


Abstract
The physiological implications of lysophosphatidic acid occupancy of individual receptors are largely unknown because selective agonists/antagonists are unavailable currently. The molecular cloning of three high-affinity lysophosphatidic acid receptors, LPA1, LPA2, and LPA3, provides a platform for developing receptor type-selective ligands. Starting with an N-acyl ethanolamide phosphate LPA analog, we made a series of substitutions at the second carbon to generate compounds with varying spatial, stereochemical, and electronic characteristics. Analysis of this series at each recombinant LPA receptor using a guanosine 5′-O-(3-[35S]thio)triphosphate (GTP[γ35S]) binding assay revealed sharp differences in activity. Our results suggest that these receptors have one spatially restrictive binding pocket that interacts with the 2-substituted moieties and prefers small hydrophobic groups and hydrogen bonding functionalities. The agonist activity predicted by the GTP[γ35S] binding assay was reflected in the activity of a subset of compounds in increasing arterial pressure in anesthetized rats. One compound with a bulky hydrophobic group (VPC12249) was a dual LPA1/LPA3 competitive antagonist. Several compounds that had smaller side chains were found to be LPA1-selective agonists.
Lysophosphatidic acid (LPA; 1-acyl, 2-hydroxyl-sn-glycerol-3-phosphate) is a family of lysophospholipid mediators that elicit diverse biological responses including calcium mobilization, cytoskeletal rearrangements, and mitogenesis (Moolenaar, 1994). Transient rises in blood pressure in rats and guinea pigs after intravenous LPA injection have also been documented (Tokumura et al., 1978). LPA is released by activated platelets and accumulates in serum to low micromolar levels (Schumacher et al., 1979; Simon et al., 1982; Watson et al., 1985). The induction of platelet aggregation and fibroblast recruitment along with its mitogenic capabilities implicate this lipid as a wound healing hormone (Moolenaar, 1995). Another pathologic fluid containing substantial amounts of LPA is the malignant ascites characteristic of ovarian cancer (Xu et al., 1995). Interestingly, the LPA found in these two fluids differs in that LPA from ascitic fluid is reportedly enriched in 2-acyl LPA species (Xu et al., 1995). Study of this 2-acyl LPA isoform is made difficult, however, by its chemical instability (i.e., the rapid migration of the acyl chain to the thermodynamically favored 1 position in an aqueous environment).
LPA signals cells in part via a set of G protein-coupled receptors named LPA1, LPA2, and LPA3 (formerly Edg-2, Edg-4, and Edg-7)1 (Hecht et al., 1996; An et al., 1997b; Bandoh et al., 1999; Im et al., 2000b). These receptors share 50 to 55% identical amino acids and cluster with five other receptors (S1P1, S1P2, S1P3, S1P4, and S1P5, formerly Edg-1, Edg-5, Edg-3, Edg-6, and Edg-8) for the structurally-related lipid sphingosine 1-phosphate (S1P) (An et al., 1997a; Lee et al., 1998; van Brocklyn et al., 2000; Im et al., 2000a; Yamazaki et al., 2000). LPA1 is most associated with activation of Giα pathways and is expressed in oligodendrocytes (Allard et al., 1998; Weiner et al., 1998) and peripheral tissues (An et al., 1998), whereas LPA2 and LPA3 are associated most prominently with Gq/11α pathways (Bandoh et al., 1999; Im et al., 2000b). LPA2 mRNA is found in testis and peripheral blood leukocytes (An et al., 1998) whereas LPA3 mRNA has been localized to prostate, testes, pancreas, kidney, and heart (Bandoh et al., 1999; Im et al., 2000b).
The physiologic implications of occupation of individual LPA receptors are largely unknown, partly because of a lack of receptor subtype selective ligands. This paucity led us to design and test a series of 2-substituted ethanolamide derivatives varying in degrees of size, hydrophobicity, and stereochemistry. The parent compound of our series,N-acyl ethanolamide phosphate (NAEPA) has been shown to be nearly indistinguishable from LPA in evoking platelet aggregation (Sugiura et al., 1994) and GTP[γ35S] binding at LPA1 and LPA2 containing membranes (Im et al., 2000b) but is distinctly less active than LPA at recombinant LPA3 (Im et al., 2000b) or in depolarizing Xenopus laevis oocytes (Santos et al., 2000). A pair of 2-substituted NAEPA compounds have already been reported. The 2-carboxyl-containing compound (N-acyl serine phosphoric acid) has been documented to antagonize both LPA-driven platelet aggregation (Sugiura et al., 1994) and oocyte depolarization (Liliom et al., 1996) and is a partial agonist at mammalian LPA receptors (Hooks et al., 1998). The 2-methylene hydroxy-containing compound, which is an analog of 2-acyl LPA wherein the ester is replaced by an amide, was shown by us to activate recombinant LPA receptors in a stereoselective fashion, whereas mitogenic responses and platelet aggregation did not show this stereoselectivity (Hooks et al., 2001). Because of the interesting properties of the existing 2-substituted NAEPA compounds, we synthesized and examined an expanded series of these compounds.
In this article, we present an analysis of several more compounds from this series. In addition to confirming the strong preference of each recombinant LPA receptor for a single enantiomer, we found compounds that exhibited increased potency at the LPA1receptor relative to LPA. The predicted enantiomeric selectivity was observed in a variety of assays, including the hypertensive response in anesthetized rats. Additionally, a dual LPA1/LPA3 competitive antagonist was identified and characterized.
Experimental Procedures
Transient Expression in HEK293T Cells.
The appropriate receptor plasmid DNA (encoding mouse LPA1, human LPA2 or human LPA3) was mixed with equal amounts of expression plasmids (pcDNA3) encoding a mutated (C351F) rat Gi2α, cow β1, and γ2 proteins, and these DNAs were used to transfect monolayers of HEK293T cells (where ‘T’ indicates expression of the SV-40 virus large T antigen) using the calcium phosphate precipitate method (Wigler et al., 1977). After about 60 h, cells were harvested, and membranes were prepared, aliquoted, and stored at −70°C until use (Im et al., 2000b).
GTP[γ35S] Binding.
The GTP[γ35S] assay was performed as described previously (Im et al., 2000b). Membranes containing 5 μg of protein were incubated in 0.1 ml of GTP-binding buffer (50 mM HEPES, 100 mM NaCl, 10 mM MgCl2, pH 7.5) containing 5 μg of saponin, 10 μM GDP, 0.1 nM GTP[γ35S] (1200 Ci/mmol), and indicated lipid(s) for 30 min at 30°C. Samples were analyzed for membrane-bound radionuclide using a Brandel Cell Harvester (Gaithersburg, MD). The C351F mutation renders the Gi2α protein resistant to inactivation by pertussis toxin or the alkylating agent N-ethylmaleimide; in practice, however, background binding was sufficiently low to obviate these maneuvers.
Measurement of cAMP Accumulation.
Assays for cAMP accumulation were conducted on populations of 5 × 105 cells stimulated with 10 μM forskolin in the presence of the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine for 15 min at 30°C. cAMP was measured by automated radioimmunoassay.
Measurement of Intracellular Calcium.
We used a FLIPR (Molecular Devices, Inc., Menlo Park, CA) to measure intracellular calcium in A431 and HEK293T cells. A431 cells were seeded (∼50,000 cells/well) in 96-well, clear bottom black microplates (Corning Costar Corp., Cambridge, MA) and left overnight in CO2incubator at 37°C. HEK293T cells were treated likewise, but seeded onto poly(d-lysine) coated microplates (Becton Dickinson, Franklin Lakes, NJ). A431 cells were dye-loaded with 4 μM Fluo-3 AM ester (Molecular Probes Inc., Eugene, OR) in a loading buffer (1× HEPES-buffered saline, pH 7.4, containing 20 mM HEPES, 0.1% BSA, and 2.5 mM probenecid) for 1 h at 37°C. Cells were then washed four times with the loading buffer and exposed in the FLIPR to sets of compounds. HEK293T cells were loaded with 2 μM Fluo-4 AM ester (Molecular Probes Inc., Eugene, OR) in the same loading buffer without probenecid for 30 min and washed four times before being exposed to compounds in the FLIPR. In all cases, each concentration of each compound was tested in at least quintuplicate.
Determination of KI.
K I values for VPC12249 in experiments were determined by plotting the log of Dose Ratio-1 at each concentration of inhibitor against the log concentration of inhibitor. Thex-intercept of the linear transformation is equal to the inverse log of the K I.
Stable Expression in RH7777 Cells.
Rat hepatoma RH7777 cell monolayers were transfected with the mLPA1plasmid DNA using the calcium phosphate precipitate method and clonal populations expressing the neomycin phosphotransferase gene were selected by addition of Geneticin (G418) to the culture media. The RH7777 cells were grown in monolayers at 37°C in a 5% CO2/95% air atmosphere in growth media consisting of: 90% minimal essential medium, 10% fetal bovine serum, 2 mM glutamine, and 1 mM sodium pyruvate.
Cardiovascular Measurements.
All procedures were performed on male Wistar rats in accordance with National Institutes of Health and University of Virginia animal care and usage guidelines. Anesthesia was induced by 5% halothane (in 100% O2). Rats were intubated and artificially ventilated with 1.5 to 1.8% halothane in 100% O2 for surgical procedures. A femoral artery was cannulated to record mean arterial pressure (MAP) and heart rate (HR), and a femoral vein was cannulated to administer anesthetic agents. A femoral vein was cannulated for administration of lipids. The left splanchnic nerve was isolated via a retroperitoneal approach, and the segment distal to the suprarenal ganglion was placed on two Teflon-coated silver wires that had been bared at the tip (250 μm bare diameter; A-M Systems, Everett, WA). The nerve and wires were embedded in a dental impression material (polyvinysiloxane; Darby Dental Supply, Westbury, NY), and the wound was closed around the exiting recording wires.
On completion of surgery, the halothane anesthesia was terminated and was replaced by a α-chloralose (30 mg/kg solution in 3% sodium borate; 70 mg/kg initial bolus followed by hourly supplements of 20 mg/kg i.v.; Fisher Scientific, Pittsburgh, PA). Rats were allowed to stabilize for 45 min before tests began. End-tidal CO2 was monitored by infrared spectroscopy and was maintained between 3.5 and 4.0%. Body temperature (measured rectally) was maintained at 37°C.
All physiological variables were monitored on a chart recorder (model RS 3600; Gould, Valley View, OH) and simultaneously stored on a videocassette recorder via a digitizer interface (model 3000A; frequency range: DC-22 kHz; Vetter Digital, Rebersburg, VA) for off-line computer analysis. Data were analyzed with Spike 2 (Cambridge Electronics). The MAP was calculated from the pulse pressure measured by a transducer (Statham P10 EZ; Gould) connected to the brachial arterial catheter. The HR was determined by triggering from the pulse pressure (Biotach; Gould). Splanchnic nerve activity was filtered (10 Hz-3 kHz band pass with a 60-Hz notch filter), full-wave rectified, and averaged in 1-s bins. The femoral venous catheter (dead space, 100 μl) was loaded with each lipid and was flushed with 200 μl of saline to expel the drug.
Materials.
Chemicals for syntheses were purchased from Aldrich Chemical Company (Milwaukee, WI), Sigma Chemicals (St. Louis, MO), Advanced ChemTech Chemical Company (Louisville, KY), and/or NovaBiochem Chemical Company (Laufelfingen, Switzerland) and were used without further purification. GTP[γ35S] was purchased from Amersham Pharmacia Biotech (Piscataway, NJ), Fura-3 and Fura-4 AM were purchased from Molecular Probes (Eugene, OR), A431 and RH7777 cells were purchased from the American Type Culture Collection (Manassas, VA), and tissue culture media and serum was from Invitrogen (Carlsbad, CA). HEK293T cells were a gift from Dr. Judy White's laboratory (Dept. of Cell Biology, University of Virginia, Charlottesville, VA) while G protein β and γ DNAs were a gift from Dr. Doug Bayliss (Dept. of Pharmacology, University of Virginia). LPAs (1-oleoyl and 1-palmitoyl) and dioctyl glyceryl pyrophosphate were purchased from Avanti Polar Lipids (Alabaster, AL).
Results
Using N-oleoyl ethanolamide phosphoric acid (NAEPA) as a lead structure, we synthesized a series of 2-substituted LPA analogs (Fig. 1). The details of the synthesis and analysis of the full set of compounds in this series are to be provided in a separate publication (W. L. Santos, C. E. Heise, K. R. Lynch, T. L. Macdonald, in preparation). Each compound was characterized by 1H NMR,13C NMR, and mass spectrometry.

Structures of 2 substituted ethanolamide phosphoric acid compounds. Compounds in the left column contain the denoted functional group at R 1 with a hydrogen atom at the R 2 position; those in the right column contain indicated functional group atR 2 with a hydrogen atom at theR 1 position. Compounds were synthesized, purified and analyzed as will be described in Santos et al. (in preparation).
The differential coupling of LPA1 versus LPA2 and LPA3, the lack of a reliable radioligand binding assay, and the near ubiquity of endogenous LPA responses prohibited the use of most common receptor assay techniques (i.e., measurements of adenylyl cyclase activity, calcium mobilization, and radioligand binding) to assess each compound's activity. Therefore, we adapted a GTP[γ35S] binding assay to measure the relative efficacies and potencies of each compound compared with LPA as described previously (Im et al., 2000b; McAllister et al., 2000). This assay isolates each recombinant LPA receptor and allows analysis of all three receptors using the same system. Note that membranes from HEK293T cells transfected with only G protein DNAs (ie, no receptor DNA) were devoid of LPA-stimulated GTP binding despite expressing endogenous LPA receptors (Fig. 2D).

GTP[γ35S] binding to HEK293T cell membranes in response to LPA and analogs with hydrophilic constituents. HEK293T cells were transfected with LPA1 (A), LPA2 (B), or LPA3 (C) receptor DNAs plus G protein DNAs or G protein DNAs alone (D) (see Experimental Procedures for details). Lipids were dissolved at 1 mM in aqueous 1.0% fatty acid-free BSA and diluted in aqueous 0.1% BSA. LPA analogs contained oleoyl (18:1) fatty acid chains. Closed characters represent R 1 enantiomers and open characters represent R 2 enantiomers; all sets of curves include a 1-oleoyl-(S)-LPA curve as a reference. Points are in triplicate and are representative of at least two experiments. Typical values for zero and 100% binding were 300 and 800 dpm/tube for LPA1, 300 and 1000 dpm/tube for LPA2, and 300 and 2000 dpm/tube for LPA3, respectively.
Many NAEPA compounds with various 2-substituents were synthesized and examined; those with methyl, ethyl, isopropyl, benzyl, methylene hydroxy, carbomethyl, methylene amino, and benzyl-4-oxybenzyl functionalities (Fig. 1) are reported herein. Because the 2 position is a prochiral site, both enantiomers (at R 1and R 2) of the eight compounds were synthesized. Three patterns were revealed when we tested the agonist compounds in this series at the three LPA receptors in the broken cell assay. First, each LPA receptor showed a marked selectivity (1 log order or more) for one enantiomer. This confirms and extends our previously reported observation of stereoselectivity by LPA receptors for NAEPA compounds containing the 2-carboxyl (Hooks et al., 1998) or 2-methylene hydroxy groups (Hooks et al., 2001). Second, those compounds with substitutions at the R1 position were invariably the more potent agonists (Fig. 2,3). Third, agonist potency decreases as the bulk of the substituent increases.

GTP[γ35S] binding to HEK293T cell membranes in response to LPA and analogs with hydrophobic substituents. HEK293T cells were transfected with LPA1 (A), LPA2 (B), or LPA3 DNAs (C) and G protein DNAs. Lipids were dissolved at 1 mM in aqueous 1.0% fatty acid-free BSA and diluted in aqueous 0.1% BSA. Open characters representR 1 enantiomers and closed characters represent 1-oleoyl-(S)-LPA curve as a reference. All compounds contain oleoyl (18:1) fatty acid chains. Points are in triplicate and are representative of at least two experiments. Typical values for zero and 100% binding were 300 and 800 dpm/tube for LPA1, 300 and 1000 dpm/tube for LPA2, and 300 and 2000 dpm/tube for LPA3, respectively.
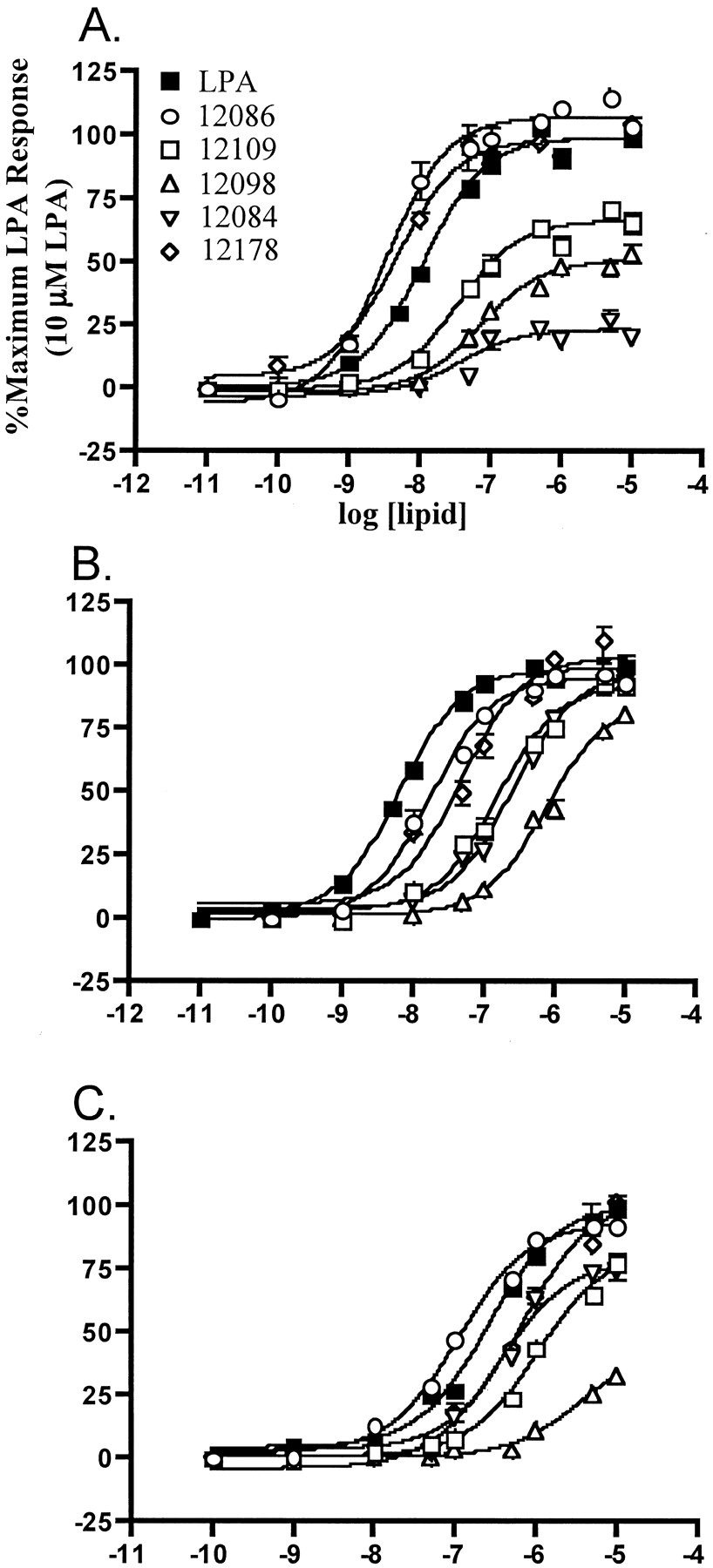
The 2-substituted NAEPA compounds containing either hydrophilic (methylene hydroxy, carbomethyl, methylene amino) or hydrophobic moieties (methyl, ethyl, isopropyl, benzyl) exhibited agonist activity in the GTP[γ35S] binding assay (Figs. 2 and3). The smaller groups conferred greater potency, with the methyl (VPC12086), methylene hydroxy (VPC31143), and methylene amino (VPC12178) compounds being more potent than 1-oleoyl LPA at LPA1 (Figs. 2 and 3 and Table1). In addition, because the 2-substituent becomes bulkier, the efficacy was noticeably reduced at this receptor. In contrast, bulkier hydrophobic side chains, although less potent, were fully efficacious at the LPA2receptor (Fig. 3B and Table 1). As was observed with the LPA1 receptor, the small methyl and methylene amino groups conferred the highest potency at the LPA2 receptor, but none of these compounds proved more potent than 1-oleoyl LPA at this site. The LPA3 receptor exhibited much the same profile as the LPA2 receptor with regard to efficacies and potencies of compounds relative to LPA. However, the LPA3 receptor characteristically exhibited higher (1–2 log order) EC50 values for all compounds, including LPA. Presumably, the LPA3 receptor has an intrinsically lower affinity for LPA and LPA analogs (Fig. 3C and Table 1). Like the hydrophilic compounds, each LPA receptor showed strong stereoselectivity for a hydrophobic substituent in the R1 position (Fig. 1 and Table 1).
Agonist activities of 2-substituted N-oleoyl ethanolamide phosphoric acid compounds
We wished to determine whether the stereoselectivity predicted by the broken cell assay at recombinant LPA receptors extends to endogenous LPA responses. We have reported previously that this is not the case for LPA stimulation of tritiated thymidine incorporation as an index of mitogenesis (Hooks et al., 2001). In contrast, the rank order potencies of the compounds predicted by the GTP[γ35S] binding assay are mimicked by calcium mobilizing activities in a variety of cell lines including A431, HEK293, and MDA MB231 (data not shown). To investigate an LPA response in a physiologic context, we monitored MAP, heart rate, and postganglionic sympathetic tone in anesthetized adult rats as a function of LPA or LPA analog administration. LPA has been shown previously to increase blood pressure transiently in this model (Tokumura et al., 1978). As shown in Fig. 4, intravenous injection of three enantiomeric pairs of compounds resulted in a transient increase in MAP with the same pattern of stereoselectivity as observed with the in vitro assays. Concomitant with this rise in MAP was a decrease in heart rate and sympathetic output indicative of baroreceptor reflex response (Fig. 4).

Blood pressure changes in response to intravenous injection of LPA analogs to ventilated anesthetized rats. A, representative trace of change in MAP, HR, and sympathetic outflow [splanchnic nerve activity (SNA)] on injection of 50 pmol of compound VPC12178. B, same representative traces on administration of 50 pmol of the enantiomeric compound, VPC12048. C to E, measurement of change in MAP in response to administration of LPA analogs at varying concentrations. Points are in triplicate from three rats (C), or duplicate from two rats (D, E). VPC31143 and VPC31144 in (D) contain palmitoyl (16:0) fatty acid chains.
The compounds that were only slightly efficacious at LPA1 (e.g., the benzyl-containing VPC12084) were assayed for their ability to antagonize LPA-induced GTP[γ35S] binding. Although this compound did block LPA activity in the GTP[γ35S] binding assay (not shown), the benzyl compound (VPC12084) was revealed to posses appreciable agonist activity in assays with greater levels of amplification (e.g., whole cell assays of calcium mobilization or inhibition of cAMP accumulation) (Fig.5). In the course of exploring variations of the benzyl substituent, we found that a benzyl-4-oxybenzyl substituent in the same relative configuration (R 1, VPC12204) had reduced, but still measurable, agonist activity in whole cell assays (Fig. 5). However, its enantiomer [i.e., VPC12249, the compound with the benzyl-4-oxybenzyl substituent in the S(R 2; see Fig. 1) configuration] was completely devoid of agonist activity in the whole-cell assays (Fig. 5) and in the GTP[γ35S] binding assay (not shown).
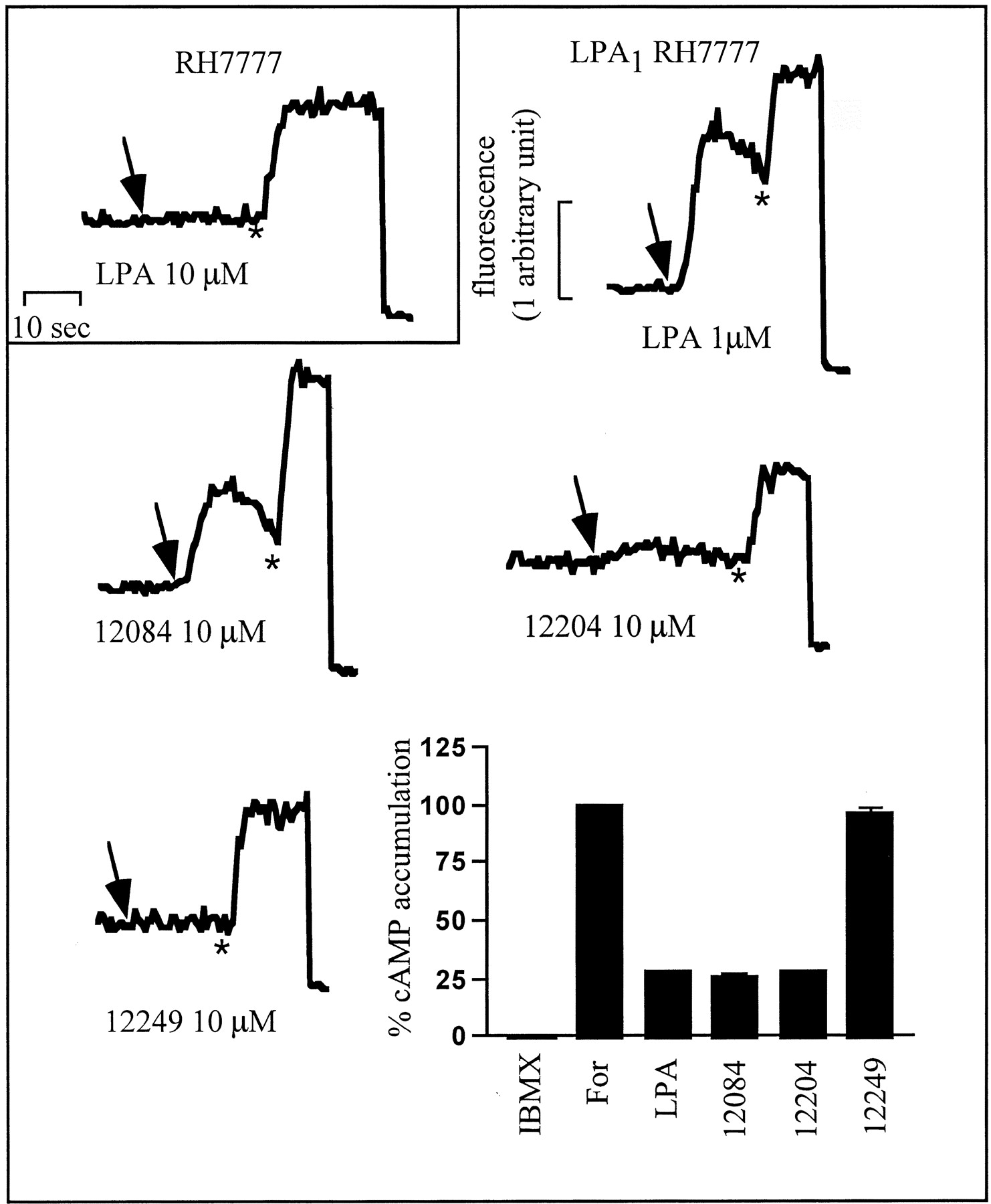
Calcium mobilization traces and inhibition of cAMP accumulation assay using mouse LPA1 expressing RH7777 cells. Upper left shows a calcium transient in response to LPA in untransfected cells. Remaining calcium mobilization traces are from LPA1 receptor stably expressing RH7777 cells. Arrow indicates addition of denoted compound; ∗ indicates addition of digitonin (to 75 μM) followed by EDTA (to 5 μM) to measure total intracellular calcium. Bar graph is also from LPA1 receptor stably expressing RH7777 cells showing accumulation of cAMP data.
We tested VPC12249 for its ability to block LPA-induced GTP[γ35S] binding at each recombinant LPA receptor. As shown by the rightward, parallel shifts in the concentration response curves as a function of VPC12249 concentration, this compound is a surmountable antagonist at the LPA1 and LPA3 but not the LPA2, receptors (Fig.6). The K Ivalues for VPC12249 determined by Schild regression are 137 and 428 nM at the LPA1 and LPA3receptors, respectively, in this assay. The same activity was determined with human LPA1 using a recombinant baculovirus-infected insect Sf9 cell membrane preparation (M. D. Davis and K. R. Lynch, unpublished observations).

Effect of the benzyl 4-oxybenzyl compounds, VPC12204 and VPC12249, on GTP[γ35S] binding. GTP[γ35S] binding assays of LPA1- (A), LPA2- (B), and LPA3- (C, D) transfected HEK293T cell membranes showing LPA concentration response curves with increasing concentrations of VPC12204 or VPC12249. Points are in triplicate and are representative of at least two experiments. Insert represents Schild regression analysis of concentration response curves.
The antagonist activity measured in the broken cell assays was confirmed in whole-cell experiments wherein LPA-induced increases in free intracellular calcium in HEK293T cells were blocked. This cell type expresses the LPA1 and LPA3 but not LPA2 receptor genes as determined by RT-PCR (not shown). As documented by the concentration response curves shown in Fig.7A, increasing concentrations of VPC12249 resulted in parallel, rightward shifts in the LPA concentration response curves (K I = 132 nM). The extent of rightward shift observed in the same experimental protocol with A431 cells, which express the LPA2 as well as the LPA1 and LPA3 genes (RT-PCR not shown), was much smaller (K I = 1970 nM; Fig. 7C) as predicted from the lack of antagonist activity of VPC12249 at the calcium-mobilizing LPA2 receptor in the GTP-binding assay (see Fig. 6). The blocking action of VPC12249 was not a general postreceptor event, as shown by the lack of antagonism of ATP-evoked calcium transients (Fig. 7B). Inhibition of forskolin-induced increases in cAMP levels in RH7777 cells stably expressing LPA1 was also inhibited by VPC12249 (Fig. 7D).
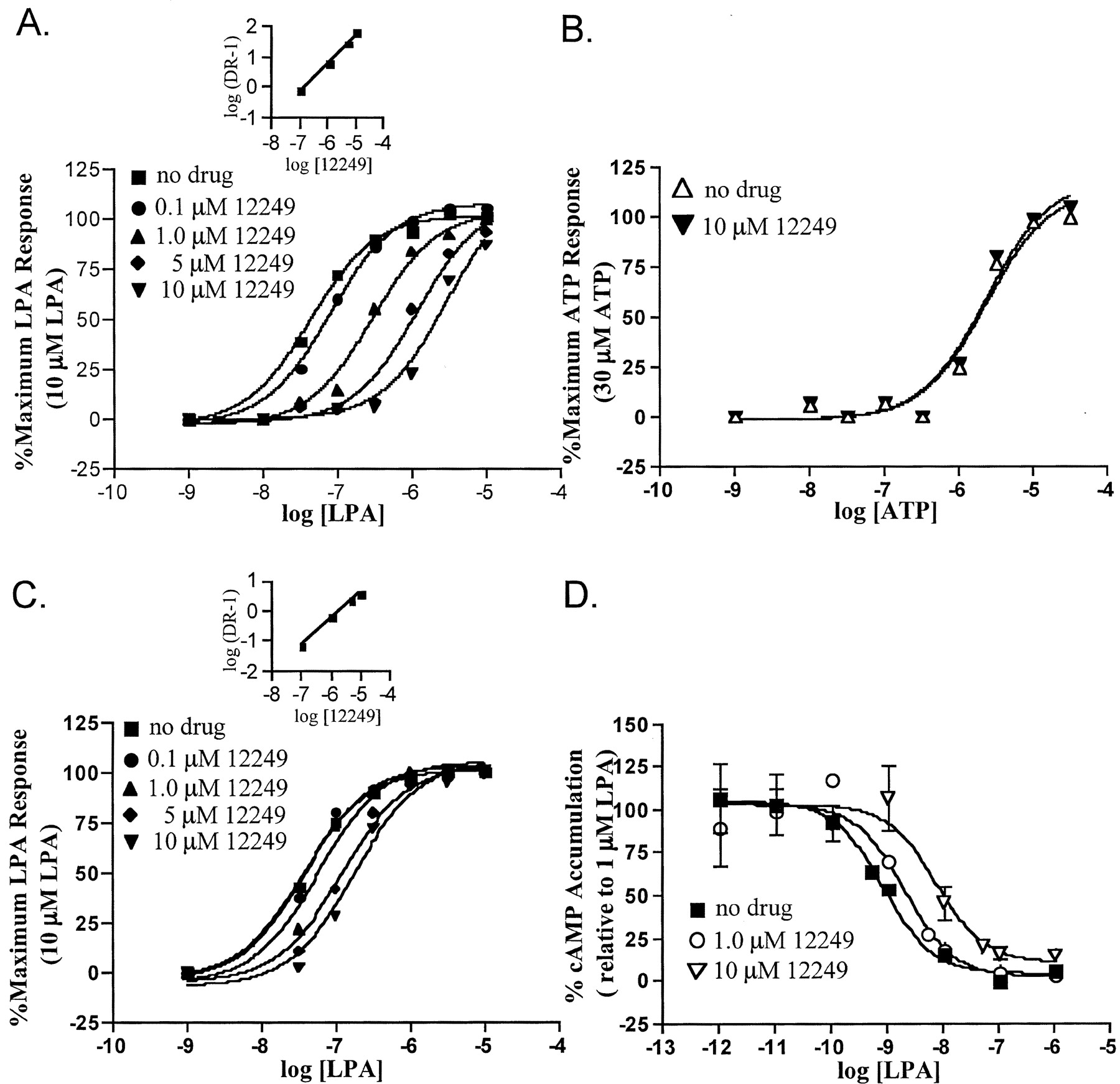
Effect of the benzyl 4-oxybenzyl compound VPC12249 on calcium mobilization and inhibition of cAMP accumulation. A, calcium mobilization of HEK293T cells showing LPA concentration response curves with increasing concentrations of VPC12249. B, calcium mobilization of HEK293T cells showing ATP dose-response curve with and without 10 μM VPC12249. C, calcium mobilization of A431 cells showing dose response curves with increasing concentrations of VPC12249. D, inhibition of forskolin-driven rises in cAMP on LPA1 receptor stably expressing RH7777 cells showing LPA concentration response curve with increasing concentrations of VPC12249. Calcium data points are replicates of at least five and representative of two experiments. Inhibition of cAMP accumulation data points are in duplicate with 0% averaging 3 pmol/tube and 100% averaging 80 pmol/tube cAMP. Inset, Schild regression analysis of concentration response curves.
Discussion
The lack of medicinal chemistry associated with the LPA1, LPA2, and LPA3 receptors prompted us to develop LPA receptor selective agonists and antagonists. Among the most fruitful series that we have synthesized are 2-substituted N-acyl ethanolamide-phosphates. The lead compound, N-palmitoyl ethanolamide phosphate (Sugiura et al., 1994), in which the glycerol moiety of LPA is replaced with ethanolamine, has been shown to be indistinguishable from LPA in potency and efficacy at the mammalian LPA1 and LPA2 receptors (Im et al., 2000b) but is less potent than LPA at the LPA3 receptor (Im et al., 2000b). Although the synthetic routes to the compounds in our series did not proceed through NAEPA, they all contain this backbone (Fig. 1).
We have adapted a GTP[γ35S] binding assay to analyze directly the activation of individual recombinant LPA receptors, which allowed determination of relative efficacies and potencies at each receptor with a common assay platform. The same concentration response curves were obtained regardless of whether the recombinant receptor used exogenous G proteins from various mammalian species (HEK293T cells; see Experimental Procedures) or endogenous G proteins (RH7777 cells, data not shown). More importantly, the rank-order potencies established with the broken-cell, membrane-based GTP[γ35S] binding assay were maintained in whole-cell assays of calcium mobilization and inhibition of cAMP accumulation as well as in blood pressure responses in whole animals. Thus, our primary assay for compound potency and efficacy is a valid predictor of activity at endogenous LPA receptors.
In our exploration of various 2-substituents of the NAEPA backbone, several trends became apparent. First, each LPA receptor showed a marked (1 log order or more) preference for one enantiomer. We first reported this differential activity (albeit at undefined LPA receptors) using the 2-carboxyl-containing compound N-acyl serine phosphoric acid (Hooks et al., 1998) and later with the 2-methylene hydroxy compound at LPA1, LPA2, and LPA3 (Hooks et al., 2001). This stereoselectivity was evidenced further in vivo by inducing transient increases in blood pressure in ventilated, anesthetized rats in a dose-dependent manner. The lack of stereoselectivity of mitogenic responses to LPA mimetics such as the 2-methylene hydroxy compound is central to our argument that such responses are, at least in some cell types, independent of LPA1–3 receptors (Hooks et al., 2001).
Second, most substitutions were well tolerated in that they resulted in agonist ligands (Table 1). Although most active compounds were partial agonists with reduced potency (relative to LPA), the methyl (VPC12086), methylene hydroxy (VPC31143), and methylene amino (VPC12178) compounds are notable in that they are more potent than LPA at the LPA1 receptor. A pattern observed with all three LPA receptors was that activity is conferred for hydrophobic or hydrophilic compounds only when the substituent is at theR 1 position and theR 2 position is a hydrogen atom (refer to Fig. 1 for structures; see also Figs. 2 and 3). In aggregate, these data indicate to us that each LPA receptor has one spatial region within the ligand binding site for hydrophobic or hydrophilic functionalities and is restrictive in nature by recognizing only smaller substituents as potent agonists.
Third, we note again that the LPA3 receptor, unlike the LPA1 and LPA2receptors, discriminates between unsaturated and saturated acyl groups (Bandoh et al., 1999; Im et al., 2000b). Although this article focuses on oleoyl compounds, we have examined a number of these compounds as palmitoyl forms and find consistently that the saturated analogs are less potent at only the LPA3 receptor. Furthermore, LPA3 seems to have a lower affinity for LPA and LPA analogs, as indicated by a rightward shift in all concentration response curves.
Several of the compounds described deserve special mention. The first of these is the methylene hydroxy-containing compound (VPC31143). This compound is an analog of 2-acyl LPA in which the ester is replaced by an amide, thus conferring chemical stability (i.e., chain migration is prevented). The LPA in malignant ascites has been reported to consist of substantial amounts of the 2-acyl species; this is the form of LPA that reportedly confers a greater biological activity on ovarian cancer cells (Xu et al., 1995). The 2-acyl LPA analog (VPC31143) was equipotent to 1-acyl LPA at the LPA3 receptor, less potent at the LPA2 receptor, but more potent at the LPA1 receptor, which supports the notion that 2-acyl LPA has a different biologic activity than 1-acyl LPA.
A second pair of compounds worth noting exhibit significantly greater potency at the LPA1 receptor compared with either the LPA2 or LPA3 receptors: the methylene hydroxy- and methylene amino-containing compounds (VPC31143 and VPC12178, respectively) are both fully efficacious and highly potent at the LPA1 receptor with at least a 10-fold lower EC50 value at this receptor type than at either the LPA2 or LPA3 sites (Table 1). To our knowledge, these compounds represent the first receptor type selective agonists and as such might prove useful for elucidating the receptor entities responsible for various effects in cultured cells and tissues.
A third interesting pair of compounds contain a benzyl-4-oxybenzyl substituent at the 2 position in either the R orS configuration (VPC12204 and VPC12249, respectively). These compounds are potent, dual LPA1/LPA3 receptor antagonists. VPC12204 is more potent at inhibiting LPA induced activation of LPA3 but retains the partial agonist activity of the parent benzyl compound (VPC12084) at LPA1 (Fig. 5). The enantiomer of this compound (VPC12249) is entirely devoid of agonist activity in our most sensitive assays. We do not know if VPC12249 is an inverse agonist, but its blockade is surmountable, and thus can be considered a competitive antagonist. The K I value calculated for VPC12249 at inhibiting calcium mobilization in HEK293T cells is ∼130 nM. These cells express the LPA1 and LPA3 receptors as judged by RT-PCR analysis; the calcium response to LPA probably flows through both receptors because pretreatment with pertussis toxin partially blunts but does not completely inhibit the resultant calcium transient (our unpublished observations). In addition, the calculatedK I value of VPC12249 at the LPA1 receptor from the GTP[γ35S] binding assay is nearly identical to that obtained for these whole cells (137 versus 132 nM). The greater than 10-fold higher K I value measured with A431 cells is expected in view of the low potency of VPC12249 in antagonizing LPA2 mediated responses in assays of the recombinant LPA2 receptor. Furthermore, the agreement in data from the membrane based GTP[γ35S] assay and the whole cell assays of VPC12249 lends further credence to the broken cell assay system for rapidly assessing the activity of novel chemical entities at recombinant LPA receptors.
A recent report from Tigyi and associates describes a dual LPA1/LPA3 receptor antagonist, dioctyl glycerol pyrophosphate (Fisher et al., 2001), although this compound is a much less potent blocker of the LPA1 site. Our laboratory is in the process of comparing this compound with VPC12249 in our assay systems. Our preliminary results support the contention that dioctyl glycerol pyrophosphate is a LPA3 receptor blocker, although we find it less potent than VPC12249.
In summary, we have continued to explore variations of theN-acyl ethanolamide phosphate backbone in search of novel chemical entities that might prove useful in probing LPA biology. We have found that compounds with small substituents at the second carbon atom are in general potent and efficacious agonists; indeed, in some cases, these compounds are more potent and efficacious than LPA, providing two LPA1 receptor type selective agonists. Our observation that efficacy decreased sharply with the bulk of hydrophobic 2-substituents led to the discovery of a dual LPA1/LPA3 competitive antagonist, VPC12249. We are profiling this molecule in a variety of in vitro and in vivo experimental systems as well as attempting to develop similar compounds with enhanced potency and receptor type selectivity.
Acknowledgments
We thank Dr. Shelley B. Hooks (Dept. of Pharmacology, University of North Carolina, Chapel Hill, NC) and Michael D. Davis (Dept. of Biochemistry, University of Virginia) for sharing unpublished data. We thank also Dr. Gabor Tigyi (Dept. of Physiology, University of Tennessee at Memphis) for sharing his manuscript before publication inMolecular Pharmacology. We are grateful to Dr. Patrice Guyenet (Dept. of Pharmacology, University of Virginia) for use of his laboratory's equipment to make the blood pressure measurements reported herein and to Dr. John Raymond (Dept. of Medicine/Nephrology, Medical University of South Carolina, Charleston, SC) for allowing us time on the FLIPR instrument.
Footnotes
- Received August 7, 2001.
- Accepted September 25, 2001.
-
↵1 The IUPHAR subcommittee on lysophospholipid receptor nomenclature has recommended that henceforth the colloquial ‘Edg’ nomenclature be replaced with LPA (or S1P) subscript number, where the number indicates order of molecular cloning. Thus Edg-2 becomes LPA1, Edg-4 becomes LPA2, and Edg-7 becomes LPA3.
-
This work was supported by National Institutes of Health research grants R01-GM52722 and R01-CA88994 (to K.R.L. and T.L.M.) and predoctoral traineeship T32-GM07055 (to C.E.H. and W.L.S.) and by American Heart Association postdoctoral fellowship AHA-130274N (to A.M.S.). The FLIPR was purchased with National Institutes of Health shared equipment grant S10-RR13005 (to John R. Raymond, Medical University of South Carolina).
Abbreviations
- LPA
- lysophosphatidic acid
- NAEPA
- N-acyl ethanolamide phosphoric acid
- Edg
- endothelial differentiation gene
- HEK
- human embryonic kidney
- GTP[γ35S]
- guanosine 5′-O-(3-[35S]thio)triphosphate
- FLIPR
- fluorimetric imaging plate reader
- BSA
- bovine serum albumin
- MAP
- mean arterial pressure
- HR
- heart rate
- RT-PCR
- reverse transcription-polymerase chain reaction
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}