

Abstract
The hyperpolarization-activated Ih current, coded for by hyperpolarization-activated, cyclic nucleotide-gated (HCN) channels, controls synaptic integration and intrinsic excitability in many brain areas. Because of their role in pacemaker function, defective HCN channels are natural candidates for contributing to epileptogenesis. Indeed, Ih is pathologically altered after experimentally induced seizures, and several independent data indicate a link between dysfunctional HCN channels and different forms of epilepsy. However, direct evidence for functional changes of defective HCN channels correlating with the disease in human patients is still elusive.
By screening families with epilepsy for mutations in Hcn1 and Hcn2 genes, we found a recessive loss-of-function point mutation in the gene coding for the HCN2 channel in a patient with sporadic idiopathic generalized epilepsy. Of 17 screened members of the same family, the proband was the only one affected and homozygous for the mutation.
The mutation (E515K) is located in the C-linker, a region known to affect channel gating. Functional analysis revealed that homomeric mutant, but not heteromeric wild-type/mutant channels, have a strongly inhibited function caused by a large negative shift of activation range and slowed activation kinetics, effectively abolishing the HCN2 contribution to activity. After transfection into acutely isolated newborn rat cortical neurons, homomeric mutant, but not heteromeric wild type/mutant channels, lowered the threshold of action potential firing and strongly increased cell excitability and firing frequency when compared with wild-type channels.
This is the first evidence in humans for a single-point, homozygous loss-of-function mutation in HCN2 potentially associated with generalized epilepsy with recessive inheritance.
Introduction
Several forms of inheritable idiopathic epilepsy syndrome are ion channelopathies, i.e., pathologies associated with dysfunctional ion channels (Mulley et al., 2003; Steinlein, 2004; Hirose et al., 2005; Lerche et al., 2005; Turnbull et al., 2005; Avanzini et al., 2007; Catterall et al., 2008).
Among the ion channels involved, the hyperpolarization-activated, cyclic nucleotide-gated (HCN) channels have received special attention recently. HCN channels code for a mixed Na+–K+ inward current activated on hyperpolarization below a threshold of ∼−50 mV; this “funny” (If) current was originally described in the heart, where it underlies pacemaker activity (Brown et al., 1979; DiFrancesco, 1993). Widely expressed in neurons, HCN channels have important functions such as the modulation of excitability and rhythmicity, signal integration, and plasticity (Ih current) (Pape, 1996; Robinson and Siegelbaum, 2003; Biel et al., 2009). Because of their role in neuronal excitability, defective HCN1/HCN2 channels, the HCN isoforms expressed in distal dendrites of pyramidal cells in the hippocampus and neocortex (Notomi and Shigemoto, 2004), are considered potential contributors to pathological firing in specific forms of epilepsy (Bender and Baram, 2008; Dubé et al., 2009; Dyhrfjeld-Johnsen et al., 2009; Reid et al., 2009; Lewis and Chetkovich, 2011).
Evidence supporting a link between functional alteration of HCN channels and epileptogenesis has resulted typically from analysis of Hcn1 and Hcn2 knock-out mouse models. Cortical excitability and epileptogenesis are enhanced by loss of HCN1 expression and abolishment of Ih in HCN1-null mice (Huang et al., 2009). Lack of HCN1 channels increases the dendritic input resistance in cortical neurons, leading to greater synaptic integration and firing (Huang et al., 2009). In the same HCN1-null mouse model, greater dendritic excitability and temporal summation was also observed in hippocampal CA1 pyramidal neurons (Nolan et al., 2004; Tsay et al., 2007). In agreement with a role for Ih downregulation in epileptogenesis, HCN2-deficient mice generated by global Hcn2 knock-out exhibit spontaneous absence seizures (Ludwig et al., 2003). Generalized spike–wave absence seizures were also observed in spontaneous Hcn2 mutant mice (apathetic), where channel proteins are truncated at the C terminus and express poorly (Chung et al., 2009).
HCN1 and HCN2 variants have been identified in familial or sporadic febrile seizure patients, although significant mutation-dependent functional changes were not apparent (Tang et al., 2008). A recent study identified an HCN2 variant consisting of a deletion of three consecutive prolines (delPPP) in the C terminus in patients with febrile seizures and genetic epilepsy with febrile seizures plus, which, according to statistical analysis, increases current magnitude in mutant channels (Dibbens et al., 2010). However, how this mutation affects the functional channel properties and increases neuronal excitability was not established.
While existing data therefore clearly show a link between HCN channel dysfunction and epileptogenesis, no specific mutation-induced HCN channel modification has been correlated functionally with increased neuronal excitability. To investigate this, we used a candidate gene approach and screened a panel of partial and generalized idiopathic epileptic patients and related families for mutations in the Hcn1 and Hcn2 genes.
Materials and Methods
Patient recruitment
All protocols used in this research project were reviewed and approved by the Ethical Committees of the San Gerardo Hospital, Monza and of the University of Milan.
We selected adult and adolescent patients presenting either generalized or partial idiopathic epilepsy. Patients were recruited from the Department of Neurology and Neuroscience, Center for Epilepsy and the Clinic of Infantile Neuropsychiatry, Pediatric Neurophysiology, San Gerardo Hospital, Monza, Italy. All subjects included in this study gave informed consent before recruitment. After recruitment, 5 ml of blood in sodium-citrate or a saliva sample was obtained from patients. Diagnosis of epilepsy was made according to the International League Against Epilepsy's International Classification of Epileptic Seizures (Berg et al., 2010). Patients with symptomatic epilepsy or other known channelopathies were not included in this study.
DNA extraction and mutation analysis
Screening for mutations in human Hcn1 and Hcn2 genes was performed on genomic DNA extracted from whole blood and/or saliva samples (QIAamp DNA Blood Mini Kit; Qiagen). This led to the identification of a proband with the homozygous G→A mutation in position 1543 in the exon 5 of the Hcn2 gene (chromosome 19p13.3) (Vaccari et al., 1999).
Primers selected to amplify Hcn1 and Hcn2 coding sequences in various amplicons using FastStart Taq polymerase (Roche) are listed in Table 1.
Primers used for screening Hcn1 and Hcn2
PCR products were analyzed by single-strand conformational polymorphism and/or by denaturing high-performance liquid chromatography; final mutagenesis identification was accomplished by DNA sequencing (Bio-Fab Research).
A control group of 350 nonepileptic subjects was also investigated for the presence of the hHcn2 1543G→A mutation, which was found in one allele of one control individual. In the case of deceased individuals (Patients II-2, II-3, II-4, II-5, and III-4; Fig. 1A), the DNA was extracted from personal items (handkerchief, tooth, and eyeglasses; Patients II-2, II-3, II-4), a paraffin-embedded sample from gastric bioptic tissue (Patient II-5), and a kidney stone (Patient III-4). DNA extraction procedures are described below.
Kidney stone.
The external surface of the kidney stone was scraped off and discarded, 5 g of the internal material was pulverized in liquid nitrogen, and demineralization was obtained by resuspending the sample for 3 h in EDTA (0.5 m). After centrifugation (at 4000 rpm for 3 min), the pellet was resuspended in the stain extraction buffer with proteinase K and kept overnight at 56°C. The sample was processed the following day with a commercial kit (QIAamp DNA Maxi Kit) for DNA purification.
Paraffin-embedded tissue.
The sample was first scraped to eliminate possible external contamination; two tissue slices were then manually cut with a scalpel and processed according to the protocol provided by the QIAamp DNA mini kit.
Handkerchief.
We first scanned the handkerchief with the Crime-lite tool (Crimesight Technologies) to verify whether a region of the fabric containing organic material was present. The region of interest was then cut away and processed according to the protocol described in the QIAamp DNA mini kit.
Tooth and eyeglasses.
Organic material was collected from the tooth and eyeglasses with wet cotton swabs that were then processed for DNA extraction using the QIAamp DNA mini kit.
The low quality/high fragmentation of the DNA extracted from personal items required the shortening of the region to be amplified by PCR to an 86-bp-long amplicon. The primers used were 5′-ACTTCCGCCAGAAGATCCAC-3′ (forward) and 5′-GTTGAGCTCGCCCAGGAT-3′ (reverse). The genetic profiles of the DNA extracted from these specific samples were compared with those of the living offspring using the AmpFlSTR Profiler and AmpFlSTR MiniFiler (Applied Biosystems). The sequence of the fragments obtained was resolved by analysis with an ABI 310 Genetic Analyzer, followed by Genescan 3.1.2 and Genotyper 2.1 analysis (Applied Biosystems). The results excluded exogenous human DNA contamination.
Mutagenesis
We used hHCN2 cDNA cloned in the pcDNA 3.1/Hygro vector. To perform mutagenesis and functional expression studies, we introduced the point mutation 1543 G→A into wild-type hHCN2 cDNA using the QuikChange II XL site-directed mutagenesis kit (Stratagene) with the primers 5′-AGGGCAAGATGTTTGACAAGGACAGCATCCTGGGC-3′ (forward) and 5′-GCCCAGGATGCTGTCCTTGTCAAACATCTTGCCCT-3′ (reverse). The correct insertion of the desired mutation was then further confirmed by direct sequencing.
Neonatal rat cortical neuron cultures
Neocortical neurons were isolated from postnatal day 3 (P3) CD rat pups (Charles River). Rats were anesthetized (isoflurane) and decapitated; brains were quickly removed and placed in ice-cold dissociation medium containing the following (in mm): 134 Na-isethionic acid, 23 glucose, 15 HEPES, 2 KCl, 4 MgCl2, 0.1 CaCl2, and 10 kynurenic acid, pH 7.2. The cerebral cortex was isolated using fine tweezers, chopped into small pieces, and digested in a dissociation medium containing 1.3 mg/ml protease (type XIV; Sigma) for 20 min at 37°C. The tissue was rinsed twice in dissociation medium and mechanically dissociated using a series of fire-polished Pasteur pipettes. The dissociated neurons were plated onto poly-d-lysine-coated 35 mm dishes at a density of 1.5 × 106 cells. After 1 h at 37°C and 5% CO2, the dissociation medium was replaced with Neurobasal A culture medium (Invitrogen) supplemented with B27 (Invitrogen), 1 mm Glutamax-I (Invitrogen), 10 ng/ml β-FGF (Invitrogen), 50 U/ml penicillin G (Sigma), and 50 μg/ml streptomycin (Sigma). Neuronal cultures were kept at 37°C in 5%CO2 and transfected the same day, as detailed below.
In vitro expression studies
Transient transfection of wild-type and/or mutated human HCN2 cDNA was performed with commercial transfection kits (StoS Transfection KIT, GeneSpin; and Lipofectamine 2000, Invitrogen). Either a GFP- or YFP-containing vector (pmaxGFP, Amaxa Biosystems or pEYFP–N1, Clontech) was cotransfected with HCN2 cDNA in homomeric and heteromeric transfections in CHO cells or neurons. For each 35 mm Petri dish, for CHO cells, we used 1.5 μg of the HCN2-containing vector and 0.5 μg of GFP-containing plasmid (for neurons: 3 μg and 0.1 μg of EYFP-containing plasmid, respectively). In heteromeric studies involving transfection of equal amounts of wild-type and mutated cDNAs, the amount of each HCN2 plasmid was 0.75 μg for CHO cells (1.5 μg for neurons) per Petri dish.
CHO cells were cultured in a 1:1 mixture composed of DMEM (Invitrogen) and Ham's F12 mediums supplemented with 10% fetal calf serum (Invitrogen) and antibiotics (Sigma).
Immunofluorescence
Samples were fixed in 2% formaldehyde, permeabilized, and blocked in PBS with 1% bovine serum albimin, 0.3% Triton X-100, and 10% donkey serum, then incubated overnight at 4°C with primary antibodies (anti-HCN2, 1:100, Alomone Labs; and anti-YFP, 1:500, AbCam). Samples were then rinsed in PBS and incubated for 1 h with Alexa-488- and Alexa-594-conjugated secondary antibodies (1:1000). After a final rinse, coverslips were mounted with Vectashield mounting medium with DAPI (Vector). Fluorescence staining was analyzed by video confocal microscopy (ViCo; Nikon).
Electrophysiology
The current elicited in single CHO cells/neurons by expression of HCN2 (referred to as Ih throughout) was investigated by patch-clamp analysis 48 h after transfection.
CHO cells were dispersed by trypsin and plated onto 35 mm plastic Petri dishes. A dish was placed on the stage of an inverted microscope at room temperature (25–26°C) and GFP-expressing cells were selected for patch-clamp analysis in whole-cell configuration. Cells were initially superfused with a Tyrode solution containing the following (in mm): 140 NaCl, 5.4 KCl, 1.8 CaCl2, 5.5 d-glucose, and 5 HEPES-NaOH, pH 7.4. Pipettes contained the following (in mm): 10 NaCl, 130 KCl, 1 EGTA, 5 HEPES-KOH, 0.5 MgCl2, 2 ATP (Na salt), 0.1 GTP (Na salt), and 5 phosphocreatine, pH 7.2. For Ih recordings, the extracellular solution contained the following (in mm): 110 NaCl, 30 KCl, 1.8 CaCl2, 0.5 MgCl2, and 5 HEPES-NaOH, pH 7.4. To improve dissection of the Ih current, 1 mm BaCl2, 2 mm MnCl2, and 100 μm NiCl2 were added. To evaluate the effects of saturating concentrations of intracellular cAMP on the activation curve of the Ih current (see below), cAMP 10 μm (DiFrancesco and Tortora, 1991) was added to the pipette solution.
Neurons were superfused with an external solution containing the following (in mm): 129 NaCl, 1.25 NaH2PO4, 1.8 MgSO4, 1.6 CaCl2, 3 KCl, 10 Na-HEPES, 35 glucose, pH 7.4. The pipette solution contained the following (in mm): 120 K-gluconate, 15 KCl, 2 MgCl2, 0.2 EGTA, 20 phosphocreatine, 2 ATP-Na2, 0.2 GTP-Na2, 0.1 leupeptin, 10 K-HEPES, pH 7.2. In current-clamp recordings, the membrane potential was kept at −70 mV.
In both CHO cells and neurons, experiments comparing properties of mutated versus control channels were always performed on day-matched cells. Activation curves for Ih current were obtained by standard activation/deactivation protocols and analyzed with the Boltzmann equation y = 1/(1 + exp[(V − V½)/s]), where y is fractional activation, V is voltage (mV), V½ is half-activation voltage (mV), and s is the slope factor (mV). The holding potential was −35 mV in all experiments. Chord conductances of the Ih current in CHO cells (gh) were obtained using the equation gh = Ih/(V − Vh), where V is the test membrane potential and Vh is the reversal potential of the Ih current (−17.2 ± 1.5 mV, n = 9). Conductance and current densities were calculated by normalizing to cell capacitance.
Statistical analysis
Comparison of data was made with independent Student's t test or one-way ANOVA followed by Fisher test for means comparison, and significance level was set to p = 0.05.
Results
Patients (n = 113; 67 adults and 46 children) with idiopathic partial and generalized epilepsy, either sporadic or familial, were screened for mutations in the coding regions of the Hcn1 and Hcn2 channel genes. We identified several single-nucleotide polymorphisms (SNPs), all of which previously reported (http://www.ncbi.nlm.nih.gov/projects/SNP) except one (Table 2).
SNPs in the epilepsy cohort with corresponding synonymous/nonsynonymous alterations and allele frequencies
The novel SNP identified was a homozygous missense mutation in exon 5 of the Hcn2 gene leading to the protein mutation E515K, and was identified in one patient with sporadic generalized epilepsy (Fig. 1A, proband IV-5). We then extended the collection and screening of DNA to a total of 17 related family members. The E515K Hcn2 mutation was found in heterozygous conditions in two deceased grandparents (II-3 and II-4), the parents (III-4 and III-5), and sister (IV-6) of the proband, none of whom had the disease; all other relatives investigated were wild-type in both alleles.

Idiopathic generalized epilepsy in a patient with mutated HCN2 pacemaker channel. A, Four-generation family pedigree of proband's relatives. The proband (black square, IV-5) was the only family member with the disease and expressing the homozygous E515K mutation, while four more members expressing the heterozygous mutation were disease-free (crossed symbols; II-3, II-4, III-4, III-5, and IV-6). Open symbols represent individuals with wild-type HCN2 channels. Deceased individuals are indicated by a slash, and individuals whose DNA could not be obtained are marked with a dot. Gray symbols are unrelated individuals. B, EEG recording (amplitude, 100 μV/cm) showing the presence of generalized, synchronous spike–waves with prevalence on the anterior derivations (Fp1–F7 and Fp2–F8).
None of the deceased individuals investigated had shown any sign of epileptic disease in life. The proband suffered from a severe form of generalized epilepsy characterized by absence- and tonic–clonic seizures. The earliest manifestations appeared at the age of 12 years, with no record of febrile seizures during infancy. Brain MRI was within normal limits and EEG analysis revealed the presence of generalized sharp-wave complexes (Fig. 1B). Use of Valproate therapy alone did not control symptoms satisfactorily and the patient, at the age of 28 years, started an add-on treatment with lamotrigine, which substantially decreased the frequency and severity of seizures. After 14 months, however, the patient discontinued use of lamotrigine due to persistence of mild symptoms and the appearance of side effects (myoclonic twitches) and replaced it first with Topiramate, soon discontinued because of inefficacy on seizure control, and ultimately with Levetiracetam.
The presence of either the homozygous or the heterozygous E515K mutations in the individuals investigated was confirmed by electropherograms (Fig. 2A).

Recessive HCN2 mutation found in a generalized epilepsy patient. A, Right, Sequence identification of a G-to-A substitution (arrows) of two alleles at position 1543 in the epileptic patient IV-5 of Figure 1A), causing the replacement of glutamic acid 515 by lysine. Middle, The same substitution was present in one allele only in his healthy sister, individual IV-6 of Figure 1A. Left, Wild-type sequence. B, topology and sequence alignment of the C termini of the four hHCN channel isoforms, showing the C-linker region and the CNBD. A′–F′ and A–C are α-helices (P is pore), and 1–8 are β-sheets (Zagotta et al., 2003). Glutamic acid 515 (red background in hHCN2) is conserved in all isoforms and is part of the C′ helix. C, 3D ribbon representation of the tetrameric assembly of C termini of hHCN2 viewed perpendicular (left) and parallel (right) to fourfold axis (Zagotta et al., 2003). E515 residues are drawn as space filling plots. D, Diagram of one hHCN2 subunit showing the approximate position of E515 in the C-linker.
The mutation E515K is an acidic-to-basic residue mutation involving a Glu residue located in the C-linker of the HCN2 channel protein, i.e., the region connecting the S6 transmembrane domain to the cyclic nucleotide binding domain (CNBD; Fig. 2B–D); residue Glu 515 is highly conserved within the HCN channel family (Fig. 2B), suggesting functional relevance. Indeed, published data indicate that charged residues in this region are involved in the control of the HCN2 channel gating mechanism (Decher et al., 2004). More specifically, E515 is part of a ring of contiguous, negatively charged residues (D514, E515, and D516 in hHCN2) that form a narrow tunnel lining the inner part of the channel mouth (Fig. 2C) and can affect gating (Johnson and Zagotta, 2005). Also, interestingly, mutations in the C terminus of the HCN4 isoform are known to modify the channel function and to cause arrhythmias (Schulze-Bahr et al., 2003; Ueda et al., 2004; Milanesi et al., 2006).
We therefore sought to investigate, first, whether the E515K mutation modifies the biophysical properties of HCN2 channels, and second, whether the functional contribution of HCN2 channels to neuronal activity is altered by the mutation. To verify whether the E515K mutation modifies channel properties, we separately transfected wild-type (wt) and mutated hHcn2 cDNA into CHO cells; we also cotransfected identical amounts of wt and mutated cDNA to mimic the conditions of heterozygotes.
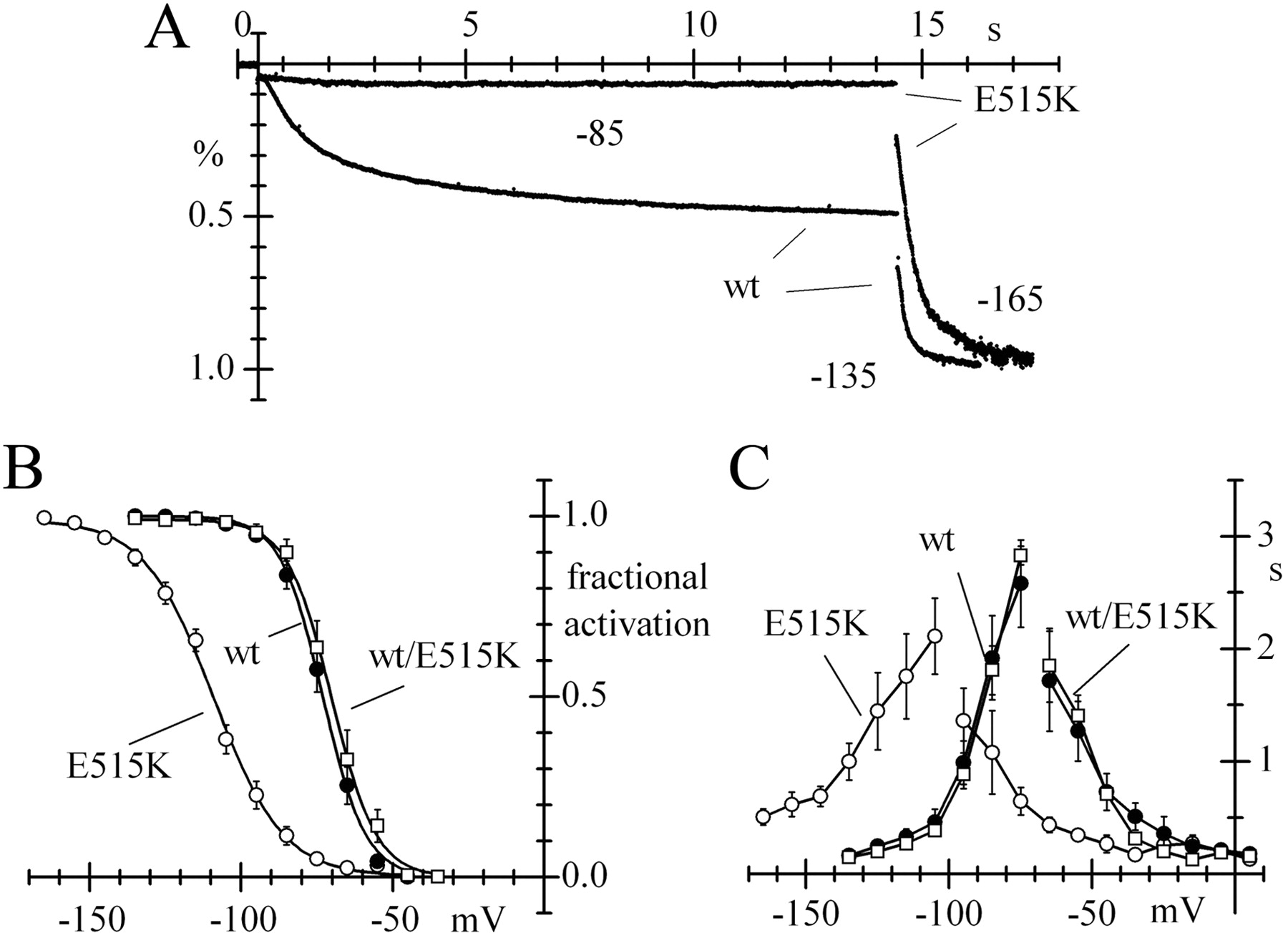
CHO expression efficiencies of wt, heteromeric wt/E515K, and homomeric E515K mutant channels were not significantly different according to one-way ANOVA and Fisher test. The normalized fully activated conductances, measured at −135 mV for wt and wt/E515K and at −165 mV for E515K channels, were as follows: 16.1 ± 3.8, n = 8; 15.7 ± 5.5, n = 7; and 11.0 ± 3.7 pS/pF, n = 12, respectively. However, E515K mutant channels were activated at much more negative voltages than wt channels (Fig. 3A) due to a shift of −30.6 mV in the activation curve, while the activation curves of wt/E515K and wt channels coincided (V½ was −71.9, −70.5, and −102.5 mV for wt, wt/E515K, and E515K channels, respectively; Fig. 3B).

Kinetic properties of wt and homomeric E515K mutant HCN2 channels expressed in CHO cells. A, A double-step protocol shows that large Ih changes are recorded at −85/−135 mV from a holding potential of −35 mV in cells expressing wt, but not mutated E515K channels, where little or no current is present at −85 mV and full activation occurs only at highly negative voltages (−165 mV in this case), demonstrating a large hyperpolarizing shift of the current activation range. B, Mean Ih activation curves measured for wt, heteromeric wt/E515K, and homomeric E515K channels showing a large hyperpolarizing shift for homomeric E515K, but not heteromeric wt/E515K channels. Half-activation voltages (V1/2) and slope factors (s) were −71.9 ± 2.4 and 6.6 ± 0.9 mV (n = 8), −70.5 ± 2.0 and 6.3 ± 0.3 mV (n = 8), and −109.5 ± 1.8 and 11.0 ± 0.5 mV (n = 13) for wt, wt/E515K, and E515K channels, respectively. C, Mean time constant curves showing large negative shifts of both activation and deactivation curves of E515K relative to wt and wt/E515 channels; curves drawn through points.
It should be noted that, when wt and mutant subunits are coexpressed, a mixture of heteromeric, homomeric wt, and homomeric mutant channels are bound to form. However, since the two subunits have similar expression rates, the distribution of homomeric and heteromeric constructs can be calculated using simple combinatory rules. The total number of possible tetrameric constructs is 24 = 16, and the only construct where all subunits are mutant will therefore contribute a tiny fraction (1/16th = 6.25%) of the whole recorded current. Relative to wt channels, this fraction could be expected to contribute a negative shift of the order of 30.6/16 = 1.9 mV in the activation curve from coexpression experiments, a value too small to be detectable. This observation supports the view that the E515K mutation is functionally recessive and alters channel properties only in homomeric constructs.
The time-constant curve for homomeric E515K channels was also shifted to the negative direction, indicating slower activation and faster deactivation relative to wt channels, while wt/E515K channel kinetics were unaltered (Fig. 3C).
To verify that the large shift in Figure 3 was not due to different cAMP sensitivities of homozygous mutant and wild-type channels, we measured the open probability curves of Ih in the presence of saturating concentrations of intracellular cAMP (DiFrancesco and Tortora, 1991). Fitting data with the Boltzmann equation yielded half-activation voltages V1/2 = −64.7 ± 2.2 and −102.2 ± 2.7 mV and slope factors s = 8.2 ± 0.5 and 11.4 ± 0.7 mV for wt (n = 6) and E515K channels (n = 4), respectively. cAMP therefore shifted the activation curves of the two types of channels similarly (7.2 and 7.3 mV, respectively). This result rules in favor of the large negative shift of the homomeric E515K mutant curve being a constitutive new property induced by the mutation.
The highly negative voltage range of activation and the slower current activation at resting voltages suggest that the HCN2 current contribution to activity of homomeric E515K mutant channels is strongly reduced, if not completely abolished, at physiological voltages. Further, since heteromeric wt/E515K channels have the same properties of wt channels, our expression data indicate that the E515K mutation is recessive in relation to the biophysical properties of HCN2.
We next proceeded to verify whether the abnormal kinetic properties of E515K channels contribute to alter functionally neuronal activity and excitability. To this aim, we transfected wt and mutant HCN2 DNA into primary cultures of cortical neurons acutely isolated from newborn rats at P3. These cultures were adopted because it is known that early during development (<P10), newborn rat cortical neurons express a tiny Ih current, which grows dramatically (>10-fold), particularly at distal apical dendritic sites, during development from P10 to >P40 (Atkinson and Williams, 2009). P3 neurons can therefore be considered as a viable substrate to evaluate functional changes associated with increasing expression of HCN channels. Using immunofluorescent staining with anti-HCN2 antibodies in confocal microscopy experiments, we verified that wt, heteromeric, and homomeric mutant channels were all readily expressed and detectable in transfected neurons, while the HCN2 signal from control neurons was below detection level (Fig. 4).

Transfection of human HCN2 into neonatal rat neurons. Single confocal sections of neuronal cultures cotransfected with the EYFP plasmid and with the empty pcDNA 3.1 plasmid (control) or the HCN2-containing pcDNA 3.1 plasmid as indicated. The HCN2 signal (red) was detected in HCN2-transfected cells also expressing EYFP (green) but not in control cells, suggesting that the signal from endogenous HCN2 channels was below detection level. Nuclei counterstained with DAPI (blue). Scale bar, 20 μm.
The changes induced by the E515K mutation in the HCN2 kinetic properties following expression in cortical neurons were similar to those seen in CHO cells.
In Figure 5A, representative current traces recorded during two-step protocols to −75/−135 mV show that, while the first step activates a substantial fraction of current in wt and heteromeric wt/E515K channels, more negative voltages are required to activate homomeric E515K channels, indicating a negative shift of the activation curve of the latter. Compared with the activation curve of wt channels (V1/2 = −74.0 mV), that of homomeric E515K channels was shifted by −24.6 mV (V1/2 = −98.6 mV), while that of heteromeric wt/E515K channels was essentially unchanged (V1/2 = −75.5 mV; Fig. 5B). A negative shift of some 10–15 mV, resulting in slower activation, was also observed in the activation time constant curve of E515K versus wt and wt/E515K channels, which were instead superimposable (Fig. 5C). The presence of a large shift of the E515K activation curve was also reflected in the plot of current density, obtained by averaging records during voltage-clamp steps of fixed duration (3 s) after normalization to cell capacitance. As shown in Figure 5D, the mean current density for E515K channels was comparable to that measured in neurons transfected with the empty plasmid, corresponding to the endogenous Ih current, at physiological voltages (−55/−75 mV), while it converged toward wt and wt/E515K values at high negative voltages (−135 mV), in accordance with saturation of activation at this voltage. At −135 mV, current densities were −0.6 ± 0.3 (control, n = 6), −5.4 ± 1.9 (wt, n = 6), −4.9 ± 1.6 (wt/E515K, n = 6), and −4.5 ± 1.0 pA (E515K, n = 10); the latter three mean values are nonsignificantly different among them, and different from the mean of control density (one-way ANOVA with Fisher test). This indicates that the efficiency of transfection in neuronal cultures was similar for all types of channels and that the lower Ih density in E515K-expressing cells at physiological voltages is entirely attributable to the negative shift of the activation curve. Resting potentials were significantly more depolarized in wt- (−47.5 ± 1.4 mV, n = 24) and in wt/E515K-transfected neurons (−50.8 ± 1.5 mV, n = 13) than in either E515K mutant-transfected (−55.3 ± 1.0 mV, n = 27) or control (−55.8 ± 1.9 mV, n = 12) neurons (one-way ANOVA with Fisher test), in agreement with a contribution to the resting level of wt and heteromeric wt/E151K, but not of homomeric E515K mutant channels.

Properties of Ih from wt and E515K mutant HCN2 channels expressed in rat neonatal cortical neurons. A, Normalized current traces recorded with a double-step protocol to −75/−135 mV from a holding potential of −35 mV, showing that mutant channels activate at more negative voltages than wt channels. B, Plots of mean Ih activation curves of wt, wt/E515K, and E515K channels; a large negative shift of the E515K curve is apparent, while wt and wt/E515K curves essentially superimpose. Half-activation voltages (V1/2) and slope factors (s) from Boltzmann curve fitting were −74.0 ± 3.1 and 8.7 ± 0.7 mV (wt, n = 6), −75.5 ± 1.1 and 7.6 ± 1.1 mV (wt/E515K, n = 4), and −98.6 ± 2.6 and 14.7 ± 1.1 mV (E515K, n = 4). C, Mean activation time constant curves of wt (n = 12), wt/E515K (n = 7), and E515K channels (n = 11) showing superimposition of the first two and a shift to more negative voltages of the latter; curves drawn through points. D, Mean isochronal I-V curves, normalized to cell capacitance, for Ih recorded from cells transfected with empty plasmid, reflecting endogenous current (control, n = 11) and with wt (n = 14), wt/E515K (n = 9), and E515K channels (n = 18) upon application of 3 s steps in the range of −55 to −135 mV from a holding potential of −35 mV. Note that current densities from wt and wt/E515K channels are similar in the whole voltage range, and that the density from E515K channels approaches wt and wt/E515K densities at high negative voltages.
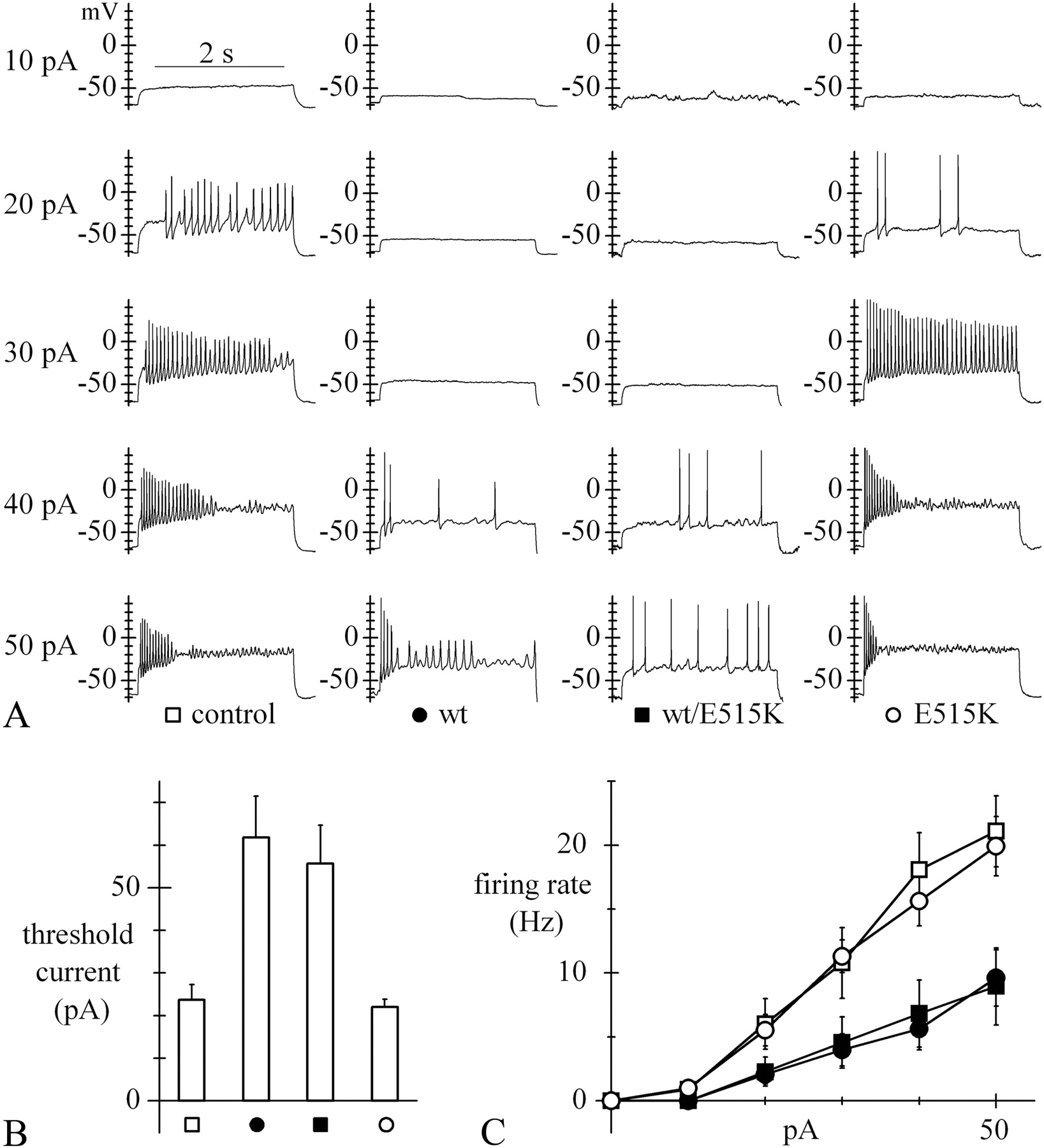
Further effects of the expression of wt or E515K channels on neuronal properties are shown in Figures 6 and 7. A depolarizing sag developing during injection of hyperpolarizing current indicates the presence of a rectifying component attributable to Ih (Maccaferri et al., 1993). In Figure 6, injection of hyperpolarizing current steps (10–40 pA) elicited marked sag depolarizations in wt- but not in E515K-transfected neurons (Fig. 6A). The mean normalized sag amplitude was significantly larger in wt than in E515K neurons for all current steps applied (Fig. 6B). These data further support the view that the E515K mutation reduces the contribution of Ih current to activity.

Effect of the E515K mutation on sag depolarization. A, Sample voltage traces recorded during injection of 2.5 s, 10–40 pA hyperpolarizing current steps into cells transfected with wt (left) or homomeric E515K mutated channels (right). B, Mean sag, measured as sag size normalized to peak hyperpolarization, from wt- (n = 4) and E515K-expressing (n = 4) cells. Values were significantly different at all levels of injected current (p < 0.05). All measurements were made with resting voltage held at −70 mV.

Effect of the E515K mutation on neuronal excitability. A, Sample voltage traces recorded during injection of 2.5 s, 10–50 pA depolarizing current steps into the same types of cells as in Figure 5D. All measurements were performed with resting voltage held at −70 mV. B, Bar plot of mean threshold current required to trigger action potential firing; values were 23.6 ± 3.6 (n = 11), 61.8 ± 9.7 (n = 22), 55.7 ± 9.0 (n = 14), and 22.0 ± 1.9 pA (n = 20) for control, wt, wt/E515K, and E515K cells, respectively. According to one-way ANOVA, wt and wt/E515K curves, like control and E515K curves, were nonsignificantly different among themselves, while each of the former pair was significantly different from each of the latter. C, Mean rate of firing recorded upon current injection in the various cell types (symbols as in A); wt (n = 19) and wt/E515K-expressing cells (n = 14) are clearly less excitable than control (n = 11) and E515K-expressing cells (n = 20).
To investigate whether the E515K mutation affects neuronal excitability, we injected depolarizing current steps of variable amplitude (10–50 pA) into control, wt-, wt/E515K-, and E515K-transfected neurons (Fig. 7). As apparent in the sample traces in Figure 7A, transfection of wt or wt/E515K channels led to a strong reduction of activity relative to control neurons, due to both an increase in threshold current and a reduced firing rate, while E515K-transfected neurons behaved like control neurons. The mean firing threshold current was twofold to threefold higher in wt and wt/E515K neurons than in control neurons, while it was similar to control in E515K neurons (Fig. 7B). The mean firing rate measured during application of current steps from experiments as in Figure 7A and plotted against injected current (Fig. 7C) showed that, while E515K neurons were as responsive to current injection as control neurons, wt and wt/E515K neurons were substantially less excitable in the whole range investigated (significantly different from control and E515K neurons in the range 20–50 pA, p < 0.05).
The results of Figures 6 and 7 show that a reduced function of HCN2 channels, such as the one due to the E515K mutation, is associated with higher excitability.
Discussion
Our results show that the homozygous E515K mutation in human HCN2 channels causes a large negative shift of the activation curve and slowing of activation, and a consequent strong reduction of Ih availability near resting voltages; the changes induced by the mutation therefore result in a physiological loss of function. These changes cause a substantial increase of neuronal excitability, a condition predisposing to epileptogenesis, and are associated with a recessive type of inheritance compatible with the idiopathic generalized epilepsy of the proband in the family pedigree of Figure 1.
HCN2 channels are expressed ubiquitously in most brain areas including the hippocampus, thalamus, cerebellum, and cortex (Notomi and Shigemoto, 2004; Biel et al., 2009). Although we investigated excitability in cortical neurons, where HCN2 channels are a minority isoform, a preferential locus of action of the mutation could be the thalamus, where HCN2 is the majority HCN isoform and channel density is particularly high (Santoro et al., 2000; Biel et al., 2009).
The Ih current, coded for by HCN channels, is normally activated at resting voltage levels (Maccaferri et al., 1993) and can therefore contribute importantly to control of excitability and plasticity phenomena (Robinson and Siegelbaum, 2003; Lewis and Chetkovich, 2011). HCN channels affect rhythmic activity in firing neurons and their expression in dendrites represents an important factor modulating integrative properties of the neuron (Magee, 1999). These properties naturally imply a potential involvement in pathological network oscillations typical of epilepsy.
Pathological alteration of Ih has been shown in several models of seizure-inducing epileptogenesis or increased susceptibility to seizure (Shah et al., 2004; Zhang et al., 2006; Dugladze et al., 2007). Several studies suggest that downregulation of Ih can be proepileptogenic (Peng et al., 2010). In a rat chemoconvulsant (pilocarpine) model of epilepsy, dendritic HCN channels of CA1 hippocampal neurons were substantially downregulated (Jung et al., 2007); interestingly, the activation curve of Ih recorded from postpilocarpine CA1 pyramidal neuron dendrites was more negative than in sham animals, similar to the effect we have observed with E515K channels (Figs. 3, 5). In a genetic rat model of absence epilepsy (WAG/Rij), rapid loss of dendritic HCN1 channels preceded seizure onset and had a direct role in initiating and propagating spontaneous generalized seizures (Kole et al., 2007).
The antiepileptic drug lamotrigine (LTG) increases Ih in hippocampal CA1 pyramidal neurons by a depolarizing shift of the Ih activation curve (Poolos et al., 2002) and/or increased Ih conductance (Peng et al., 2010). This action of LTG leads to a depression of dendritic synaptic excitability (Poolos et al., 2002), supporting the view that the opposite action, i.e., Ih downregulation, underlies amplification of EPSP summation and hyperexcitability.
Hyperexcitability following Ih downregulation is attributable to an increased input resistance of dendritic trees, where HCN channels are generally more densely expressed than in the soma (Magee, 1999); the action on input resistance in this case compensates for the more hyperpolarized membrane voltage associated with Ih inhibition, an effect that should by itself decrease excitability. It should be noted that opposite results have also been reported, such as upregulation of Ih in hyperexcitable CA1 pyramidal neurons following febrile seizures (Chen et al., 2001; Dyhrfjeld-Johnsen et al., 2009). Ih may therefore be antiepileptogenic in some settings when enhanced, and in others when depressed (Peng et al., 2010). This dichotomy likely results from the balance between the contrasting effects on input resistance and membrane resting voltage level, which may vary as a function of neuron type, developmental stage, distribution of channel expression, channel modulation, and other factors (Dyhrfjeld-Johnsen et al., 2009; Noam and Baram, 2010). In agreement with this view, recent work has shown that patients with febrile seizure syndromes have a higher probability (2.4%) of carrying delPPP in HCN2 than wild-type individuals do (0.2%), and that the Ih current from mutated channels is ∼35% larger than control (Dibbens et al., 2010). Interestingly, whether Ih upregulation enhances or inhibits firing depends on the interaction of Ih with the delayed-rectifier M-type K+ current (George et al., 2009).
As discussed above, indications for a link between hyperexcitability and abnormal function of HCN channels are numerous; however, there is often no clear-cut way to distinguish which is the cause and which the effect, and direct evidence for a causal role of dysfunctional HCN channels in epileptogenesis in humans is lacking. In a previous search for HCN-dependent, genetically determined epilepsies, a screening of 84 partly familial and partly sporadic febrile seizure patients revealed the expression of several HCN1 and HCN2 variants, including the mutation R527Q in HCN2 (Tang et al., 2008). However, the mechanism by which this mutation alters the channel contribution to activity was not identified. In contrast with our results with E515K, the mutation R527Q did not affect the position of the channel activation curve (Tang et al., 2008, their Fig. 3). The lack of functional effects of the R527Q mutation is surprising since R527Q and the mutation E515K are both located in the same C-linker region and both affect charged residues, though only the latter involves a charge reversal (from a negatively to a positively charged residue). This suggests that electrical charge reversal in E515K may have a role in the strong effect on HCN2 kinetics (Craven and Zagotta, 2004; Decher et al., 2004; Tang et al., 2008).
According to our data, the large negative shift of the HCN2 activation curve, accompanied by slower activation, essentially abolishes the contribution of homomeric E515K mutant HCN2 channels to activity. It is therefore interesting to compare our data with those from Hcn2 knock-out models. HCN2-deficient mice do indeed exhibit spontaneous absence epilepsy seizures (Ludwig et al., 2003). In vitro analysis has shown that when stimulated by current injection, thalamocortical neurons from HCN2-deficient mice exhibit high-frequency burst firing, rather than the lower frequency tonic firing typical of wild-type neurons, compatible with increased susceptibility to synchronized oscillations in the thalamocortical network and generation of spike-and-wave discharges typical of absence seizures (Ludwig et al., 2003). Also interesting is the comparison of our results with data from a spontaneous Hcn2 mutant mouse (apathetic) where truncation of the channel protein at a location within the CNBD (ap mutation) causes failure of HCN2 membrane expression, thereby mimicking HCN2 knock-out (Chung et al., 2009). Mice homozygous for the ap mutation exhibit ataxia, generalized spike–wave absence seizures, and rare generalized tonic–clonic seizures, while heterozygous mutant mice have a normal gait and show no overt behavioral arrest episodes, although careful EEG analysis revealed occasional absence seizures and infrequent episodes of spike–wave discharges in 20% of mice. Some of the observations made in apathetic mice are strikingly similar to our findings: in both cases, generalized seizures are associated with spontaneous HCN2 mutations resulting in lack of channel contribution to activity and loss of function in homozygous, but not heterozygous mutant conditions.
In summary, we describe for the first time a homologous single-point, loss-of-function mutation in HCN2 in a patient with idiopathic generalized epilepsy that can be functionally correlated with the pathology and is compatible with recessive inheritance. Previously reported HCN2 mutants were either not clearly associated with the disease (R527Q; Tang et al., 2008) or were not associated with idiopathic generalized epilepsy (delPPP; Dibbens et al., 2010). The association of the E515K mutation with generalized epilepsy is thus unique.
Since the E515K mutation was identified in a sporadic patient, cosegregation with the disease could not be verified; however, investigation of the family pedigree showed that of 17 related members, the proband was the only one affected and homozygous for the mutation. It is known that autosomal recessive inheritance is common in epilepsies with early age onset and progressive development, as the case of the proband, while monogenic idiopathic epilepsies of autosomal dominance inheritance have normally a more benign course (Steinlein, 2004). In agreement with an autosomal recessive type of inheritance, functional analysis of the variant, expressed in either CHO cells or acutely dissociated rat cortical neurons, showed lack of contribution to activity of homomeric mutant, but not heteromeric wt/mutant channels. Correspondingly, homomeric mutant, but not heteromeric wt/mutant channels, led to a strong increase in excitability when transfected into rat neurons.
E515 is located in the C-linker and belongs to a ring of negatively charged residues (D514, E515, and D516; Fig. 2C) affecting gating but not permeation of HCN2 and CNG channels (Johnson and Zagotta, 2005). The large negative shift of the activation curve and time constant curve of homomeric E515K channels we found is in agreement with this evidence. Because of the large negative shift of activation threshold and slowing of activation kinetics, effectively abolishing the contribution of the variant HCN2 channels to activity, the homozygous E515K behaves as a physiological loss-of-function mutation simulating Hcn2 knock-out. E515K may represent a specific case of a broader mechanism correlating HCN loss-of-function modifications and epilepsy, and may suggest perspective therapeutic interventions targeting HCN channels.
Footnotes
We thank Drs D. Grioni, L. Dalprà, M. Contri, E. Beghi and G. Bogliun for providing biological samples. This work was supported by grants from the Italian Ministry of Education, University and Research (PRIN 2003052919 and FIRB RBLA035A4X) to D.D.
The authors report no financial conflicts of interest.
- Correspondence should be addressed to Dario DiFrancesco, Dipartimento di Scienze Biomolecolari e Biotecnologie, via Celoria 26, 20133 Milano, Italy. dario.difrancesco{at}unimi.it





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}