


Abstract
Kinins are involved in a variety of physiological and pathophysiological processes related to cardiovascular homeostasis, inflammation, blood flow, and nociception. Under physiological conditions, the bradykinin B2 (BKB2) receptor is constitutively expressed and mediates most of kinins' actions. However, the mechanisms regulating BKB2 receptor gene expression are still poorly understood. In this study, 4.6 kilobases of the 5′-flanking region from the rat BKB2 receptor gene were sequenced, and computer analysis revealed several sites for transcriptional factors. Nine promoter mutants were cloned in luciferase reporter gene vectors and transfected in NG108-15 cells and rat aorta vascular smooth muscle cells (VSMCs), showing several positive and negative regulatory elements. A classical silencer with 56 base pairs (bp) caused a decrease in reporter gene activity in NG108-15 cells and VSMCs and was able to inhibit the thymidine kinase promoter. Using electrophoretic mobility shift assay and surface plasmon resonance assay, protein-DNA interactions in the silencer region were determined and specific sets of protein-silencer complexes were detected in both cell types. More intense complexes were observed in the central 21 bp of the silencer and mutation in a putative SRE-1 site strongly impaired the protein-DNA binding. Down-regulation of the BKB2 receptor population in NG108-15 cells promoted by N 6, 2′-O-dibutyryladenosine 3′:5′-cyclic monophosphate was paralleled by an increase in the amount of nuclear proteins bound to the silencer sequence showing an inverse relationship between protein-silencer complexes and the transcription of the BKB2 receptor gene. In summary, these data highlight the cell-specific regulation of the BKB2 receptor and the importance of a silencer element present in the regulatory region of the gene.
Bradykinin (BK) is a vasoactive nonapeptide released from kininogens by the proteolytic activity of kallikreins. This member of the kinin family has been implicated in a variety of physiological and pathological processes related to cardiovascular homeostasis, inflammation, and cell proliferation. The diverse biological responses to BK are mediated by two specific receptors, B1 and B2, that possess the putative seven transmembrane domain characteristic of G protein-coupled receptors (Bhoola et al., 1992). Under physiological conditions, most of the biological effects of kinins are mediated through the bradykinin B2 (BKB2) receptor, which is constitutively expressed at normal conditions and present in a variety of tissues (Bhoola et al., 1992). Stimulation of this receptor at physiological and pathological states such as hypertension, asthma, and vascular injury results in intense vasodilation, increased blood flow, and hypotension (Regoli and Barabé, 1980), and its genetic ablation leads to the loss of bradykinin responsiveness of most cell types (Borkowski et al., 1995). The kallikrein-kinin system is present in rat hearts (Nolly et al., 1994). Several studies have shown that cardioprotective effects of angiotensin-converting enzyme inhibitors on ventricular hypertrophy (Schölkens et al., 1991; Linz et al., 1993), on cardiac function in genetically hypertensive and transgenic rats overexpressing human tissue kallikrein (Silva et al., 2000), on reperfusion damage after myocardial ischemia (Tio et al., 1991), and on neointima formation after endothelial injury (Fahry et al., 1992) are also mediated via stimulation of B2 receptors. It is suggested that BKB2 receptor plays an important role in cell proliferation processes during tumorigenesis mediating the kinins' mitogenic actions and increasing the blood flow leading to angiogenesis, which is a crucial process for the formation of solid tumors (Roberts and Gullick, 1989). Moreover, transformation of cells in vitro by dbl andras oncogene is associated with increased BKB2 receptor number (Parries et al., 1987; Roberts, 1989; Ruggiero et al., 1989;Pesquero et al., 1996).
In vitro studies have demonstrated an increase in the number of BKB2 receptors expressed in the cell membrane in different cell types by bradykinin (Pesquero et al., 1996), platelet-derived growth factor (Dixon et al., 1996), estrogen (Madeddu et al., 1997), cAMP (Pesquero et al., 1996; Marin-Castaño et al., 1998), interleukin-1β (Schmidlin et al., 1998), glucocorticoids (Scherrer et al., 1999), tumor necrosis factor-α (Haddad et al., 2000), and glucose (Christopher et al., 2001). Down-regulation of the BKB2 receptor was observed in differentiated NG108-15 cells treated with cAMP. In these cells, BK causes a transient Ca2+-activated K+ outward and the differentiation decreases its sensibility to the BK (Tokutomi et al., 1994). However, little is known about the mechanisms involved in the down-regulation of BK effect.
Despite the importance of BKB2 receptor in cardioprotection, tumorigenesis, and other physiological events, to date, little is known about the transcriptional mechanisms that regulate BKB2 receptor expression in animal or human cells under physiological conditions.
The molecular structure and the genomic sequence of the rat BKB2 receptor gene were characterized, including part of the 5′-flanking region (Pesquero et al., 1994). The gene consists of four exons and spans more than 32 kb of genomic DNA and contains no typical transcription initiation motif-sequences TATA or CCAAT boxes, like most of the G protein-coupled receptors (Hosoda et al., 1992). Our group demonstrated that after transfection in NG108-15 cells the first 1143 bp of the putative promoter sequence were shown to be functional after activation of the luciferase reporter gene (Pesquero et al., 1994). Transcription regulatory elements were identified in the first 1143 bp of the putative promoter sequence, including the cAMP-responsive element (CRE), which showed to be functional upon the addition of 8-bromo-cAMP, causing significant activation of the reporter gene (Pesquero et al., 1996). In a recent study, Saifudeen et al. (2000)demonstrated that the rat BKB2 receptor gene is transcriptionally regulated by the tumor suppressor protein p53. Two p53 binding elements were detected in the gene promoter; however, the protein binding resulted in enhancement or inhibition of the transcription of the gene, depending on the sequence activated (Saifudeen et al., 2000).
To understand the molecular mechanisms underlying the regulation of the rat BKB2 receptor gene, in this study, we characterized the functional promoter and presented evidence for trans-acting factors binding to a silencer motif in the BKB2 promoter, which may have important implications in the regulation of gene expression.
Materials and Methods
BKB2 Receptor Promoter Constructs.
A genomic clone containing approximately 6 kb of the 5′-flanking region, the exon 1, and approximately 7 kb of the intron 1 of the rat BKB2 receptor gene was digested with the restriction enzymesEcoRI/XbaI, resulting in a fragment of 3487 bp that was cloned in the pBluescript II SK vector containing a smaller fragment of 1142 bp obtained by the previous digestion of the genomic clone with XbaI. The entire sequence of 4629 bp was released from the vector by the digestion with BamHI/NotI and cloned in the vector pΔLUX (BamHI/NotI) at the 5′-flanking region of the luciferase reporter gene derived from the vector pUHC13-1 (Pesquero et al., 1994). The construct was named pB(−4629) and consisted of the longest regulatory sequence and 100 bp of the exon 1 of the BKB2 receptor gene. The restriction sitesEcoRI and XbaI were maintained in the 5′ and 3′ ends, respectively, in the construct. The eight remaining constructs containing smaller sequences of the regulatory region of the receptor gene were named according to the distance of their 5′ ends to the transcriptional start site of the gene and were generated by deletion of the 5′ region of the pB(−4629) construct as follows (restriction enzymes shown in parentheses): pB(−2639) (HindIII/XbaI), pB(−1142) (XbaI), pB(−1086) (XhoI/XbaI), pB(−931) (StuI/XbaI), pB(−827) (NcoI/XbaI), pB(−634) (BglII/XbaI), pB(−514) (BalI/XbaI), and pB(−287) (BsmI/XbaI).
Cell Culture Transfection and Luciferase Reporter Gene Assay.
The luciferase reporter gene assay was carried out to determine promoter regulatory-specific elements and cell type-specific features. Promoter constructs were transiently transfected into two cell types that are known to express the BKB2 receptor gene (Oza et al., 1990; Yokoyama et al., 1994; Douillet et al., 2000). The rat aorta vascular smooth muscle cells (VSMCs) were grown in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA) supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin, and the cells were used in passages P4 to P10. The mouse neuroblastoma and rat glioma hybrid cells (NG108-15) were grown in the same conditions as VSMCs but the medium was supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.1 mM hypoxanthine, 400 nM aminopterin, and 0.016 mM thymidine, and the cells were used in passages P10 to P20. Both cell lines were transiently transfected in triplicate with 5 μg of each of the nine promoter constructs and the negative control pΔLux, using the calcium phosphate coprecipitation method, as described previously (Pesquero et al., 1994). For Heterologous Promoter Assay two reporter gene vectors containing the silencer sequence in sense and antisense orientations flanking the 5′ end of the herpes simplex virus-TK promoter and luciferase gene were transfected in both cell lines. Gene reporter assay was carried out to compare promoter activity in differentiated and undifferentiated NG108-15 cells. For this purpose, NG108-15 cells were treated withN 6-2′-O-dibutyryladenosine 3′,5′-cyclic monophosphate (dBcAMP) and transfected as described above, with reporter promoter constructs pB(−1142) and pB(−1086), with and without the silencer element, respectively. In all experiments, as internal control for transfection efficiency, the cells were cotransfected with 2 μg of pCH110 vector (Pharmacia, Peapack, NJ) containing lacZ gene under the control of the simian virus 40 early gene promoter. Cellular extracts were prepared and the luciferase and galactosidase activities were detected in a luminometer (Auto Lumat LB 953, EG&E Berthold, Evry, France). The values for luciferase activity were divided by the lacZ activity and statistically analyzed using analysis of variance.
Preparation of Nuclear Extracts.
Nuclear extracts for electrophoretic mobility shift assay (EMSA) were prepared according toAvellar et al. (1997) with some modifications. Briefly, pelleted VSMCs and differentiated and undifferentiated NG108-15 cells (5 × 105) were resuspended in 400 μl of cold lysis buffer (10 mM Tris-HCl, pH 7.9, 10 mM KCl, 0.1 mM EDTA, pH 8.0, 10% glycerol, 1 mM dithiothreitol, 0.1 mM phenylmethylsulfonyl fluoride, 1.0 μg/ml leupeptin, 1.0 μg/ml pepstatin, and 0.5 μg/ml aprotinin) and kept on ice for 15 min. Nonidet P-40 (25 μl, 10%) was added and cells were vortexed for 10 s. Pelleted nuclei were obtained by a 2500g centrifugation at 4°C for 30 s. The supernatants were removed and the pellets washed in 100 μl of cold lysis buffer, centrifuged for 10 s at 2500g at 4°C, and resuspended in 100 μl of nuclear extract buffer (10 mM Tris-HCl, pH 7.5, 0.5 M KCl, 1 mM EDTA, and 10% glycerol and protease inhibitors). The suspensions were gently agitated in a shaker plate for 15 min at 4°C and centrifuged for 5 min at 2500g at 4°C. The supernatants (nuclear extracts) were dialyzed (0.025-μm membrane; Millipore Corporation, Bedford, MA) against 50 volumes of DNA binding buffer (10 mM Tris-HCl, pH 7.5, 25 mM KCl, 0.1 mM EDTA, and 10% glycerol and protease inhibitors) for 4 h at 4°C. The proteins were quantified by using the Protein Assay kit (Bio-Rad, Hercules, CA) and frozen at −80°C until the EMSA.
Oligonucleotides and Probes.
Eight double-stranded oligonucleotides were used in the EMSA experiments. The specific 56-bp motif was obtained from digestion of the BKB2 receptor promoter clone pB(−1142) with the restriction enzymes XbaI/XhoI and labeled by filling-in reaction using [α-32P]dCTP and Klenow fragment (Prime-It Random Primer kit; Stratagene, La Jolla, CA). Three oligonucleotidic fragments of the 56-bp element, named F1, F2, and F3, were chemically synthesized. The F1 fragment (5′-CTA GAG CCT GCA GCC TCG G-3′) spans the 5′ end of the silencer, the F2 fragment (5′-TGT CTG TGG ATC ACC GCC CAG-3′) the central silencer region, and the F3 fragment (5′-CTT CTC TGT CTG CTC C-3′) the 3′ end. Annealing of the sense and reverse complementary oligonucleotides after incubation at 95°C for 5 min and cooling at room temperature generated the double-stranded fragments. After running on 20% acrylamide nondenaturing gel, the double-stranded oligonucleotides were isolated from the gel by diffusion. The fragments were labeled using [γ-32P]ATP and T4 polynucleotide kinase for the EMSA. Mutation analysis of the F2 fragment was carried out to identify the elements required for transcription factors binding in the silencer and to identify putative transcription factors. Two mutants were designed based on putative binding sites for transcriptional factors detected previously in this study by computer analysis. In the F2-M1 mutant the SL3-3 enhancer factor-1 (SEF-1) binding site (Thornell et al., 1993) was altered from CTG to AAA and in the F2-M2 mutant the sterol-responsive element-1 (SRE-1) (Briggs et al., 1993) was altered from TCA to GGG. The F2-M3 mutant was randomly altered at the 3′ end, from CCC to TTT. The mutants were synthesized and labeled as described above and sense strand sequences are as follows: F2-M1, 5′-TGT AAA TGG ATC ACC GCC CAG-3′; F2-M2, 5′-TGT CTG TGG AGG GCC GCC CAG-3′; and F2-M3, 5′-TGT CTG TGG ATC ACC GTT TAG-3′). For competition studies, unlabeled double-stranded oligonucleotides, including the 56-bp motif; F1, F2 and F3 fragments; F2-M1, F2-M2, and F2-M3 mutants; and nonspecific 45 bp (5′-CTA CTA ATG GTG ATC ATT AGG TGA TAA AAC CAG CCT GAA ACC TTT-3′) were used as competitors.
EMSA of Nuclear Protein Binding.
Four sets of EMSA were carried out. In the first assay, nuclear proteins from NG108-15 cells and VSMCs (15 and 20 μg, respectively) were preincubated for 15 min on ice with 0.1 μg of poly(dI-dC), DNA binding buffer, with or without the unlabeled oligonucleotide competitors 56 bp and nonspecific 45 bp in 10-, 25-, 50-, and 100-fold molar excess and water until a final volume of 20 μl. The 56-bp 32P-labeled probe (35,000 cpm) was added and samples were incubated for 30 min on ice. Protein-DNA complexes were resolved on a 5% polyacrylamide gel (acryl/bisacrylamide ratio 37.5:1) buffered with 0.5× Tris borate-EDTA (10 mM Tris-HCl, 10 mM boric acid, and 0.2 mM EDTA) for 3 h at 160 V, and the gel were transferred to 3MM paper (Whatman, Maidstone, UK), dried, and autoradiographed with an intensifying screen at −80°C. In the second assay, nuclear proteins were preincubated under the same protocol as described above with and without unlabeled oligonucleotide competitors F1, F2, and F3 in 50-fold molar excess and incubated for 30 min with 32P-F1, 32P-F2, and 32P-F3 probes. In the third assay, 20 μg of NG108-15 nuclear proteins was preincubated with and without unlabeled F2 mutant competitors (F2-M1, F2-M2, and F2-M3) in 50-fold molar excess and incubated with 32P-F2-M1,32P-F2-M2, and 32P-F2-M3 probes in the same conditions as described above. In the fourth assay, to compare protein-DNA complexes formation between dBcAMP-treated and nontreated NG108-15 cells, increasing amounts of nuclear proteins (2.5, 5.0, and 10 μg) were incubated with 32P-labeled full-length 56-bp element and EMSA experiments were performed as described above. All the experiments were made in triplicate.
Surface Plasmon Resonance (SPR).
SPR was used to study the interactions under real-time conditions between nuclear proteins from VSMCs, NG108-15 cells, and heart and the core part (F2 fragment) of the BKB2 receptor silencer. Experiments were performed on a BIAcore 3000 biosensor instrument (BIAcore AB, Uppsala, Sweden) at 25°C. Two different biotinylated oligonucleotides (Genset SA, Paris, France) were used, one containing a 15-bp spacer sequence (5′-biotin-GTT CAC AGA GTC CAT-3′) followed by the F2 silencer sequence and another oligonucleotide containing the 15-bp spacer sequence followed by a scrambled F2 silencer sequence (5′-CAC TGG CAG TCG AGC TCT TGC-3′). These oligonucleotides were immobilized on a streptavidin-coated sensor-chip (BIAcore AB) and hybridized to obtain double-stranded F2 and F2 scrambled oligonucleotides. The biotinylated oligonucleotides were immobilized in HBS buffer (10 mM HEPES, pH 7.4, and 150 mM NaCl) at a flow rate of 10 μl/min to obtain binding of 700 to 900 resonance units (RUs) in the different experiments. The complementary strands homologous to the F2 and F2 scrambled sequences were hybridized with a flow rate of 2 μl/min in HBS buffer containing 300 mM NaCl to obtain a 250- to 350-fold increase in RUs, indicating significant double-strand formation. The difference in the amount of double-stranded F2 and F2 scrambled immobilized within an experiment was always less than 6%. Nuclear extracts proteins (1–3 μg) were injected in 30 μl in triplicate at a flow rate of 10 μl/min in HBS buffer containing 4 mM CHAPS and 100 μg/ml poly(dI-dC). Regeneration of the chip after each cycle was done using 5 μl of 0.02% SDS at a flow rate of 10 μl/min (data not shown).
Differentiation of NG108-15 Cells.
To correlate the number of BKB2 receptors at the cell membrane with its expression at transcriptional level, a model for modulation of the receptor was produced. The NG108-15 cells are a good model because its differentiation causes down-regulation of the BKB2 receptor (Tokutomi et al., 1994). Initially, NG108-15 cells were grown as described above until the treatment for differentiation. The cells were used from passage P17 to P20. Differentiation was induced by addition of 1 mM dBcAMP (Sigma-Aldrich, St. Louis, MO) into the medium and by reducing the FBS to 5% for a period of 4 days. The medium was replaced by fresh dBcAMP medium on the 2nd day of the treatment.
Extracellular Acidification Rate Measurements.
To study the membrane BKB2 receptor expression on NG108-15 cells, extracellular acidification rate was measured using the Cytosensor microphysiometer system (Molecular Devices, Sunnyvale, CA) (McConnell et al., 1992) after stimulation with different agonists. Briefly, treated and nontreated NG108-15 cells were removed from the culture flask with 1× phosphate-buffered saline, mixed with 7 μl of agarose (Molecular Devices), and plated into sterile capsule-cups (seated in 12-well plates) at a density of 5 × 105 cells/cup. The cells and agarose (final volume 10 μl) were placed at 4°C for 20 min to allow complete polymerization of the mixture. Spacers (100 μm in thickness) were added to the center of the cell capsules followed by capsule inserts. The assembled cup was then transferred to sensor chambers containing 1 ml of low-buffered (1 mM sodium phosphate) Dulbecco's modified Eagle's medium with 0.1% bovine serum albumin and without bicarbonate but containing 2 mM glutamine and additional 44.4 mM NaCl to replace bicarbonate and adjust osmolarity. The sensor chambers were placed on the Cytosensor microphysiometer and allowed to equilibrate for more than 30 min before the beginning of the experiment. The medium was run through the chambers at a rate of 100 μl/min at 37°C. To assess shifts in the extracellular acidification rate, cells were stimulated over a period of 20 s with BK at concentrations of 10−6 and 10−7 M. The agonists Met- and Leu-enkephalin were added as control for the specificity of the dBcAMP effect (Brandt et al., 1976; Klee and Nirenberg, 1976). The experiment was conducted in triplicate.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR).
To evaluate BKB2 receptor down-regulation in treated NG108-15 cells caused by an inhibition of transcription, BKB2 receptor mRNA from treated and nontreated cells was extracted by TRIzol reagent (Invitrogen) and amplified in triplicate by RT-PCR as follows: 1 μg of extracted RNA from treated and nontreated cells was reverse transcribed in the presence of 1 μg of random hexanucleotides, 1.25 mM of each dNTP, 40 units of RNasin (Promega, Madison, WI), and 200 units of Moloney murine leukemia virus reverse transcriptase (Invitrogen) in 20 μl of PCR buffer containing 50 mM KCl, 20 mM Tris-HCl, pH 8.4, 2.5 mM MgCl2, and 1 mg/ml bovine serum albumin. A PCR reaction was performed with 10 μl of the reverse transcription reaction in a 100-μl reaction volume of PCR buffer containing 50 ng of BKB2 receptor sense BKPro3 (5′ATC CTC ACT CGT CTT TGT CC3′) and antisense Race1 (3′GTC TGG GCA GTT GAC CTC TG5′) primers and β-actin sense HBc5 (5′CCT CGC CTT TGC CGA TCC3′) and antisense Rbac3 (3′AGG AAG AGG ATG CGG CAG TGG5′) primers as control, 0.5 mM of each dNTP, and 2 units of Taq polymerase (Invitrogen) for 25 to 35 cycles (95°C/45 s, 58°C/1 min, and 72°C/1 min) followed by a final 7-min extension at 72°C. Amplification products were analyzed in a 3% (w/v) agarose gel electrophoresis and stained with ethidium bromide.
Results
BKB2 Receptor Gene Promoter Is Regulated by Both Positive and Negative Elements in NG108-15 Cells and Rat Aorta VSMCs.
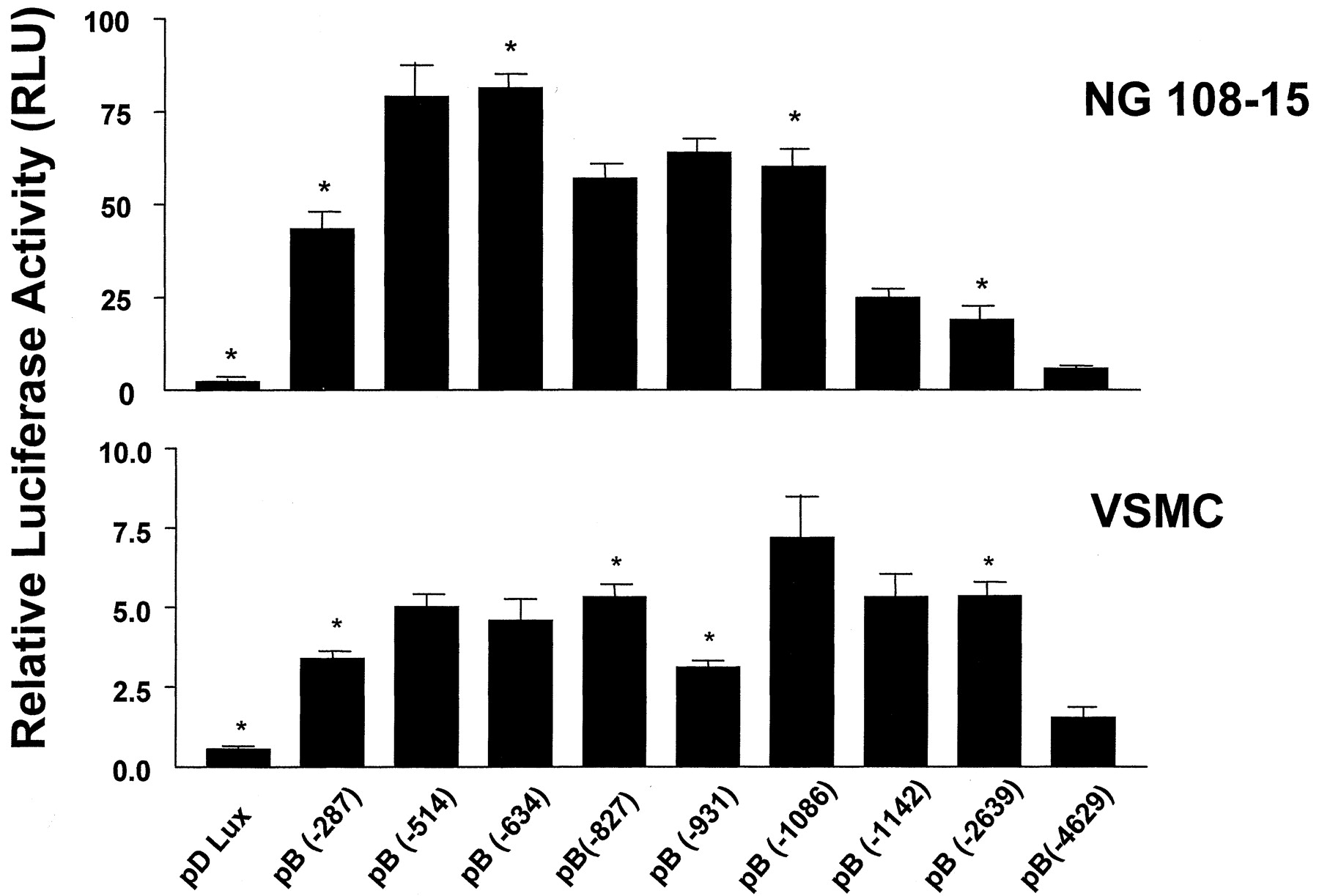
To identify the sequences essential for transcription in the rat BKB2 receptor gene and other important regulatory elements, we cloned and sequenced a 4.6-kb fragment of the 5′-flanking sequence from a previously isolated rat genomic BKB2 clone (Pesquero et al., 1994) (Fig. 1). Computer analysis (Matinspektor; Genomatix Software GmbH, Munich, Germany) of this sequence identified more than 300 putative transcription factor sites, which potentially bind to the promoter receptor. The sites for the factors GATA-1, CCAAT displacement protein (CDP), E2F, Egr-2, interleukin-6, activator protein-1, SP-1, ras, nuclear factor-κB, p53, SEF-1, cAMP responsive element binding protein, sterol regulatory element binding protein, serum, glucocorticoid, interferon, and estrogen appeared repeatedly along the sequence. A series of nine 5′-end deletion mutants were generated as shown in Fig. 1. These constructs were transfected into NG108-15 and rat aorta VSMCs, and relative luciferase activity expressed as relative light units (RLUs) was measured in the cellular extracts. Several positive and negative elements were identified in both cell lines (Fig.2). The luciferase activity was approximately 10 times higher in the NG108-15 cells than in VSMCs. The first 287 bp of the regulatory region were able to increase luciferase activity significantly in both cell lines, being 20- and 7-fold higher in NG108-15 cells and VSMCs, respectively, compared with the negative control vector pΔLux. The insertion of 227 bp at position −287 caused a significant increase in the luciferase activity in NG108-15 cells (47%) and VSMCs (33%), suggesting the presence of a positive regulatory element (PRE) in this sequence. Another PRE was detected only in VSMCs at position −931 to −1086, causing an increase in luciferase activity of 57%. A negative regulatory element (NRE), located at position −1086 to −1142 proved to be of particular interest in both cell lines because the insertion of only 56 bp caused a 50% decrease in the luciferase reporter activity in NG108-15 cells and a 26% decrease in VSMCs. Other NREs were found in each cell line, in NG108-15 cells at position −634 to −827 and in VSMCs at position −827 to −931, which decreased by 30 and 44% the reporter activity, respectively. The insertion of the longest regulatory sequence containing 1990 bp from position −2639 to −4629 caused a markedly decrease in luciferase activity in both cell types (70%). These observations point to the presence of several enhancer and silencer motifs in the 5′-flanking region of the constitutive BKB2 receptor gene.

Nucleotide sequence of the rat BKB2 receptor regulatory region. The nucleotide sequence (GenBank data bank accession number AF374005) of the 5′-flanking region of the rat BKB2 receptor gene is shown. Nucleotide numbering starts at the transcriptional start site (+1), as defined by primer extension analysis, and proceeds as indicated in the right-hand margin. The nine promoter mutants are shown in bold letters and an arrow indicates the 5′ end of each mutant. The silencer element is shown in italics and doubly underlined. Bold letters indicate the boundaries of the silencer fragments F1, F2, and F3. Exon 1 is underlined.

Luciferase reporter gene assay for the rat BKB2 receptor promoter deletion mutants. Illustration shows luciferase activity in NG108-15 cells and VSMCs transfected with reporter constructs containing various 5′ deletions. Luciferase activities are expressed as RLUs and normalized for transfection efficiency with β-galactosidase activity. The values represent the means ± S.E. of three independent assays. ∗, p < 0.05 in relation to the next longer deletion mutant.
BKB2 Receptor Expression Is Regulated by a Classical Silencer in the 5′-Flanking Region in Different Cell Types.
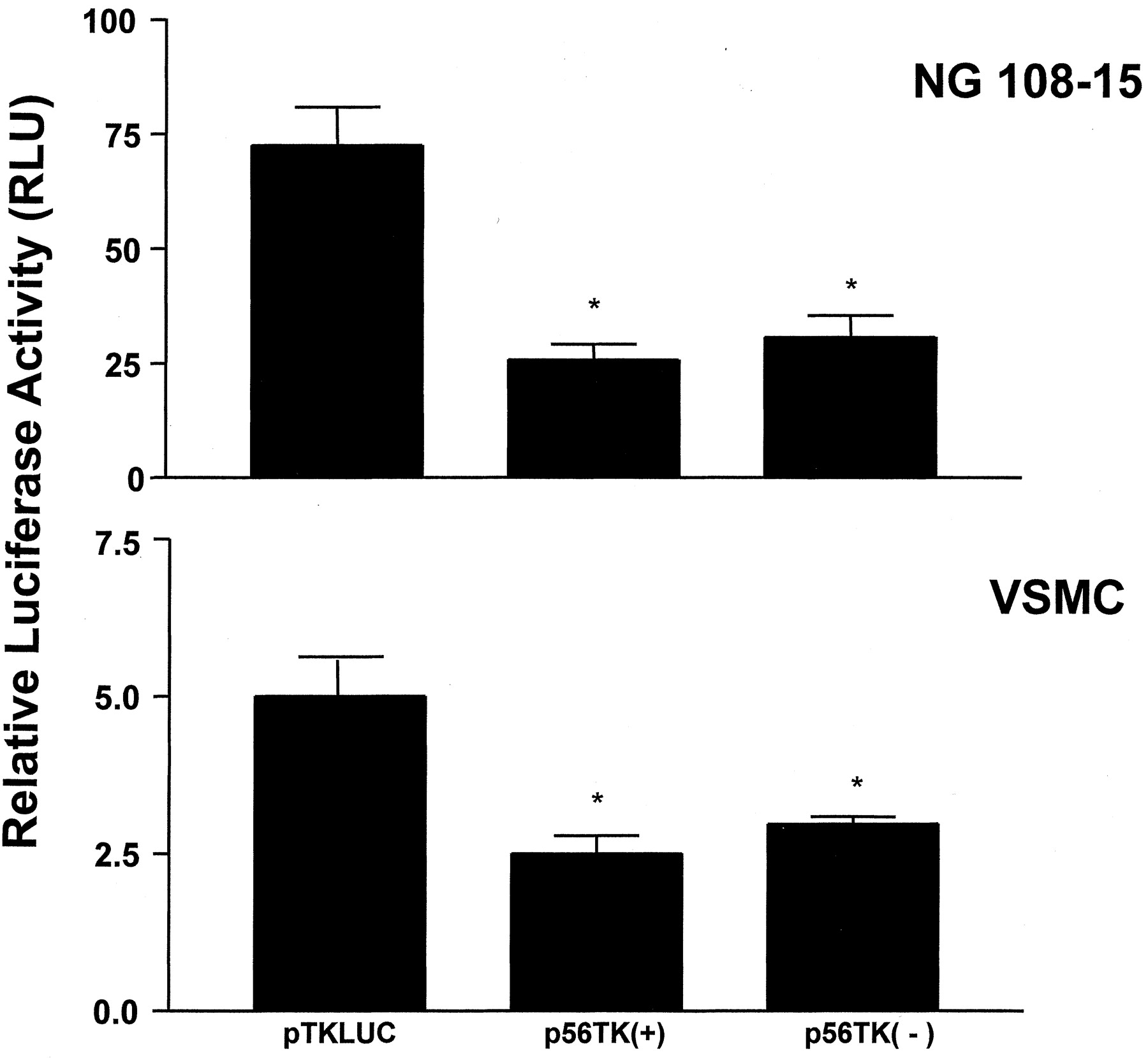
The 56 bp located at position −1086 to −1142 in the 5′-flanking region of the BKB2 receptor gene were able to decrease the luciferase activity in both cells, suggesting its function as a silencer element. Accordingly, we tested whether this fragment fulfills the characteristics of a classical silencer by generating two heterologous promoter vectors in which the 56-bp fragment was inserted in either the sense or antisense orientations 5′ to the herpes simplex virus-TK promoter into a TK luciferase reporter vector. The heterologous promoter assay results showed a 3- and 2-fold decrease in the luciferase activity in NG108-15 and rat VSMC, respectively (Fig. 3). As in the previous experiment, the relative luciferase activity was approximately 10 times higher in the NG108-15 cells compared with the VSMCs.

Inhibition of the heterologous thymidine kinase (TK) promoter by the BKB2 receptor silencer element. Luciferase activity in NG108-15 cells or aorta VSMCs transfected with the 56-bp fragment inserted in the sense (+) or antisense (−) orientation 5′ to the heterologous TK promoter followed by the luciferase reporter gene. Luciferase activities are expressed as RLUs and normalized for transfection efficiency with β-galactosidase activity. The values represent the means ± S.E. of three independent assays. ∗,p < 0.05 in relation to the control vector pTKLUC.
Specific Protein-DNA Interactions within the Silencer Region of the Rat BKB2 Receptor Gene.
To verify the ability of the silencer element to interact with transcription factors, nuclear extracts from both cell lines were incubated with the32P-labeled 56-bp silencer element with and without oligonucleotide competitors and EMSAs were performed. The EMSA revealed specific sets of protein-DNA complexes in both cell types (Fig. 4). In NG 108-15 cells, two intense complexes (A1 and A2) were formed when 15 μg of nuclear protein was incubated with the silencer probe (Fig. 4A). The presence of a 10-, 25-, 50-, and 100-fold molar excess of the unlabeled 56-bp silencer, which gradually promoted the complete inhibition of nuclear protein-silencer binding, confirmed the specificity of these protein-DNA complexes. The addition of a nonspecific unlabeled 45-bp oligonucleotide competitor did not alter the protein-DNA interaction pattern. The incubation of the silencer with 20 μg of nuclear proteins from rat aorta VSMCs resulted in the formation of two protein-DNA complexes (B1 and B2) (Fig. 4B). Like in the NG 108-15 cells, formation of these complexes was completely inhibited by the addition of 10-, 25-, 50-, and 100-fold unlabeled silencer and the nonspecific 45-bp competitor did not influence the complex protein-DNA formation.
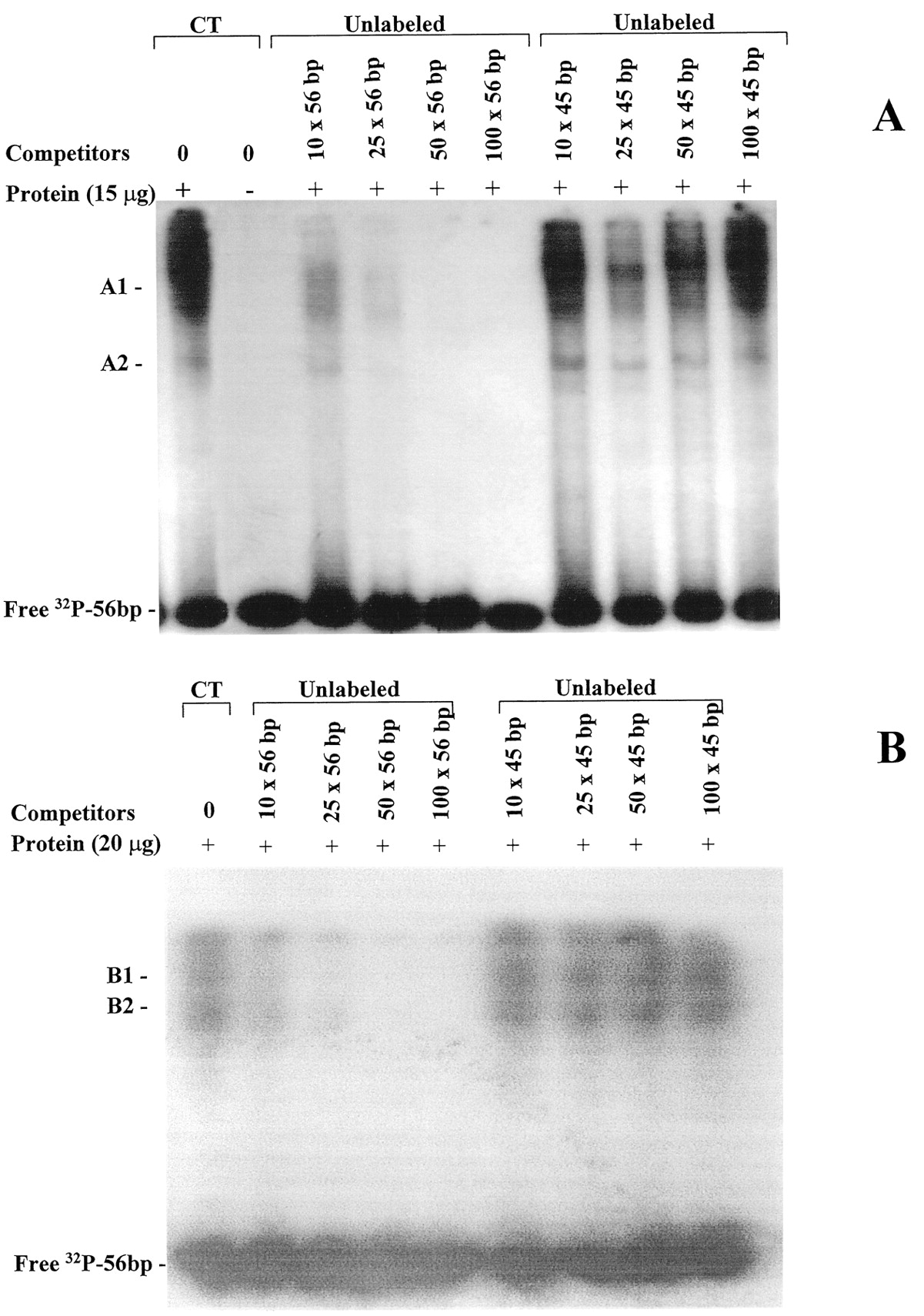
Binding of nuclear proteins to double-stranded 56-bp BKB2 receptor silencer. Electrophoretic mobility shift assays were performed with nuclear extracts from NG108-15 cells (A) and aorta VSMCs (B) and 32P-labeled 56-bp silencer. Nuclear proteins were incubated with the labeled silencer in the absence (CT, 0) and presence of 10-, 25-, 50-, and 100-fold molar excess of unlabeled double-stranded 56-bp or nonspecific 45-bp oligonucleotide used as competitors as indicated in each lane. In NG108-15 cells and aorta VSMCs, two specific silencer-protein complexes are present (A1 and A2 and B1 and B2, respectively). The results are representative of three independent experiments.
Specific Protein-DNA Interactions within Silencer Fragments.
To assess the minimum silencer motif that could interact with transcription factors, three double-stranded32P-labeled oligonucleotides, named F1, F2, and F3 (Fig. 1), were synthesized based on the silencer sequence and used in EMSA, and the oligonucleotide F2 was used in SPR with NG108-15 cells and VSMCs (Fig. 5). The specificity of the nuclear protein binding to which sequence was tested using 50-fold molar excess of unlabeled double-stranded F1, F2, F3, and nonspecific 45-bp oligonucleotides as competitors. In NG108-15 cells several specific and nonspecific nuclear protein-DNA interactions were revealed when 15 μg of protein was used in EMSA (Fig. 5A). With the F1 probe, many specific protein-DNA complexes were detected. Several minor complexes and one major complex (C1) were eliminated by an excess of unlabeled double-stranded F1 fragment but not by random 45-bp oligonucleotide. A nonspecific complex (C2) did not disappear even when F1 competitor was added. With F2 probe, three specific complexes (D1, D2, and D3) and with F3 probe, two specific complexes (E1 and E2) were observed. However, the highest amount of specific protein-DNA interaction was detected with the F2 probe as evidenced by the presence of more intense bands in the gel bound to the fragment F2 compared with the other fragments. In VSMCs (Fig. 5B) the addition of 20 μg of nuclear proteins generated two specific protein-DNA complexes (G1 and G2), which were exclusives to the F2 fragment, indicating that probably this sequence is responsible for the interactions between the transcription factors and the silencer. To further evaluate the interaction between the F2 fragment and nuclear proteins from NG108-15 cells, VSMCs, and heart, SPR was performed. SPR monitors, in real time, the interactions between macromolecules. Biotinylated double-stranded F2 and scrambled F2 fragment were immobilized and nuclear proteins were injected separately by constant flow. Sensorgram shows significant binding of nuclear proteins to the intact F2 oligonucleotide from both cell lines evidenced by the increase in the RUs; however, a very low association of nuclear proteins from the heart to the F2 fragment was observed (Fig. 5C). Although the same amounts of nuclear proteins from both cell lines were injected, more intense association was observed with nuclear proteins from NG108-15 cells than VSMCs. Real-time binding of nuclear proteins was specific because increasing concentrations of NG108-15 nuclear proteins resulted in increasing responses (Fig. 5C, inset, shown for NG108-15 cell nuclear extract only).

Binding of nuclear proteins to the fragments of the BKB2 receptor silencer. Electrophoretic mobility shift assays were performed with nuclear extracts from NG108-15 cells (A) and aorta VSMCs (B) and 32P-labeled fragments of the silencer (F1, F2, and F3). Nuclear proteins were incubated with each labeled oligonucleotide in the absence (CT, 0) and in the presence of 50-fold molar excess of unlabeled fragments and 45-bp nonspecific oligonucleotide used as competitors. A, in NG108-15 cells several major specific complexes (C1, D1, D2, D3, E1, and E2) and a nonspecific complex (C2) are formed. B, in VSMCs, two specific silencer-protein complexes are present (G1 and G2). The results are representative of three different experiments. C, sensorgram showing the real-time interaction of the nuclear proteins to the double-stranded F2 oligonucleotide using surface plasmon resonance analysis. Represented is the response in RUs of the flow cell with immobilized double-stranded F2 silencer oligonucleotide minus the response of the flow cell with immobilized double-stranded F2 scrambled silencer oligonucleotide. Inset, dose response (0.02–0.2 μg) using NG108-15 cell nuclear extracts.
Silencer F2 Fragment Mutation Analysis: Protein-DNA Interactions.
To identify the most important elements required for the interaction of transcription factors in the silencer, EMSA was carried out using the intact F2 silencer fragment and three mutants and NG108-15 nuclear proteins. In the F2-M1 and F2-M2, mutations were generated in two putative binding sites (SEF-1 and SRE-1 binding sites, respectively), which were previously identified by computer analysis. In the F2-M3 mutant, the F2 fragment was randomly altered at the 3′ end. EMSA results revealed that the three mutants altered the protein-DNA complex formation pattern at different levels (Fig.6). F2-M1 mutant did not impair the formation of preexisting complexes but favored the formation of a new protein-DNA complex, and F2-M3 mutant decreased the protein-DNA complexes intensity, suggesting that the mutation caused a protein-DNA affinity loss. The mutation of the central silencer sequence in F2-M2 mutant, however, impaired most of complex formation. Competition assays with labeled and unlabeled mutants showed that F2-M1 and F2-M3 strongly impaired the formation of protein-DNA complex formation, whereas unlabeled F2-M2 slightly impaired complex formation showing to be an important element for transcription factors interactions in the silencer.
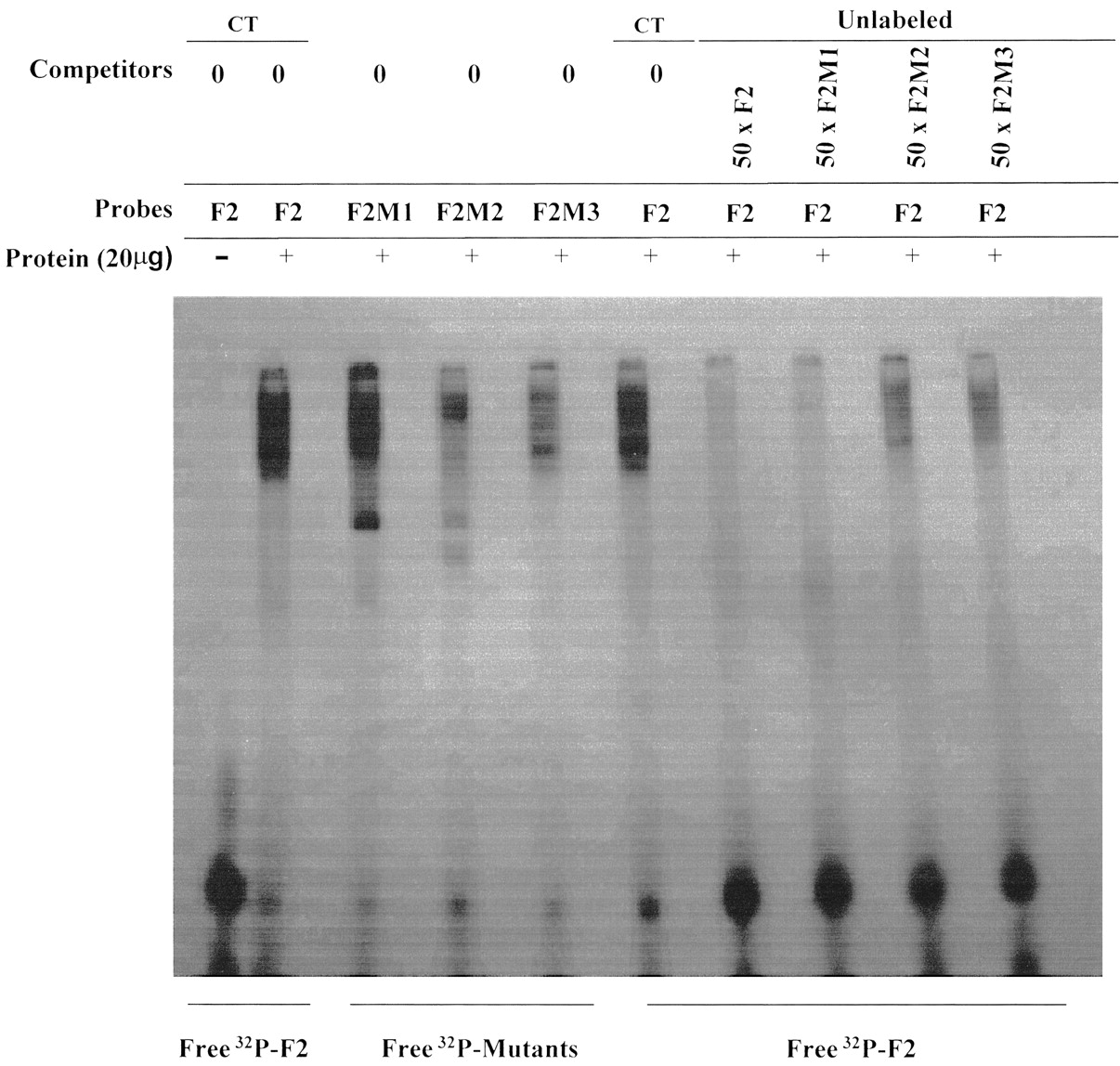
Binding of NG108-15 nuclear proteins to F2 fragment mutants. Electrophoretic mobility shift assays were performed with nuclear extracts from NG108-15 cells and 32P-labeled mutants F2-M1, F2-M2, and F2-M3. Nuclear proteins were incubated with each labeled oligonucleotide in the absence (CT, 0) and in the presence of 50-fold molar excess of unlabeled F2 fragment and mutants used as competitors. The results are representative of three different experiments.
BKB2 Receptor Down-Regulation in NG108-15 Cells by dBcAMP Treatment.
To obtain a BKB2 receptor down-regulation model, NG108-15 cells were differentiated by treatment for 4 days with dBcAMP and by reducing the serum concentration to 5%. Similarly to Battaini et al. (1994), we observed some morphological changes in the cells, which stopped dividing and exhibited rounded bodies with extensive neurite outgrowth. The effect of differentiation on the BKB2 receptor was evaluated by the analysis of its functional coupling with the agonist BK in the Cytosensor microphysiometer system, which measures the changes in the extracellular acidification rate. After administration of 10−7 and 10−6 M BK, significantly lower acidification rates were observed in dBcAMP-treated cells compared with nontreated cells (200 and 290% versus 370 and 640%, respectively, of the basal rate), confirming a BKB2 receptor down-regulation (Fig.7A). BK effect on the acidification rate could be blunted by prior administration of HOE-140, a specific B2 receptor antagonist (data not shown). The opioid receptors agonists Met- and Leu-enkephalin were used as controls, and their effects were not significantly altered in dBcAMP-treated cells compared with control cells (150 versus140% of basal level, respectively; data not shown).

BKB2 receptor down-regulation in dBcAMP treated NG108-15 cells. A, effect of BK (10−7 and 10−6 M) on the extracellular acidification rate (ECAR) measured by the Cytosensor system of nontreated (circles) and dBcAMP-treated (squares) NG108-15 cells. Bars depict the time of BK application. Inset shows the means ± S.E. of the effect of bradykinin on the ECAR of control and treated cells. Cytosensor assays were performed in triplicate. B, RT-PCR of total RNA extracted from dBcAMP-treated (T) and nontreated (NT) NG108-15 cells. RNA was isolated and 25, 30, and 35 cycles RT-PCR were performed using BKB2 receptor and β-actin-specific primers. Result is representative of three independent experiments. Bar graph represents means ± S.E. of the intensities of the bands and is expressed as fold increase above control. ∗, p < 0.05 in relation to the next amplified band. C, electrophoretic mobility shift assays were performed with nuclear extracts from differentiated and undifferentiated NG108-15 cells and the 56-bp BKB2 receptor silencer. Increasing amounts of nuclear proteins (2.5, 5.0, and 10 μg) were incubated with the labeled silencer oligonucleotide and two specific protein-DNA complexes (H1 and H2) were observed. The results are representative of three different experiments.
Inhibition of Rat BKB2 Receptor Gene Transcription in Treated NG108-15 Cells.
RT-PCRs with dBcAMP-treated and nontreated NG108-15 cells were performed using rat BKB2 receptor primers. The results showed that the receptor mRNA content in treated NG108-15 cells decreased compared with nontreated cells (p < 0.05) as evidenced by the intensity of the generated bands normalized to β-actin mRNA (Fig. 7B). These results strongly suggest that the decrease in the number of the BKB2 receptors in the membrane of treated NG108-15 cells was caused by inhibition of gene transcription.
EMSA results (Fig. 7C) using nuclear extracts from differentiated and undifferentiated cells and the 56-bp silencer probe showed the formation of more intense protein-DNA complexes (H1 and H2) with nuclear proteins from treated cells compared with nontreated cells, suggesting an increase in the protein-silencer interaction in differentiated cells.
Gene reporter assay results using cellular extracts from differentiated and undifferentiated NG108-15 cells transfected with the control (pΔLux) and two promoter constructs containing or not the silencer sequence [pB(−1142) and pB(−1086), respectively] showed a significant decrease in the luciferase activity of differentiated cells transfected with the construct pB(−1142) containing the silencer element compared with the construct pB(−1086) without the silencer (33%, p < 0.05). Undifferentiated cells transfected with the same constructs showed no significant decrease in the luciferase activity (data not shown).
Discussion
Bradykinin B2 receptors are constitutively expressed and widely distributed in the majority of mammalian tissues. However, recent studies have revealed that the expression of the BKB2 receptor gene is developmentally regulated in the kidney and in the cardiovascular system (El-Dahr, 1997) and induced by mitogenic growth factors, cytokines (Dixon et al., 1996; Schmidlin et al., 1998), and agents such as cAMP (Pesquero et al., 1996; Marin-Castaño et al., 1998), estrogen (Madeddu et al., 1997), glucocorticoids (Scherrer et al., 1999), and glucose (Christopher et al., 2001) in physiological and pathological conditions.
Despite its physiological importance in cardioprotection and tumorigenesis the expression of the BKB2 receptor gene, including the characterization of the functional promoter and transcriptional factors involved in gene regulation, is poorly understood. Previous studies showed that the gene promoter contains no typical transcription initiation motifs, and the first 1143 bp of the promoter are capable of activating the reporter gene luciferase and contain several binding sites for transcription factors, including a CRE andras-responsive element that have proven to be important in the regulation of the gene expression (Pesquero et al., 1994, 1996).
To define more precisely the regulation of B2 receptor gene expression we extended the 5′-flanking region of the gene to 4629 bp and sequenced and characterized the functional promoter, including potential domains (cis-factors) and transcription factors (trans-factors) involved in the regulation of the gene. Computer analysis (MatInspector; Genomatix) of the promoter sequence detected more than 300 putative binding elements for transcription factors such as GATA-1, CDP, E2F, Egr-2, interleukin-6, activator protein-1, SP-1, nuclear factor-κB, p53, SEF-1, SRE-1, cAMP responsive element-binding protein, ras, serum, glucocorticoids, interferon, and estrogen. The GATA-1 binding site and ras responsive element are repeated 13 and six times, respectively, along the sequence. Although these factors are potentially important in the BKB2 receptor gene regulation, further studies are necessary to demonstrate the specific mechanisms by which these factors regulate the promoter of the gene.
To identify specific regulatory elements of the BKB2 promoter, we generated chimerical constructs containing 100 bp of the exon 1 followed by nine different promoter fragments cloned upstream to the luciferase reporter gene and transfected into NG108-15 cells and VSMCs. Reporter gene results revealed a cell type-specific regulation for the BKB2 receptor gene. Luciferase activity in the NG108-15 cell was 10 times higher compared with the VSMCs for all constructs, which is in agreement with several studies showing a higher expression of BKB2 receptor in the membrane of NG108-15 cells (Lee et al., 1989) compared with VSMCs (Oza et al., 1990; Douillet et al., 2000).
The presences of several PREs and NREs were found within the 4629-bp sequence in both cell lines. The construct pB(−287), containing the first 287 bp of the regulatory region and 100 bp of the exon 1, significantly activated the reporter gene expression in both cell lines. This result suggests that the core promoter of the gene is present in this sequence, playing a critical role in determining the initiation and maintenance of the basal transcription level. The basal transcription apparatus might be formed close to the pyrimidine-rich repeat sequence CTCCAGCTCC, which is the main transcription start site of the gene (Pesquero et al., 1994), conserved in several TATA-less promoters (Weis and Reinberg, 1992). A short NRE of 56 bp located at position −1086 to −1142 present in the construct pB(−1142) decreased the reporter gene expression in both cell lines. The ability of this region to significantly repress homologous and a TK heterologous promoter not dependent on its orientation, and in unrelated cell lines it characterized the sequence as a classical silencer (Ogbourne and Antalis, 1998). The longest cloned regulatory sequence spanning from −1 to −4629 bp, construct pB(−4629), significantly decreased the reporter gene in both cell types. This sequence activated the luciferase expression at the lowest level compared with the other promoter fragments. This result could be explained by the inhibitory activity of the CDP localized between −2716 and −2919 bp in the promoter and detected by computer analysis. The CDP is a well known transcriptional repressor that has been shown to down-regulate the expression of several genes, including the gp91-phox gene that is highly expressed in macrophages and granulocytes (Catt et al., 1999).
Differences between the cell types were also observed in the EMSA, which revealed that transcription factors bound to the silencer element in NG108-15 and VSMCs resulted in specific protein-DNA complexes. More intense complexes were observed in the NG108-15 cells, indicating that a major quantity of proteins is activating the silencer element, thus inhibiting the gene expression. Because this cell line presents a higher expression of BKB2 receptor in the membrane compared with VSMCs, a smaller amount of protein bound to the silencer was expected to keep the expression level. This result suggests that to maintain the basal constitutive expression of the BKB2 receptor in the NG108-15 cells it is necessary that a greater quantity of proteins bind to the silencer element. The amount and specificity of such proteins, nevertheless, is variable from cell to cell to modulate the gene expression in specific states and tissues.
To determine the minimum silencer sequence capable of binding transcriptional factors, the 56 bp were divided into three smaller fragments. EMSA showed that most of the protein complexes were bound to the F2 fragment in both cell lines, and even being the unique binding site in VSMCs. This is in agreement with the SPR results in which the amount of protein immobilized to the F2 fragment in the NG108-15 cells was higher than in VSMCs and heart, which presents a very low protein binding to F2. These results indicate that this central 21-bp sequence is the main target for transcriptional factors binding and essential for full silencer activity in these cells. Putative binding sites for the transcription factor SEF-1 and the SRE-1 were detected in the F2 fragment sequence by computer analysis. Mutations on the F2 fragment showed that the replacement of three nucleotides in the core string of the SRE-1 resulted in strong impairment of protein-DNA binding in NG108-15 cells, indicating that this motif is probably required for transcription factors binding. Competition assays with the intact F2 sequence probe and unlabeled SRE-1 mutant hardly impaired intact F2 complex formation, indicating that this element is important for transcription factors interactions and a candidate to activate the silencer element regulating the BKB2 receptor gene.
The NG108-15 cell line is a model for the study of the BKB2 receptor. This cell line has served as a model to study molecular mechanisms of BK receptor mediated modulation of neuronal excitability (Yokoyama et al., 1994). Treatment of these cells with dBcAMP and serum reduction to 5% lead to a cell differentiation and down-regulation of the BKB2 receptor (Tokutomi et al., 1994). In the present work, the differentiated NG108-15 model was used to verify the BKB2 receptor down-regulation in the membrane as a consequence of transcription inhibition via silencer. Bradykinin generated a much smaller biological response in the differentiated cells as evaluated by the extracellular acidification rate, indicating a down-regulation of BKB2 receptors. Furthermore, a significant decrease in the receptor mRNA in differentiated cells indicated transcription inhibition of the gene. In fact, EMSA confirmed that a larger quantity of proteins from differentiated cells was bound to the silencer, demonstrating an inverse relationship between the BKB2 receptor expression in the membrane and complexes formation with the silencer. These results suggest that the BKB2 receptor down-regulation in the differentiation process in NG108-15 cells could be through an inhibition of the transcription rate. In addition, silencer inhibition capacity was confirmed by gene reporter assay where differentiated cells transfected with the construct containing the silencer significantly decreased the luciferase activity compared with undifferentiated cells. Therefore, the silencer element was shown to be more active in differentiated cells as a consequence of the larger quantity of protein binding observed in the EMSA experiment.
Despite the BKB2 receptor is constitutively expressed in a large number of cells, the gene expression is regulated during development in the kidney, cardiovascular system, and later in some pathological states such as diabetes and cardiac hypertrophy by substances such as cAMP, glucose, glucocorticoids, and estrogen, among others, which modulate the mRNA expression, protein levels, and cell surface BKB2 receptors in tissues. Our results strongly indicate that the classical silencer element present in the regulatory region of the BKB2 promoter could be one of the mechanisms by which the gene is down-regulated at the transcriptional level either in pathophysiological or physiological conditions. The understanding of the molecular mechanisms involved in the receptor regulation give rise to new therapies in the treatment of hypertension, cardiac hypertrophy, and inflammatory diseases.
Taken together, these results are the first demonstration of the cell-specific regulation of the rat BKB2 receptor gene and the existence of a classical silencer in the promoter of the gene. Future studies, however, will be necessary to identify the specific transcriptional factors involved in the gene regulation in disease states where the receptor expression is altered.
Acknowledgments
We are grateful to Nelson A. Mora, Ivan H. Cordeiro, and Josefa M. Casemiro for excellent technical assistance.
Footnotes
- Received July 2, 2002.
- Accepted August 23, 2002.
-
This work was supported by grants from Fundação de Amparo à Pesquisa no Estado de São Paulo (Processo 96/10659-2), Volkswagenstiftung (I/77209), and a Programa de Cooperação Internacional Brasil-Alemanha grant from Coordenação de Aperfeiçoamento de Pessoal de Nı́vel Superior and Deutscher Akademischer Austauschdienst.
Abbreviations
- BK
- bradykinin
- BKB2
- bradykinin B2 receptor
- bp
- base pair(s)
- kb
- kilobases(s)
- CRE
- cAMP-responsive element
- VSMC
- vascular smooth muscle cell
- FBS
- fetal bovine serum
- dBcAMP
- N6-2′-O-dibutyryladenosine 3′,5′-cyclic monophosphate
- EMSA
- electrophoretic mobility shift assay
- GATA-1
- globin transcription factor-1
- SEF-1
- SL3-3 enhancer factor-1
- SRE-1
- sterol-responsive element-1
- SPR
- surface plasmon resonance
- HBS
- HEPES-buffered saline
- RU
- resonance unit
- CHAPS
- 3-[(3-cholamidopropyl)dimethylammonio]propanesulfonate
- RT-PCR
- reverse transcription-polymerase chain reaction
- RLU
- relative light unit
- PRE
- positive regulatory element
- NRE
- negative regulatory element
- HOE-140
- d-Arg[Hyp3,Thi5,d-Tic7,Oic8]bradykinin
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}