


Abstract
Loss of response on repetitive drug exposure (i.e., tachyphylaxis) is a particular problem for the vasoconstrictor effects of medications containing oxymetazoline (OXY), an α1-adrenoceptor (AR) agonist of the imidazoline class. One cause of tachyphylaxis is receptor desensitization, usually accompanied by phosphorylation and internalization. It is well established that α1A-ARs are less phosphorylated, desensitized, and internalized on exposure to the phenethylamines norepinephrine (NE), epinephrine, or phenylephrine (PE) than are the α1B and α1D subtypes. However, here we show in human embryonic kidney-293 cells that the low-efficacy agonist OXY induces G protein–coupled receptor kinase 2–dependent α1A-AR phosphorylation, followed by rapid desensitization and internalization (∼40% internalization after 5 minutes of stimulation), whereas phosphorylation of α1A-ARs exposed to NE depends to a large extent on protein kinase C activity and is not followed by desensitization, and the receptors undergo delayed internalization (∼35% after 60 minutes of stimulation). Native α1A-ARs from rat tail artery and vas deferens are also desensitized by OXY, but not by NE or PE, indicating that this property of OXY is not limited to recombinant receptors expressed in cell systems. The results of the present study are clearly indicative of agonist-directed α1A-AR regulation. OXY shows functional selectivity relative to NE and PE at α1A-ARs, leading to significant receptor desensitization and internalization, which is important in view of the therapeutic vasoconstrictor effects of this drug and the varied biologic process regulated by α1A-ARs.
Introduction
The neurotransmitter in the central and peripheral (sympathetic) nervous system norepinephrine (NE) and the adrenal gland hormone epinephrine (EPI) regulate important biologic processes through activation of α1-adrenoceptors (α1-ARs), including behavioral responses (Doze et al., 2011; Berridge et al., 2012), male reproductive performance (Sanbe et al., 2007; de Almeida Kiguti and Pupo, 2012), and contraction of vascular and nonvascular smooth muscles (Docherty, 2010). The products encoded by the three α1-AR genes are named α1A-, α1B-, and α1D-ARs. All three α1-ARs are seven-transmembrane domain receptors that activate Gq/11 heterotrimeric proteins, leading to stimulation of phospholipase C, inositol, and diacylglycerol production and increases in cytosolic Ca2+ concentrations (Chen and Minneman, 2005).
The relatively large array of drugs that target α1-ARs either mimic (agonists) or inhibit (antagonists) the effects of the endogenous catecholamines NE and EPI and are part of the therapeutic arsenal available for the relief of hypertension, prostatic hyperplasia, and shock. In addition to reverting hypotension in shock, α1-AR agonists are vasoconstrictors when locally applied to the nasal mucous membrane or the eye and are also components of over-the-counter oral medications for the common cold and influenza.
Repeated exposure to an agonist may lead to diminished responses, a process known as tachyphylaxis [as defined in Neubig et al. (2003)]. One cause of tachyphylaxis is receptor desensitization, usually accompanied by receptor phosphorylation and internalization. Tachyphylaxis is a particular problem for vasoconstrictors and nasal decongestants containing imidazoline α1-AR agonists such as oxymetazoline (OXY), naphazoline, and xylomethazoline, and the rebound hyperemia and rhinitis medicamentosa observed with some of these drugs has been suggested to be linked to α1-AR desensitization (Vaidyanathan et al., 2010). The α1-ARs are known to be subject to phosphorylation and internalization on exposure to NE, EPI, or phenylephrine (PE). However, there are substantial differences in the degree of phosphorylation, internalization, and desensitization among the α1-AR subtypes. For instance, α1Β-ARs are phosphorylated to a greater extent in response to either NE or tetradecanoyl phorbol acetate [TPA, a protein kinase C activator (PKC)] than α1A-ARs (Vazquez-Prado et al., 2000; Garcia-Sainz et al., 2004; Cabrera-Wrooman et al., 2010). Also, α1A-ARs are less internalized on exposure to NE, EPI, or PE than are the α1Β- or α1D-ARs (Chalothorn et al., 2002; Wang et al., 2007; Stanasila et al., 2008; Cabrera-Wrooman et al., 2010). When compared in the same cellular background, recombinant α1A-ARs are much more resistant to desensitization induced by NE or TPA than are α1Β- and α1D-ARs (Vazquez-Prado and Garcia-Sainz, 1996; Vazquez-Prado et al., 2000; Cabrera-Wrooman et al., 2010). Therefore, as far as receptor phosphorylation, desensitization, and internalization are concerned, the view that the α1A-ARs are less tightly regulated than the other two subtypes is acceptable [reviewed in Cotecchia (2010)]. In this context, the tachyphylaxis observed for the vasoconstrictor effects of medications containing imidazoline agonists is intriguing because these drugs are often selective ligands for α1A-ARs (Minneman et al., 1994) given that this receptor subtype is much less susceptible to phosphorylation, desensitization, and internalization than are the α1Β- and α1D-ARs.
The present investigation extends early studies describing tachyphylaxis in responses of rat smooth muscle tissues to the α1A-AR–selective and low efficacy agonist OXY (Ruffolo et al., 1977; Rice et al., 1991) and shows in human embryonic kidney (HEK-293) cells that OXY induces rapid G protein–coupled receptor kinase 2 (GRK2)-dependent phosphorylation, desensitization, and internalization of human recombinant α1A-ARs, whereas NE caused phosphorylation of receptors that is largely dependent on PKC and is not followed by rapid desensitization and internalization. In addition to shedding light on the molecular mechanisms of the tachyphylaxis of the vasopressor and nasal decongestant effects of OXY and the resulting rebound hyperemia and rhinitis medicamentosa, these findings show clear evidence of distinct agonist-directed receptor phosphorylation, desensitization, and internalization mechanisms. These findings are important in view of the varied biologic functions regulated by these receptors.
Materials and Methods
Experiments in Rat Smooth Muscle Tissues.
The experimental procedures were approved by the Ethics Committee for the Use of Experimental Animals from Universidade Estadual Paulista (UNESP)—Botucatu and are in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Male Wistar rats (16–20 weeks old, 260–380 g, provided by the Central Bioterium from UNESP, Botucatu, Brazil) were killed by decapitation, and selected tissues were excised and prepared for digital recording of isometric contractions as follows: the vas deferens (epididymal portion) and tail artery (distal segments) were cleaned of adherent tissues and mounted in organ baths under 9.8 mN (vas deferens) or 14.7 mN (tail artery) tension in a nutrient solution with the following composition (mM): NaCl 138, KCl 5.7, CaCl2 1.8, NaH2PO4 0.36, NaHCO3 15, dextrose 5.5 (for vas deferens); NaCl 119, KCl 4.7, CaCl2 2.5, KH2PO4 1.2, MgSO4 1.2, NaHCO3 25, dextrose 5.5 (tail artery) prepared in glass-distilled, deionized water, maintained at 30°C (vas deferens) or 37°C (tail artery), pH 7.4, and continuously bubbled with 95%O2/5%CO2. All experiments were done in the presence of a cocktail of inhibitors containing cocaine (6 μM), corticosterone (10 μM), idazoxan (1 μM), and propranolol (0.1 μM) to block neuronal uptake, extraneuronal uptake, and α2- and β-adrenergic receptors, respectively. After an equilibration period of 60 minutes with adjustments of basal tension and changes of physiologic solution at each 20 minutes, the tissues were repeatedly challenged with KCl 80 mM at 20-minute intervals until contractions of similar magnitude were obtained. Then consecutive concentration-response curves to the agonists in the vas deferens (NE or OXY) and tail artery (PE or OXY) were obtained with 45-minute intervals between each one. In some experiments, the vas deferens and tail artery were treated with 10 µM OXY or 10 µM NE for 5 minutes, extensively washed, and 45 minutes later, a concentration-response curve to PE or NE was constructed. PE was chosen as an agonist in the tail artery because there is a significant participation of α2-ARs in the contractions induced by NE in this vessel (Kamikihara et al., 2005). Curve fitting and pEC50 calculation was performed with the software package GraphPad Prism (version 4.00; GraphPad Software, San Diego, CA). All values are shown as means ± S.E.M. of n experiments. Differences between mean values were tested for statistical significance (P < 0.05) using Student’s paired or unpaired t tests.
Cell Culture and Transfections.
HEK-293 cells were propagated in 100-mm dishes in Dulbecco’s modified Eagle’s medium with sodium pyruvate, supplemented with 10% heat-inactivated fetal bovine serum, 10 mg/ml of streptomycin, and 100 U/ml of penicillin in a humidified atmosphere with 5% CO2 at 37°C. Confluent plates were subcultured at a ratio of 1:3. Cells were transfected with 15 µg of cDNA of the mammalian expression plasmid pDT containing N-terminal sequential hexahistidine and FLAG epitope-tagged human α1A (α1A-1 splice variant), α1B, or N-terminal truncated mutant of α1D-ARs (Δ1-79α1D-AR) by Lipofectamine (Invitrogen, Carlsbad, CA), and when specified, stably transfected cells were selected with Geneticin (400 µg/ml). The constructs were generously provided by Dr. K. P. Minneman and are described elsewhere (Vicentic et al., 2002; Pupo et al., 2003; Hague et al., 2004). In addition, HEK-293 cells were transfected with 20 µg of cDNA of the expression plasmid pcDNA1.1 encoding the sequence for a kinase-deficient mutant of bovine GRK2 in which lysine 220 was replaced by a methionine [C20-GRK2-K220M3, described in Ferguson et al. (1995)], generously donated by Dr. Maria de Fatima M. Lazari, from UNIFESP, and stably transfected cells were selected with Geneticin (400 µg/ml) and designated DNGRK2 cells.
[3H]Prazosin Binding in Membrane Preparations from HEK-293 Cells.
For radioligand binding measurements, confluent 100-mm plates were washed with phosphate-buffered saline (PBS; 20 mM NaPO4, 154 mM NaCl, pH 7.6), and cells were harvested by scraping. HEK-293 cells expressing recombinant human α1A-, α1B-, or Δ1-79α1D-adrenoceptors were collected by centrifugation and homogenized with a Polytron; membranes were collected by centrifugation at 30,000g for 20 minutes and resuspended in 1× buffer A (25 mM HEPES, 150 mM NaCl, pH 7.4). Radioligand binding sites were measured by saturation analysis of specific binding of the α1-adrenoceptor radioligand [3H]prazosin (20–2000 pM). Nonspecific binding was defined as binding in the presence of 100 μM phentolamine. The pharmacological specificity of radioligand binding sites was determined by displacement of [3H]prazosin (350 pM) by selected ligands, and data were analyzed by nonlinear regression analysis using the software package GraphPad Prism (version 4.00).
[3H]Prazosin Binding in Intact HEK-293 Cells.
HEK-293 cells growing in 100-mm plates were transiently transfected with the expression plasmid pDT containing N-terminal sequential hexahistidine and FLAG epitope-tagged α1A-AR and 24 hours later were transferred to poly-D-lysine treated 24-well microplates (100,000 cells/well). Approximately 48 hours post transfection, the cells were incubated with 10 µM NE or 10 µM OXY for different times as indicated at 37°C in Dulbecco’s modified Eagle’s medium or with increasing concentrations of NE and OXY for 30 minutes and then washed twice with ice cold PBS (1.0 ml, pH 7.2). After washing, the cells were incubated with 1 nM [3H]prazosin (80 Ci/mmole; Amersham Biosciences, Sunnyvale, CA) at 4°C in PBS to reduce the partioning of the radioligand for 10–12 hours (Leeb-Lundberg et al., 1987). Radioligand nonspecific binding was defined in presence of 100 µM phentolamine or 100 µM EPI. After the incubation with the radioligand, the cells were washed twice with 500 µl of ice-cold PBS containing 0.1% bovine serum albumin and then scrapped in 500 µl of MilliQ water. To each sample, 4 ml of scintillation cocktail was added (OptiPhase HiSafe 3; PerkinElmer, Waltham, MA), and 3H was counted in a liquid scintillation analyzer (1900 TR; PACKARD, Canberra, ACT, Australia).
Intracellular Ca2+ Measurements.
For intracellular Ca2+ (iCa2+) mobilization assays, HEK-293 cells expressing recombinant human α1A-ARs were cultivated overnight in black-walled, clear-bottom 96-well microplates covered with poly-D-lysine (30,000 cells/well). The next day, 100 μl of a modified Hanks’ balanced salt solution (HBSS; 137 mM NaCl, 5.36 mM KCl, 1.18 mM, 1.26 mM CaCl2·2H2O, 0.49 MgCl2·6H2O, 0.41 MgSO4.7H2O, 0.44 KH2PO4, 4.17 NaHCO3, 0.35 NaHPO4, 5.5 mM D-glucose, 20 mM HEPES, and 2.5 mM probenecid, pH 7.4) containing the fluorescent Ca2+ indicator FLUO-4 NW (0.1% v/v; Invitrogen) was added to each well according to the manufacturer’s instructions for 1 hour at 37°C. Before loading the fluorescent Ca2+ indicator, some wells were incubated with NE or OXY (both at 10 µM for 5 minutes), carefully washed at least three times with 200 µl of PBS (without Ca2+ and Mg2+), and then incubated with 100 μl of modified HBSS containing the fluorescent indicator. After 1 hour of incubation with the fluorescent indicator, 90 μl of modified HBSS (without probenecid) was added to each well, and basal fluorescence (excitation 485 nm, filter 485/20 nm, emission 525 nm, filter 516/20 nm) was measured in a microplate reader (Synergy 4; BioTek, Winooski, VT) every 2 seconds for 10 seconds. For the construction of concentration-response curves, 10 μl of assay buffer containing increasing concentrations of NE or OXY (final concentration in the well from 0.1 nM to 3 µM) was added through onboard dispensers at minimal speed (225 μl.s−1), and the effect on fluorescence was recorded every 2 seconds for 50 seconds. Data were collected online in a microcomputer, analyzed by the software Gen5 (Biotek), and the ratios between the peak minus basal fluorescence were taken and expressed as percentage of the maximal increase induced by NE 10 µM. The fittings for the concentration-response curves were calculated using the software package GraphPad Prism (version 4.00). In some experiments, cells were seeded in a six-well plate (106 cells/well) and at least 12 hours later incubated with Fura-2 AM (5 μM) for 1 hour at 37°C in a Krebs-Ringer HEPES buffer supplemented with 0.05% bovine serum albumin. After loading the calcium indicator, the cells were trypsinized, washed, and resuspended in 2 ml of the above-mentioned buffer supplemented with 1.2 mM CaCl2. Fluorescence readings were taken in an Aminco-Bowman Series 2 spectrometer (SLM Instruments Inc., Urbana, IL) with the excitation monochromator set at 340 and 380 nm and emission monochromator set at 510 nm (Fura2-AM).
Confocal Microscopy of HEK-293 Cells.
HEK-293 cells transiently transfected with the p enhanced green fluorescent protein (eGFP)-N3 plasmid encoding recombinant human α1A-ARs fused at the C-terminus with enhanced green fluorescent protein [α1A/eGFP, described in Hague et al. (2004) and kindly provided by Dr. Chris Hague from University of Washington, Seattle, WA] were cultivated in quartz coverslips covered with poly-d-lysine for 24 to 36 hours and imaged in a laser scanning confocal microscope (LSM510 META; Carl Zeiss, Jena, Germany) with Plan-Neofluar 40× oil immersion objective lens (Leica, Wetzlar, Germany) before and during the treatment with 10 µM NE or 10 µM OXY for 30 minutes at 37°C. In some experiments, the cells in the coverslips were treated with 30 nM prazosin 30 minutes before the incubation of 10 µM OXY. eGFP fluorescence was excited using an argon laser at a wavelength of 488 nm, and the emitted fluorescence was detected at 505 nm. Changes in fluorescence intensity were estimated using Image J 1.38x software (NIH, Bethesda, MD; http://rsb.info.nih.gov/ij/) in areas just below the cell surface before (basal) and during the incubation with drugs. Data were normalized to a percentage of the fluorescence obtained before agonist treatment, and the increases in fluorescence intensity above that observed before drug treatments were taken as measures of receptor internalization.
Phosphorylation of α1A-ARs.
HEK-293 cells transiently transfected with the peGFP-N3 plasmid encoding recombinant human α1A-ARs fused at the C-terminus with enhanced green fluorescent protein eGFP (α1A/eGFP) were serum-starved for 24 hours, maintained in phosphate-free Dulbecco’s modified Eagle’s medium for 1 hour, and then incubated in 1 ml of the same medium containing [32P]Pi (150 μCi/ml) for 3–5 hours at 37°C, as previously described (Vazquez-Prado et al., 2000). Labeled cells were stimulated as indicated and at the end of the incubation washed with ice-cold phosphate-buffered saline and solubilized with 1.0 ml of ice-cold solubilization buffer containing the following: 100 mM NaCl, Tris 10 mM, Triton X-100 0.1% (Sigma-Aldrich Brasil Ltda., São Paulo, Brazil), Nonidet P40 0.1%, SDS 0.05%, 10 mM NaF, 1 mM Na3VO4, 10 mM β-glycerophosphate, 10 mM sodium pyrophosphate, 1 mM p-serine, 1 mM p-threonine, and 1 mM p-tyrosine. The plates were maintained on ice for 1 hour. The extracts were centrifuged at 12,700g for 15 minutes at 4°C, and the supernatants were collected. Receptors were immunoprecipitated using rabbit antibodies generated in our laboratory and directed against eGFP, as described (Avendano-Vazquez et al., 2005; Cabrera-Wrooman et al., 2010). Receptor phosphorylation was detected with a Molecular Dynamics PhosphorImager and quantified with ImageQuant software (Amersham Biosciences). Data fell within the apparatus’s linear detection range and were plotted using Prism 4.0 (GraphPad Software).
Drugs.
Drugs were obtained from the following sources: bisindolylmaleimide I (3-(N-[dimethylamino]propyl-3-indolyl)-4-(3-indolyl)maleimide, 3-[1-[3-(dimethylamino)propyl]1H-indol-3-yl]-4-(1H-indol-3-yl)1H-pyrrole-2,5dione) and rottlerin (1-[6-[(3-acetyl-2,4,6-trihydroxy-5-methylphenyl)methyl]-5,7-dihydroxy-2,2-dimethyl-2H-1-benzopyran-8-yl]-3-phenyl-2-propen-1-one) (Calbiochem, La Jolla, CA); corticosterone, hispidin (6-(3,4-dihydroxystyrl)-4-hydroxy-2-pyrone), L-norepinephrine bitartrate salt, oxymetazoline HCl, phenylephrine HCl, and oxymetazoline HCl from Sigma-Aldrich Brasil Ltda.; cocaine (Cocainum hydrochloricum puriss) C. H. Boehringer (Germany); FURA2-AM and fluo-4 NW (Invitrogen); BMY-7378 (8-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-8-azaspiro[4.5]decane-7,9-dione dihydrochloride), 5-methylurapidil HCl, prazosin HCl, (±)-propranolol HCl, and idazoxan HCl from Research Biochemicals Inc., Natick, MA; [3H]prazosin (PerkinElmer); Protein A_Agarose (Upstate Biotechnology, Lake Placid, NY); nitrocellulose membranes (Bio-Rad, Hercules, CA); 32P[Pi] (8500-9120 Ci/mmol; PerkinElmer); chemiluminiscence kits (Pierce, Rockford, IL).
Results
Tachyphylaxis in Contractions of the Rat Tail Artery and Vas Deferens Induced by OXY.
The contractions of the rat vas deferens to NE (Pupo, 1998) and rat tail artery to PE (Kamikihara et al., 2005) under conditions of α2- and β-ARs blockade are mediated by the activation of α1A-ARs. At least three consecutive concentration-response curves to PE in the rat tail artery (Fig. 1A) or NE in the vas deferens (Fig. 1B) produced similar maximal effects (Emax) and pEC50 values (Table 1), showing no evidence for tachyphylaxis in the contractions induced by these agonists under the conditions investigated. OXY behaved as a partial agonist in the rat tail artery (Fig. 1C) and vas deferens (Fig. 1D) in relation to PE and NE, respectively. However, in contrast to the consecutive concentration-response curves for NE and PE, there was intense tachyphylaxis to contractions induced by OXY in the tail artery (Fig. 1C) as reflected by about a 30-fold rightward shift and about a 45% reduction in Emax (Table 1) and also in the vas deferens, where OXY failed to contract the tissue upon a second or third agonist exposure (Fig. 1D).

Consecutive concentration-response curves for contraction of the rat tail artery to PE (A) and OXY (C) and vas deferens to NE (B) and OXY (D). The interval between the consecutive concentration-response curves (CRC) was 45 minutes. Values are means ± S.E.M. of four to six independent experiments performed with tissues from different rats.
Maximal contractions (g of tension) and pEC50 values of consecutive concentration-response curves for NE, PE, and OXY in the rat tail artery and vas deferens
One-way analysis of variance followed by Dunnett’s test indicates differences from the respective value determined in the first concentration-response curve.
To check whether OXY induced tachyphylaxis in the contractions of the rat tail artery and vas deferens in response to PE and NE, the tissues were incubated with OXY (10 µM) for 5 minutes, extensively washed, and after a 45-minute recovery interval exposed to PE or NE. In rat tail artery (Fig. 2A) and vas deferens (Fig. 2B) pre-exposed to OXY (10 µM/5 minutes), PE and NE were 20-fold less potent than in time controls that had been treated with vehicle (Table 2). OXY (10 µM/5 minutes) was unable to induce tachyphylaxis to PE or NE if the treatment was performed in the presence of prazosin (30 nM), indicating that the effect depends on α1-ARs activation (unpublished data).

Concentration-response curves for the contraction of the rat tail artery (A) and vas deferens (B) in response to PE and NE, respectively, 45 minutes after treatment of the tissues with 10 µM OXY for 5 minutes or vehicle (matched control). Values are means ± S.E.M. of four independent experiments performed with tissues from different rats.
Maximal contractions (g of tension) and pEC50 values of concentration-response curves for PE and NE 45 minutes after treatment of tissues with 10 µM OXY for 5 minutes or with vehicle
Binding Characteristics of α1A-ARs Expressed in HEK-293 Cells.
Saturation binding characteristics of [3H]prazosin to membrane preparations from HEK-293 cells stably transfected with α1A-ARs revealed a maximum binding capacity (Bmax) of 2042 ± 158 fmol/mg of protein and an equilibrium dissociation constant (KD) of 371 ± 83 pM (mean ± S.E.M. of four separate experiments). There was no specific binding of [3H]prazosin to membrane preparations from nontransfected or mock-transfected (empty pDT vector) HEK-293 cells (unpublished data). Competition binding experiments were carried out using OXY (α1A-selective ligand), 5-methyl urapidil (α1A-selective), and BMY7378 (α1D-selective) in membrane preparations from HEK-293 cells expressing human α1A, α1Β, and an N-terminal truncated mutant of the human α1D-ARs (Δ1-79α1D-AR, which traffics efficiently to the cell membrane) (Pupo et al., 2003; Hague et al., 2004; Nojimoto et al., 2010). All the ligands competed for the binding of [3H]prazosin, with the curves fitting a single-site isotherm. As expected, 5-methyl urapidil [-log equilibrium dissociation constant (pKi)]: α1A = 8.9 ± 0.2; α1B = 7.1 ± 0.2 ; Δ1-79α1D = 7.7 ± 0.2, n = 4) and OXY (pKi: α1A = 6.8 ± 0.2; α1B = 5.2 ± 0.1; Δ1-79α1D = 5.5 ± 0.1, n = 3 to 4) inhibited the binding of [3H]prazosin with high relative affinities consistent with an interaction with α1A ARs, whereas BMY7378 (pKi: α1A = 7.0 ± 0.1 ; α1B = 6.8 ± 0.2; Δ1-79α1D = 8.6 ± 0.1) showed higher affinity in membrane preparations from cells expressing Δ1-79α1D-ARs. Note that OXY was ∼20- to 40-fold more potent at α1A-ARs than at the other two receptors.
Tachyphylaxis in HEK-293 Cells Expressing α1A ARs on Responses to NE after Pretreatment with OXY.
OXY increased iCa2+ concentrations in HEK-293 cells stably transfected with human recombinant α1A ARs and was a partial agonist relative to NE (Fig. 3A; Table 3). There was intense tachyphylaxis in the iCa2+ response to NE in HEK-293 cells expressing human α1A-AR that were pretreated with OXY (10 μM/5 minnutes), but not in cells pretreated with NE (10 μM/5 minute) (Fig. 3B; Table 4). OXY did not induce tachyphylaxis in the iCa2+ increases induced by carbachol through activation of endogenous muscarinic receptors, suggesting that the desensitization is specific for α1A-ARs (results not shown).

(A) Concentration-response curves for iCa2+ increases induced by NE and OXY in HEK-293 cells stably expressing α1A-ARs and loaded with the Ca2+ indicator Fluo-4 at 37°C. (B) Concentration-response curves for NE in cells treated with 10 µM NE or 10 µM OXY for 5 minutes at 37°C, washed twice, and then loaded with Fluo-4. Real-time fluorescence was recorded every 2 seconds, with agonist additions after 10 seconds. Responses represent the difference between basal and peak fluorescence and are expressed as a percentage of the response to 10 µM NE. Values are means ± S.E.M. of four independent experiments performed in duplicate.
Parameters of agonism (Emax and pEC50) for NE and OXY in the increase of intracellular Ca2+ in HEK-293 cells expressing α1A-ARs
Parameters of agonism (Emax and pEC50) for NE in the increase of intracellular Ca2+ in HEK-293 cells expressing α1A-ARs treated with vehicle, 10 µM NE/5 minutes, or 10 µM OXY/5 minutes
One-way analysis of variance followed by Dunnett’s test indicates differences from the respective value determined in cells treated with vehicle.
Rapid Internalization of α1-ARs Activated by OXY.
The localization of α1A-ARs in HEK-293 cells in the absence and presence of NE or OXY was assessed by confocal microscopy of cells expressing α1A ARs tagged with eGFP at the C-terminus (α1A/eGFP, Fig. 4). In nonstimulated cells, the α1A/eGFP were localized mainly at the plasma membrane (time 0` in Fig. 4). OXY (10 µM) induced a loss of fluorescence at the plasma membrane, which was already measurable after 10 minutes of exposure (Fig. 4, A and B). In contrast, loss of fluorescence in the plasma membrane of cells treated with NE (10 µM) was modest and observed ony after 60 minutes of exposure (unpublished data). Treatment of HEK-293 cells expressing α1A/eGFP with prazosin (30 nM/30 minutes) inhibited the loss of fluorescence in the plasma membrane induced by OXY (10 µM), indicating that this effect depends on α1A/eGFP activation (Fig. 4).

Localization of the α1A/eGFP monitored by confocal microscopy in HEK-293 cells in the absence (time 0`) and in the presence of 10 µM N, 10 µM OXY, or 30 nM prazosin plus 10 µM OXY at 37°C for 30 minutes (A). The images were taken at different times and are representative of three to four independent transfections. Changes in intracellular fluorescence intensity in cells treated with NE or OXY and in cells treated with OXY in presence of prazosin (B). Data represent the mean ± S.E.M. of three to four independent transfections. Two-way analysis of variance with Bonferroni post-test indicates differences from the effect of NE in the respective time (*P < 0.05; **P < 0.01;***P < 0.001) and differences from the effect of OXY plus 30 nM prazosin in the respective time (#P < 0.05; ##P < 0.01).
The efficacies of OXY and NE in internalizing α1A ARs (N-terminally FLAG-tagged receptors) were further compared in HEK-293 cells transiently expressing the receptors and treated with these agonists for various times, washed twice with PBS, and incubated with 1 nM [3H]prazosin for 10 to 12 hours at 4°C (to reduce the partitioning of the radioligand) to quantify receptors at the cell surface. Figure 5A shows the total and nonspecific binding of [3H]prazosin defined in presence of 100 µM phentolamine (lipophilic ligand) or 100 µM EPI (hydrophilic ligand). In these experiments, phentolamine and EPI revealed similar amounts of nonspecific binding, indicating that under these experimental conditions, the radioligand labels cell surface receptors. The time course of α1A-AR internalization following NE (10 µM) or OXY (10 µM) addition is shown in Fig. 5B. OXY, but not NE, induced rapid loss of cell surface binding detectable within 5 minutes of stimulation and maximal after about 30 minutes. On the other hand, loss of cell surface binding following the addition of NE was detected only after at least 45 minutes of stimulation. A full concentration-response curve for the effect of NE and OXY on cell surface binding after 30 minutes of stimulation is shown in Fig. 5; within 30 minutes of stimulation, NE up to 10 μM was unable to reduce cell surface α1A-ARs (n = 3), whereas OXY internalized about 50% of the receptors with potency (pEC50) of 6.9 ± 0.2 (n = 3). To determine whether the reduction of the binding of [3H]prazosin in OXY-treated cells was due to incomplete washing of the agonist, cells were treated with OXY (10 µM for 5 minutes), washed, and then incubated with increasing concentrations of [3H]prazosin at 4°C for 10 hours (Fig. 5D); saturation analysis of [3H]prazosin binding showed that OXY treatment (10 µM for 5 minutes) reduced the Bmax (248 ± 25 versus 183±13 fmol/well, P < 0.05 in Student’s t test; n = 3) without changing affinity (KD = 294 ± 50 versus 360 ± 69 pM, n = 3). As expected, treatment with NE (10 µM for 5 minutes) had no effect on [3H]prazosin binding (Bmax = 255 ± 14 fmol/well and KD = 286 ± 47 pM, n = 3; P > 0.05 compared with nonstimulated cells). The lack of reduction in affinity indicates that OXY was readily washed out and that the reduction of [3H]prazosin binding was due to loss of binding sites (α1A-ARs) and not related to “competition” resulting from incomplete removal of the agonist.

[3H]Prazosin binding in intact HEK-293 cells transiently transfected with α1A-ARs to access cell surface receptors. (A) The total and nonspecific binding of [3H]prazosin displaced by 100 µM epinephrine or 100 µM phentolamine in intact HEK-293 cells to check for the labeling of cell surface receptors. (B) Cells were treated with either 10 µM NE or 10 µM OXY for various periods of time at 37°C, and the receptor populations at the cell surface were determined by [3H]prazosin specific binding at 4°C for 10 hours and compared with the specific binding in cells not treated with the agonists (time 0`). (C) Concentration-response curves for NE and OXY for the loss of cell surface [3H]prazosin specific binding after 30 minutes of stimulation with the agonists. (D) The saturation of specific binding of [3H]prazosin (defined by 100 µM phentolamine) in intact HEK-293 cells previously treated with either 10 µM NE or 10 µM OXY for 5 minutes and washed twice at the end of the treatment. Data represent the mean ± S.E.M. of three to four independent experiments performed in duplicate from different transfections. (A) Student’s t test indicates that the nonspecific binding of [3H]prazosin in presence of epinephrine is not different from that in presence of phentolamine. Two-way analysis of variance with Bonferroni post-test indicates differences from the effect of NE in the respective time (**P < 0.01;***P < 0.001).
Phosphorylation of α1A-AR after Activation by NE and OXY.
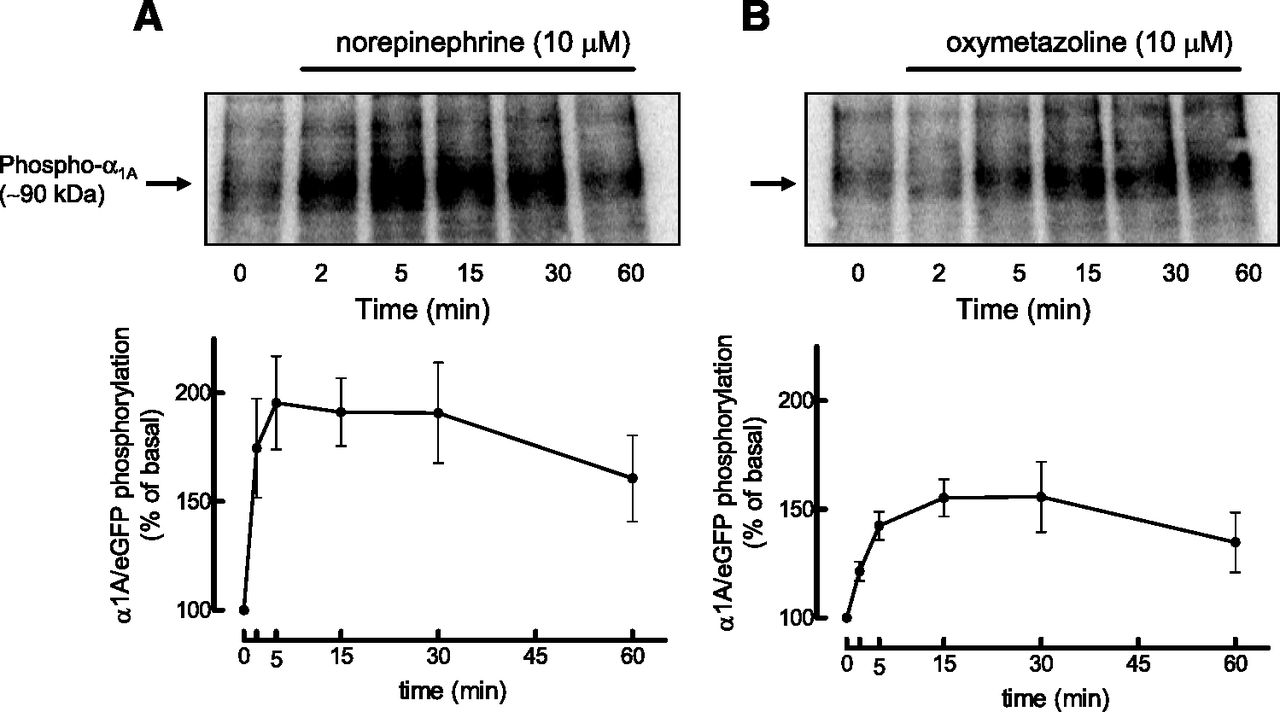
The phosphorylation of α1A/eGFP activated by NE or OXY was determined in HEK-293 cells transiently transfected with the receptor and metabolically labeled with inorganic phosphate ([32P]Pi); cells were treated for various times with the agonists and the α1A/eGFP-ARs were immunoprecipitated with an antiserum against eGFP. Previous studies have shown that the phosphorylation pattern of α1A/eGFP in response to NE or TPA is similar to that of nontagged α1A AR (Cabrera-Wrooman et al., 2010). Autoradiograms showing the time courses of these phosphorylations are shown in Fig. 6. Increase in phosphorylation of α1A ARs was detected after 2 and 5 minutes of stimulation with NE and OXY, respectively, and was maximal after about 5 to 15 minutes. However, the receptors activated by NE displayed almost twice the levels of phosphorylation as those activated by OXY.
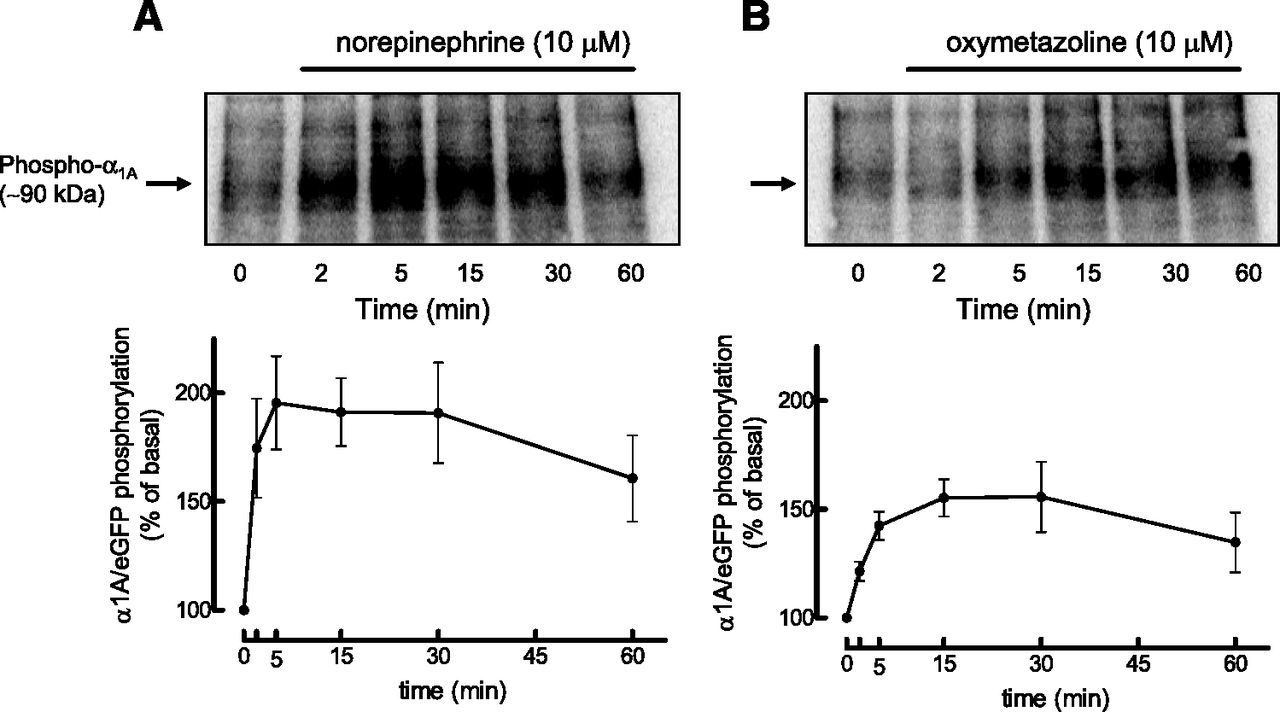
Time courses of the effects of NE (A) and OXY (B) on phosphorylation of α1A/eGFP in HEK-293 cells. HEK-293 cells transiently transfected with α1A/eGFP were metabolically labeled with 150 μCi/ml [32P]Pi and treated for the times indicated with 10 µM NE or 10 µM OXY. [32P]-labeled α1A/eGFP was immunoprecipitated and visualized by SDS-PAGE and autoradiography. The phosphorylated receptors are expressed as percentage above basal values determined in nonstimulated cells (time 0`). Data represent the mean ± S.E.M. of three to four independent experiments performed with different transfections.
Effects of a Dominant Negative Mutant of GRK2 and of PKC Inhibitors on α1A AR Phosphorylation, Internalization, and Desensitization.
HEK-293 cells were transfected with a dominant negative mutant of GRK2, in which lysine 220 was replaced by a methionine to disrupt the kinase activity of the enzyme (Ferguson et al., 1995) and a stable cell line was selected (DNGRK2 cells). NE (10 µM) caused an approximate doubling of phosphorylation of transiently transfected α1A/eGFP above basal in DNGRK2 cells (Fig. 7A), only slightly less than that observed in HEK-293 cells. In addition, in DNGRK2 cells, α1A ARs activated by NE also underwent modest and delayed internalization (Fig. 7B). However, in sharp contrast, phosphorylation of α1A/eGFP activated by OXY in DNGRK2 cells was greatly reduced (Fig. 7C). Internalization of α1A-ARs by OXY in DNGRK2 cells was greatly reduced and occurred at a slower rate than that observed in HEK-293 cells (Fig. 7D). However, OXY did not induce tachyphylaxis for the iCa2+ response to NE in DNGRK2 cells (Fig. 8), indicating an important role for GRK2 in the phosphorylation, desensitization, and internalization of α1A-ARs to OXY.

Time courses of the effects of NE and OXY on receptor phosphorylation and internalization in HEK-293 cells stably expressing a dominant-negative mutant of GRK-2 (DNGRK2 cells). In (A) and (C) are shown representative autoradiograms and the plot of phosphorylated α1A/eGFP receptors immunoprecipitated from DNGRK2 cells treated for the times indicated with 10 µM NE and 10 µM OXY, respectively. The specific binding of [3H]prazosin to the surface of DNGRK2 cells transiently expressing α1A-ARs treated for the times indicated with 10 µM NE and 10 µM OXY are shown in (B) and (D), respectively. For comparison, the NE and OXY curves for receptor phosphorylation and internalization in HEK-293 cells treated with NE or OXY are shown as dashed lines in the respective graphs. Data represent the mean ± S.E.M. of three to four independent experiments performed with different transfections.

Effects of the pretreatment of HEK-293 and DNGRK2 cells with 10 µM OXY for 5 minutes in the iCa2+ increases induced by 10 µM NE. Cells transiently expressing α1A-ARs were treated or not with 10 µM OXY for 5 minutes, washed, loaded with the fluorescent calcium indicator FURA-2 AM for 1 hour at 37°C, and then challenged with 10 µM NE. iCa2+ increases are expressed as percentage of the peak response to 10 µM thapsigargin in the respective cell line not pretreated with OXY. Data represent the mean ± S.E.M. of three independent experiments performed with different transfections. P < 0.05 in unpaired Student’s t test compared with the respective iCa2+ response in cells not pretreated with OXY. ns, not significant, P > 0.05.
To check for the participation of PKC isoforms, phosphorylation of α1A ARs activated by NE or OXY (both at 10 μM/15 minutes) was determined in DNGRK2 cells treated with bisindolylmaleimide I (1 µM, non-subtype selective PKC inhibitor) (Toullec et al., 1991), Gö6976 (12-(2-cyanoethyl)-6,7,12,13-tetrahydro-13-methyl-5-oxo-5H-indolo[2,3-a]pyrrolo[3,4-c]carbazole) (0.1 µM, PKCα, and β1 selective inhibitor (Martiny-Baron et al., 1993), hispidin (1 µM, PKCβ selective inhibitor (Gonindard et al., 1997), and Rottlerin (1 µM, PKCδ selective inhibitor (Gschwendt et al., 1994). The phosphorylation of α1A-ARs in response to NE was greatly reduced by bisindolylmaleimide I and Gö6976, but not by hispidin or rottlerin, suggesting the involvement of PKCα (Fig. 9A). On the other hand, the small phosphorylation of α1A-ARs in response to OXY in DNGRK2 was not affected by any of the PKC inhibitors tested (Fig. 9B). The effect of the nonselective protein kinase inhibitor staurosporine (100 nM/30 minutes) on α1A-AR internalization in HEK-293 cells was investigated. Staurosporine completely inhibited the delayed internalization of α1A-ARs caused by NE (Fig. 9C), but it had little effect on the internalization of receptors to OXY (Fig. 9D).

Effects of PKC inhibitors on receptor phosphorylation and internalization. (A) and (B) show representative autoradiograms and the plot of phosphorylated α1A/eGFP immunoprecipatetd from DNGRK2 cells treated with 10 µM NE and 10 µM OXY for 15 minutes, respectively, in the absence (basal) and presence of the PKC inhibitors bisindolylmaleimide I (1 µM, nonselective); Gö6976 (0.1 µM, PKCα-selective); hispidin (1 µM, PKCβ-selective); and rottlerin (1 µM, PKCδ-selective) incubated 30 minutes before stimulation with agonists. The effects of the nonselective PKC inhibitor staurosporine (100 nM) in the specific binding of [3H]prazosin to the surface of HEK-293 cells transiently expressing α1A-ARs treated for the times indicated with 10 µM NE and 10 µM OXY are shown in (B) and (D), respectively. For comparison, the NE and OXY curves for receptor phosphorylation and internalization in HEK-293 cells treated with NE or OXY in the absence of staurosporine are shown as dashed lines in the respective graphs. Data represent the mean ± S.E.M. of three to four independent experiments performed with different transfections. In (A) and (B), one-way analysis of variance followed by Dunnett’s test indicates differences from the effect of NE alone (*P < 0.05; **P < 0.01).
Discussion
The present study investigated the tachyphylaxis resulting from the low-efficacy partial agonist OXY in tissues and cells expressing rat native and human recombinant α1A-ARs. The patterns of phosphorylation, desensitization, and internalization of human recombinant α1A-ARs exposed to OXY in HEK-293 cells sharply contrasted with the pattern observed for NE: α1A-ARs activated by OXY are phosphorylated by GRK2, are desensitized, and undergo rapid internalization, whereas α1A-ARs activated by NE are phosphorylated largely by PKC and internalize at a much slower rate.
Although OXY is a partial agonist relative to PE or NE in contractions of the rat tail artery and vas deferens (α1A-AR–mediated effects) and in increases in iCa2+ in HEK-293 cells expressing human recombinant α1A-ARs, it caused intense tachyphylaxis, whereas no tachyphylaxis was observed to the responses induced by the full agonists NE and PE. As partial agonists require higher receptor occupancies than full agonists to produce their effects, one could argue that tachyphylaxis may be an expected feature of responses to partial agonists, as they would be more affected by reduction in receptor density than full agonists. However, responses to NE and PE also showed tachyphylaxys in tissues or cells previously exposed to OXY, but not to NE or PE, unveiling a distinctive property of this imidazoline. This ability of OXY to cause tachyphylaxis in contractions of the rat vas deferens to other adrenergic agonists was previously described (Ruffolo et al., 1977; Rice et al., 1991), and it was concluded that it resulted either from slow dissociation of OXY from the receptor causing “pseudoirreversible” antagonism or to a unique ability of this drug to desensitize/internalize α1-ARs (Rice et al., 1991). The present and previous studies do not support receptor inactivation resulting from “pseudo-irreversible” antagonism as an explanation. A full Schild analysis showed that OXY (0.1–30 µM) behaves as a simple reversible competitive antagonist of contractions of the rat vas deferens and anococcygeous smooth muscle induced by NE (Kenakin, 1984; Campos et al., 2003). Saturation binding studies of [3H]prazosin to intact HEK-293 cells expressing α1A-ARs previously exposed to OXY (Fig. 5C) showed a reduction in the maximal binding without alteration in the pKD; this effect of OXY is not seen in the binding of [3H]prazosin to membrane preparations of HEK-293 cells expressing α1A-ARs. In addition, the activation of α1A/eGFP in HEK-293 cells by OXY caused its translocation from the cell membrane to the intracellular compartment (Fig. 4).
Confocal microscopy and [3H]prazosin binding experiments in intact cells showed that OXY caused rapid internalization of α1A-ARs within about 5 minutes of addition, whereas internalization of α1A-ARs by NE was much smaller and observed only after about 45–60 minutes of exposure. These findings can be compared with several studies using a variety of techniques to localize receptors that have reported only small and delayed internalization of α1A-ARs, and all of these studies employed phenethylamine agonists to activate the receptor. For instance, confocal microscopy of HEK-293 cells expressing α1-AR subtypes tagged at the C-terminus with GFP showed that the α1A-AR exposed to PE internalizes only after 50 minutes of stimulation (Chalothorn et al., 2002). Similarly, in HEK-293 cells expressing hemagglutinin-tagged α1A-ARs, EPI induced modest α1A-AR internalization after 90 minutes, whereas precipitation of cell surface α1A-ARs labeled with biotin does not reveal significant receptor internalization (Stanasila et al., 2008). [3H]Prazosin binding assays in intact rat-1 fibroblasts expressing human hemagglutinin-tagged α1A-AR (Price et al., 2002) and flow cytometry of receptor localization (Morris et al., 2004) also confirm that NE induces HA-tagged α1A-AR internalization after about 30–50 minutes. Thus, the present results with OXY show that the rate and extent of α1A-ARs internalization depend on the nature of the agonist that is activating the receptor. This suggests biased agonism for OXY compared with NE toward the internalization pathway, and full concentration-response curves show that OXY was indeed more potent in internalizing α1A-ARs than in increasing iCa2+ (Vanessa Lima, manuscript in preparation).
Although α1A-ARs activated by NE were phosphorylated to a greater extent than were those activated by OXY, the kinase involved in the phosphorylation of the α1A-AR was agonist dependent. Phosphorylation of α1A-ARs exposed to OXY in cells expressing a dominant-negative mutant of GRK2 was drastically reduced, whereas the phosphorylation of receptors activated by NE was little affected. Conversely, the inhibition of PKC in DNGRK2 cells virtually abolished phosphorylation of α1A-ARs exposed to NE but had no clear effect on phosphorylation of receptors exposed to OXY. The involvement of PKC in the phosphorylation of α1A-ARs by NE is well described (Vazquez-Prado and Garcia-Sainz, 1996; Vazquez-Prado et al., 2000; Price et al., 2002). In the present study, the phosphorylation of α1A-ARs by NE was inhibited by bisindolylmaleimide I and Gö6976, but not by hispidin or rottlerin, suggesting that PKCα is the main isoform involved. However, it is important to mention that the selectivity of most protein kinase inhibitors has been recently questioned (Davies et al., 2000; Bain et al., 2007) and that caution should be taken to define the role of a particular kinase by the use of these compounds. There is indirect evidence for the involvement of GRK2 in the phosphorylation of α1A-ARs in rat-1 fibroblasts, as the overexpression of GRK2, but not of GRK6, reduced maximal inositol phosphate accumulation in response to NE (Price et al., 2002). However, overexpression of GRK2 in HEK-293 cells had no effect on the internalization of α1A-ARs activated by EPI (Stanasila et al., 2008). Interestingly, the modest and delayed internalization of α1A-ARs after NE was inhibited by the protein kinase inhibitor staurosporine, whereas internalization of α1A-ARs after OXY in HEK-293 cells expressing a dominant negative mutant of GRK2 was smaller and slower than in HEK-293 cells not expressing the dominant negative mutant. These results suggest that a specific protein kinase is recruited depending on the agonist activating the α1A-AR, which might generate different patterns of receptor phosphorylation that in turn causes interactions with specific intracellular proteins leading to receptor desensitization/internalization. Similar agonist-dependent recruitment of kinases is well described for CCR7 and µ-opioid receptors; CCR7 receptors are phosphorylated by GRK3 and GRK6 on exposure to the endogenous chemokine CCL19 and undergo internalization, whereas receptors exposed to the chemokine CCL21 are phosphorylated only by GRK6 and this is not followed by internalization (Zidar et al., 2009). Also, µ-opioid receptors activated by DAMGO are phosphorylated by GRK2 and undergo internalization, whereas PKC phosphorylates the receptors activated by morphine or fentanyl, which in turn are not internalized (Johnson et al., 2006; Hull et al., 2010). Such barcoding in receptor phosphorylation and the respective kinases involved have been elucidated for the µ-opioid receptor (Doll et al., 2011, 2012).
An important piece of information that remains to be clarified is the role of β-arrestins in the internalization of α1A-ARs activated by OXY and NE, as biased agonism at β-arrestins is a well established concept (Reiter et al., 2012). Recent studies have shown that α1A-ARs activated by EPI do not interact with β-arrestin 1 and 2 (Stanasila et al., 2008) and that the receptor activated by NE or labetalol recruits β-arrestin 2 only when heteromerizing with CXCR2 receptors (Mustafa et al., 2012). However, experiments using confocal microscopy and coimmunoprecipitations of Flag-tagged α1A-ARs and β-arrestin 2 failed to detect this recruitment in response to activation by NE and OXY (Pupo and Akinaga, unpublished observations), indicating that this will be a difficult task.
It was recently shown that transgenic mice expressing a constitutively active mutant of the α1A, but not of the α1B-AR, have an increased life span, mainly as a result of cardiac and neuroprotection (Perez and Doze, 2011), broadening the potential therapeutic applications of drugs that selectively interfere with the signaling that results from activation of each of these α1-AR subtypes. To accomplish this, an understanding of the signaling profiles of α1-AR ligands in native and recombinant receptor systems is important. Functional selectivity for α1-AR ligands at α1A-ARs expressed in Chinese hamster ovary cells was recently described, and OXY was a full agonist in the extracellular acidification rate, a partial agonist for iCa2+ increase, but it failed to stimulate cAMP production (Evans et al., 2011). The results of the present study indicate that manipulation of the functional selectivity/biased agonism of α1A-AR ligands aiming for drugs that stabilize a receptor conformation less prone to desensitization would be advantageous for the maintenance of the vasoconstriction that underlies the therapeutic decongestant effects of these drugs.
In conclusion, our findings demonstrate the differential involvement of PKC and GRK2 in the phosphorylation of α1A-ARs activated by NE and OXY, respectively, and the functional repercussion of this distinct recruitment on α1A-AR desensitization and internalization. This is clear evidence of ligand-directed signaling for OXY at α1A-AR toward the internalization pathway.
Acknowledgments
The authors thank Drs. Roger Summers, Bronwyn A. Evans, and Dana Hutchinson for helpful discussions, and Drs. Soraya Smaili and Rodrigo Ureshino for assistance in the confocal microscopy.
Authorship Contributions
Participated in research design: Akinaga, Lima, Kiguti, García-Sáinz, Pupo.
Conducted experiments: Akinaga, Lima, Hébeler-Barbosa, Alcántara-Hernández.
Performed data analysis: Akinaga, Lima, Alcántara-Hernández, García-Sáinz, Pupo.
Wrote or contributed to the writing of the manuscript: Akinaga, Kiguti, García-Sáinz, Pupo.
Footnotes
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) [Grant 08/50423-7], Coordenação de Aperfeiçoamento de Pessoal de Nível Superior CAPES [Grant 9150/11-0], Dirección General de Asuntos de Personal Académico-Universidad Nacional Autónoma de México DGAPA-UNAM [Grant IN200812], and Consejo Nacional de Ciencia y Tecnología CONACYT [Grant 177556].
Abbreviations
- AR
- adrenoceptor
- bisindolylmaleimide I
- 3-(N-[dimethylamino]propyl-3-indolyl)-4-(3-indolyl)maleimide, 3-[1-[3-(dimethylamino)propyl]1H-indol-3-yl]-4-(1H-indol-3-yl)1H-pyrrole-2,5dione
- Bmax
- maximum binding capacity
- BMY-7378 (8-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-8-azaspiro[4.5]decane-7
- 9-dione dihydrochloride)
- eGFP
- enhanced green fluorescent protein
- Emax
- maximal effect
- EPI
- epinephrine
- Gö6976
- 12-(2-cyanoethyl)-6,7,12,13-tetrahydro-13-methyl-5-oxo-5H-indolo[2,3-a]pyrrolo[3,4-c]carbazole
- GRK
- G-protein receptor kinase
- HBSS
- Hanks’ balanced salt solution
- HEK
- human embryonic kidney cells
- hispidin
- 6-(3,4-dihydroxystyrl)-4-hydroxy-2-pyrone
- iCa2+
- intracellular Ca2+
- NE
- norepinephrine
- OXY
- oxymetazoline
- PBS
- phosphate-buffered saline
- PE
- phenylephrine
- PKC
- protein kinase C
- rottlerin
- 1-[6-[(3-acetyl-2,4,6-trihydroxy-5-methylphenyl)methyl]-5,7-dihydroxy-2,2-dimethyl-2H-1-benzopyran-8-yl]-3-phenyl-2-propen-1-one
- Received September 10, 2012.
- Accepted January 30, 2013.
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}