


Abstract
Adenosine deaminase (ADA) is an enzyme of the purine metabolism that has been largely considered to be cytosolic. Recently, it has been demonstrated that the enzyme appears on the surface of lymphocytes where it interacts with the T-cell activation antigen CD26. ADA also appears on the surface of nonlymphoid cells anchored to adenosine A1 receptors. Here it is demonstrated that cell surface ADA in ADA+/CD26− T lymphocytes anchors to adenosine receptors of the A2B subtype (A2BR). An interaction between A2BR and cell surface ADA has been demonstrated in transfected Chinese hamster ovary cells and Jurkat J32 T lymphocytes. This has been proved by coimmunoprecipitation, binding of exogenous ADA to A2BR+ cells, and coimmunolocalization. The specificity of the interaction has also been demonstrated by the lack of interaction with other members of the G protein-coupled receptor superfamily. Binding of ADA to A2BR increases the affinity of the agonist 5′-N-ethylcarboxamidoadenosine and cAMP production. This effect occurs even when ADA devoid of enzyme activity is used. Therefore, in lymphocytes, cell surface ADA, apart from degrading extracellular adenosine, regulates those actions of adenosine that are mediated via adenosine receptors of the A2B subtype.
The key role of adenosine in the development and function of the immune system is demonstrated in the severe combined immunodeficiency syndrome (SCID) associated with the congenital defect of adenosine deaminase, the enzyme that degrades the nucleoside [reviewed by Hershfield and Mitchell (1989)]. Adenosine deaminase (ADA; E.C. 3.5.4.4) catalyzes the hydrolytic deamination of adenosine and 2′-deoxyadenosine to inosine and 2′-deoxyinosine, respectively. Although the location of the enzyme is mainly cytosolic, ADA is also found associated to the outer face of the plasma membrane. ADA has been cloned, and the amino acidic sequence does not contain any signal peptide or transmembrane domain [see Franco et al. (1997a,b) for review]. In lymphocytes, Kameoka et al. (1993) have reported that the activation marker CD26, also known as dipeptidyl peptidase IV, binds ADA to the cell surface. In nonlymphoid cells, we have identified the A1 adenosine receptor (A1R) as another cell surface ADA-anchoring protein (Ciruela et al., 1996).
The mechanism whereby the ADA-associated SCID affects lymphoid cells is not completely understood, but recently it has been suggested that signaling through adenosine receptors in lymphocytes and lymphocyte precursors contributes to the pathogenesis (Apasov et al., 1997; Resta and Thompson, 1997). Four adenosine receptors, which belong to the superfamily of heptaspanning receptors coupled to G proteins have been cloned (A1, A2A, A2B, and A3) (Ralevic and Burnstock, 1998). In lymphocytes, adenosine analogs produce an accumulation of intracellular cAMP, suggesting that the adenosine receptors expressed are of the A2 subtype and not of the A1 subtype (Nordstedt et al., 1987). Adenosine, acting through A2 adenosine receptors (A2Rs), regulates the T cell receptor-triggered activation-related events such as cell proliferation, interleukin-2 production, up-regulation of interleukin-2 receptor α-chain (CD25), and lymphocyte-mediated cytolysis (Wolberg et al., 1975; Dos Reis et al., 1986; Antonysamy et al., 1995; Huang et al., 1997). A2ARs are involved in adenosine-mediated inhibition of murine T cell activation and expansion (Huang et al., 1997). A2BRs are up-regulated, elicit significant reductions in interleukin-2 production in activated human T cell (Mirabet et al., 1999), and contribute to the deactivation of macrophages, because they reduce the interferon-γ-induced up-regulation of major histocompatibility complex class II molecules, the activity of nitric-oxide synthase, and the production of proinflammatory cytokines (Xaus et al., 1999).
In this report, a close interaction between cell surface ADA and the A2B adenosine receptor present on Jurkat J32 T cells and Chinese hamster ovary (CHO) cells transfected with the cDNA coding for the human A2BR, is demonstrated by ADA binding, confocal microscopy, and coimmunoprecipitation. Moreover, it is shown that this novel protein-protein interaction regulates the binding of agonists to A2BRs by increasing the ligand-binding affinity and the 5′-N-ethylcarboxamidoadenosine (NECA)-induced second messenger production.
Experimental Procedures
Materials.
Fluorescein isothiocyanate (FITC), tetramethylrhodamine isothiocyanate (TRITC), saponin, paraformaldehyde, bovine serum albumin, polyethylenimine (50%), propidium iodide, NECA, and RNase were purchased from Sigma Chemical Co. (St. Louis, MO). [3H]N-NECA (15.1 Ci/mmol) was purchased from Perkin Elmer Life Sciences (Boston, MA). Glycine and electrophoresis reagents were obtained from Boehringer Mannheim (Barcelona, Spain). Nonidet P-40 was from Calbiochem (La Jolla, CA). Sephadex G-25 fine grade columns and protein A-Sepharose CL-4B were obtained from Amersham Pharmacia Biotech (Uppsala, Sweden). Deionized water further purified with a Millipore Milli-Q system (Bedford, MA) was used throughout. Bovine ADA (type VIII, 200 IU/mg of protein; Sigma), filtered through Sephadex G-25, was used before all assays. Bovine ADA activity (30 IU/ml) was completely abolished by preincubation with 0.1 mM HgCl2 (2 h); removal of free Hg2+ was accomplished by gel filtration using Sephadex G-25 (Ciruela et al., 1996).
Cells and Transfections.
Chinese hamster ovary (CHO) cells stably transfected with cDNA coding for the human A2BR have been previously characterized (Pierce et al., 1992). CHO cells stably transfected with cDNA coding for the human dopamine D2 receptor were kindly provided by Dr. Kjell Fuxe (Karolinska Institutet, Stockholm, Sweden). Transfected CHO cells were grown in Dulbecco's modified Eagle's medium (DMEM)/Ham's F-12 (1:1) (Life Technologies, Inc., Gaithersburg, MD) containing 10% fetal calf serum, 2 mM l-glutamine, antibiotics (100 U/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml Fungizone), and a 1.6 mg/ml concentration of the neomycin analog G418. Wild-type CHO cells were cultured in the same conditions described for transfected cells but in the absence of G418. For transient expression, HEK 293 cells were used. Cells growing in 75-cm3 dishes were transiently transfected with 10 μg of DNA encoding for human metabotropic glutamate receptors mGluR1α or mGluR1β by calcium phosphate precipitation (Jordan et al., 1996). Cells were grown in DMEM supplemented with 1 mM sodium pyruvate, 2 mM l-glutamine, 100 U/ml penicillin/streptomycin, and 10% (v/v) fetal calf serum (37°, 5% CO2). Cells were harvested 48 h after transfection.
The human T cell line Jurkat J32 was grown in RPMI 1640 medium (Life Technologies) supplemented with 10% inactivated fetal bovine serum, 2 mM l-glutamine, and antibiotics. Peripheral blood mononuclear cells from healthy donors were isolated from buffy coat cells using the Ficoll gradient method (Boyum, 1968); further purification of lymphocytes from peripheral blood mononuclear cells was performed by depletion of contaminating cells by adherence to plastic plates. In vitro activation of cells was carried out with 1 μg/ml phytohemagglutinin (PHA) for 65 h or was conducted on well plates coated with OKT3 antibody. For OKT3 antibody immobilization, 300 μl of 10 mM PBS, pH 7.4, containing 2.5 μg/ml purified OKT3 antibody were placed for 3 h at 37°C in 24-well, flat-bottomed plates. After two washes with 2 ml of cold PBS, cells were added to wells at a density of 106 cells/ml to begin the activation experiments.
Antibodies.
Rabbit anti-ADA antibody (Serotek, Oxford, UK) has been developed in our laboratory previously (Arán et al., 1991). The antibody against A2B receptor, MPE1, is an affinity-purified (chromatographed through specific peptide coupled to Sepharose) version of the antipeptide antisera ME developed and characterized as described elsewhere (Mirabet et al., 1999). The fluorescein-conjugated anti-CD26 monoclonal antibody (mAb) Ta1 was from Coulter Clone (Izasa, Barcelona, Spain). Affinity-purified polyclonal antibody F1 against an extracellular epitope of metabotropic mGluR1 was characterized elsewhere (Ciruela and McIlhinney, 1997). Polyclonal antibodies against human D2 dopamine receptors were purchased from Santa Cruz Biotechnologies (Santa Cruz, CA). The fluorescein-conjugated rabbit IgG was from Sigma, and donkey anti-rabbit IgG-peroxidase was from Boehringer Mannheim.
Protein Content Determination and Conjugation to Fluorochromes.
Protein was quantified by the bicinchoninic acid method (Pierce Chemical Co., Rockford, IL) as described by Sorensen and Brodbeck (1986) and using bovine serum albumin as standard. Antibodies and the enzyme ADA were conjugated to fluorescein isothiocyanate or tetramethylrhodamine isothiocyanate as described elsewhere (Mirabet et al., 1999).
Immunostaining Assays and Cell Sorting.
Transfected or wild-type CHO cells and transfected HEK 293 cells, grown on glass coverslips, and peripheral blood lymphocytes or Jurkat J32 cells (4 × 106) were fixed with 2% (w/v) paraformaldehyde and stained as described by Mirabet et al. (1999). When necessary, cells were fixed and permeabilized adding 0.05% saponin for CHO cells or 0.2% Triton for HEK 293 cells (included in the fixation solution). For nuclear staining with propidium iodide, cells were incubated with 50 μl of 5 μg/ml propidium iodide and 2 μg/ml RNase for 1 min at room temperature. For confocal microscopy analysis, a Leica TCS 4D confocal scanning laser microscope adapted to an inverted Leitz DMIRBE microscope (Leica Lasertechnik GmbH, Heidelberg, Germany) was used. The colocalization analysis was made by means of Multi Color software (version 2.0; Leica Lasertechnik GmbH). Flow cytometry analysis was done using an EPICS Profile flow cytometer (Coulter, Hialeah, FL). The parameters used to select cell populations for analysis were forward and side light scattering. Cell sorting was performed using the EPICS Profile equipment. For this purpose, Jurkat J32 cells were fixed and labeled with Ta1-FITC anti-CD26 mAb to select CD26− cells. A minimum number of 250,000 lymphocytes were selected, recovered in PBS, and processed for further analysis.
125I-Labeled ADA Binding.
Bovine ADA was iodinated by using the Bolton-Hunter reagent (Amersham Pharmacia Biotech) according to the instructions of the supplier.
For 125I-labeled ADA binding, cells were washed twice with PBS buffer and suspended at 3 × 105 cells/ml. Aliquots were incubated for 2 h with 125I-ADA in the presence or absence of unlabeled ADA in a final volume of 300 μl. Then cells were washed in cold PBS, resuspended in 0.2% SDS, and placed in 10-ml vials containing Formula 989 mixture. Vials were shaken overnight and counted using a 1600 Tri-Carb scintillation counter (Packard Instrument Co., Inc., Meriden, CT) with 50% efficiency.
Ligand Binding Experiments.
Membrane suspensions from A2B-transfected CHO cells were obtained according to Casadó et al. (1990). 38 nM [3H]NECA binding to membrane suspensions (0.5 mg of protein/ml) were carried out at 25°C in 50 mM Tris-HCl buffer, pH 7.4, in the absence or presence of the indicated amount of ADA. After 2 h of radioligand incubation, free and membrane-bound radioligand were separated by rapid filtration of 500-μl aliquots in a Brandel (Gaithersburg, MD) cell harvester through Whatman (Clifton, NJ) GF/C filters embedded in polyethyleneimine. Nonspecific binding was defined as the binding remaining in the presence of an excess of displacer (100 μM NECA). For competition experiments radioligand binding was performed as described above in the presence of increasing amounts of NECA. Filters were transferred to scintillation vials containing 10 ml of Formula 989 (Perkin Elmer Life Sciences). Radioactivity was counted using a Packard 1600 Tri-Carb scintillation counter with 50% of efficiency. In all cases five replicates of each point were performed. Competition data were fitted using a nonlinear regression program as previously described (Casadó et al., 1990, 1992).
Immunoprecipitation and Immunoblotting.
Cell extracts and cell membranes were obtained as described by Ciruela et al. (1999). Cell membranes were solubilized (Ciruela et al., 1999) and immunoprecipitated with anti-ADA, or irrelevant rabbit anti-goat IgG antibodies covalently coupled to a protein A matrix (Schneider et al., 1982) (4°C, overnight). Each immunoprecipitate was washed and resuspended in 60 μl of SDS-polyacrylamide gel electrophoresis nonreducing sample buffer (0.125 M Tris-HCl, pH 6.8, 4% SDS, 20% v/v glycerol, 0.02% bromphenol blue). Samples were heated 15 min at 37°C, and after centrifugation at 1200g, immunoprecipitated proteins were resolved by SDS-polyacrylamide gel electrophoresis in 7.5% gels (Laemmli, 1970). Immunoblotting was performed using the anti-ADA antibody (10 μg/ml) or the anti-A2BR MPE1 antibody (2 μg/ml) and anti-rabbit IgG-peroxidase (Boehringer Mannheim). Polyvinyl difluoride membranes (Immobilon-P; Millipore) were incubated in equal volumes of SuperSignal Chemiluminescent substrates 1 and 2 (Pierce Chemical Co.). The detection reagent was drained off, and the filters were placed in contact with a film (Hyperfilm ECL), which was developed by chemiluminescence.
cAMP Determination.
A2B-transfected CHO cells were grown as confluent monolayers in 24-well cluster dishes (approximately 130,000 cells/well). Cells were washed five times with 2.5 ml of Hanks' balanced salt solution buffer (140 mM NaCl, 5 mM KCl, 1 mM magnesium chloride, 1 mM magnesium sulfate, 1.2 mM calcium chloride, 10 mM HEPES, 5 mM glucose, 0.3 mM potassium dihydrogen phosphate, and 2 mM sodium hydrogen phosphate), pH 7.2 at 37°C. Cells were then incubated (15 min at 37°C) in 0.4 ml of Hanks' balanced salt solution buffer containing 30 μM cAMP phosphodiesterase inhibitor RO-20-1724 (Calbiochem); when included, the concentration of ADA was 0.5 U/ml. cAMP production was induced by addition of 1 μM NECA. After 15 min in the presence of the ligand, reaction was stopped by diluting with 2 volumes of cold ethanol. Samples were centrifuged at 2000g for 15 min at 4°C, and the supernatants were transferred to a clean tube. Samples were dried by gassing with nitrogen at 60°C, and the pellets were resuspended in water. cAMP was quantified by use of an enzyme immunoassay kit from Amersham Pharmacia Biotech following the manufacturer's protocol without acetylation.
Results
Expression of ADA on the Cell Surface of CHO Cells Overexpressing A2BRs.
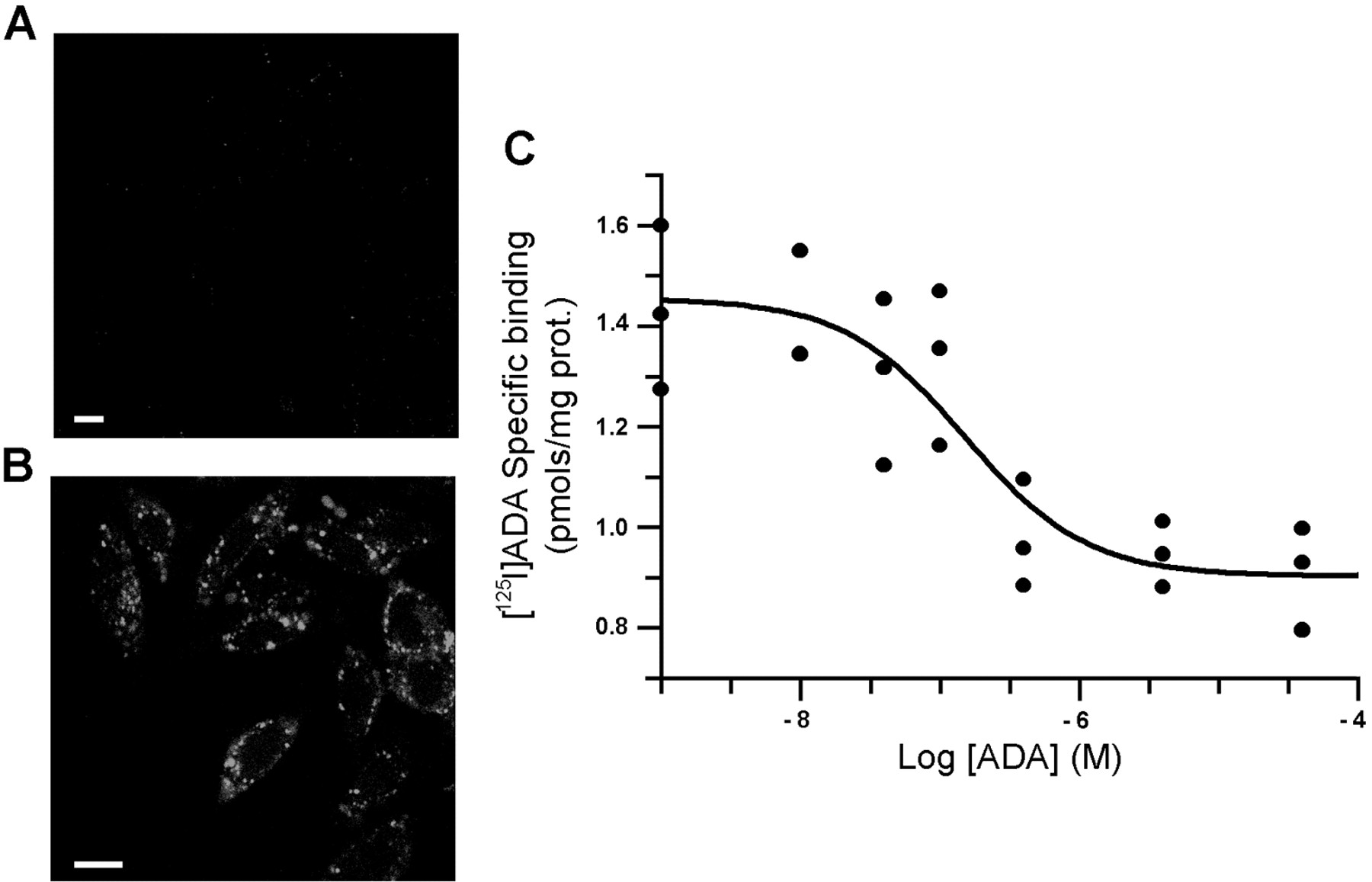
The FITC-MPE1 antibody, which specifically recognizes an extracellular epitope of A2BR, was used to detect the expression of these receptors in CHO cells transfected with the cDNA coding for the human receptor (CHO-A2B). Parental CHO cells could not be labeled with the MPE1 antibody (Fig. 1A), whereas CHO-A2B cells showed a marked staining (Fig. 1C, in green). Parental CHO cells had cytoplasmic ADA, which was detected using permeabilized cells, but did not express cell surface ADA (Fig. 1B). However, when functional A2BR was overexpressed in CHO cells, cell surface ADA could be detected (Fig.1C, in red) and the analysis by confocal microscopy demonstrated a very high degree of colocalization (in yellow) between ADA (red fluorescence) and A2BR (green fluorescence). This colocalization indicates that ADA and A2BR are close on the surface of transfected cells. Binding of FITC-conjugated ADA to parental CHO cell surface could not be detected by confocal microscopy (Fig. 2A), but was readily measurable in the case of with CHO-A2B cells (Fig. 2B). To quantify the binding of ADA to the surface of CHO-A2B cells, we used125I-labeled ADA. As shown in Fig. 2C, the binding of 125I-ADA was dose-dependent and saturable.

Immunodetection of A2BR and ADA in wild-type CHO and CHO-A2B cells. Wild-type CHO (A and B) and CHO-A2B cells (C) were grown on coverslips. A, cells were fixed (left) or permeabilized (right) and stained with 100 μg/ml FITC-anti-A2BR MPE1. B, cells were fixed (left) or permeabilized (right) and stained with 50 μg/ml FITC-anti-ADA (in green) and treated with 5 μg/ml propidium iodide (in red) to show the cell nucleus. A vertical section of a single cell is shown. C, cells were fixed and stained with 100 μg/ml FITC-anti-A2BR MPE1 (in green) and 50 μg/ml TRITC-anti-ADA (in red). Cells were processed for confocal microscopy analysis as described under Experimental Procedures. Scale bar, 10 μm.

Binding of exogenous ADA to CHO cells. Wild-type CHO (A) and CHO-A2B cells (B) grown on coverslips were incubated with 2 U/ml bovine FITC-ADA (30 min, 37°C), washed, and analyzed by confocal microscopy as described underExperimental Procedures. Bar, 10 μm. C, binding of125I-ADA to CHO-A2B cell surface in absence or presence of unlabeled ADA.
Colocalization of Cell Surface ADA and A2BR in T Lymphocytes.
To know whether these results obtained in a heterologous model are physiologically relevant, the localization of the cell surface ADA and the A2BR was studied in human cells expressing the two molecules. Jurkat J32 T lymphocytes naturally express the two proteins but also express CD26, which is an ADA-anchoring protein (see the introduction). The labeling of Jurkat J32 cells (Fig. 3A) with FITC-anti-A2BR MPE1 (green fluorescence) and TRITC-anti-ADA (red fluorescence) indicated a high degree of colocalization (in yellow) between ADA and A2BR. As reported previously, there was also a high degree of colocalization between ADA (detected with TRITC-anti-ADA, red fluorescence) and CD26 (detected with FITC-Ta1 mAb, green fluorescence) in Jurkat J32 cells (Fig. 3B). The distribution of ADA, CD26, and A2BR in cells was studied by flow cytometry. ADA+ Jurkat J32 cells were heterogenous, because there were ADA+CD26+ and also ADA+CD26− cells. These results are in agreement with the existence of ADA+CD26− cells in peripheral blood lymphocytes (data in preparation). To analyze whether A2BR were expressed in ADA+CD26− cells, a sorting procedure was devised. Jurkat J32 cells were stained with the FITC-conjugated Ta1, and only CD26− cells were selected. The sorted cells were analyzed by confocal microscopy to confirm that no CD26 was expressed. These CD26−cells were then labeled with FITC-MPE1 and TRITC-anti-ADA. Confocal microscopy images showed that cell surface ADA colocalized with A2BRs in ADA+CD26−-sorted cells (Fig. 3C).

Cell surface expression of A2BR, ADA, and CD26 on Jurkat J32 cells. Fixed Jurkat J32 cells (4 × 106) were stained with 100 μg/ml FITC-anti-A2BR MPE1 (in green) and 50 μg/ml TRITC-anti-ADA (in red) (A), or with 100 μg/ml FITC-anti-CD26 Ta1 mAb (in green) and 50 μg/ml TRITC-anti-ADA (in red) (B). The 20% of Jurkat J32 CD26− cells, separated by sorting according to the staining with 100 μg/ml FITC-anti-CD26 Ta1 mAb, were labeled with 100 μg/ml FITC-anti-A2BR MPE1 (in green) and 50 μg/ml TRITC-anti-ADA (in red) (C). Colocalization by confocal microscopy is shown in yellow. Bar, 10 μm.
The ability of ADA to interact with A2BR was confirmed by immunoprecipitation using the anti-ADA antibody or an irrelevant antibody. Membranes from parental CHO, CHO-A2B, and Jurkat J32 cells were used for these experiments. Immunoprecipitates were analyzed by blotting with anti-ADA antibody and with anti-A2B MPE1 (Fig.4). The results show that membranes from parental CHO cells lacked ADA. In contrast, ADA was present in membranes from CHO-A2B and Jurkat J32 cells (Fig.4A). When the immunoblotting was developed with anti-A2BR MPE1 antibody (Fig. 4B), a band of 42 kDa was detected in membranes from CHO-A2B and Jurkat J32 cells but not in membranes from parental CHO cells. This band, which did not appear when using an irrelevant antibody, corresponds to A2B adenosine receptors. A second band of higher molecular mass, which has been also detected in A2BR-transfected human embryonic kidney cells or in some human tissues (Puffinbarger et al., 1995; Linden et al., 1999), may correspond to receptor dimers. These results also indicate the existence of an interaction between cell surface ADA and A2BR on the surface of CHO-A2B and Jurkat J32 cells.

Immunoprecipitation of A2BR by the anti-ADA antibody. Solubilized cell membranes were immunoprecipitated as indicated under Experimental Procedures with anti-ADA antibody or with an irrelevant rabbit anti-goat IgG antibody, both covalently coupled to a protein A matrix. Immunoblotting of immunoprecipitates was performed using anti-ADA antibody to detect cell surface ADA or anti-A2BR MPE1 to detect A2BR.
Previous studies in our laboratory have shown an increase of CD26 and cell surface ADA expression in response to lymphocyte activation (Martı́n et al., 1995b). Activation signals delivered by either phytohemagglutinin or anti-T cell receptor/CD3 complex antibodies (OKT3) led also to a significant increase in both the percentage of cells expressing the A2BR and the intensity of labeling (Mirabet et al., 1999). Hence we determined whether ADA and A2BR colocalized in activated peripheral blood lymphocytes. In fact, the higher level of expression of A2BR on the cell surface of OKT3-activated peripheral blood lymphocytes, which was detected with FITC-MPE1 (Fig.5, left), correlated with the increase of cell surface ADA (Fig. 5, middle), and both labels showed a high degree of colocalization (in the images in Fig. 5, right, the colocalization is shown in white). Similar results were obtained with PHA-activated lymphocytes (data not shown).

Immunodetection of A2BR and cell surface ADA in resting and activated peripheral blood lymphocytes. Resting (4 × 106) (A) or OKT3-activated peripheral blood lymphocytes (B) were fixed and labeled with 100 μg/ml FITC-anti-A2BR MPE1 (left) and 50 μg/ml TRITC-anti-ADA (middle). Superimposition of images shows colocalization (right) of A2BR and cell surface ADA. Bar, 10 μm.
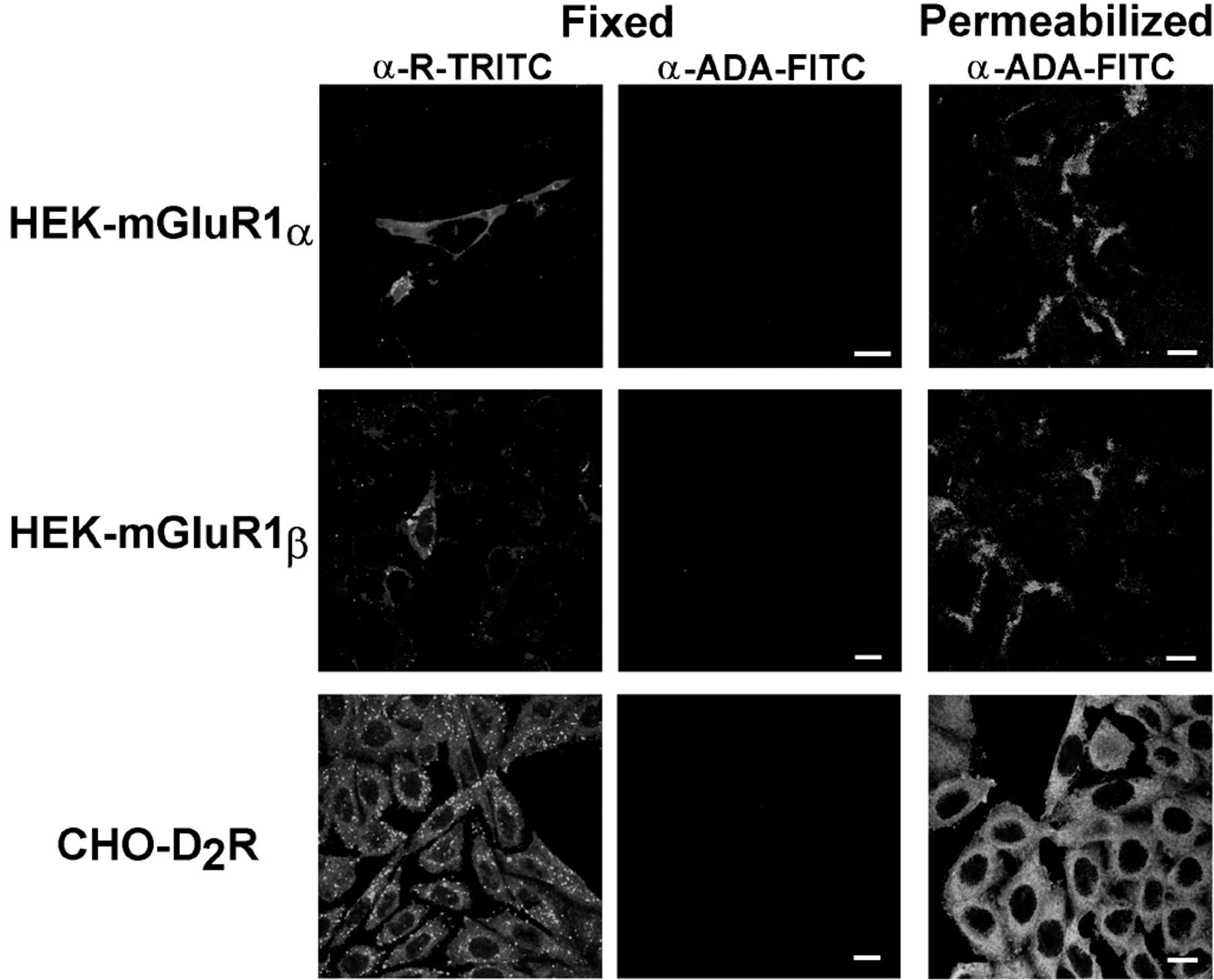
The specificity of the interaction was also assayed by immunocytochemical assays in cells transfected with cDNA encoding for metabotropic glutamate and dopamine receptors, which also belong to the superfamily of G protein-coupled receptors. Both A2BR and dopamine D2receptors are coupled positively to the adenylate cyclase, whereas metabotropic glutamate 1α receptors and adenosine A1 receptors are coupled to phospholipase C. Although the cells transfected with the cDNA for all these receptors contain ADA in the cytoplasm, ADA is not expressed on the surface of the transfected cells (Fig.6).

Lack of expression of cell surface ADA in cells overexpressing metabotropic glutamate or dopamine receptors. Transfected cells (fixed or permeabilized as indicated underExperimental Procedures) were labeled with 50 μg/ml FITC-anti-ADA and with TRITC-conjugated antibodies against the transfected receptors α-R-TRITC. Bar, 10 μm.
Effect of ADA upon NECA Binding to A2BR and upon NECA-Induced cAMP Production.
The effect of ADA upon the binding of NECA to A2BR was analyzed by means of radioligand binding assays as described under Materials and Methods. The binding of [3H]NECA to CHO-A2BR cells was enhanced in a dose-dependent fashion by exogenous ADA (Fig. 7A). This enhancement was similar when ADA, devoid of enzymatic activity, was used. This indicates that the effect is caused by the interaction with A2BR rather than an inactivation of endogenous adenosine, which could be present in the preparation. In displacement experiments performed in the presence and absence of ADA, it is shown that ADA leads to a reduction (from 2.1 ± 0.5 to 0.4 ± 0.1 μM) in the K i value for unlabeled NECA (Fig. 7B) without affecting the unspecific binding. Therefore, the affinity of NECA for A2BR in transfected CHO-A2BR cells increases when ADA interacts with the receptor. In agreement to this potentiation in the NECA binding, ADA led to a 36% increase in the NECA-induced cAMP levels (Fig. 7C).

Effect of ADA upon NECA binding and NECA-induced cAMP increases. A2BR-transfected CHO cells were used for experiments of cAMP production, and A2BR-transfected CHO cell membranes were used for radioligand binding experiments. A, the effect of active ADA (black bars) or Hg2+-inactivated ADA (gray bars) was analyzed in experiments of 38 nM [3H]NECA binding to CHO-A2B cell membranes. Data from a representative experiment in quadruplicates (mean ± S.D.) are given in percentages with respect to the value obtained in an assay performed in the absence of ADA (100%). B, displacement curves of 38 nM [3H]NECA binding by unlabeled NECA in the absence (●) or in the presence (▵) of 5 IU/ml ADA. Data from a representative experiment in quadruplicates (mean ± S.D.) are given. C, production of cAMP in the presence and absence of 5 IU/ml ADA. The concentration of NECA used in the assays was 1 μM. Data from a representative experiment in quadruplicates (mean ± S.D.) are given. *p < 0.005 respect to the value with NECA.
Discussion
Cell surface ADA needs to be anchored to the plasma membrane by means of specific receptors. In lymphocytes, CD26 has been described as an ADA-anchoring protein. In nonlymphoid cells it has been reported that A1 adenosine receptors can anchor ADA to the cell surface. Both ADA/CD26 and ADA/A1R interactions are functionally important, as ADA/CD26 have a costimulatory role in the lymphocytes and ADA facilitates ligand binding to A1R and regulates signal transduction (Franco et al., 1997a,b). In this article, we report a third protein able to interact with ADA on the cell surface, the A2BR. The use of CHO cells transfected with the human cDNA for the A2BR was important to demonstrate the differences between the interactions of ADA/CD26 and ADA/A2BR. Wild-type CHO cells express hamster CD26, and it is well-reported (Dong et al., 1997) that rodent CD26 is unable to bind ADA (either human or rodent ADA). Therefore, cell surface ADA cannot be detected in CHO cells despite the fact that these cells have cytoplasmic ADA. This is also why wild-type CHO cells are a good negative control for cell surface ADA expression. When the human A2BR is expressed in these cells, ADA appears on the cell surface, because it can bind to this anchoring protein. It should be noted that we find colocalization between ADA and A2BR in human lymphocytes and that bovine ADA can be bound to human A2BR. These results indicate that human A2BR is able to interact with human, bovine, and hamster ADA. It has been demonstrated that CD26 does not participate in the transport of ADA toward the cell surface but anchors ADA after the protein has been released from homologous or heterologous cells (Dong et al., 1996). The fact that we find no differences in the level of ADA exported toward the medium in parental and CHO-A2BR cells (not shown) suggest that ADA is released from the cell by a mechanism also independent of the expression of A2BR and, afterwards, it is bound to the ADA-anchoring protein.
Apart from these CD26, A1R, and A2BR proteins, which behave as cell surface receptors for ADA, ADA is able to interact in the cytoplasm with the Grb2 isoform Grb3-3 (Ramos-Morales et al., 1997). Grb2 plays an essential role in cell growth and differentiation by connecting tyrosine kinase receptors to activation of the Ras pathway. Grb3-3 is a naturally occurring isoform of Grb2 that carries a 41-amino acid deletion in the SH2 domain, which abolishes the binding to tyrosine-phosphorylated proteins but retains functional SH3 domains, which allow interaction with other proteins through binding to proline-rich sequences. Grb3-3 may be involved in apoptosis (Fath et al., 1994), but the role of this molecule has not been fully elucidated. The variety of interactions in which ADA participates can be explained from a structural point of view. The three-dimensional structure, which is known for the murine enzyme expressed inEscherichia coli (Wilson et al., 1991), indicates that ADA possesses several hydrophobic zones that are unusual for soluble enzymes and that are likely involved in different protein-protein interactions [see Franco et al. (1997b) for review].
There is some controversy related to the percentage of cells expressing ADA on the cell surface of human lymphocytes and of human T cell lines. The expression of cell surface ADA is usually determined by incubation with anti-ADA antibodies before fixation of the cells (Kameoka et al., 1993; Martı́n et al., 1993, 1995a,b; De Meester et al., 1994;Dong and Morimoto, 1996; Dong et al., 1996, 1997; Morimoto and Schlossman, 1998). Fixation before incubation with antibodies is a key step. The percentage of Jurkat J32 T cells expressing cell surface ADA can be 20% if the incubation is performed in nonfixed cells or 100% if fixed cells are treated with the antibodies. The use of fixed cells has allowed us to detect ADA+CD26− populations in Jurkat J32 T cells (see Results). In this ADA+CD26− population, ADA is bound to the cell surface through A2BR. The relevance of the existence of these two different populations is at present unknown. However, the fact that ADA and A2BR are coordinately up-regulated in response to activating signals indicates that both A2BR and the ADA/A2BR interaction have a role in T cell activation. We have previously described how adenosine, acting through A2BR, contributes to deactivation of macrophages, because it reduces the up-regulation of major histocompatibility complex class II molecules, the activity of nitric-oxide synthase or the production of proinflammatory cytokines (Xaus et al., 1999). In human lymphocytes, adenosine, acting through A2BR, elicits a significant reduction in interleukin-2 production (Mirabet et al., 1999). The role of ADA bound to A2BR, apart from degrading the extracellular adenosine, is to modulate the binding of agonists. In fact, the presence of ADA, irrespective of its catalytic activity, leads to an increase in the affinity of NECA for A2BR (see Fig. 7) and, accordingly, to an increase in the cAMP levels induced by activation of A2BR. This is similar to what has been reported for the ADA/A1R module in a smooth muscle cell line (Ciruela et al., 1996). There are no obvious homologies between A2BR or A1R and CD26, which is the third protein able to anchor ADA to the cell surface. This suggests that the interaction of ADA with CD26 or with A2BR or A1R occurs via different epitopes in the ADA molecule [see Franco et al. (1997a) for details on ADA epitopes].
Acknowledgments
We acknowledge the technical help received from Jaume Comas and M. del Rosario González (flow cytometry) and from Susana Castel (confocal microscopy section).
Footnotes
- Received January 5, 2000.
- Accepted August 11, 2000.
-
Send reprint requests to: Prof. Rafael Franco, Dept. Bioquı́mica i Biologia Molecular, Universitat de Barcelona, Martı́ i Franquès 1, 08028 Barcelona, Spain. E-mail:r.franco{at}sun.bq.ub.es
-
This research was supported by Spanish Comisión Interministerial de Ciencia y Tecnologia's Investigation and Development programs Salud y Farmacia (SAF97/0066) and Biotechnology (BIO1999/0601/C02).
Abbreviations
- SCID
- severe combined immunodeficiency syndrome
- ADA
- adenosine deaminase
- A2BR
- adenosine A2B receptor
- CHO
- Chinese hamster ovary
- NECA
- 5′-N-ethylcarboxamidoadenosine
- mAb
- monoclonal antibody
- FITC
- fluorescein isothiocyanate
- TRITC
- tetramethylrhodamine isothiocyanate
- DMEM
- Dulbecco's modified Eagle's medium
- HEK
- human embryonic kidney
- PHA
- phytohemagglutinin
- mAb
- monoclonal antibody
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}