Article Text

Abstract
A substantial body of evidence has accumulated to suggest a role for the xanthine oxidase metabolic pathway in the pathophysiology of chronic heart failure and other cardiovascular diseases
- BNP, brain natriuretic peptide
- CHF, chronic heart failure
- NO, nitric oxide
- XO, xanthine oxidase
- allopurinol
- heart failure
- xanthine oxidoreductase
Statistics from Altmetric.com
Biochemical and pharmacologic studies implicate xanthine oxidoreductase (XO) as a major source of reactive oxygen species in the cardiovascular system. XO derived oxygen-free radicals lead to impaired endothelium dependent vasodilator capacity via scavenging and premature degradation of endothelium derived nitric oxide (NO) to peroxynitrate within a cascade of further radical formation. Impaired vasodilator capacity, the hallmark of endothelium dysfunction, leads to impaired tissue perfusion and hereby constitutes an important aspect of the pathophysiology of diabetes, hypertension, arteriosclerosis, and chronic heart failure (CHF). Hence, blocking the XO generated oxygen radical accumulation emerged as an intriguing novel treatment option to prevent oxygen radical accumulation and its adverse effects. An increasing number of clinical studies have been initiated to test whether this therapeutic approach translates into meaningful beneficial pathophysiological changes. In aortocoronary bypass surgery, treatment with allopurinol has been shown to counteract effects of ischaemia–reperfusion injury1 leading to reduced need for inotropic support2 and to increased immediate and late (six month) left ventricular contractility3
In this issue of the Heart, Gavin and Struthers report on a treatment trial using allopurinol in patients with CHF.4 The authors tested the hypothesis that XO inhibition in CHF may improve exercise capacity and also prognostic markers such as brain natriuretic peptide (BNP). This is a very timely hypothesis to test. The most important pathophysiologic findings that have led to the design of the study are: XO activity is up-regulated in patients with CHF,5–7 leading to increased oxygen radical load and hyperuricaemia,8 independently of renal impairment or of the effects of diuretics.9 Endothelial dysfunction is a constant feature in CHF and the XO pathway has been identified as one important underlying mechanism. Increased vascular resistance in CHF is related to the degree of hyperuricaemia.8 Hyperuricaemia has been shown to be a strong and independent marker of poor prognosis in CHF.10 Finally, several short term treatment studies showed allopurinol attenuated endothelial dysfunction in these patients.11–13
DIRECT MYOCARDIAL EFFECTS OF ALLOPURINOL
Independently of the beneficial effects on vasodilator capacity, direct myocardial effects have been observed for allopurinol. Recent studies in both CHF patients and models of experimental heart failure showed that xanthine oxidase inhibition increased contractile capacity14 due to a calcium (Ca2+) sensitising mechanism15 and improved myocardial efficiency6 by reducing myocardial oxygen consumption.16 After myocardial infarction in a mouse model, allopurinol treatment significantly attenuated left ventricular dilatation and dysfunction17
In contrast to the aforementioned results, Gavin and Struthers did not find an improvement of exercise capacity at maximum or sub-maximum exercise level.4 How do we explain this unexpected neutral outcome? If the results by Gavin and Struthers are the correct and repeatable reflection of the fact that allopurinol treatment does not improve exercise capacity, an important surrogate end point for treatment effects in CHF is not met. This in itself may not be a problem as a number of studies on angiotensin converting enzyme inhibitors or β blockers failed to show benefits on exercise capacity in CHF patients,18 and nobody would want to treat CHF patients without these drugs today.
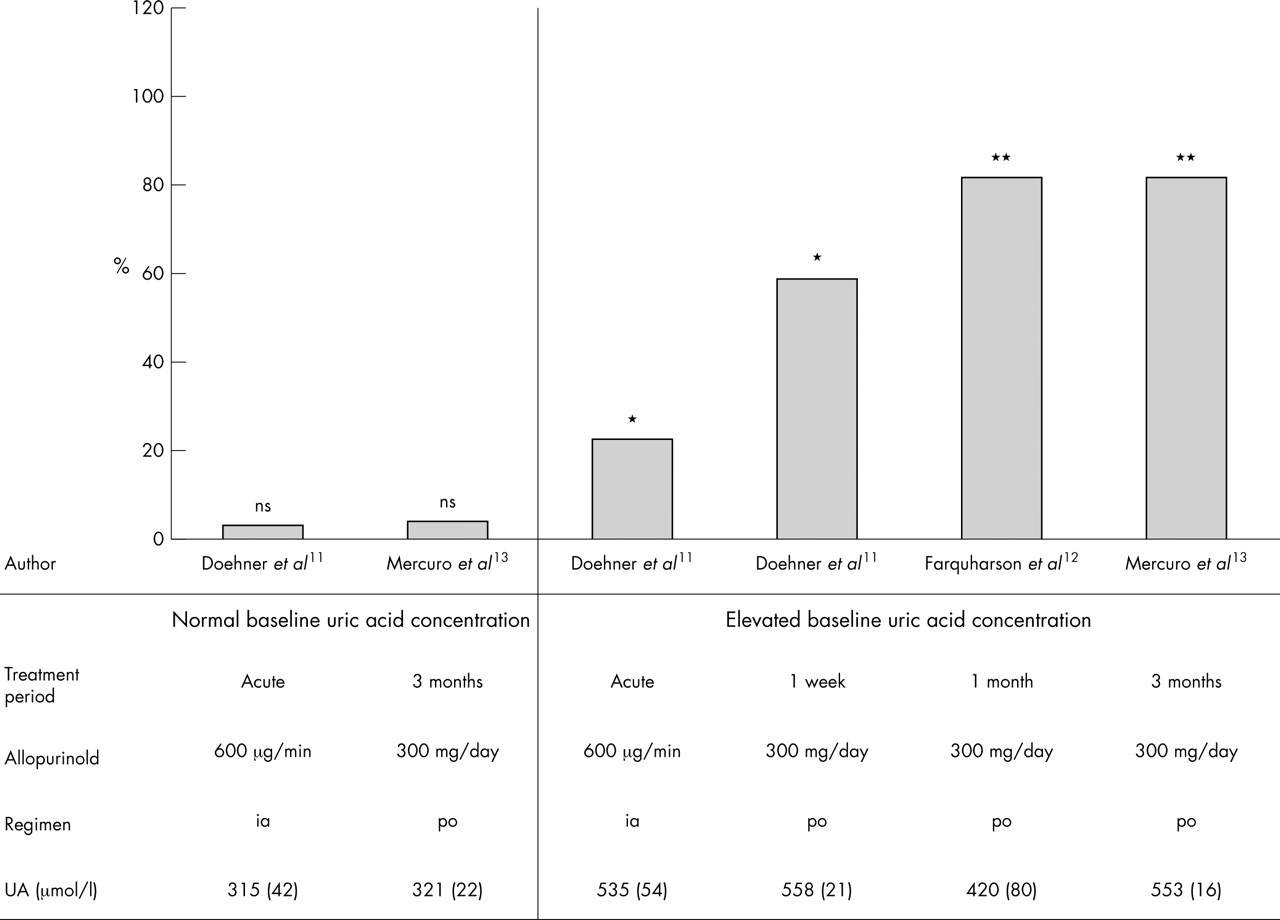
However, there are some aspects of the study by Gavin and Struthers4 that may explain, to some degree, the neutral results. Most importantly, a pronounced effect of XO inhibition by allopurinol may be observed only if XO activity is indeed up-regulated. Accordingly, in previous treatment trials, the improvement of endothelium dysfunction in CHF patients was particularly tested in hyperuricaemic patients.11–13 However, no effect on vasodilator capacity has been observed following allopurinol treatment in patients with normal uric acid concentrations11,13 (fig 1). The conclusion might be that targeted XO inhibition as a novel treatment approach might be suitable only in those patients where increased uric acid concentrations are an indicator of up-regulated XO activity. Extending this approach to patients with normal uric acid values might increase the number of non-responders. From the current study as presented we are not able to say whether baseline uric acid concentrations were raised in the treatment group nor whether baseline uric acid values were related to the observed changes in exercise capacity or BNP concentrations. Although baseline concentrations of uric acid are not presented it appears that patients with raised and with normal uric acid concentrations were included. It would be interesting to know whether the effect of treatment on exercise capacity was different in subgroups with high and with normal uric acid.
{kind=link}
Allopurinol treatment in patients with chronic heart failure and its effect on vasodilator capacity. Allopurinol improved endothelium dependent vasodilator capacity in hyperuricaemic patients, but not in patients with normal uric acid (UA) concentrations (sorted by normal versus raised uric acid concentrations and duration of treatment; data presented as percentage change from baseline).
ASSESSING EXERCISE CAPACITY
Another aspect is the use of the six minute walk test to assess exercise capacity. This test is simple and hence has been used in a considerable number of intervention studies in CHF patients. The present study was powered to detect an improvement in exercise capacity of 23%. There are a number of trials that failed to show a treatment effect employing this method. Exercise training improves exercise capacity by 12–32% when assessed by peak oxygen consumption (Vo2) measurement,19 but only by about 5% when assessed by the six minute walk test.20 In several studies six minute walk testing failed to have prognostic value in CHF whereas there is agreement that the assessment of peak Vo2 is the gold standard for assessing prognosis in CHF patients.21 Hence, it appears that the six minute walk test is not very sensitive to clinical changes in CHF patients, particularly when patients are in mild disease states as in the present study (walking distance > 410 m, BNP near normal, mortality 2% in six months).4
So is the concept of XO inhibition in CHF to be rejected following the findings of this study? Certainly not. First of all, in the present study BNP was lowered by allopurinol treatment (p = 0.03) which is very encouraging. There is a substantial body of evidence suggesting that the xanthine oxidase metabolic pathway is not merely the final step of purine degradation, with the formation of uric acid as a metabolically inert waste product. In humans the organs with the highest XO activity are the intestine and the liver, with low or undetectable levels of activity in the brain, kidney, lung, and muscle.22 The localisation of XO primarily in the endothelial cells of the capillaries suggests that XO is involved in specific functions of the vascular system.23 Given the capacity to generate free oxygen radicals, this enzyme may also have a role in bactericidal defence mechanisms,24 especially at the barrier between the intestinal lumen and the body tissues. This physiologic mechanism may provide an acute adaptive response to environmental factors.
LONG TERM STIMULATION OF XO
One could hypothesise that, analogous to other regulatory systems in CHF, long term stimulation of XO may result in chronic activation of this mechanism leading to maladaptive processes and eventually harmful effects. The latter provides the pathophysiologic link of uric acid with a large variety of detrimental processes, including increased cytokine production,25 cell apoptosis,26 and endothelial dysfunction,27 all of which occur in CHF patients. There is increasing evidence that the XO metabolic pathway is embedded within a complex web of cross linked metabolic and immune regulatory mechanisms and abnormally increased XO activity significantly contributes to pathophysiologic processes in CHF (reviewed in more detail by Anker and colleagues10). New studies are underway using allopurinol but also other XO inhibitors like oxopurinol.28 CHF is a metabolic illness, but no metabolic treatment is available yet. We believe that XO inhibition is a useful metabolic treatment for hyperuricaemic CHF patients.
Beyond XO dependent radical accumulation, recent data suggest that also uric acid itself may contribute to cardiovascular pathophysiology. This might be surprising in view of the antioxidant capacity that has been attributed to uric acid. Uric acid has been identified as a principal endogenous danger signal mediating immune response upon cell injury.29 Accordingly, in a mouse model uric acid infusion caused increased endotoxin stimulated tumour necrosis factor-α production and hence proinflammatory immune activation.30 Also, promotion of low density lipoprotein oxidation31 and smooth muscle cell proliferation by uric acid have been shown in vitro.32
CONCLUSION
A substantial body of evidence has accumulated to suggest a role for the XO metabolic pathway in the pathophysiology of CHF and other cardiovascular diseases. A beneficial effect of targeted inhibition of XO, and hence reduction of radical load and uric acid production, has been suggested in a number of studies. Despite a mostly neutral result in the current study by Gavin and Struthers4 on exercise capacity in CHF, more work is warranted to elucidate further the potential of this novel treatment option. In those proof of concept studies, we suggest the tailored treatment option of XO inhibition might be restricted to hyperuricaemic CHF patients.
REFERENCES
Footnotes
-
↵* Also Department of Clinical Cardiology, National Heart & Lung Institute, Imperial College, London, UK
Linked Articles
- Miscellanea