 ± 0.14; contralateral, 0.63 ± 0.17; ipsilateral, 0.44 ± 0.17; F(2,25) = 7.6; p < 0.01) (Fig. 7E). Differences in mEPSC amplitude were not detected between groups by one-way ANOVA (control, 14.3 ± 7.0 pA; contralateral, 13.4 ± 3.9 pA; ipsilateral, 14.8 ± 4.3 pA; F(2,25) = 0.18; p = 0.83) (Fig. 7F). Similarly, we did not detect a difference in mean event 10–90% rise times (control, 1.7 ± 0.5 ms; contralateral, 1.9 ± 0.3 ms; ipsilateral, 1.8 ± 0.4 ms; F(2,52) = 0.7; p = 0.5) or decay time constants (control, 6.0 ± 2.8 ms; contralateral, 7.0 ± 1.7 ms; ipsilateral, 6.5 ± 1.4 ms; F(2,52) = 0.6; p = 0.6).

Increased mEPSC frequency in hilar eGFP-GABA interneurons in slices from the ipsilateral dentate gyrus. A–C, Example whole-cell patch-clamp recordings from eGFP-GABA neurons in a control slice (A), a slice contralateral to the injury (B), and a slice ipsilateral to the injury (C). The line above each trace indicates area expanded below (arrows). D, Average mEPSC frequency for cells in each treatment group. E, Average mEPSC/sEPSC ratio for each treatment group. F, Average mEPSC amplitude for each treatment group. Significance is indicated by an asterisk (*). Error bars indicate SD.
Increased photostimulation-evoked activity in eGFP-GABA neurons after TBI
Based on our analysis of mEPSC/sEPSC ratios, we hypothesized that nearly 60% of EPSCs in hilar eGFP-GABA neurons were action potential mediated versus nearly 30% in cells from control slices. This suggests that excitatory synaptic input to eGFP-GABA neurons is enhanced because of greater local network activation of these neurons after injury, in addition to increased synaptic contacts. However, even in the presence of substantial mossy fiber sprouting, granule cells in slices ipsilateral to the injury rarely, if ever, fired spontaneous action potentials at rest in “normal” ACSF (n = 12) because entorhinal inputs to granule cells were removed and recurrent excitatory circuits are normally masked by recurrent inhibition (Hunt et al., 2009, 2010). In contrast, CA3 pyramidal cells were spontaneously active at rest in control animals (n = 3 of 4) as previously described (Spencer and Kandel, 1961; Wong et al., 1979; Wong and Prince, 1981) and sometimes form functional synaptic connections with hilar GABAergic interneurons (Kneisler and Dingledine, 1995; Doherty and Dingledine, 2001; Wittner et al., 2006; Mangin et al., 2008). Therefore, we hypothesized that eGFP-GABA neurons receive increased excitatory inputs because of activity in both granule cells and CA3 pyramidal neurons after TBI.
Glutamate photostimulation applied to granule cells and CA3 pyramidal cells allowed for focal activation of cell bodies and dendrites but not axons of passage (Callaway and Katz, 1993; Hunt et al., 2010). After photostimulation of granule cells, an increase in evoked synaptic activity was observed in two of six eGFP-GABA cells from controls (3 of 71 stimulation sites) and three of six eGFP cells contralateral to the injury (3 of 50 stimulation sites). After photostimulation of CA3 pyramidal neurons, an increase in synaptic activity was observed in two of five eGFP-GABA cells from controls (4 of 20 stimulation sites) and one of six eGFP-GABA cells contralateral to the injury (1 of 20 stimulation sites). Positive evoked responses were rarely observed in cells from controls and contralateral to the injury. Examples of such responses are shown in Figure 8.

Responses in hilar eGFP-GABA neurons after glutamate photostimulation of the granule cell and pyramidal cell layers in control slices. A, Example responses to photostimulation of the granule cell and CA3 pyramidal cell layers in a control slice. The numbers to the left of each trace indicate corresponding numbered stimulus position shown in the inset. The arrowheads above each trace indicate time of stimulation. A positive response was observed after stimulation at site 7. Shaded regions of the responses at sites 2 and 7 are expanded in B and C. B, Three consecutive overlapping responses to photostimulation of the granule cell layer at site 2 shown in A. A positive response was not detected at this stimulation site. C, Three consecutive overlapping responses to photostimulation of the CA3 pyramidal cell layer before (top traces) and after (bottom traces) TTX application at site 7 shown in A. A positive response of two to three EPSCs was evoked in each trial. The time between photostimulation (1) and eEPSC onset (2) was 21 ms. Note that eEPSC onset is time-locked to the stimulus. D, Example responses to photostimulation of the granule cell and CA3 pyramidal cell layers in a slice contralateral to the injury. A positive response was observed after stimulation at site 5. Application of 2 μm TTX blocked synaptic responses but not the direct current (*).
Unlike the sparse local innervation detected in control slices, photostimulation of granule cells or CA3 pyramidal cells resulted in robust eEPSCs in hilar eGFP-GABA neurons ipsilateral to the injury (Fig. 9). A synaptically mediated response was observed in each of seven cells after photostimulation applied to the granule cell layer (28 of 85 stimulation sites; 1–8 sites per cell) and in five of seven cells after photostimulation applied to area CA3 (12 of 36 stimulation sites; 1–4 sites per cell). Because relatively few positive responses were observed in cells from slices of controls and contralateral to the injury, data in these cells were combined for analysis against responses from cells ipsilateral to the injury. A frequency histogram was constructed to show the distribution of the average number of eEPSCs for each group (Fig. 9B,C). The total number of sites that had a positive versus negative response was greater in cells ipsilateral to the injury (p < 0.001 for granule cell layer stimulation; p < 0.05 for CA3 stimulation; Fisher's exact test). The range of the number of eEPSCs was increased in eGFP-GABA neurons ipsilateral to the injury after photostimulation applied to granule cells (p < 0.05; two-tailed Mann–Whitney U) and to CA3 pyramidal neurons (p < 0.05; two-tailed Mann–Whitney U). Therefore, both the number of stimulation sites with a positive response and the number of EPSCs evoked at each site were increased in eGFP-GABA cells ipsilateral to the injury.
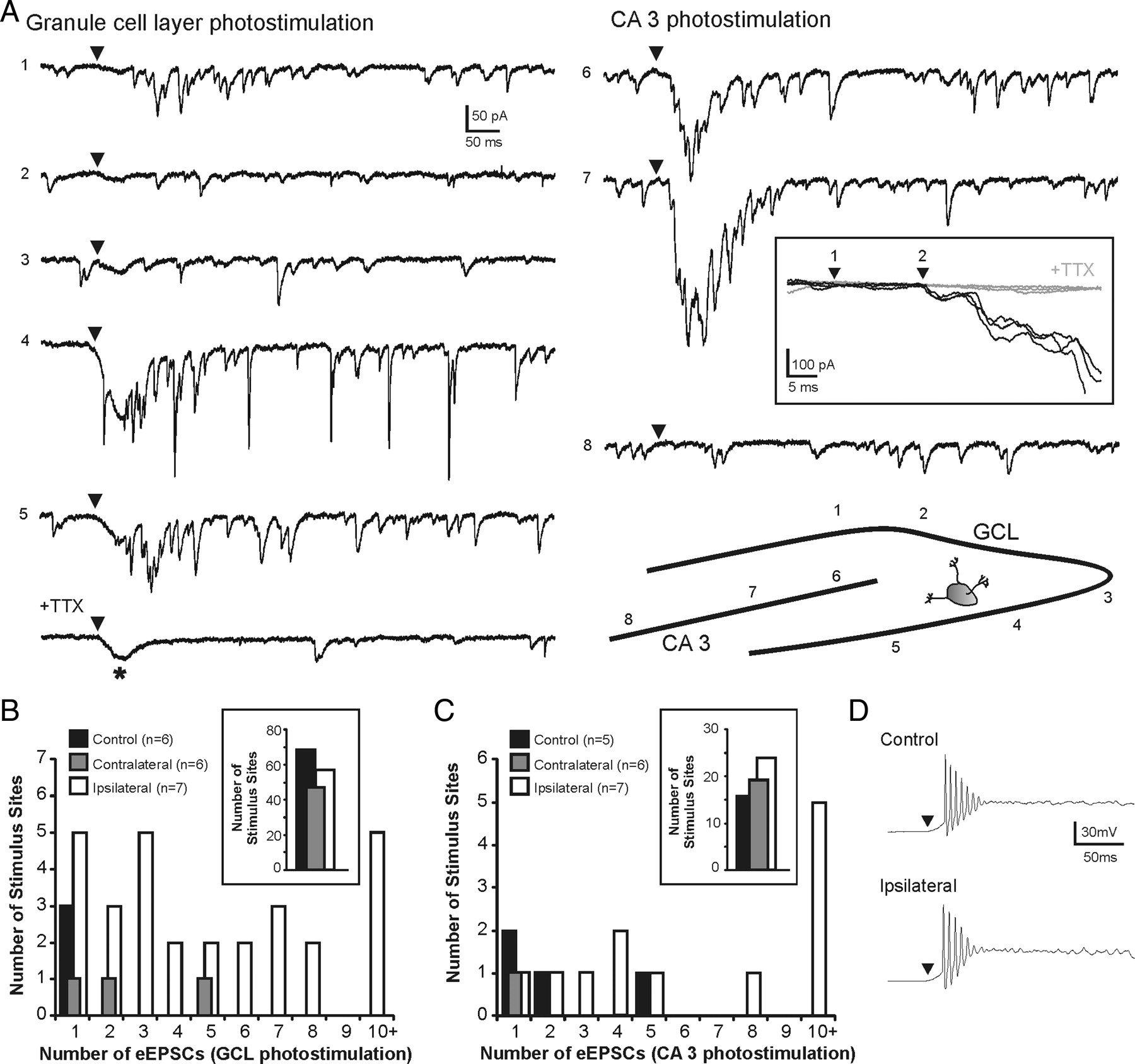
Increased synaptic connections to hilar eGFP-GABA interneurons from granule cells and CA3 pyramidal cells detected by glutamate photostimulation in a slice ipsilateral to the injury. A, Example responses to photostimulation of the granule cell and CA3 pyramidal cell layers. The numbers to the left of each trace indicate corresponding numbered stimulus position shown in the inset. The arrowheads above each trace indicate time of stimulation. Positive responses were observed at stimulation sites 1 and 4–7. Note: Addition of 2 μm TTX blocked photostimulation-evoked synaptic activity but not the current because of direct activation of the recorded neuron, indicated by the asterisk (*). The boxed inset shows three consecutive overlapping responses to photostimulation of the CA3 pyramidal cell layer at site 7 before (black traces) and after (gray traces) TTX application. The time between photostimulation (1) and eEPSC onset (2) was 15 ms. Note that eEPSC onset is time-locked to the stimulus. B, C, Frequency histograms show the distribution of the average number of eEPSCs after photostimulation of the granule cell layer (B) and the CA3 pyramidal cell layer (C). The insets show number of stimulus sites that had a negative response to photostimulation (i.e., 0 eEPSCs). D, Example response to direct photostimulation of recorded CA3 pyramidal cells in control (top) and ipsilateral to the injury (bottom).
Changes in the intrinsic properties of principal cells after TBI might make them more responsive to glutamate photostimulation and result in greater action potential firing. To rule out this possibility, we obtained whole-cell current-clamp recordings from CA3 pyramidal neurons. All cells fired a series of four to nine action potentials after direct glutamate stimulation. There were no significant differences between cells from controls (n = 4) and ipsilateral to the injury (n = 4) in total spike number evoked per cell (6.2 ± 1.5 in controls; 5.0 ± 1.0 ipsilateral to the injury; p = 0.48) (Fig. 9D). This is consistent with our previous findings that showed the number of action potentials directly evoked by photostimulation in granule cells also does not change after TBI (Hunt et al., 2010). Therefore, under these experimental conditions, there was no significant change in evoked excitability of individual principal neurons that would affect responses of presynaptic cells to glutamate photoactivation.
Discussion
Intuitively, inhibitory neuron loss and the subsequent loss of synaptic contacts after TBI might explain the reduction in synaptic inhibition of granule cells. Parvalbumin-, cholecystokinin-, and somatostatin-immunoreactive cells are reduced in number shortly after moderate TBI (Lowenstein et al., 1992; Toth et al., 1997; Santhakumar et al., 2000), and numbers of parvalbumin- and GAD-67-immunoreactive hilar interneurons remain reduced 5 months after injury (Santhakumar et al., 2000). Whether certain subtypes of hilar neurons are preferentially lost after TBI, as is often observed in temporal lobe epilepsy, remains unresolved. Previous studies assessing population responses to paired-pulse stimulation suggested that overall inhibition of granule cells increases after TBI (Reeves et al., 1997; Hunt et al., 2009). However, the relationship between extracellular paired-pulse responses and overall synaptic inhibition in granule cells is indirect and may rely on other circuit characteristics (Waldbaum and Dudek, 2009). Recording IPSCs is a more sensitive measure of synaptic inhibition; our finding that IPSC frequency is reduced after TBI is similar to results in epilepsy models (Kobayashi and Buckmaster, 2003; Shao and Dudek, 2005). Surprisingly, we did not detect changes in PPRs as reported in the pilocarpine model of epilepsy (Kobayashi and Buckmaster, 2003). This may be attributable to model-specific differences, stimulator position, stimulus intensity, or other technical differences. Importantly, hilar-evoked IPSC PPRs reflect overall strength of inhibition and not necessarily changes occurring in specific GABAergic subtypes after TBI, such as the eGFP-GABAergic subpopulation. In contrast to our findings, a recent study suggested reduced mIPSC frequency in granule cells contralateral to CCI in rats, where the ipsilateral dentate gyrus was reportedly unsuitable for recordings after severe CCI (Mtchedlishvili et al., 2010). The cortical cavity produced in our model can occasionally extend into the hippocampus and dentate gyrus (Hall et al., 2005; Saatman et al., 2006; Hunt et al., 2009, 2010), but it does not ablate the ipsilateral hippocampus.
The slower decay time for sIPSCs recorded at the granule cell soma is consistent with increased activity or axon sprouting of dendritically projecting GABA neurons after injury (Pearce, 1993; Kobayashi and Buckmaster, 2003; Zhang et al., 2009, Halabisky et al., 2010). IPSC kinetics returned to control levels after TTX application. This implies that a change in the activity of dendritically projecting GABA neurons may be involved because mIPSCs originate mostly from synapses proximal to the granule cell somata (Soltesz et al., 1995), whereas sIPSCs represent a mixture of somatic and dendritic inputs. However, TBI may affect pools of synaptic vesicles for spontaneous and miniature GABA release differently (Mathew et al., 2008). Other possibilities include changes in GABAA receptor subunits (Brooks-Kayal et al., 1998), the ratio of dendritic versus perisomatic synapses (Cossart et al., 2001), or passive properties of granule cells after TBI. It is unlikely that dendritic sIPSCs went undetected, because unitary IPSCs can be detected in granule cells after presynaptic action potential firing in hilar eGFP-GABA neurons, which project to dendrites (Zhang et al., 2009). Altered IPSC kinetics in granule cells have also been observed in epileptic animals (Kobayashi and Buckmaster, 2003; Shao and Dudek, 2005; Sun et al., 2007).
Anatomical evidence suggests hilar somatostatin GABAergic interneurons normally receive synaptic contacts from mossy fibers (Leranth et al., 1990; Katona et al., 1999) and occasionally from CA3 pyramidal cells (Wittner et al., 2006). We found that hilar eGFP-GABA interneurons receive excitatory input from both cell groups. These inputs are increased after TBI, independent of whether mossy fiber sprouting is present in the slice. An increase in CA3 output to hilar GABA neurons has not previously been shown in any model of TBI or epilepsy, and this may be an important compensatory mechanism for reestablishing feedback inhibition in the dentate gyrus after injury. Consistent with this, an increase in the excitability and number of excitatory inputs in hilar eGFP-GABA neurons after pilocarpine-induced seizures has been reported (Halabisky et al., 2010). Although granule cell input to hilar interneurons can be strong when granule cells are depolarized experimentally (Scharfman et al., 1990), action potential-driven sEPSCs in eGFP-GABA neurons seem likely to arise mostly from CA3 pyramidal cells, because granule cells were rarely active in our preparation, whereas CA3 neurons fire spontaneously at rest. Whether CA3 network-driven activity is attributable to increased recurrent excitatory connections among CA3 neurons or another means of network activation is unknown. Although nearly 50% of hilar mossy cells die after even moderate TBI (Toth et al., 1997; Santhakumar et al., 2000), these cells could also contribute to enhanced excitability of inhibitory neurons and cannot be ruled out.
Synaptic connections between local principal cells and eGFP-GABA neurons identified by photostimulation were relatively sparse in control slices. Slice geometry, septotemporal location, depth within the slice, and the distance of recorded eGFP-GABA neuron from afferent principal neurons could all influence the ability to identify synaptic connections in slices. For consistency, recordings were obtained from transverse slices of the dorsal dentate gyrus near the injury site. Responses in contralateral slices acted as internal controls and were nearly identical with responses in slices from control animals. Individual granule cells often make only a single contact onto putative hilar somatostatin interneurons (Acsády et al., 1998), and the probability of detecting granule cell input to hilar GABA neurons in paired recordings is normally low (Scharfman et al., 1990). Regardless, evoked activity reflecting intact local synaptic inputs to eGFP-GABA neurons was increased after TBI and could be elicited from relatively distant sites. This was consistent with our findings that interneurons are more active and receive more spontaneous excitatory input after TBI. Increased CA3 network activation or involvement of secondary excitatory connections (e.g., with mossy cells) or extrasynaptic mechanisms (e.g., altered glutamate clearance) after photostimulation could underlie some of the increased input to eGFP-GABA neurons over several milliseconds and cannot be ruled out. However, the relatively short onset latencies of eEPSCs were time-locked to the photostimulus and were often similar to evoked granule cell-to-granule cell synaptic connections after TBI (Hunt et al., 2010), consistent with a monosynaptic response at the onset of evoked activity. Increased synaptic input to eGFP-GABA neurons after TBI likely arises from activity of both granule cells and CA3 pyramidal cells, with CA3 input being predominant under normal conditions because of the intrinsic activity of pyramidal cells and granule cells contributing during states of heightened activity.
The functional implications of enhanced excitability of hilar eGFP-GABA neurons on dentate gyrus network function after TBI remains speculative. One possibility is that enhanced input–output relationships of eGFP-GABA neurons partially compensate for the selective loss of these neurons after injury (Zhang et al., 2009). However, injury-induced modifications of GABAergic networks appear to reflect a target domain-specific shift of inhibitory control over granule cells. Increased connectivity of eGFP-GABA neurons likely augments inhibitory shunting of excessive excitatory perforant pathway inputs into the dendrites of granule cells (Oliva et al., 2000), which would tend to prevent the activation of new recurrent excitatory circuits formed by mossy fiber sprouting. Alternatively, perisomatic inhibition provided by basket cells is important for controlling action potential firing and synchronizing principal cell output (Lytton and Sejnowski, 1991; Cobb et al., 1995; Miles et al., 1996). These cells receive fewer excitatory synaptic inputs, and a higher basket cell-to-granule cell transmission failure rate is observed in epilepsy (Zhang and Buckmaster, 2009). An interesting, albeit speculative, possibility is that the inhibitory control provided by dentate basket cells is diminished after injury, because granule cell synchrony is augmented by the emergence of new recurrent mossy fiber circuits (Dudek and Spitz, 1997; Hunt et al., 2009). Regardless, the global loss, preservation, or enhancement of synaptic inhibition after TBI may not be as critical to principal cell function as GABA neuron subtype-specific modifications of inhibitory circuits.
It is possible that injury-induced changes in the connectivity and excitability of hilar eGFP-GABA interneurons reflect a pathogenic response. These neurons have extensive axonal arborizations and large terminal fields in the dentate molecular layer (Leranth et al., 1990; Freund and Buzsáki, 1996; Katona et al., 1999; Buckmaster et al., 2002). Increased input–output relationships of surviving GABA cells might allow for a single neuron to exert greater control over a larger number of principal cells. Inhibitory axon sprouting has long been a proposed mechanism for network synchronization in epilepsy (Babb et al., 1989), and modeling suggests that highly interconnected neurons promote this activity (Traub and Wong, 1982; Morgan and Soltesz, 2008). Dendritically projecting GABA neurons have been proposed to time rhythmic cortical cell discharges (Szabadics et al., 2001) and to drive ictogenesis (Wendling et al., 2002). Moreover, Bonifazi et al. (2009) recently reported that GABAergic interneurons with high numbers of inputs and outputs can act as “superconnected hubs” to coordinate synchronous activation of principal neurons. A depolarizing shift in the GABA reversal potential might also contribute to enhanced firing frequency of hilar eGFP-GABA neurons after TBI (van den Pol et al., 1996). Fewer inhibitory neurons, each with greater influence over more principal cells, implies that periodic failure of these neurons (e.g., transient inactivation during excessive or sustained excitation) may be more detrimental to restraining excitatory inputs to granule cells after injury than in controls.
Footnotes
-
This work was supported by an Epilepsy Foundation predoctoral fellowship (R.F.H.) and funding from National Institutes of Health Grants AG21981 (S.W.S.) and NS052302 (B.N.S.). Thanks to Dr. Peter Gyarmati for helpful discussion during the preparation of this manuscript.
- Correspondence should be addressed to Dr. Bret N. Smith, Department of Physiology, University of Kentucky College of Medicine, MS508 Chandler Medical Center, 800 Rose Street, Lexington, KY 40536. bret.smith{at}uky.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}