

Abstract
With a worldwide incidence as high as 6.7% of children, febrile seizures are one of the most common reasons for seeking pediatric care, but the mechanisms underlying generation of febrile seizures are poorly understood. Febrile seizures have been suspected to have a genetic basis, and recently, mutations in GABAA receptor and sodium channel genes have been identified that are associated with febrile seizures and generalized seizures with febrile seizures plus pedigrees. Pentameric GABAA receptors mediate the majority of fast synaptic inhibition in the brain and are composed of combinations of α(1–6), β(1–3), and γ(1–3) subunits. In αβγ2 GABAA receptors, the γ2 subunit is critical for receptor trafficking, clustering, and synaptic maintenance, and mutations in the γ2 subunit have been monogenically associated with autosomal dominant transmission of febrile seizures. Here, we report that whereas trafficking of wild-type α1β2γ2 receptors was slightly temperature dependent, trafficking of mutant α1β2γ2 receptors containing γ2 subunit mutations [γ2(R43Q), γ2(K289M), and γ2(Q351X)] associated with febrile seizures was highly temperature dependent. In contrast, trafficking of mutant α1β2γ2 receptors containing an α1 subunit mutation [α1(A322D)] not associated with febrile seizures was not highly temperature dependent. Brief increases in temperature from 37 to 40°C rapidly (<10 min) impaired trafficking and/or accelerated endocytosis of heterozygous mutant α1β2γ2 receptors containing γ2 subunit mutations associated with febrile seizures but not of wild-type α1β2γ2 receptors or heterozygous mutant α1(A322D)β2γ2 receptors, suggesting that febrile seizures may be produced by a temperature-induced dynamic reduction of susceptible mutant surface GABAA receptors in response to fever.
Introduction
Febrile seizures are one of the most common childhood neurological disorders, with a worldwide incidence of 1–14% (Hauser, 1994). It has been suggested that the height of the fever and the rapidity of the elevation of temperature are both involved in triggering a seizure, and thus typical treatment of a febrile seizure is to reduce temperature by antipyretics and passive cooling. Although most are self-limited, complex prolonged febrile seizures have been proposed to lead to hippocampal mesiotemporal sclerosis and complex partial epilepsy, and thus understanding the mechanisms of febrile seizures has substantial clinical importance.
The specific causes of febrile seizures and the role of fever in provoking febrile seizures are unclear. Previous studies have suggested that interleukin-1β, a pyrogenic proinflammatory cytokine, and hyperpolarization-activated cyclic nucleotide-gated cation channels are involved in the generation of febrile seizures or enhanced seizure susceptibility in animals, whereas neuropeptide Y could prevent febrile seizures by increasing seizure threshold (Bender et al., 2003; Dube et al., 2005a,b). In addition, it is believed that febrile seizures have a major genetic component with dominant inheritance in some families, but complex inheritance is probably operative in the majority of cases (Rich et al., 1987). Multiple loci have been proposed for febrile seizures (chromosomes 8q13-q21, 19p, 2q23–24, 6q22–24, and 5q14-q15) (Baulac et al., 2004). Recently, several mutations in the GABAA receptor γ2 subunit gene were reported to be associated with febrile seizures [γ2(R43Q), γ2(K289M), and γ2Q(351X), γ2(IVS6 + 2T − >G)], although there was often an extended phenotypic spectrum in these pedigrees (Baulac et al., 2001; Wallace et al., 2001; Harkin et al., 2002). All patients with febrile seizures or generalized seizures with febrile seizures plus (GEFS+) in these pedigrees carried the γ2 subunit mutation, strongly suggesting a correlation between febrile seizures and impaired GABAA receptor γ2 subunit function. Quite interestingly, in a pedigree with a mutation of the GABAA receptor α1 subunit, α1(A322D), all affected family members had the homogenous phenotype of juvenile myoclonic epilepsy (Cossette et al., 2002) without a history of febrile seizures, suggesting that the phenotype resulting from this mutation of the GABAA receptor α1 subunits may be not temperature related.
The above γ2 subunit missense and truncation mutations are located in different locations, suggesting that each mutation may have a different functional consequence that may produce disinhibition and nonfebrile seizures. However, what is the basis for the common febrile seizure phenotype? The γ2 subunit is critical for receptor trafficking (Keller et al., 2004; Rathenberg et al., 2004), clustering (Essrich et al., 1998), and synaptic maintenance (Schweizer et al., 2003), suggesting that mutant receptors might have temperature-dependent effects on surface receptor stability. Therefore, we determined the role of elevated temperature on the function of α1β2γ2S receptors with or without γ2S subunit mutations identified in febrile seizure or GEFS+ pedigrees.
Materials and Methods
Expression vectors with GABAA receptor subunits.
The cDNAs encoding human α1, β2, and γ2S GABAA receptor subunit subtypes were subcloned into the expression vector pcDNA3.1(+) and the cDNAs encoding rat α1, β2, and γ2L subunits were subcloned into the expression vector pCMVNeo. Enhanced yellow fluorescent protein (EYFP) or enhanced cyan fluorescent protein (ECFP) was inserted between amino acids 4 and 5 of the mature human α1 and γ2S subunit cDNAs. The ecliptic pHluorin [a pH-sensitive green fluorescent protein (GFP) variant]-tagged rat γ2L GABAA receptor subunit was kindly provided by Dr. Stephen J. Moss (University of Pennsylvania, Philadelphia, PA). All point mutations in both fluorescence-tagged and untagged human α1 and γ2S and rat γ2L subunit constructs were made using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) and were confirmed by DNA sequencing.
Hippocampal neuron culture.
Hippocampi were dissected from the brains of embryonic day 19 (E19) Sprague Dawley rat pups. Dissociation of cells and culturing procedures have been described previously (Chong et al., 2003). The neurons were plated on 35 mm dishes or coverslips at a density of 0.5–1 × 105 cells/ml and first maintained in DMEM (Invitrogen, Carlsbad, CA) supplemented with 6% fetal bovine serum for the first 3 d and then maintained with Neurobal and B-27 supplement. The experiments were initiated on days 5–7 in culture. Neurons were transfected with rat α1, β2, and pHluorin-tagged γ2L wild-type subunit cDNAs (cDNA ratio of 1:1:1 for wild type) and γ2L(Q351X) mutant subunit cDNAs (cDNA ratio of 1:1:0.5:0.5 for heterozygous) for 6 d with Fugene reagents per the suggestions of the manufacturer before imaging.
Electrophysiology.
Lifted whole-cell recordings were performed as reported previously (Kang and Macdonald, 2004). Briefly, human embryonic kidney 293T (HEK293T) cells were cotransfected with 4 μg of each subunit plasmid and 2 μg of the pHook-1 cDNA (Invitrogen) using a modified calcium phosphate precipitation method and selected 24 h after transfection by magnetic hapten-coated beads. Whole cells were voltage clamped at −50 mV.
Live cell confocal microscopy and fluorescence quantification.
Live cell confocal microscopy and data quantification were performed as described previously with minor modifications (Kang and Macdonald, 2004). The temperature-controlled confocal microscopy was performed using CTI-Control 3700 digital plus TempControl 37-2 digital system. COS-7 cells were plated on poly-d-lysine-coated, glass-bottom imaging dishes at the density of 1–2 × 105 cells and cotransfected with 1 μg of each human subunit plasmid with either calcium phosphate precipitation or Lipofectamine Plus reagents according to the suggestions of the manufacturer. Fluorescence-tagged heterozygous α1(A322D)β2γ2S receptors were formed by coexpression of α1-ECFP, α1(A322D)-EYFP, β2, and γ2S subunit cDNAs at a ratio of 0.5:0.5:1:1, and fluorescence-tagged heterozygous α1β2γ2S(R43Q, K289M, and Q351X) receptors were formed by coexpression of α1, β2, γ2S-ECFP, and γ2S(R43Q, K289M, or Q351X)-EYFP subunit cDNAs at ratio of 1:1:0.5:0.5, giving an equal gene dose of each subunit for electrophysiological recording. Cell membrane dye FM4-64 (10 μm) (Invitrogen, Eugene, OR) was applied to label the cell plasma membrane. Cells were examined with excitation at 458 nm for ECFP, 514 nm for EYFP, and 543 nm for FM4-64. ECFP emission was detected with a 475–525 nm bandpass (bp) filter, FM4-64 emission was detected with 560 long-pass filter. EYFP emission was separated from ECFP and FM4-64 by reflecting the emitted light off a NFT545 dichroic mirror and filtering it via a 530–600 bp filter. PHluorin was examined with excitation at 488 nm and emission with 505 nm long-pass filter. The digital images were taken with 63× 1.4 objective and acquired with 1.8–2× zoom and 512 × 512 pixel resolution. Identical image acquisition settings were used for the same cells over a series of time courses. The fluorescence pixel intensity values of cell surface areas were determined using MetaMorph imaging software by colocalizing each specific area with FM4-64 for the plasma membrane, and the fluorescence intensities were measured in both EYFP and ECFP channels through color combination, which included both ECFP-tagged wild-type receptors and EYFP-tagged mutant receptors. All images were first background detected and then thresholded and background subtracted. The threshold value was determined for each experiment based on the image background and used for all images from that experiment. The fluorescent clusters in hippocampal neurons were quantified by randomly choosing 10 × 500 μm2 nonoverlapping fields with fluorescent puncta in dishes transfected with either wild-type or mutant receptors. After incubation at 40°C, the fluorescence puncta of both wild-type and mutant receptors were counted in the same regions at different time points using the same image acquisition settings.
Biotinylation and Western blot analysis.
Cell surface receptor biotinylation and Western blot analysis were performed as described previously (Kang and Macdonald, 2004). For temperature challenges, after transfection for 48 h, HEK293T cells were transferred from a 37°C incubator to a 40°C incubator for 1 h. The cells were then washed with cold PBS buffer and incubated with sulfo-N-hydroxysuccinimide biotin for 1 h at 4°C as described previously. After SDS-PAGE, the membranes were incubated with GABAA receptor α1 subunit-specific antibody (bd24) or an antibody to GFP (Chemicon, Temecula, CA) overnight at 4°C with gentle rotation. After washing, the membranes were incubated with horseradish peroxidase-conjugated secondary antibody (goat anti-mouse IgG; 1:2000; Upstate Biotechnology, Lake Placid, NY). The antibody-reactive bands were revealed by chemiluminescence. The Western blots were quantified with ChemiImager AlphaEaseFC.
Data analysis.
Macroscopic currents were low-pass filtered at 2 kHz, digitized at 10 kHz, and analyzed using pClamp9 software suite (Molecular Devices, Union City, CA). Numerical data were expressed as mean ± SEM. Statistical significance, using Student’s unpaired t test (GraphPad Prism; Graph Pad, San Diego, CA), was taken as p < 0.05.
Results
Heterozygous mutant γ2S subunit-containing α1β2γ2S receptors displayed reduced surface receptor expression with temperature elevation
Because all pedigrees with γ2 subunit mutations are associated with febrile seizures and previous studies indicated that one of the mechanisms underlying the afebrile seizures associated with these mutations is reduced receptor surface expression and endoplasmic reticulum (ER) retention (Harkin et al., 2002; Kang and Macdonald, 2004; Sancar and Czajkowski, 2004; Hales et al., 2005), it is possible that elevated temperature reduces receptor surface expression. To explore this, we expressed wild-type α1β2γ2S and heterozygous α1β2γ2S(R43Q), α1β2γ2S(K289M), and α1β2γ2S(Q351X) receptors in HEK293T cells and varied temperature between 37 and 40°C.
We used biotinylation and Western blotting with anti-GFP antibody of coexpressed α1, β2, and EYFP-tagged γ2S subunits to determine surface expression of wild-type and heterozygous mutant receptors incubated at 37°C (Fig. 1A). We found that γ2S subunit surface expression was significantly reduced for all heterozygous receptors relative to wild-type receptors (Fig. 1B, filled bars). After incubation of wild-type receptors at 40°C for 1 h, there was no significant change in γ2S subunit surface expression relative to that at 37°C (Fig. 1A,B). However, after incubation of heterozygous receptors at 40°C for 1 h, surface expression of each mutant γ2S subunit was significantly reduced relative to wild-type γ2S subunit incubated at 40°C for 1 h or to mutant γ2S subunits at 37°C (Fig. 1A,B). After a 2 h incubation at 40°C, wild-type receptors also showed reduced surface expression, but total protein was not changed compared with control dishes maintained at 37°C (data not shown).

Elevated temperature further reduced surface protein expression of mutant γ2S subunit-containing GABAA receptors. A, Western blot analysis of biotinylated γ2S and α1 subunit surface proteins from HEK293T cells expressing wild-type and heterozygous mutant α1β2γ2S-EYFP receptors incubated at 37 or 40°C for 1 h. The cell membranes were biotinylated, and equal amounts of protein were conjugated with beads and loaded, and the membranes were immunoblotted with mouse monoclonal anti-GFP and anti-α1 antibodies. B, Quantification of Western blots of biotinylated EYFP-tagged γ2S subunit surface protein from HEK293T cells expressing wild-type and mutant α1β2γ2S-EYFP receptors incubated at different temperatures. C, Quantification of Western blots of biotinylated α1 subunit surface protein from HEK293T cells expressing wild-type and mutant GABAA α1β2γ2S-EYFP receptors incubated at different temperatures. B, C, In all groups, data represent the mean ± SEM (n = 4; *p < 0.05, **p < 0.01 vs wild type at 37°C; ††p < 0.01 vs wild type at 40°C; §p < 0.05 vs the same mutation at 37°C; two-tailed unpaired Student’s t test).
Because a previous study suggested that γ2 subunits could traffic to the cell surface alone (Connolly et al., 1999), we also detected α1 subunit surface expression at 37°C after expression of wild-type and heterozygous receptors to confirm that there was a temperature-dependent alteration of surface expression of heterozygous α1β2γ2 receptors, not just γ2 subunits (Fig. 1A). We found that α1 subunit surface expression was also reduced for each mutant receptor relative to receptors containing wild-type γ2S subunits (Fig. 1C, filled bars), suggesting that surface expression of pentameric mutant α1β2γ2 receptors was reduced instead of γ2 subunits alone. After incubation of heterozygous receptors at 40°C for 1 h, surface expression of α1 subunits was significantly reduced relative to surface expression in wild-type receptors or in mutant receptors at 37°C (Fig. 1A,C).
To control for a nonspecific effect of elevated temperature on receptor surface expression, heterozygous α1(A322D)β2γ2S receptors were expressed also, because the α1(A322D) mutation is not associated with febrile seizures. In contrast to the results obtained with heterozygous expression of receptors containing mutant γ2S subunits, after heterozygous expression of α1(A322D)β2γ2 receptors, α1 subunit surface reduction was the same at 37°C and after incubation at 40°C for 1 h (data not shown).
Heterozygous mutant γ2S subunit-containing, but not mutant α1(A322D) subunit-containing, α1β2γ2S receptors displayed decreased surface and increased intracellular localization with temperature elevation
Consistent with the reduction of receptor surface expression demonstrated using immunoblotting, we also found that after a 30 min incubation at 40°C, heterozygous α1β2γ2S(R43Q), α1β2γ2S(K289M), and α1β2γ2S(Q351X) receptors had reduced membrane surface and increased intracellular expression in COS-7 cells (Fig. 2). Wild-type γ2S subunits tagged with ECFP (blue) and EYFP (green) and the plasma membrane marker FM4-64 (red) were primarily colocalized (white) on the membrane surface (Fig. 2A, top row) and expressed a small amount of intracellular receptor (aqua) with incubation at 37°C or after a 30 min incubation at 40°C (Fig. 2A). For heterozygous expression, wild-type and mutant γ2S subunits were tagged with ECFP and EYFP, respectively. Membrane surface fluorescence intensities of all heterozygous mutant γ2S subunits were significantly reduced compared with wild-type γ2S subunits (Fig. 2B, filled bars). After a 30 min incubation at 40°C, there was reduced membrane surface expression of receptor (loss of white), and both mutant and coexpressed wild-type γ2S subunits were localized intracellularly (cyan) (Fig. 2A, middle three rows). Consistent with this, membrane surface fluorescence of each mutant subunit was decreased after a 30 min incubation at 40°C compared with incubation at 37°C and to wild-type subunits at 37 or 40°C (Fig. 2B).
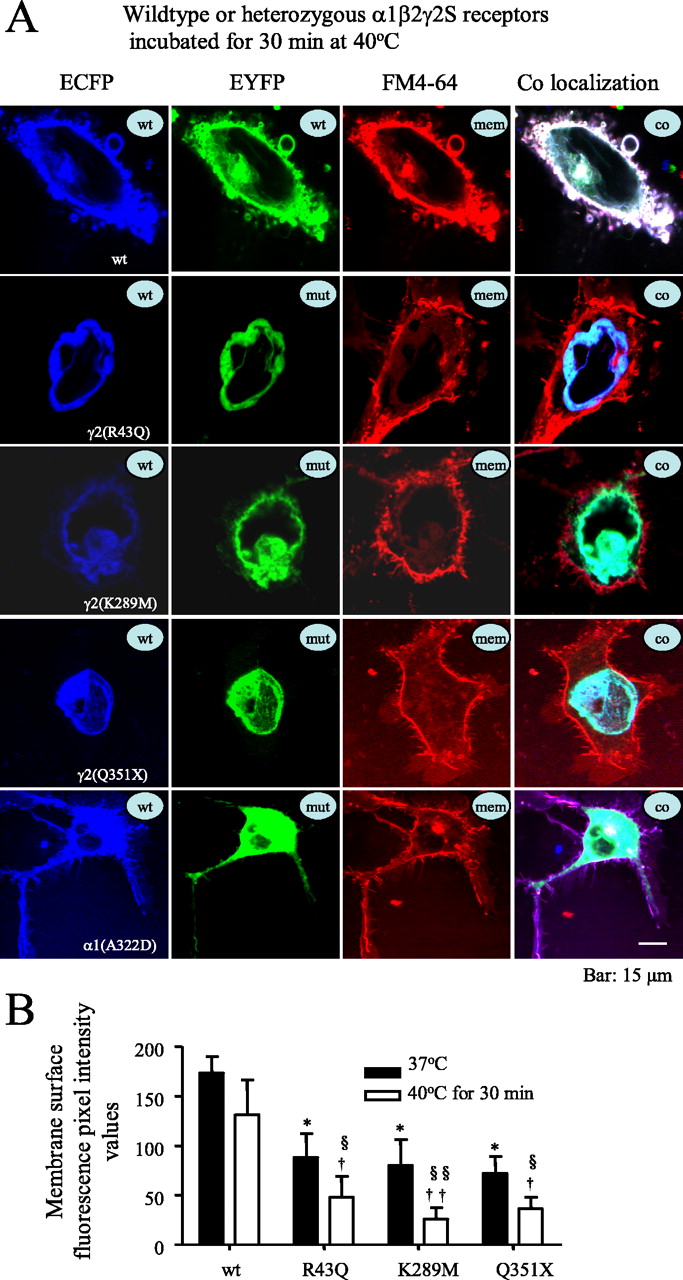
Elevated temperature rapidly increased intracellular retention of mutant γ2S subunit-containing GABAA receptors. A, Confocal microscopy images of fluorescence-tagged wild-type and mutant γ2S subunit-containing α1β2γ2S receptors and α1(A322D) subunit-containing α1β2γ2S receptors in COS-7 cells after a 30 min incubation at 40°C. wt, ECFP-tagged wild-type subunits; mut, EYFP-tagged mutant subunits; mem, membrane marker FM4-64; co, colocalization of all ECFP, EYFP, and FM4-64 channels. With heterozygous expression, mutant and wild-type γ2S(R43Q), γ2S(K289M), and γ2S(Q351X) subunits and mutant α1(A322D), but not wild-type α1, subunits were localized intracellularly in compartments that had the morphology of the ER and colocalized with the ECFP-ER marker (data not shown). The loss of surface receptor illustrated here was an extreme example to illustrate the point. Other cells showed less extensive loss of surface receptor. B, Total membrane surface receptor fluorescence pixel intensity values of heterozygous α1β2γ2S(R43Q), α1β2γ2S(K289M), and α1β2γ2S(Q351X) receptors were reduced after incubation at 37°C compared with wild-type receptors (filled bars) and were further reduced after a 30 min incubation at 40°C (open bars). In all groups, data represent the mean ± SEM (n = 19–23 cells from 5 transfections; *p < 0.05 vs wild type at 37°C; †p < 0.05, ††p < 0.01 vs wild type at 40°C; §p < 0.05, §§p < 0.01 vs the same mutation at 37°C; two-tailed unpaired Student’s t test).
In contrast to the results obtained with the mutant γ2S subunits, with heterozygous expression, wild-type α1-ECFP subunits coregistered with the cell membrane marker FM4-64 (purple) and were also distributed intracellularly (Gallagher et al., 2005), but mutant α1(A322D)-EYFP subunits did not coregister with wild-type subunits and surface membrane (absence of white) and were mainly localized intracellularly with wild-type receptors (cyan) (Fig. 2A, bottom row). These results are consistent with the report that at room temperature or at 37°C with heterozygous expression of α1(A322D)β2γ2S receptors, there was intracellular ER retention and ER associated degradation of mutant subunits before receptor assembly (Gallagher et al., 2005) and also suggests that attachment of EYFP or ECFP to the γ2S subunit did not alter its temperature-dependent trafficking or folding.
Decrease of surface receptor and increase of intracellular receptor with elevated temperature was rapid in heterozygous mutant γ2 subunit-containing α1β2γ2 receptors in heterologous cells
Because all of the heterozygous α1β2γ2S(R43Q), α1β2γ2S(K289M), and α1β2γ2S(Q351X) receptors developed extensive intracellular localization with a 30 min incubation at elevated temperature (40°C), we determined how soon the intracellular localization occurred after temperature elevation. Using heterozygous α1β2γ2S(K289M) receptors, we detected a rapid change in membrane surface and intracellular receptor localization. COS-7 cells were cotransfected with heterozygous α1β2γ2S(K289M) receptors with the wild-type γ2S subunits tagged with ECFP and the mutant γ2S subunit tagged with EYFP (Fig. 3) and maintained at 37°C. The culture dish was transferred to a microscope stage that was heated to 40°C. After 5 min on the heated stage, the receptors displayed a merged aqua color both on the surface and intracellularly (Fig. 3, 5 min, bottom, red box). Over the next 6 min at 40°C, the intensity of the surface receptors rapidly diminished, and the intensity and amount of intracellular receptor rapidly increased (Fig. 3, 7 min and 11 min, bottom, red boxes).

Reduction of surface receptor and increased intracellular retention with elevated temperature in mutant γ2(K289M) subunit-containing α1β2γ2 receptors was rapid and dynamic in heterologous cells. The representative images illustrate the rapid time course of the reduction of heterozygous α1β2γ2S-ECFP/γ2S(K289M)-EYFP surface receptors in COS-7 cells. Cells were cotransfected with heterozygous α1β2γ2S/γ2S(K289M) receptors with the wild-type γ2S tagged with ECFP and the mutant γ2S subunit tagged with EYFP. The receptors displayed a merged aqua color both on the surface and intracellularly after incubation at 40°C for 5 min (boxed area). With incubation at 40°C over a rapid time course, the fluorescence intensity of the surface receptors progressively diminished (7 and 11 min, bottom panel, red boxes) with loss of cyan color on the cell surface (changes of cyan color in the red box, red arrow), and the intracellular fluorescence intensity progressively increased with the accumulation of intracellular receptors (changes of cyan color of the coregistered image in the red box; red double arrow). wt, ECFP-tagged wild-type subunits; mut, EYFP-tagged mutant subunits; co, colocalization of all ECFP and EYFP channels.
Decrease of surface heterozygous mutant γ2 subunit-containing α1β2γ2 receptors was rapid at elevated temperature in hippocampal neurons
Because neurons may have different trafficking machinery, the effects of elevated temperature on surface expression of wild-type and mutant receptors must be determined. To explore the receptor surface dynamics in neurons, pHluorin-tagged rat wild-type γ2L and mutant γ2L(Q351X) subunits were coexpressed with rat α1 and β2 subunits in hippocampal neurons for 6 d. PHluorin should only effectively generate fluorescence when at the cell surface and should produce no or minimal fluorescence at acidic pH levels (pH < 6.5) characteristic of vesicular compartments. After 6 d, γ2L subunit-coupled fluorescence was visible and appeared as puncta on neurons expressing both wild-type and mutant receptors (Fig. 4A) (see supplemental Figs. 1 and 2 for enlarged views, arrows, available at www.jneurosci.org as supplemental material). Because pHluorin only fluoresces at the surface, the puncta on neurons were assumed to be surface receptors. Neurons transfected with heterozygous mutant receptors had fewer fluorescent puncta compared with wild-type receptors from the same areas (Fig. 4B), indicating there were fewer receptors trafficked to the surface in neurons. The fluorescent puncta with wild-type receptors showed minimal reduction in fluorescence after 40 min at 40°C, whereas fluorescent puncta with heterozygous mutant receptors showed substantial reduction in fluorescence within 20 min at 40°C. After incubation at 40°C for 30 min, there was more loss of puncta expressing mutant than wild-type receptors by comparing the total number of puncta in the same regions of chosen neurons (Fig. 4B). This loss was not likely caused by the experimental manipulations such as photobleaching, because there was no appreciable loss of puncta after incubation at 37°C over the same time course (data not shown). The rapid reduction of surface receptor expression and increased intracellular localization in both heterologous cells and neurons suggested that receptor trafficking deficiency and/or accelerated endocytosis was very dynamic and rapid.

Rapid reduction of heterozygous α1β2γ2/α1β2γ2(Q351X) receptors with elevated temperature on the membrane surface of hippocampal neurons. A, Heterozygous α1β2γ2L-pHluorin/α1β2γ2L(Q351X)-pHluorin receptor on the surface of rat hippocampal neurons was reduced rapidly by temperature elevation to 40°C. As illustrated in the left panels, neurons were cotransfected with heterozygous α1β2γ2L-pHluorin/α1β2γ2L(Q351X)-pHluorin receptors, and receptors were imaged as puncta on the surface of neurons. With incubation at 40°C, the fluorescent puncta were reduced, with loss or fading of fluorescence on the cell surface (red arrows). TI, Transmitted image; wt, wild-type α1β2γ2L-pHluorin receptors; mut, heterozygous mutant α1β2γ2L-pHluorin/α1β2γ2L(Q351X)-pHluorin receptors. B, Membrane surface fluorescence clusters of heterozygous mutant α1β2γ2L-pHluorin/α1β2γ2L(Q351X)-pHluorin receptors were reduced after incubation at 40°C at different times compared with wild-type receptors measured over equivalent areas and were further reduced after a 20 min incubation at 40°C compared with the receptor fluorescence puncta in the same regions. In all groups, data represent the mean ± SEM (n = 10–14 neurons from 4 transfections; *p < 0.05 vs wild type at the same time points; †p < 0.05 vs the same mutation after incubation at 40°C for 3 min; two-tailed unpaired Student’s t test).
Wild-type α1β2γ2S receptor currents were reduced reversibly by elevated temperature
We next determined the temperature dependence of wild-type α1β2γ2S receptor currents. Wild-type receptor peak amplitude was significantly reduced after incubation at 40°C for 2.5 h (Fig. 5A,B) but was not reduced after incubation at 40°C for 30 min (data not shown) to 1 h (Fig. 5C,D). After incubation at 40°C for 2.5 h, only minimal current was evoked by a saturating GABA concentration (1 mm) (Fig. 5A,B). However, during recovery at 25°C, peak current amplitude of the same cells increased 30-fold in 45 min and was then stable for at least 2 h, suggesting that at 25°C, ∼45 min was required for full recovery of α1β2γ2S receptors from an elevated temperature-induced reduction in surface receptors. The results also demonstrate that the reduction in surface wild-type α1β2γ2S receptors produced by decreased trafficking or the accelerated endocytosis at elevated temperature was reversible.
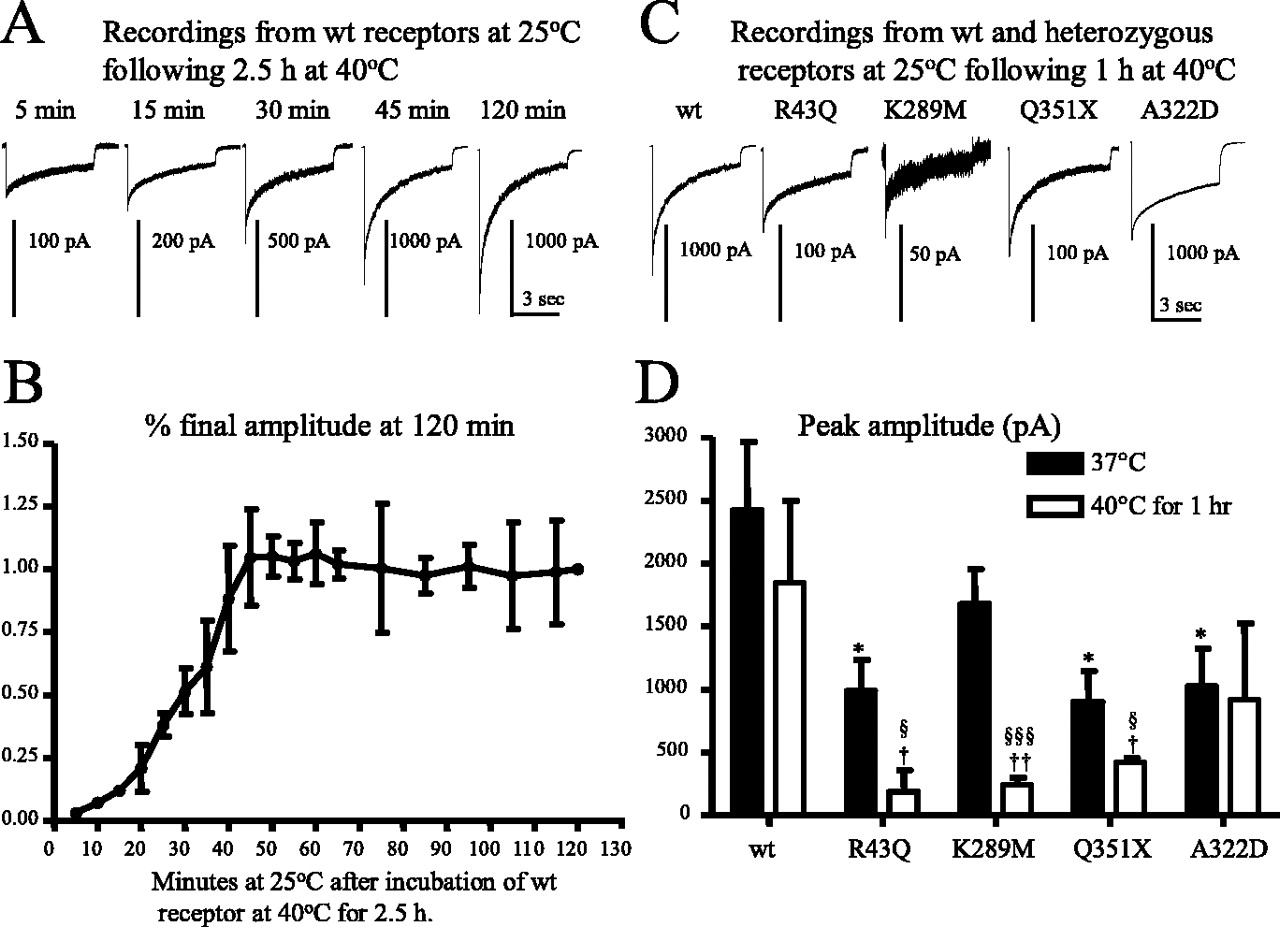
Both wild-type and mutant γ2S subunit-containing GABAA receptors demonstrated a trafficking/recycling deficiency with elevated temperature. A, Representative currents are presented from a single cell expressing wild-type α1β2γ2S receptors recovering at 25°C from 2.5 h of incubation at 40°C. B, Wild-type receptor peak current amplitudes increased over a 45 min time course at 25°C and then stabilized after incubation at 40°C for 2.5 h. The ordinate denotes current amplitudes at each time point over the current amplitude obtained at 120 min after incubation at 25°C. Data were averaged from four cells. C, Representative currents are presented that were recorded at room temperature from cells that were incubated at 40°C for 1 h. D, Mutant γ2S subunit-containing receptors challenged with 40°C had reduced peak currents compared with wild-type and with the same mutant γ2S subunit-containing receptors at 37°C. In each group, data represent the mean ± SEM (n = 12–17; *p < 0.05 vs wild type at 37°C; †p < 0.05 and ††p < 0.01 vs wild type at 40°C; §p < 0.05 and §§§p < 0.001 vs the same mutations at 37°C; two-tailed unpaired Student’s t test). All currents were recorded in HEK293T cells under lifted whole-cell configurations and were voltage clamped at −50 mV.
Mutant γ2S subunit-containing, but not α1(A322D) subunit-containing, α1β2γ2S receptor currents were reduced additionally by elevated temperature
To determine the temperature sensitivity of heterozygous α1β2γ2S receptors containing γ2S subunit mutations, we recorded from cells expressing each mutant receptor at room temperature (25°C) after incubation at 37°C or within 30 min after a 1 h incubation at 40°C (Fig. 5C). Peak α1β2γ2S(R43Q), α1β2γ2S(K289M), and α1β2γ2S(Q351X) currents were substantially reduced relative to wild-type currents after incubation at 37°C (Fig. 5D, filled bars). Peak heterozygous α1β2γ2S(R43Q), α1β2γ2S(K289M), and α1β2γ2S(Q351X) currents were reduced substantially after 1 h at 40°C (Fig. 5C) compared with wild-type currents after incubation at 37°C or a 1 h incubation at 40°C or compared with heterozygous currents after incubation at 37°C (Fig. 5D).
Heterozygous α1(A322D)β2γ2S receptor currents were significantly reduced compared with wild-type currents recorded at 25°C after incubation at 37°C (Fig. 5D, wt and A322D, filled bars). However, in contrast to the results obtained with receptors containing mutant γ2S subunits, currents obtained from heterozygous α1(A322D)β2γ2S receptors after a 1 h incubation at 40°C were not further decreased relative to currents obtained after incubation at 37°C (Fig. 5D, A322D, filled and open bars).
Discussion
Variations in temperature have effects on most cellular events, and several neurological disorders are provoked by elevated temperature, including febrile seizures and febrile episodic ataxia (calcium channels, CACN1A) (Subramony et al., 2003). Temperature changes have been shown to affect plasma membrane states (Thompson et al., 1985) and synaptic transmission (Volgushev et al., 2000). For example, synaptic vesicle recycling has been shown to be temperature dependent. The size of recycling vesicles is twice as large, and the speeds of both endocytosis and exocytosis are faster at physiological temperature than at room temperature (Micheva and Smith, 2005). Although the dynamic temperature dependence of turnover of GABAA receptors is unclear, there is evidence that inhibitory synaptic strength can be modulated within 10 min through recruiting more functional GABAA receptor to the postsynaptic plasma membrane (Wan et al., 1997).
The basis for the common febrile seizure phenotype resulting from mutations in GABAA receptor γ2(R43Q), γ2(K289M), and γ2(Q351X) subunits has been unknown. Previous studies indicated that each of the γ2 subunit epilepsy mutations produced different alterations in receptor function that would lead to disinhibition and thus afebrile seizures. The γ2 subunit mutation, γ2(R43Q), associated with autosomal dominant childhood absence seizures and febrile seizures is located in the N terminus and was suggested to impair diazepam sensitivity (Wallace et al., 2001) or to alter receptor kinetics or to reduce peak amplitude (Bianchi et al., 2002) of α1β2γ2 receptor currents caused by receptor ER retention and degradation leading to impaired receptor surface expression (Kang and Macdonald, 2004; Sancar and Czajkowski, 2004; Hales et al., 2005). The autosomal dominant γ2 subunit mutation, γ2(K289M), associated with GEFS+ is located in the extracellular TM2-TM3 loop, and it was reported that the mutation caused reduced α1β2γ2 receptor peak current in oocytes (Baulac et al., 2001). However, we reported that the mutation caused accelerated channel deactivation and reduced single-channel open time (Bianchi et al., 2002) with normal peak amplitude when recorded at room temperature (25°C), suggesting a gating defect. In another small autosomal dominant GEFS+ pedigree, a γ2 subunit mutation, γ2(Q351X), introduces a premature stop codon with the loss of 78 C-terminal amino acids. It was reported that homozygous expression of the γ2(Q351X) subunit truncation totally abolished α1β2γ2 currents, and use of a fluorescent epitope-tagged γ2(Q351X) subunit revealed ER retention of homozygous α1β2γ2(Q351X) receptors.
Our results demonstrated that with heterozygous expression, each of these γ2S subunit mutation-containing receptors had reduced γ2S subunit protein surface expression at 37°C and additional reduced surface expression with elevated temperature, suggesting a reduced complement of α1β2γ2S receptors on the cell surface. In addition, we demonstrated that all three heterozygous receptors had reduced α1 subunit protein surface expression at 37°C and additionally reduced surface expression with elevated temperature. From these results, we conclude that pentameric α1β2γ2S receptors were reduced on the surface, and that there was no compensatory increase in cell surface α1β2 receptors when the mutation-containing receptors were challenged with increased temperature.
In cystic fibrosis transmembrane conductance regulator (CFTR), intracellular mutant protein trafficking has been studied extensively. The most common cystic fibrosis mutation in the CFTR gene (ΔF508) results in a protein that fails to mature conformationally and does not exit from the ER to the cell surface (Gelman and Kopito, 2002). At a lower temperature, however, substantial mutant protein is folded and transported to the plasma membrane surface (Kopito, 1999). Consequently, both wild-type and mutant receptors reached the surface and were both internalized by endocytosis, but the mutant protein was much more subject to endocytosis than the wild-type protein (Heda et al., 2001). Similarly, understanding the temperature dependence and speed and magnitude of dynamic cycling of membrane surface wild-type and mutant heterozygous GABAA receptors will be critical for understanding the rapid change in surface GABAA receptors produced by elevated temperature.
Consistent with the fact that all of the heterozygous γ2S subunit mutation-containing receptors had further reduction of surface expression with elevated temperature, our confocal microscopy and electrophysiological data also supported the conclusion that elevated temperature resulted in additional receptor localized to intracellular compartments, including the ER. Interestingly, the increased intracellular receptors were not only the mutant but also the coexpressed wild-type receptors, suggesting there might be cotrafficking of wild-type and mutant receptors or a dominant-negative effect of the mutant receptors. Importantly, the dynamic receptor surface reduction caused by accelerated endocytosis and/or increased retention was detected <10 min after the rise in temperature in heterozygous γ2(K289M) subunit-containing α1β2γ2S receptors. Our previous study of γ2(R43Q) receptors revealed enhanced ER retention of mutant receptors at 37°C, but a dynamic temperature-dependent turnover caused by endocytosis of surface receptors has not been described. In neurons, the reduction of surface receptor could be detected within 20 min after the rise in temperature in heterozygous γ2(Q351X) subunit-containing α1β2γ2L receptors (Fig. 4). In neurons, the γ2 subunit is important for receptor and gephyrin clustering (Wang et al., 1999; Alldred et al., 2005). It is possible that any disease-causing mutation in γ2 subunits would compromise receptor trafficking and/or endocytotic recycling and that this trafficking defect could be worsened by elevated temperature. However, it is unknown whether and how elevated temperature would affect receptor internalization and/or forward trafficking or receptors to the surface membrane. Nevertheless, the rapid reduction in surface receptors observed in both heterologous cells and neurons might explain why children harboring one of the γ2 subunit mutations might have seizures associated with temperature elevation, a conclusion that must be confirmed in vivo.
In contrast to heterozygous mutant γ2S subunit-containing receptors, heterozygous α1(A322D) subunit-containing α1β2γ2S receptors did not have temperature-dependent reduction in current. This may be attributable to the specific effect of the α1(A322D) subunit mutation on α1β2γ2S receptor expression. We demonstrated that the α1(A322D) subunit mutation reduced α1(A322D), but not α1, subunit expression after translation but before receptor assembly, resulting in ER-associated degradation (Gallagher et al., 2005). Thus, mutant α1 subunit-containing α1β2γ2S receptors are not assembled, and membrane α1β2γ2S receptors almost exclusively contain wild-type α1 subunits and are not subject to temperature-dependent effects. In contrast, we demonstrated that although trafficking of the mutant γ2 subunit-containing α1β2γ2S receptors to the surface is deficient, both mutant and wild-type receptors are present on the surface (Macdonald et al., 2004). In addition, we have evidence that heterozygous wild-type and mutant α1β2γ2S(Q351X) receptors have trafficking interactions, suggesting that the temperature-sensitive mutant γ2 subunit-containing receptors interact with wild-type receptors to alter trafficking/endocytosis of both wild-type and mutant receptors.
Our data also suggest that wild-type α1β2γ2S receptors have impaired trafficking during prolonged temperature elevation. After incubation for 2.5 h at 40°C, peak whole-cell current amplitudes from cells expressing wild-type receptors increased 30-fold over 45 min at room temperature and were then stable for the next 2 h, suggesting that ∼45 min was required for full recovery of α1β2γ2S receptors from an elevated temperature-induced trafficking deficiency. Our data also suggested that the trafficking deficiency of wild-type α1β2γ2S receptors at elevated temperature was reversible.
GABAA receptors containing mutant γ2 subunits associated with febrile seizures and GEFS+ have impaired αβγ2 channel function, thus lowering the threshold for nonfebrile seizures. We have shown that wild-type α1β2γ2S receptor trafficking is also vulnerable to high temperature but to a lesser degree. Because the γ2 subunit is critical for receptor trafficking, clustering, and synaptic maintenance, any mutations causing misfolding and impaired assembly and ER retention, increased degradation, or rapid endocytosis would result in reduced surface expression and inhibitory synaptic strength in neurons that would be worsened by fever resulting in febrile seizures. Our finding that surface expression of GABAA receptors containing mutant γ2S subunits, but not containing the α1(A322D) subunit mutation, is vulnerable to elevated temperature and that all three GABAA receptor γ2S subunit mutations showed rapidly compromised receptor trafficking or accelerated endocytosis when challenged with elevated temperature may explain why febrile seizures are provoked by fever in individuals harboring the γ2S, but not the α1(A322D), subunit mutations.
Footnotes
This work was supported by National Institutes of Health Grant R01 NS33300.
- Correspondence should be addressed to Dr. Robert L. Macdonald, Vanderbilt University Medical Center, 6140 Medical Research Building III, 465 21st Avenue, South, Nashville, TN 37232-8552. Email: robert.macdonald{at}vanderbilt.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}