

Abstract
Synaptotagmin-1, the Ca2+ sensor for fast neurotransmitter release, was proposed to function by Ca2+-dependent phospholipid binding and/or by Ca2+-dependent soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex binding. Extensive in vivo data support the first hypothesis, but testing the second hypothesis has been difficult because no synaptotagmin-1 mutation is known that selectively interferes with SNARE complex binding. Using knock-in mice that carry aspartate-to-asparagine substitutions in a Ca2+-binding site of synaptotagmin-1 (the D232N or D238N substitutions), we now show that the D232N mutation dramatically increases Ca2+-dependent SNARE complex binding by native synaptotagmin-1, but leaves phospholipid binding unchanged. In contrast, the adjacent D238N mutation does not significantly affect SNARE complex binding, but decreases phospholipid binding. Electrophysiological recordings revealed that the D232N mutation increased Ca2+-triggered release, whereas the D238N mutation decreased release. These data establish that fast vesicle exocytosis is driven by a dual Ca2+-dependent activity of synaptotagmin-1, namely Ca2+-dependent binding both to SNARE complexes and to phospholipids.
Introduction
Neurotransmitter release is mediated by synaptic vesicle exocytosis in a reaction that is catalyzed by soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex assembly, and is triggered by Ca2+ binding to the two C2 domains of the synaptic vesicle protein synaptotagmin-1 (Südhof, 2004; Brunger, 2005; Sorensen, 2005). In vitro, Ca2+ binding causes synaptotagmin-1 to form complexes with both phospholipids and with SNARE complexes (Bennett et al., 1992; Yoshida et al., 1992; Davletov and Südhof, 1993; Sollner et al., 1993; Chapman and Jahn, 1994; Chapman et al., 1995; Li et al., 1995; Banerjee et al., 1996; Kee and Scheller, 1996; Schiavo et al., 1997; Fernandez et al., 2001; Rickman and Davletov, 2003; Rickman et al., 2004; Bowen et al., 2005). In vivo, mutations that selectively decrease synaptotagmin-1 phospholipid binding cause a corresponding decrease of release (Fernandez-Chacon et al., 2001; Li et al., 2006), whereas mutations that enhance phospholipid binding produce an increase of release (Rhee et al., 2005). These data demonstrated that Ca2+ binding to synaptotagmin-1 triggers neurotransmitter release and that Ca2+-dependent phospholipid binding is an essential component of the Ca2+-triggering reaction.
In contrast to phospholipid binding, however, SNARE binding by synaptotagmin-1 has been difficult to examine. No synaptotagmin-1 mutation is known that selectively enhances or depresses SNARE binding without changing phospholipid binding, precluding an in vivo analysis of SNARE binding by synaptotagmin-1. A large body of indirect evidence, primarily based on transfected cells, supports the importance of SNARE binding by synaptotagmin-1 (Wang et al., 2001; Zhang et al., 2002; Shin et al., 2003; Bai et al., 2004), but no in vivo data confirming this importance are available. Thus, a major question is now whether synaptotagmin-1 acts primarily as a Ca2+-dependent phospholipid binding protein in triggering exocytosis, with its SNARE binding having a separate role (possibly in priming vesicles for subsequent release), or whether the interactions of synaptotagmin-1 with both SNAREs and phospholipids collaborate in Ca2+ triggering of neurotransmitter release.
We previously analyzed two aspartate-to-asparagine substitutions in the Ca2+-binding site of the C2A domain (D232N and D238N) that interfere with Ca2+ binding to the C2A domain in slightly different ways, but do not alter its atomic structure, and have little effect on Ca2+-dependent phospholipid binding by synaptotagmin-1 (Fernandez-Chacon et al., 2002). In knock-in mice, the D238N mutation produced no major change in synaptic function, but the D232N mutation caused an unexplained increase of synaptic depression during repetitive stimulation (Fernandez-Chacon et al. 2002). Moreover, a separate study using rescue experiments of synaptotagmin-1 knock-out neurons with D232N-mutant synaptotagmin-1 suggested that the D232N mutation may increase the Ca2+ sensitivity of evoked responses (Stevens and Sullivan, 2003), although again no biochemical basis for the changes was identified.
To clarify these puzzling observations and to search for a potential mechanism to account for the unexplained electro physiological phenotype of the D232N mutation, we analyzed the effect of the D232N and D238N mutations on the biochemical properties and physiological functions of native synapto tagmin-1 expressed in knock-in mice at physiological levels. This approach avoids artifacts produced by recombinant proteins and/or by overexpression experiments. Our current data demonstrate that, in addition to Ca2+-dependent phospholipid binding, Ca2+-triggered SNARE interactions by synaptotagmin-1 are rate-limiting in stimulating fast neurotransmitter release.
Materials and Methods
Mouse breedings.
All analyses were performed on littermate offspring from heterozygous breedings. For the biochemical experiments and for cultures of cortical neurons, offsprings of matings between standard D232N- or D238N-mutant heterozygotes were used. For the hippocampal neuronal cultures, offspring from D232N/D238N-mutant compound heterozygotes were used. All genotyping was performed as described previously (Fernandez-Chacon et al., 2002).
Coimmunoprecipitations.
One gram of mouse brain was homogenized with a tissue homogenizer (Thomas Scientific, Philadelphia, PA) in 20 ml of 50 mm HEPES-NaOH, pH 6.8, 0.1 m NaCl, 4 mm EGTA, 5 μg/ml leupeptin, 2 μg/ml aprotinin, 1 mm PMSF, and 1 mm DTT. Triton X-100 (1%) was added, proteins were extracted for 1 h at 4°C with rocking, insoluble proteins were removed by centrifugation (150,000 × g for 1 h), and the supernatant was used for experiments. Coimmunoprecipitation reactions were performed with polyclonal syntaxin-1 antibodies, U6251 (15 μl of serum) or monoclonal synaptobrevin 2 antibody Cl69.1 (10 μl of ascite) attached to protein A- or G-Sepharose beads (10 μl; Amersham Biosciences, Piscataway, NJ) in a 1 ml volume of immunoprecipitation buffer (50 mm HEPES-NaOH, pH 6.8, 0.1 m NaCl, 4 mm EGTA, 2 mm MgCl2, 0.5% Triton X-100 in the presence of different concentrations of free Ca2+). Free Ca2+ concentrations were calculated with EqCal software (Biosoft, Ferguson, MO). Coimmunoprecipitation reactions were incubated at 4°C for 2 h with rocking, beads were washed five times with 1 ml of corresponding immunoprecipitation buffer, and bound proteins were eluted with SDS-PAGE sample buffer. Synaptotagmin-1 (Cl41.1 or V216), synaptosomal-associated protein of 25 kDa (SNAP-25) (Cl71.2 or P913), synataxin-1 (HPC-1 or U6251), synaptobrevin 2 (Cl69.1 or P939), synaptophysin 1 (Cl43.1), Munc-18 (purified mouse anti-Munc-18 monoclonal antibody; catalog #610337; BD Transduction Laboratories, Lexington, KY), and complexins 1 and 2 (L668 and L669) were quantified by immunoblotting using 125I-labeled secondary antibodies.
Phospholipid binding.
Total membranes were isolated from mouse brain homogenates (1 g of mouse brain/20 ml of 50 mm HEPES-NaOH, pH 6.8, 0.1 m NaCl, 4 mm EGTA) after centrifugation at 150,000 × g for 1 h. Samples were rehomogenized in 20 ml of 50 mm HEPES-NaOH, pH 6.8, 0.1 m NaCl, 4 mm EGTA, and for partial digestion of synaptotagmin-1, 0.005% trypsin and 0.053 mm EDTA were added, incubated for 1 h at room temperature with rotation, and 25 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mm PMSF, 1 mm DTT, and 2% goat serum were added. The trypsinized membranes were then centrifuged at 150,000 × g for 1 h, and the cytosolic domain of synaptotagmin-1 was recovered in the supernatant and stored at −80°C. Phospholipid binding assays were performed using heavy liposomes with a synaptic phospholipid composition and phosphatidylinositol phosphate (PIP) and phosphatidylinositol bisphosphate (PIP2) as indicated previously (Deutsch and Kelly, 1981; Benfenati et al., 1989; Rhee et al., 2005; Li et al., 2006). After SDS-PAGE, synaptotagmin-1 associated with heavy liposome was quantified by immunoblotting using Cl41.1 and 125I-labeled secondary antibody.
Electrophysiological recordings from cortical neurons cultured at high density.
Primary cortical neurons were isolated from postnatal day 1 (P1) mice of wild-type or synaptotagmin-1 D232N-mutant mice, dissociated by trypsin digestion, and plated on poly-d-lysine-coated glass coverslips (Maximov and Südhof, 2005). Neurons were cultured in vitro for 14–18 d in MEM (Invitrogen, San Diego, CA) supplemented with B27 (Invitrogen), glucose, transferrin, fetal bovine serum, and Ara-C (Sigma, St. Louis, MO). Synaptic responses were triggered by 1 ms current injection (900 μA) through a local extracellular electrode (FHC concentric bipolar electrode; catalog #CBAEC75) with a model 2100 Isolated Pulse Stimulator (A-M Systems, Carlsborg, WA), and recorded in whole-cell mode using a Multiclamp 700A amplifier (Molecular Devices, Union City, CA). Data were digitized at 10 kHz with a 2 kHz low-pass filter. The whole-cell pipette solution contained the following (in mm): 135 CsCl2, 10 HEPES, 1 EGTA, 4 Mg-ATP, 0.4 Na-GTP, and 10 QX-314 (lidocaine N-ethyl bromide), pH 7.4. The bath solution contained the following (in mm): 140 NaCl, 5 KCl, 2 CaCl2, 0.8 MgCl2, 10 HEPES, and 10 glucose, pH 7.4. IPSCs were pharmacologically isolated by addition of 50 μm d-AP5 and 20 μm CNQX to the bath solution. Hypertonic sucrose (0.5 m) solution was applied for 20 s through a perfusion system at a speed of 4 ml/min. Series resistance was compensated to 60–70%, and recordings with series resistances of >15 MΩ were not included. Data were analyzed using Clampfit 9.02 (Molecular Devices) or Igor 4.0 (Wavemetrics, Lake Oswego, OR).
Synaptic responses were recorded 2 min after obtaining whole-cell patch before recording of evoked synaptic responses, allowing the internal pipette solution to diffuse into the patched neuron. In almost all cases, we obtained robust evoked responses from neurons we recorded. Stimulation intensity of 900 μA was suprathreshold in most of the cases. We maintained the stimulation at this level for all recordings to reduce the variability among cells. Most, if not all, of axons passing in a close vicinity of the stimulation electrode were activated. The major source of variability in our experimental condition was attributable to the synaptic density. To minimize this variation, we plated wild-type and mutant neurons at similar densities and recorded on the same day between two genotypes.
Electrophysiology from microisland cultures of hippocampal neurons.
Cultures of hippocampal neurons from D232N- or D238N-mutant mice were prepared at P0 or P1 on microislands of glia cells (preplated in 10% fetal bovine serum), under conditions favoring formation of autapses, and used for experiments after 10–20 d in culture (Rhee et al., 2005; Li et al., 2006). Before seeding neurons in a density of 500 per cm2, the medium was exchanged to Neurobasal medium A (Invitrogen) with supplement B27 (Invitrogen). Only dots containing single neurons were used. Excitatory EPSCs were analyzed in extracellular medium containing the following (in mm): 140 NaCl, 2.4 KCl, 10 HEPES, 10 glucose, 4 CaCl2, 4 MgCl2, pH 7.3, 300 mOsm. Synaptic transmission was recorded in whole-cell configuration under voltage clamp using 1–2 ms depolarizations from −75 to 0 mV to induce action potentials. Hypertonic sucrose solutions contained 0.5 m sucrose in addition to the regular external solution. Patch pipette solutions included the following: 125 mm K-gluconate, 10 mm NaCl, 4.6 mm MgCl2, 4 mm ATP-Na2, 15 mm creatine phosphate, 20 U ml−1 phosphocreatine kinase, 1 mm EGTA, 300 mOsm and adjusted to pH 7.3. All analyses procedures were performed as described previously (Fernandez-Chacon et al., 2001).
Statistics.
All data are presented as means ± SEMs. Unpaired or paired Student's t tests or two-way ANOVA tests were used as indicated to assess significance.
Results
The D232N mutation increases Ca2+-dependent but not Ca2+-independent SNARE binding by synaptotagmin-1
We first examined the effect of the D232N and D238N mutations on Ca2+-dependent and Ca2+-independent interactions of synaptotagmin-1 with SNARE complexes using immunoprecipitations of brain proteins from knock-in mice. In initial experiments, we probed these interactions at different ionic strength because the relative degree of Ca2+-dependent versus Ca2+-independent binding of wild-type synaptotagmin-1 to SNARE complexes is strongly dependent on the ionic strength (Fig. 1A) (Tang et al., 2006). Unexpectedly, the D232N mutation strongly increased Ca2+-dependent binding of synaptotagmin-1 to SNARE complexes, but had no effect on Ca2+-independent binding (Fig. 1B). The D238N mutation, in contrast, did not alter Ca2+-dependent SNARE binding by synaptotagmin-1.

Immunoprecipitation analysis of native wild-type and mutant synaptotagmin-1 binding to SNARE complexes using polyclonal syntaxin-1 antibodies. A, Representative immunoblots of syntaxin-1 immunoprecipitates. Brain proteins from synaptotagmin-1 D232N-mutant and wild-type mice (WT) were solubilized with 1% Triton X-100 to promote SNARE complex formation, and immunoprecipitated with polyclonal syntaxin-1 antibodies at increasing NaCl concentrations, with or without 1 mm free Ca2+. Immunoprecipitates were blotted with monoclonal antibodies to synaptotagmin-1 (Syt-1), SNAP-25 (Cl 71.1), and syntaxin-1 (HPC-1); bands were probed with 125I-labeled secondary antibodies and visualized in a phosphorimager. B, Quantitations of synaptotagmin-1 and SNAP-25 coimmunoprecipitated with syntaxin-1 at increasing NaCl concentrations in the presence or absence of 1 mm Ca2+. Quantitations were performed with 125I-labeled secondary antibodies; amounts of coimmunoprecipitated proteins in this and all other immunoprecipitation experiments are normalized for the amount of the immunoprecipitated protein [i.e., syntaxin-1 (Fig. 1) or synaptobrevin-2 (Fig. 2); n = 3]. In this and all following figures, gray symbols indicate WT, red symbols indicate D232N mutant, and blue symbols indicate D238N mutant. Data shown are means ± SEMs. C, Quantitations of synaptotagmin-1, Munc18-1, and complexins 1/2 (Cpx 1/2) coimmunoprecipitated with syntaxin-1 in the presence of increasing Ca2+ concentrations in 100 mm NaCl (n = 3). Statistical analyses in B and C were performed with a two-way ANOVA test. n.s., Nonsignificant.

Immunoprecipitation analysis of synaptotagmin-1 binding to SNARE complexes using monoclonal synaptobrevin-2 antibodies. A, Representative immunoblots of immunoprecipitations performed in 100 mm NaCl as described in Figure 1, except that monoclonal synaptobrevin-2 antibodies were used (Syt-1, synaptotagmin-1; Syp 1, synaptophysin-1; Syb 2, synaptobrevin-2). Bands were visualized by enhanced chemiluminescence. B, Quantitations of synaptotagmin-1 coimmunoprecipitated with synaptobrevin-2 in the presence or absence of 1 mm free Ca2+. Quantitations were performed with 125I-labeled secondary antibodies (means ± SEMs; n = 3).
We next measured the Ca2+ dependence of SNARE binding by synaptotagmin-1 in buffer of approximately physiological ionic strength (100 mm NaCl, 50 mm HEPES-NaOH). The D232N mutation increased SNARE binding at all Ca2+ concentrations tested (Fig. 1C). Again, the D238N mutation had no obvious effect on SNARE binding. Ca2+ did not change the amount of other coimmunoprecipitated proteins (complexins 1/2 and Munc18-1). Quantitations showed that, in the absence of Ca2+, ∼0.6% of the total synaptotagmin-1 was stably bound to syntaxin-1, whereas Ca2+ increased this to nearly 10% of the total synaptotagmin-1 in D232N-mutant brains. In contrast, ∼4% of the total Munc18-1, SNAP-25, synaptobrevin 2, and complexin 1/2 are stably bound to syntaxin-1 in the presence or absence of Ca2+. The fact that complexin, a protein that only binds to assembled SNARE complexes (McMahon et al., 1995), is coimmunoprecipitated with the SNARE antibodies in these experiments demonstrates that synaptotagmin-1 binding to SNARE complexes is studied. Nevertheless, to ensure that the effect of the D232N mutation on the Ca2+-dependent interaction of synaptotagmin-1 with SNARE complexes was not an artifact of the syntaxin-1 immunoprecipitations, we immunoprecipitated SNARE complexes with antibodies to synaptobrevin/VAMP (vesicle-associated membrane protein) and measured the binding of D232N-mutant synaptotagmin-1 as a function of Ca2+ (Fig. 2). Again, we observed an approximate twofold increase in binding by the D232N mutation.
Viewed together, these data demonstrate that the Ca2+-dependent interaction of native synaptotagmin-1 with native SNARE complexes is selectively enhanced by the D232N mutation, whereas the similar D238N substitution does not significantly alter SNARE complex binding. Although the lack of an atomic structure of the synaptotagmin-1/SNARE complex makes it difficult to understand the structural basis for the strikingly differential effect of two very similar mutations, it seems likely that Ca2+-dependent binding of SNARE complexes to synaptotagmin-1 involves negatively charged SNARE residues that are normally repelled by aspartate 232, thereby increasing the binding strength in the absence of aspartate 232.
Effect of the D232N and D238N mutations on Ca2+-dependent phospholipid binding
To investigate the effects of the D232N and D238N mutations on Ca2+-dependent phospholipid binding by synaptotagmin-1 in greater detail than we had done previously (Fernandez-Chacon et al., 2002), we decided to focus on native synaptotagmin-1 because there may be differences between recombinant and native proteins. To optimize the binding reactions, we first examined the effect of increasing the concentrations of phosphatidylinositols in the phospholipid bilayer on the Ca2+-dependent and Ca2+-indepenent binding of native synaptotagmin-1 (Fig. 3A,B). When we measured the binding of native synapto tagmin-1 to vesicles with a lipid composition resembling that of synaptic vesicles but with increasing concentrations of PIP and PIP2, we found that, as expected (Li et al., 2006), phosphatidylinositol phosphates strongly coactivated Ca2+-dependent phospholipid binding by synaptotagmin-1 (Fig. 3A,B). Surprisingly, however, we did not observe any Ca2+-independent binding of native synaptotagmin-1 to phospholipids containing even high concentrations of PIP and PIP2, a result that differs from previous studies using recombinant proteins (Bai et al., 2004; Li et al., 2006). To resolve this discrepancy, we directly compared phospholipid binding by native and recombinant synaptotagmin-1 fragments with identical sequences (Fig. 3C,D). Binding reactions at different ionic strengths revealed that, even at low ionic strengths, only recombinant but not native synaptotagmin-1 exhibited Ca2+-independent binding to phospholipid membranes containing 0.5% PIP and 0.1% PIP2 (Fig. 3C,D). Thus, for native synaptotagmin-1, PIP and PIP2 are coactivators of Ca2+-dependent phospholipid binding but are not sufficient to mediate Ca2+-independent binding.

Comparison of Ca2+-induced phospholipid binding by equivalent fragments of recombinant and native synaptotagmin-1. A, B, Analysis of the effect of incorporating phosphatidylinositides (PIP, PIP2) into liposomes on the Ca2+-independent and Ca2+-dependent binding of native synaptotagmin-1. Panels show representative immunoblots visualized with 125I-labeled secondary antibodies (A) and quantitations of binding (B) in the presence or absence of 0.1 mm Ca2+ (means ± SEMs; n = 3). Experiments were performed with the cytosolic region of synaptotagmin-1 obtained by mild trypsin digestion of total brain membranes, and with heavy liposome with a “synaptic” composition (41% phosphatidylcholine, 32% phosphatidylethanolamine, 12% phosphatidylserine, 5% phosphatidylinositol, and 10% cholesterol by weight). Binding was performed with a centrifugation assay in which synaptotagmin-1 bound to liposomes is measured by immunoblotting. C, D, Recombinant (C) and native (D) synaptotagmin-1 fragments were prepared as indicated (C1, D1). C2, D2, Representative immunoblots visualized with 125I-labeled secondary antibodies of recombinant (C2) and native (D2) synaptotagmin-1. The recombinant glutathione S-transferase (GST)-rat synaptotagmin-1 (GST-rSyt1; residue 86–421) includes the trypsin-hypersensitive site that is cleaved in native synaptotagmin-1; thus, the native and recombinant trypsin-produced fragments contain identical sequences. Fragments were bound at different NaCl concentrations with and without 0.1 mm Ca2+ to liposomes with a phospholipid synaptic vesicle composition (phosphatidylcholine, 41%; phosphatidylethanolamine, 32%; phosphatidylserine, 12%; phosphatidylinositol, 5%; cholesterol, 10%), and with 0.1% PIP and 0.5% PIP2. C3, D3, Binding was quantified for both recombinant (C3) and native (D3) synaptotagmin-1 using immunoblotting with 125I-labeled secondary antibodies and phosphorimager detection, and is expressed as percentage of the maximum. Data are presented as means ± SEMs (n = 3). Statistical analyses were performed with a two-way ANOVA test.
We next tested the effects of the DN mutations on Ca2+-dependent phospholipid binding. Consistent with previous results (Fernandez-Chacon et al., 2002), the D232N mutation did not alter the apparent Ca2+ affinity or extent of phospholipid binding by native synaptotagmin-1 (Fig. 4A,B). In contrast, the D238N mutation caused a small but significant decrease in phospholipid binding (Fig. 4B). Both mutations destabilized the synaptotagmin-1/Ca2+/phospholipid complex at high NaCl concentrations (Fig. 4C). Viewed together, these experiments establish that two closely spaced mutations in the Ca2+-binding sites of the C2A domain have distinct effects on the biochemical properties of synaptotagmin-1 at physiological ionic strengths: whereas the D232N mutation selectively enhances Ca2+-dependent interactions of synaptotagmin-1 with SNARE complexes without altering phospholipid binding, the D238N mutation does not alter SNARE binding but decreases the apparent Ca2+ affinity of phospholipid binding.

Ca2+-dependent binding of native wild-type or mutant synaptotagmin-1 to liposomes. All experiments were performed as in Figure 3, A and B. A, B, Analysis of the Ca2+ concentration dependence of the binding of native wild-type (WT) or D232N- and D238N-mutant synaptotagmin-1 to liposomes containing 0.5% PIP/0.1% PIP2 in 100 mm NaCl. Panels show representative immunoblots (A) and quantitations of binding (B). C, NaCl concentration dependence of the binding of native WT or D232N- and D238N-mutant synaptotagmin-1 to liposomes containing 0.5% PIP/0.1% PIP2 in the absence or presence of 1 mm free Ca2+. B and C show means ± SEMs (n = 3 for D232N; n = 4 for D238N). Statistical analyses were performed by two-way ANOVA test.
The D232N mutation increases fast synchronous Ca2+-triggered release in inter-neuronal synapses
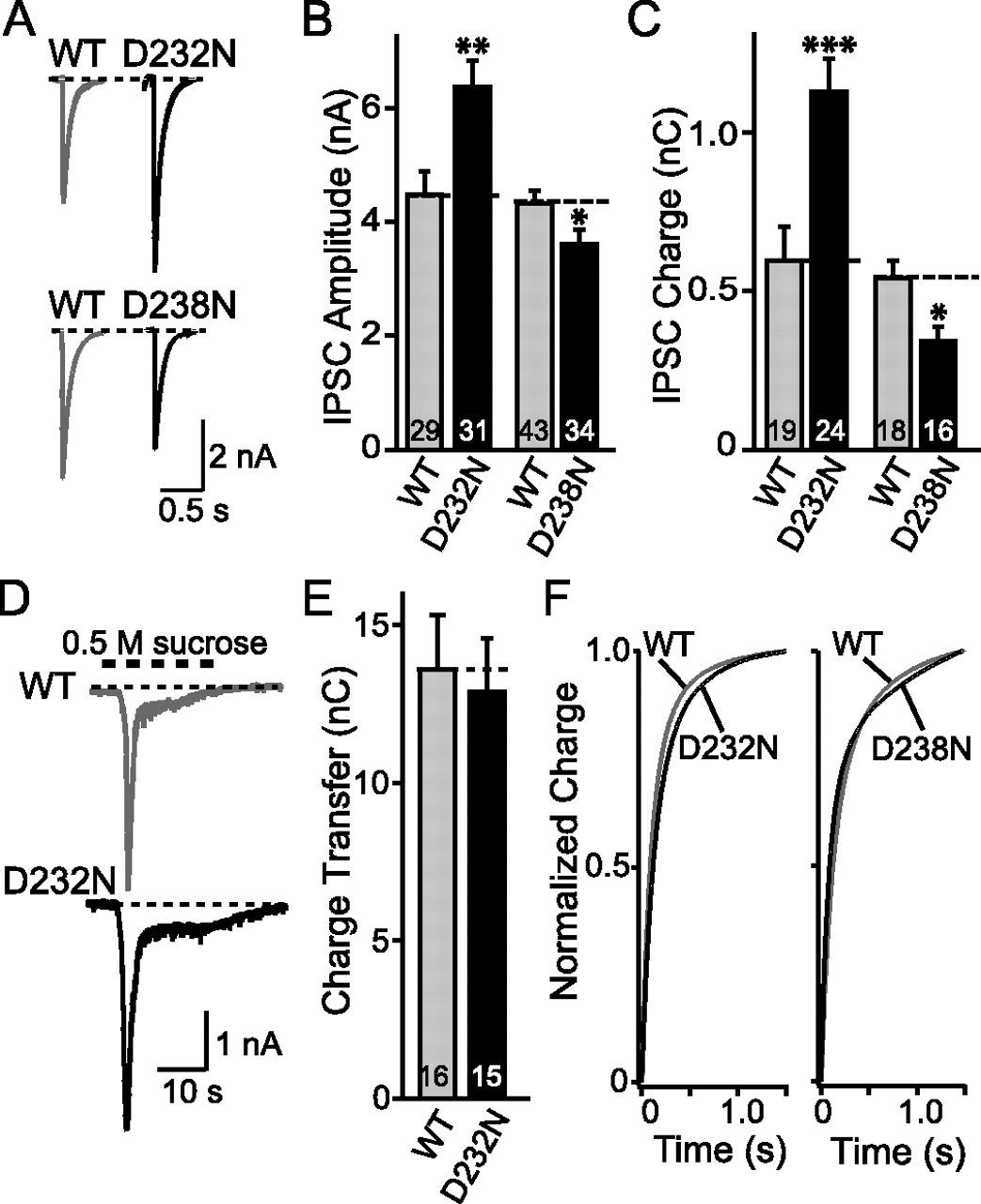
To test the effect of the aspartate mutations in the Ca2+-binding site on neurotransmitter release, we analyzed synaptic responses in cultured cortical neurons from littermate wild-type and D232N- or D238N-mutant mice. We studied evoked IPSCs by stimulating presynaptic neurons with a focal electrode (Maximov and Südhof, 2005) (Fig. 5A). We found that, in absolute terms, the D232N mutation caused a ∼1.5-fold increase in the IPSC amplitude, and a ∼2.0-fold increase in the synaptic charge transferred per action potential (Fig. 5B,C). The D238N mutation, in contrast, caused moderate, statistically significant decreases in IPSC amplitude and synaptic charge transfer. Thus, the two point mutations alter release in a manner that precisely correlates with their effects on the biochemical phospholipid- and SNARE-binding activities of synaptotagmin-1.

Evoked IPSCs in synapses containing wild-type or mutant synaptotagmin-1. A, Representative traces of IPSCs evoked by focal stimulation in cortical neurons cultured from littermate wild-type (WT) and D232N- or D238N-mutant mice. Recordings were made in 2 Ca2+/0.8 Mg2+; stimulation artifacts were truncated for display purposes. B, C, Summary graphs of the amplitudes and total charge transfer during IPSCs evoked at low frequency (<1 Hz) in neurons from littermate WT and D232N- (B) or D238N-mutant mice (C). D, Representative traces of IPSCs induced by hypertonic sucrose (0.5 m). E, Total synaptic charge transfer integrated over 30 s in response to a 0.5 m sucrose application for 20 s. F, Time course of synaptic responses to isolated action potentials in cultured cortical neurons from littermate WT and D232N- or D238N-mutant neurons. Graphs show the integrated synaptic charge transfer plotted as a function of time for D232N-mutant (left panel) and D238N-mutant neurons (right panel) compared with their littermate control cultures (D232N WT, n = 18; D232N, n = 21; D238N WT, n = 23; D238N, n = 20). Data shown are means ± SEMs. Statistical significance was assessed by unpaired t test (*p < 0.05; **p < 0.01; ***p < 0.001; numbers of recorded neurons are indicated in the bars).
Several potential causes for the large increase in synaptic responses in D232N-mutant synapses can be suggested, for example, changes in the size of the readily releasable pool (RRP) of vesicles, the mode of release (synchronous vs asynchronous), or the Ca2+ sensitivity of release. We tested these possibilities by first measuring the size of the RRP using an application of hypertonic sucrose (Rosenmund and Stevens, 1996), but failed to detect a significant change in D232N-mutant synapses in either the size of the RRP (Fig. 5D,E) or the kinetic properties (data not shown) of hypertonic sucrose-induced release. We next examined the possibility that the D232N mutation may have shifted the mode of release from synchronous to asynchronous by measuring the time course of release, but again did not observe a significant difference between wild-type and D232N- or D238N-mutant synapses (Fig. 5F). Finally, we assessed the apparent Ca2+ sensitivity of release by titrating the amount of release as a function of the extracellular Ca2+ concentration (Fig. 6). We found that the D232N mutation caused a ∼1.5-fold increase in the apparent Ca2+ affinity of release, whereas the D238N mutation had no significant effect (Fig. 6C,D; Table 1). Note that, in all of these studies, we measure absolute response sizes in multiple independent cultures obtained from littermate knock-in and control mice. These data are in agreement with rescue studies in autapses (Stevens and Sullivan, 2003). Interestingly, the apparent Ca2+ affinity of release for extracellular Ca2+ in inter-neuronal wild-type synapses measured here closely parallels that observed previously in autapses (Fernandez-Chacon et al., 2001).

Ca2+ dependency of synaptic responses in D232N- and D238N-mutant synapses. A, B, Representative traces of evoked IPSCs at the indicated concentrations of free Ca2+. Stimulation artifacts are removed for display purposes. C, D, Dose–response curves of evoked IPSCs in neurons from littermate wild-type (WT) and D232N- (C) or D238N-mutant mice (D). Data shown are means ± SEMs (n = 4–22 neurons depending on Ca2+ concentration). Statistical significance was assessed by a two-way ANOVA test. The curve shown represents the result of a fit of the data to a Hill function that is described in Table 1.
Ca2+ affinities in excitatory and inhibitory synapses
These data indicate that the D232N mutation greatly increases the release probability of synaptic vesicles by enhancing their Ca2+ sensitivity, whereas the D238N mutation slightly decreases the release probability. To obtain independent evidence for this conclusion, we examined paired-pulse responses in the mutant synapses because a decrease in release probability usually causes an increased paired-pulse ratio (i.e., facilitation), whereas an increase in release probability causes a decreased paired-pulse ratio (i.e., depression) (Zucker and Regehr, 2002). Indeed, application of two closely spaced action potentials with different interstimulus intervals revealed that D232N-mutant synapses exhibited increased depression compared with wild-type control neurons, whereas D238N-mutant synapses exhibited decreased depression (i.e., facilitation) relative to wild-type neurons (Fig. 7A,B). Moreover, we monitored short-term plasticity during repetitive train stimulation in D232N- and D238N-mutant synapses. Again, we observed increased depression in D232N-mutant and decreased depression in D238N-mutant neurons (Fig. 7C,D). These results are consistent with an increase of release probability in D232N-mutant, and a decrease of release probability in D238N-mutant synapses, and are in agreement to previous data obtained in excitatory autapses (Fernandez-Chacon et al., 2002). In the synaptotagmin-1 mutant synapses examined here, just as in the synaptotagmin-1-deficient synapses we studied previously (Geppert et al., 1994; Maximov and Südhof, 2005), the effect was restricted to fast synchronous release, and no significant effect on asynchronous release during stimulus trains was observed (Fig. 8).
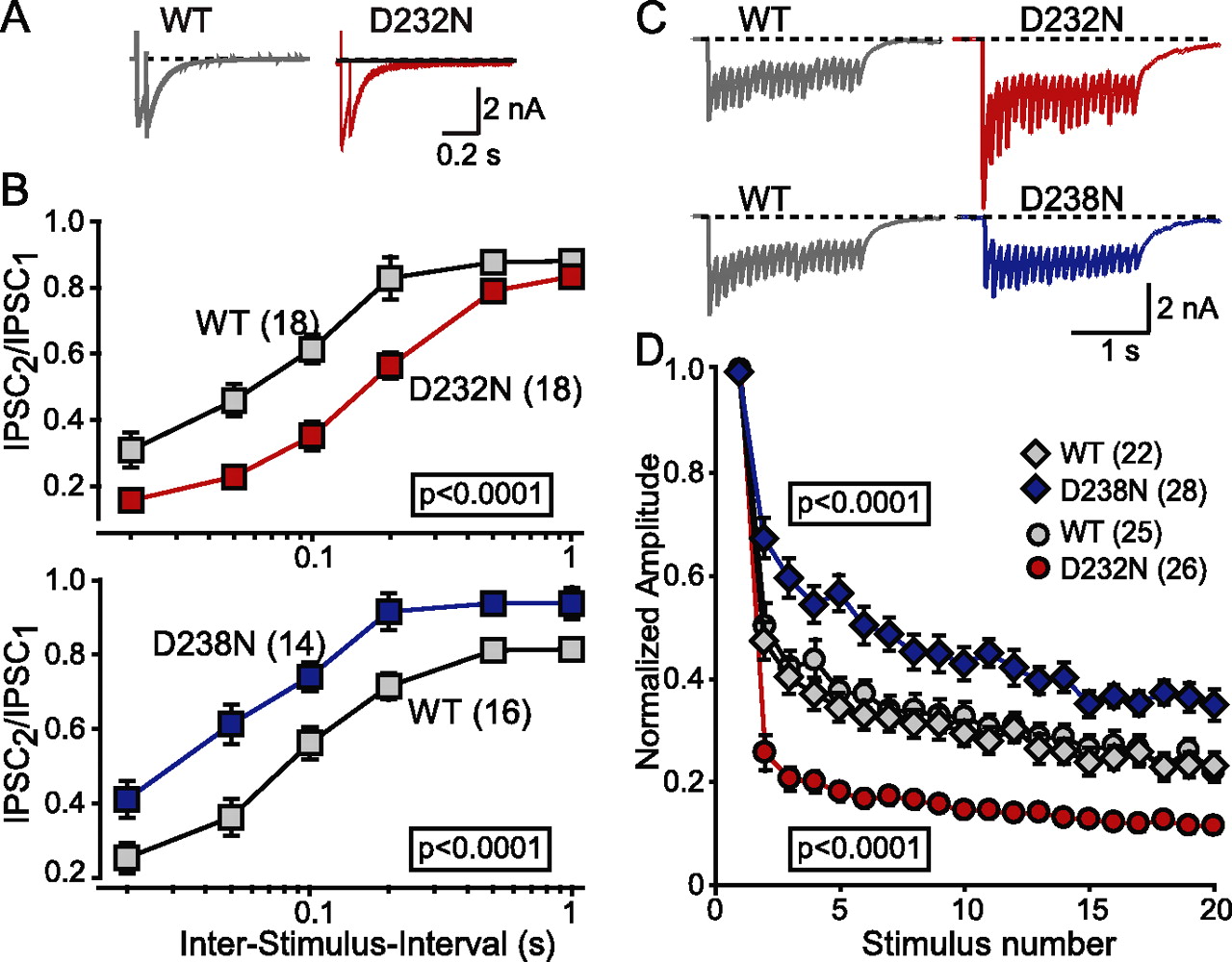
Short-term synaptic plasticity in synapses containing wild-type (WT) or mutant synaptotagmin-1. A, B, Representative IPSCs (A; interstimulus interval, 50 ms) and summary graphs (B) of paired-pulse stimulation experiments. IPSCs were examined in responses to two closely spaced action potentials elicited by focal stimulation in cultured cortical neurons. The summary graph (B) displays the ratio of the second to the first IPSC amplitude as a function of the interstimulus interval. C, Representative traces of IPSCs during a 2 s, 10 Hz stimulus train. D, Summary graphs of IPSC amplitudes during 10 Hz stimulus trains. IPSCs were normalized to the first response. Note that stimulation artifacts are removed for display purposes. In B and D, data shown are means ± SEMs (number of analyzed neurons are shown in brackets). Statistical significance was assessed by a two-way ANOVA test.

Effect of D232N and D238N mutations on asynchronous release during and after a 10 Hz stimulus train. A, Representative traces of inhibitory synaptic responses in D232N- or D238N-mutant neurons and littermate wild-type (WT) control neurons when 10 stimuli at 10 Hz were elicited by focal stimulation. Delayed asynchronous release was defined as the synaptic response observed 100 ms after the last stimulus (shaded area). Recordings were from cortical neurons cultured at high density from littermate wild-type and D232N- or D238N-mutant mice. B–G, Total charge transfer during the first response (B, E), total delayed release (C, F), and total amount of release integrated over the entire stimulus train (D, G) in D232N- and D238N-mutant neurons, respectively. Note that stimulation artifacts are removed for better presentation. Data shown are means ± SEMs. *p < 0.05; **p < 0.01.
The D232N mutation increases the apparent Ca2+ sensitivity of release in excitatory autapses
All physiological experiments reported here up to this point were performed in inhibitory inter-neuronal synapses. To test whether the phenotype observed is applicable to excitatory responses, and to validate the conclusions with a different approach, we monitored synaptic transmission in autapses. In these experiments, we crossed compound heterozygous D232N/D238N-mutant mice with each other, and cultured neurons from littermate offspring that were homozygous for the D232N or the D238N mutation. In this manner, we compared the two mutants directly to each other without a wild-type control. Neurons were cultured under conditions that favor formation of autapses and were analyzed by whole-cell recordings.
Measurements of synaptic responses evoked by action potentials or by hypertonic sucrose in the same neuron demonstrated that the size of these two types of synaptic responses was not significantly different between the D232N- and D238N-mutant neurons (Fig. 9A–C). However, when we determined for each individual neuron the vesicular release probability Pvr (calculated as the ratio of action potential- to sucrose-evoked synaptic response in a given neuron), we noted that D232N-mutant neurons displayed an almost twofold increase in the Pvr compared with D238N-mutant neurons (Fig. 9D).

Evoked EPSCs in autapses formed by D232N- and D238N-mutant neurons. A, Representative traces of EPSCs evoked in the same neuron by consecutive stimulation with an action potential (AP) (induced by somatic depolarization from −75 to 0 mV for 2 ms) and with hypertonic sucrose (0.5 m for 3 s). EPSCs were recorded in isolated hippocampal neurons cultured on glial microislands where they form hundreds of autapses. Neurons were cultured from littermate D232N- and D238N-mutant mice derived from crossings of compound heterozygous mutant mice. Capacitative and somatic currents are blanked for display purposes. B–D, Summary graphs of the synaptic charge integrals of action potential-induced EPSCs (B) and sucrose-induced EPSCs (C; corresponds to the RRP), and of the ratio of the charge integrals of action potential-induced to sucrose-induced EPSCs (D; defined as the vesicular release probability Pvr). Data shown are means ± SEMs. Statistical significance was assessed by unpaired t test (**p < 0.01; numbers of recorded neurons are indicated in the bars).
To test whether the difference in Pvr between the two synaptotagmin-1 mutants is attributable to a difference in apparent Ca2+ sensitivity (which might lead to a failure to observe differences in the absolute synaptic responses if the Ca2+ concentration used for those measurements falls into a range of relative lower sensitivity to external Ca2+), we titrated the magnitude of synaptic responses to isolated action potentials at different ambient Ca2+ concentrations (Fig. 10A,B). We plotted the results normalized either for synaptic responses under control conditions (Fig. 10C), or for the maximal synaptic response in a titration (Fig. 10D). When fitted with a Hill function, the apparent Ca2+ affinity of release in D232N-mutant neurons was found to be ∼1.8-fold higher than in D238N-mutant neurons (i.e., the apparent Kd value was ∼0.6-fold lower) (Table 1). The data confirm [as described above and consistent with data from rescue experiments (Stevens and Sullivan, 2003)] that the D232N mutation causes a relative shift to higher apparent Ca2+ sensitivities of release compared with the D238N mutation. Thus, in autapses similar to inter-neuronal synapses, the D232N mutation induces a relative increase in the apparent Ca2+ sensitivity of release.

Determination of the apparent Ca2+ affinity of release from excitatory synapses in autapses from D232N- and D238N-mutant mice. Recordings were made from autapses in hippocampal neurons cultured from littermate offspring of matings between compound heterozygous mutant mice (i.e., mice carrying one D232N and one D238N mutant allele). Ca2+ titrations were performed by interleafing EPSC measurements at the indicated test Ca2+ concentration in 1 mm Mg2+ with measurements under the standard conditions for autapses (with 4 mm Ca2+ and 4 mm Mg2+) to control for possible rundown of synaptic responses. A, Representative superimposed traces of action potential-evoked EPSCs recorded in D232N- (red) and D238N-mutant neurons (blue) during low-frequency stimulation (0.2 Hz for 30–60 s) at different Ca2+ concentrations as indicated. B, Same traces as shown in A, but normalized to the maximal amplitudes of the EPSCs (i.e., to the responses obtained in 10 mm Ca2+/1 mm Mg2+). Note that stimulus artifacts are removed for display purposes. C, D, Summary graphs of the Ca2+ dependence of synaptic responses in autapses. EPSC amplitudes were normalized to the EPSC obtained in standard extracellular medium containing 4 mm Ca2+/4 mm Mg2+ (C), or to the maximal EPSC amplitude (D). Data in C and D were fitted to a Hill function (Table 1). Data shown are means ± SEMs (number of analyzed neurons are shown in brackets). Statistical significance was assessed by a two-way ANOVA test.
Discussion
Our data demonstrate that a single amino acid substitution in synaptotagmin-1 (D232N) selectively increases its Ca2+-dependent binding to SNARE complexes but not to phospholipids, and that the same mutation increases the amount of release triggered by Ca2+ influx into nerve terminals in response to an action potential. Moreover, our results show that a different single amino acid substitution in a nearby residue (D238N) selectively decreases the apparent Ca2+ affinity of synaptotagmin-1 during phospholipid binding (although its effect is smaller than that of the D232N mutation on SNARE binding), and decreases the amount of release triggered by Ca2+ accordingly. The effects of these substitutions on release were limited to fast synchronous release, because asynchronous release was not significantly altered. Our data thus suggest that synaptotagmin-1 functions by dual Ca2+-dependent activities in triggering fast synchronous release, namely by binding to both phospholipids and SNARE complexes, raising the tantalizing possibility that synaptotagmin-1 triggers fast neurotransmitter release by the Ca2+-dependent coupling of phospholipid membranes to SNARE complexes that were assembled during priming. With Ca2+ influx triggered by action potentials, this coupling could occur by simultaneous binding of a single synaptotagmin-1 molecule to both SNAREs and phospholipids, or by separate binding of the two synaptotagmin-1 molecules in a constitutive synaptotagmin-1 dimer (Perin et al., 1991) to SNAREs or phospholipids.
The following evidence supports these conclusions and shows that the effects we observed are specific.
-
Biophysical studies previously demonstrated that the D232N and D238N mutations selectively but differentially alter intrinsic Ca2+ binding by the C2A domain but have no significant effect on its atomic structure (Fernandez-Chacon et al. 2002). Thus, these mutations do not introduce nonspecific structural changes into synaptotagmin-1.
-
The D232N and D238N mutations are very similar [both neutralize a negatively charged amino acid in the second Ca2+-binding loop (and the third top loop overall) (Ubach et al., 1998) of the C2A domain of synaptotagmin-1], and the amino acids involved are only five residues apart. Thus, these mutations control for each other and do not mediate their very different biochemical and physiological effects by nonspecific electrostatic mechanisms.
-
Although the phospholipid-binding and the synaptic phenotype of the D238N mutation are subtle, the SNARE complex binding and the synaptic phenotype of the D232N mutation are robust, suggesting that these changes are central to the function of synaptotagmin-1.
-
Our experiments were performed with knock-in mice in which mutant synaptotagmin-1 is expressed at normal levels, and analyzed native synaptotagmin-1 from knock-in mice instead of recombinant proteins. The former avoids artifacts induced by overexpression in rescue experiments (or worse, by overexpression experiments in cells such as PC12 cells containing endogenous synaptotagmin-1), whereas the latter prevents the problems with recombinant proteins that may lack crucial modifications. The potential problems with recombinant proteins are illustrated in the Ca2+-independent binding of recombinant synaptotagmin-1 to membranes containing phosphatidylinositol phosphates, binding that is not observed for native synaptotagmin-1 (Fig. 3B–D). Thus, we are studying native proteins in a physiological context.
-
Finally, we analyzed the D232N- and D238N-mutant synapses with two different electrophysiological approaches, inter-neuronal inhibitory and autaptic excitatory synapses. The central finding that the D232N mutation increases the apparent Ca2+ affinity of release was confirmed for both preparations, and the changes of short-term plasticity during stimulation trains (a faster depression in D232N-mutant synapses) are essentially the same in both preparations. Thus, the phenotype is not dependent on a particular type of synapse or a particular experimental approach, although the effects of the mutations are more pronounced in inter-neuronal synapses.
Our current results add to previous studies that had demonstrated that Ca2+-dependent binding of synaptotagmin-1 to phospholipid membranes is a crucial step in neurotransmitter release in vivo. Specifically, it was shown that mutations that selectively decrease phospholipid binding by synaptotagmin-1 cause a corresponding decrease of release (Fernandez-Chacon et al., 2001), whereas mutations that enhance phospholipid binding produce an increase of release (Rhee et al., 2005). In addition, a large number of previous studies supported the notion that SNARE binding by synaptotagmin-1 is important, but these results were mostly obtained with transfected cells and/or recombinant proteins, and often presented contradictory conclusions (Bennett et al., 1992; Yoshida et al., 1992; Davletov and Südhof, 1993; Sollner et al., 1993; Chapman and Jahn, 1994; Chapman et al., 1995; Li et al., 1995; Banerjee et al., 1996; Kee and Scheller, 1996; Schiavo et al., 1997; Fernandez et al., 2001; Wang et al., 2001; Zhang et al., 2002; Rickman and Davletov, 2003; Shin et al., 2003; Bai et al., 2004; Rickman et al., 2004; Bowen et al., 2005). These previous studies had two limitations. First and most importantly, no synaptotagmin-1 mutation was known that selectively alters SNARE binding, whereas such mutations are available for phospholipid binding. In the absence of such a selectively acting mutation, no in vivo analysis of SNARE binding by synaptotagmin-1 was possible. Second, these studies mostly relied on transfected neuroendocrine cells that represent an artificial and potentially misleading methods to study neurotransmitter release. This problem is illustrated by the controversy regarding the role of synaptotagmin-1 (and other proteins) in fusion pores. Overexpression experiments suggested that the synaptotagmin-1/SNARE interaction shapes the fusion pore (Wang et al., 2001; Bai et al., 2004). However, knock-in experiments that alter synaptotagmin-1 function without changing its expression levels do not reveal a role in fusion pores (Sorensen et al., 2003). Moreover, overexpression of many other proteins also alters fusion pore dynamics in transfected cells, suggesting that sheer overexpression of a protein can change the membrane tension and thereby secondarily affect fusion pores (for review, see Jahn et al., 2003). We tried in a previous study to circumvent the difficulty associated with testing the importance of synaptotagmin-1/SNARE interactions using an alternative approach that uses Sr2+ instead of Ca2+ to trigger release, because Sr2+ is relatively ineffective in stimulating the interaction of synaptotagmin-1 with SNARE complexes (Shin et al., 2003). However, Sr2+-triggered release is also mostly asynchronous, limiting its use in probing synaptotagmin-1/SNARE interactions in release (Xu-Friedman and Regehr, 2000). Given the previous difficulty in directly testing the importance of the SNARE/synaptotagmin-1 interaction, the finding reported here that a single amino acid substitution in synaptotagmin-1 selectively alters this interaction may provide a useful general tool for future studies.
Why did we not observe in our initial study (Fernandez-Chacon et al. 2002) the large increase of synaptotagmin-1/SNARE interactions that we report here? Several potential explanations can be advanced. Only recent results uncovered the extreme sensitivity of the synaptotagmin/SNARE interaction to ionic strength (Tang et al., 2006), and thus we were not previously able to analyze the mutant mice properly for changes in SNARE binding. Moreover, we were misled by the problems inherent with recombinant proteins, as opposed to analyzing native synaptotagmin-1. The differences between the native and recombinant proteins are probably not attributable to a modification of native synaptotagmin-1, but rather caused by the contamination of recombinant synaptotagmin-1 with bacterial acidic small molecules that is extremely difficult to remove (Ubach et al., 2001).
A major remaining question regards the atomic mechanism by which synaptotagmin-1 binds to both phospholipids and SNARE complexes via its top Ca2+-binding loops. It is unexpected that two similar mutations, D232N and D238N, with comparable although not identical effects on intrinsic Ca2+ binding (Fernandez-Chacon et al., 2002) have dramatically different effects on the Ca2+-dependent interactions of synaptotagmin-1 with SNARE complexes and phospholipids. This observation implies that, although these interactions involve the same Ca2+-binding sites of synaptotagmin-1, their precise atomic constraints differ. Functionally, this observation suggests that SNARE complex and phospholipid binding are indeed tightly coupled in synaptotagmin-1, consistent with the notion that pulling these two components of the membrane fusion machinery together represents the mechanism of action of synaptotagmin-1. A better biophysical description of these interactions will be required to clarify this hypothesis.
Footnotes
-
This study was supported by Boehringer Ingelheim Fonds (A.C.M.), German Research Council Grant Ro1296 5/3 (C.R.), and National Institutes of Health Grant NS050655 (C.R.). We thank I. Leznicki, J. Mitchell, L. Fan, N. Hamlin, I. Herfort, and A. Roth for excellent technical assistance and Dr. Jose Rizo for insightful discussions.
- Correspondence should be addressed to Thomas C. Südhof at the above address. thomas.sudhof{at}utsouthwestern.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}