


Abstract
The I1 subtype of imidazoline receptors (I1R) is a plasma membrane protein that is involved in diverse physiological functions. Available radioligands used so far to characterize the I1R were able to bind with similar affinities to α2-adrenergic receptors (α2-ARs) and to I1R. This feature was a major drawback for an adequate characterization of this receptor subtype. New imidazoline analogs were therefore synthesized and the present study describes one of these compounds, 2-(2-chloro-4-iodo-phenylamino)-5-methyl-pyrroline (LNP 911), which was of high affinity and selectivity for the I1R. LNP 911 was radioiodinated and its binding properties characterized in different membrane preparations. Saturation experiments with [125I]LNP 911 revealed a single high affinity binding site in PC-12 cell membranes (K D = 1.4 nM;B max = 398 fmol/mg protein) with low nonspecific binding. [125I]LNP 911 specific binding was inhibited by various imidazolines and analogs but was insensitive to guanosine-5′-O-(3-thio)triphosphate. The rank order of potency of some competing ligands [LNP 911, PIC, rilmenidine, 4-chloro-2-(imidazolin-2-ylamino)-isoindoline (BDF 6143), lofexidine, and clonidine] was consistent with the definition of [125I]LNP 911 binding sites as I1R. However, other high-affinity I1R ligands (moxonidine, efaroxan, and benazoline) exhibited low affinities for these binding sites in standard binding assays. In contrast, when [125I]LNP 911 was preincubated at 4°C, competition curves of moxonidine became biphasic. In this case, moxonidine exhibited similar high affinities on [125I]LNP 911 binding sites as on I1R defined with [125I]PIC. Moxonidine proved also able to accelerate the dissociation of [125I]LNP 911 from its binding sites. These results suggest the existence of an allosteric modulation at the level of the I1R, which seems to be corroborated by the dose-dependent enhancement by LNP 911 of the agonist effects on the adenylate cyclase pathway associated to I1R. Because [125I]LNP 911 was unable to bind to the I2binding site and α2AR, our data indicate that [125I]LNP 911 is the first highly selective radioiodinated probe for I1R with a nanomolar affinity. This new tool should facilitate the molecular characterization of the I1 imidazoline receptor.
Most of the imidazoline ligands, such as clonidine, idazoxan, and related compounds, are known to bind to α2-adrenergic receptors as well as to imidazoline receptors (Ruffolo et al., 1995). Extensive biochemical and physiological studies led to the subclassification of imidazoline receptors in three main classes: I1, I2, and I3 (Regunathan and Reis, 1996; Ernsberger, 1999;Bousquet et al., 2000). Cloning strategies and biochemical studies have assigned I2 binding sites (I2BS) to a modulatory site on monoamine oxidases A and B (Tesson et al., 1995). The existence of I3 receptors (non-I1/non-I2) has been suggested according to insulin release properties of some imidazolines in β pancreatic cells (Chan et al., 1991; Zaitsev et al., 1996;Rustenbeck et al., 1997). Although KATP channel closure has been implicated in these effects, recent results (Efanov et al., 2001a,b; Chan et al., 2001) have characterized a KATP independent pathway activation leading to enhanced insulin release by imidazolines. However, attempts to characterize the associated imidazoline binding sites have been unsuccessful because of the lack of specific radioligands.
Conversely, the I1 subtype of imidazoline receptors has been amply characterized by binding assays using radiolabeled clonidine or analogs (Molderings et al., 1993; Piletz et al., 1996; Ernsberger et al., 1997) and its pharmacological selectivity assessed. I1 receptor (I1R) is a plasma membrane receptor protein (Heemskerk et al., 1998) that has been shown to be coupled to a G protein in human platelets, bovine chromaffin cells, and PC-12 cells (Molderings et al., 1993; Piletz et al., 1996; Greney et al., 2000). Transduction pathways have already been associated with this receptor in the PC-12 cells: activation of a phosphocholine-specific phospholipase C (Separovic et al., 1996) and inhibition of an adenylate cyclase (Greney et al., 2000). Moreover, pharmacological studies have shown that this receptor is involved in several functions such as regulation of the cardiovascular function (Bousquet et al., 1984; Ernsberger et al., 1990), modulation of the ocular pressure (Ogidigben et al., 2001), control of the catecholamine release from chromaffin cells (Nguyen and De Lean, 1987), and renal sodium excretion (Smyth and Penner, 1999).
So far, all the radioligands used to characterize the I1 receptors were “hybrid” molecules able to bind with similar affinities both to I1 receptors and to α2-adrenoceptors (Molderings et al., 1993; Piletz et al., 1996; Ernsberger et al., 1997). The lack of selective ligands hindered the use of these radioligands in binding assays on membranes bearing the two types of receptors. In such membrane preparations, α2-adrenoceptor blocking conditions were essential to reveal and characterize the I1 receptors. In an effort to fully characterize these receptors, we developed a series of new imidazolines or analogs to obtain selective and high-affinity I1 receptor ligands. We recently synthesized a series of pyrroline analogs with no detectable affinities for I2BS as well as for α2-adrenoceptors (Schann et al., 2001). Among the available pyrroline compounds, LNP 911 was found to be highly selective for I1R and interestingly exhibited nanomolar affinity for these receptors.
As unlabeled LNP 911 exhibited high-affinity for I1 receptors detected by [125I]para-iodoclonidine (PIC) in PC-12 cell membranes and high selectivity for I1receptors compared with α2-adrenoceptors and I2 binding sites (S. Schann, H. Greney, M. Dontenwill, D. Urosevic, C. Rascente, G. Lacroix, L. Monassier, V. Bruban, J. Feldman, B. Pfeiffer, et al. Methylation of imidazoline related compounds leads to loss of α2-adrenoceptor affinity. Synthesis and biological evaluation of new selective I1 imidazoline receptor tools, manuscript in preparation), we decided to radioiodinate this drug and to investigate the resulting radioligand as a selective probe for the I1 receptors. This report describes the properties of [125I]LNP 911 as the first selective I1 receptor radioligand and, hence, for the first time, an allosteric modulation of the I1 receptor binding site has been suggested by the use of this ligand. In addition, LNP 911 behaves as an allosteric enhancer of I1 receptor transduction pathway.
Experimental Procedures
Materials.
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), penicillin, and streptomycin were obtained from Invitrogen (Cergy-Pontoise, France). Benazoline was synthesized by Prof. Pigini (Camerino, Italy), BDF6143 was kindly provided by Beiersdorf-Lilly (Hamburg, Germany). Moxonidine was kindly provided by Solvay Pharmaceuticals GmbH (Hannover, Germany). Cirazoline was a gift from Synthelabo (Bagneux, France). Rilmenidine was a gift from Laboratoires Servier (Courbevoie, France). All other compounds were purchased from Sigma (L'Isle d'Abeau Chesnes, Saint-Quentin Fallavier, France). [3H]Idazoxan and [3H]RX 821002 was obtained from Amersham Biosciences (Orsay, France). [125I]PIC was purchased from PerkinElmer Life Sciences (Paris, France). The chemical structures of the imidazoline and pyrroline compounds are shown in Fig.1.

Chemical structures of imidazoline and pyroline compounds.
LNP 911 Synthesis.
The LNP 911 [2-(2-chloro-4-iodo-phenylamino)-5-methyl-pyrroline] synthesis will be described in detail elsewhere. Briefly, 2-(2-chloro-4-bromo-phenylamino)-5-methyl-pyrroline was obtained by reaction of 4-bromo-2-chloro-aniline with 5-methyl-pyrrolidinone in the presence of POCl3. This intermediate was then transformed into the stannous compound 2-(2-chloro-4-tributylstannyl-phenylamino)-5-methyl-pyrroline. The radioiodination step was made as follow: 2-(2-chloro-4-tributylstannyl-phenylamino)-5-methyl-pyrroline (50 μg) in MeOH (50 μl) was mixed with HCl (0.5 M; 42 μl) and [125I]NaI (IMS30; 185 MBq; 5 mCi; 50 μl; Amersham Biosciences, Little Chalfont, Buckinghamshire, England). Reaction was initiated by addition of chloramine-T (50 μl; 1 mg/ml) and was allowed to react for 10 min. The reaction mixture was loaded onto a Jupiter C-18 reversed phase–high-performance liquid chromatography column (250 × 4.6 mm; Phenomenex, Macclesfield, Cheshire, England) and purified to obtain 70% yield of [125I]LNP 911 (74 TBq/mmol; 2000 Ci/mmol) using a linear gradient with water, methanol, and trifluoroacetic acid. This product was diluted with ethanol to 100 μCi/ml and stored at 4°C. The cold marker [127I]LNP 911 was demonstrated to coelute with the same retention time as [125I]LNP 911. The radiochemical purity of [125I]LNP 911 was shown to be >95% with less than 1% free iodide at initial analyses on a C-18 column using a linear gradient with water, propanol, and trifluoroacetic acid. The synthesis of intermediates was made in our laboratory by S.S. and J.D.E. and the custom radioiodination step was carried out by Iodine Ligand Development, Amersham Biosciences.
Cell Cultures.
PC-12 cells were obtained from Dr. G. Rebel (IRCAD, Strasbourg, France). They were cultured in 75-cm2 flasks in DMEM (1000 mg/l glucose) supplemented with 10% heat-inactivated FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. When the cells reached confluence (3 to 4 days after plating), they were harvested by 2-min exposure to 0.25% trypsin at 37°C. For binding assays, after removing the medium, cells at confluence were frozen in the flasks at −20°C until use to prepare membranes. HT29 cells were obtained from Dr. H. Paris (Institut National de la Santé et de la Recherche Médicale U338, Toulouse, France) and cultured in 75-cm2 flasks in DMEM (4500 mg/l glucose) supplemented with 10% heat-inactivated FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were harvested at confluence after 48-h incubation in fresh DMEM without FBS, and membranes were prepared immediately. CHO-α2A, CHO-α2B, and CHO-α2c cell lines expressing the human recombinant adrenoceptors (provided by Prof. A. D. Strosberg, Paris, France) were grown in Ham's F12 medium supplemented with 2 mM glutamine, 10% fetal bovine serum, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 400 μg/ml G418, in 5% CO2 at 37°C. Cells were passaged every 3 to 4 days. These CHO-α2A, CHO-α2B, and CHO-α2ccell lines expressed 1.8, 10, and 1.3 pmol of receptors/mg of protein, respectively, as determined by saturation experiments with [3H]RX 821002.
Membrane Preparations.
Frozen PC-12 cells were scraped into cold Tris-HEPES buffer (5 mM Tris-HEPES, pH 7.7, 0.5 mM EDTA, 0.5 mM EGTA, and 0.5 mM MgCl2) and homogenized with a Potter homogenizer. After centrifugation at 75,000g for 20 min, the pellet was washed twice in cold Tris-HEPES buffer and centrifuged. Pellets were resuspended in the same buffer at 2 to 4 mg of protein/ml. Membrane preparations were stored at −80°C until use. HT29 cell membrane preparations were obtained after homogenisation of the cells in 50 mM cold Tris-HCl buffer, pH 7.5, containing 5 mM EDTA with a Polytron homogenizer (Kinematica, Basel, Switzerland). The homogenate was then centrifuged at 25,000g for 25 min and the pellet was washed twice with Tris-HCl buffer without EDTA. Membrane preparations were stored at −80°C until use. Rabbit kidney membranes were prepared as follows: renal cortex from male New Zealand rabbits were homogenized in ice-cold Tris-HCl buffer (50 mM Tris-HCl, 250 mM sucrose, pH 7.4) The homogenate was centrifuged at 500g for 10 min at 4°C. The resulting supernatant was centrifuged at 28,000g for 30 min and the pellet was washed twice in binding buffer (50 mM Tris HCl, pH 7.4). The membrane was stored at −80°C until used.
Transfected CHO cells, grown to confluence, were harvested in PBS/2 mM EDTA. The suspension was centrifuged (1000g, 10 min) and the resulting pellet homogenized in 20 mM HEPES, pH 7.4 using a Polytron homogenizer (Kinematica). The homogenate was then centrifuged (43,000g, 30 min, 4°C) and the membrane pellet was resuspended in the same buffer with 10 strokes in a Potter homogenizer and then sonicated for 15 s. Aliquots of membrane preparations were stored at −80°C.
Binding Assays.
Binding assays with [125I]PIC on PC-12 cell membranes were performed at 25°C as described elsewhere (Greney et al., 2000). Binding assays with [125I]LNP 911 on PC-12 cell membranes were all performed at 25°C, except as specified otherwise, and were as follows. Incubation was initiated by the addition of membranes (20 to 50 μg of protein) in a final volume of 250 μl of Tris-HEPES buffer (50 mM Tris-HEPES, pH 7.7, 0.5 mM EDTA, 0.5 mM EGTA, and 0.5 mM MgCl2) and were carried out at 25°C during 60 min (equilibrium conditions). Specific binding of [125I]LNP 911 increased linearly with increasing protein from 8 to 85 μg of proteins. The reaction was stopped by rapid vacuum filtration through GF/B glass fiber filters with a Brandel harvester (Gaithersburg, MD), followed by three rapid washes of the filters with 3 ml of ice-cold 50 mM Tris-HCl buffer, pH 7.4. Radioactivity retained on the dried filters was determined in a Minaxi gamma counter (Packard BioScience, Meriden, CT). Nonspecific binding was defined as [125I]LNP 911 binding in the presence of 100 μM PIC, and accounted for about 10% of the total radioactivity when 0.2 nM [125I]LNP 911 was used. The choice of 100 μM PIC came from pilot experiments showing that at this concentration, the residual binding obtained with PIC was similar to that obtained with all the other drugs tested (clonidine,para-aminoclonidine, and LNP 911). For saturation experiments, seven concentrations of [125I]LNP 911, ranging from 0.05 to 6.6 nM, were used. Competition studies were performed using 0.2 nM [125I]LNP 911 (0.1K d) and 11 to 13 different concentrations of the unlabeled ligand under investigation, ranging from 10−10 to 10−3 M in the absence or presence of 100 μM GTPγS.
Kinetic association experiments were carried out with 0.2 nM [125I]LNP 911. Association was initiated by addition of membranes; dissociation was initiated by the addition of 100 μM LNP 911 after 60 min incubation of the membranes with the radioligand. Reactions were stopped at time points between 10 s and 60 min by rapid filtration and washing with ice-cold buffer.
For preassociation experiments, membranes were preincubated or not with the radioligand (0.2 nM) for 120 min at 4°C; then increasing concentrations of moxonidine (maintained at 4°C) were added for an additional 120-min incubation at 4°C. Kinetic pilot experiments allowed us to determine that specific [125I]LNP 911 (0.2 nM) binding in PC-12 cell membranes reached equilibrium at about 90 min at 4°C and remained stable for at least 240 min.
HT29 membrane binding assays were performed as described elsewhere (Devedjian et al., 1991). Incubation was initiated by the addition of membranes (50 μg protein/assay) with 1 nM [125I]LNP 911 (corresponding to theK d value of the radioligand in PC-12 cell membranes) and was carried out at 25°C during 60 min. Assays were then processed as described above. Nonspecific binding was defined with 10−4 M PIC.
For rabbit kidney membrane binding assays, membranes (100 μg/250 μl) were incubated for 60 min at 25°C with 5 nM [3H]idazoxan (in the presence of 10 μM (−)-norepinephrine in 0.005% ascorbic acid, to avoid binding of the radioligand to α2ARs) or 1 nM [125I]LNP 911 and increasing concentrations of drugs (10−10 to 10−3 M). Nonspecific binding was determined with 10 μM Cirazoline for [3H]idazoxan binding or 100 μM PIC for [125I]LNP 911 binding. Radioactivity retained on the filters was determined in a beta TriCarb counter (Packard BioScience) or a Minaxi gamma counter for [3H]idazoxan and [125I]LNP 911 bindings, respectively.
For [ 3H]RX 821002 binding assays in CHO cells, membranes (30 μg/ml for CHO-α2A, CHO-α2B, and 100 μg/ml for CHO-α2c) were incubated 1 h at room temperature in binding buffer (33 mM Tris, pH 7.5 containing l mM EDTA) in a final volume of 500 μl containing 0.8, l, or 2 nM [3H]RX821002, respectively, for hα2A-, hα2B-, hα2c-AR. At these concentrations, [3H]RX821002 was shown to label α2AR exclusively (Chan et al., 1994). Nonspecific binding was defined with 10 μM phentolamine. Incubations were stopped by rapid filtration through GF/B unifilters, followed by three successive washes with ice-cold buffer. Protein concentrations were measured by the method of Bradford et al. (1976) using bovine serum albumin as standard.
cAMP Experiments.
cAMP experiments were conducted as described previously (Greney et al., 2000). In antagonism experiments, LNP 911 (10−5 M) was added to the medium for 10 min before the addition of other drugs. The radioreceptor assay kit for dosage of cAMP (Amersham Biosciences) was used according to the instructions of the manufacturer.
Data Analysis.
Data from kinetic, saturation, and competition experiments were analyzed using the least-square fitting program Prism (GraphPad Software Inc., San Diego, CA).K i values were calculated according to the Cheng and Prusoff (1973) equation. The significance of the improvement of fit obtained by the two-site equation over the one-site equation was analyzed by F-statistics (partial F-test). Results are expressed as mean values ± S.E.M. The statistical significance of differences was analyzed by Student's t test.
Results
Binding Characteristics of Unlabeled LNP 911.
The affinity of LNP 911 for α2-adrenergic receptors were measured in human α2A-, α2B-, or α2C-adrenoceptor transfected CHO cell membranes. [3H]RX 821002 specific binding to α2A-adrenoceptor-transfected CHO cell membranes was totally displaced by phentolamine (K i = 8.2 ± 0.5 nM,n = 7) a well-known α2-AR ligand (Fig. 2A). On the other hand, LNP 911 displaced the [3H]RX 821002 specific binding with a low affinity (K i = 2500 ± 348 nM, n = 2). The affinities of LNP 911 for [3H]RX 821002 specific binding to α2BAR and α2CAR expressed in CHO cells were, respectively of 1,750 ± 30 nM (n = 2) and >10,000 nM (n = 2).

Competition curves of unlabeled LNP 911 on I1 receptors, I2 binding sites, and α2A adrenergic receptors. A, inhibition of [3H]RX 821002 binding on α2A-adrenergic receptor-transfected CHO cell membranes. Membranes (15 μg) were incubated for 60 min with [3H]RX 821002 (0.8 nM) and increasing concentrations of the drugs, LNP 911 (▴) or phentolamine (▵). Nonspecific binding was defined by 10 μM phentolamine. B, inhibition of [3H]idazoxan binding on rabbit kidney membrane I2 binding sites. Membranes (100 μg) were incubated for 60 min with [3H]idazoxan (5 nM) and increasing concentrations of the drugs, Cirazoline (⋄) or LNP 911 (♦). Nonspecific binding was defined by 10 μM Cirazoline. C, inhibition of [125I]PIC binding on PC-12 cell membrane I1 receptors. Membranes (100–200 μg) were incubated for 30 min at 25°C with [125I]PIC (0.5 nM) and increasing concentrations of the drug. Nonspecific binding was defined by 100 μM benazoline. Competition curves with LNP 911 (▪) and moxonidine (■) were shallow and were best fit to a two-site model (LNP 911,p = 0.003; moxonidine, p = 0.03). The data are presented as the average of triplicate determinations of at least three independent experiments.
[3H]Idazoxan specific binding to I2BS, in rabbit kidney membrane preparations, was totally displaced by Cirazoline, a selective I2BS ligand, with a K i of 11.1 ± 1.4 nM (n = 6) (Fig. 2B). In this model, LNP 911 displaced the [3H]idazoxan specific binding with a low affinity (K i = 25,428 ± 2,737 nM, n = 3).
In contrast with these results, LNP 911 proved able to displace the [125I]PIC-specific binding to I1R in PC-12 cell membrane preparations with two affinities [K i = 0.2 ± 0.1 nM (38% of total sites) and 10,000 ± 6,031 nM, n = 3] as was the case for unlabeled moxonidine [K i = 34 ± 5 nM (58% of total sites) and 24,000 ± 10,458 nM, n = 5] (Fig. 2C). These results showed that LNP 911 is a high-affinity and selective ligand for I1 receptors. To further investigate the binding properties of LNP 911, the compound was radioiodinated and investigated as a potential radioligand for I1receptors.
Binding Properties of [125I]LNP 911 in PC-12 Cell Membrane Preparations.
Kinetic studies indicated that specific [125I]LNP 911 (0.2 nM) binding in PC-12 cell membranes reached equilibrium at about 30 min at 25°C and remained stable for at least 120 min (Fig. 3). The apparent association rate constant (kobs) was 0.49 ± 0.01 min−1 (n = 3) and the half-time of the bound complex (t 1/2) was 1.38 ± 0.03 min (n = 3). The binding of 0.2 nM [125I]LNP 911 in these membranes was reversed at 25°C by the addition of 100 μM cold LNP 911 and the dissociation was complete by 900 s. The dissociation rate constant (k −1) determined by non linear analysis of the binding data were 0.026 ± 0.002 min−1 (n = 13) and the half-time dissociation time (t 1/2) of the bound complex was 26.6 ± 2.3 s (n = 13). From the calculated k+1 (k+1 = 2.32 min−1 nM−1) and the k−1 values, the apparent equilibrium dissociation constant (K d) was estimated to be 0.01 nM.

Kinetics of [125I]LNP 911 binding to PC-12 cell membranes at 25°C. Time course of association (▪). Membranes were incubated as described in experimental procedures with [125I] LNP 911 (0.2 nM) for different periods of time at 25°C. The constant rate kobs was calculated to be 0.5 min−1. The rate constant (k+1) was calculated according to the equation k+1 = (kobs− k−1) / ([125I]LNP 911) and was determined to be 2.39 min−1· nM−1. Total specific binding (100%) corresponded to 5767 cpm and nonspecific binding determined by 100 μM PIC represented 10% of the total binding. The results shown are representative of three separate experiments. Time course of dissociation (▴): membranes were incubated for 60 min at 25°C with 0.2 nM [125I] LNP 911, and thereafter a large excess of LNP 911 (100 μM) was added (t = 0; i.e., 60 min after the start of incubation). The apparent rate constant k−1 value in this experiment was 0.022 min−1(determined by a nonlinear regression analysis). Binding att = 0 (100%) represented 3943 cpm. Nonspecific binding of [125I] LNP 911 was determined by 100 μM PIC in triplicate at the different times indicated. The results shown are representative of 13 separate experiments.
[125I]LNP 911 binding at 25°C in PC-12 cell membranes was saturable and of high affinity. Nonlinear regression analysis of saturation binding isotherms indicated that [125I]LNP 911 bound to an apparently homogeneous population of sites with aK d value of 1.4 nM (1.2–1.6, two experiments) and a B max = 398 fmol/mg protein (356–440, two experiments) (Fig.4). The percentage of nonspecific binding defined with 100 μM PIC increased linearly with increasing radioligand concentrations and ranged from 11% at 0.2 nM to 34% at 6.6 nM of [125I]LNP 911.

Equilibrium binding studies with [125I] LNP 911 in PC-12 cell membranes. Saturation binding experiments at 25°C were performed as described under Experimental Procedures, using increasing concentrations of [125I]LNP 911 (0.05- 6.6 nM) with 20- to 50-μg membrane proteins. Specific binding at different concentrations of [125I]LNP 911 was determined with 100 μM PIC. The calculated K d value in this experiment was 1.65 nM and the B max was 356 fmol/mg proteins. The results shown are representative of two separate experiments. ▪, total binding; ▴, nonspecific binding; ▵, specific binding.
We evaluated the saturation binding isotherms of [125I]PIC in the same batches of PC-12 cell membranes and thus confirmed that this radioligand bound also to an apparent homogenous population of sites with aK d of 0.44 ± 0.04 nM and aB max of 50.7 ± 3.7 fmol/mg protein (n = 3).
Specificity of [125I]LNP 911 Binding.
To characterize the ligand recognition properties of the [125I]LNP 911 binding sites, competition experiments with a series of imidazolines and related compounds were performed at 25°C on PC-12 cell membranes. Competition binding curves of some drugs could be resolved into two sites of high and low affinity, respectively, whereas others displaced the radioligand with a low affinity only (Fig. 5 and Table1). In the first group of compounds, the rank order of potency for the high-affinity binding sites was LNP 911 > PIC > rilmenidine > BDF 6143 > lofexidine > clonidine. The second group of compounds included moxonidine, efaroxan, benazoline, para-aminoclonidine, and idazoxan. Intriguingly, the rank order of potency obtained in these competition experiments did not fit exactly with the definition of I1 receptors. Some I1receptor ligands were found to displace [125I]LNP 911 either with high affinities (PIC, rilmenidine, BDF 6143) or with low affinities (moxonidine, efaroxan, benazoline). When pK i values for the high affinity [125I]LNP 911 displacing drugs were compared with their pK i values for [125I]PIC binding in the same membrane preparations, a statistically significant correlation was obtained (r = 0.87, p = 0.02) (Fig.6). These results suggest the existence of, at least, a relationship between I1 receptors defined by [125I]PIC and the [125I]LNP 911 binding sites in PC-12 cell membranes. [125I]LNP 911 specific binding to PC-12 cell membranes was not inhibited by endogenous ligands such as noradrenaline, serotonin, or histamine at concentrations up to 10−5 M (Table 1).
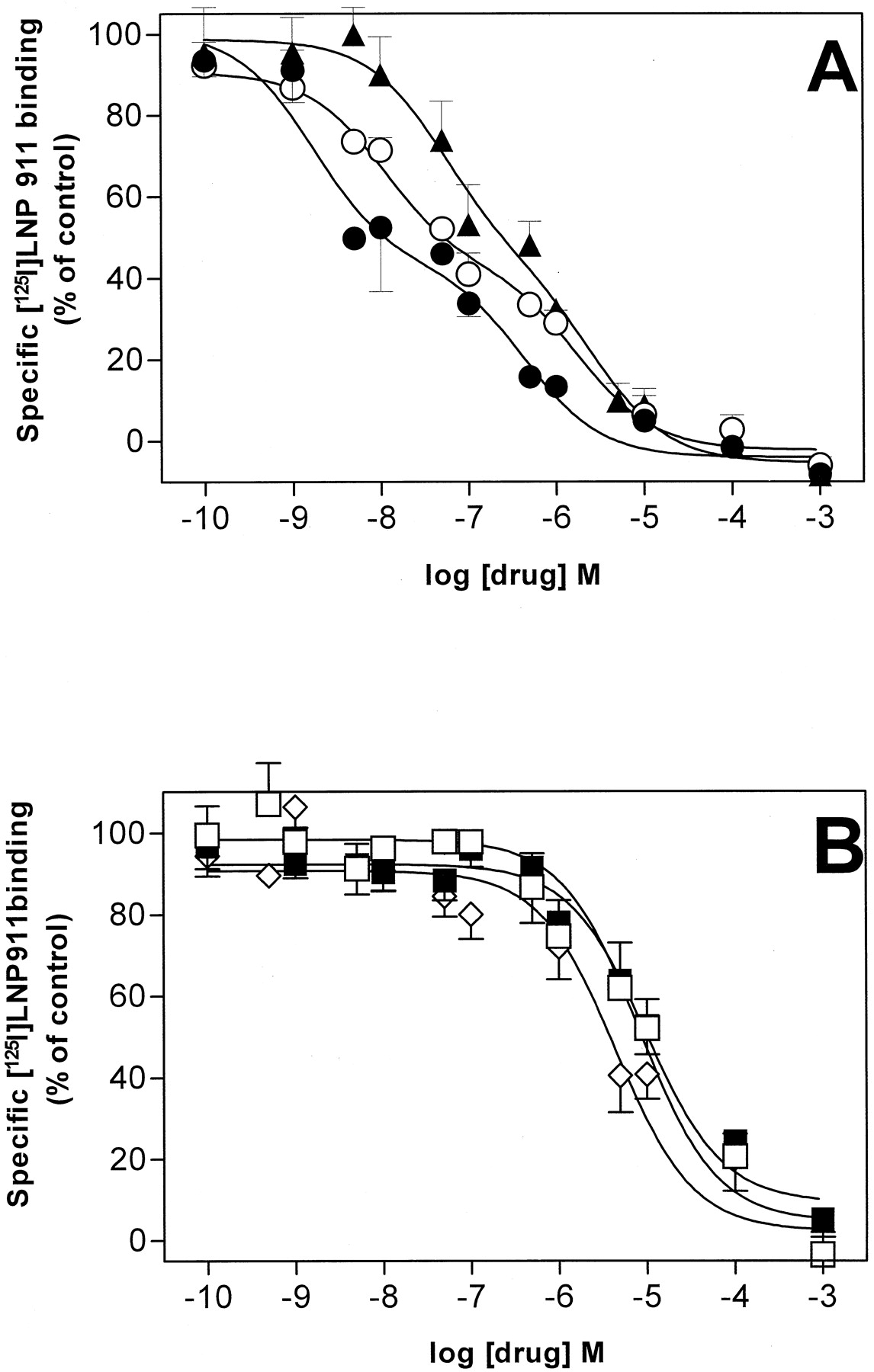
Competition studies for [125I]LNP 911 binding in PC-12 cell membranes. Membranes (20–50 μg) were incubated for 60 min at 25°C with [125I]LNP 911 (0.2 nM) and increasing concentrations of the competitors. Nonspecific binding (defined by 100 μM PIC) represented 10% of total counts bound. Total specific binding (100%) represented 4900 cpm. A, competition curves with LNP 911 (●), PIC (○), and rilmenidine (▴) were shallow and were best fit to a two-site model (LNP 911, p = 0.012; PIC, p = 0.0004; rilmenidine,p = 0.009). B, competition curves with moxonidine (■), efaroxan (▪) and benazoline ⋄ were best fit to a one-site model (moxonidine, p = 0.16; efaroxan,p = 0.66; benazoline, p = 0.79). Each point is the mean of 6 to 8 experiments performed in triplicate and using different membrane preparations.
Inhibitory binding constants (K i) for ligands in competition binding studies with [125I]LNP 911
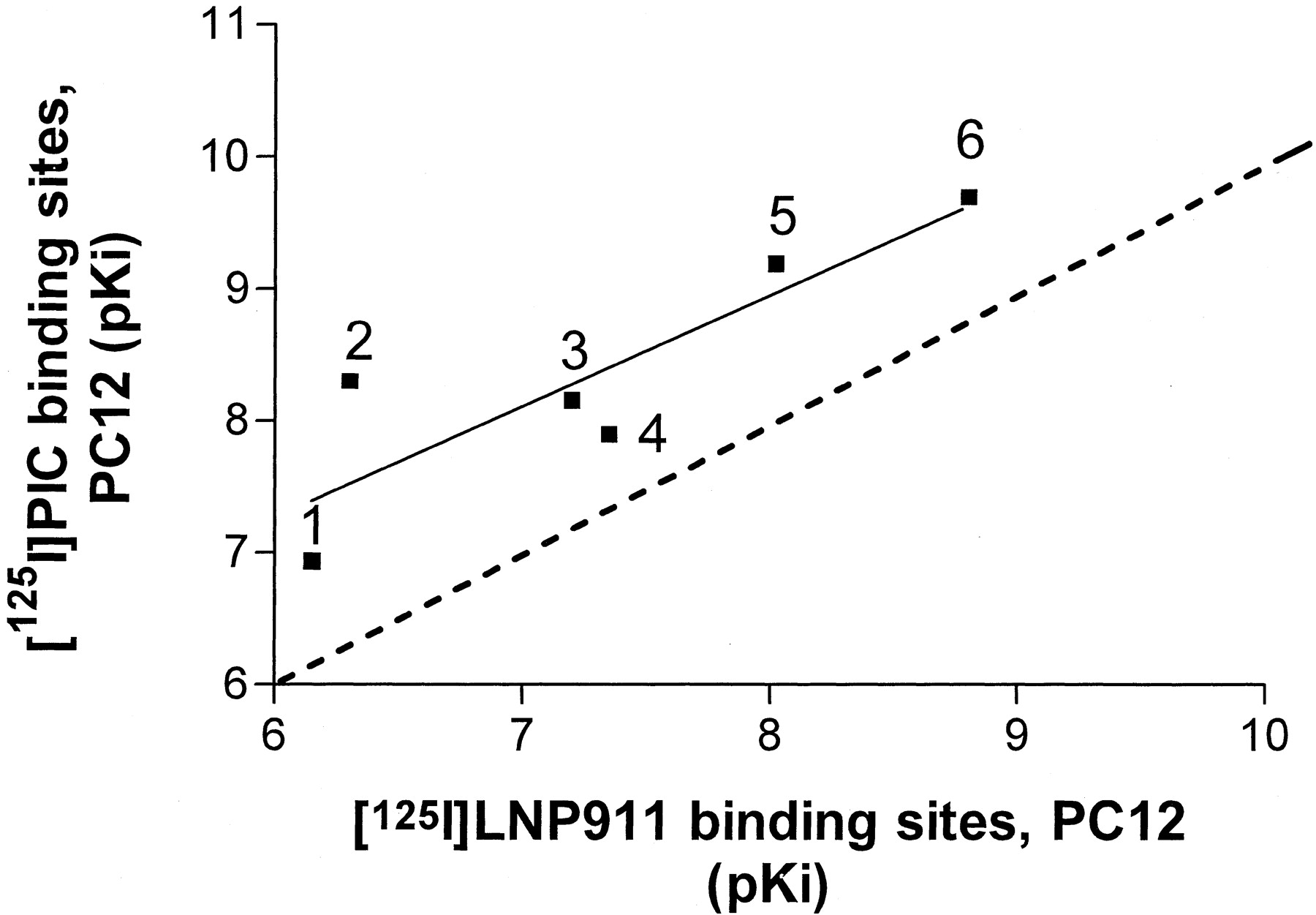
Correlation plot of high affinity constants (pK i) for [125I]LNP 911 binding sites and [125I] PIC binding sites in PC-12 cell membranes. Competition studies with [125I]PIC and with [125I]LNP 911 in PC-12 cell membranes were performed as described in Figs. 2C and 5, respectively. Compound codes are as follows: 1, clonidine; 2, lofexidine; 3, BDF 6143; 4, rilmenidine; 5,p-iodo-clonidine; 6, LNP 911. The correlation line was drawn by linear regression analysis (GraphPad Prism);r = 0.87; P = 0.02. pK i values were calculated according toCheng and Prusoff (1973).
Allosteric Effect of Moxonidine on [125I]LNP 911 Binding Sites.
Because moxonidine was described as an high-affinity I1R ligand in different models but displaced [125I]LNP 911 with rather low affinity, relationships between moxonidine and [125I]LNP 911 were studied in details. It was first hypothesized that optimal binding conditions might differ from one drug to the others. In an effort to optimize these binding conditions, different experimental protocols were compared. Changing either the temperature of the binding assays from 25°C to 4°C or the buffer composition from Tris-HEPES to HME (5 mM HEPES; 0.5 mM MgCl2; 0.5 mM EGTA, pH 7.4) did not improve the IC50 of moxonidine for [125I]LNP 911 binding sites: IC50 in Tris-HEPES buffer at 4°C = 9100 ± 690 (n = 4), IC50 in HME buffer at 25°C = 20,670 ± 11,200 (n = 3) compared with IC50 in standard binding conditions (Tris-HEPES buffer at 25°C) (8,225 ± 525,n = 6). However, competition curves of moxonidine appeared complex in particular in Tris-HEPES buffer at 4°C. In fact, at concentrations of the drug ranging from 10−8to 10−6 M, an increase of [125I]LNP 911 specific binding was observed at this temperature rather than a decrease suggesting the existence of an allosteric modulation on [125I]LNP 911 binding sites as was observed for other receptors (Hejnova et al; 1995; Jakubik et al., 1997). We next performed displacement experiments at 4°C of preassociated [125I]LNP 911 (0.2 nM) during 120 min (equilibrium conditions) at 4°C in Tris-HEPES buffer with moxonidine. In this case, biphasic competition curves were obtained for moxonidine (Fig. 7A) allowing the determination of a high-affinity site (IC50 = 74 ± 11 nM; 29% of sites) and a low-affinity site (IC50 = 234 ± 107 μM) (n= 3) for this drug. As reported above, competition curves of moxonidine on [125I]LNP 911 binding sites performed at 4°C in Tris-HEPES buffer without preincubation of the radioligand did not allow to detect the high affinity site (Fig. 7A).

Allosteric transition occurring during [125I]LNP 911 binding in PC-12 cell membranes. A, effect of temperature and preincubation of the radioligand on the competition curves of moxonidine on [125I]LNP 911 binding in PC-12 cell membranes. Membranes were preincubated (♦) or not (⋄) with the radioligand (0.2 nM) for 120 min at 4°C then increasing concentrations of moxonidine were added for an additional 120 min incubation at 4°C. Competition curves without preincubation were best fit to a one-site model (p = 0.7) although competition curves with preincubation were shallow and were best fit to a two-site model (p = 0.0004). Each point is the mean of three to four experiments performed in triplicate. Total specific binding (100%) represented about 3660 cpm. B, kinetic dissociation at 25°C of the radioligand was performed in the absence (●) or the presence (○) of 50 μM moxonidine. Dissociation was started with the addition of LNP 911 (100 μM) after 60 min incubation at 25°C (as described under Experimental Procedures). The apparent rate constants of dissociation (k−1) were 0.019 and 0.029 for the dissociation with LNP 911 or with LNP 911 in the presence of moxonidine, respectively. Binding att = 0 (100%) represented 3917 and 4485 cpm in the absence and presence of moxonidine, respectively. The result shown is representative of four separate experiments.
The question of whether moxonidine could allosterically modulate the binding of [125I]LNP 911 has next been addressed. To investigate this hypothesis, the effects of 50 μM of this drug on the [125I]LNP 911 dissociation kinetic at 25°C were studied (Fig. 7B). Compared with the k−1 value measured in the absence of drug [k−1 = 0.021 ± 0.003 min−1 (n = 4)], moxonidine led to a significant increase of the k−1[0.032 ± 0.001 min−1 (n = 4) (p < 0.05, t test)]. In order to extent this result, we next tested idazoxan and PIC. Idazoxan proved also able to increase the k−1 [0.039 ± 0.003 min−1 (n = 3) (p< 0.05, t test)]. At the other hand, PIC had no significant effect on the k−1 [0.030 ± 0.009 min−1 (n = 12)]. This indicated that 50 μM moxonidine or idazoxan accelerated the dissociation rate of about 1.5- and 1.8-fold, respectively.
[125I]LNP 911 Recognizes Neither α2a-Adrenergic Receptors nor I2 Binding Sites.
PC-12 cells do not endogenously express α2-adrenergic receptors (Duzic and Lanier, 1992; Williams et al., 1998). To confirm the selectivity of [125I]LNP 911 for I1R versus α2AR, already suggested by the data obtained with the unlabeled ligand (see above), the ability of [125I]LNP 911 to bind to α2-adrenergic receptors was checked in HT29 cell membrane preparations. This cell line endogenously expresses the α2A-subtype of adrenergic receptors. In this cell membrane preparation, the total binding of [125I]LNP 911 (1 nM) was not displaced at all by rauwolscine, a reference α2AR antagonist, indicating that [125I]LNP 911 does not bind to α2A-AR (data not shown).
To investigate the capability of [125I]LNP 911 to bind to I2BS, we used rabbit kidney membrane preparations. In these membrane preparations, [125I]LNP 911 (1 nM) specific binding was displaced by idazoxan only with a low affinity (IC50 = 16,000 ± 7,437 nM,n = 3) indicating that, at this concentration, [125I]LNP 911 did not bind to I2BS (Fig. 8). In this model, competition binding curves with PIC were biphasic and could be resolved into two sites of high (IC50 = 3.4 ± 0.6 nM; 70% of sites) and low affinity (IC50 = 3,600 ± 678 nM) (n= 3) indicating that [125I]LNP 911 proved able to selectively reveal the I1R present in the rabbit kidney membrane preparations.

Selectivity of [125I]LNP 911 binding in rabbit kidney membranes. Competition assays were carried out with [125I]LNP 911 (1 nM) and with increasing concentrations of the competitor. Competition curves for PIC (○) were shallow and best fit to a two-site model (p = 0.003) and those for idazoxan (●) were best fit to a one-site model. Each point is the mean of three experiments performed in triplicate and using different membrane preparations.
Is LNP 911 an Agonist or an Antagonist for the I1Receptors?
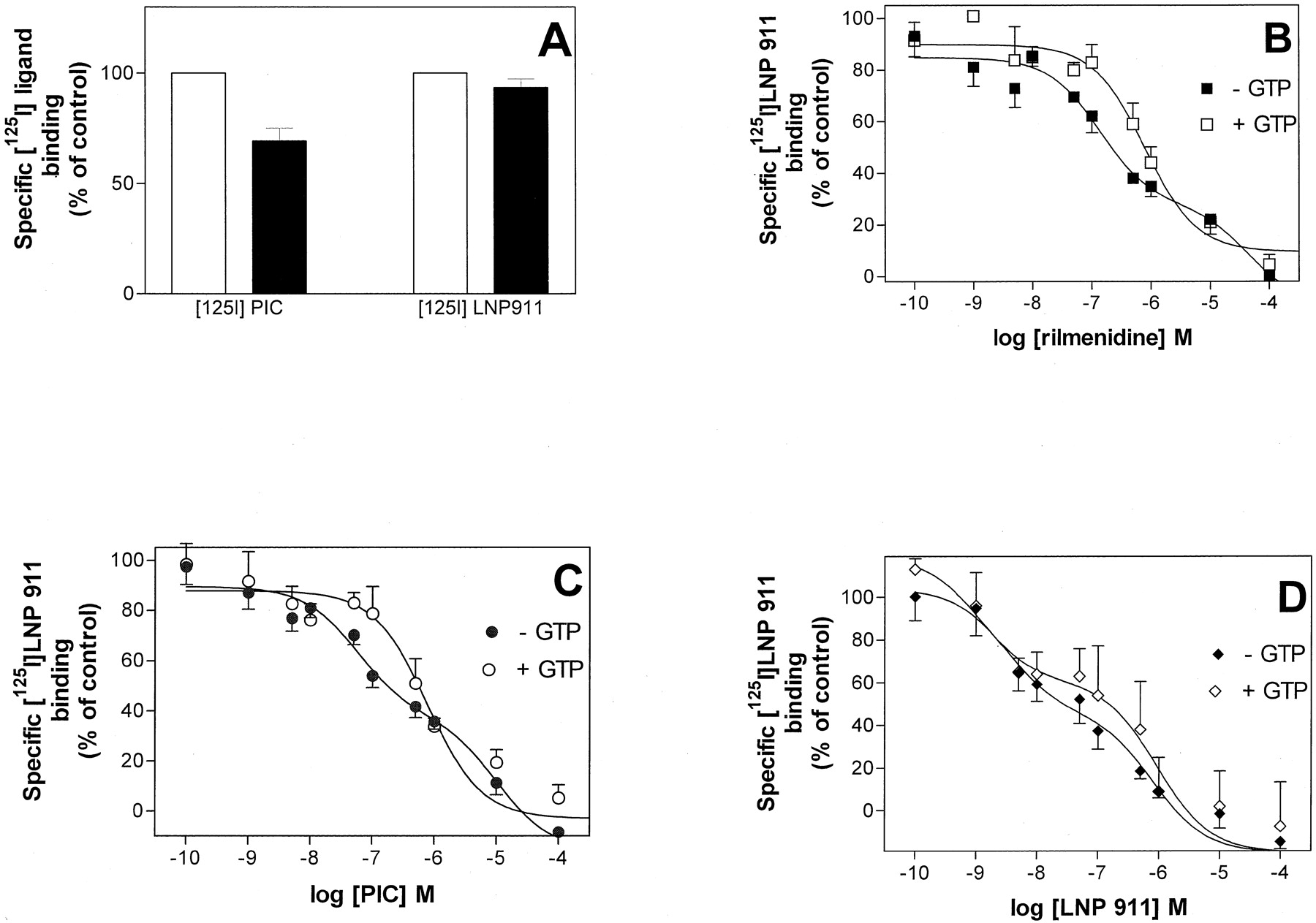
We next determined whether LNP 911 behaves as an agonist or an antagonist for the I1 receptors. Because I1 receptors are known to belong to the G protein-coupled receptor family, we first checked the effect of a nonhydrolyzable analog of GTP, GTPγS, on the binding characteristics of [125I]LNP 911. As shown in the Fig.9A, 100 μM GTPγS did not affect the specific binding of [125I]LNP 911, although the specific binding of [125I]PIC was inhibited by 31 ± 6% (n = 6). The competition curves of cold LNP 911 on [125I]LNP 911 binding sites in the absence and presence of 100 μM GTPγS were next recorded. As shown in Fig. 9D, this competition curve was not affected by the addition of GTPγS [K i1 = 2.8 ± 0.8 (46% of sites) and K i2 = 933 ± 68 nM in the absence of GTPγS; K i1 = 1.0 ± 1.3 (42% of sites) and K i2 = 830 ± 69 nM in the presence of GTPγS]. On the other hand, competition curves of rilmenidine and PIC on [125I]LNP 911 binding sites, which were biphasic in the absence of GTPγS (Figs. 5A and 9, B and C), became monophasic in the presence of GTPγS [K i = 914 ± 683 nM and 688 ± 73 nM for rilmenidine and PIC, respectively (n = 3) (Fig. 9, B and C)]. According to these results, LNP 911 behaves as an antagonist and PIC and rilmenidine as agonists of the I1receptors.

Effect of GTPγS on [125I] LNP 911 and [125I] PIC binding to PC-12 cell membranes. A, specific binding of [125I] LNP 911 (0.2 nM) and [125I] PIC (0.5 nM) in the absence (■) or in the presence of 100 μM GTPγS (▪). The data are presented as the average of triplicate determinations of six experiments for [125I] PIC binding and 11 experiments for [125I] LNP 911. Total specific binding (100%) for [125I] PIC represented 3502 cpm and for [125I] LNP 911 represented 2393 cpm. B, C, and D, competition curves of rimenidine, PIC, and LNP 911, respectively on [125I] LNP 911 (0.2 nM) binding sites. The experiments were carried out as described under Materials and Methods. Competition curves with rilmenidine (▪), PIC (●) and LNP 911 (♦) in the absence of GTPγS were shallow and best fitted to a two-site model (rilmenidine, p = 0.009; PIC, p = 0.0004; LNP 911, p = 0.02). Competition curves with rilmenidine (▪) or PIC (●) in the presence of 100 μM GTPγS were best fitted to a one-site model (rilmenidine, p = 0.48; PIC, p= 0.478). Competition curve with LNP 911 (⋄) in the presence of 100 μM GTPγS were shallow and best-fitted to a two-site model (LNP 911,p = 0.04). Each point is the mean of three experiments performed in triplicate.
To confirm these results, we tested the effect of LNP 911 on the cAMP transduction pathway associated with the I1receptors in the PC-12 cells (Greney et al., 2000). In a first series of experiments, we tested the effects of LNP 911 at 10−5 M alone or in combination with three I1 ligands, moxonidine, rilmenidine, and PIC (Fig. 10A). LNP 911 (10−5 M) had no effect on its own on the basal (82 ± 7 and 81 ± 7 fmol/min/105 cells in the absence and presence of LNP 911, respectively) and forskolin-stimulated cAMP level (222 ± 19 and 226 ± 21 fmoles/min/105 cells in the absence and presence of LNP 911, respectively) in the cells as expected for an antagonist. On the other hand, moxonidine (as shown previously in Greney et al., 2000), PIC, and rilmenidine (10−5 M) proved able to decrease slightly but significantly the forskolin-stimulated accumulation of cAMP in the cells acting thus as agonists on this pathway (Fig. 10A). However, when LNP 911 (10−5M) was added 10 min before the three other drugs (10−5 M), it did not inhibit their effect but rather enhanced the decrease in forskolin-stimulated cAMP level obtained with the three agonists (11 ± 2, 12 ± 3, and 14 ± 6% inhibition for moxonidine, rilmenidine, and PIC, respectively, in the absence of LNP 911; 24 ± 8, 33 ± 7, and 26 ± 6% inhibition, respectively, in the presence of LNP 911; Fig. 10A). In the case of moxonidine however, the difference between its effect without or with LNP 911 did not reach statistical significance although when paired experiments were compared, the latter was always greater than the former.

Effect of LNP 911 on forskolin-stimulated cAMP level response of I1R agonists in PC-12 cells. A, cells (105) were preincubated in the absence or presence of 10 μM LNP 911 for 10 min at 37°C before the addition of I1R agonists (10−5 M) and 1 μM forskolin. cAMP level, after a subsequent incubation of 30 min at 37°C, was measured as described under Materials and Methods. Results represent the average of at least three experiments performed in triplicate and are expressed as percentage of inhibition ± S.E.M. of the control (0% = in the presence of forskolin only). In the absence of LNP 911, basal cAMP production was 82 ± 7 fmol/min/105 cells; forskolin-stimulated cAMP production was 222 ± 18.6 fmol/min/105 cells. In the presence of LNP 911, basal cAMP production was 81.7 ± 7.6 fmol/min/105 cells; forskolin-stimulated cAMP production was 226 ± 21 fmol/min/105cells. ★,p < 0.05, paired t test; statistically different from values with the corresponding agonist. B, C, and D, same methods as in A except that increasing concentrations of LNP 911 (10−9 to 10−3 M) were preincubated for 10 min at 37°C before the addition of the I1R agonists. B, moxonidine (10 μM); C, rilmenidine (10 μM); D, PIC (10 μM). Results represent the average of at least three experiments performed in triplicate and are expressed as percentage of inhibition ± S.E.M. of the control (0% = in the presence of forskolin only).
According to these unexpected results for an putative antagonist, we next checked the dose dependence of the effects of LNP 911. LNP 911 had no effect on its own over the entire concentration range of 10−9 to 10−3 M. But LNP 911 dose dependently enhanced the effects of moxonidine (10−5 M), rilmenidine (10−5 M), and PIC (10−5M). At high concentration of LNP 911 (10−3 M), the enhancement was of 4.7, 4.3, and 3.8 fold, respectively (Fig. 10, B-D).
Discussion
Since the demonstration that the centrally mediated hypotensive effect of some imidazoline drugs was dissociated from their capability to activate the α2-adrenergic receptors, the existence of new receptor entities has been widely documented (Bousquet et al., 1984). These so-called I1 receptors have been characterized by binding studies (Molderings et al., 1993; Piletz et al., 1996; Ernsberger et al., 1997) and by cellular (Separovic et al., 1996; Greney et al., 2000) and “in vivo” effects (Bousquet et al., 1984; Ernsberger et al., 1990; Smyth and Penner, 1999; Bruban et al., 2001; Ogidigben et al., 2001). At the time, the available radioligands, used to perform binding studies on the I1R, exhibit low selectivity versus α2AR (Molderings et al., 1993; Piletz et al., 1996; Ernsberger et al., 1997). In an effort to facilitate the identification and characterization of the I1receptors, we attempted to develop high-affinity compounds that selectively recognize these sites. Our line of investigation was already successful with the synthesis of LNP 509, which exhibited a high selectivity for the I1R compared with α2-AR and decreased blood pressure (Schann et al., 2001). In a series of pyrroline compounds, the LNP 911 has been selected as a high affinity I1 receptor ligand. The radioiodinated derivative of LNP 911 was fully characterized and the data presented are in agreement with [125I]LNP 911 being the first highly selective radioligand for the I1 receptors.
Unlabeled LNP 911 proved able to displace the specific binding of [125I]PIC to I1R on PC-12 cell membranes with a high affinity (K i = 0.2 nM). The specific binding of [125I]LNP 911, in the same membrane preparations, was saturable, reversible, and of high affinity (K d = 1.4 nM). Importantly, nonspecific binding of [125I]LNP 911 determined with PIC, the I1R reference ligand, was very low (∼10% at the K d of [125I]LNP 911) compared with other I1R radioligands (50% for [125I]PIC (Piletz et al., 1996; Separovic et al., 1996); 55% for [3H]clonidine (Molderings et al., 1993). Moreover, all the imidazolines tested in competition assays displaced the same amount of [125I]LNP 911 total binding. In particular, unlabeled LNP 911 and PIC defined the same amount of nonspecific binding when used at 100 μM, indicating that these two drugs bound to identical sites in these experimental conditions.
Analysis of saturation isotherms indicated that [125I]LNP 911 apparently recognized only one homogenous population of sites with a high affinity. Comparison of the densities of [125I]PIC binding sites (this work; Greney et al., 2000) and [125I]LNP 911 binding indicates that [125I]LNP 911 bound to about 7- to 9-fold more sites than [125I]PIC. One explanation may be that [125I]LNP 911 could be an antagonist where [125I]PIC may be an agonist. Such discrepancies betweenB max values obtained either with an agonist or with an antagonist have already been observed [e.g., for the A3 adenosine receptors (Varani et al., 2000)]. The results on binding assays agree with this hypothesis, because [125I]LNP 911 binding sites were not affected by GTPγS although [125I]PIC binding sites were clearly decreased. On the other hand, competition curves of unlabeled LNP 911 for [125I]LNP 911 binding were unaffected by GTPγS although those of PIC and rilmenidine were changed from biphasic to monophasic curves. The biphasic competition curves on [125I] LNP 911 binding sites obtained with agonists refer to the coupled-uncoupled form of G protein-coupled receptors as shown in the Fig. 9, B and C. On the other hand, the biphasic competition curves obtained with nonradiolabeled LNP 911 on [125I] LNP 911 binding sites (Fig. 9D) are related to the complex combination of competitive binding sites and allosteric binding sites (see below). Thus, from a binding point of view, LNP 911 might be an antagonist, whereas PIC and rilmenidine behave as agonists.
This point was also addressed in functional experiments in which PIC, rilmenidine, and moxonidine (this work; Greney et al., 2000) exhibit agonist properties. However, although being inactive by itself on the transduction pathway as should be observed with an antagonist, LNP 911 increased rather than decreased the agonist effects. This could denote an allosteric enhancer property of LNP 911 (see below).
Kinetic analysis of [125I]LNP 911 binding also indicated that [125I]LNP 911 bound to an apparently homogenous population of sites. The constants of association and dissociation were very fast and therefore consistent with the identification by [125I]LNP 911 of a real receptor (Limbird et al., 1976). The fast dissociation rate observed for [125I]LNP 911 binding sites is in agreement with previous kinetic results for I1 receptors (Piletz et al., 1991).
Thus far, almost all of the binding data obtained in different membrane preparations on I1R with [3H]clonidine or [125I]PIC revealed that competition curves with imidazolines were better resolved in two compartments (Molderings et al., 1993; Piletz et al., 1996; Greney et al., 2000). Our results with [125I]LNP 911 are in agreement with such results in that some imidazolines displayed high and low affinity compartments in competition experiments. However, other drugs exhibited only a low affinity for [125I]LNP 911 binding sites in standard binding assays (without preincubation of the radioligand, using Tris-HEPES buffer and incubation at 25°C) although they proved able to displace [125I]PIC binding to I1 receptors with a high affinity in different I1 receptor binding models (Piletz et al., 1996;Separovic et al., 1996; Greney et al., 2000). In fact, the correlation between the pK i of the high affinity I1R ligands on [125I]LNP 911 binding sites and the pK i of the same drugs on [125I]PIC binding sites and the fact that PIC and LNP 911 were able to displace the same amount of [125I]LNP 911 total binding indicate that identical binding sites/I1R are probably concerned.
In this context, our observation that moxonidine, efaroxan and benazoline exhibited low affinities (in the micromolar range) for [125I]LNP 911 binding sites was unexpected because these three ligands were known as high-affinity I1R ligands (Separovic et al., 1996; Greney et al., 2000). This apparent discrepancy might be the result of complex allosteric regulation at the level of the I1R binding sites. Three lines of evidence confirm that an allosteric modulation may exist for the I1R: 1) moxonidine proved able to significantly increase the dissociation parameter (k−1) of [125I]LNP 911. 2) A biphasic competition curve for moxonidine was obtained when [125I]LNP 911 was allowed to preassociate with its binding sites during 2 h at 4°C. These particular binding conditions revealed a high-affinity binding compartment for moxonidine (IC50 = 74 ± 11 nM), which seems similar to the one obtained in [125I]PIC binding experiments (IC50 = 68 ± 5 nM; Greney et al., 2000). 3) LNP 911, although inactive on its own, on the cAMP pathway, clearly potentate the effects obtained with I1 receptor agonists, moxonidine, PIC, and rilmenidine. According to these results, we suggest that I1R are submitted to a complex allosteric modulation which is underlined, for the first time, by the use of our new ligand LNP 911. LNP 911 behaves as an allosteric enhancer according to its physiological properties; conversely, an agonist (moxonidine) was shown to change the binding parameters of LNP 911. Such an agonist effect on the allosteric ligand binding has already been demonstrated on the muscarinic receptor (Trankle et al., 1999). LNP 911 might combine competitive binding and allosteric enhancement properties that look like the characteristics of allosteric modulators for adenosine or metabotropic glutamate 1 receptors (Gao et al., 2001; Knoflach et al., 2001). The fact that LNP 911 inhibits binding of agonists and yet enhances the functional responses to agonists remains unclear. Nevertheless, Gao et al. (2001) have shown that some allosteric enhancers not only bind to allosteric sites but can also compete with agonists at the orthosteric sites and even potentiate the functional responses to agonists.
The selectivity of LNP 911 for I1R versus I2BS and α2-adrenoceptors was checked in different ways: 1) In rabbit kidney membranes, where I2BS are largely expressed (Coupry et al., 1990), unlabeled LNP 911 proved unable to displace [3H]idazoxan with a high affinity (K i >10−5 M) and inversely [125I]LNP 911 binding was not displaced by idazoxan (K i>10−5 M). 2) In HT29 cell membranes, which are known to contain the α2A-adrenoceptor subtype (Devedjian et al., 1991) or in α2A,B,C-adrenoceptor transfected CHO cell membranes, unlabeled LNP 911 displaced [3H]RX 821002 with K i>10−6 M. Inversely, [125I]LNP 911 binding sites were not displaced by rauwolscine in the HT29 cell membrane preparations. These results confirm the high selectivity of LNP 911 for I1R over the other imidazoline binding proteins, α2-adrenergic receptors, and I2BS.
This new I1R selective radioligand has already proved its usefulness for studying these receptors in tissues containing different imidazoline binding proteins. The binding results obtained in rabbit kidney membranes are in agreement with the existence, in this tissue, of I1R besides I2BS, as suggested already (Hamilton et al., 1991; Gargalidis-Moudanos and Parini, 1995). In fact, [125I]LNP 911 total binding, although not displaced by idazoxan, a high-affinity I2BS ligand, was totally displaced by PIC (an I1R high-affinity ligand) with an IC50 of 3 nM. These results confirm the high selectivity of this new radioligand and open the possibility to investigate in depth the interaction between different imidazoline binding proteins (in particular the α2-adrenergic receptors), which was already suggested by physiological studies in our group (Bruban et al., 2001).
In conclusion, [125I]LNP 911 is the first highly selective radioligand exhibiting a nanomolar affinity for I1R. Its properties allows the characterization of these receptors even in tissues or cells expressing different imidazoline binding proteins (α2-adrenoceptors or I2BS). [125I]LNP 911 also reveals that I1R are submitted to complex allosteric modulation largely depending on both the radioligand and the competitor used in binding studies. In addition, LNP 911 shares functional properties with allosteric enhancers. This new tool will be essential in further studies concerning the I1R in depth characterization.
Acknowledgments
We are grateful to I.R.I.S./Servier Laboratories for their financial support and their technical help. We thank Gabriel Lacroix for his help in the synthesis of LNP 911.
Footnotes
- Received August 9, 2001.
- Accepted April 9, 2002.
-
G.H. and U.D. contributed equally to this work.
Abbreviations
- I2BS
- I2 binding site(s)
- I1R
- I1 receptor(s)
- PIC
- para-iodoclonidine
- LNP 911
- 2-(2-chloro-4-iodo-phenylamino)-5-methyl-pyrroline
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- HME
- HEPES/MgCl2/EGTA
- α2AR
- α2-adrenoceptor(s)
- PBS
- phosphate-buffered saline
- CHO
- Chinese hamster ovary
- BDF6143
- 4-chloro-2-(imidazolin-2-ylamino)-isoindoline
- GTPγS
- guanosine-5′-O-(3-thio)triphosphate
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}