


Abstract
Nifedipine and FPL 64176 (FPL), which block and potentiate L-type voltage-gated Ca2+ channels, respectively, modulate Cav1.2 more potently than Cav1.3. To identify potential strategies for developing subtype-selective inhibitors, we investigated the role of divergent amino acid residues in transmembrane domains IIIS5 and the extracellular IIIS5-3P loop region in modulation of these channels by nifedipine and FPL. Insertion of the extracellular IIIS5-3P loop from Cav1.2 into Cav1.3 (Cav1.3+) reduced the IC50 of nifedipine from 289 to 101 nM, and substitution of S1100 with an A residue, as in Cav1.2, accounted for this difference. Substituting M1030 in IIIS5 to V in Cav1.3+ (Cav1.3+V) further reduced the IC50 of nifedipine to 42 nM. FPL increased current amplitude with an EC50 of 854 nM in Cav1.3, 103 nM in Cav1.2, and 99 nM in Cav1.3+V. In contrast to nifedipine block, substitution of M1030 to V in Cav1.3 had no effect on potency of FPL potentiation of current amplitude, but slowed deactivation in the presence and absence of 10 μM FPL. FPL had no effect on deactivation of Cav1.3/dihydropyridine-insensitive (DHPi), a channel with very low sensitivity to nifedipine block (IC50 ∼93 μM), but did shift the voltage-dependence of activation by ∼−10 mV. We conclude that the M/V variation in IIIS5 and the S/A variation in the IIIS5-3P loop of Cav1.2 and Cav1.3 largely determine the difference in nifedipine potency between these two channels, but the difference in FPL potency is determined by divergent amino acids in the IIIS5-3P loop.
Introduction
Inhibitors of L-type voltage-gated Ca2+ channels have long been used in the treatment of cardiovascular diseases such as hypertension and angina pectoris. In these indications, the specific target is inhibition of Cav1.2, the predominant L-type channel in vascular smooth muscle, to induce vasodilation (Catterall, 2000). However, the closely related L-type channel Cav1.3 is expressed in SA and AV nodal tissue (Platzer et al., 2000), and is probably an important target for suppression of supraventricular arrhythmias. None of the three chemical classes of L-type channels blockers currently in clinical use [dihydropyridines (DHPs), phenylalkylamines (PAAs), or benzothiazepines (BTZs) (Hockerman et al., 1997b)] have a high degree of discrimination between Cav1.2 and Cav1.3. Outside of the cardiovascular system, Cav1.2 and Cav1.3 are expressed in various types of neurons (Hell et al., 1993) and endocrine cells (Seino et al., 1992), where they are thought to play distinct roles in cellular regulation. For example, Cav1.3 has been implicated in mediating Ca2+ oscillations in dopaminergic neurons of the substantia nigra that may lead to Ca2+ overload and contribute to the selective loss of these neurons in Parkinson’s disease (Guzman et al., 2009, 2010; Surmeier and Schumacker, 2013). In addition, autoantibodies that activate Cav1.3 have been detected in serum from patients with type 1 diabetes (Juntti-Berggren et al., 1993; Bason et al., 2013), suggesting a role for excessive Cav1.3 activation in autoimmune-mediated β cell death. These observations have driven the search for selective inhibitors of Cav1.3 as potential therapeutics for Parkinson disease and type 1 diabetes.
Given the attractiveness of Cav1.3 as therapeutic targets, several efforts to develop subtype-selective L-type channel blockers have been published. One study examined dozens of derivatives of the DHP scaffold but reported only modest degrees of selectivity for Cav1.3 over Cav1.2 (Chang et al., 2010), and another study examining 5-unsubstituted DHPs reported compounds with better Cav1.3 selectivity (Tenti et al., 2014). A screen of over 60,000 compounds identified a class of compounds, pyrimidine-2,4,6-triones, as moderately selective inhibitors of Cav1.3 over Cav1.2 (Kang et al., 2012, 2013). However, one follow-up study concluded that the selectivity of the lead pyrimidine-2,4,6-trione (compound 8) was dependent on the subtype of the auxiliary β subunit expressed with Cav1.3 (Huang et al., 2014), and another concluded that compound 8 was an activator of L-type channels (Ortner et al., 2014).
The mixed results reported in studies using derivatives of DHPs or screens of chemical libraries suggest the need for more insight into differences between Cav1.2 and Cav1.3 that might be exploited in selective-drug development. The molecular pharmacology of Cav1.2 is well studied. The molecular determinants of Cav1.2 modulation by DHPs (Hockerman et al., 1997c; Sinnegger et al., 1997; Yamaguchi et al., 2003; Lin et al., 2011), PAAs (Hockerman et al., 1995, 1997a; Dilmac et al., 2004), and BTZs (Hering et al., 1996; Hockerman et al., 2000; Dilmac et al., 2003) have been identified, and homology models of the binding sites have been developed (Cosconati et al., 2007; Cheng et al., 2009; Tikhonov and Zhorov, 2009). On the other hand, the molecular pharmacology of Cav1.3 has not been extensively studied. One reason for this disparity may be that the critical residues for drug block of Cav1.2 are highly conserved in Cav1.3, leading to the perception that the drug binding site in both channels is identical. However, Cav1.3 is reported to be less sensitive to block by some DHPs than Cav1.2 (Xu and Lipscombe, 2001; Huang et al., 2013), but the molecular determinants that mediate this difference in DHP affinity are not known.
The transmembrane domains of Cav1.2 and Cav1.3 that compose the drug binding pockets are nearly identical, but two subtle differences, one each in IIIS5 and IIIS6, exist. In addition, the extracellular IIIS5-3P domains of these channels are highly divergent. The IIIS5-3P domain contains two amino acid residues that are critical for DHP block of Cav1.2 (Yamaguchi et al., 2000, 2003), yet these residues are conserved between Cav1.2 and Cav1.3. However, another cluster of amino acids, closer to IIIS5 and not conserved between Cav1.2 and Cav1.3 is reported to influence DHP binding affinity (Wang et al., 2007). Therefore, we examined if substitution of these key divergent amino acids from Cav1.2 into Cav1.3 could reduce the IC50 for nifedipine (1,4-dihydro-2,6-dimethyl-4-(2-nitrophenyl)-3,5-pyridinecarboxylic acid dimethyl ester) and EC50 for the L-type channel agonist FPL 64176 compared with wild-type Cav1.3.
Materials and Methods
Chemicals and Reagents.
All reagents, unless otherwise indicated, were purchased from MilliporeSigma (St. Louis, MO). Oligonucleotides used for site-directed mutagenesis were obtained from GenScript (Nanjing, People’s Republic of China). The Cav1.342 (AF370010) and Cav1.342a (AF370009) clones (Xu and Lipscombe, 2001) with three cloning errors repaired (Huang et al., 2013) were the gift of Dr. Tuk-Wah Soong, University of Singapore. The Cav1.2 clone (M67515) (Snutch et al., 1991) was the gift of Dr. Terrance Snutch, University of British Columbia.
Cell Culture.
The tsA201 variant of the human embryonic kidney 293 cell line was grown at 37°C, 5% CO2, in Dulbecco’s modified Eagle’s medium (Life Technologies/Thermo Fisher Scientific, Grand Island, NY) supplemented with 10% fetal bovine serum (Atlanta Biologic, Lawrenceville, GA), 100 IU/ml penicillin, and 100 μg/ml streptomycin.
Site-Directed Mutagenesis.
All mutant Cav1.342 (in pcDNA6) and Cav1.2 (in pcDNA3) α1 subunits (except Cav1.3+) were constructed as described previously (Dilmac et al., 2003). To construct Cav1.3+, an oligonucleotide encoding amino acids 1058–1118 of Cav1.2 was ligated into Cav1.342 in pSPORT6 after excision of the IIIS5-3P encoding DNA with BamH1 and BstB1. The final version in Cav1.342 pcDNA6 was created by ligation of the BamH1/EcoRV fragment containing the chimeric region from pSPORT6. All mutant constructs were verified by DNA sequencing and restriction digest analysis.
Electrophysiological Recordings.
Mutant and wild type Cav1.2 or Cav1.3 α1 subunits were coexpressed with α2δ1 (Williams et al., 1992) and β3 (Castellano et al., 1993) subunits (both in pcDNA3), and pEGFPN1 (Clontech, Mountain View, CA) by transfection of tsA201 cells, as described previously (Dilmac et al., 2003). Transfected cells were identified by green fluorescent protein fluorescence. Micropipettes were pulled from borosilicate capillaries to an inside diameter of approximately 3–5 μm using a Sutter P-87 pipette puller (Sutter Instruments, Novato, CA), and polished with a Narishige MF 830 micro forge (Narishige, Amityville, NY). The pipette solution contained: (in millimolars) 180 NMDG, 40 HEPES, 4 MgCl2, 12 phosphocreatine, 5 BAPTA, 2 Na2ATP, 0.5 Na3GTP, 0.1 leupeptin, and pH was adjusted to 7.3. The extracellular solution contained (in millimolars): 140 NaCl, 20 CsCl2, 10 BaCl2, 10 HEPES, 10 glucose, 10 sucrose, 1 MgCl2, and pH was adjusted to 7.4. In experiments with balanced NMDG, the extracellular solution was altered to contain 30 mM NMDG, with a corresponding reduction in NaCl concentration. Whole-cell voltage clamp recordings were made at room temperature using an Axopatch 200B amplifier (Axon Instrument, Sunnyvale, CA). Data were sampled at 10 kHz and filtered at 1 kHz. Drugs were applied in the extracellular solution with a Biologic RSC 160 perfusion system (BioLogic, Sayssinet-Pariset, France). logIC50 values for nifedipine block were determined by fitting the fraction of current blocked at each drug concentration to the equation: Fraction Blocked = a − (a/(1 + ([nifedipine]/IC50)b)), where a = maximum fraction blocked, b = slope. logEC50 values for FPL potentiation were determined by normalizing the increase in current with each concentration of FPL to the increase in current observed with 10 μM FPL. When fitting equations to the nifedipine dose-response data (logIC50), we set the minimum at zero, and let the slope and maximal block vary. This reflects the experimental observation that current block is often incomplete even at maximally effective concentrations. When fitting equations to the FPL 64176 dose-response data (logEC50), we set the minimum at zero and the maximum at 1 (maximal current stimulation) but allowed slope to vary. The range of N values for dose-response curves represent the number of data points for each drug concentration. The number of separate experiments performed (i.e., cells clamped) to obtain a given dose-response curve is equal to or greater than the highest number of replicates indicated for any single drug concentration. The basis of the logIC50 and logEC50 values ± S.E. of the fit shown in Table 1 is the fit of all of the data for a given channel construct. V1/2 activation values were determined by plotting normalized tail-current amplitudes versus the corresponding 100-millisecond depolarizing voltage steps from −50 to +60 mV, in 10 mV-increments, from a holding potential of −80 mV. The data were fit to the equation, I = 1/(1 + exp((V1/2 − V)/k)), where k is a slope factor. The steady-state inactivation protocol used 10-second conditioning pulses from −80 to +20 mV in 10-mV increments from a holding potential of −90 mV, followed by a 100-millisecond test pulse to +10 mV. V1/2 inactivation was determined by plotting the normalized test pulse amplitude versus the conditioning pulse potential, and fitting the data to the equation I = 1/(1 + exp(−(V − V1/2)/k)), where k is a slope factor. When fitting equations to the data for voltage-dependence of activation and inactivation, we set curves to start at 0 or 1, respectively, and force the curves to plateau at 1 or 0, respectively. Slopes were allowed to vary. The time course of channel deactivation was determined by fitting tail-current decay to either a single or double exponential function.
Pharmacology and voltage-dependence of Cav1.2, Cav1.3, and mutant channels
Homology Models of Cav1.2 and Cav1.3 on the Basis of the Structure of Cav1.1.
Homology models of Cav1.2 and Cav1.3 were generated using SWISS-MODEL (Guex et al., 2009; Benkert et al., 2011; Bertoni et al., 2017; Bienert et al., 2017; Waterhouse et al., 2018). The structure of Cav1.1 (PDB-code: 5gjw) was used as template for modeling (Wu et al., 2016). Cav1.2 and Cav1.3 share sequence identities of 72% and 71% with Cav1.1, respectively.
Data Analysis and Statistics.
Data were analyzed using Clampfit 10.6 (Axon Instruments) and SigmaPlot 11 (Systat Software, San Jose, CA). logIC50 and logEC50 values were determined using GraphPad Prism 7.04 (GraphPad Software, La Jolla, CA). Comparisons of two means were made with Student’s unpaired t test. Comparisons of three or more means were made using one-way analysis of variance. P < 0.05 was considered significant. Data shown are means ± S.E. Lines are fits of the equations indicated for each type of experiment to the data.
Results
Characterization of Ba2+ Current Conducted by Cav1.2 or Cav1.3 Coexpressed with the β3 and α2δ1 Subunits in tsA201 Cells.
We assessed the biophysical and pharmacological properties of Cav1.2 and Cav1.3 in our expression system. As expected, Cav1.3 activated at more negative voltages than Cav1.2 (P < 0.001) (Fig. 1A; Table 1), and Cav1.2 inactivated at slightly more negative voltages than Cav1.3 (P < 0.01) (Fig. 1B; Table 1). We next examined the potency of nifedipine block of both channel types. We chose nifedipine because it is the most compact of the dihydropyridine Ca2+ channel antagonists (Supplemental Fig. 1), and our preliminary screen of several structurally distinct dihydropyridines revealed a substantial difference in nifedipine potency in blocking Cav1.2 compared with Cav1.3 (Supplemental Fig. 1). Channels were activated with 100-millisecond steps to +10 mV at a frequency of 0.033 Hz from a holding potential of −80 mV. After a baseline current was established, increasing concentrations of nifedipine were applied via a perfusion capillary in the bath solution. Figure 1C shows sample traces and the compiled dose-response curves for both channel subtypes. As expected, Cav1.2 was blocked more potently by nifedipine than Cav1.3, with IC50 values of 22 ± 2 nM and 289 ± 30 nM, respectively (P < 0.001). The truncated splice variant Cav1.342a (Xu and Lipscombe, 2001) had been reported to be less sensitive to nifedipine than the full-length Cav1.342 variant (Huang et al., 2013). Therefore, we examined the dose-dependence of nifedipine block of Cav1.342a, and determined the IC50 for nifedipine to be 436 ± 24 nM, greater than that of Cav1.342 (P < 0.01) (Fig. 1C). We chose to use the full-length Cav1.342 variant in the subsequent experiments, since it is structurally more similar to the Cav1.2 variant used in this study.

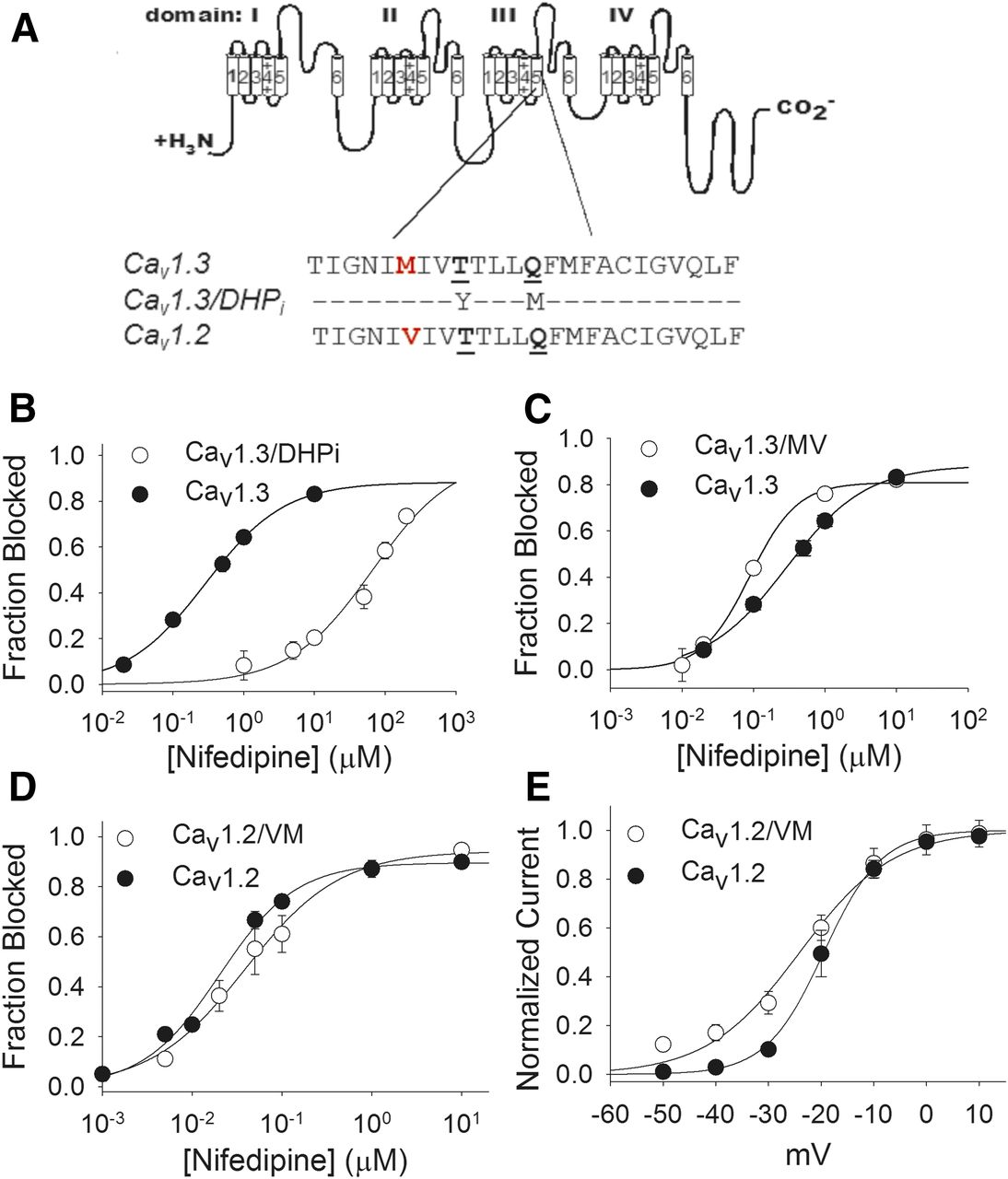
Characterization of Cav1.2 and Cav1.3 biophysical properties and nifedipine block. (A) Determination of the voltage-dependence of activation in Cav1.2 and Cav1.3. V1/2 activation values were −20 ± 0.5 mV for Cav1.2 (N = 6) and −30 ± 1.5 mV for Cav1.3 (N = 9) (P < 0.001). (B) Determination of the voltage-dependence of inactivation in Cav1.2 and Cav1.3. V1/2 inactivation values were −41 ± 0.6 mV for Cav1.2 (N = 6) and −36 ± 1.3 mV (N = 5) for Cav1.3 (P < 0.01). (C) Determination of potency of nifedipine block of Cav1.2 and Cav1.3. The IC50 values of nifedipine block were 22 ± 2 nM (N = 3–12) for Cav1.2 and 289 ± 30 nM (N = 7) for Cav1.3 (P < 0.001). Example traces from experiments are shown at right. The IC50 for nifedipine block of Cav1.342a, a truncated splice variant, was 436 ± 24 nM (N = 5 to 6), statistically significantly greater than that of Cav1.3 (P < 0.01).
The IIIS5 Transmembrane Domain Plays a Key Role in Nifedipine Block of Cav1.3.
Studies in Cav1.2 have established transmembrane domain IIIS5 as a key component of the DHP binding pocket (Mitterdorfer et al., 1996). Specifically, mutations of T1039 and Q1043 (underlined in Fig. 2A) to the corresponding residues in DHP-insensitive voltage-gated Ca2+ channels results in a Cav1.2 mutant channel (termed Cav1.2/DHPi) that is markedly less sensitive to DHPs but normally sensitive to diltiazem (Hockerman et al., 2000; Lin et al., 2011). We made the corresponding Cav1.3/DHPi mutant, and as expected, it was substantially less sensitive to nifedipine than Cav1.3 (Fig. 2B). In fact, we were unable to determine the maximum percent of Cav1.3/DHPi current blocked because the nifedipine concentrations at the high end of the range (>200 μM) were at the limit of aqueous solubility (Ran et al., 2002). Assuming maximal inhibition of 90% of current, we estimated the IC50 of nifedipine block of Cav1.3/DHPi to be ∼93 μM, more than 300 times that for Cav1.3. As with the corresponding mutation in Cav1.2 (Hockerman et al., 2000), the sensitivity of Cav1.3/DHPi to block by the BTZ diltiazem was not reduced compared with Cav1.3 (Supplemental Fig. 2).

Contribution of transmembrane domain IIIS5 to nifedipine block of Cav1.3. (A) Amino acid sequence alignment of the IIIS5 transmembrane domains in Cav1.3, Cav1.2, and the mutant Cav1.3/DHPi. The only difference is the M to V switch at position 1030/1036 (in red). The underlined residues were mutated to create Cav1.3/DHPi and are critical for dihydropyridine block of Cav1.2. (B) Nifedipine dose-response curve for block of Cav1.3/DHPi. The IC50 of nifedipine for Cav1.3/DHPi was estimated at ∼93 μM. (C) Dose-response curve for nifedipine block of Cav1.3/MV. The IC50 value was 89 ± 7 nM (N = 5–7), less than the IC50 of nifedipine block of Cav1.3 (P < 0.001). (D) Dose-response curve for nifedipine block of Cav1.2/VM. The IC50 value was 39 ± 5 nM (N = 4–6), greater than the IC50 for block of Cav1.2 (P < 0.05). (E) Voltage-dependent activation of Cav1.2/VM. The V1/2 activation for Cav1.2/VM was −24 ± 1 mV (N = 8), more negative than that for Cav1.2 (P < 0.05).
Given that transmembrane domain IIIS5 clearly contributes to the DHP binding pocket in Cav1.3, we next examined the single amino acid in this domain that is not conserved between Cav1.2 and Cav1.3, M1030 (Fig. 2A). The corresponding position in Cav1.2 (1036) is occupied by a V residue, so we constructed the mutant channel Cav1.3/MV, to determine if this conservative change could contribute to the difference in nifedipine potency between Cav1.2 and Cav1.3. The V1/2 inactivation of Cav1.3/MV was not different from that of Cav1.3; however, the V1/2 activation of Cav1.3/MV (−26 ± 1.1) (Table 1) was more positive than that for Cav1.3 (P < 0.05). The M1030V mutation increased the potency of nifedipine block of Cav1.3, reducing the IC50 from 289 to 89 ± 7 nM (P < 0.001) (Fig. 2C). Given that this relatively conservative change in structure shifted the potency of nifedipine block of Cav1.3 toward those of Cav1.2, we asked if the reciprocal change in Cav1.2 (Cav1.2/VM) would shift the potency of nifedipine block toward that of Cav1.3. Indeed, we found that the V1036M mutation increased the IC50 of nifedipine for block of current compared with Cav1.2 (39 ± 6 nM) (P < 0.05) (Fig. 2D). In addition, the V1/2 activation of Cav1.2/VM was −24 ± 1 mV, more negative than that for Cav1.2 (P < 0.01) (Fig. 2E; Table 1). Thus, this single, conservative difference between Cav1.2 and Cav1.3 in transmembrane segment IIIS5 contributes to differences in both voltage-dependence of activation and nifedipine potency between these two channels.
The Extracellular Domain IIIS5-3P Contributes to the Difference in Nifedipine Potency Between Cav1.2 and Cav1.3.
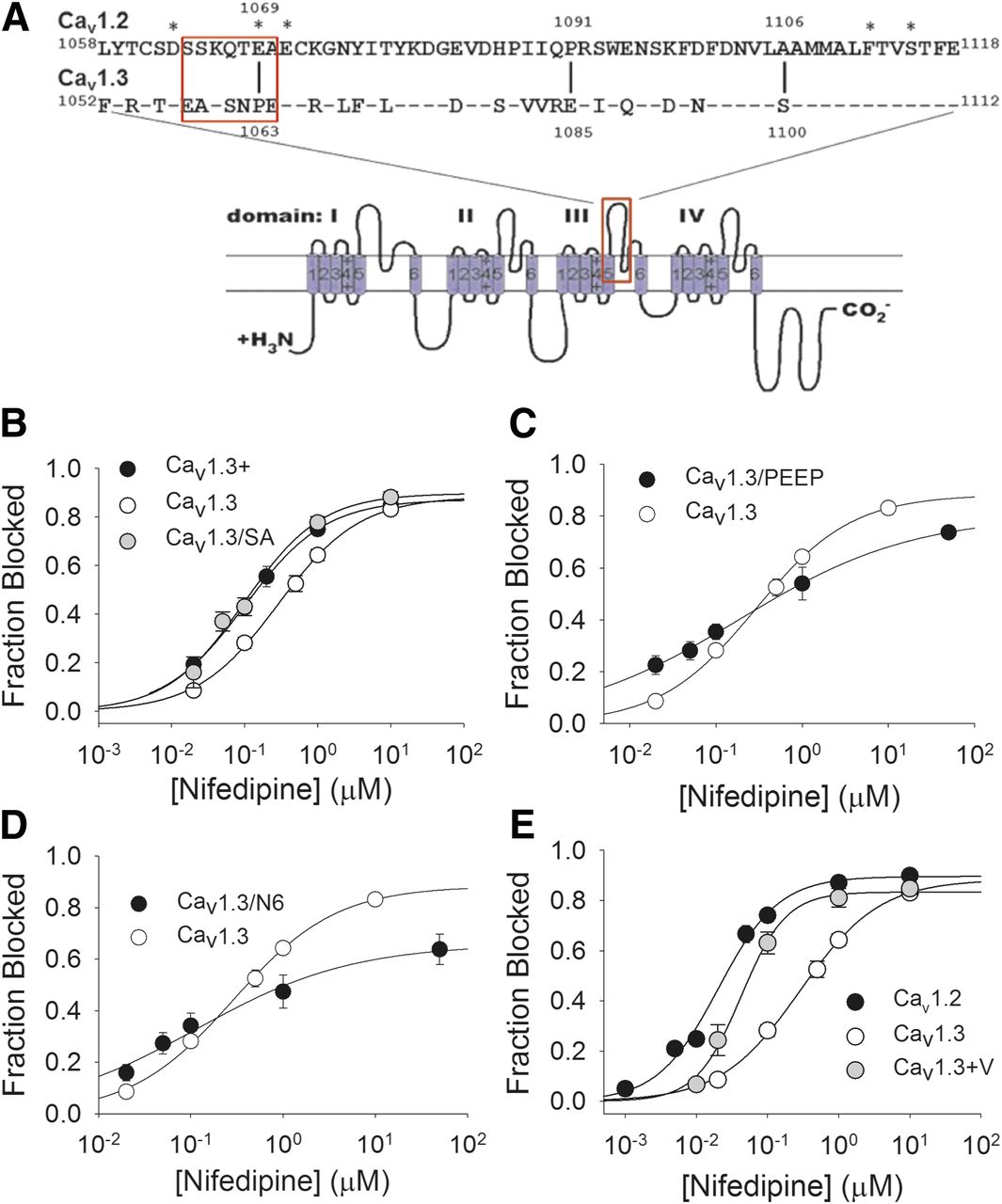
Given that the small difference in amino acid sequence between Cav1.2 and Cav1.3 in IIIS5 only partially accounts for the difference in nifedipine potency, we next examined the role of the extracellular domain just downstream of IIIS5, the IIIS5-3P loop. This region is an area of relatively high amino acid sequence divergence between Cav1.2 and Cav1.3 (Fig. 3A), and some determinants of DHP potency/affinity have been identified in this region. Therefore, we created a chimeric channel, Cav1.3+ that incorporates the Cav1.2 IIIS5-3P loop into the Cav1.3 background, to determine the effect of this region on the potency of nifedipine block. The voltage-dependence of activation and inactivation were both essentially unchanged in Cav1.3+ compared with Cav1.3 (see Table 1). However, the IC50 for nifedipine block of Cav1.3+ (101 ± 4 nM) was reduced compared with that for Cav1.3 (P < 0.001) (Fig. 3B).

Contribution of the IIIS5-3P loop to nifedipine block of Cav1.3. (A) Amino acid sequence alignment of the extracellular IIIS5-3P loops of Cav1.2 (aa 1058–1118) and Cav1.3 (aa 1052–1112). Twenty-four of the 60 amino acids in this segment are not conserved. Identities are indicated with a dash. Asterisks indicate amino acid residues previously reported to influence dihydropyridine modulation of Cav1.2. The Cav1.3+ mutant incorporated all of the Cav1.2-specific amino acids in this segment into Cav1.3. The Cav1.3+V mutant is Cav1.3+ combined with the substitution of V for M at position 1030. The Cav1.3/SA mutant incorporated only the S to A substitution at position 1100. The Cav1.3/PEEP mutation incorporated the substitution of P for E and E for P at positions 1063 and 1085, respectively. The Cav1.3/N6 mutation incorporated the six Cav1.2-specific amino acid residues from position 1064 to 1070 (boxed residues) into Cav1.3. (B) Dose-response curve for block of Cav1.3+ and Cav1.3/SA by nifedipine. Cav1.3+ (black circles) and Cav1.3/SA (gray circles) were both more sensitive to block by nifedipine than Cav1.3 (P < 0.001; P < 0.01, respectively). IC50 for nifedipine block of: Cav1.3+ = 101 ± 4 nM (N = 6–8); Cav1.3/SA = 99 ± 24 nM (N = 4 to 5). (C) Dose-response curve for block of Cav1.3/PEEP by nifedipine. The IC50 for nifedipine block of Cav1.3/PEEP was 188 ± 28 nM (N = 3–7), not different from that of Cav1.3; however, the Hill slope (0.43 ± 0.02), was shallower than that for Cav1.3 (0.78 ± 0.04) (P < 0.001). (D) Dose-response curve for block of Cav1.3/N6 by nifedipine. The IC50 for nifedipine block of Cav1.3/N6 was 116 ± 53 nM (N = 5–9), lower than that of Cav1.3 (P < 0.05). The Hill slope was (0.52 ± 0.10), shallower than that for Cav1.3 (0.78 ± 0.04) (P < 0.05). (E) Dose-response curve for nifedipine block of Cav1.3+V compared with those for Cav1.2 and Cav1.3. The IC50 for nifedipine block of Cav1.3+V was 42 ± 5 nM (N = 4–10), lower than that for Cav1.3 (P < 0.001).
We next asked if a particular region of the IIIS5-3P loop could account for the increase in nifedipine potency in block of Cav1.3+ versus Cav1.3. The IIIS5-3P loop extends from the end of IIIS5 to the conserved E residue in the domain III selectivity filter (Fig. 3A). The region just upstream of the conserved selectivity filter E residue of homologous domain IIII (Yang et al., 1993) (1118 in Cav1.2, 1112 in Cav1.3; Fig. 3A) is known to be involved in DHP modulation of Cav1.2 (Yamaguchi et al., 2000, 2003) but is highly conserved between Cav1.2 and Cav1.3. Mutation of the nearest nonconserved residue upstream of E1112 in Cav1.3 (S1100) resulted in a channel (Cav1.3/SA) with V1/2 activation not different from Cav1.3 but with markedly left-shifted V1/2 inactivation (see Table 1). The IC50 for nifedipine block of Cav1.3/SA was 99 ± 24 nM, indistinguishable from that for Cav1.3+ (Fig. 3B). The IIIS5-3P loop of both Cav1.2 and Cav1.3 contain two P residues, one of which is conserved (1081/1087) and another that differs significantly in position relative to the conserved P residue (P1063 in Cav1.3 and P1091 in Cav1.2) (see Fig. 3A). We reasoned that this difference in P configuration could affect the conformation of the conserved, distal portion of the IIIS5-3P loop, and thus DHP affinity. Therefore, we created Cav1.3/PEEP, with P residues at position 1081 and 1085, but a P to E switch at position 1063, mimicking the P configuration of the Cav1.2 IIIS5-3P loop. The voltage-dependence of inactivation was not different from Cav1.3, and the voltage-dependence of activation was ∼3 mV more negative than Cav1.3 (see Table 1). The IC50 for nifedipine block of Cav1.3/PEEP was 188 ± 28 nM, not statistically significantly lower than the IC50 for block of Cav1.3 (Fig. 3C; Table 1). However, the Hill slope for the dose-response curve for nifedipine block of Cav1.3PEEP (0.43 ± 0.02) was shallower than Cav1.3 (P < 0.001). We next turned our attention to a region of the IIIS5-3P loop proximal to IIIS5 that contains a cluster of three negatively charged residues in Cav1.2 (D1063, E1069, E1071), reported to affect DHP binding affinity (Wang et al., 2007). Only two of these negative charges are conserved in Cav1.3 (D1057 and E1065); moreover, the amino acid sequence surrounding these residues is highly divergent between Cav1.2 and Cav1.3 (see Fig. 3A). Therefore, we created the mutant Cav1.3/N6 with the Cav1.2 sequence from amino acid 1064–1070 (SSKQTEA) inserted into the corresponding position (1058–1064) in Cav1.3. We found that expression of Cav1.3/N6 yielded functional channels, but the current was outward with 180 mM NMDG in the intracellular solution and no NMDG in the extracellular solution. Therefore, we used NMDG-balanced solutions in recordings with Cav1.3/N6, which restored inward barium current. The voltage-dependence of activation of Cav1.3/N6 under these conditions was −17 ± 0.8 mV, and the voltage-dependence of inactivation was −34 ± 0.6 mV (Table 1). We found that the IC50 for nifedipine block of Cav1.3/N6 (116 ± 53 nM) was slightly (P < 0.05) lower than that for Cav1.3, but the Hill slope of the dose-response curve (0.52 ± 0.1) was also less than Cav1.3 (P < 0.05) (Fig. 3D).
Given that the decreases in nifedipine IC50 for both Cav1.3/MV and Cav1.3+ were relatively modest, we asked if combining these mutations would further increase the potency of nifedipine block. The V1/2 activation of the resulting mutant channel, Cav1.3+V, was not different from Cav1.3, but the V1/2 inactivation was shifted by −6 mV (see Table 1). However, the IC50 for nifedipine block of Cav1.3+V was reduced to 42 ± 5 nM (Fig. 3E), compared with 289 ± 30 nM for Cav1.3 (P < 0.001), but was still greater than the IC50 of nifedipine for Cav1.2 (P < 0.05). Thus, amino acid differences in the IIIS5-3P loop, along with the single amino acid divergence in IIIS5, account for the vast majority of the difference in potency of nifedipine block of Cav1.2 and Cav1.3.
We next asked if the small remaining gap in nifedipine potency between Cav1.3+V and Cav1.2 could be closed. Besides IIIS5 and the IIIS5-3P loop, transmembrane domain IIIS6 also contributes to the DHP binding pocket in Cav1.2 (Hockerman et al., 1997b). The only amino acid residue in IIIS6 not conserved between Cav1.2 and Cav1.3 is an I/V divergence at position 1156/1150; moreover, mutation of I1156 in Cav1.2 to A resulted in a significant decrease in DHP binding affinity (Peterson et al., 1997). Unfortunately, we found that substitution of V for I at position 1150 in IIIS6 of either Cav1.3+V or Cav1.3/MV resulted in channels that yielded little to no current upon expression in tsA201 cells.
Differences in the IIIS5-3P Loop Are Responsible for the Difference in Potency of FPL in Cav1.2 and Cav1.3.
The nondihydropyridine compound FPL 64176 (FPL) (Ginap et al., 1993) is a well characterized potentiator of Cav1.2 current (Liu et al., 2003). Reconstruction of the DHP binding site in the P/Q-type channel Cav2.1 conferred potentiation of current by FPL, as well as potent block by DHP antagonists (Sinnegger et al., 1997). However, very little is known about FPL modulation of Cav1.3. Therefore, we compared the potency of FPL potentiation of current in Cav1.2 and Cav1.3. The experiments with Cav1.3 used balanced NMDG solutions because we found that application of FPL frequently induced outward current when the extracellular solution contained no NMDG (Supplemental Fig. 3A), suggesting that FPL binding substantially affects the permeability of Cav1.3 to NMDG. We found that the EC50 for potentiation of current amplitude in Cav1.2 by FPL was 103 ± 40 nM (Fig. 4, A and D). In contrast, the EC50 for potentiation of Cav1.3 current amplitude by FPL was 854 ± 236 nM (P < 0.05) (Fig. 4, B and D). Thus, as with nifedipine, Cav1.3 is less sensitive to FPL than Cav1.2.

Potency of FPL 64176 potentiation of Cav1.2, Cav1.3, and mutant channels. (A–C) Example traces showing FPL potentiation of Cav1.2, Cav1.3, and Cav1.3+V, respectively. Note the marked slowing of the tail current in Cav1.2 that is absent in Cav1.3. (D) Dose-response curves for FPL 64176 potentiation of Cav1.2, Cav1.3, and mutant channels. The EC50 values for FPL potentiation of current for Cav1.2 and Cav1.3 were 103 ± 40 nM (N = 3–8) and 854 ± 236 nM (N = 3–7), respectively (P < 0.05). The EC50 for FPL potentiation of the mutant Cav1.3+V (99 ± 5 nM) (N = 3–7) was not different from that of Cav1.2 but was different from that of Cav1.3 (P < 0.05). In contrast, the EC50 for FPL potentiation of the mutant Cav1.3/MV was 737 ± 20 nM (N = 5), not different from that of Cav1.3. Data are shown as the mean fractional increase in current compared with 10 μM FPL 64176 ± S.E.
We next asked if some of the same differences between Cav1.2 and Cav1.3 that account for the difference in nifedipine potency could also account for the difference in the potency of FPL in these two channel subtypes. We first measured the potency of FPL potentiation of current in Cav1.3+V, since this mutant had nearly the same sensitivity to nifedipine as Cav1.2. We were able to perform these experiments in the standard solution set, since FPL did not induce outward current in Cav1.3+V. The EC50 for potentiation of current amplitude by FPL in Cav1.3+V was 99 ± 5 nM (Fig. 4, C and D), indistinguishable from the EC50 of FPL for potentiation of Cav1.2. We measured the EC50 for FPL potentiation of Cav1.3/MV current amplitude in the standard solution set since we did not observe outward currents in the presence of FPL in this mutant. The EC50 of FPL for Cav1.3/MV was 737 ± 20 nM, not different from the EC50 for Cav1.3 (Fig. 4D). Taken together, these results suggest that the molecular determinants of the difference in potency of FPL lie within the IIIS5-3P loop. Cav1.3+ exhibited outward current in the presence of FPL, similar to Cav1.3 (Supplemental Fig. 3A). However, we were unable to measure the potency of FPL potentiation of this mutant because, even in the NMDG-balanced solution set, FPL induced erratic changes in current amplitude (Supplemental Fig. 3B). We were able to measure the potency of FPL potentiation of the IIIS5-3P loop mutants Cav1.3/PEEP, Cav1.3/N6, and Cav1.3/SA, and found that none of these mutants displayed increased sensitivity to potentiation of current by FPL compared with Cav1.3 (Table 1). Thus, we have identified two regions of amino acid divergence between Cav1.2 and Cav1.3 within the IIIS5-3P loop, Cav1.2 1106/Cav1.3 1100 and Cav1.2 1064–1070/Cav1.3 1058–64, that appear to confer differences in sensitivity to nifedipine block but not in FPL potentiation of these two channels.
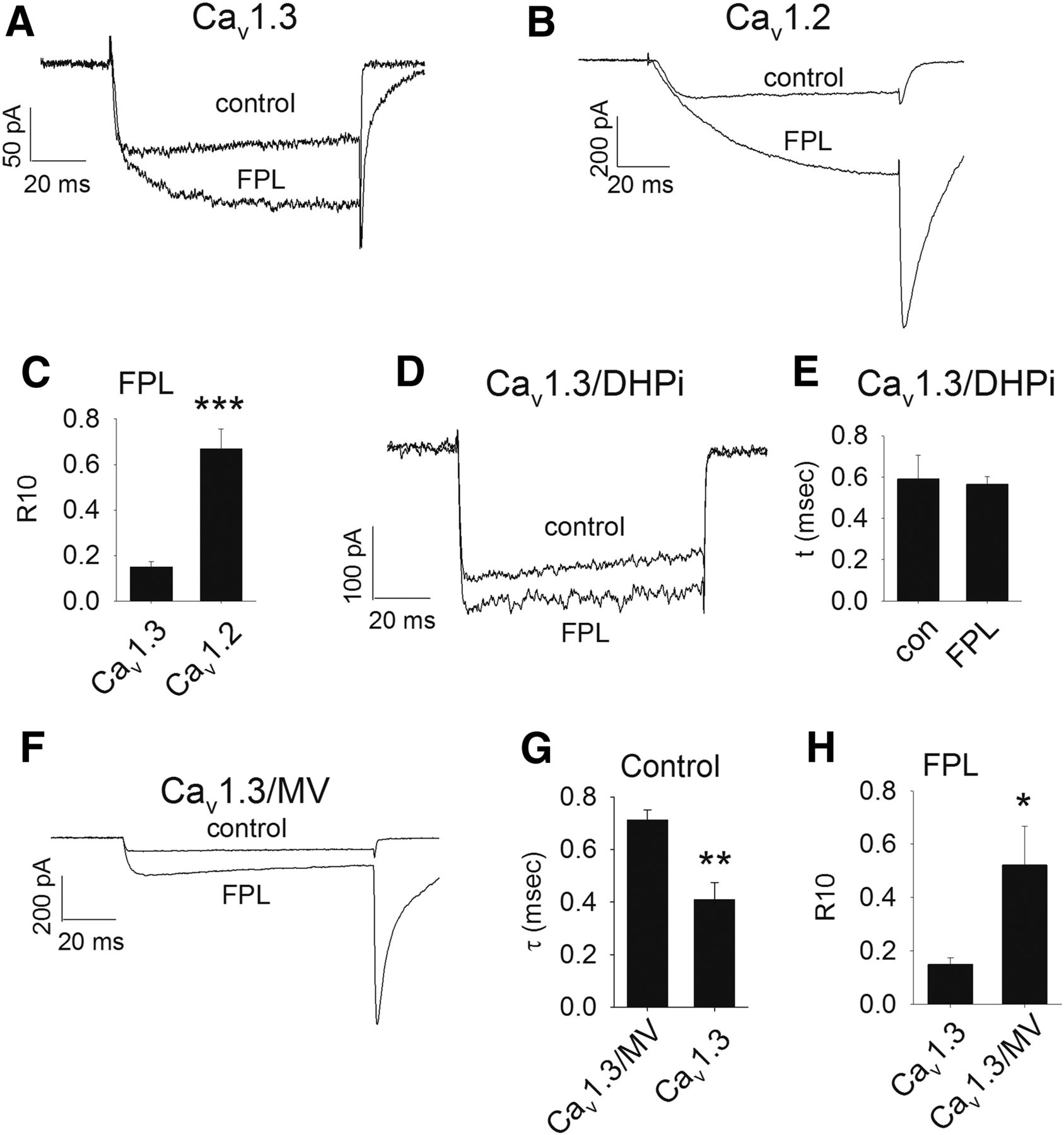
FPL has a strong effect on the kinetics of deactivation as well as the voltage-dependence of activation of Cav1.2 and Cav1.3. Figure 5 shows the effect of 10 μM FPL on tail-current kinetics, a measure of the rate of deactivation. Cav1.3 displays a fast rate of closing with a single time constant (τ) in the absence of FPL, but a second, slower τ is observed in the presence of FPL (Fig. 5A; Table 2). In contrast, deactivation in Cav1.2 in the absence of FPL follows two τs. However, a single slow τ is principally observed in the presence of FPL that is greater than both τs in the absence of FPL (Fig. 5B; Table 2). Given the differences in the kinetics of deactivation in Cav1.3 versus Cav1.2, we compared the FPL-induced slowing of deactivation in these channels by measuring the fraction of the tail current remaining 10 milliseconds after reaching peak (R10). The R10 for both Cav1.2 and Cav1.3 in the absence of FPL was negligible. Figure 5C shows that the R10 of Cav1.2 in the presence of 10 μM FPL (0.67 ± 0.09) was greater than that of Cav1.3 (0.15 ± 0.02) (P < 0.001), indicating a greater slowing of deactivation by FPL in Cav1.2. We also found that FPL shifted the V1/2 activation of Cav1.2 by −26 mV but only −10 mV in Cav1.3 (Table 2). Thus, FPL is not only more potent in stimulating current amplitude in Cav1.2 compared with Cav1.3 but also has stronger effects on deactivation kinetics and the voltage-dependence of activation in Cav1.2 at a maximally effective concentration (10 μM).

Kinetics of tail-current decay in the presence and absence of FPL 64176 in Cav1.2, Cav1.3, and mutant channels. (A) Example 100-millisecond depolarization demonstrating tail-current decay in Cav1.3 in the presence or absence of 10 μM FPL 64176. (B) Example 100-millisecond depolarization demonstrating tail-current decay in Cav1.2 in the presence or absence of 10 μM FPL 64176. (C) The R10 value (fraction of tail current remaining 10 millisecond after peak) in the presence of FPL was greater in Cav1.2 (0.67 ± 0.09, N = 6) compared with that of Cav1.3 (0.15 ± 0.02) (N = 5) (***P < 0.001). (D) Example 100-millisecond depolarization demonstrating tail-current decay in Cav1.3/DHPi in the presence or absence of 10 μM FPL 64176. (E) The time constant for deactivation of Cav1.3/DHPi (τ = 0.59 ± 0.11 milliseconds, N = 5) was not affected by the presence of 10 μM FPL (τ = 0.60 ± 0.04 milliseconds, N = 5). (F) Example 100-millisecond depolarization demonstrating tail-current decay in Cav1.3/MV in the presence or absence of 10 μM FPL 64176. (G) The time constant for deactivation of Cav1.3/MV in the absence of FPL followed a single time constant (τ = 0.70 ± 0.13 milliseconds, N = 5) that was slower than that of Cav1.3 (**P < 0.01). (H) The R10 value for Cav1.3/MV tail current in the presence of 10 μM FPL (0.51 ± 0.15, N = 5) was greater than that of Cav1.3 (*P < 0.05).
Kinetics of tail current decay in the presence and absence of FPL 64176
We next asked if the Cav1.3/DHPi channel was less sensitive to FPL than Cav1.3. Deactivation of Cav1.3/DHPi followed a single τ that was not different from that of Cav1.3 (Table 2) but was not altered by 10 μM FPL (Fig. 5, D and E). Not surprisingly, no significant increase in current was observed upon application of 10 μM FPL to Cav1.3DHPi. Interestingly, 10 μM FPL did shift the V1/2 activation of Cav1.3DHPi by −9 mV (Table 2). Since IIIS5 is clearly crucial for the action of FPL, we examined the kinetics of deactivation in Cav1.3/MV (Fig. 5F). In the absence of FPL, Cav1.3/MV deactivation followed a single τ (0.71 ± 0.04 milliseconds) that was slightly, but statistically significantly greater than Cav1.3 (0.41 ± 0.07 milliseconds) (P < 0.01) (Fig. 5G). In the presence of 10 μM FPL, the R10 was greater in Cav1.3/MV (0.52 ± 0.15 milliseconds) (P < 0.05) compared with Cav1.3 (Fig. 5H). However, deactivation of Cav1.2/VM was not different from that of Cav1.2 either in the absence or presence of FPL (Table 2). Thus, the M to V switch at position 1030 of Cav1.3 does not affect FPL potency but does affect both deactivation and slowing of deactivation by FPL.
Discussion
The voltage-dependence and sensitivity to nimodipine (a DHP antagonist) of the Cav1.3 cDNA used in this study (Cav1.342) was previously characterized (Xu and Lipscombe, 2001). The V1/2 activation reported here is indistinguishable from that initial characterization. Further, Xu and Lipscombe reported an ∼20-fold higher IC50 for nimodipine block of Cav1.3 compared with Cav1.2. For our comparison of DHP antagonist potency, we chose nifedipine since it is the most compact molecule in this class, with no extended side chains that might interact with amino acids outside of the canonical DHP binding site, yet it retains excellent potency. Our results indicating an ∼13-fold higher IC50 for nifedipine block of Cav1.3 compared with Cav1.2 is in line with the decreased potency of nimodipine in block of Cav1.3 compared with Cav1.2 reported by Xu and Lipscombe. Though they did not report an EC50 for agonist potentiation of Cav1.3, Xu and Lipscombe did report a modest shift in V1/2 activation of Cav1.3 by 1 μM concentration of DHP agonist Bay K 8644 (∼ −7 mV), similar to the modest leftward shift in V1/2 activation we observed in Cav1.3 in the presence of 10 μM FPL. Thus, our data show that our expression system recapitulates the primary differences between Cav1.2 and Cav1.3, most notably, the left-shifted activation and lower sensitivity of current to block by DHP antagonists of Cav1.3 compared with Cav1.2.
Another study examined both the binding affinity and block potency of the DHP antagonist PN200-110 (isradipine) for a Cav1.3 clone from human pancreas (Cav1.38A) (Koschak et al., 2001). Interestingly, the KD for [3H]PN200-110 binding was not significantly different between Cav1.38A and Cav1.2 cloned from rabbit cardiac muscle (Tanabe et al., 1987). However, the IC50 for block of current by PN200-110 was reported to be 8.5-fold higher for Cav1.38A than Cav1.2, in excellent agreement with the difference in nifedipine potency in blocking Cav1.3 and Cav1.2 in this study. KD values for binding of DHPs to L-type channels in isolated membranes are invariably lower than IC50 values for current block. For example, the KD for binding of [3H]PN200-110 to the Cav1.2 clone used in this study is 55 pM, whereas the IC50 for PN200-110 block is 7 nM (Peterson et al., 1997). Binding isotherms in both studies clearly indicated a single [3H]PN200-110 binding site, which probably reflects the open, inactivated state of the channel at 0 mV. Thus, it is probable that the Hill slopes different from 1 that we observed for nifedipine block of Cav1.3 and some of the mutant channels used in this study reflect the presence of distinct voltage-dependent channel conformations that regulate DHP affinity.
Though the DHP binding pockets of Cav1.2 and Cav1.3 are highly conserved, our results suggest that relatively minor differences in transmembrane segment IIIS5 and the IIIS5-3P loop can largely account for the difference in potency of nifedipine in block of Cav1.2 and Cav1.3. The IIIS5 helix is clearly a critical component of the Cav1.3 DHP binding pocket, as mutation of T1033 and Q1037 in Cav1.3/DHPi results in a marked loss of nifedipine potency. The side chains of M1030/V1036 in Cav1.3 and Cav1.2 are projected to align to the same face of the IIIS5 helix as the T and Q residues required for high-potency DHP block (Mitterdorfer et al., 1996), supporting our finding that swapping the Cav1.3-specific residue at this position into Cav1.2 (V1036M) shifts nifedipine potency toward that of Cav1.3, and vice versa. Interestingly, the swap of channel subtype-specific residues in this position also results in small reciprocal shifts in V1/2 activation (Table 1). However, only the Cav1.3/MV mutant exhibited slower deactivation, both in the presence and absence of FPL (Table 2). This observation, that decreasing the bulk of the amino acid side-chain at position 1030 in Cav1.3 affects voltage-dependence of activation and the rate of tail-current decay, suggests that position 1030 in IIIS5 (outer pore helix) may interact with IIIS6 (inner pore helix) in a manner that regulates channel gating. Previously published models of DHP binding in Cav1.2 suggest that amino acid residues directly interacting with DHP drugs are conserved between Cav1.2 and Cav1.3 (Cosconati et al., 2007; Tikhonov and Zhorov, 2009). To understand how subtle differences in amino acid sequence might account for a significant difference in nifedipine potency, we constructed homology models of Cav1.3 and Cav1.2 (Fig. 6) on the basis of the recently published high-resolution cryo-EM structure of Cav1.1 (Wu et al., 2016). The models suggest that the increase in side-chain bulk between Cav1.3 and Cav1.2 at position 1030/1036 (M vs. V) could potentially decrease accessibility of nifedipine to the critical Q1037 and F1106 residues (Fig. 6A). In addition, the model predicts that S1100 in Cav1.3 can form a hydrogen bond with N1094, an interaction that could potentially constrain the movement of the 3P helix during nifedipine binding (Fig. 6B). The corresponding positions in Cav1.2 are occupied by an alanine residue (1106) and a glutamate (1100), precluding such an interaction (Fig. 6C). Interestingly, S1100 of Cav1.3 is conserved in the corresponding position of Cav1.1 (S1002) and the position corresponding to N1094 of Cav1.3 is a histidine in Cav1.1 (H996). These residues, with the assistance of D998, may form a hydrogen bond in Cav1.1 (Fig. 6D), which may contribute to the lower binding affinity of Cav1.1 for [3H]PN200-110 (270 pM) (Peterson et al., 1996) compared with Cav1.2 (55 pM) (Peterson et al., 1997). Thus, our model suggests that the effect of the Cav1.3S/A mutation on nifedipine potency is indirect, and that the displacement of the 3P helix may be required for high potency block of Cav1.2 by DHP drugs.

Influence of Cav1.3-specific amino acid residues on the DHP binding pocket. Homology models of Cav1.2 and Cav1.3 were created on the basis of the high-resolution cryoEM structure of Cav1.1. (A) View of the DHP binding pocket of Cav1.3 framed by the IIIS5 helix (bottom), IVS6 helix (top), and the 3P helix (right) with V1036 from Cav1.2 superimposed on M1030. (B) View of the backside of the 3P helix in Cav1.3 showing a potential H-bond between the Cav1.3-specific residues S1100 and N1094. (C) View of the backside of the 3P helix in Cav1.2 with the positions of A1106 and D1100 indicated. (D) View of the backside of the 3P helix in Cav1.1 showing potential H-bond between S1002 and H996 facilitated by D998.
Our studies of FPL potentiation of Cav1.2, Cav1.3, and the various mutant channels also yielded some novel results. First, Fig. 4 clearly shows that FPL is much more potent in potentiating current conducted by Cav1.2 compared with Cav1.3. This difference can be ascribed completely to amino acid differences in the IIIS5-3P loop between these two channels. Nevertheless, the conserved T and Q residues in IIIS5 are clearly important for FPL action on Cav1.3 even though the nearby M1030V mutation did not increase the potency of FPL action in isolation. However, the inclusion of V1030 in Cav1.3+V was critical for stabilizing FPL potentiation of current and revealing the increased sensitivity of this mutant to FPL. Interestingly, despite a complete loss of slowing of deactivation by FPL, the FPL-induced shift in V1/2 activation in Cav1.3/DHPi was not different from that of Cav1.3, suggesting distinct sites of action on Cav1.3 for these two characteristic effects of FPL on L-type channel gating. Unfortunately, we were not able to further resolve the amino acid resides that confer the difference in sensitivity to FPL between Cav1.2 and Cav1.3 beyond the IIIS5-3P loop, as none of the mutations within this domain that increased nifedipine potency improved FPL potency at Cav1.3. It is possible that these determinants may be among the sixteen other amino acid differences between Cav1.2 and Cav1.3 within this domain that we did not examine.
In our studies of the Cav1.3/N6 mutant, we made the unexpected observation that outward current often developed during the course of an experiment. The standard solution set used in this study sets up a large NMDG gradient across the membrane. Mutations in the pore region of Cav1.2 were previously reported to lead to enhanced permeability of NMDG, as evidenced by a marked shift in reversal potential that was abolished by equalizing the NMDG concentration in the extracellular and intracellular solutions (Hockerman et al., 1995). Indeed, we found that, by equalizing the NMDG concentration in the intra- and extracellular solutions, the outward current observed in the Cav1.3/N6 mutant was abolished, and we were able to complete the biophysical and pharmacological measurements reported in Table 1. Likewise, we found that Cav1.3 and the Cav1.3+ mutant tended to undergo current reversal upon FPL application (Supplemental Fig. 3A) that was abolished in Cav1.3 by equalizing the NMDG concentrations. However, even this maneuver left unstable current when FPL was applied to Cav1.3+, and we were unable to determine an EC50 for FPL stimulation of this mutant (Supplemental Fig. 3B). FPL was previously reported to alter the permeability of Cav1.2 (Fan et al., 2001), such that Cd2+ became a permeant ion, rather than a pore blocker, in the absence of Ca2+. Thus, our observation that FPL can induce NMDG permeability in Cav1.3 is consistent with the notion that FPL binding may induce conformational changes in the IIIS5-3P loop that affect the ion selectivity of Cav1.3. Interestingly, neither the Cav1.3+V nor the Cav1.3M/V mutant conducted outward current in the presence of FPL in the standard solution set, suggesting that the M1030 residue may play a role in the observed permeability changes in Cav1.3.
In summary, this study demonstrates that the reduced sensitivity of Cav1.3 to both nifedipine and FPL compared with Cav1.2 can be attributed largely to amino acid differences within the previously defined DHP binding pocket. In the case of nifedipine, this difference can be attributed to the M/V divergence in transmembrane domain IIIS5, and an S/A divergence in the IIIS5-3P loop. Our homology models suggest that divergence in IIIS5 results in distinct stearic effects on drug binding, whereas the divergence in the IIIS5-3P loop may regulate displacement of the 3P helix upon ligand binding.
Acknowledgments
We gratefully acknowledge Dr. Terrance Snutch (University of British Columbia) and Dr. Tuck-Wah Soong (National University of Singapore) for generously providing cDNAs encoding Cav1.2 and Cav1.3 subunits, respectively.
Authorship Contributions
Participated in research design: Wang, Hockerman.
Conducted experiments: Wang, Tang, Harvey, Hockerman.
Contributed new reagents or analytic tools: Salyer, Li, Rantz, Lill.
Performed data analysis: Wang, Tang, Harvey, Hockerman.
Wrote or contributed to the writing of the manuscript: Wang, Hockerman.
Footnotes
- Received March 9, 2018.
- Accepted June 28, 2018.
↵1 Y.W. and S.T. contributed equally to this work.
This work was supported by a grant from the American Heart Association Midwest Affiliate [15GRNT25750021] to G.H.H.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- Bay K 8644
- methyl 2,6-dimethyl-5-nitro-4-[2-(trifluoromethyl)phenyl]-1,4-dihydropyridine-3-carboxylate
- BTZ
- benzothiazepines
- DHP
- dihydropyridines
- FPL 64176
- 2,5-dimethyl-4-[2-(phenylmethyl)benzoyl]-1H-pyrrole-3-carboxylic acid methyl ester
- HEPES
- 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid
- nifedipine
- 1,4-dihydro-2,6-dimethyl-4-(2-nitrophenyl)-3,5-pyridinecarboxylic acid dimethyl ester
- NMDG
- N-methyl-d-glucamine
- PAA
- phenylalkylamine
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}