


Abstract
By general consensus, the omnipresent purine nucleoside adenosine is considered a major regulator of local tissue function, especially when energy supply fails to meet cellular energy demand. Adenosine mediation involves activation of a family of four G protein–coupled adenosine receptors (ARs): A1, A2A, A2B, and A3. The A3 adenosine receptor (A3AR) is the only adenosine subtype to be overexpressed in inflammatory and cancer cells, thus making it a potential target for therapy. Originally isolated as an orphan receptor, A3AR presented a twofold nature under different pathophysiologic conditions: it appeared to be protective/harmful under ischemic conditions, pro/anti-inflammatory, and pro/antitumoral depending on the systems investigated. Until recently, the greatest and most intriguing challenge has been to understand whether, and in which cases, selective A3 agonists or antagonists would be the best choice. Today, the choice has been made and A3AR agonists are now under clinical development for some disorders including rheumatoid arthritis, psoriasis, glaucoma, and hepatocellular carcinoma. More specifically, the interest and relevance of these new agents derives from clinical data demonstrating that A3AR agonists are both effective and safe. Thus, it will become apparent in the present review that purine scientists do seem to be getting closer to their goal: the incorporation of adenosine ligands into drugs with the ability to save lives and improve human health.
I. Introduction
The purine nucleoside adenosine has been identified as a major local tissue function regulator, particularly when cellular energy supply fails to meet the demand. Given its ability to equalize energy intake to metabolic demand, in the 1980s it was reputed to be a “retaliatory metabolite” (Fredholm et al., 2011; Fredholm, 2014). Adenosine is omnipresent, it is released by nearly all cells and is generated in the extracellular space through ATP breakdown by a series of ectoenzymes, including apyrase (CD39) and 5′-nucleotidase (CD73) (Zimmermann, 2000). The latter dephosphorylates extracellular AMP to adenosine, thus regulating the step that limits its formation. Extracellularly, adenosine concentration equilibrium is maintained by reuptake mechanisms operated through the action of specific transporters. Then, inside the cell, it is phosphorylated to AMP by adenosine kinase or degraded to inosine by adenosine deaminase. Intracellularly, adenosine formation is dependent upon the hydrolysis of AMP by an intracellular 5-nucleotidase or by hydrolysis of S-adenosyl-homocysteine. It is estimated that the levels of adenosine in the interstitial fluid fall within the 30–300 nM range (Fredholm et al., 2001).
Adenosine concentrations increase under metabolically unfavorable conditions. Tissue hypoxia, for example, leads to enhanced breakdown of ATP and increased generation of adenosine. In addition to this route, the release of adenosine might be potentiated by hypoxia-dependent inhibition of the salvage enzyme, adenosine kinase, which rephosphorylates the nucleoside to AMP (Decking et al., 1997). As adenosine is unstable, its half-life limited by deamination or cellular reuptake, a hypoxia-induced increase typically affects only local adenosine receptor signaling. Adenosine most likely belongs to the group of autacoids because it is not released in a transmitter or hormone-like fashion.
Adenosine mediates its effects by activation of a family of four G protein–coupled receptors (GPCRs): the A1, A2A, A2B, and A3 adenosine receptors (ARs) (Ralevic and Burnstock, 1998). These receptors differ in 1) their affinity for adenosine, 2) the type of G proteins they recruit and, finally, 3) the downstream signaling pathways activated in the target cells. A1 and A3ARs inhibit the regulation of adenylyl cyclase (AC) activity, whereas activation of the A2A and A2BAR subtypes stimulates AC, which leads to increases in cAMP levels. Early pharmacologic evidence for the existence of ARs was provided by specific antagonism—exerted by methylxanthines, caffeine, and theophylline—of adenosine-induced effects in the heart and brain (Sattin and Rall, 1970).
ARs are widely distributed throughout the body and the fact they are present in basically all cells makes them an interesting target for pharmacologic intervention in many pathophysiologic conditions linked to increased adenosine levels. Development of AR agonists/antagonists would, therefore, seem opportune, but the challenge is to ensure they are devoid of side effects.
In particular, A3AR is now recognized as a potential therapeutic target and biologic marker given its overexpression in inflammatory and cancer cells, compared with low levels found in healthy cells. Fortunately, recent developments in the field of AR agonists and antagonists have helped scientists design and develop safer, more specific lead and back-up candidates for clinical development (Gessi et al., 2011a). The agonists are now considered protective agents in some therapeutic areas and drug candidates in both preclinical and clinical studies. The goal of this review is to cover both the basic science and relevant therapeutic applications of A3AR ligands and provide an authoritative account of the current status of the field.
II. The Discovery of the A3 Adenosine Receptor
The existence of the A3AR was hypothesized about 30 years ago in an attempt to characterize the type of ARs involved in the inhibitory action of adenosine at the frog neuromuscular junction (Ribeiro and Sebastião, 1984). A distinct AR was claimed to exist in the brain that was coupled to Ca2+ metabolism (Ribeiro and Sebastião, 1986). However, this was not the same A3AR finally cloned. Another milestone on the way to the definition of the A3AR was found in antigen-stimulated RBL-2H3 cells (Ali et al., 1990). This receptor was not given a name, but subsequent work identified it as what we now know as the A3AR. Therefore the receptor functionally coupled, via a G protein, to phospholipase C (PLC) and Ca2+ in a stimulatory manner was distinguished from the putative A3AR, which inhibited Ca2+-dependent responses in electrically excitable tissues independently of AC (Ribeiro and Sebastião, 1986).
Then, isolation of a cDNA clone encoding a novel putative GPCR from a rat testis cDNA library was reported. Although the ligand for this receptor was not identified, the authors understandably speculated that the receptor, designated tgpcr1, could play a role in male reproduction (Meyerhof et al., 1991). In 1992, several cDNA sequences from rat striatum encoding GPCRs were reported, one of which (designated R226) was identical to tgpcr1 (Zhou et al., 1992). On the basis of the transmembrane domain sequence homology with adenosine A1 (58%) and A2AARs (57%) and its particular ability to bind AR ligands, it was concluded that R226 encoded a novel AR designated as A3AR (Fozard, 2010). The high expression of the receptor in the testis was confirmed, but, more importantly, low level mRNAs were also shown to be present in the lungs, kidneys, heart, and parts of the central nervous system (CNS), implying that A3AR could have more widespread biologic significance than simply modulating testicular function. Therefore, A3AR is the only AR subtype to be cloned before its pharmacologic identification.
III. Molecular Characterization of the A3 Adenosine Receptor
Homologs of the rat striatal A3AR have been cloned from sheep and humans, thus revealing large interspecies differences in A3AR structure. For example, rat A3AR presents only a 74% sequence homology with sheep and human A3AR, whereas between sheep and humans, this homology is 85%; moreover equine A3AR has also shown a high degree of sequence similarity with that of humans and sheep (Brandon et al., 2006). This is reflected in the very different pharmacologic profiles of the species homologs, especially in terms of antagonist binding, which has made characterization of this AR subtype difficult.
A3AR has been mapped on human chromosome 1p21–p13 (Atkinson et al., 1997) and consists of 318 amino acid residues. It has been determined that the A3AR gene contains two exons separated by a single intron of about 2.2 kb (Murrison et al., 1996). The upstream sequence does not contain a TATA-like motif, but it does have a CCAAT sequence and consensus binding sites for SP1, NF-IL6, GATA1, and GATA3 transcription factors. Involvement of the latter in transcriptional control of this gene would be consistent with the receptor playing a role in immune function.
A3AR is a GPCR characterized by its C-terminal portion, which faces the intracellular compartment and seven transmembrane spanning domains. This region presents multiple Ser and Thr residues that may serve as potential phosphorylation sites of importance for rapid receptor desensitization upon agonist application (Palmer and Stiles, 2000). Phosphorylation leads to a decreased number of receptors in the high-affinity state and decreased agonist potency to inhibit AC activity. In human astrocytoma and murine melanoma cells, a short agonist exposure time results in rapid A3AR internalization and functional desensitization, whereas prolonged treatment with the A3AR agonists induces receptor uncoupling with receptor downregulation (Trincavelli et al., 2002a; Madi et al., 2003). This event was suggested to be mediated by mitogen-activated protein kinases (MAPKs) responsible for a feedback mechanism that controls GPCR kinase activity and receptor phosphorylation in Chinese hamster ovary (CHO) cells transfected with A3AR (Trincavelli et al., 2002b). An A3AR desensitization mechanism has also been proposed in rat hippocampal slices during oxygen and glucose deprivation (Pugliese et al., 2007).
To gain an insight into the molecular characteristics of A3AR ligand interaction, a thermodynamic analysis of A3AR binding site has been addressed. This original approach has shown that agonist binding is always totally entropy driven, whereas antagonist binding is driven by both enthalpy and entropy (Merighi et al., 2002b). Interestingly, the similarity between the thermodynamic parameters of all ARs most likely reflects a common ligand receptor interaction mechanism for ARs (Borea et al., 2000). This may explain the difficulty in obtaining selective adenosine ligands. Therefore, the availability of thermodynamic data adds important information to the decision-making process in drug development (Gessi et al., 2008a).
IV. Medicinal Chemistry and Pharmacology of A3 Adenosine Receptor Ligands
A. Agonists
Potent and selective A3AR agonists stem from multiple substitutions of the parent nucleoside, adenosine (Fig. 1). The structural modifications implicate N6-, C2-, and 5′-substitutions combined with the modification of ribose moiety.
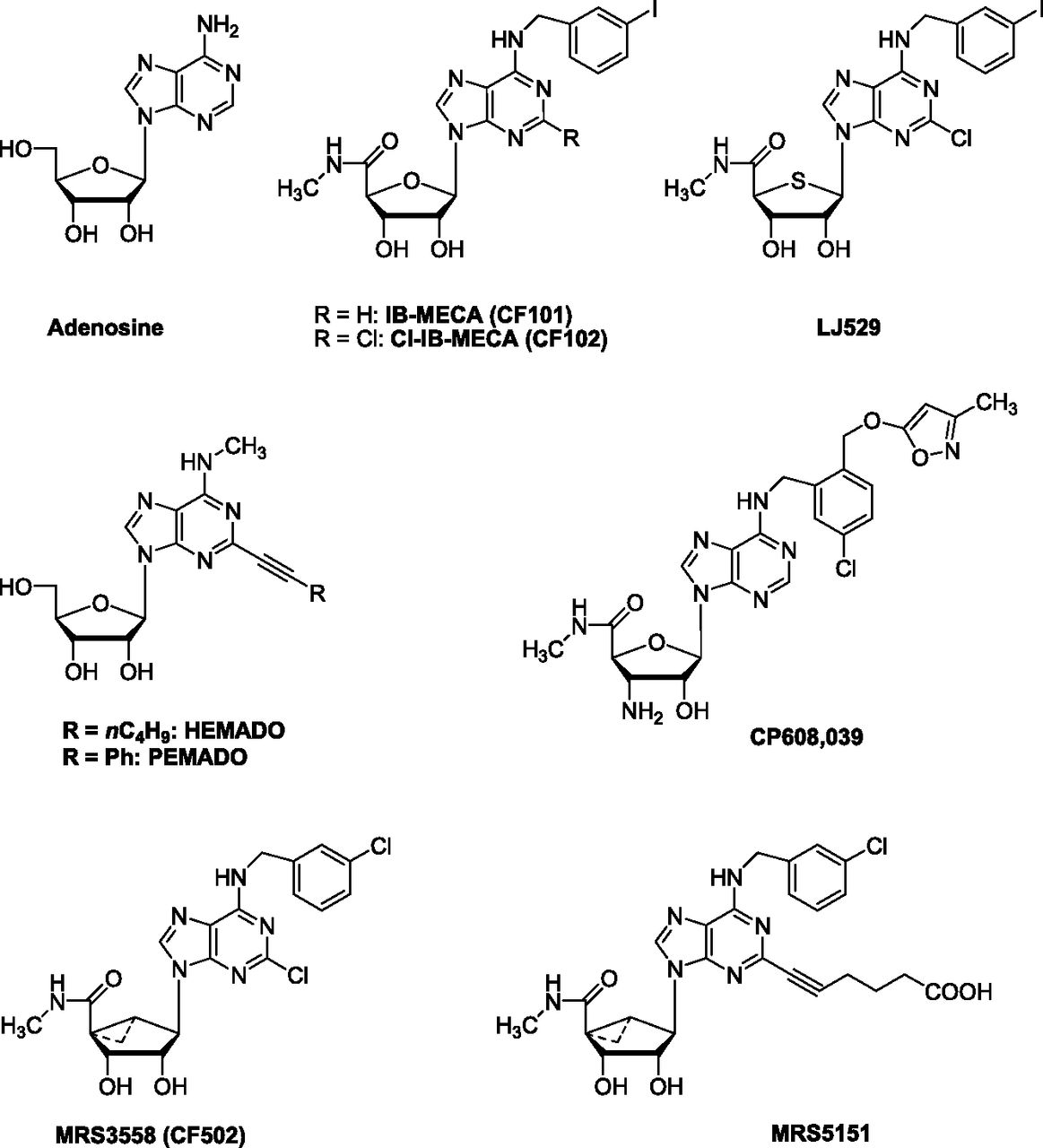
Chemical structures of typical adenosine derivatives as A3AR agonists.
Prototypical A3AR agonists, such as N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide (IB-MECA, CF101), derive from a combined modification of adenosine at the 5′- and at N6-positions (Fig. 1) (Gallo-Rodriguez et al., 1994). In receptor binding studies, IB-MECA has displayed constant inhibitory binding (Ki) values of 51, 2900, and 1.8 nM for human (h) A1, A2A, and A3ARs, respectively, being 28- and 1611-fold selective against A1 and A2A ARs (Table 1). The introduction of small groups at the C2 position of IB-MECA generally increased both A3AR affinity and selectivity, thus leading to the discovery of 2-chloro-N6-(3-iodobenzyl)-adenosine-5′-N-methyluronamide (Cl-IB-MECA, CF102), a potent A3AR agonist with a Ki value of 1.4 nM at hA3AR and with good selectivity for the other ARs (Fig. 1; Table 1) (Kim et al., 1994). On the basis of the bioisosteric rationale, the 4′-thio analogs of Cl-IB-MECA were synthesized as A3AR ligands (Jeong et al., 2003). Among them, 2-chloro-N6-(3-iodobenzyl)-5-N-methylcarbamoyl-4-thioadenosine (LJ529) displayed a Ki value of 0.38 nM at the hA3AR (Fig. 1). LJ529 is reported to have a higher binding affinity to hA3AR than Cl-IB-MECA (Ki = 0.38 versus 1.4 nM; Table 1) (Jeong et al., 2003).
Affinity and selectivity values of selected A3AR agonists
Affinity values are derived from Fredholm et al., 2011, unless otherwise indicated.
Another class of analogs structurally related to the adenosine core consists of derivatives having 2-(ar)-alkynyl chains combined with various substituents at the 6-position. In particular, 2-phenylethynyl-N6-methyladenosine (PEMADO) has shown hA3AR affinity in the low nanomolar range (Ki = 0.44 nM), with A1/A3 and A2A/A3 selectivity of about 74,000 and 94,000, respectively (Fig. 1; Table 1) (Volpini et al., 2002). The ability to inhibit forskolin-stimulated AC has been tested and all these derivatives have proved to be partial A3AR agonists; their efficacy is not significantly modified by the introduction of small alkyl substituents in the N6-position (Volpini et al., 2009). One compound in this series, 2-hexyn-1-yl-N6-methyladenosine (HEMADO), has shown high affinity (Ki = 1.1 nM) and 300- and 1091-fold selectivity versus the A1 and A2AAR subtypes, respectively (Fig. 1; Table 1). The tritium-labeled form [3H]HEMADO binds to hA3AR with an affinity equilibrium binding constant (KD) of 1.1 nM (Klotz et al., 2007). Efforts to identify A3AR agonists that are both potent and selective have led to the discovery of 3′-amino analogs properly modified at the 5′- and N6- positions. One of these, N6-[2-(3-methylisoxazol-5-ylmethoxy)-5-chloro]benzyl-3′-amino-adenosine-5′-N-methylcarboxamide, coded CP608,039, binds to hA3AR with a Ki of 5.8 nM and possesses over 1000- and 8000-fold selectivity versus hA1 and hA2AAR, respectively (Fig. 1; Table 1). Compound CP608,039 displays full agonist activity at the hA3AR, inhibiting the isoproterenol-stimulated cAMP increase with an EC50 of 3.4 nM (DeNinno et al., 2003).
Other selective A3AR agonists have been reported based on modification of the ribose ring. The analogs contain the (N)-methanocarba (bicyclo[3.1.0]hexane) ring system, a rigid ribose substitute lacking the ether oxygen (Tchilibon et al., 2005). In this series, 4-[6-(3-chlorobenzylamino)-2-chloro-9H-purin-9-yl]-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (MRS3558, CF502) has the pharmacologic profile of a full agonist with subnanomolar affinity (Ki = 0.29 nM for the hA3AR; Table 1) (Fig. 1). This compound shows an 897-fold selectivity versus the hA1AR subtype, whereas it is greatly reduced (11-fold) in the mouse due to an increased tolerance of this ring system at A1AR. As in the 2-alkynyl (N)-methanocarba derivative (MRS5151), the introduction of 2-alkynyl chains of varying lengths tends to increase A3AR selectivity in the mouse (m) (up to 430-fold) and preserve it in humans (6261-fold) (Fig. 1; Table 1). Molecular modeling predicted that the sulfonate groups on C2-phenylethynyl substituents would provide high affinity at both m/hA3AR, whereas a N6-p-sulfophenylethyl substituent would determine higher hA3AR versus mA3AR affinity. N6-3-Chlorobenzyl-2-(3-sulfophenylethynyl) derivative (MRS5841) binds selectively to human and mouse A3ARs (Ki hA3AR = 1.9 nM) as an agonist, whereas the corresponding p-sulfo isomer (MRS5701) displays mixed A1/A3AR agonism (Paoletta et al., 2013).
In this context, different chemically functionalized alkynyl chains (esters, amino groups, or carboxylic acid) potentially useful for making conjugates as receptor probes have been synthesized (Melman et al., 2008). Recently, macromolecular conjugates [e.g., polyamidoamine (PAMAM) dendrimers], a particularly versatile and biocompatible class of polymeric drug carriers of chemically functionalized agonists, have been synthesized as potent polyvalent activators of A3AR (Tosh et al., 2010).
B. Antagonists
In an initial attempt, a large number of heterocyclic compounds were synthesized and evaluated as A3AR antagonists (Jacobson et al., 1995; Siddiqi et al., 1995; Ji et al., 1996). In particular a triazoloquinazoline derivative 9-chloro-2-(2-furanyl)-5-[(phenylacetyl)amino][1,2,4]-triazolo[1,5-c]quinazoline (MRS1220) was developed as a very potent compound at the hA3AR, with Ki of 0.59 nM (Fig. 2; Table 2) (Kim et al., 1996).

Chemical structures of typical A3AR antagonists.
Affinity and selectivity values of selected A3AR antagonists
Affinity values are derived from Fredholm et al., 2011, unless otherwise indicated.
Several xanthine or purine analogs were examined first, but none showed significant affinity or selectivity to rat (r) A3AR (Jacobson et al., 2009).
An approach to designing dihydropyridines that bind to ARs without binding to L-type calcium channels has been described. For example, a trisubstituted 1,4-dihydro-6-phenylpyridine analog 3-ethyl-5-benzyl-2-methyl-4-phenylethynyl-6-phenyl-1,4-dihydropyridine-3,5-dicarboxylate (MRS1191), has been found to inhibit radioligand binding at the hA3AR with a Ki value of 31.4 nM (Fig. 2; Table 2), whereas the same derivative is nearly inactive in binding at A1 and A2AARs (Jacobson et al., 1997). One of the first heterocyclic, selective, and competitive A3AR antagonists was 3-propyl-6-ethyl-5[(ethylthio)carbonyl]-2-phenyl-4-propyl-3-pyridine-carboxylate (MRS1523), a pyridine derivative that acts as a highly selective antagonist of A3AR with good potency in both humans and rodents, with Ki values of 18.9 nM for hA3AR and 113 nM for rA3AR (Fig. 2; Table 2) (Li et al., 1998). MRS1523 exhibits only a weaker antagonistic activity toward A1 and A2AARs (Ki = 15.6 and 2.05 μM for rA1 and A2AARs, respectively) (Li et al., 1998).
Another class of analogs, structurally related to isoquinoline and quinazoline urea derivatives, was found as adenosine A3AR antagonists. The combination of the optimal substituents in the two series led to the potent hA3AR antagonist N-(2-methoxyphenyl)-N′-(2-(3-pyridyl)quinazolin-4-yl)urea VUF-5574, with a Ki value of 4.03 nM and over 2400-fold selectivity versus A1 and A2AARs (Fig. 2; Table 2). In an in vitro functional assay, the compound competitively antagonized the inhibition of cAMP production induced by N6-ethylcarboxamidoadenosine in CHO cells expressing hA3AR with a pA2 value of 8.1 (van Muijlwijk-Koezen et al., 2000).
The first example of an AR antagonist containing the pyrazolo-triazolo-pyrimidine scaffold was reported in 1993 (Gatta et al., 1993). Intensive efforts in the chemical synthesis of compounds based on the systematic substitution at the C2-, C5-, C9-, N-7, and N8-positions of the tricyclic template led to the MRE series (Baraldi et al., 1999, 2000, 2002, 2003). This innovative series of compounds includes N-[2-(2-furanyl)-8-propyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-yl]-N′-(4-methoxyphenyl)urea (MRE 3008F20) an antagonist with high affinity at hA3AR (Ki = 0.82 nM; Table 2) and high selectivity (1463- and 172-fold) over human A1 and A2AARs, respectively (Fig. 2) (Varani et al., 2000). In a functional assay MRE 3008F20 showed antagonist activity capable of blocking the effect of IB-MECA on cAMP production in CHO cells (IC50 = 4.5 nM). The tritium-labeled compound was able to bind the hA3AR expressed in CHO cells with a KD value of 0.82 nM and a Bmax value of 297 fmol/mg protein (Varani et al., 2000). The isosteric replacement of the phenyl with a 4-pyridyl moiety led to the water-soluble hA3AR antagonist 5N(4-methoxyphenylcarbamoyl)amino-8-phenylethyl-2-(2-furyl)-pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine (MRE 3005F20) with subnanomolar affinity versus hA3AR (Ki = 0.01 nM) and high selectivity (35,000- and 10,000-fold versus A1 and A2AARs, respectively), suggesting it as an ideal candidate for the pharmacologic and clinical investigation of the hA3AR subtype (Fig. 2; Table 2) (Maconi et al., 2002).
The synthesis of 1,2,4-triazolo[5,1-i]purine derivatives by the modified method of pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidines has been reported as showing high affinity and selectivity for the hA3AR, such as the 5-n-butyl-8-(4-trifluoromethylphenyl)-3H-[1,2,4]triazolo-[5,1-i]purine compound (OT-7999) (Fig. 2). In receptor binding assays, OT-7999 displayed high affinity for the hA3AR (Ki = 0.95 nM) and >10,526-fold selectivity relative to other AR subtypes (Table 2). The ring annelation of xanthine derivatives for the development of AR antagonists has been investigated in depth (Okamura et al., 2002). The pyrido[2,1-f]purine-2,4-diones, which could be considered tricyclic xanthine derivatives, have been reported to exert subnanomolar affinity to hA3AR (Drabczyńska et al., 2003). An important innovation of such a series, compared with xanthines, is the significant increase in water solubility, achieved by introducing a basic nitrogen atom, which could be protonated under physiologic conditions.
The imidazopurinone ring-enlarged 8(R)-ethyl-4-methyl-2-(2,3,5-trichlorophenyl)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-5-one (PSB-10) has shown high affinity for hA3ARs (Ki = 0.44 nM) with high selectivity over A1 and A2AARs (3864- and 6136-fold, respectively; Fig. 2; Table 2) (Muller et al., 2002). PSB-10 has demonstrated inverse agonist activity in binding studies in CHO cells expressing recombinant hA3AR (IC50 = 4 nM). Another similar compound of this series is 8(R)-ethyl-7,8-dihydro-4-methyl-2-phenyl-1H-imidazo[1,2-g]purin-5-one (PSB-11), exhibiting a Ki value of 2.34 nM for the hA3AR and good selectivity versus all other AR subtypes (Ozola et al., 2003) (Fig. 2; Table 2).
The 2-(4-bromophenyl)-7,8-dihydro-4-propyl-1H-imidazo[2,1-i]purin-5(4H)-one derivative KF26777 has revealed subnanomolar affinity to hA3AR (Ki 0.20 nM) and high selectivity over A1 and A2A subtypes (9000- and 2350-fold, respectively) (Fig. 2; Table 2). It inhibits Cl-IB-MECA-induced [35S]guanosine-5′-O-(3-thiotriphosphate) binding to human embryonic kidney 293 cells (IC50 = 270 nM) and enhances intracellular Ca2+ concentrations in human promyelocytic cells (Saki et al., 2002).
Interestingly, the 5-(2-fluoroethyl) 2,4-diethyl-3-(ethylsulfanylcarbonyl)-6-phenylpyridine-5-carboxylate compound was presented as a ligand with high affinity, able to serve as the first positron emission tomography tracer for the A3AR (Wadsak et al., 2008).
Moreover, a very recent study has obtained a novel series of A3AR partial agonists and antagonists as truncated 2-hexynyl-N6-substituted-(N)-methanocarba nucleosides, with Ki values of 7.8−16.0 nM (Nayak et al., 2014). These compounds were screened for renoprotective effects in a human kidney fibrosis model. Most compounds exhibited antifibrotic effects, with (1R,2R,3S,4R,5S)-4-(2-(hex-1-ynyl)-6-(methylamino)-9H-purin-9-bicyclo[3.1.0]hexane-2,3-diol (Structure A) being the most potent, indicating its potential as a good therapeutic candidate for treating renal fibrosis (Fig. 2; Table 2).
V. Distribution of the A3 Adenosine Receptor
The generation of cDNA for A3AR has made the identification of the tissue distribution of this receptor subtype possible. A3AR is widely expressed, its mRNA being revealed in the testis, lung, kidneys, placenta, heart, brain, spleen, liver, uterus, bladder, jejunum, proximal colon, and eye of rats, sheep, and humans (Zhou et al., 1992; Salvatore et al., 1993; Linden, 1994; Rivkees, 1994; Dixon et al., 1996; Burnett et al., 2010). However, marked differences in expression levels do exist within and between species. In particular, rat testis and mast cells express high concentrations of A3 mRNA, whereas low levels have been detected in most other rat tissues (Linden et al., 1993; Salvatore et al., 1993). Human lung and liver are the organs that express high amounts of A3 mRNA, whereas levels in the aorta and brain have been found to be low (Salvatore et al., 1993). The lung, spleen, pars tuberalis, and pineal gland express the highest concentrations of A3 mRNA in sheep.
The presence of the A3AR protein has been evaluated through radioligand binding, immuno- or functional assays in a variety of primary cells, tissues (Table 3), and cell lines (Table 4). In the brains of mouse, rat, gerbil, and rabbit, a widespread, relatively low level of A3AR binding sites has been found (Jacobson et al., 1993; Ji et al., 1994). Owing to this very low expression, other authors have reported the impossibility of detecting either the A3AR gene or binding site in the CNS from in situ hybridization experiments (Rivkees et al., 2000), and others have described A3AR expression in the thalamus and hypothalamus (Yaar et al., 2002). However, electrophysiologic and biochemical evidence suggests the presence of A3AR in the rat hippocampus (Dunwiddie et al., 1997; Macek et al., 1998; Lopes et al., 2003) and cortex (Brand et al., 2001); functional studies have also indicated its presence in the brain and retinal ganglion cells (Jacobson et al., 1993; von Lubitz et al., 1994; Haskó et al., 2005; Zhang et al., 2006a, 2010). The proposed presence of A3AR at motor nerve terminals (Ribeiro and Sebastião 1986) was recently demonstrated through pharmacologic and immunohistochemical studies (Cinalli et al., 2013). At cellular level, A3AR expression has been observed in microglia and astrocytes, the resident immune cells of the CNS (Hammarberg et al., 2003; Björklund et al., 2008a,b; van der Putten et al., 2009; Ohsawa et al., 2012; Gessi et al., 2013). Overall these data iron out the controversy as to whether A3AR are in fact in the CNS.
Distribution and effects of A3ARs in primary tissue/cells
Distribution and effects of A3ARs in cell lines
In cardiomyocytes no direct evidence of the presence of A3AR has been found (Peart and Headrick, 2007); however, a plethora of studies has reported it as being responsible for cardioprotection in a variety of species and models, including isolated cardiomyocytes and isolated myocardial muscle preparations (Tracey et al., 1997; Shneyvays et al., 1998, 2001; Thourani et al., 1999a,b; Cross et al., 2002; Harrison et al., 2002; Germack and Dickenson, 2004; Headrick and Peart, 2005; Xu et al., 2006). High A3AR expression has been revealed in the human coronary and carotid artery (Hinze et al., 2012; Grandoch et al., 2013).
In enteric neurons and epithelial cells, the A3AR was first evidenced by immunohistochemical studies (Christofi et al., 2001; Antonioli et al., 2010; Ren et al., 2011) and subsequently quantified in colonic mucosa by radioligand binding experiments (Gessi et al., 2004a). The presence of functional A3AR has also been demonstrated in human lung parenchyma, in lung type 2 alveolar-like cells (A549), and in bronchi through radioligand binding and immunohistochemical assays (Varani et al., 2006; Calzetta et al., 2011).
It is worth noting that A3AR has been revealed in a variety of primary cells involved in inflammatory responses. In rat basophilic leukemia cells (RBL-2H3), binding experiments have detected a high A3AR density (Ramkumar et al., 1993; Olah et al., 1994) and different studies have reported that A3AR plays a role in rat mast cell degranulation (Carruthers and Fozard, 1993; Fozard and Carruthers, 1993; Ramkumar et al., 1993; Hannon et al., 1995; el-Hashim et al., 1996; Fozard et al., 1996; Hua et al., 2008; Gomez et al., 2011). Recently, A3AR stimulating degranulation has been demonstrated also in LAD2 bone marrow-derived human mast cells (Leung et al., 2014). Human eosinophils were the first cells in which native hA3AR was detected by using radioligand binding (Kohno et al., 1996a; Morschl et al., 2008), and this was then followed by human neutrophils (Bouma et al., 1997; Gessi et al., 2002; Chen et al., 2006; van der Hoeven et al., 2008; Corriden et al., 2013; Mulloy et al., 2013), monocytes (Broussas et al., 1999, 2002; Thiele et al., 2004), macrophages (McWhinney et al., 1996; Szabo et al., 1998; Gessi et al., 2010a), foam cells (Gessi et al., 2010a), dendritic cells (Panther et al., 2001; Fossetta et al., 2003; Dickenson et al., 2003; Hofer et al., 2003), lymphocytes (Gessi et al., 2004b; Varani et al., 2009, 2010b), splenocytes, bone marrow cells, lymphonodes (Bar-Yehuda et al., 2011), and synoviocytes (Varani et al., 2008, 2010c; Stamp et al., 2012). Human chondrocytes and osteoblasts, two key cell types in the skeletal system, were recently found to express A3AR (Vincenzi et al., 2013).
Finally, very high A3AR protein expression was observed in a variety of cancer cell lines (Gessi et al., 2001, 2007, 2010b; Merighi et al., 2001, 2009; Suh et al., 2001; Morello et al., 2009; Jajoo et al., 2009; Cohen et al., 2011; Hofer et al., 2011; Varani et al., 2011a, 2013; Kanno et al., 2012; Nogi et al., 2012; Kamiya et al., 2012; Otsuki et al., 2012; Vincenzi et al., 2012; Nagaya et al., 2013; Sakowicz-Burkiewicz et al., 2013; Madi et al., 2013) and in cancer tissues (Gessi et al., 2004a; Madi et al., 2004; Bar-Yehuda et al., 2008; Varani et al., 2011a), thus suggesting a role for this subtype as a tumoral marker.
VI. Intracellular Pathways Regulated by the A3 Adenosine Receptor
A3ARs have been shown to couple to classic or G protein–dependent second messenger pathways through activation of both Gi and Gq family G proteins (Palmer et al., 1995; Merighi et al., 2003; Haskò and Cronstein, 2004) (Fig. 3). In transfected CHO cells, the ability of both recombinant hA3ARs to inhibit cAMP accumulation and endogenous A3ARs in RBL-2H3 to stimulate PLC is abolished by pretreatment with pertussis toxin, suggesting a functional coupling of this Gi protein receptor (Ali et al., 1990; Zhou et al., 1992; Varani et al., 2000). Furthermore, A3ARs signaling could increase phosphatidylinositol-specific PLC activity (Ali et al., 1990; Ramkumar et al., 1993; Abbracchio et al., 1995; Zheng et al., 2007) and cause the release of Ca2+ from intracellular stores in different cellular models (Gessi et al., 2001, 2002; Merighi et al., 2001; Englert et al., 2002; Fossetta et al., 2003; Shneyvays et al., 2004, 2005; Kim et al., 2012). In a broad study of site-directed mutagenesis of the A3AR, the mutation of the highly conserved tryptophan (W6.48) in the transmembrane domain 6 of GPCRs was first characterized (Gao et al., 2002). Recently, it was reported that this residue plays an important role in determining the structural basis of agonist efficacy and ligand bias in activating A3AR intracellular signaling (Stoddart et al., 2014).

Schematic representation of second messengers and intracellular signaling pathways, downstream targets, mediated by A3ARs stimulation. Upon activation of the A3AR by adenosine 1) the Gα subunit is dissociated from the receptor and Gβγ and decreases AC catalytic activity and cAMP production; 2) PLC is activated leading to Ca2+ increase or PI3K, Akt phosphorylation; 3) G protein RhoA and PLD are stimulated; 4) MAPK family, ERKs, JNK, p38, is modulated; and 5) KATP channels are opened. The final targets downstream these pathways are a series of transcription factors, such as NF-κB, CREB, HIF-1α, c-myc. Thus, A3AR can elicit multiple signaling pathways within a cell.
In addition to GPCR recruitment, it has been reported that A3AR stimulation activates other important pathways, such as the monomeric G protein RhoA and phospholipase D, thus inducing cardioprotection (Lee et al., 2001; Mozzicato et al., 2004). In cardiac cells, A3AR triggers sarcolemmal KATP channels, mediating A3AR-dependent protection from ischemia/reperfusion (IR) injury (Tracey et al., 1998; Wan et al., 2008). However, the opening of a mitochondrial KATP channel has also been proposed as the end effector of the late preconditioning obtained through A3AR stimulation in mice (Zhao and Kukreja, 2002).
Considerable evidence supports the involvement of protein kinase C (PKC) in both early and delayed preconditioning (Liu et al., 1994). In particular ischemic preconditioning-mediated cardioprotection has been achieved through A2B/A3AR stimulation of PKCɛ-triggered aldehyde dehydrogenase type-2 activation in cardiac mast cells (Koda et al., 2010). As for delayed preconditioning, PKCδ has been shown to play an essential role in the cellular signaling cascade that produces the protective effect of A3AR stimulation in the mouse heart (Zhao and Kukreja, 2003). Cl− channel activation by ischemic preconditioning and a nonselective A1/A3AR agonist has also been revealed as a mechanism inducing protection against ischemia/reperfusion injury by enhancing cell volume regulation (Diaz et al., 2010).
Moreover PLC, PKC, or chelation of intracellular Ca2+ has been implicated in the reduction of excitotoxicity and in the increase of neuroprotection through the activation of metabotropic glutamate receptor 1 and A3AR (Dennis et al., 2011). PKC is also involved in the stimulatory effect induced by Cl-IB-MECA on tumor necrosis factor α (TNF-α) release in lipopolysaccharide (LPS)-stimulated macrophages (Forte et al., 2011).
There is considerable evidence for A3AR-mediated effects on MAPKs in a multitude of different cellular models (Merighi et al., 2010). The first example of A3AR-mediated activation of extracellular signal-regulated kinases (ERK1/2) and the modulation of mitogenesis was described in human fetal astrocytes (Neary et al., 1998). Subsequent and more detailed studies have been performed in CHO cells stably expressing A3AR (Schulte and Fredholm, 2000). A3AR signaling to ERK1/2 in CHO cells proved to be dependent on βγ release from pertussis toxin–sensitive G proteins, phosphoinositide 3-kinase (PI3K), Ras, and MAP kinase kinase (MEK) (Schulte and Fredholm, 2002). Importantly, there are several examples of ERK1/2 phosphorylation mediated by endogenously expressed A3AR, e.g., in both primary mouse microglia cells and the N13 microglia cell line, in colon carcinoma, glioblastoma, and in foam cells (Hammarberg et al., 2003; Merighi et al., 2006, 2007a,b; Gessi et al., 2010a,b). In contrast, in melanoma cells, A3AR stimulation is unable to activate ERK phosphorylation, whereas A3AR antagonists are able to improve MEK activity (Fishman et al., 2002b; Merighi et al., 2002a). Indeed, it was demonstrated that A3AR inhibits A375 melanoma cell proliferation by impairment of ERK activation, a discrepancy that may be due to the presence of different signaling pathways in different cell lines, possibly due to crosstalk between the PI3K/AKT and ERK1/2 pathways (Merighi et al., 2005b). In addition, in prostate cancer cells, A3AR inhibits ERK1/2 activity through the reduction of AC and protein kinase A (Jajoo et al., 2009). In glioma, Cl-IB-MECA mediates suppression of ERK1/2, thus inducing caspase-dependent cell death (Kim et al., 2012). Inhibition of ERK1/2 is also associated with the A3AR inhibition of LPS-stimulated TNF-α release in mouse RAW 264.7 cells (Martin et al., 2006). Importantly, MAPK activation has been implicated in IR injury where ERK1/2 exerts a cytoprotective effect, whereas p38 and c-Jun amino-terminal kinase promote cell injury and death. It has been reported that pretreatment with A3AR agonists upregulates phosphorylated ERK1/2 levels, inducing a marked improvement in lung injury and attenuation of apoptosis after reperfusion (Matot et al., 2006). Interestingly, A3AR activation in rat cardiomyocytes has proved to increase ERK1/2 phosphorylation by involving Gi/o proteins, PKC, and Tyr kinase–dependent/independent pathways (Germack and Dickenson, 2004).
In addition to ERK1/2, there is experimental evidence that A3ARs also activate p38 MAPKs in several cellular models, e.g., hCHO-A3, hypoxic melanoma, glioblastoma, and colon carcinoma cells (Hammarberg et al., 2004; Merighi et al., 2005a, 2006, 2007b). This pathway has also been observed in A3AR-stimulated activity of antidepressant-sensitive serotonin transporters (Zhu et al., 2011). A3AR activation protects cardiomyocytes from hypoxia via phosphorylation of p38 MAPK, located downstream of the mitochondrial KATP channel opening (Leshem-Lev et al., 2010). Accordingly, increased phosphorylation of p38 has been found after Cl-IB-MECA treatment, which proves beneficial, protecting the rat heart subjected to ischemia (Hochhauser et al., 2007). In contrast, activation of A2A and A3ARs inhibits p38 MAPK and nuclear factor-κB (NF-κB) pathways in human synoviocytes (Varani et al., 2010c). As for c-Jun amino-terminal kinase, activation by A3AR has been retrieved in microglia, leading to cell migration (Ohsawa et al., 2012), and in glioblastoma cells, mediating an increase in matrix metalloproteinase-9 (MMP-9) (Gessi et al., 2010b).
Another relevant pathway associated with A3ARs is the PI3K/Akt (Merighi et al., 2003). A3AR activation triggers phosphorylation of Akt, protecting rat basophilic leukemia 2H3 mast cells from apoptosis by signaling, which involves the βγ subunits of Gi and PI3K-β (Gao et al., 2001). Moreover, it was demonstrated that A3AR increases Akt phosphorylation in rat cardiomyocytes (Germack et al., 2004). Recently, transient catecholamine administration was found to trigger preconditioning via generation of adenosine and oxygen free radicals (ROS), thus activating the A3AR, PI3K/Akt and ERK in rat hearts and leading to cardioprotection (Salie et al., 2012). In glioblastoma cells, activation of PI3K-Akt-pBad by A3AR stimulation was demonstrated to trigger inhibition of paclitaxel-induced apoptosis (Merighi et al., 2007a). This pathway was also observed in cardiac myocytes subjected to ischemia/hypoxia and reperfusion/reoxygenation (Hussain et al., 2014). In human melanoma A375 and glioblastoma cells, A3AR stimulation has produced PI3K-dependent phosphorylation of Akt with antiproliferative effect and MMP-9 increase, respectively (Merighi et al., 2005b; Gessi et al., 2010b).
There is accumulating evidence that the anti-inflammatory A3AR-mediated activity uses the PI3K/Akt and NF-κB signaling pathways. In LPS-treated BV2 microglial cells and in monocytes, A3AR activation suppresses TNF-α and interleukin (IL)-12 production, respectively, by inhibiting PI3K/Akt and NF-κB activation (Hasko et al., 1998; la Sala et al., 2005; Lee et al., 2006a, 2011). Inhibition of Akt is triggered by A3AR for the reduction of LPS-mediated hypoxia-inducible-factor 1α (HIF-1α) accumulation in murine astrocytes (Gessi et al., 2013). Downregulation of the PI3K/Akt-NF-κB signaling pathway has also been observed in the inhibitory effect of IB-MECA in adjuvant-induced arthritis and in mesothelioma (Fishman et al., 2006; Madi et al., 2007; Varani et al., 2011a). Recently, it was reported that A3AR suppresses angiogenesis by inhibiting PI3K/Akt/mammalian target of rapamycin signaling in endothelial cells (Kim et al., 2013). On the other hand, activation of the PI3K/Akt signaling pathway is triggered by A3AR in B16 melanoma cells and in human skin explants, thus enhancing pigmentation (Madi et al., 2013).
Increasing evidence highlights a crucial involvement of protein kinase A and Akt in the inactivation of glycogen synthase kinase 3β, a key element in the Wnt signaling pathway, generally active during embryogenesis and tumorigenesis to increase cell cycle progression and cell proliferation. This effect has been induced by IB-MECA in melanoma, in hepatocellular carcinoma, in synoviocytes from rheumatoid arthritis (RA) patients, and in adjuvant-induced arthritis rats (Fishman et al., 2002a; Bar-Yehuda et al., 2008; Ochaion et al., 2008).
The number of pathways seen to be triggered by A3AR makes an in-depth understanding of the complexity of such signaling in different cellular elements and pathologies of paramount importance (Fig. 3).
VII. Biologic Functions and Therapeutic Applications of the A3 Adenosine Receptor
A. Central Nervous System
There is interest in understanding A3AR involvement in normal and pathologic conditions of the CNS despite its low expression in the brain (Rivkees et al., 2000; Burnstock et al., 2011). The role of A3ARs in several diseases is often controversial, depending on acute and chronic agonist administration (Jacobson, 1998; Von Lubitz, 1999). It has been hypothesized that A3ARs play a protective role in the first phase of ischemia by decreasing synaptic transmission (Pugliese et al., 2003; Rivera-Oliver and Diaz-Rios, 2014). However, with prolonged A3AR stimulation the effects transform from protective to injurious, increasing excitotoxicity (Pugliese et al., 2007).
A proconvulsant effect of A3ARs has been observed in the immature brain, suggesting that it may facilitate seizure-induced neuronal damage (Boison, 2008). Accordingly, it has also been reported that A3ARs decrease the stability of currents generated by gamma aminobutyric acid in different epileptic tissues, thus suggesting that adenosine antagonists may offer therapeutic opportunities in various forms of human epilepsy (Roseti et al., 2009).
A3AR agonists have been reported to depress locomotor activity, thus suggesting a possible inhibition of excitatory neurotransmission in cortical neurons (Boison, 2007). Similarly, an increased motor activity has been revealed in A3AR knockout (KO) mice (Björklund et al., 2008a). It was recently demonstrated that A3AR receptors are present in the nerve terminal and muscle cells at the neuromuscular junctions. The presence of these receptors in the neuromuscular synapse allows the receptors to be involved in the modulation of transmitter release (Garcia et al., 2014).
A3AR is able to reduce excitotoxicity and promote neuroprotection in hippocampal CA3 pyramidal neurons, after ischemic damage, through coactivation of mGluR1; the mechanism involved is endocytosis and subsequent degradation of AMPAR protein levels (Dennis et al., 2011; Sebastiao et al., 2012). Neuroprotection has also been observed through inhibition of P2X7 receptor-induced death of rat retinal ganglion cells (Hu et al., 2010).
An upregulation of A3ARs has been reported in the hippocampus of a transgenic mouse model of Alzheimer’s disease where altered oxidative phosphorylation was detected before amyloid deposition, whereas no change has been observed in the brains of Parkinson’s patients (von Arnim et al., 2006; Varani et al., 2010b). Interestingly investigations have shown that A3AR stimulation rapidly enhances the activity of antidepressant-sensitive serotonin transporters and that the stimulation of SERT activity is lost in A3AR KO mice (Zhu et al., 2011). A3AR-stimulated SERT activity is primarily mediated by p38 MAPK-linked pathways, thus supporting the idea that the use of agents that selectively block A3ARs may diminish SERT surface expression and activation. This evidence suggests the use of A3AR antagonists for the treatment of mood disorders characterized by hyposerotonergic states (Zhu et al., 2007, 2011).
Glial A3AR activation by high adenosine levels subsequent to brain injury may be implicated in neuroinflammatory tissue responses (Hammarberg et al., 2003, 2004). A role for A3AR as dynamically regulated suppressors of A2A-mediated inhibition of proinflammatory cytokine responses was reported in microglial cells where A3AR signaling may also be involved in the ADP-induced process extension and migration (van der Putten et al., 2009; Ohsawa et al., 2012). In contrast, an anti-inflammatory effect, leading to a reduction in LPS-stimulated TNF-α production and cell migration, was observed in the same cells (Lee et al., 2006a; Choi et al., 2011).
A3AR stimulation of neuroprotective substances was also shown in mouse astrocytes (Wittendorp et al., 2004). As regards neuroprotection, A3ARs inhibit LPS-induced HIF-1α accumulation in murine astrocytes, thus resulting in downregulation of the genes involved in inflammation and hypoxic injury, i.e., inducible nitric oxide synthase and A2BARs receptors, in both normoxic and hypoxic conditions, thus adding a novel mechanism responsible for protective effects against brain injury (Gessi et al., 2013).
To date, several A3AR neuroprotective and anti-inflammatory effects have been demonstrated although further studies are needed before clinical utility can be achieved.
B. Cardiovascular System
Several studies in neonatal rat cardiomyocytes suggest that A3AR stimulation produces direct cardioprotective effects (Germack and Dickenson, 2005; McIntosh and Lasley, 2012). Although there is low A3AR expression in myocardial tissue, a number of works demonstrated that acute treatment with agonists induces protective “anti-ischemic” effects (Auchampach et al., 1997; Tracey et al., 1997; Thourani et al., 1999a,b; Ge et al., 2006, 2010; Xu et al., 2006; Chanyshev et al., 2012). These findings have also been confirmed in A3AR-overexpressing mice, where infarct size was smaller than in wild-type mice after in vivo regional IR (Shneyvays et al., 2004). Similarly, anti-ischemic effects have been found with positive allosteric modulators as well as with dendrimeric A3AR agonists (Wan et al., 2011; Du et al., 2012; Chanyshev et al., 2012).
As for the timing of cardioprotection, there is significant evidence that A3AR activation exerts a cardioprotective effect both before ischemia and during reperfusion (Gessi et al., 2008b; McIntosh and Lasley, 2012). Interestingly, some studies have indicated that protection occurs postischemia through inhibition of either neutrophil-induced reperfusion injury or myocyte apoptotic cell death (Jordan et al., 1999; Maddock et al., 2002); others, instead, have found that preischemic A3AR activation is effective and necessary for cardioprotection (Thourani et al., 1999a,b). It has been demonstrated that A3AR agonism is able to trigger an anti-infarct response with either pre-or postischemic treatment (Auchampach et al., 2003). Moreover, A3AR activation is able to mimic or induce myocardial preconditioning, meaning that transient stimulation of the A3AR before induction of ischemia leads both to early and to delayed protection (Peart and Headrick, 2007). Preconditioning through AR modulation may have clinical relevance (for example, in cardiac surgery) although pretreatment is rarely permitted in acute myocardial infarction. For this reason, achieving protection from IR injury through administration of the drug after ischemia or during reperfusion would be more useful (Nishat et al., 2012).
A novel prosurvival pathway by which A3ARs ameliorate myocardial ischemia/reperfusion injury is through its effect on caspase-3 activity, MEK1/2-ERK1/2, and PI3K/AKT (Hussain et al., 2014). There is also evidence that A3ARs enhance cellular antioxidant capacity, thus contributing to vasoprotection and reduced cardiac myocyte death and strongly supporting an A3AR-dependent cardioprotective response. These effects have reduced infarct size, inhibited apoptosis, and improved postischemic contractile function (Zhai et al., 2011). According to powerful cardioprotective effects against myocardial injury, A3AR activation has also been found to improve myocardial survival against cardiotoxic side-effects induced by chemotherapy (Sandhu et al., 2014).
A3AR has, moreover, been seen to play a role in the modulation of blood vessel function (Burnstock and Ralevic, 2014). Activation of A3AR leads to endothelium-dependent aortic contraction through cyclooxygenase-1, which may play a role in cardiovascular inflammation, including hypertension and atherosclerosis (Ansari et al., 2007). A3AR has also been shown to 1) inhibit or negatively modulate coronary flow in isolated mouse heart (Talukder et al., 2002), 2) cause vasoconstriction in hamster arterioles (Shepherd et al., 1996), and 3) reverse vascular hyporeactivity after hemorrhagic shock in rats (Zhou et al., 2010). A3AR-mediated vasoconstriction may involve indirect signaling through nonvascular cell types, such as mast cells that may reside within the vascular wall, by releasing such factors as histamine and thromboxane (Tilley et al., 2000; Zhao and Kukreja, 2002). Indeed, A3AR-mediated vasoconstriction has been proved to depend on the inhibition of cAMP accumulation in smooth muscle and in cultured aorta (Talukder et al., 2002). Another pathway linking A3AR to contraction of the mouse aorta is through ROS generation, via activation of NADPH oxidase 2 (El-Awady et al., 2011). It has recently been reported that, via A2B and A2A/A3ARs, adenosine induces hyaluronan matrix modulation in the smooth muscle cells of the human coronary artery, thereby increasing smooth muscle cells proliferation, migration, and monocyte adhesion. For this reason, adenosine has been proposed as a regulator of plaque stability and inflammatory cell trafficking in atherosclerosis (Reiss and Cronstein, 2012; Grandoch et al., 2013). However, although a genetic deficiency in the A3AR subtype has been demonstrated as significantly reducing the proliferative potential of aortas in organ culture, it does not attenuate the development of atherosclerotic lesions in response to a high-fat diet or vascular injury in vivo (Jones et al., 2004). On the other hand, it has been shown that, in hypoxic foam cells, adenosine stimulates HIF-1α accumulation, vascular endothelial growth factor secretion, and foam cell formation, suggesting the potential use of A3AR antagonists in blocking major steps in atherosclerotic plaque development (Gessi et al., 2010a).
In conclusion, in the cardiovascular system, A3AR modulation appears to have an important protective function. However, this area needs greater exploration through clinical studies to confirm the evidence for preclinical models.
C. Pulmonary System
The role of adenosine in regulating the respiratory system is well known and elevated levels of the nucleoside have been found in bronchoalveolar lavage, blood, and exhaled breath condensate of patients with asthma and chronic obstructive pulmonary disease (COPD).
It is known that A3ARs may be involved in both pro- or anti-inflammatory responses depending on the cell type involved (Salvatore et al., 2000). In particular, the strongest evidence for a functional role of A3AR in mast cell activation comes from the use of genetic A3AR and mast cell KO mice where degranulation appears to depend on A3AR activation, thus suggesting that adenosine exposure can result in A3AR-dependent airway inflammation (Tilley et al., 2003). Likewise, adenosine fails to induce histamine release from lung mast cells obtained from A3AR KO mice and mediated airway hyperresponsiveness by both A3AR-dependent and -independent mechanisms in rodents (Zhong et al., 2003). Accordingly, in A3AR KO mice the reconstitution of wild-type mast cells restores the airway hyperresponsiveness (Hua et al., 2008). In contrast, in a bleomycin model of pulmonary inflammation and fibrosis, A3ARs have provided anti-inflammatory functions, regulating production of the mediators involved in fibrosis (Morschl et al., 2008; Burnstock et al., 2012). Although A3AR protein has not been found in human lung mast cells, it is highly present in human eosinophils with anti-inflammatory functions (Reeves et al., 2000; Hasko et al., 2008). As A3ARs inhibit degranulation of eosinophils, it has been speculated that specific A3AR agonists might be useful in eosinophil-dependent allergic disorders, such as asthma and rhinitis (Spicuzza et al., 2006). However, adenosine deaminase KO mice treated with selective A3AR antagonists have shown a marked attenuation of pulmonary inflammation, decreased eosinophil infiltration, and reduced airway mucus production (Young et al., 2004).
Interestingly, COPD patients have shown decreased A2BAR density and increases in A2A and A3ARs in peripheral lung compared with the levels found in smokers with normal lung function (Varani et al., 2006). An increase in A3ARs found in bronchoalveolar lavage macrophages from COPD patients appears to be closely associated with the presence of high levels of proinflammatory cytokines. In a human leukemic monocyte lymphoma cell line (U937), IL-1β, and TNF-α are both able to bring about significant increases in A2A and A3AR density (Varani et al., 2010). In contrast, A3AR agonists have been suggested as a potential effective therapy for IR-induced lung injury (Matot et al., 2006). Indeed, inosine has been seen to have anti-inflammatory effects in allergic lung inflammation by recruiting A3ARs (da Rocha Lapa et al., 2013). Accordingly, A3AR activation was recently found to attenuate lung IR injury through a neutrophil-dependent mechanism, suggesting the use of A3AR agonists as a novel therapeutic strategy to prevent lung IR injury and primary graft dysfunction after transplantation (Gazoni et al., 2010; Mulloy et al., 2013).
The role of A3AR in the human lung has still to be clarified, although data in recent literature would appear to lean toward a protective effect.
D. Immune System and Inflammation
A3ARs are present in immune cells and are involved in the physiopathologic regulation of inflammatory and immune processes mediated by adenosine (Antonioli et al., 2010; Hasko and Cronstein, 2013).
Neutrophil behavior is strongly affected by the A3AR mediating inhibition of the oxidative burst and chemotaxis with anti-inflammatory activity (Bouma et al., 1997; Gessi et al., 2002; van der Hoeven et al., 2008). Accordingly, these effects have also been observed in a model of severe IR injury after lung transplantation (Mulloy et al., 2013). On the other hand, it was also reported that, together with P2Y2, A3AR guides neutrophil chemotaxis after ATP release (Chen et al., 2006) and also plays a role in neutrophil migration by positively affecting innate immune response (Butler et al., 2012). In particular, this adenosine subtype aggregates in immunomodulatory microdomains on human neutrophil membranes and promotes the formation of bacteria-tethering cytonemes, which are important for phagocytosis, thus suggesting a key role in innate immune response (Corriden et al., 2013).
Adenosine has been proposed as a possible inhibitor of killer T-cell activation in the microenvironment of solid tumors (Hoskin et al., 1994a,b, 2002). The nucleoside interferes with activation-induced expression of the costimulatory molecules CD2 and CD28, possibly through A3AR recruitment (Butler et al., 2003). However, the adenosine-mediated inhibitory effect on the ability of lymphokine-activated killer cells to kill tumor cells has essentially been attributed to the cAMP-elevating A2AAR (Raskovalova et al., 2005). In contrast to the immunosuppressive role of adenosine in the environment of solid tumors, A3AR stimulates murine bone marrow cell proliferation in vitro through the production of the granulocyte colony-stimulating factor by human peripheral blood mononuclear cells (PBMC). Accordingly, when administered before chemotherapy, adenosine increases leukocyte and neutrophil numbers (Fishman et al., 2000). In addition, A3AR activation has been found to enhance natural killer (NK) cell activity and most likely the NK cell-mediated destruction of tumor cells (Harish et al., 2003). Therefore, it is well established that, within the solid tumor microenvironment, adenosine is an important inhibitor of tumor cell destruction by NK and lymphokine-activated killer cell signaling, primarily through the A2A and A3ARs on the surface of T cells (Hoskin et al., 2008). Indeed, ex vivo activation of CD8+ T cells with the A3AR agonist improves adoptive immunotherapy for melanoma (Montinaro et al., 2012). We emphasize that the identification of signal transduction pathways, through which adenosine exerts its inhibitory effects on cell-mediated antitumor immune responses, may allow for the development of focused pharmacologic strategies to reduce, or ablate, the impact of adenosine-mediated immune suppression in cancer patients (Kumar, 2013).
Adenosine is an endogenous regulator of monocyte-macrophage functions, first producing high amounts of inflammatory mediators during the early stages of inflammation and later participating in the resolution of this process. As for the role A3ARs play in the inhibition of macrophage production of TNF-α, discrepant results have been obtained. For example, some studies attributed reduction of TNF-α to A3AR in both human and mouse species (Sajjadi et al., 1996; McWhinney et al., 1996; Lee et al., 2006a), whereas others, using ARs KO mice, found this effect to be mediated essentially by A2AAR and, to a lesser extent by A2BAR, without A3AR involvement (Kreckler et al., 2006). Still others found that Cl-IB-MECA is able to enhance TNF-α production in LPS-treated macrophages in an NF-κB–dependent manner (Forte et al., 2011). Further macrophage functions regulated by A3AR include the reduction of the chemokine macrophage inflammatory protein (MIP) 1α and the inhibition of interferon regulatory factor 1, inducible nitric oxide synthase, and CD36 gene expression in RAW264.7 murine and THP-1 human cells (Hasko et al., 1996; Barnholt et al., 2009). In addition, IB-MECA inhibits the respiratory burst of human monocytes by blocking NADPH oxidase activity (Broussas et al., 1999; Thiele et al., 2004). In addition to inflammatory mediator modulation, A3AR may be involved in human ventricular remodeling by stimulating MMP-9 production, relevant for revascularization, supporting the use of A3AR agonists during the remodeling phase (Velot et al., 2008). Although most studies indicate a role for A3AR agonists as inhibitors of inflammation, a recent novel application of A3AR antagonists was discovered for a novel series of truncated nucleosides, i.e., that of inhibiting TGF-β1–induced collagen I upregulation, making them appear as good therapeutic candidates for treating renal fibrosis (Nayak et al., 2014).
Dendritic cells are antigen-presenting cells that are specialized to activate naive T lymphocytes and initiate primary immune responses (Gessi et al., 2009). The role of A3ARs in regulating mature dendritic cell function is less well established (Koscso et al., 2011). It has been reported that A1 and A3ARs are predominantly expressed in immature human dendritic cells and that their stimulation induces Ca2+ mobilization from intracellular stores, actin polymerization, and chemotaxis (Panther et al., 2001). Mature dendritic cells, however, downregulate A3ARs and mainly present A2AARs, and both these subtypes inhibit TNF-α release in the mouse dendritic cell line XS-106 (Dickenson et al., 2003).
A3AR activation affects numerous mast cell functions in rodents and humans including degranulation, apoptosis, and regulation of vasopermeability (Gao et al., 2001; Feoktistov et al., 2003). Studies performed on cellular and animal models have shown that A3AR increases degranulation in rodents (Ramkumar et al., 1993; Reeves et al., 1997; Salvatore et al., 2000; Smith et al., 2002; Zhong et al., 2003). Although in the past there was no evidence that the A3AR played a similar role in human mast cells, recently this effect was reported in human LAD2 mast cells (Feoktistov and Biaggioni, 1995; Ryzhov et al., 2004; Leung et al., 2014). A3ARs are also emerging in the treatment of bowel inflammation, because IB-MECA markedly reduces colon levels of such proinflammatory cytokines as IL-1, IL-6, and IL-12 and decreases local production of MIP-1α or MIP-2, thus providing a powerful leukocyte downregulation in bowel inflammation (Mabley et al., 2003). Different studies have evaluated the effects of A3AR agonists on gene dysregulation and tissue injury in rat models of colitis. A3AR agonists prevent the induction of various cytokine/chemokine/inflammatory genes and promote marked suppression of ROS production, which leads to significant amelioration of intestinal injury. In the stomach, jejunum, colon ileum, cecum, and liver, A3AR upregulation has been observed during colitis to provide anti-inflammatory processes in the intestine and liver (Mabley et al., 2003; Lee et al., 2006b). Inhibition of IL-2, TNF-α, and IFN-γ production and upregulation of IL-10 have been observed in cultured splenocytes derived from CF101-treated animals, thus pointing toward a marked anti-inflammatory effect (Bar-Yehuda et al., 2011).
More recently, A3ARs were found to be overexpressed in different autoimmune disorders such as Crohn’s disease and psoriasis. The upregulation observed in these pathologies could be attributed to adenosine, which, under conditions of stress, accumulates in the extracellular environment. Most transcription factors, such as NF-κB and CREB, have been revealed as promoting inflammation and being inversely associated with A3AR upregulation (Ochaion et al., 2009). Accordingly, CF101 was tested in a phase II, multicenter, randomized, double-blind, dose-ranging, placebo-controlled trial in patients with moderate to severe chronic plaque-type psoriasis. In this study the drug was found to be safe and well tolerated, and the improvement was progressive and linear throughout the period examined (David et al., 2012). As a consequence, CF101 has entered a phase II/III randomized, double-blind, placebo-controlled, dose-finding study of the efficacy and safety of daily CF101 administered orally in patients with moderate to severe plaque psoriasis (ClinicalTrials.gov).
Overall, the data in the literature suggest that A3AR activation can induce important anti-inflammatory effects in several cellular models. The results achieved thus far with A3AR agonists in clinical studies on such major inflammatory conditions as arthritis and psoriasis are quite promising, and we are confident that they will be translated into treatments for other flogosis-related pathologies.
E. Rheumatoid Arthritis and Ostheoarthritis
Rheumatoid Arthritis is a chronic autoimmune inflammatory disorder of unknown etiology that affects approximately 1% of the population worldwide (Varani et al., 2010a). It is widely accepted that, to prevent unfavorable outcome, RA must be treated early with effective therapy (Flogel et al., 2012). Although, in recent years, great progress has been made in this direction, particularly using biologic drugs, to date no univocal, effective, and safe pharmacologic treatment is available.
Knowledge of the effects of adenosine has revealed that ARs could be a useful target for RA therapy. Adenosine production has emerged as an important cell mechanism to regulate inflammation due to an increase in receptor density and/or functionality. A3ARs are upregulated in RA, psoriasis, and Crohn's disease (Ochaion et al., 2009). Interestingly they are also overexpressed in untreated RA patients and in methotrexate-treated RA patients, and administration of anti–TNF-α drugs normalizes their number and functionality (Varani et al., 2009). Furthermore, A3AR density inversely correlates with Disease Activity Score in 28 or 44 joints, suggesting a direct role of the endogenous activation of these receptors in the control of RA inflammation (Varani et al., 2011b). The overexpression of A3ARs in RA has been directly linked to the increase in NF-κB, a key player in the pathogenesis of arthritic diseases (Bar-Yehuda et al., 2007; Ochaion et al., 2009).
In RA patients, adenosine suppresses the elevated levels of proinflammatory cytokines such as TNF-α and IL-1β, and clinical evidence has shown that A3AR agonists modulate an improvement in signs and symptoms of the disease (Forrest et al., 2005; Silverman et al., 2008). Furthermore, A3AR agonists prevent cartilage damage, osteoclast/osteophyte formation, bone destruction, and markedly reduce pannus and lymphocyte formation in rat models (Rath-Wolfson et al., 2006; Bar-Yehuda et al., 2009). The anti-inflammatory effect of A3ARs has also been observed in fibroblast-like synoviocytes derived from the synovial fluid of RA patients and is closely associated with a decrease in NF-κB and TNF-α release (Ochaion et al., 2008). Oral treatment with CF101 has led to amelioration and a marked decrease in clinical manifestations of the disease. In a phase I study in healthy subjects, CF101 proved safe and well tolerated, offering linear pharmacokinetic activity (van Troostenburg et al., 2004). This was confirmed in a now successfully concluded phase II study in RA patients in which CF101 mediated improvement in signs and symptoms, thus suggesting an opportunity for its development as an antirheumatic agent (Silverman et al., 2008; ClinicalTrials.gov).
The most common form of arthritis is osteoarthritis (OA), which is the most important cause of disability in older adults. The current recommended treatment of OA involves weight loss, physical therapy, and the use of pain relievers. However, these drugs do not reverse the degenerative process in OA and show some adverse effects on cartilage metabolism (Zhang et al., 2008). It is well known that p38 MAPKs are involved in controlling such cellular responses as adenosine-mediated proinflammatory cytokine release (Fotheringham et al., 2004). Accordingly, a pathway involving the reduction in p38 MAPKs, NF-κB, TNF-α, and IL-8 by A3AR activation was observed in human synoviocytes (Varani et al., 2010c). It was also reported that the NF-κB signaling pathway is deregulated by the presence of IB-MECA and is involved in OA pathogenesis. In addition, CF101 induces inflammatory cell apoptosis and acts as a cartilage protective agent, suggesting that it may be a suitable candidate for the treatment of OA (Bar-Yehuda et al., 2009). The safety and efficacy of CF101 has also been evaluated in a phase II clinical study with patients suffering from OA of the knee (Fishman et al., 2012).
Interestingly, a link has been found between ARs and their modulation by such physical agents as pulsed electromagnetic fields (PEMFs). In vitro studies on joint cells have suggested that exposure to PEMFs mediates a significant protection against the catabolic effect of proinflammatory cytokines and an anabolic action increasing matrix synthesis and cell proliferation (Fini et al., 2013). Moreover, PEMFs are able to mediate the upregulation of A3ARs in bovine chondrocytes and synoviocytes (Varani et al., 2008; De Mattei et al., 2009). These data have been further confirmed in human synoviocytes where the copresence of A3ARs and PEMFs reduces the release of PGE2, IL-6, and IL-8, whereas it increases IL-10 release (Ongaro et al., 2012). Recently, in human T/C-28a2 chondrocytes and human FOB 1.19 osteoblasts, PEMFs and A3AR stimulation have been seen to reduce PGE2, IL-6, and IL-8 production, suggesting their potential in the treatment of inflammatory bone and joint disorders (Vincenzi et al., 2013). Therefore, PEMFs could be an innovative physiologic alternative to the use of agonists as they can meditate the tissue-specific agonist effects without any desensitization and downregulation. All the above indicates that PEMFs could be a very interesting example of “soft pharmacology.”
Overall, in joint diseases, the behavior of A3AR strongly suggests that it could play a therapeutic role in this area and underlines the need for further pharmacologic investigation into how the inflammatory conditions closely associated with these diseases are modulated.
F. Muscle System
It is a well reported fact that IR can cause significant injury in skeletal muscle, because this is the most vulnerable tissue in the extremities (Blaisdell, 2002). Therefore protecting skeletal muscle from IR insult is an important aim of therapy to ameliorate muscle and organ injury (Tsuchida et al., 2003).
A3ARs have been found to protect the skeletal muscle against IR damage. Interestingly, the A3AR agonist is a prime candidate for pharmacologic intervention in the first few hours postinjury based on several working hypotheses concerning its mechanism of protection. The pathway involves PLC β2/β3, which in turn activates PKC via diacylglycerol (Zheng et al., 2007). PKC has been shown to activate KATP channels and cause protection. That adenosine and ischemic preconditioning of the skeletal muscle have an energy-sparing effect is consistent with a consequence of sarcolemmal KATP channel activation. The latter can cause an abbreviation of the muscle action potential duration, reduce Ca2+ influx and overload, and thus ameliorate ischemic injury (Liang et al., 2010). Furthermore, the role of the A3AR agonist in mitigating Ca2+ influx and overload, through its effect on PKC signaling, can potentially decrease MMP activation and subsequent upregulation of downstream proteolytic cascades (Liang et al., 2010).
Another mechanism of A3AR protection is its anti-inflammatory effect, exerted not at the skeletal muscle level but at the immune cell level. In this signaling pathway, A3AR activation on circulating immune cells suppresses their function and decreases immune cell-mediated damage to the skeletal muscle. This concept is supported by the finding that activated mast cells and neutrophils are important contributors to, if not mediators of, skeletal muscle IR damage (Fishman et al., 2012).
G. Eye Diseases
A3ARs have been widely implicated in many ocular diseases including dry eyes, glaucoma, and uveitis. When compared with normal eyes, A3AR mRNA and protein levels are found to be consistently increased in the nonpigmented ciliary epithelium in pseudoexfoliation syndrome with glaucoma (Schlotzer-Schrehardt et al., 2005).
In the past, A3AR KO mice showed lower intracellular pressure, thus suggesting that A3AR antagonists could play a role in the treatment of glaucoma (Yang et al., 2005). In addition, the use of A3AR antagonists appears to be a specific, alternate approach for treating ocular hypertension in patients affected by the pseudoexfoliation syndrome in open angle glaucoma, which is typically associated with anterior chamber hypoxia and elevated intraocular pressure (IOP) (Yang et al., 2005). Accordingly, a series of nucleoside-derived antagonists have been found to lower IOP across species (Wang et al., 2010). Recently, it was reported that adenosine may trigger oligodendrocyte death via activation of A3AR, suggesting that this mechanism contributes to optic nerve and white matter ischemic damage (González-Fernández et al., 2014).
On the other hand, it was shown that, in retinal ganglion cells, A3AR prevents activation of the P2X7/NMDA receptors responsible for the rise in Ca2+ and apoptosis, thus suggesting the neuroprotective potential of A3AR agonists in glaucoma treatment. These findings were confirmed in in vivo experiments (Zhang et al., 2006b, 2010; Hu et al., 2010). CF101 entered a clinical phase of drug development. Studies from a phase II clinical trial revealed that it was well tolerated and induced a statistically significant improvement in patients with moderate to severe dry eye syndrome. Interestingly, in the same clinical trial, CF101 decreased IOP, thus demonstrating its efficacy as an IOP-lowering agent. These data, and the anti-inflammatory characteristics of CF-101, support pursuing study of this drug as a potential treatment of the signs and symptoms of dry eye syndrome and glaucoma (Avni et al., 2010; Fishman et al., 2013). Indeed now CF101 has entered a phase II, randomized, double-masked, placebo-controlled, parallel-group study of the safety and efficacy of daily CF101 administered orally in subjects with elevated intraocular pressure (ClinicalTrials.gov).
CF101 has also been effective in an experimental model of autoimmune uveitis, supporting further exploration of this molecule for the treatment of uveitis (Bar-Yehuda et al., 2011). Oral treatment with CF101, initiated upon disease onset, improves clinical uveitis fundoscopy score and ameliorates the pathologic manifestations of the disease. Now CF101 has entered a phase II, randomized, double-masked, placebo-controlled study of the safety and efficacy of daily CF101 administered orally in subjects with active, sight-threatening, noninfectious intermediate or posterior uveitis (ClinicalTrials.gov).
H. Cancer
A very interesting area of possible application for A3AR ligands is in cancer therapy. The possibility that A3AR may play a role in the development of cancer was suggested several years ago (Fishman et al., 1998, 2000; Merighi et al., 2003; Gessi et al., 2011b).
Adenosine is present at high levels in cancer tissues and in the interstitial fluid of several tumors, in sufficient concentration to interact with ARs (Blay et al., 1997; Antonioli et al., 2013). In particular, A3ARs are present in different types of tumor cells, such as HL60 and K562 human leukemia (Gessi et al., 2002), Jurkat (Gessi et al., 2001), and U937 human lymphoma (Gessi et al., 2010a), Nb2 rat lymphoma (Fishman et al., 2000), A375 human melanoma (Merighi et al., 2001), PGT-beta mouse tumor pineal gland (Suh et al., 2001), human glioblastoma (Merighi et al., 2006; Gessi et al., 2010b), human prostatic (Jajoo et al., 2009), and mesothelioma cells (Varani et al., 2011a). A3AR subtype overexpression has also been demonstrated in colon cancer tissues, compared with normal mucosa, obtained from patients undergoing surgery (Gessi et al., 2004a; Madi et al., 2004). Upregulation in tissues is reflected in peripheral blood cells, thus making this adenosine subtype a possible marker for cancer (Gessi et al., 2004a). Another study showed upregulated A3AR mRNA expression in hepatocellular carcinoma (HCC) tissues compared with adjacent normal tissues (BarYehuda et al., 2008). Remarkably, upregulation of A3AR was noted in PBMCs derived from HCC patients compared with healthy subjects. Interestingly, these results suggest that, in PBMCs, A3AR reflects receptor status in remote tumor tissue (Gessi et al., 2004a). Similarly, in malignant mesothelioma pleura, mRNA, and protein expression of A3ARs have been found statistically increased with respect to healthy mesothelial pleura (Varani et al., 2011a).
Clinical investigations have clearly shown that the prevalence of hypoxic tissue areas is a specific property of solid tumors (Melillo et al., 2004). Furthermore, hypoxia appears to induce increased intracellular adenosine levels and stabilize the most important factors involved in hypoxia, such as HIF-1α. A3ARs stimulation induces HIF-1α accumulation in different cancer cell lines (Merighi et al., 2005a, 2006). This has been seen to lead to an increase in angiopoietin-2 and/or vascular endothelial growth factor, depending on the cell model investigated (Merighi et al., 2007b).
As for the role the A3AR subtype plays in cancer cells, several other functions have been described. Both pro- and antiapoptotic as well as pro- and antiproliferative effects have been reported depending on the level of receptor activation (Jacobson, 1998; Merighi et al., 2005b; Gessi et al., 2007; Kim et al., 2010; Taliani et al., 2010; Varani et al., 2011a; Sakowicz-Burkiewicz et al., 2013). At first, telomerase activity inhibition and cytostatic effects were observed in tumor cells (Fishman et al., 2000, 2001). Then the intracellular pathway involved in A3AR-mediated tumor growth inhibition was identified (Fishman et al., 2004, 2012). In contrast, some studies showed that A3AR agonist inhibition of cell proliferation was only obtained by micromolar concentrations (Lu et al., 2003; Merighi et al., 2005b; Nakamura et al., 2006). Furthermore, in U87MG cells, A3AR stimulation induced an increase in MMP-9, through the AP-1 activation responsible for increased glioblastoma cell invasion, as observed in macrophages (Velot et al., 2008; Gessi et al., 2010b). However, other studies reported that A3ARs reduces the ability of prostate cancer cells to migrate in vitro and metastasize in vivo (Jajoo et al., 2009). Accordingly, in the same cells, IB-MECA inhibits cell proliferation and induces G1 cell cycle arrest, apoptosis, and migration (Morello et al., 2009; Aghaei et al., 2011). Stimulation of A3ARs exerts a cytotoxic and proapoptotic effect on malignant mesothelioma cells (Varani et al., 2011a). Interestingly, the antitumor effect of A3ARs is potentiated by PEMFs in cultured neural cancer cells such as PC12 and U87MG glioblastoma cells, thus decreasing NF-κB activation and cell proliferation. Moreover, PEMF and A3AR stimulation are able to significantly increase p53 levels, cytotoxicity, and apoptosis in tumor cells (Vincenzi et al., 2012).
A3AR agonists have also been investigated in in vivo studies. In all the experimental models, given their stability and bioavailability profile, the drugs were administered orally. The studies included syngeneic, xenograft, orthotopic, and metastatic experimental animal models utilizing IB-MECA and Cl-IB-MECA in melanoma, colon, prostate, and hepatocellular carcinomas. A3AR agonists prevented the growth of primary B16-F10 murine melanoma tumors in syngeneic models. Moreover, in an artificial metastatic model, IB-MECA inhibited the development of B16-F10 murine melanoma lung metastases. Response specificity was demonstrated by A3AR antagonist administration which reversed the effect of the agonist. Furthermore, in combination with the chemotherapeutic agent cyclophosphamide, IB-MECA, or Cl-IB-MECA induced an additive antitumor effect on the development of B16-F10 melanoma lung metastatic foci (Fishman et al., 2009). In addition, the combination of Cl-IB-MECA and paclitaxel caused significant cytotoxicity on two melanoma cell lines through multiple mechanisms of cell death (Soares et al., 2014). Furthermore, in xenograft models, IB-MECA counteracted the development of human colon and prostate carcinoma in nude mice. In these studies, the combined treatment with IB-MECA and 5-fluorouracil or taxol, respectively, resulted in an enhanced antitumor effect. IB-MECA prevented the growth of primary and liver metastases of CT-26 colon carcinoma cells inoculated in the spleen. Finally, Cl-IB-MECA treatment dose dependently inhibited hepatocellular tumor growth and reduced liver inflammation (Gao and Jacobson, 2007; Bar-Yehuda et al., 2008; Fishman et al., 2009; Cohen et al., 2011). Accordingly, Cl-IB-MECA significantly reduced tumor growth and cancer pain in rat bone-residing breast cancer (Varani et al., 2013).
The safety and clinical effects of CF102 were assessed in patients with advanced unresectable HCC, in a phase I/II trial. This study included 18 hepatocellular carcinoma patients treated with three different doses of CF102 to determine the pharmacokinetic behavior and safety profile of long-term administration (Stemmer et al., 2013). Owing to its safety and favorable pharmacokinetic characteristics, phase II trial for advanced liver cancer is ongoing (ClinicalTrials.gov).
Overall, these data suggest that A3ARs might be a biologic tumor marker and that A3AR modulation could be used to treat cancer.
I. Pain
Chronic neuropathic pain is a major unresolved health care issue with global human and socioeconomic impact (5–10% occurrence in Europe and the United States). Its general incidence is augmented by pain from chemotherapy-induced peripheral neuropathy. Therefore, the identification of novel therapeutic targets that can effectively resolve this condition would be necessary.
The contribution of A3AR in pain states has been evaluated in different studies. A nociceptive and proinflammatory response resulting in edema formation was first demonstrated after activation of A3AR; this effect was mediated by both serotonin and histamine release, most likely from mast cells and subsequent actions on the sensory nerve terminal (Sawynok et al., 1997). Then, there was less heat hyperalgesia after carrageenan in A3ARKO mice probably due to reduced inflammation (Wu et al., 2002). Furthermore, investigation of the behavioral effects of A3ARKO mice showed a decreased sensitivity to some painful stimuli (Fedorova et al., 2003). However an opposite activity in pain modulation was observed in further studies. Intrathecal delivery of IB-MECA attenuated the inflammatory component of the formalin test (Yoon et al., 2005). In addition, recently, the treatment of chronic neuropathic pain was proposed as a novel potential application of A3AR agonists (Paoletta et al., 2013). A3AR agonists blocked the development of mechanically and chemotherapy-induced neuropathic pain in a dose-dependent fashion and significantly augmented the analgesic effects of currently used analgesics (Chen et al., 2012). Accordingly, in an animal model of surgery-induced metastasis Cl-IB-MECA significantly reduced bone cancer pain, providing the pharmacologic rationale for using selective A3AR agonists as a new approach in chronic neuropathic pain (Varani et al., 2013). Interestingly, very recently, the first mechanistic insight into beneficial effects of A3AR agonists against neuropathic pain has been explored (Janes et al., 2014a,b). In particular it has been found that these effects were due to the inhibition of an astrocyte-associated neuroinflammatory response in a model of oxaliplatin-induced peripheral neuropathy (Janes et al., 2014b). Furthermore A3AR agonists were found to prevent the development of paclitaxel-induced neuropathic pain by modulating spinal glial-restricted redox-dependent signaling pathways (Janes et al., 2014a).
Importantly, the activation of A3AR in humans by potent, selective, and orally bioavailable A3AR agonists is not associated with cardiac or hemodynamic side effects at variance with A1 and A2A ARs and could represent a viable therapeutic strategy for chronic pain of distinct etiologies. There is evidence for both central and peripheral sites of action of A3AR agonists in protection against chronic neuropathic pain, and there is a need for new pharmacologic tools to explore this phenomenon. The biochemical basis underlying the protective and beneficial effects of A3AR agonists in chronic neuropathic pain is unknown and deserves further in depth investigation. It has been hypothesized that mitoprotective effects and/or attenuation of neuroinflammatory processes in spinal cord mediate the underlying protective A3AR role.
From a translational perspective, this could hopefully lead to a rapid investigation of A3AR agonists, already in clinical trials, for neuropathic pain treatment.
VIII. Recent Drug Development Efforts
Allosteric modulation of GPCRs now represents one of the most exciting areas in modern drug discovery. One GPCR system that is of particular interest with respect to allosteric modulation is adenosine and its A3AR subtype.
Today our knowledge of the GPCR structure has been substantially improved by providing greater understanding of the conformational changes that occur when an agonist binds to the receptor at the same site as the endogenous ligand, the so-called orthosteric site (Chung et al., 2011; Rasmussen et al., 2011; Hill et al., 2014). However, in recent years it has become evident that drugs can also bind to a topographically distinct site (allosteric) on the GPCR protein and induce conformational changes that modify the affinity, or efficacy, of a ligand occupying the classic orthosteric binding site (Kenakin, 2009, 2012; Keov et al., 2011). Key molecular scaffolds that support allosteric modulation at the A3AR have been identified, 3-(2-pyridinyl)isoquinolines (e.g., VUF5455) and [3H]imidazo-[4,5-c]quinolin-4-amines (e.g., LUF5999, LUF6000, and LUF6001), with differing effects on the affinity and efficacy of a selective A3AR agonist (Goblyos et al., 2009; Can-fite Biopharma, 2010; Gao et al., 2011) (Fig. 4). LUF6000 was shown to enhance agonist efficacy in a functional assay and decrease agonist dissociation rate without affecting agonist potency.

Representative A3AR allosteric modulators and dendrimers.
In an attempt to overcome the issues associated with the orthosteric antagonism shown by 3-(2-pyridinyl)isoquinolines, a series of ring opened imidazoquinolinamines were synthesized to afford a range of 2,4-disubstituted quinolines as a new class of A3AR allosteric enhancers (Heitman et al., 2009). Notably, the best compound (LUF6096) was not only able to allosterically enhance the binding of Cl-IB-MECA to a similar level as LUF6000, but also displayed negligible orthosteric affinity for any of the AR subtypes. The ubiquitous distribution of ARs and the potential for serious side effects via the target receptor in a different organ or cell type can limit their utility. Allosteric enhancers may represent a strategy to overcome these limitations. However, although in vitro studies have provided convincing evidence for allosteric mechanisms of action for a number of ligands, therapeutic application of these mechanisms relies on their successful translation into whole animal physiology. The efficacy of LUF6096 in an in vivo model of infarction has been shown to be a good starting point (Du et al., 2012). Indeed, LUF6096 had no effect on baseline hemodynamic parameters; however, pretreatment with LUF6096 before coronary occlusion and during reperfusion produced a marked reduction in infarct size (Du et al., 2012). An equivalent reduction was also demonstrated if LUF6096 was administered immediately before reperfusion (Du et al., 2012). It would therefore appear that allosteric enhancers of A3AR may prove to be highly useful therapeutic strategies, selectively augmenting the actions of adenosine released locally under conditions of disease and stress. However, more in-depth investigation is required into the potential of these molecules to increase selective actions of adenosine in specific organs and cell types in whole animal.
Another original effort to improve A3AR activation has been the attachment of methanocarba adenosine derivatives to PAMAM dendrimers (Jacobson and Melman, 2010) (Fig. 4). Dendrimers are tree-like polymers widely used in in vivo drug delivery that can serve as biocompatible polymeric nanocarriers of bioactive molecules of interest (Menjoge et al., 2010). These multivalent GPCR ligand-dendrimer conjugates display pharmacologic properties that are qualitatively different from those of monomeric drugs and might be useful as novel tools to study GPCR homo- and heterodimers and higher aggregates. Assuming proper functionalization of a ligand for covalent conjugation, the resulting GPCR ligand-dendrimer conjugates have displayed dramatically increased potency or selectivity in comparison with the monomeric, small molecular ligands (Kim et al., 2008a; Jacobson, 2010). The use of nanocarriers for stably conjugated drugs that act at the cell surface may provide pharmacokinetic and pharmacodynamic advantages, such as the prevention of premature degradation (Kim et al., 2008a; Jacobson, 2010). These derivatives have been claimed for the treatment of inflammation, cardiac ischemia, stroke, asthma, diabetes, and cardiac arrhythmias (Jacobson and Melman, 2009). Interestingly, it has been demonstrated that the PAMAM dendrimer conjugate specifically activates A3ARs to improve postischemic/reperfusion function in isolated mouse hearts (Wan et al., 2011). Similar results were confirmed in rat primary cardiac cell cultures and in the isolated heart where these strategically derivatized nucleosides display enhanced cardioprotective potency (Chanyshev et al., 2012). In vivo testing will now be needed to explore the effects on pharmacokinetics and tissue targeting. The potential of this approach for use in animal models of disease is still to be proven, and such important points as cellular uptake, pharmacologic effects of ligands binding to adjacent GPCR proteins, changes in selectivity according to conjugate composition, and side effects all need to be addressed (Jacobson, 2010).
IX. Concluding Remarks
Investigation of A3ARs and their ligands is rapidly growing and having an increasing impact on the drug discovery process. There is now extensive evidence that A3ARs are involved in the physiologic regulation of several homeostatic processes and in the etiology of many diseases (Fig. 5). The present review documents the state of knowledge of the adenosine A3AR. It covers a wide range of information, including data from studies of theoretical, molecular and cellular pharmacology, signal transduction, new drug discoveries, and clinical applications. Detailed understanding of the physicochemical aspects and molecular biology of the A3AR provides a solid basis for its future development as a target for adenosine-based pharmacotherapies. Recognition and characterization of intracellular pathways modulated by A3AR activation support the belief that modulating these signaling routes is likely to lead to considerable advances in the management of many diseases. The identification of new, potent, and selective A3AR ligands opens new frontiers for the elucidation of the therapeutic potential arising from stimulating or blocking the A3AR. The A3AR appears to play a prominent role under ischemic conditions and remains a promising target for treating neurodegenerative diseases associated with acute ischemia. In terms of clinical utility, it will be critical to explore in greater detail the efficacy of the A3AR-mediated protective response in diseased hearts and skeletal muscle. There is emerging evidence that the A3AR may be a good target in neuropathic pain. A3ARs are present in all immune cells and are involved in the regulation of inflammatory and immune processes, suggesting new therapeutic strategies may emerge for inflammatory conditions such as sepsis, asthma, and autoimmune disorders including rheumatoid arthritis, Crohn’s disease, and psoriasis. The oral bioavailability of certain A3AR agonists together with encouraging data from clinical studies support the development of these agents as antirheumatic drugs. The effectiveness of the A3AR agonist Cl-IB-MECA in several animal tumor models has led to the introduction of this molecule into a program of clinical studies. The excellent safety profile has brought about the initiation of phase II clinical studies in patients with hepatocellular carcinoma, which are currently ongoing. Based on the important scientific and clinical advances overviewed in this review, it would seem that purine scientists are definitely getting closer to their goal of transforming adenosine ligands into drugs with the ability to save lives and improve human health.

Schematic diagram illustrating the functional role of A3ARs. Activation of the A3AR pathway by adenosine can affect a number of organs, including pulmonary, immune, cardiovascular, muscle and central nervous system, and pathologies like eye, joint diseases, and cancer, thus regulating various biologic outcomes.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Borea, Varani, Vincenzi, Baraldi, Tabrizi, Merighi, Gessi.
Footnotes
Abbreviations
- AC
- adenylyl cyclase
- AR
- adenosine receptors
- CHO
- Chinese hamster ovary
- Cl-IB-MECA/CF102
- 2-chloro-N6-(3-iodobenzyl)-adenosine-5′-N-methyluronamide
- CNS
- central nervous system
- COPD
- chronic obstructive pulmonary disease
- CP608,039
- N6-[2-(3-methylisoxazol-5-ylmethoxy)-5-chloro]benzyl-3′-amino-adenosine-5′-N-methylcarboxamide
- ERK1/2
- extracellular signal-regulated kinases
- GPCR
- G protein–coupled receptors
- HCC
- hepatocellular carcinoma
- HEMADO
- 2-hexyn-1-yl-N6-methyladenosine
- HIF-1α
- hypoxia-inducible-factor 1α
- IB-MECA/CF101
- N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide
- IL
- interleukin
- IOP
- intraocular pressure
- IR
- ischemia/reperfusion
- KF26777
- 2-(4-bromophenyl)-7,8-dihydro-4-propyl-1H-imidazo[2,1-i]purin-5(4H)-one
- Ki
- inhibitory binding constant
- KO
- knock-out
- LPS
- lipopolysaccharide
- LJ529
- 2-chloro-N6-(3-iodobenzyl)-5-N-methylcarbamoyl-4-thioadenosine
- MAPKs
- mitogen-activated protein kinases
- MEK
- MAP kinase kinase
- MIP
- macrophage inflammatory protein
- MMP-9
- metalloproteinase-9
- MRE 3005F20
- 5N(4-methoxyphenylcarbamoyl)amino-8-phenylethyl-2-(2-furyl)-pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine
- MRE 3008F20
- N-[2-(2-furanyl)-8-propyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-yl]-N'-(4-methoxyphenyl)urea
- MRS1220
- 9-chloro-2-(2-furanyl)-5-[(phenylacetyl)amino][1,2,4]-triazolo[1,5-c]quinazoline
- MRS1191
- 3-ethyl-5-benzyl-2-methyl-4-phenylethynyl-6-phenyl-1,4-dihydropyridine-3,5-dicarboxylate
- MRS1523
- 3-propyl-6-ethyl-5[(ethylthio)carbonyl]-2-phenyl-4-propyl-3-pyridine-carboxylate
- MRS3558/CF502
- 4-(6-(3-chlorobenzylamino)-2-chloro-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide
- MRS5151
- 2-alkynyl (N)-methanocarba
- MRS5701
- p-sulfo isomer
- MRS5841
- N6-3-chlorobenzyl-2-(3-sulfophenylethynyl)
- NF-κB
- nuclear factor-κB
- NK
- natural killer
- OA
- osteoarthritis
- OT-7999
- 5-n-butyl-8-(4-trifluoromethylphenyl)-3H-[1,2,4]triazolo-[5,1-i]purine
- PAMAM
- polyamidoamine
- PBMC
- peripheral blood mononuclear cells
- PEMADO
- 2-phenylethynyl-N6-methyladenosine
- PEMFs
- pulsed electromagnetic fields
- PI3K
- phosphoinositide 3-kinase
- PLC
- phospholipase C
- PSB-10
- 8(R)-ethyl-4-methyl-2-(2,3,5-trichlorophenyl)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-5-one
- PSB-11
- 8(R)-ethyl-7,8-dihydro-4-methyl-2-phenyl-1H-imidazo[1,2-g]purin-5-one
- RA
- rheumatoid arthritis
- ROS
- oxygen free radicals
- Structure A
- (1R,2R,3S,4R,5S)-4-(2-(hex-1-ynyl)-6-(methylamino)-9H-purin-9-bicyclo[3.1.0]hexane-2,3-diol
- TNF-α
- tumor necrosis factor α
- VUF-5574
- N-(2-methoxyphenyl)-N′-(2-(3-pyridyl)quinazolin-4-yl)urea
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





{kind=link}
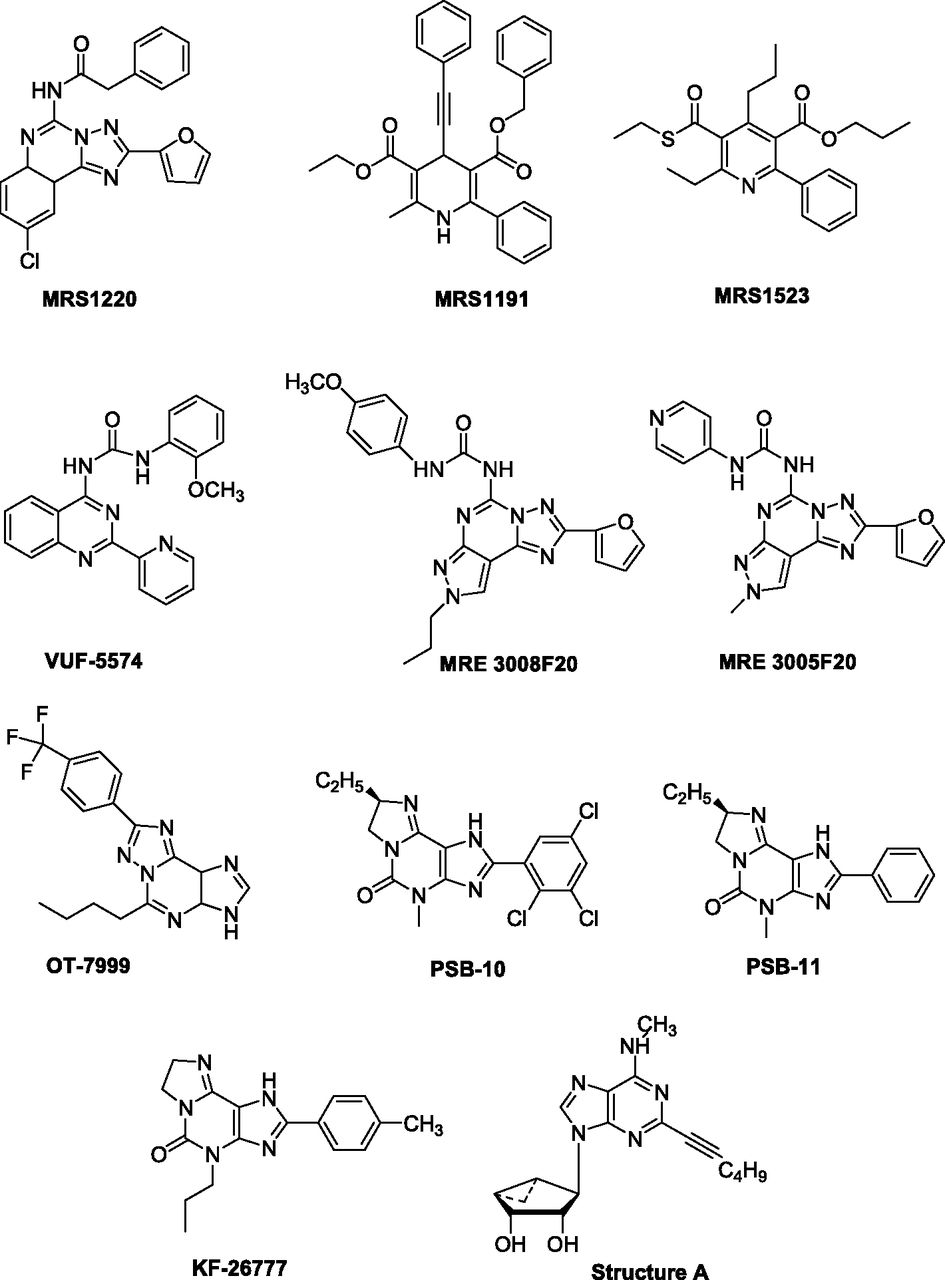{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. The Discovery of the A3 Adenosine Receptor
- III. Molecular Characterization of the A3 Adenosine Receptor
- IV. Medicinal Chemistry and Pharmacology of A3 Adenosine Receptor Ligands
- V. Distribution of the A3 Adenosine Receptor
- VI. Intracellular Pathways Regulated by the A3 Adenosine Receptor
- VII. Biologic Functions and Therapeutic Applications of the A3 Adenosine Receptor
- VIII. Recent Drug Development Efforts
- IX. Concluding Remarks
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters