


Abstract
In this work, we evidenced characteristic features of agonist-induced trafficking of receptor stimulus for the rat neurotensin receptor 1 (NTS1). Thus, reverse potency orders between two agonists, EISAI-1 and neuromedin N, were observed in inositol 1,4,5-trisphosphate and cAMP assays in Chinese hamster ovary cells transfected with this receptor. Indeed, compared with other agonists, EISAI-1 presented lower relative potency toward inositol 1,4,5-trisphosphate production than toward cAMP accumulation, guanosine 5′-O -(3-[35 S]thio)triphosphate binding, and [3H]arachidonic acid production. These results indicated pathway-dependent differences in EISAI-1 intrinsic efficacies, favoring activations of Gs- and Gi/o-related pathways over the Gq/11-related pathway. Moreover, although coupling to Gq/11 and Gi/o involved the third intracellular loop and the C-terminal domain of the NTS1 receptor, respectively, we demonstrated that deletion of the latter domain suppressed agonist-induced cAMP accumulation, suggesting that this domain also mediated coupling to Gs. Together, these results indicated that, unlike other agonists, EISAI-1 discriminated between the pathways involving the receptor C-terminal domain and that involving the third intracellular loop. These properties of EISAI-1 were also observed in cortical neurons endogenously expressing the NTS1 receptor. They were further attributed to the functionalization of its COOH end by an ethyl group, because the unesterified analog EISAI-2 presented normal behavior on inositol 1,4,5-trisphosphate production.
These findings support the hypothesis of agonist-selective receptor states with distinct conformations or accessibilities of intracellular domains. They also suggest that the differential involvement of these domains in coupling to G proteins might represent a molecular basis for agonist-selective responses through G protein-coupled receptors.
It is now well established that a single G protein-coupled receptor (GPCR) can interact with several G proteins or other transducing molecules, thereby activating multiple signaling pathways. Moreover, several studies revealed that a ligand could show differential abilities to trigger these pathways (Kenakin, 1996, 2001; Berg et al., 1998; Brink et al., 2000; Wenzel-Seifert and Seifert, 2000). One important implication of this finding is the possibility of developing drugs acting selectively on one of the responses associated to a receptor.
Essentially two mechanisms could lead to agonist-selective activation of effector pathways: differential strength of signaling and agonist-directed trafficking of receptor stimulus (Kenakin, 1995). The first mechanism relies on differential tightness of coupling between the receptor and the various transducing molecules. Agonists of high efficacy will thus induce a pleiotropic response, whereas agonists of low efficacy will trigger only the most efficiently coupled pathway. The second mechanism relies on pathway-dependent differences in agonist intrinsic efficacies. This mechanism was most efficiently illustrated by reverse agonist potency or efficacy orders among different pathways (Kenakin, 1996, 2001). Such a phenomenon cannot be explained by differential strength of signaling-based mechanism and strongly supports the hypothesis of agonist-specific active receptor states.
The occurrence of multiple active receptor states theoretically strengthens the possibility to design pathway-selective agonists. However, for any given receptor, achieving this purpose requires to determine both the differences among active states that could account for the differential responses and the determinants of the ligand-receptor interactions that govern orientation toward one of these states. In this respect, because interactions with G proteins involve the receptor intracellular domains, knowledge of the respective role of these domains in the activation of each pathway is a prerequisite for determining the functionally relevant differences among receptor states. The aim of our article was to investigate these different aspects using the NTS1 neurotensin receptor as a model.
Neurotensin (NT) is a brain-gut tridecapeptide acting as a neuromodulator in the brain and a paracrine or circulating hormone in periphery (Kaskow and Nemeroff, 1991; Rostene and Alexander, 1997). NT agonists or antagonists have been suggested to be of potential use for the treatment of pain, eating behavior, psychotic troubles, drug abuse, and stress (Rostene and Alexander, 1997; Berod and Rostene, 2002; Kinkead and Nemeroff, 2002; Kitabgi, 2002). Moreover, overexpression of NT receptors in various tumors suggested that NT-related ligands could represent valuable tools for tumor targeting (Reubi et al., 1999; Hillairet de Boisferon et al., 2002).
Three NT receptors, NTS1, NTS2, and NTS3, have been cloned to date. NTS1 and NTS2 receptors belong to the GPCR family, whereas NTS3 receptor belongs to the family of sorting receptors (for review, see Vincent et al., 1999; Kitabgi, 2002). NTS1 receptor mediates most of the physiological functions ascribed to NT, whereas NTS2 receptor might be involved in NT-induced analgesia (Kitabgi, 2002). The functions of NTS3 receptor still remain to be established (Mazella, 2001). However, recent data evidenced its involvement in cell migration (Martin et al., 2003).
Previous studies suggested a major coupling of NTS1 receptor to Gq/11 (Hermans and Maloteaux, 1998; Vincent et al., 1999). However, stimulation of this receptor also activated other pathways such as production of arachidonic acid or cAMP, through Gi/o or Gs, respectively (Yamada et al., 1993; Gailly et al., 2000). Although coupling to Gq/11 was previously reported to involve the third intracellular loop of NTS1 receptor (Yamada et al., 1994), we recently demonstrated that coupling to Gi/o involved the carboxy-terminal portion of this receptor (Najimi et al., 2002). However, the domain mediating coupling to Gs has not yet been identified.
In the present study, we evidenced characteristic features of agonist-directed trafficking of receptor stimulus for the NTS1 receptor. Thus, reverse potency orders between two agonists, EISAI-1 and neuromedin N (NN), were observed in inositol 1,4,5-trisphosphate (InsP3) and cAMP assays in CHO cells transfected with this receptor. We further demonstrated that, in contrast with other agonists, EISAI-1 preferentially activated cAMP and [3H]arachidonic acid production over InsP3 production. Moreover, whereas the latter pathway involves the receptor third intracellular loop, we showed that agonist-induced cAMP accumulation, like [3H]arachidonic acid production, involved the C-terminal domain. Together, these results indicated that, unlike other agonists, EISAI-1 discriminated among pathways involving different intracellular domains of the NTS1 receptor. This property of EISAI-1 could be attributed to the functionalization of its COOH end.
These data support the hypothesis of multiple agonist-selective receptor states. They further suggest that differential involvement of receptor intracellular domains in coupling to G proteins might represent a molecular basis for agonist-selective responses through GPCRs.
Materials and Methods
Materials. All culture media were from Invitrogen (Cergy Pontoise, France). NT, neuromedin N, JMV449 (Doulut et al., 1992), and EISAI-2 (Machida et al., 1993) were from Neosystem (Strasbourg, France). EISAI-1 (Machida et al., 1993) was a generous gift of Eisai Co., Ltd. (Tokyo, Japan). Primary structures of these agonists are presented in Table 1. [35S]GTPγS (specific activity, 1000 Ci/mmol) was from PerkinElmer Life Sciences (Boston, MA) and [3H]arachidonic acid (specific activity, 212 Ci/mmol) was from PerkinElmer Life Sciences (Paris, France). [3H]InsP3 assay kit and [3H]cAMP assay kit were purchased from Amersham Biosciences Inc. (Saclay, France). All other reagents were from Sigma (St-Quentin Fallavier, France).
Structure of NT agonists
Receptor Expression and Cell Cultures. Expression of the wild-type NTS1 receptor in transfected CHO cells was performed as described previously (Boudin et al., 1995). In the present study, we used two clones expressing different amounts of the NTS1 receptor. The first clone, called CHO-NTR-H cells, expressed 2.9 ± 0.8 pmol of 125I-NT binding sites/mg of protein (n = 5); the second one, called CHO-NTR-L cells, expressed 0.65 ± 0.12 pmol of 125I-NT binding sites/mg of protein (n = 5). The cloning and functional expression of the NTS1 receptor lacking its COOH terminus (NTRdel372) was reported previously (Hermans et al., 1996). The CHO cells transfected with this truncated receptor, termed CHO-NTRdel372 cells, expressed 0.35 ± 0.08 pmol of 125I-NT binding sites/mg of protein (n = 3).
CHO cells were cultured in α-minimal essential medium (without nucleosides and deoxyribonucleosides), supplemented with 10% fetal calf serum, 2 mM glutamine, and 250 μg/ml G418. Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2, 95% air. For InsP3 production measurement, 1.6 × 106 cells/well were seeded in six-well plates and were grown for 2 days before use. For measurement of [3H]arachidonic acid release, 4 × 105 cells/well were seeded in 24-well plates and were grown for 2 days before use.
Primary cultures of rat cortical neurons were prepared from embryonic day 17 Wistar rats. Cerebral cortices were dissected under sterile conditions in phosphate-buffered saline (PBS; 137 mM NaCl, 21 mM NaHPO4, 29 mM KH2PO4, and 1.2 mM KCl, pH 7.3). Cells were mechanically dissociated in culture medium (minimal essential medium) supplemented with 19.8 mM glucose, 5 mM HEPES, 50 U/ml penicillin, 50 μg/ml streptomycin, 5% fetal calf serum, 5 μg/ml insulin, 20 nM progesterone, 100 μg/ml human transferrin, 30 nM sodium selenite, 0.1 mM putrescine, and 10 μg/ml BSA), collected by centrifugation (500g, 5 min), resuspended in culture medium at a concentration of 2 × 106 cells/ml, and plated at a density of 5 × 105 cells/cm2 in 6- or 24-well Costar multiwell plastic culture plates previously coated with poly-d-lysine (10 min at room temperature with 20 μg of poly-d-lysine/ml H2O, rinsed twice with water and once with PBS). Cultures were maintained at 37°C in a moist atmosphere consisting of 95% air and 5% CO2. Culture medium was renewed after 5 days, and cells were used for experiments after 8 days in vitro. All experiments were performed in accordance with the European Communities Council Directives for the care and use of laboratory animals.
Membrane Preparation. CHO cells or cortical neurons grown in six-well culture plates were scraped in ice-cold PBS, collected by centrifugation at 500g for 5 min, and lysed in ice-cold 5 mM Tris-HCl, pH 7.4, by successive passages through a syringe with a 26-gauge1/2 needle. The suspension was centrifuged for 10 min at 17,000g at 4°C, passed again through the syringe, and centrifuged as described above. Finally, the membranes were resuspended in 50 mM Tris-HCl, pH 7.4, at approximately 10 mg of protein/ml and stored at -80°C. Protein content was measured by the Bio-Rad protein assay (Bio-Rad, Munich, Germany).
[125I]NT Binding Assays. Binding experiments were performed in 50 mM Tris-HCl, pH 7.4, 0.2% BSA, 5 mM MgCl2, and 0.8 mM 1,10-ortho-phenanthroline. For competition experiments, membranes of transfected CHO cells expressing either wild-type NTS1 or NTRdel372 (10 and 15 μg of protein, respectively), or membranes from primary cortical cultures (15 μg of protein) were incubated for 30 min at 25°C with 50 pM 125I-Tyr3NT (125I-NT, 2000 Ci/mmol) prepared as described previously (Sadoul et al., 1984) and increasing concentrations of competitors in a final volume of 500 μl. For saturation experiments, membranes were incubated for 30 min at 25°C with increasing concentrations (10–400 pM) of 125I-NT. Nonspecific binding was determined in the presence of 10-6 M unlabeled NT. The assay was terminated by adding 5 ml of ice-cold rising buffer (50 mM Tris-HCl buffer, pH 7.4, supplemented with 0.2% BSA), free radio-ligand was eliminated by filtration under vacuum through GF/B glass filters (Whatman, Maidstone,. UK) presoaked for 1 h with polyethylenimine (0.2% in water), and membranes were rinsed twice with 5 ml of rinsing buffer. Radioactivity remaining on the membranes was measured in a gamma counter (PerkinElmer Life Sciences). The competition data were analyzed using the EBDA-LIGAND program (Munson and Rodbard, 1980).
Measurement of InsP3 Levels. CHO cells or cortical neurons grown in six-well plates were incubated for 30 min at 37°C in 2 ml of their respective culture medium from which serum was omitted, supplemented with 20 mM LiCl and 0.1% BSA. Stimulation with the different peptides was then performed during 20 s, medium was aspirated, 500 μl of ice-cold 7% perchloric acid was added, and plates were put on ice. The resulting cell lysate was collected and centrifuged for 3 min at 11,000g at 4°C. After centrifugation, supernatant (430 μl) was neutralized with potassium bicarbonate. Precipitate was removed by centrifugation (11,000g, 4°C, 5 min), and supernatant was stored at -20°C. The InsP3 content in the supernatant was measured with a [3H]InsP3 assay kit (Amersham Biosciences Inc.), according to the instructions of the manufacturer. Data were analyzed using Prism software (GraphPad Software Inc., San Diego, CA).
Measurement of Cyclic AMP Levels. Cell membranes (50 μg) were incubated for 15 min at 32°C with NT agonists in a final volume of 150 μl of reaction buffer (50 mM Tris-HCl, pH 7.4, supplemented with 1.5 mM 3-isobutyl-1-methylxanthine, 1.6 mM ATP, 0.5 mM GTP, 5 mM MgCl2, 1 mM EGTA, 0.1% bovine serum albumin, 0.8 mM 1,10-ortho-phenanthroline, 67 U/ml creatine phosphokinase, and 2.5 mM creatine phosphate). At the end of incubation, the tubes were placed in a boiling water bath for 5 min, put on ice, and centrifuged for 10 min at 9000g. cAMP content of the supernatant was determined using [3H]cAMP assay kit (Amersham Biosciences Inc.) according to the instructions of the manufacturer. Data were analyzed using Prism software (GraphPad Software Inc.).
Measurement of [3H]Arachidonic Acid Production. Cells grown in 24-well plates were incubated overnight at 37°C in culture medium supplemented with 0.5 μCi/ml [3H]arachidonic acid, allowing a progressive incorporation of labeled arachidonic acid into membrane phospholipids. Cells were then rinsed three times for 5 min with prewarmed Krebs-Ringer-HEPES buffer (125 mM NaCl, 4.8 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 2.2 mM CaCl2, 5.6 mM glucose, and 25 mM HEPES, pH 7.4) supplemented with 0.2% fatty acid-free BSA. Stimulation with the peptides was then performed during 15 min, medium was collected, centrifuged for 10 min at 11,000g at 4°C, and radioactivity present in the supernatant was counted by liquid scintillation. Cells were lysed in 0.1 N NaOH and radioactivity was counted by liquid scintillation. [3H]Arachidonic acid production was expressed as the percentage of incorporated radioactivity. Data were analyzed using Prism software (GraphPad Software Inc.).
Binding of [35S]GTPγS. The specific binding of [35S]GTPγS was measured on cell membranes as described previously (Gailly et al., 2000). Briefly, cell membranes (80 μg of protein) were resuspended in binding buffer (50 mM Tris-HCl, pH 7.4, containing 5 mM MgCl2, 1 μM 1,10-ortho-phenanthroline, 0.1% BSA, 1 μM GDP, 150 mM NaCl, and 1 mM dithiothreitol). After incubation for 30 min at 37°C, the suspension was immediately filtered through GF/B glass fiber filters (presoaked for 1 h in 0.5% polyethylenimine) and washed twice with ice-cold binding buffer using a 24-channel harvester (Semat; Brandel Inc., Gaithersburg, MD). Radioactivity was estimated by scintillation counting. The nonspecific binding was measured in the presence of 0.1 mM guanosine 5′-(β,γ-imido)triphosphate. The binding data were analyzed by nonlinear regression using the Prism software (GraphPad Software Inc.).
Statistics. Experiments were performed at least three times each. All values are expressed as means ± S.E.M. Statistical comparisons between two experimental groups were performed using Student's t test. Experiments comprising more than two experimental groups were analyzed through ANOVA variance analysis followed by Dunnett's or Newman-Keuls tests. Differences at p < 0.05 were considered as significant.
Results
Pharmacological Profiles of InsP3 and cAMP Production through Stimulation of the NTS1 Receptor Expressed in CHO Cells. We first compared the ability of two natural agonists, NT and NN, and three synthetic agonists, JMV449, EISAI-1, and EISAI-2 (Table 1), to bind to the NTS1 receptor and to stimulate InsP3 and cAMP production in CHO-NTR-H cells, expressing high amounts of the NTS1 receptor (2.9 pmol of 125I NT binding sites/mg of protein).
As indicated in Table 2, JMV449 and NT competed with high-affinity 125I-NT specific binding on homogenates of CHO-NTR-H cells with similar inhibition constant (Ki) values, EISAI-2 was almost 2 times less potent, whereas EISAI-1 and NN showed 3- and 18-fold lower affinities, respectively. Regarding second messenger measurements, different potency orders were observed for these agonists toward cAMP and InsP3 production, with reversal of potency orders between EISAI-1 and NN in the two assays (Fig. 1; Table 2).
Pharmacological profile of InsP3 and cAMP production in CHO cells expressing the NTS1 receptor
Inhibition constants (Ki) were determined from data obtained in competition experiments with 125I-NT (50 pM) by using the Cheng-Prusoff equation, with a Kd value for 125I-NT (obtained in saturation binding experiments) of 0.12 nM. Maximal binding capacities were 2.9 and 0.65 pmol of 125I-NT binding sites/mg of protein for CHO-NTR-H and CHO-NTR-L cells, respectively. Potency (EC50) of each agonist to increase cAMP or InsP3 production was determined from concentration-response curves as described under Materials and Methods. Values represent the means ± S.E.M. of three to four independent experiments.

Concentration-response curves for cAMP (A) and InsP3 production (B) by JMV449 (□), EISAI-2 (▴), NT (○), NN (⋄), and EISAI-1 (▾) in CHO-NTR-H cells. Data represent means ± S.E.M. of three to four independent experiments, each performed in sextuplicate, in which the average of basal value (performed in sextuplicate) has been subtracted. Basal cAMP production represented 50 to 70 pmol of cAMP/mg of protein, and basal InsP3 production represented 9 to 20 pmol of InsP3/mg of protein.
Indeed, the rank of potencies found for stimulation of cAMP production by all five agonists followed their affinity order (Fig. 1A; Table 2). Similarly, the relative potencies of JMV449, NT, EISAI-2, and NN to increase InsP3 production were consistent with their respective Ki values (Fig. 1B; Table 2). In contrast, EISAI-1 showed an almost 100-fold lower relative potency for InsP3 production than expected (Fig. 1B; Table 2). This compound was around 13 times less potent than NN in this assay, whereas it was 7 times more potent than NN toward cAMP production. These results indicate that EISAI-1 presented discriminative properties at the expense of InsP3 production. Such properties were clearly not shared by the unesterified analog EISAI-2, because this compound showed consistent relative potencies among the two pathways. No increase in cAMP accumulation or InsP3 production was found when agonists were tested on parent untransfected CHO cells (not shown).
As shown in Fig. 1B, the maximal InsP3 production induced by EISAI-1 in CHO-NTR-H cells was similar to that elicited by other agonists. Under our experimental conditions, EISAI-1 therefore behaved as a full agonist of low relative potency for InsP3 production. However, it is well documented that the apparent potency or efficacy of agonists is directly influenced by the expression level of the receptor (Hermans et al., 1999). Because CHO-NTR-H cells expressed the NTS1 receptor at relatively high density, the functional properties of EISAI-1 were further characterized using another clone, CHO-NTR-L cells, expressing around 5 times lower NTS1 receptor density (0.65 pmol of 125I-NT binding sites/mg of protein). As shown in Fig. 2A and Table 2, EISAI-1 still behaved as a full agonist of high relative potency on cAMP accumulation. In contrast, this agonist was 950-fold less potent than JMV449 on InsP3 production (Fig. 2B; Table 2). It was unpractical to run the assay at more than 300 μM EISAI-1. At this concentration, the effect of this compound was significantly lower than the maximal effect induced by JMV449 [300 μM EISAI-1: 164 ± 12 pmol of InsP3/mg of protein (n = 3); 1 μM JMV449: 257 ± 16 pmol of InsP3/mg of protein (n = 3); p < 0.01]. However, due to the low potency of EISAI-1 in this assay, it remains unsure that the maximal effect of this agonist on InsP3 production was actually attained at this concentration.

Concentration-response curves of JMV449 (□) and EISAI-1 (▾)on cAMP (A) and InsP3 (B) production in CHO-NTR-L cells. Data represent means ± S.E.M. of three independent experiments, each performed in sextuplicate, in which the average of basal value (performed in sextuplicate) has been subtracted. Basal cAMP production represented 40 to 70 pmol of cAMP/mg of protein, and basal InsP3 production represented 10 to 15 pmol of InsP3/mg of protein.
Because EISAI-1 was a potent competitor of 125I-NT binding to NTS1 receptor but increased InsP3 production only at high concentrations, we investigated whether this compound could antagonize the action of NT on InsP3 production in CHO-NTR-L cells. As shown in Fig. 3, the increase in InsP3 production induced by a 3 × 10-8 M concentration of NT was strongly inhibited in the presence of 10-6 M EISAI-1. In three independent experiments, EISAI-1 decreased by 63.7 ± 0.7% (mean ± S.E.M.) the effect of NT on InsP3 production. This compound similarly antagonized the action of JMV449 in this assay (not shown).
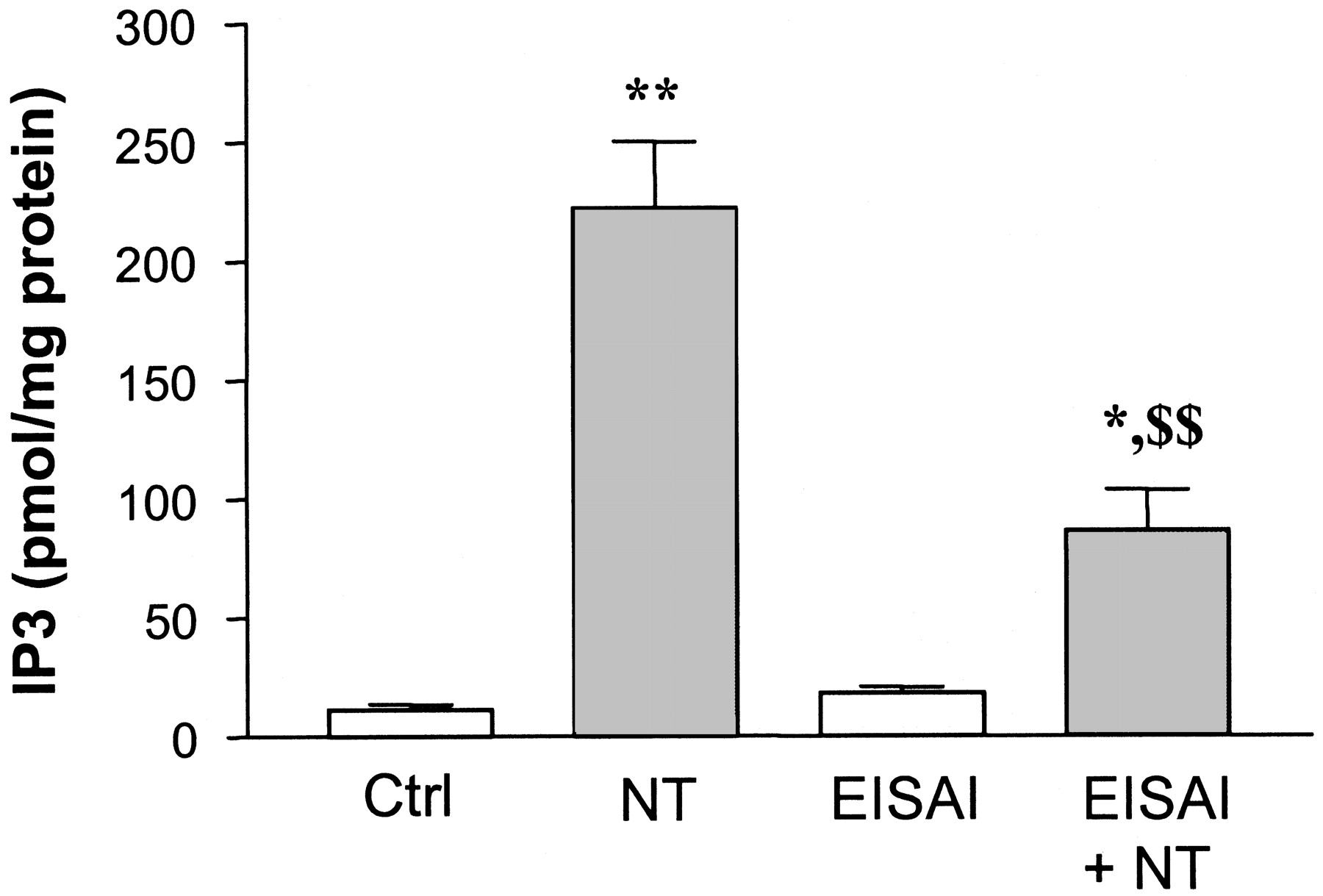
Inhibition by EISAI-1 (10-6 M) of the increase in InsP3 production induced by NT (3 × 10-8 M) in CHO-NTR-L cells. Data represent means ± S.E.M. of one representative experiment, over three independent experiments, performed in sextuplicate. *,**, p < 0.05, p < 0.01 versus the corresponding control; $$, p < 0.01 versus NT alone (ANOVA followed by Newman-Keuls test).
Characteristics of EISAI-1-Induced Arachidonic Acid Production and Binding of [35S]GTPγS in CHONTR-L Cells. In addition to InsP3 production and cAMP accumulation, which are related to the activation of Gq/11 and Gs, respectively, we also evaluated on CHO-NTR-L cells the potency of EISAI-1 to increase [3H]arachidonic acid production and specific binding of [35S]GTPγS, two intracellular events that we previously found to be mediated through coupling of the NTS1 receptor to Gi/o (Gailly et al., 2000). As indicated in Fig. 4, EISAI-1 behaved as a full agonist of high potency in these two assays. Respective EC50 values for JMV449 and EISAI-1 were 0.23 ± 0.05 and 2.5 ± 0.5 nM for [3H]arachidonic acid production (Fig. 4A) and 3.8 ± 0.8 and 48.0 ± 7.6 nM for binding of [35S]GTPγS (Fig. 4B). EISAI-1 was thus around 10-fold less potent than JMV449 for [3H]arachidonic acid production and binding of [35S]GTPγS.

Concentration-response curves of JMV449 (□) and EISAI-1 (▾)on [3H]arachidonic acid production (A) or specific binding of [35S]GTPγS (B) in CHO-NTR-L cells. A, data represent means ± S.E.M. of three independent experiments, each performed in sextuplicate. The average of basal [3H]arachidonic acid production (performed in sextuplicate) has been subtracted and represented 0.15 to 0.40% of the incorporated radioactivity. B, data represent means ± S.E.M. of three independent experiments, each performed in duplicate. Basal [35S]GTPγS, taken as 100%, represented 15,000 to 18,000 dpm.
Effect of NT Agonists on Second Messenger Production in Primary Cultures of Rat Cortical Neurons. To know whether the low relative ability of EISAI-1 to induce InsP3 production in CHO cells revealed an intrinsic property of this compound on rat NTS1 receptor or merely reflected an artifactual observation generated by the overexpression of this receptor in heterologous cells, the responses to EISAI-1 and JMV449 were measured on the NTS1 receptor naturally expressed in primary cultures of rat cortical neurons (Lépée-Lorgeoux et al., 2000).
At variance with the data obtained with CHO cells, no significant increase in cAMP accumulation was observed in membranes prepared from cortical neurons upon stimulation by JMV449 at concentrations ranging from 10-10 to 10-6 M (not shown). In contrast, both JMV449 and EISAI-1 increased InsP3 production in this model, and the maximal effects of the two peptides were similar (Table 3). However, EISAI-1 was 120-fold less potent than JMV449 for InsP3 production in these cells. EISAI-1 behaved therefore as a full agonist of low relative potency for InsP3 production in rat cortical neurons.
Comparative effect of JMV449 and EISAI-1 on InsP3 production in primary cultures of rat cortical neurons
Inhibition constants (Ki) were determined from data obtained in competition experiments with 125I-NT (50 pM) by using the Cheng-Prusoff equation, with a Kd value for 125I-NT (obtained in saturation binding experiments) of 0.18 nM. Maximal binding capacity was 0.21 pmol of 125I-NT binding sites/mg protein. Potency (EC50) and maximal effect (Emax) of agonists on InsP3 production were determined from concentration-response curves as described under Materials and Methods. Values represent the means ± S.E.M. of three independent experiments.
As shown in Fig. 5, both JMV449 and EISAI-1 induced a moderate increase in [3H]arachidonic acid production in the primary cultures. Both peptides led to similar maximal levels (0.86 ± 0.06 and 0.88 ± 0.03% of incorporated radioactivity for 10-8 M JMV449 and 10-8 M EISAI-1, respectively). Although the amplitude of this stimulation was not sufficient to draw EC50 values with sufficient precision, values around 2 nM could be estimated for both agonists. EISAI-1 therefore acted as a full agonist of high relative potency for [3H] arachidonic acid production in these cultures.

Effect of JMV449 and EISAI-1 on [3H]arachidonic acid production in primary cultures of rat cortical neurons. Data represent means ± S.E.M. of three independent experiments, each performed in sextuplicate. **, p < 0.01 versus control without agonist (ANOVA followed by Dunnett's test).
These results indicate that the difference between relative potencies of EISAI-1 toward InsP3 and [3H]arachidonic acid production, observed with the NTS1 receptor expressed in CHO cells, is also obtained with the receptor endogenously expressed in rat cortical neurons.
Effect of Deleting the COOH Terminus of the NTS1 Receptor on cAMP Production in CHO Cells. Our results on InsP3 production suggested that EISAI-1 presented low intrinsic efficacy to activate coupling of the NTS1 receptor to Gq/11, which was previously shown to involve the third intracellular loop of the receptor (Yamada et al., 1994). However, EISAI-1 seemed to readily induce [3H]arachidonic acid production, [35S]GTPγS binding and cAMP accumulation, suggesting that this agonist efficiently activated both Gi/o and Gs. Because we recently demonstrated that coupling of the NTS1 receptor to Gi/o involved the C-terminal portion of this receptor (Najimi et al., 2002), we investigated whether this domain could also be involved in the coupling to Gs.
JMV449-mediated accumulation of cAMP was thus evaluated in cells expressing a truncated NTS1 receptor (termed NTRdel372 receptor) lacking the 52 amino acids that constitute its COOH-terminal intracellular portion. We previously reported that this receptor was still coupled to Gq/11, but not to Gi/o activation (Hermans et al., 1996; Najimi et al., 2002).
As shown in Fig. 6A, a complete lack of response on cAMP accumulation was obtained when JMV449 was tested on the NTRdel372 receptor, suggesting that the C-terminal portion of the rat NTS1 receptor was crucial for the coupling of this receptor to Gs. Control experiments showed that the lack of effect of JMV449 was not due to an impairment of adenylyl cyclase activity in the CHO cells expressing the NTRdel372 receptor, because 10-5 M forskolin induced similar increases in cAMP levels in membranes expressing the wild-type or the truncated NTS1 receptor (Fig. 6B).

A, concentration-response curves of JMV449 on cyclic AMP production in CHO-NTR-H cells expressing the wild-type NTS1 receptor (wt, □) or in CHO-NTRdel372 cells expressing the NTS1 receptor deleted of its C-terminal portion (del372, ▪); data represent means ± S.E.M. of three independent experiments, in which the average of basal value (performed in sextuplicate) has been subtracted and represented around 40 to 60 pmol of cAMP/mg of protein. B, cyclic AMP production by 10-5 M forskolin in CHO-NTR-H cells (wt) or in CHO-NTRdel372 cells (del372). Data represent one representative experiment, over three independent experiments, performed in sextuplicate. ***, p < 0.001 versus the corresponding control (Student's t test). C, inhibition constants (Ki) and effect of JMV449 and EISAI-1 on InsP3 production in CHO-NTRdel372 cells. Ki values were determined from data obtained in competition experiments with 125I-NT (50 pM) by using the Cheng-Prusoff equation, with a Kd value for 125I-NT (obtained in saturation binding experiments) of 0.23 nM. Maximal binding capacity was 0.35 pmol of 125I-NT binding sites/mg of protein. Potency (EC50) and maximal effect (Emax) of agonists on InsP3 production were determined from concentration-response curves as described under Materials and Methods. Data represent means ± S.E.M. of three to five independent experiments.
As presented in Fig. 6C, the discrepancy between the binding property of EISAI-1 and its potency to produce InsP3 was also found when this compound was tested on the NTRdel372 receptor. On this truncated receptor, the Ki value obtained for EISAI-1 was 4 times lower than that observed for JMV449. In contrast, in the InsP3 production assay, a 1,400-fold difference was found between the EC50 values obtained for the two agonists. Maximal effects of the two peptides were similar. Therefore, EISAI-1 acted as a full agonist of low relative potency for InsP3 production on CHO cells transfected with the truncated NTS1 receptor.
Discussion
In the present work, we first evidenced differential activation by agonists of signaling pathways associated to the NTS1 receptor. In particular, we demonstrated the occurrence of two phenomena that represent the most illustrative characteristics of agonist-directed trafficking of receptor stimulus: reverse agonist potency orders among pathways associated to a receptor and agonist-directed preferential activation of pathways usually considered as less efficiently coupled to this receptor.
As mentioned in the Introduction, two mechanisms could lead to differential activation of signaling pathways by agonists: strength of signaling and agonist-directed trafficking of receptor stimulus (Kenakin, 1995, 2001). The latter designation was sometimes used to describe phenomena that could indeed involve either of these mechanisms (Cussac et al., 2002; Manning, 2002). However, such a distinction is of fundamental importance not only at a theoretical level but also in terms of potential design of drugs with targeted pathway selectivity. Strength of signaling-based mechanism relies on stabilization of different amounts of a same active receptor state by agonists of various efficacies. In that case, only one kind of pathway-selective drug can be expected: low-efficacy agonists that induce only the most tightly coupled pathway. On the contrary, consistent with a probabilistic view of receptor function (Kenakin and Onaran, 2002), agonist-induced trafficking of receptor stimulus introduces the hypothesis of different agonist-selective receptor states that may favor activation of either of the pathways. In such a situation, tightness of coupling to G proteins or other transducing molecules is no longer a characteristic of the sole receptor but also depends on the agonist. Selective drugs may therefore be potentially evidenced toward either of the pathways.
Like reverse orders of agonist efficacies, reverse orders of potencies such as observed here between EISAI-1 and neuromedin N are indicative of pathway-dependent differences in ligand intrinsic efficacies. Such a phenomenon cannot be accounted for by simple strength of signaling-based mechanism. It was therefore suggested to represent one of the observations best supporting the hypothesis of multiple agonist-selective receptor states (Kenakin, 1996; 2001). Our results extend previous reports in that field (Spengler et al., 1993; Robb et al., 1994; Perez et al., 1996; Berg et al., 1998; Sagan et al., 1999; Brink et al., 2000; Wenzel-Seifert and Seifert, 2000) and suggest that EISAI-1 stabilizes receptor states that are functionally different from those stabilized by the other agonists.
Another point of interest was the pathways favored by EISAI-1. As illustrated with pituitary adenylate cyclase-activating polypeptide receptors (Spengler et al., 1993), agonists tested in previous studies still activated preferentially the pathway usually associated to the receptor. In contrast, in the present work, EISAI-1 clearly favored cAMP and [3H]arachidonic acid production versus InsP3 production. To our knowledge, this is the first observation of a drug preferentially activating pathways considered as being less efficiently coupled to a receptor, illustrating the potential interest of agonist-induced trafficking of receptor stimulus in the development of pathway-selective drugs.
The difference between relative potencies of EISAI-1 to induce InsP3 and [3H]arachidonic acid production was also observed in rat cortical neurons, suggesting that it did not arise from the overexpression of NTS1 receptor in CHO cells. The lack of agonist-induced cAMP accumulation and the lower [3H]arachidonic acid production observed in neuronal cultures are consistent with the preferential coupling of NTS1 receptor to Gq/11 in many natural systems (Hermans and Maloteaux, 1998; Vincent et al., 1999). This coupling selectivity could result, for instance, from the relative expression of the receptor and G proteins, lesser coupling efficiency to rat than to Chinese hamster GS or Gi/o, or masking of the C-terminal receptor tail by proteins absent from the CHO cells. However, activation of adenylyl cyclase by NT agonists was observed in some other cases such as prostate cancer cells (Ishizuka et al., 1993), indicating that it occurs not only in CHO cells but also in selected systems endogenously expressing NTS1 receptor.
The second aim of our study was to gain further insight on the functionally relevant differences between the active receptor states stabilized by EISAI-1 and by other agonists. In this respect, because G proteins interact with the receptor intracellular domains, functionally different receptor states should at least differ in the conformation or accessibility of one of these domains. Previous studies showed that the third intracellular loop of the NTS1 receptor was required for coupling to Gq/11-but not to Gs-type proteins (Yamada et al., 1994). We further demonstrated that the C-terminal portion of the NTS1 receptor was involved in coupling to Gi/o (Najimi et al., 2002), mediating both [3H]arachidonic acid release and binding of [35S]GTPγS. In the present study, we show that deletion of the receptor C-terminal domain suppresses agonist-induced cAMP accumulation, indicating that this domain is also essential for activation of Gs.
Therefore, unlike other agonists, EISAI-1 seems to discriminate between the pathways involving the C-terminal domain and that involving the third intracellular loop of the NTS1 receptor. Because other agonists efficiently activate all these pathways, it can be concluded that the corresponding receptor states present favorable conformations of both intracellular domains for efficacious activation of Gs, Gi/o, and Gq/11. Similarly, the high relative efficacy of EISAI-1 toward cAMP accumulation, [3H]arachidonic acid release and binding of [35S]GTPγS indicates that the receptor states stabilized by this peptide present a conformation of the C-terminal domain suitable for activation of Gs and Gi/o. One of the hypotheses to explain the lower relative potency and efficacy of EISAI-1 toward InsP3 production could therefore be that these receptor states present a conformation or accessibility of the third intracellular loop less favorable for activation of Gq/11 than those obtained with the other agonists.
Such a differential involvement of intracellular domains in coupling to distinct G proteins, as seen here with the NTS1 receptor, might underlie the possibility of discrimination among the corresponding pathways by agonists. Indeed, there is no a priori reason that the complexes formed between a receptor and different agonists should present conformations of distinct receptor domains leading to parallel extents of activation of the corresponding G proteins. Separation of parameters at the level of the receptor molecule therefore gives the best potential to find agonist-selective patterns of response. However, one cannot exclude that recruitment of different G proteins interacting with the same receptor domain might also be differentially altered by agonists. For instance, some different conformational requirements of the interactions between the receptor COOH terminus and Gi/o or Gs could further open the possibility of agonist selectivity toward one of these G proteins.
The last point addressed in our study concerned the determinants of the EISAI-1 molecule underlying its discriminative properties. Both EISAI compounds differ from the original C-terminal hexapeptide of NT by methylation of their N-terminal end and three amino acid substitutions: Arg8-to-Lys, Tyr11-to-Trp, and Ile12-to-tert-Leu (Table 1). These modifications alone cannot account for the lower ability of EISAI-1 to induce InsP3 production, because EISAI-2 retains a normal behavior in this test. The functional characteristics of EISAI-1 may thus be brought by functionalization of its COOH end by an ethyl group.
Interestingly, recent data (Barroso et al., 2000) indicated that the free carboxyl group of the NT C-terminal end interacted with Arg327 of the NTS1 receptor. This residue is located in the receptor sixth transmembrane domain, connected to the third intracellular loop. Disruption of this interaction through amidation of the NT carboxyl group or mutation of the Arg327 residue led to an impairment of the NT-induced InsP3 production, suggesting that this interaction played a major role in positioning the neighboring third intracellular loop for an efficacious coupling to Gq/11 (Barroso et al., 2000). The lower efficacy to stimulate InsP3 production brought by the esterification of the carboxyl group in EISAI-1 further supports this hypothesis. Our data also indicate that the loss of this interaction does not alter coupling to Gi/o and Gs, mediated by the more distal C-terminal domain of the receptor. Evaluation of the binding and functional characteristics of EISAI-1 and EISAI-2 on the Arg327 receptor mutant expressed in CHO cells would be of particular importance to gain more information on the molecular correlates of the differential properties of these agonists.
Together, these results provide further insights on the mechanisms underlying agonist-induced trafficking of receptor stimulus, at the level of both the receptor and the ligand. They also illustrate the possibility to consider pathway-dependent selectivity in the search of new therapeutic agents. In this respect, it should be noticed that NT was shown to have a dual action on the proliferation of prostate cancer cells, inducing proliferation through InsP3 production and reducing proliferation through cAMP production (Ishizuka et al., 1993). Due to overexpression of the NTS1 receptor in several types of cancer, NT-related ligands were recently developed for tumor targeting (Hillairet de Boisferon et al., 2002). Therefore, development of cAMP-selective NT agonists endowed with preferential antimitogenic properties would provide valuable tools in that field.
Acknowledgments
We thank Dr. Neil Insdorf for precious help in the writing of the manuscript and fruitful discussions. We also thank Dr. P. Kitabgi for fruitful discussions, and V. Raphael, S. Millerioux, and C. Binet for skillful technical assistance. We are grateful to Eisai Company for the gift of EISAI-1 hexapeptide.
Footnotes
-
This research was supported by Institut National de la Santé et de la Recherche Médicale (INSERM) and the National Fund for Scientific Research (Belgium). D.S. was supported by Poste d'Accueil INSERM pour Ingénieur des Grandes Ecoles.
-
ABBREVIATIONS: GPCR, G protein-coupled receptor; NTS1, neurotensin receptor 1; NT, neurotensin; NN, neuromedin N; InsP3, inositol 1,4,5-trisphosphate; CHO, Chinese hamster ovary; GTPγS, guanosine 5′-O-(3-thio)triphosphate; PBS, phosphate-buffered saline; BSA, bovine serum albumin; ANOVA, analysis of variance.
-
↵1 Current address: Department of Pharmacology, School of Pharmaceutical Sciences, Showa University, 1-5-8 Hatanodai, Shinagawa, Tokyo 142-8555, Japan.
- Received December 16, 2002.
- Accepted April 30, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}