


Abstract
Activation of the A3 adenosine receptor (A3AR) contributes to the cardioprotective, bronchoconstrictive, and hypotensive effects of adenosine. Agonist occupation of the A3AR results in a rapid desensitization of receptor function, which is associated with the phosphorylation of the receptor protein by one or more members of the G protein-coupled receptor kinase family of protein kinases. Although we demonstrated previously that phosphorylation of the C-terminal 14 amino acids of the rat A3AR is crucial for rapid desensitization to occur, the identity of the critical phosphorylation sites has remained unknown. Here, we demonstrate that the simultaneous mutation of Thr307, Thr318, and Thr319 to Ala residues dramatically reduces agonist-stimulated phosphorylation and rapid desensitization of the rat A3AR. Individual mutation of each residue demonstrated that Thr318 and Thr319 are the major sites of phosphorylation. Phosphorylation at Thr318 appeared to be necessary to observe phosphorylation at Thr319, but not vice versa. However, the replacement of Thr318 with a glutamate residue demonstrated that the simple addition of negative charge at position 318 was not sufficient to rescue phosphorylation at position 319. In addition, the mutation of two predicted palmitoylation-site cysteine residues proximal to the regulatory domain resulted in the appearance of an agonist-independent basal phosphorylation. Therefore, G protein-coupled receptor kinase-mediated phosphorylation of the C-terminal tail of the A3AR in situ appears to follow a sequential mechanism, perhaps involving receptor depalmitoylation, with phosphorylation at Thr318 being particularly important.
Adenosine is a ubiquitous regulator of cellular function. Its effects on target tissues are mediated by binding to four adenosine receptor (AR) subtypes termed A1, A2A, A2B, and A3 (Ralevic and Burnstock, 1998). Importantly, even though it is the most recently identified of the AR family, the A3AR has already been shown to play a crucial role in some of the most important physiological effects of adenosine. These include cardioprotection from ischemia-reperfusion injury (Dougherty et al., 1998; Liang and Jacobson, 1998), eosinophil activation (Kohno et al., 1996), and neuroprotection (Von Lubitz et al., 1994). These effects are initiated by interaction of agonist-occupied A3ARs with members of the Gi family of guanine nucleotide-binding regulatory proteins (G proteins) (Gilman, 1987). Like almost all G protein-coupled receptor (GPCRs), the A3AR is predicted to consist of an extracellular N-terminal domain linked to a cytoplasmic C-terminal tail by seven transmembrane-spanning α-helices (Ji et al., 1998).
Like many other GPCRs, intracellular signals initiated by agonist-occupied rat A3ARs are subject to a rapid homologous desensitization (Ali et al., 1990; Ramkumar et al., 1993;Apgar, 1994). Rapid termination of GPCR signaling is initiated typically by receptor phosphorylation events catalyzed by either second messenger-activated kinases or GPCR kinases (GRKs) (Hausdorff et al., 1990). The latter constitute a family of six enzymes that specifically phosphorylate agonist-occupied receptors (Pitcher et al., 1998). We have shown previously that rapid functional desensitization of the rat A3AR expressed in Chinese hamster ovary (CHO) cells is associated temporally with the phosphorylation of the receptor by one or more members of the GRK family (Palmer et al., 1995). This is in contrast to the A1AR, which is not phosphorylated and desensitizes much more slowly than the A3AR in this system (Ramkumar et al., 1991;Palmer et al., 1996; Gao et al., 1999). By generating a chimeric A1-A3AR, termed A1CT3AR, we demonstrated that the A3AR C-terminal domain was responsible for conferring sensitivity to GRK phosphorylation and rapid desensitization kinetics (Palmer et al., 1996). However, the identity of the residues phosphorylated by GRKs has remained unknown.
To accurately determine the phosphorylation sites within the C-terminal tail of the A3AR phosphorylated by GRKs in response to agonist exposure, we assessed the consequences of specific mutations on agonist-stimulated A3AR phosphorylation. In light of these experiments, we propose a sequential model of A3AR phosphorylation that may be controlled by palmitoylation of the receptor within its C-terminal domain.
Experimental Procedures
Materials.
N 6-(3-Iodobenzyl)-5′-N-methylcarboxamidoadenosine (IBMECA) was the generous gift of Dr. Kenneth Jacobson (National Institutes of Health, Bethesda, MD). Cell culture supplies were obtained from Life Technologies Europe (Paisley, Scotland, UK). Radiochemicals were obtained from DuPont-New England Nuclear (Boston, MA).125I-N 6-(4-Aminobenzyl)-5′-N-methylcarboxamidoadenosine (ABMECA) was synthesized and purified as described previously (Olah et al., 1994). Sources of other materials have been described elsewhere (Palmer et al., 1995, 1996).
Receptor cDNA Constructs and Expression.
Site-directed mutagenesis of the rat A3AR was performed using a two-step polymerase chain reaction-based protocol with pCMV5/hemagglutinin epitope-tagged rat A3AR cDNA as a template (Palmer et al., 1995). The presence of the indicated mutations was verified by dideoxynucleotide sequencing.
CHO cell lines stably expressing the indicated A3AR cDNAs were generated by cotransfecting cells with mutant A3AR cDNA in a pCMV5 expression vector and pSV2Neo (conferring neomycin resistance) according to a modified calcium phosphate precipitation-glycerol shock procedure described previously (Palmer et al., 1995). After selection in G418, resistant clones were isolated, expanded, and screened for recombinant A3AR expression by radioligand binding using125I-ABMECA (Olah et al., 1994). Cells were propagated at 37°C in T-75 flasks in Ham's F-12 medium supplemented with 10% (w/v) fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin in a humidified atmosphere containing 5% CO2.
Receptor Phosphorylation.
A3AR-expressing CHO cells were plated onto 6-well dishes at a density of approximately 1 × 106 cells/well and cultured overnight in regular medium. The next day, the cells were washed twice with phosphate-free Dulbecco's modified Eagle's medium and incubated for 90 min in the same medium supplemented with 1 U/ml adenosine deaminase and 0.2 mCi/ml [32P]orthophosphate. After stimulation with or without the A3AR agonists (R)-N 6-(phenylisopropyl)adenosine [(R)-PIA] and 5′-N-ethylcarboxamidoadenosine (NECA), reactions were terminated by placing the cells in ice and washing them three times with ice-cold PBS solution. All subsequent procedures were performed at 4°C unless stated otherwise. Cells were solubilized by the addition of 0.5 ml of immunoprecipitation buffer (50 mM sodium HEPES, pH 7.5, 5 mM EDTA, 10 mM sodium phosphate, 10 mM sodium fluoride, 0.1 mM phenylmethylsulfonyl fluoride, 0.7 μg/ml pepstatin A, and 10 μg/ml concentration each of soybean trypsin inhibitor and leupeptin). After a 60-min incubation on a rotating wheel, insoluble material was removed by centrifugation (14,000g for 15 min). Extracts were then equalized by protein assay and precleared of nonspecific binding proteins by incubation with protein A-Sepharose in the presence of 0.2% (w/v) IgG-free BSA. Receptors were then immunoprecipitated from precleared supernatants by incubation for 2 h with protein A-Sepharose and 1 μg of 12CA5. Immune complexes were isolated by centrifugation, washed twice with immunoprecipitation buffer supplemented with 0.2 M ammonium sulfate and once with immunoprecipitation buffer alone, and eluted from the protein A-Sepharose by the addition of electrophoresis sample buffer and incubation at 37°C for 1 h. Analysis was by SDS-polyacrylamide gel electrophoresis (PAGE) using 10% (w/v) polyacrylamide resolving gels and autoradiography. Quantification of phosphorylation experiments was by excision of bands from the dried gel and Cerenkov counting.
Because A3AR phosphorylation was being compared between CHO cell lines expressing different receptor levels (Table1), it was critical that the amount of receptor loaded for SDS-PAGE from each cell line was equivalent. Therefore, A3AR expression levels were determined on the day of phosphorylation assays by radioligand binding using a saturating concentration (approximately 10 nM) of125I-ABMECA. The amount of receptor in each transfected cell population (in pmol/mg protein) multiplied by the protein content of the solubilized fraction taken for immunoprecipitation (mg protein/sample) produced a value for the level of receptors in each immunoprecipitation sample (pmol/sample). For SDS-PAGE analysis, the amount of receptor loaded was normalized to that of the sample with the least receptor and loading volumes were equalized by the addition of electrophoresis sample buffer.
Characterization of cell lines stably expressing WT and mutant A3ARs
Cell Surface Biotin Labeling.
This was performed exactly as described in Palmer et al. (1995) except that the loading of immunoprecipitates derived from different A3AR-expressing cell lines was normalized with respect to receptor number as described for the phosphorylation experiments.
Radioligand Binding and Adenylyl Cyclase Assays.
Radioligand binding experiments using 125I-ABMECA were performed and analyzed as described previously (Olah et al., 1994). Adenylyl cyclase assays were performed exactly as described previously using the A3AR agonist IBMECA (Palmer et al., 1995). Dose-response curves were analyzed using a noniterative curve-fitting program (Prism version 2.0; GraphPad, La Jolla, CA).
Data Analysis.
Data are presented as mean ± S.D. for the number of experiments indicated. Statistical significance was determined by two-tailed Student's t tests with significance assessed at P < .05.
Results
Generation of Cell Lines Expressing Thr → Ala Mutant A3ARs.
Previous studies have demonstrated that agonist occupation of the A3AR results in the phosphorylation of Thr residues within the C-terminal 14-amino acid region of the receptor protein (Palmer et al., 1996). To assess which of these residues was most important in determining susceptibility to receptor phosphorylation, a panel of CHO cell lines was generated expressing mutant A3ARs in which candidate Thr residues within the C-terminal domain were mutated to nonphosphorylatable Ala and Glu residues (Fig.1A). In addition, two potential sites of palmitate attachment proximal to the regulatory C-terminal domain (Cys302 and Cys305) were mutated to Ala to assess any role for these residues in controlling A3AR phosphorylation. The radioligand binding parameters for each of the cell lines used in this study are given in Table 1. Importantly, each mutant A3AR displays a similar affinity for the A3AR agonist radioligand125I-ABMECA as the wild-type (WT) A3AR (Table 1).

Predicted membrane-spanning topography of the rat A3AR. A, predicted seven transmembrane-spanning topography of the rat A3AR is shown along with the primary sequence of the 14-amino acid C-terminal regulatory domain. Candidate GRK phosphorylation sites (Thr307, Thr318, and Thr319) and palmitoylation sites (Cys302 and Cys305) are highlighted. B, nontransfected and transfected CHO cells expressing the indicated A3ARs were labeled with biotin-long alkyl spacer chain-hydrazide as described inExperimental Procedures. After immunoprecipitation with 12CA5 and protein A-Sepharose, samples were normalized for receptor expression before fractionation by SDS-PAGE. After transfer of fractionated proteins to nitrocellulose, biotin-labeled A3ARs were visualized by probing with horseradish peroxidase-conjugated streptavidin. This is one of three experiments that produced qualitatively similar results.
To account for the fact that the cell lines we used displayed a range of B max values in radioligand binding assays (Table 1), volumes of receptor immunoprecipitates loaded for SDS-PAGE were normalized with respect to receptor number as determined by radioligand binding assays using 125I-ABMECA as described in Experimental Procedures. The validity of this approach is demonstrated in Fig. 1B. After the labeling of cell surface glycoproteins with biotin, hemagglutinin epitope-tagged A3ARs were immunoprecipitated from equivalent amounts of cell extract from each of the indicated cell lines. Taking into account the different B max values obtained for each cell line, the loading volumes of the immunoprecipitates used for SDS-PAGE were then adjusted so that each lane was predicted to contain the same amount of receptor. Subsequent development of the blots after incubation with peroxidase-conjugated streptavidin proved that equalization of receptor loading in this manner was sufficient to ensure equivalent immunoprecipitation of cell-surface A3ARs from distinct cell lines.
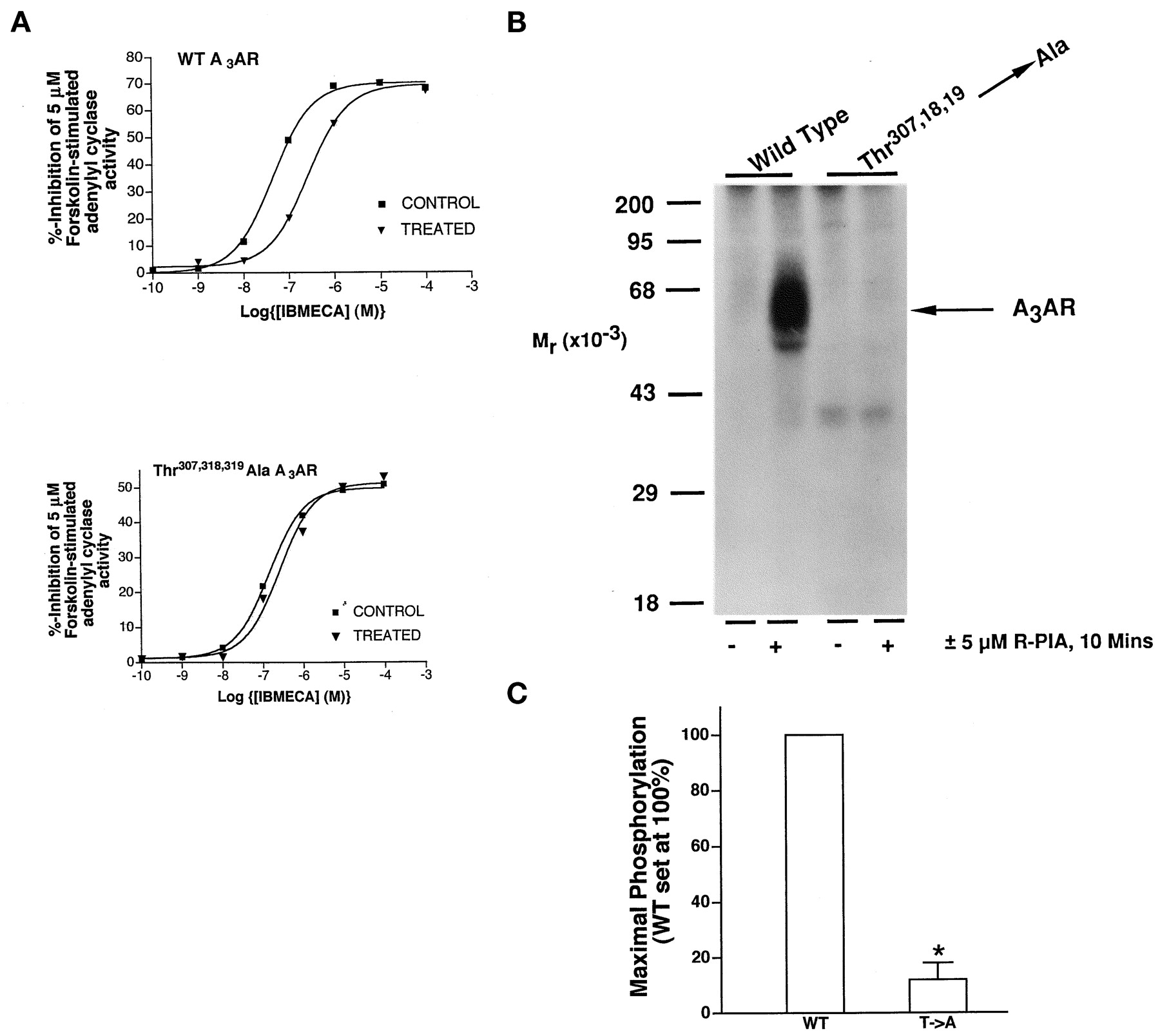
Simultaneous Mutation of Thr307, Thr318, and Thr319 → Ala Produces a Nonphosphorylated, Nondesensitizing A3AR.
Because the A3AR is phosphorylated predominantly on Thr residues in response to agonist exposure in situ, each of the threonines in the C-terminal domain was mutated simultaneously to a nonphosphorylatable Ala residue. The resulting receptor was then expressed stably in CHO cells for characterization of its abilities to undergo rapid functional desensitization and phosphorylation. Exposure of WT A3AR-expressing CHO cells to a 10 μM concentration of the AR agonist NECA for 10 min resulted in a rapid desensitization of A3AR function that is manifested as a 6-fold increase in the IC50 value for the A3AR agonist IBMECA to inhibit forskolin-stimulated adenylyl cyclase activity in isolated membranes (Palmer et al., 1995, 1996; Fig. 2A and Table 2). In contrast, cells expressing the Thr → Ala-mutated A3AR displayed only a minimal functional desensitization, with only a 1.5- to 2-fold increase in the IC50 value for IBMECA being detectable after a 10-min exposure to 10 μM NECA (Fig. 2A and Table 2). Increasing the agonist exposure time to 30 min did not produce any additional functional desensitization compared with that observed after 10 min (data not shown). A severely reduced ability to undergo functional desensitization was associated with the abolition of the ability of agonist to stimulate mutant A3AR phosphorylation compared with the WT A3AR (Fig.2B).

Effect of simultaneous mutation of Thr307→ Ala, Thr318 → Ala, and Thr319 → Ala on agonist-dependent phosphorylation and desensitization of the rat A3AR. A, CHO cells stably expressing either WT or Thr307 → Ala, Thr318 → Ala, and Thr319 → Ala-mutated A3ARs were incubated without (CONTROL) or with (TREATED) 5 μM (R)-PIA for 10 min as indicated. Membranes were then prepared for immediate assay of IBMECA-mediated inhibition of 5 μM forskolin-stimulated adenylyl cyclase activity. Representative experiments are shown from three performed for each cell line; data are summarized in Table 2. B,32P-labeled CHO cells stably expressing either WT or Thr307 → Ala, Thr318 → Ala, and Thr319 → Ala-mutated A3ARs were incubated with or without 5 μM (R)-PIA for 10 min as indicated. Cells were then solubilized for receptor immunoprecipitation with 12CA5 and analysis by SDS-PAGE and autoradiography as described inExperimental Procedures. Quantitative analysis from three experiments is shown. Phosphorylation is expressed relative to that obtained for agonist-treated WT A3AR (set at 100%). *Statistically significant difference from the WT A3AR. The average stimulation of WT A3AR phosphorylation over basal in these experiments was 20- ± 6-fold.
Effects of short-term agonist exposure on the desensitization of A3AR function in CHO cells stably expressing WT and Thr307,318,319 → Ala-mutated A3ARs
Effect on A3AR Phosphorylation of Mutating Individual C-Terminal Threonines.
To determine whether one or more of the C-terminal threonines was particularly important in controlling A3AR phosphorylation, Thr307, Thr318, and Thr319 was each mutated individually to Ala and the modified receptor expressed stably in CHO cells. Mutation of Thr307 alone produced only a small decrease in receptor phosphorylation (Fig. 3C). However, the mutation of Thr318 and Thr319 to Ala resulted in a dramatic reduction in agonist-stimulated A3AR phosphorylation, which was almost equivalent to the effect of mutating all three threonines in the C-terminal tail (Fig. 3, B and C). Mutation of Thr319 alone reduced phosphorylation by approximately 50% compared with the WT A3AR (Fig. 3, B and C). However, mutation of Thr318 to Ala had the same effect as simultaneously mutating Thr318 and Thr319 (Fig. 3, B and C).

Effect of individual Thr→Ala mutations within the C-terminal regulatory domain on agonist-dependent A3AR phosphorylation. A, 32P-labeled CHO cells stably expressing either WT or different Thr→Ala-mutated A3ARs were incubated with or without 5 μM (R)-PIA for 10 min as indicated. Cells were then solubilized for receptor immunoprecipitation with 12CA5 and analysis by SDS-PAGE and autoradiography as described inExperimental Procedures. B, quantitative analysis from three such experiments. Phosphorylation is expressed relative to that obtained for agonist-treated WT A3AR (set at 100%). *Statistically significant difference from the WT A3AR. The average stimulation of WT A3AR phosphorylation over basal in these experiments was 32 ± 7-fold.
Effect on A3AR Phosphorylation of Mutating Thr318 to Glu.
One possible explanation for the distinct effects on A3AR phosphorylation of mutating Thr318 and Thr319was that phosphorylation at Thr318 was required to observe phosphorylation at position 319. To test this hypothesis, Thr318 was mutated to a Glu residue in an attempt to mimic the addition of negative charge associated with phosphorylation at this position. However, mutation of Thr318 to Glu failed to rescue agonist-stimulated A3AR phosphorylation (Fig.4).

Effect of Thr318 → Glu mutation on A3AR phosphorylation. A, 32P-labeled CHO cells stably expressing either WT or the indicated Thr318-mutated A3ARs were incubated with or without 10 μM NECA for 10 min as indicated. Cells were then solubilized for receptor immunoprecipitation with 12CA5 and analysis by SDS-PAGE and autoradiography as described in Experimental Procedures. B, quantitative analysis from three such experiments. Phosphorylation is expressed relative to that obtained for agonist-treated WT A3AR (set at 100%). *Statistically significant difference from the WT A3AR. The average stimulation of WT A3AR phosphorylation over basal in these experiments was 28 ± 12-fold.
Effect on A3AR Phosphorylation of Mutating Predicted Palmitoylation Sites (Cys302 and Cys305).
From studies performed predominantly on the human β2- adrenergic receptors, it has been suggested that C-terminal tail cysteine residues conserved among almost all GPCRs is palmitoylated and that palmitate turnover is accelerated in response to agonist exposure (Loisel et al., 1996). The A3AR contains two predicted palmitoylation sites in its C-terminal domain: Cys302 and Cys305 (Fig. 1). To determine whether these residues played a role in controlling phosphorylation of the GRK sites within the C-terminal domain of the A3AR, the two cysteine residues were simultaneously mutated to Ala, which cannot be palmitoylated, and assayed for receptor phosphorylation in the absence and presence of a maximally effective concentration of agonist. As shown in Fig. 5, the Cys → Ala mutant A3AR exhibited a significant level of basal phosphorylation, with the addition of agonist further increasing receptor phosphorylation to levels comparable with those achieved by the WT A3AR (Fig. 5).

Effect of Cys302 → Ala and Cys305 → Ala mutations on A3AR phosphorylation. A, 32P-labeled CHO cells stably expressing either WT or the Cys302 → Ala and Cys305 → Ala-mutated A3ARs were incubated with or without 5 μMR-PIA for 10 min as indicated. Cells were then solubilized for receptor immunoprecipitation with 12CA5 and analysis by SDS-PAGE and autoradiography as described in Experimental Procedures. B, quantitative analysis from three such experiments. Phosphorylation is expressed relative to that obtained for agonist-treated WT A3AR (set at 100%). *Statistically significant difference from the basal phosphorylation observed for the WT A3AR. The average stimulation of WT A3AR phosphorylation over basal in these experiments was 28 ± 6-fold.
Discussion
Although many studies have examined the ability of peptides (Onorato et al., 1991), fusion proteins containing specific GPCR cytoplasmic domains (Prossnitz et al., 1995), and recombinant GPCRs (Debburman et al., 1995) to act as substrates for defined GRKs in vitro, relatively few studies have identified sites of GRK phosphorylation within intact GPCRs in situ. A major limitation of in vitro studies is that stoichiometries of phosphorylation by GRKs are typically much greater than those observable for receptors isolated from intact cells. This has been most elegantly demonstrated for the light receptor rhodopsin, which can incorporate up to 12 mol phosphate/mol of receptor when incubated with rhodopsin kinase/GRK1 in vitro but has a stoichiometry of 1 to 2 mol phosphate/mol of receptor when isolated from irradiated rod outer segments (Zhao et al., 1995). Similar discrepancies have been observed when comparing in vitro and in situ phosphorylation stoichiometries of the human β2-adrenergic receptor (Benovic et al., 1987;Pippig et al., 1995) and chick heart m2muscarinic acetylcholine receptor (Kwatra and Hosey, 1986; Richardson et al., 1993). In light of this limitation, an accurate determination of physiologically relevant sites phosphorylated by GRKs in intact cells is vital if GPCR desensitization mechanisms are to be manipulated for therapeutic benefit.
A prerequisite for manipulating A3AR function for potential therapeutic applications is knowledge of the signaling pathways initiated on receptor activation and the molecular mechanisms that desensitize, or “turn off,” receptor function in response to sustained agonist exposure. As such, identification of the sites within the A3AR phosphorylated by GRKs would constitute a significant advance. It is well established that both recombinant (Palmer et al., 1995, 1996) and natively expressed (Ali et al., 1990;Ramkumar et al., 1993; Apgar, 1994) A3ARs undergo a rapid functional desensitization within minutes of agonist exposure. In this study, we identified three critical Thr residues (Thr-307, -318, and -319) in the C-terminal regulatory domain whose mutation dramatically reduces both agonist-dependent phosphorylation by GRKs and rapid desensitization of A3AR function. In addition, we demonstrated that two cysteine residues that represent potential sites for receptor palmitoylation may also regulate A3AR sensitivity to GRK phosphorylation.
In vitro peptide phosphorylation and fusion protein studies with recombinant GRK2 have revealed that this ubiquitously expressed GRK prefers to phosphorylate serine and Thr residues flanked on their N-terminal side by acidic amino acids. The same studies have also shown that phosphorylation by GRK2 proceeds in a sequential, or “ordered,” manner (Onorato et al., 1991; Chen et al., 1993;Prossnitz et al., 1995). Both of these features appear to be important characteristics of agonist-stimulated A3AR phosphorylation in situ, which we have previously shown to be mediated by a kinase with a similar substrate specificity to GRK2 (Palmer et al., 1995). First, the two major sites of phosphorylation (Thr318 and Thr319) are located downstream of three acidic amino acids (Asp309, Asp312, and Glu316) (Fig. 1A). Second, a comparison of the extents to which individual Thr → Ala point-mutated A3ARs were phosphorylated suggested strongly that phosphorylation at Thr318 was essential to observe phosphorylation at Thr319. However, because the simple addition of a negative charge at position 318 (by mutating the native Thr to a negatively charged Glu residue) was not sufficient to rescue receptor phosphorylation, additional complex conformational changes associated with A3AR phosphorylation may be required. Alternatively, although the addition of a phosphate group at position 318 may be the only requirement for subsequent phosphorylation at position 319, the propionate side chain of the glutamate may be sufficiently different from a phosphoThr residue to be incapable of priming phosphorylation. Despite these caveats, our experiments potentially represent the first to demonstrate an ordered phosphorylation mechanism for a GPCR in an intact cell model system. Moreover, they contrast with the observations made by Liggett and coworkers in study of the agonist-dependent phosphorylation of the α2A-adrenergic receptor by GRKs (Eason et al., 1995). For this receptor, phosphorylation occurs on four consecutive serine residues present in the third cytoplasmic loop. The mutation of each serine individually diminishes phosphorylation by approximately 25%, suggesting that phosphorylation at a given serine occurs independently of phosphorylation at the other residues (Eason et al., 1995). Thus, although GRKs are capable of phosphorylating many different GPCRs, the molecular mechanisms by which individual GPCRs are phosphorylated by a given GRK may vary considerably.
The marked increase in basal phosphorylation of a palmitoylation site-mutated A3AR suggested strongly that these cysteine residues play an important role in controlling accessibility of the A3AR C-terminal regulatory domain to activated GRKs. In the specific case of the human β2-adrenergic receptor, removal of palmitate increases the accessibility of a serine residue within a consensus cAMP-dependent protein kinase phosphorylation site within the C-terminal domain (Moffett et al., 1996). As a result, nonpalmitoylated Cys → Ala mutant β2-adrenergic receptors are constitutively phosphorylated on this site even in the absence of agonist (Moffett et al., 1993, 1996). However, agonist-occupied A3ARs are phosphorylated exclusively by GRKs under conditions in which the activations of cAMP-dependent protein kinase and other second messenger-regulated kinases are without effect (Palmer et al., 1995). Therefore, our observations would be consistent with a more general model in which agonist-stimulated depalmitoylation of Cys302 and/or Cys305induces a conformational change within the C-terminal domain of the A3AR that increases the accessibility of the kinases that catalyze A3AR phosphorylation. Nevertheless, it should be stressed that the mutation of Cys302 and Cys305 alone cannot fully reconstitute the effect of agonist on receptor phosphorylation and that agonist occupation of the mutant receptor is necessary to enhance receptor phosphorylation to the same extent as that observed for the WT A3AR. Also, it is imperative that the validity of this model is tested by assessing palmitate incorporation and turnover in response to agonist occupation of WT and Cys → Ala mutant A3ARs.
In conclusion, we have demonstrated that agonist-dependent phosphorylation of Thr residues within the C-terminal domain of the A3AR appears to proceed in an ordered fashion, with phosphorylation at position 318 being particularly important for this process. Mutational studies also suggest that the two predicted sites of receptor palmitoylation within the C-terminal domain play an important role in regulating the accessibility of the C-terminal domain to activated GRKs. Comparison of the C-terminal sequences of the various A3ARs suggests that this region may have a conserved regulatory role across different species (Fig.6). The data presented here, combined with the conclusions made from in vitro phosphorylation studies (Chen et al., 1993; Prossnitz et al., 1995), strongly suggest that the clusters of potential phosphorylation sites found in the C-terminal domains of A3ARs derived from different species have a common role as sites of receptor phosphorylation by GRKs. Experiments are under way to test this hypothesis. The generation of mutant A3ARs that are either resistant to GRK phosphorylation or constitutively phosphorylated in the absence of agonist will be invaluable tools for assessing the role of receptor phosphorylation in initiating or terminating distinct Gi-activated signaling cascades, including activation of phospholipase C-β isoforms (Ali et al., 1990; Ramkumar et al., 1993) and extracellular signal-regulated kinase (T. M. Palmer, unpublished observations).

Sequence comparison of the C-terminal domains of cloned A3AR species homologues. Amino acid sequences of the C-terminal domains of the rat, mouse, human, dog, and sheep A3ARs beginning with the conserved predicted palmitoylation site in bold. Potential sites of phosphorylation by GRKs, defined as clusters of Ser/Thr residues downstream of acidic amino acid residues, are underlined.
Footnotes
- Received July 15, 1999.
- Accepted December 14, 1999.
-
Send reprint requests to: Timothy M. Palmer, Ph.D., Room 407, Davidson Building, Division of Biochemistry and Molecular Biology, Institute of Biomedical and Life Sciences, University of Glasgow, Glasgow G12 8QQ, Scotland, UK. E-mail:T.Palmer{at}bio.gla.ac.uk
-
T.M.P. was supported by a Postdoctoral Fellowship from the American Heart Association, North Carolina Affiliate; project grants from the British Heart Foundation and Royal Society; a Medical Research Council Co-operative Group Grant in Cellular Signalling and Molecular Genetics in Metabolic and Cardiovascular Syndromes; and equipment grants from the Wellcome Trust and Tenovus-Scotland. G.L.S. was supported by a National Heart, Lung, and Blood Institute SCOR Grant (5P50-HL54314) in Ischaemic Disease.
Abbreviations
- AR
- adenosine receptor
- G protein
- guanine nucleotide-binding regulatory protein
- GPCR
- G protein-coupled receptor
- GRK
- G-protein-coupled receptor kinase
- CHO
- Chinese hamster ovary
- IBMECA
- N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine
- ABMECA
- N6-(4-aminobenzyl)-5′-N-methylcarboxamidoadenosine
- PAGE
- polyacrylamide gel electrophoresis
- (R)-PIA
- (R)-N6-(phenylisopropyl)adenosine
- NECA
- 5′-N-ethylcarboxamidoadenosine
- WT
- wild type
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}